Embed Size (px)
Citation preview

RESEARCH ARTICLE 3403
Development 140, 3403-3412 (2013) doi:10.1242/dev.095307© 2013. Published by The Company of Biologists Ltd
INTRODUCTIONBlood flow imparts mechanical forces and distributes endocrinefactors that influence vascular development and remodeling andallow maintenance of arterial-venous identity, but the molecularpathways that govern these flow-dependent processes are not fullyunderstood (for a review, see Roman and Pekkan, 2012). Amongthe physical forces imparted by blood flow, shear stress, thefrictional force that acts in the direction of blood flow, is the mostextensively studied. Laminar shear stress induces expression of thetranscription factor Klf2 which coordinates expression of numerousflow-responsive genes that promote cell cycle arrest andvasodilation, thereby favoring vascular quiescence and conferringatheroprotection (Dekker et al., 2002; Dekker et al., 2005; Dekkeret al., 2006; Parmar et al., 2006). By contrast, disturbed (low oroscillatory) shear stress activates the transcription factors NF-κBand AP-1, which promote an atherogenic response characterized byinflammation and vasoconstriction (Lan et al., 1994; Khachigian etal., 1995; Bhullar et al., 1998; Hay et al., 2003). However, therepertoire of flow responses that exist in vivo clearly extends wellbeyond these pathways.
Mounting evidence implicates the TGFβ family type I receptor,Alk1, as a key player in a flow-responsive signaling pathway thatfunctions to promote quiescence in nascent arteries. In zebrafishembryos, alk1 (acvrl1 – Zebrafish Information Network) isexpressed predominantly in arteries proximal to the heart, whichexperience relatively high magnitudes of mechanical forces;preventing heartbeat eliminates alk1 mRNA expression (Corti et al.,
2011). Furthermore, either loss of blood flow or loss of alk1 resultsin increased expression of cxcr4a, which encodes a pro-angiogenicchemokine receptor, and decreased expression of endothelin 1(edn1), which encodes a vasoconstrictive peptide (Corti et al., 2011).Both of these genes are flow responsive in cultured endothelial cells(Wang et al., 1993; Melchionna et al., 2005), suggesting that Alk1might lie upstream of cxcr4a and edn1 in a mechanosensitivesignaling pathway. In support of this hypothesis, blood flow-mediated repression of cxcr4a correlates with diminishedendothelial cell protrusive activity in nascent zebrafish arteries(Bussmann et al., 2011), and zebrafish alk1 mutants, which exhibitabnormally high levels of arterial cxcr4a, develop enlarged arteriescontaining supernumerary endothelial cells, suggestive of failedflow-induced suppression of endothelial cell migration orproliferation (Roman et al., 2002; Corti et al., 2011). Evidence frommice further supports the idea that Alk1 functions in a flow-responsive pathway to promote quiescence of nascent arteries. Inmice, Alk1 is expressed predominantly in embryonic arterialendothelial cells, with weak expression in adults (Seki et al., 2003;Seki et al., 2004). However, Alk1 expression can be induced in adultmice during periods of active angiogenesis in arterial endothelialcells exposed to high shear stress (Seki et al., 2003). Furthermore,recent mouse studies have implicated bone morphogenetic protein(BMP) signaling in general or Alk1 signaling in particular in themaintenance of a quiescent endothelial stalk cell fate (Larrivée et al.,2012; Moya et al., 2012). Together, these data from both mouse andzebrafish support the hypothesis that Alk1 signaling mediates flow-dependent arterial endothelial cell quiescence. Notably, alk1 actsindependently of klf2a in zebrafish (Corti et al., 2011), suggestingthat multiple flow-dependent pathways coordinate in vivo to controlthe activation state of the endothelium.
Alk1 signaling is crucial for normal vascular development andhomeostasis in mice and zebrafish, with loss of function resulting inembryonic lethality associated with development of direct
1Department of Biological Sciences, University of Pittsburgh, Pittsburgh, PA 15260,USA. 2Department of Medicine, University of Cambridge, Cambridge CB2 0QQ, UK.
*Author for correspondence ([email protected])
Accepted 5 June 2013
SUMMARYBlood flow plays crucial roles in vascular development, remodeling and homeostasis, but the molecular pathways required fortransducing flow signals are not well understood. In zebrafish embryos, arterial expression of activin receptor-like kinase 1 (alk1),which encodes a TGFβ family type I receptor, is dependent on blood flow, and loss of alk1 mimics lack of blood flow in terms ofdysregulation of a subset of flow-responsive arterial genes and increased arterial endothelial cell number. These data suggest thatblood flow activates Alk1 signaling to promote a flow-responsive gene expression program that limits nascent arterial caliber. Here,we demonstrate that restoration of endothelial alk1 expression to flow-deprived arteries fails to rescue Alk1 activity or normalizearterial endothelial cell gene expression or number, implying that blood flow may play an additional role in Alk1 signalingindependent of alk1 induction. To this end, we define cardiac-derived Bmp10 as the crucial ligand for endothelial Alk1 in embryonicvascular development, and provide evidence that circulating Bmp10 acts through endothelial Alk1 to limit endothelial cell numberin and thereby stabilize the caliber of nascent arteries. Thus, blood flow promotes Alk1 activity by concomitantly inducing alk1expression and distributing Bmp10, thereby reinforcing this signaling pathway, which functions to limit arterial caliber at the onsetof flow. Because mutations in ALK1 cause arteriovenous malformations (AVMs), our findings suggest that an impaired flow responseinitiates AVM development.
KEY WORDS: Bmp10, Alk1/Acvrl1, Hereditary hemorrhagic telangiectasia, Arteriovenous malformation, Zebrafish, Flow response
Circulating Bmp10 acts through endothelial Alk1 to mediateflow-dependent arterial quiescenceDerek W. Laux1, Sarah Young1, James P. Donovan1, Corrine J. Mansfield1, Paul D. Upton2 and Beth L. Roman1,*
DEVELO
PMENT
Development ePress. Posted online 17 July 2013http://dev.biologists.org/lookup/doi/10.1242/dev.095307Access the most recent version at First posted online on 17 July 2013 as 10.1242/dev.095307

3404
connections between arteries and veins, or arteriovenousmalformations (AVMs) (Oh et al., 2000; Urness et al., 2000; Romanet al., 2002). In humans, ALK1 heterozygosity results in hereditaryhemorrhagic telangiectasia type 2 (HHT2), a vascular disordercharacterized by predisposition to development of telangiectasesand AVMs (Guttmacher et al., 1995; Johnson et al., 1996). However,despite the clear link between ALK1 signaling and AVMprevention, the ALK1 signaling pathway remains poorly defined invivo. In TGFβ family signaling, ligands bind to a heterotetramericcomplex of two type II receptors and two type I receptors, both ofwhich are serine/threonine kinases. The type II receptorsphosphorylate and thus activate the type I receptors, and the type Ireceptors then phosphorylate receptor-specific Smad proteins.Phosphorylated Smads complex with the common partner Smad,Smad4, enter the nucleus and, together with a variety oftranscription factors, regulate transcription of target genes (for areview, see Xu et al., 2012). With respect to ALK1, the ligandsTGFβ1, TGFβ3, BMP9 and BMP10 can induce ALK1-dependentphosphorylation of Smad1, Smad5 and/or Smad9 (hereafter referredto as Smad1/5/9) and stimulate activity of a phospho-Smad1/5/9(pSmad1/5/9)-responsive reporter in cultured endothelial cells (tenDijke et al., 1994; Lux et al., 1999; Oh et al., 2000; Goumans et al.,2002; Goumans et al., 2003; Brown et al., 2005; David et al., 2007;Scharpfenecker et al., 2007; David et al., 2008; Mitchell et al.,2010). However, although TGFβ-mediated activation of ALK1requires ALK5 (canonical TGFβ1 type I receptor) activity incultured endothelial cells (Goumans et al., 2003), endothelial cell-specific deletion of Alk5 in mice or Alk5 inhibition in zebrafishembryos does not affect Alk1 activity (Park et al., 2008), suggestingthat TGFβ subfamily ligands are not relevant to Alk1 signaling inembryonic development. In fact, BMP9 and BMP10 are the onlyTGFβ superfamily ligands that bind to ALK1 with high affinity invitro, and they circulate at physiologically relevant concentrations(Brown et al., 2005; David et al., 2008; Mitchell et al., 2010; Ricardet al., 2012). However, neither Bmp9- (Ricard et al., 2012) norBmp10- (Chen et al., 2004) null mice phenocopy Alk1-null mice(Oh et al., 2000; Urness et al., 2000), which present with AVMs.Although the lack of AVMs could reflect ligand redundancy,interference with both ligands via blocking antibodies and/or ligandtraps impairs mouse retinal angiogenesis but does not produceretinal AVMs (Larrivée et al., 2012; Ricard et al., 2012). As such,the identity of the activating ALK1 ligand in vivo has remainedelusive.
In this work, we use zebrafish embryos to demonstrate that Alk1kinase activity, in addition to alk1 mRNA expression, requires bloodflow, and we provide evidence that this newly defined role for bloodflow stems not from mechanical force but from distribution of thecardiac-derived circulating ligand, Bmp10. Taken together, our datadefine a novel endocrine pathway in which circulating Bmp10 bindsto endothelial cell Alk1 to induce phosphorylation of Smad1/5/9,which promotes a program of gene expression that limits endothelialcell number within nascent arteries in response to blood flow.Abrogation of this flow response results in enlarged arteries andultimately AVMs.
MATERIALS AND METHODSZebrafish lines and maintenanceAdult zebrafish (Danio rerio) were maintained according to standardprotocols (Westerfield, 1995). When appropriate, embryo medium wassupplemented with 0.003% phenylthiourea (PTU) (Sigma, St Louis, MO,USA) at 24 hours post-fertilization (hpf) to prevent melanin synthesis.Mutant line alk1y6 and the alk1y6 genotyping assay have been described
previously (Roman et al., 2002). Transgenic lines Tg(fli1a:negfp)y7,Tg(kdrl:gfp)la116, Tg(gata1a:DsRed)sd2 and Tg(fli1a.ep:mrfp-CAAX)pt505
have been described previously (Roman et al., 2002; Traver et al., 2003;Choi et al., 2007; Corti et al., 2011). To drive wild-type alk1 in endothelialcells, we generated ptol-fli1a.ep:alk1-myc by Gateway cloning (Invitrogen,Carlsbad, CA, USA), recombining pDESTtol2pA2 (Kwan et al., 2007) withp5E fli1a.ep (Villefranc et al., 2007), pME-alk1 and p3E-MTpA (Kwan etal., 2007). To drive ligand- and type II receptor-independent, constitutivelyactive alk1 (Roman et al., 2002) in endothelial cells, we generated ptol-fli1a.ebs:alk1CA-mCherry, recombining pDESTtol2pA2, p5E fli1a.ebs (fli1aEts-binding site; a kind gift from N. Lawson, University of MassachusettsMedical School, Worcester, MA, USA), pME alk1CA and p3E mCherry-pA(Kwan et al., 2007). These constructs were co-injected with transposasemRNA (Kawakami et al., 2004) into one-cell embryos to generateTg(fli1a.ep:alk1-myc)pt516, hereafter referred to as Tg(fli1a:alk1-myc), anda series of mosaic founders for Tg(fli1a.ebs:alk1CA-mCherry), hereafterreferred to as Tg(fli1a:alk1CA-mCh). The constitutively active alk1 transgenecauses severe vascular defects and is embryonic lethal in F1 embryos.
MorpholinosTranslation blocking (TB) and splice blocking (SB) morpholino-modifiedantisense oligonucleotides (GeneTools, Philomath, OR, USA) were asfollows: bmp9TB, 5�-GGAGCAAATGTCCTACGCGCCACAT-3�; bmp9SB,5�-CTCTTTATGTGTACTCACCCTGAAC-3�; bmp10TB, 5�-AAAAGT -GATTTCTGCTACCAGCCAT-3�; bmp10SB, 5�-AGGAAAATATGCAG -TTACCTTCATT-3�; and bmp10-likeTB, 5�-GCAGCAGAGAATCAGC-CATGACTGC-3�. For TB morpholinos, efficacy and specificity wereevaluated by injecting into one-cell embryos CMV-driven EGFP DNA-containing morpholino-binding sites upstream of the initiator methionine,with or without cognate or non-cognate morpholino, and assessing EGFPexpression at ~6 hpf. We could not rescue bmp10 or bmp10-like morphantphenotypes by injecting morpholino-resistant mRNA because embryosventralized (data not shown). The alk1, tnnt2a and control morpholinoshave been described previously (Sehnert et al., 2002; Corti et al., 2011).
In situ hybridization and immunostainingDigoxigenin-labeled riboprobes (Roche, Indianapolis, IN, USA) for cdh5,cxcr4a, edn1 and myl7 (myosin light chain 7, previously known as cardiacmyosin light chain) have been described previously (Yelon et al., 1999; Cortiet al., 2011). Zebrafish bmp10 was amplified from cDNA using primers 5�-ACCACAGCTGAACTCCGACT-3� and 5�-TCCACACT TGGCCA -CTACCATT-3�; and bmp10-like was amplified using primers 5�-CGCAATGAAGCACCAGAGTA-3� and 5�-CCGTCCACTGTCTCTC-ATCA-3�. Both fragments were cloned into pCRII-TOPO (Invitrogen).Whole-mount in situ hybridization was performed as described previously(Roman et al., 2002). Immunohistochemistry was performed using primaryantibodies MF20 at 1:200 (sarcomeric myosin, Developmental StudiesHybridoma Bank, Iowa City, IA, USA) or 9E10 at 1:200 (myc, Covance,Princeton, NJ, USA), biotinylated horse anti-mouse IgG at 1:200, ABCreagent (Vector Laboratories, Burlingame, CA), and 3,3�-diaminobenzidine(Sigma, St Louis, MO, USA). Embryos were photographed using an MVX-10 MacroView microscope and DP71 camera (Olympus America, CenterValley, PA, USA). For sections, embryos were embedded in JB4(Polysciences, Warrington, PA, USA), sectioned at 8 μm and imaged using anOlympus BX51 microscope and DP71 camera. Images were compiled withAdobe Photoshop CS2 version 9.0.2 (Adobe Systems, San Jose, CA, USA).
For immunofluorescence, embryos were fixed in 4% paraformaldehydein PBS overnight at 4°C, embedded in 4% NuSieve GTG agarose (Lonza,Rockland, ME, USA), and sectioned at 50 μm with a VT1000S vibratome(Leica Microsystems, Buffalo Grove, IL, USA). Rabbit anti-phospho-Smad1/5/9 (also known as anti-phospho-Smad1/5/8, 9511, Cell SignalingTechnology, Beverly, MA, USA) was used at 1:100 and goat-anti-rabbitAlexa Fluor 647 at 1:200. Sections were mounted with Vectashield HardSetmounting medium (Vector) and imaged with an Olympus Fluoview 1000confocal microscope outfitted with a UPFLN 40× oil immersion objective,with scan speed 244 Hz. Two-dimensional projections were generated fromz-series (1 μm steps) and images processed for presentation and quantitationusing MetaMorph 7.7 (Molecular Devices, Sunnyvale, CA, USA). For
RESEARCH ARTICLE Development 140 (16)
DEVELO
PMENT

presentation, images were pseudocolored and colocalization highlightedusing the ‘boost colocalization’ function. For quantitation of nuclearphospho-Smad1/5/9, threshold images were created for summed projectionsof nuclear EGFP panels, alk1-positive arteries were traced with the ‘traceregion’ tool, and all nuclei were selected within the traced region using the‘create regions around objects’ tool. Traced nuclei were transferred tocorresponding pSmad1/5/9 summed projections, and average nuclear pixelintensity was measured and normalized to the average pixel intensity for anadjacent non-vascular pSmad1/5/9-positive domain. Results are expressedas percentage of corresponding controls.
Microinjection of rhBMP10 and rhBMP9Embryos were anesthetized in 160 μg/ml tricaine (Sigma) and embedded in1% NuSieve GTG agarose in 30% Danieau/PTU. Two nl of PhenolRed/KCl buffer or Qtracker 655 non-targeted quantum dots (Invitrogen)with or without 10 μM rhBMP10 or rhBMP9 protein (R&D Systems,Minneapolis, MN, USA) were injected into the base of one caudal divisionof the internal carotid artery (CaDI) at 28 hpf. Embryos were either imagedlive for cell counts (see below) or fixed at 36 hpf and assayed byimmunofluorescence or in situ hybridization as described above.
Live confocal imagingFor live imaging, embryos were anesthetized in 160 μg/ml tricaine andinserted into 500 μm troughs in 2% SeaKem LE agarose (Lonza)/30%Danieau. Z-series (0.5-2 μm steps) were collected using a TCS SP5multiphoton/confocal microscope (Leica Microsystems, Wetzlar, Germany)outfitted with an APO L 20×/1.00 water immersion objective, non-descanned detectors and spectral detectors. EGFP was excited with a MaiTai DeepSee Ti:Sapphire laser (Newport/Spectra Physics, Santa Clara, CA,USA) at 900 nm, whereas dsRed and mCherry were excited with a 561 nmdiode. Scanning was performed either with a point scanner (400 Hz) with4× frame averaging or resonant scanner (8000 Hz) with 16× line averaging.Projections were generated using MetaMorph 7.7 and endothelial cellnumbers counted as described previously (Corti et al., 2011).
C2C12 transfection and luciferase assaysOn the day prior to transfection, C2C12 cells were seeded at 4×104
cells/well in 24-well plates. Cells were washed once with Optimem I(Invitrogen) and incubated in Optimem I for 2 hours. All wells weretransfected with Lipofectamine (2 µl/well; Invitrogen) in complex withBRE-luciferase (400 ng/well) and pRL-TK (40 ng/well). In plasmid testwells, 400 ng/well of pcDNA3-hALK1, pcDNA3-hALK1(R411Q) (akinase-dead mutant), pcDNA3-hALK3-HA or pCS2-zALK1 were alsoadded. After 4 hours, lipoplexes were removed and DMEM containing 10%FBS and antibiotic-antimycotic solution (Invitrogen) added for 24 hours.Cells were washed twice with serum-free DMEM+antibiotic and incubatedfor 21 hours, then treated for 21 hour with 0.1-3000 pg/ml rhBMP9 orrhBMP10 (R&D Systems, Abingdon, UK). Cells were then lysed andassayed for Firefly and Renilla luciferase activities using the Dual-GloLuciferase Assay kit (Promega, Southampton, UK) according to themanufacturer’s instructions. Firefly luciferase activities were normalized tothe Renilla control. Data were normalized as percentage responses usingGraphPad Prism (San Diego, CA, USA) and EC50 values calculated.Plasmids were obtained as follows: BRE-luciferase from P. ten Dijke(Leiden University Medical Center, Leiden, The Netherlands); pcDNA3-hALK1 and pcDNA3-hALK1(R411Q) from R. Trembath (King’s CollegeLondon, London, UK); and pcDNA3-hALK3-HA from K. Miyazono(University of Tokyo, Tokyo, Japan).
StatisticsEndothelial cell numbers and pSmad1/5/9 intensities were compared bytwo-tailed, unpaired Student’s t-test with significance set at P<0.05.
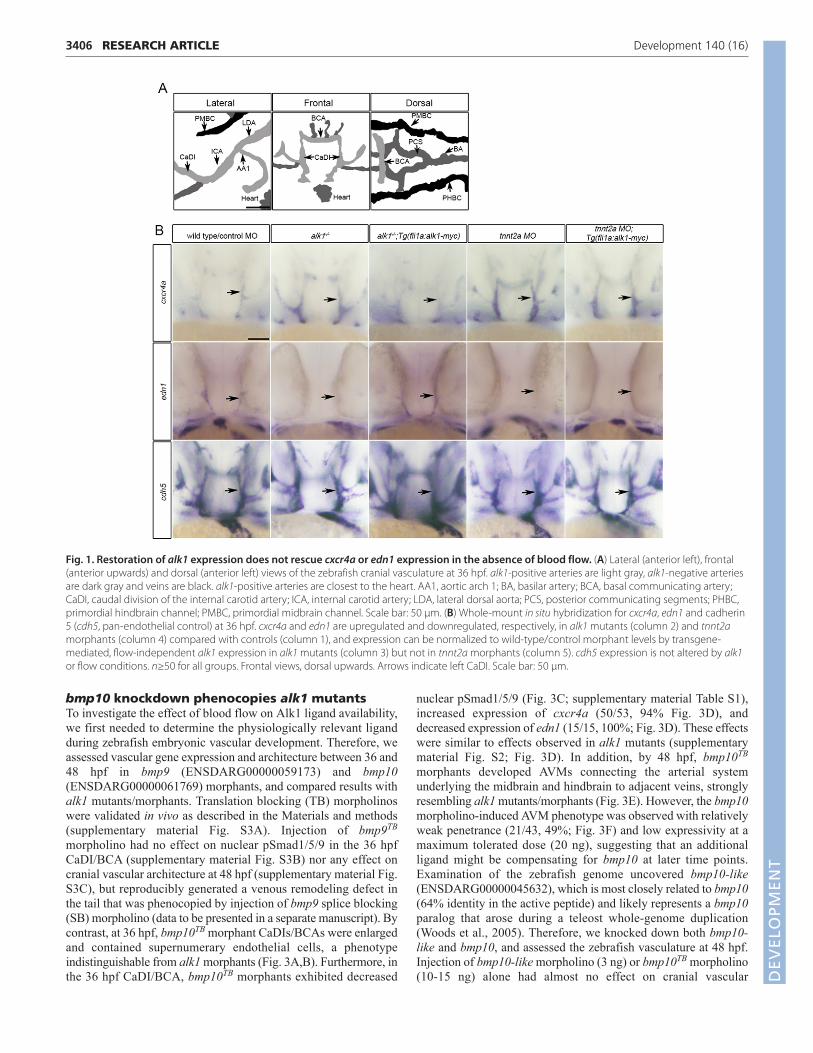
RESULTSBlood flow is required not only for alk1expression but also for Alk1 activityIn 36 hpf zebrafish embryos, alk1 is expressed predominantly inendothelial cells within arteries proximal to the heart: the first
aortic arch; the cranialward internal carotid artery, caudal divisionof the internal carotid artery (CaDI) and basal communicatingartery (BCA); and the caudalward lateral dorsal aortae (Fig. 1A).We have previously demonstrated that alk1 expression requiresblood flow, and that loss of alk1 mimics loss of blood flow interms of changes in expression of cxcr4a and edn1: both alk1mutants, which have high flow through assayed vessels, andcardiac troponin-t2a (tnnt2a) morphants, which lack heartbeat andblood flow, exhibit increased cxcr4a expression and decreasededn1 expression in alk1-expressing cranial arteries at 36 hpfcompared with corresponding controls (Fig. 1B) (Corti et al.,2011). Given that mammalian orthologs CXCR4 and EDN1 areflow responsive in cultured human endothelial cells (Wang et al.,1993; Melchionna et al., 2005), these data suggest that Alk1 mightact downstream of blood flow to control expression of thesemechanoresponsive genes. Accordingly, if Alk1 signaling issufficient downstream of blood flow, then restoration of alk1 inthe absence of flow might be expected to rescue expression ofcxcr4a and edn1. To test this hypothesis, we generated a stabletransgenic line, Tg(fli1a:alk1-myc), that expresses Alk1-myc inall endothelial cells regardless of the presence of blood flow(supplementary material Fig. S1). This transgene restores normalexpression of cxcr4a and edn1 in alk1 mutants (Fig. 1B), rescuesalk1 mutants to adulthood [n=23 alk1−/− of 130 adults fromalk1+/−;Tg(fli1a:alk1-myc) incrosses; 71% rate of rescue], and hasno untoward effects on growth and development. However, flow-independent expression of endothelial cell alk1 fails to normalizeexpression of cxcr4a or edn1 in 36 hpf tnnt2a morphants(Fig. 1B). There are two plausible explanations for thisobservation: Alk1 signaling may not be sufficient downstream ofblood flow to control expression of these genes, or flow may berequired for some aspect of Alk1 signaling in addition to alk1expression.
To determine more directly whether restoration of endothelialalk1 expression is sufficient to restore Alk1 signaling in the absenceof flow, we assessed a proximal read-out of Alk1 activity: nuclearpSmad1/5/9. pSmad1/5/9 is present in endothelial cell nuclei withinthe alk1-positive CaDIs and BCA in 36 hpf wild-type embryos, isdecreased in these arteries in alk1−/− and is restored inalk1−/−;Tg(fli1a:alk1-myc) (supplementary material Fig. S2 andTable S1), demonstrating that Alk1 signaling is necessary forSmad1/5/9 phosphorylation in these arterial endothelial cells.Because alk1 expression is dependent on blood flow (Corti et al.,2011) and Smad1/5/9 phosphorylation is dependent on alk1expression, pSmad1/5/9 in these arterial endothelial cells should bedependent on blood flow. Indeed, nuclear pSmad1/5/9 is nearlyundetectable in the CaDIs at 24 hpf, prior to the onset of flowthrough and alk1 expression in these vessels (Fig. 2A;supplementary material Table S1), and is decreased at 36 hpf intnnt2a morphants (Fig. 2B; supplementary material Table S1)compared with sibling controls (Fig. 2C; supplementary materialTable S1). However, restoration of alk1 expression via a fli1a:alk1-myc transgene in flow-deprived tnnt2a morphants fails to rescuenuclear pSmad1/5/9 (Fig. 2D; supplementary material Table S1),whereas endothelial-specific expression of a constitutively active(ligand- and type II receptor-independent) form of alk1 via afli1a:alk1CA-mCh transgene restores nuclear pSmad1/5/9 in theabsence of blood flow (Fig. 2E; supplementary material Table S1).Taken together, these data support the idea that blood flow isrequired not only for alk1 expression but also for some additionalaspect of Alk1 activity, such as type II receptor expression and/orexpression or distribution of Alk1 ligand.
3405RESEARCH ARTICLEBmp10/Alk1 mediates flow signal
DEVELO
PMENT
3406
bmp10 knockdown phenocopies alk1 mutantsTo investigate the effect of blood flow on Alk1 ligand availability,we first needed to determine the physiologically relevant ligandduring zebrafish embryonic vascular development. Therefore, weassessed vascular gene expression and architecture between 36 and48 hpf in bmp9 (ENSDARG00000059173) and bmp10(ENSDARG00000061769) morphants, and compared results withalk1 mutants/morphants. Translation blocking (TB) morpholinoswere validated in vivo as described in the Materials and methods(supplementary material Fig. S3A). Injection of bmp9TB
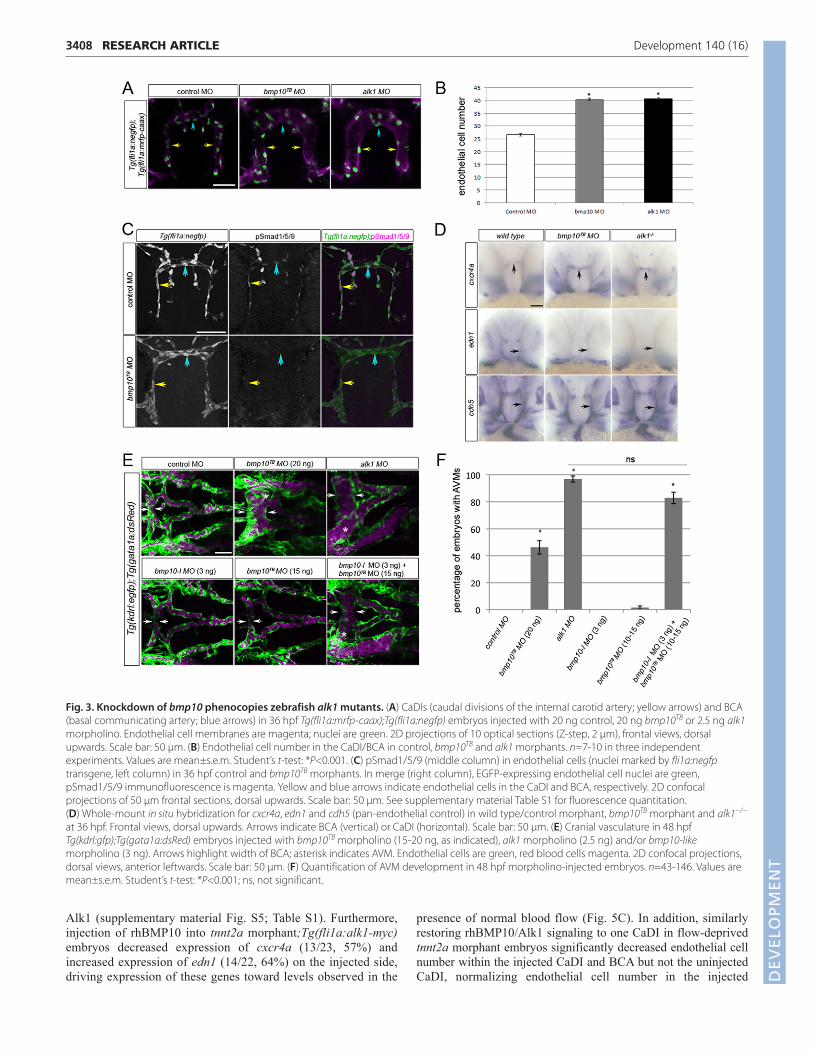
morpholino had no effect on nuclear pSmad1/5/9 in the 36 hpfCaDI/BCA (supplementary material Fig. S3B) nor any effect oncranial vascular architecture at 48 hpf (supplementary material Fig.S3C), but reproducibly generated a venous remodeling defect inthe tail that was phenocopied by injection of bmp9 splice blocking(SB) morpholino (data to be presented in a separate manuscript). Bycontrast, at 36 hpf, bmp10TB morphant CaDIs/BCAs were enlargedand contained supernumerary endothelial cells, a phenotypeindistinguishable from alk1 morphants (Fig. 3A,B). Furthermore, inthe 36 hpf CaDI/BCA, bmp10TB morphants exhibited decreased
nuclear pSmad1/5/9 (Fig. 3C; supplementary material Table S1),increased expression of cxcr4a (50/53, 94% Fig. 3D), anddecreased expression of edn1 (15/15, 100%; Fig. 3D). These effectswere similar to effects observed in alk1 mutants (supplementarymaterial Fig. S2; Fig. 3D). In addition, by 48 hpf, bmp10TB
morphants developed AVMs connecting the arterial systemunderlying the midbrain and hindbrain to adjacent veins, stronglyresembling alk1 mutants/morphants (Fig. 3E). However, the bmp10morpholino-induced AVM phenotype was observed with relativelyweak penetrance (21/43, 49%; Fig. 3F) and low expressivity at amaximum tolerated dose (20 ng), suggesting that an additionalligand might be compensating for bmp10 at later time points.Examination of the zebrafish genome uncovered bmp10-like(ENSDARG00000045632), which is most closely related to bmp10(64% identity in the active peptide) and likely represents a bmp10paralog that arose during a teleost whole-genome duplication(Woods et al., 2005). Therefore, we knocked down both bmp10-like and bmp10, and assessed the zebrafish vasculature at 48 hpf.Injection of bmp10-like morpholino (3 ng) or bmp10TB morpholino(10-15 ng) alone had almost no effect on cranial vascular
RESEARCH ARTICLE Development 140 (16)
Fig. 1. Restoration of alk1 expression does not rescue cxcr4a or edn1 expression in the absence of blood flow. (A) Lateral (anterior left), frontal(anterior upwards) and dorsal (anterior left) views of the zebrafish cranial vasculature at 36 hpf. alk1-positive arteries are light gray, alk1-negative arteriesare dark gray and veins are black. alk1-positive arteries are closest to the heart. AA1, aortic arch 1; BA, basilar artery; BCA, basal communicating artery;CaDI, caudal division of the internal carotid artery; ICA, internal carotid artery; LDA, lateral dorsal aorta; PCS, posterior communicating segments; PHBC,primordial hindbrain channel; PMBC, primordial midbrain channel. Scale bar: 50 μm. (B) Whole-mount in situ hybridization for cxcr4a, edn1 and cadherin5 (cdh5, pan-endothelial control) at 36 hpf. cxcr4a and edn1 are upregulated and downregulated, respectively, in alk1 mutants (column 2) and tnnt2amorphants (column 4) compared with controls (column 1), and expression can be normalized to wild-type/control morphant levels by transgene-mediated, flow-independent alk1 expression in alk1 mutants (column 3) but not in tnnt2a morphants (column 5). cdh5 expression is not altered by alk1or flow conditions. n≥50 for all groups. Frontal views, dorsal upwards. Arrows indicate left CaDI. Scale bar: 50 μm.
DEVELO
PMENT

architecture (n=61 and 47, respectively), whereas co-injection ofthese two morpholinos at these same doses robustly phenocopiedalk1 morphants in terms of AVM development (122/146, 84%;Fig. 3E,F), suggesting a strong genetic interaction. Similar resultswere observed with a bmp10SB morpholino, with no AVMs at 15 ng(n=48) but AVMs upon co-injection with 3 ng bmp10-likemorpholino (46/57, 81%). By contrast, co-injection of bmp9TB
morpholino (7 ng) with bmp10TB morpholino (15 ng) failed to elicitAVMs (0/29), and co-injection of bmp9TB morpholino (7 ng),bmp10TB morpholino (15 ng) and bmp10-like morpholino (3 ng)did not increase the percentage phenotype (20/31, 65%) beyond
combined injection of bmp10TB morpholino and bmp10-likemorpholino. These results demonstrate that bmp10-like actsredundantly with bmp10 and that Bmp10 but not Bmp9 is thecrucial in vivo Alk1 ligand required for arterial quiescence andAVM prevention during zebrafish embryonic vasculardevelopment.
bmp10 paralogs are expressed exclusively in theheartWe have demonstrated that concomitant knockdown of bmp10 andbmp10-like phenocopies alk1 mutants. bmp10 is undetectable at 22hpf but expressed in the developing heart as early as 24 hpf, withpredominant expression in the ventricle by 36 hpf (Fig. 4A;supplementary material Fig. S4A). By 48 hpf, bmp10 is expressedin both heart chambers but appears to be absent from theatrioventricular canal (Fig. 4A). Double staining for bmp10 mRNAand sarcomeric myosin protein revealed that bmp10 is expressedstrongly in endocardium but is largely absent from myocardium at36 hpf (Fig. 4B). bmp10-like is undetectable in the heart at 28 hpfbut is expressed at 36 and 48 hpf in distal ventricular myocardium(Fig. 4A,B). Both bmp10 and bmp10-like are expressedindependently of heartbeat (supplementary material Fig. S4B), andno discrete extracardiac expression domains of either bmp10 orbmp10-like are observed (data not shown). The temporal differencein ligand expression suggests that Bmp10-like might compensatefor Bmp10 after 36 hpf, providing a plausible explanation for theobservation that knockdown of bmp10 alone robustly phenocopiesalk1 mutants at 36 hpf, but double knockdown of bmp10 andbmp10-like is required for robust phenocopy at 48 hpf (Fig. 3).Furthermore, these data demonstrate that the heart is the most likelysource of both Bmp10 and Bmp10-like, and suggest that cardiac-derived Bmp10 ligands might be carried by blood flow to arterialendothelial Alk1.
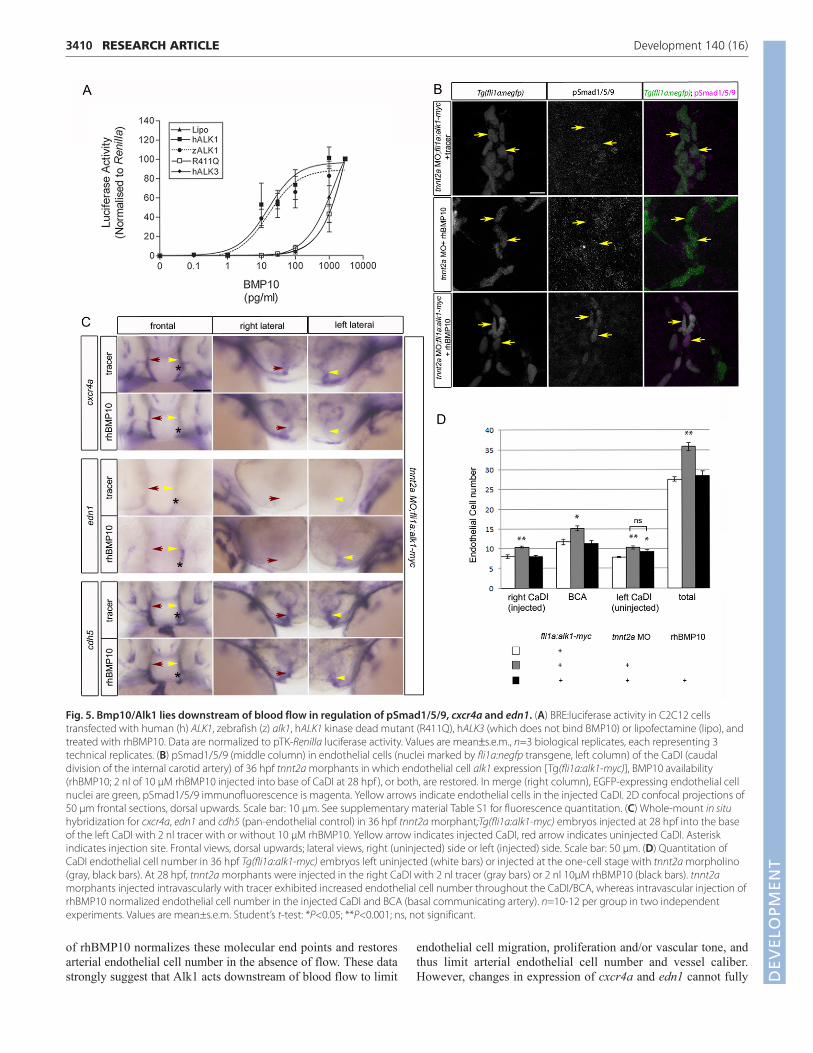
Circulating Bmp10 acts via Alk1 to limitendothelial cell number in nascent arteriesGiven that bmp10 paralogs are detectable only in the heart inzebrafish (Fig. 4) and mouse (Neuhaus et al., 1999; Chen et al.,2004), and that Bmp10 circulates in embryonic mouse plasma(Ricard et al., 2012), it stands to reason that blood flow may berequired to transport Bmp10 to arterial endothelial cell Alk1,explaining why restoration of alk1 gene expression alone isinsufficient to rescue defects in arterial endothelial cell cxcr4a, edn1and pSmad1/5/9 in the absence of flow (Figs 1, 2). To test thishypothesis, we injected tnnt2a morpholino into Tg(fli1a:alk1-myc)embryos, once again generating embryos that lack heartbeat butnonetheless express endothelial alk1. Non-transgenic siblingsinjected with tnnt2a morpholino served as blood flow deficient andtherefore alk1-deficient controls. At 28 hpf, we injected tracer(phenol red or quantum dots) alone or together with recombinanthuman (rh) BMP10 directly into the base of one CaDI. rhBMP10can act through zebrafish Alk1 to activate BMP responsive element(BRE)-driven luciferase activity in transfected C2C12 cells with anEC50 similar to that required for activation of human ALK1(Fig. 5A). Restoration of either alk1 alone [tnnt2amorphant;Tg(fli1a:alk1-myc)] or rhBMP10 alone (tnnt2a morphant+ rhBMP10) failed to rescue nuclear pSmad1/5/9 in the CaDI,whereas restoration of both alk1 and rhBMP10 increased nuclearpSmad1/5/9 ~3-fold near the site of rhBMP10 injection (Fig. 5B;supplementary material Table S1). Injection of rhBMP10 into alk1mutants failed to rescue pSmad1/5/9, supporting the idea thatrhBMP10 is not acting through other endothelial receptors besides
3407RESEARCH ARTICLEBmp10/Alk1 mediates flow signal
Fig. 2. Alk1 activity is dependent on blood flow. (A-E) pSmad1/5/9(middle column) in endothelial cell nuclei (marked by fli1a:negfptransgene, left column); in merge (right column), EGFP-expressingendothelial cell nuclei are green and pSmad1/5/9 immunofluorescence ismagenta. (A) 24 hpf wild type, prior to blood flow; (B) 36 hpf tnnt2amorphant (no flow); (C) 36 hpf wild type; (D) 36 hpf tnnt2a morphantharboring a fli1a:alk1-myc transgene; (E) 36 hpf tnnt2a morphantharboring a fli1a:alk1CA-mCh transgene. Tg(fli1a:alk1CA-mCh) embryos donot have lumenized vessels. In merge (right column), EGFP-expressingendothelial cell nuclei are green, pSmad1/5/9 immunofluorescence ismagenta. Yellow and blue arrows indicate endothelial cells in the caudaldivision of the internal carotid artery (CaDI) and basal communicatingartery (BCA), respectively. 2D confocal projections of 50 μm frontalsections, dorsal upwards. Scale bar: 50 μm. See supplementary materialTable S1 for fluorescence quantitation.
DEVELO
PMENT
3408
Alk1 (supplementary material Fig. S5; Table S1). Furthermore,injection of rhBMP10 into tnnt2a morphant;Tg(fli1a:alk1-myc)embryos decreased expression of cxcr4a (13/23, 57%) andincreased expression of edn1 (14/22, 64%) on the injected side,driving expression of these genes toward levels observed in the
presence of normal blood flow (Fig. 5C). In addition, similarlyrestoring rhBMP10/Alk1 signaling to one CaDI in flow-deprivedtnnt2a morphant embryos significantly decreased endothelial cellnumber within the injected CaDI and BCA but not the uninjectedCaDI, normalizing endothelial cell number in the injected
RESEARCH ARTICLE Development 140 (16)
Fig. 3. Knockdown of bmp10 phenocopies zebrafish alk1 mutants. (A) CaDIs (caudal divisions of the internal carotid artery; yellow arrows) and BCA(basal communicating artery; blue arrows) in 36 hpf Tg(fli1a:mrfp-caax);Tg(fli1a:negfp) embryos injected with 20 ng control, 20 ng bmp10TB or 2.5 ng alk1morpholino. Endothelial cell membranes are magenta; nuclei are green. 2D projections of 10 optical sections (Z-step, 2 μm), frontal views, dorsalupwards. Scale bar: 50 μm. (B) Endothelial cell number in the CaDI/BCA in control, bmp10TB and alk1 morphants. n=7-10 in three independentexperiments. Values are mean±s.e.m. Student’s t-test: *P<0.001. (C) pSmad1/5/9 (middle column) in endothelial cells (nuclei marked by fli1a:negfptransgene, left column) in 36 hpf control and bmp10TB morphants. In merge (right column), EGFP-expressing endothelial cell nuclei are green,pSmad1/5/9 immunofluorescence is magenta. Yellow and blue arrows indicate endothelial cells in the CaDI and BCA, respectively. 2D confocalprojections of 50 μm frontal sections, dorsal upwards. Scale bar: 50 μm. See supplementary material Table S1 for fluorescence quantitation. (D) Whole-mount in situ hybridization for cxcr4a, edn1 and cdh5 (pan-endothelial control) in wild type/control morphant, bmp10TB morphant and alk1−/−
at 36 hpf. Frontal views, dorsal upwards. Arrows indicate BCA (vertical) or CaDI (horizontal). Scale bar: 50 μm. (E) Cranial vasculature in 48 hpfTg(kdrl:gfp);Tg(gata1a:dsRed) embryos injected with bmp10TB morpholino (15-20 ng, as indicated), alk1 morpholino (2.5 ng) and/or bmp10-likemorpholino (3 ng). Arrows highlight width of BCA; asterisk indicates AVM. Endothelial cells are green, red blood cells magenta. 2D confocal projections,dorsal views, anterior leftwards. Scale bar: 50 μm. (F) Quantification of AVM development in 48 hpf morpholino-injected embryos. n=43-146. Values aremean±s.e.m. Student’s t-test: *P<0.001; ns, not significant.
DEVELO
PMENT

CaDI/BCA to that observed in the presence of normal blood flow(Fig. 5D). Because expression of CXCR4 and EDN1 can berepressed or induced, respectively, by mechanical force in culturedendothelial cells (Wang et al., 1993; Melchionna et al., 2005), theseresults support the idea that Bmp10/Alk1/pSmad1/5/9 signalingmay be crucial in transducing a mechanical signal into abiochemical response in arterial endothelial cells in vivo. In total,our results define a novel blood flow responsive signaling pathway– in which blood flow is required for alk1 expression as well as, viacirculating Bmp10, Alk1 activity – that is crucial for flow-dependent limitation of endothelial cell number in and thereforecaliber of nascent arteries.
DISCUSSIONEarly embryonic blood vessel development is controlled largely byparacrine signaling interactions that orchestrate endothelial cellmigration and proliferation, and coalescence into vascular cords, aswell as subsequent angiogenic sprouting that serves to elaborate theprimitive vascular network. However, once blood vessels form alumen, biomechanical forces and endocrine factors also come intoplay. In this work, we present evidence that blood flow is crucialfor limiting endothelial cell number within nascent arteries, and thatAlk1 acts downstream of blood flow in mediating this effect.Furthermore, we demonstrate that blood flow is required not onlyfor alk1 expression but also for Alk1 activity, and that the latterrequirement is met by provision of circulating ligand, Bmp10. Thus,flow-dependent induction of alk1 and distribution of ligandsynergize to enhance Alk1 activity.
It is well established that both BMP9 and BMP10 bind ALK1with high affinity and can induce Smad1/5/9 phosphorylation andpSmad1/5/9-dependent transcriptional responses in culturedendothelial cells (David et al., 2007; David et al., 2008; Mitchell etal., 2010). However, our work is the first to define Alk1 ligandsunequivocally in vivo. We have demonstrated in zebrafish thatbmp10 paralogs are required for Alk1 function during embryonicdevelopment, whereas bmp9 is dispensable. By contrast, neitherBmp9 nor Bmp10 mouse nulls have been reported to exhibit AVMs.Bmp9-null mice live to adulthood without apparent vascularabnormalities (Ricard et al., 2012), whereas ventricular hypoplasiaand impaired trabeculation but not AVMs have been reported inBmp10 nulls (Chen et al., 2004). There are several possibleexplanations for this discrepancy between zebrafish and mice. First,Bmp9 and Bmp10 may act redundantly in mouse embryonicvascular development, reflecting a difference in the spatial and/ortemporal ligand requirement in mouse versus zebrafish vasculature.Indeed, treatment of Bmp9-null mice with Bmp10 blockingantibodies results in early postnatal retinal vascular defects, hintingat functional redundancy, although AVMs were not noted in thesevessels (Ricard et al., 2012). Furthermore, like rhBMP10, rhBMP9was effective in restoring nuclear pSmad1/5/9 in the CaDI/BCA intnnt2a morphant;Tg(fli1a:alk1-myc) zebrafish embryos (data notshown), suggesting that Bmp9 and Bmp10 may functionredundantly in vivo if available to Alk1 contemporaneously. Inzebrafish, bmp9 is first detected in liver around 48 hpf (C.J.M. andB. Shravage, unpublished) and the liver is first vascularized around55 hpf (Korzh et al., 2008), supporting the idea that Bmp9 is notredundant with Bmp10 between 24 and 48 hpf, the time periodduring which AVMs develop in alk1 mutants (Roman et al., 2002;Corti et al., 2011). A second possible reason for the discordancebetween mice and zebrafish in terms of definition of Alk1 ligandstems from the fact that slowed heartbeat and impaired circulationprecede death of Bmp10 mouse nulls at embryonic day (E) 9.5-E10.5 (Chen et al., 2004). Thus, it is reasonable to hypothesize thatthe absence of AVMs in Bmp10 nulls is due to the earlier death ofBmp10 nulls versus Alk1 nulls (E10.5-11.5) and/or insufficientcirculation to precipitate AVM formation, which we havedemonstrated to be a flow-dependent process (Corti et al., 2011).Because trabeculation in zebrafish does not occur until 60-72 hpf inzebrafish (Liu et al., 2010; Peshkovsky et al., 2011), it is notsurprising that we noted no defects in heart development or functionthrough 48 hpf in bmp10 or bmp10-like morphants (data not shown).
In cranial arterial endothelial cells, both blood flow andBmp10/Alk1 function are required for phosphorylation ofSmad1/5/9, repression of cxcr4a and induction of edn1.Concomitant restoration of endothelial alk1 expression and injection
3409RESEARCH ARTICLEBmp10/Alk1 mediates flow signal
Fig. 4. bmp10 and bmp10-like are expressed in the heart. (A) Whole-mount in situ hybridization for bmp10, bmp10-like and myl7 at 28, 36 and48 hpf. Ventral views, anterior upwards. Scale bar: 50 μm. V, ventricle; A,atrium. Asterisk indicates atrioventricular canal. (B) Sagittal sections (8 μm)through the heart of 36 hpf embryos co-stained with bmp10, bmp10-likeor myl7 (purple) and MF20 (sarcomeric myosin; brown). Scale bar: 10 μm.DEVELO
PMENT
3410
of rhBMP10 normalizes these molecular end points and restoresarterial endothelial cell number in the absence of flow. These datastrongly suggest that Alk1 acts downstream of blood flow to limit
endothelial cell migration, proliferation and/or vascular tone, andthus limit arterial endothelial cell number and vessel caliber.However, changes in expression of cxcr4a and edn1 cannot fully
RESEARCH ARTICLE Development 140 (16)
Fig. 5. Bmp10/Alk1 lies downstream of blood flow in regulation of pSmad1/5/9, cxcr4a and edn1. (A) BRE:luciferase activity in C2C12 cellstransfected with human (h) ALK1, zebrafish (z) alk1, hALK1 kinase dead mutant (R411Q), hALK3 (which does not bind BMP10) or lipofectamine (lipo), andtreated with rhBMP10. Data are normalized to pTK-Renilla luciferase activity. Values are mean±s.e.m., n=3 biological replicates, each representing 3technical replicates. (B) pSmad1/5/9 (middle column) in endothelial cells (nuclei marked by fli1a:negfp transgene, left column) of the CaDI (caudaldivision of the internal carotid artery) of 36 hpf tnnt2a morphants in which endothelial cell alk1 expression [Tg(fli1a:alk1-myc)], BMP10 availability(rhBMP10; 2 nl of 10 μM rhBMP10 injected into base of CaDI at 28 hpf ), or both, are restored. In merge (right column), EGFP-expressing endothelial cellnuclei are green, pSmad1/5/9 immunofluorescence is magenta. Yellow arrows indicate endothelial cells in the injected CaDI. 2D confocal projections of50 μm frontal sections, dorsal upwards. Scale bar: 10 μm. See supplementary material Table S1 for fluorescence quantitation. (C) Whole-mount in situhybridization for cxcr4a, edn1 and cdh5 (pan-endothelial control) in 36 hpf tnnt2a morphant;Tg(fli1a:alk1-myc) embryos injected at 28 hpf into the baseof the left CaDI with 2 nl tracer with or without 10 μM rhBMP10. Yellow arrow indicates injected CaDI, red arrow indicates uninjected CaDI. Asteriskindicates injection site. Frontal views, dorsal upwards; lateral views, right (uninjected) side or left (injected) side. Scale bar: 50 μm. (D) Quantitation ofCaDI endothelial cell number in 36 hpf Tg(fli1a:alk1-myc) embryos left uninjected (white bars) or injected at the one-cell stage with tnnt2a morpholino(gray, black bars). At 28 hpf, tnnt2a morphants were injected in the right CaDI with 2 nl tracer (gray bars) or 2 nl 10μM rhBMP10 (black bars). tnnt2amorphants injected intravascularly with tracer exhibited increased endothelial cell number throughout the CaDI/BCA, whereas intravascular injection ofrhBMP10 normalized endothelial cell number in the injected CaDI and BCA (basal communicating artery). n=10-12 per group in two independentexperiments. Values are mean±s.e.m. Student’s t-test: *P<0.05; **P<0.001; ns, not significant.
DEVELO
PMENT

explain defects in vessel architecture resulting from loss of alk1.Although ALK1 signaling can repress CXCR4 or induce EEN1 incultured endothelial cells (Star et al., 2010; Park et al., 2012; Younget al., 2012), supporting our in vivo data, previous work hasdemonstrated that increased cxcr4a is not necessary nor is loss ofedn1 sufficient for AVM development (Corti et al., 2011), andadditional work has demonstrated that concomitant increase incxcr4a and loss of edn1 is insufficient to generate AVMs (E. Rochonand B.L.R., unpublished). Thus, further work is required to definethe molecular mechanisms and cellular behaviors that lead to arterialenlargement in the absence of alk1.
The multifaceted regulation of Alk1 signaling by blood flow isremarkable, with flow required for both alk1 expression and Alk1activity. Given that mammalian CXCR4 and EDN1 respond to shearstress and/or cyclic strain in cultured endothelial cells (Wang et al.,1993; Melchionna et al., 2005), it seems likely that Alk1 isimportant in transducing mechanical force into a biochemical signalin vivo. However, the mechanism by which blood flow upregulatesalk1 expression is currently unknown, and it remains formallypossible that the flow dependence of alk1 expression stems at leastin part from a circulating factor. Circulation of ligand clearlycontributes to the dependence of Alk1 activation on blood flow:blood flow distributes cardiac-derived circulating Bmp10 to arterialendothelial cell Alk1, thereby explaining the blood flow dependenceof Smad1/5/9 phosphorylation in these cells. However, a recentstudy reported that mechanical force induces Smad1/5/9phosphorylation in intact mouse endothelium and culturedendothelial cells in a ligand-independent manner (Zhou et al., 2012),contradicting our conclusions. This discrepancy could possibly beexplained by the fact that ligand independence in that study wasexamined via treatment with Noggin, which sequesters most BMPligands, but not BMP9 or BMP10 (Seemann et al., 2009).Alternatively, oscillatory shear stress applied in that study may havedifferent effects on BMP signaling than pulsatile laminar shearstress, which acts within zebrafish cranial arteries (Chen et al.,2011), or the type I receptor responsible for oscillatory shear-induced pSmad1/5/9 may not be Alk1. Further work is required tobetter define the roles of and probe interactions between endocrinefactors and mechanical force in the regulation of Alk1 signaling.
In summary, our data demonstrate that blood flow induces alk1expression and provides Bmp10 to arterial endothelial cell Alk1,thereby activating Smad1/5/9 phosphorylation, decreasing cxcr4aexpression and inducing edn1 expression. These changes in geneexpression, along with changes in expression of yet to be identifiedgenes, serve to dampen angiogenic behavior and to stabilize arterialendothelial cell number and caliber at the onset of blood flow. Takentogether with our previous work (Corti et al., 2011), our data suggestthat loss of Alk1 function abrogates this important flow responseand results in increased nascent arterial caliber, which in turn leadsto increased hemodynamic forces within downstream arteries. In anattempt to normalize these hemodynamic forces, downstreamvessels mount an Alk1-independent flow response that causesnormally transient conduits between this overloaded arterial systemand neighboring veins to be retained and enlarged, thereby forminghigh flow AVMs. Thus, our model suggests that in individuals withHHT, abrogation of one flow response – due to impaired ALK1signaling – leads to activation of an independent flow response thatacts to normalize hemodynamic forces, ultimately leading to AVMs.
AcknowledgementsWe thank Z. Kupchinsky and D. S. Wright for outstanding fish care, and L.Brilli, P. Corti and B. Shravage for technical contributions. We also thank N.Lawson (University of Massachusetts Medical School, Worcester, MA, USA) for
the p5E fli1a.ebs plasmid and for helpful comments on the manuscript, and C.B. Chen (University of Utah, Salt Lake City, UT, USA) and K. Kawakami(National Institute of Genetics, Shizuoka, Japan) for Gateway/tol2 vectors.
FundingThis work was supported by the National Institutes of Health (NIH) [R01HL079108 to B.L.R.]. D.W.L. was supported by the NIH [T32 HL094295-02].P.D.U. was supported by a British Heart Foundation Programme Grant[RG/08/002/24718]. Deposited in PMC for release after 12 months.
Competing interests statementThe authors declare no competing financial interests.
Author contributionsD.W.L. performed the majority of the zebrafish experiments, generated allzebrafish data figures and wrote the paper; S.Y. performed bmp9 morpholinostudies, generated the Tg(fli1a:alk1-myc)pt516 line and assisted in mostzebrafish experiments; J.P.D. analyzed bmp10 and bmp10-like morphantshunting phenotype; C.J.M. performed preliminary characterization of bmp9and bmp10 expression patterns and morphant phenotypes; P.D.U. performedluciferase assays and generated the accompanying figure; B.L.R. conceived anddirected the study, generated ptol-fli1a.ebs:alk1ca-mCherry, and wrote thepaper.
Supplementary materialSupplementary material available online athttp://dev.biologists.org/lookup/suppl/doi:10.1242/dev.095307/-/DC1
ReferencesBhullar, I. S., Li, Y. S., Miao, H., Zandi, E., Kim, M., Shyy, J. Y. and Chien, S.
(1998). Fluid shear stress activation of IkappaB kinase is integrin-dependent. J.Biol. Chem. 273, 30544-30549.
Brown, M. A., Zhao, Q., Baker, K. A., Naik, C., Chen, C., Pukac, L., Singh, M.,Tsareva, T., Parice, Y., Mahoney, A. et al. (2005). Crystal structure of BMP-9and functional interactions with pro-region and receptors. J. Biol. Chem. 280,25111-25118.
Bussmann, J., Wolfe, S. A. and Siekmann, A. F. (2011). Arterial-venous networkformation during brain vascularization involves hemodynamic regulation ofchemokine signaling. Development 138, 1717-1726.
Chang, K., Weiss, D., Suo, J., Vega, J. D., Giddens, D., Taylor, W. R. and Jo, H.(2007). Bone morphogenic protein antagonists are coexpressed with bonemorphogenic protein 4 in endothelial cells exposed to unstable flow in vitro inmouse aortas and in human coronary arteries: role of bone morphogenicprotein antagonists in inflammation and atherosclerosis. Circulation 116, 1258-1266.
Chen, H., Shi, S., Acosta, L., Li, W., Lu, J., Bao, S., Chen, Z., Yang, Z.,Schneider, M. D., Chien, K. R. et al. (2004). BMP10 is essential for maintainingcardiac growth during murine cardiogenesis. Development 131, 2219-2231.
Chen, C. Y., Patrick, M. J., Corti, P., Kowalski, W., Roman, B. L. and Pekkan, K.(2011). Analysis of early embryonic great-vessel microcirculation in zebrafishusing high-speed confocal μPIV. Biorheology 48, 305-321.
Choi, J., Dong, L., Ahn, J., Dao, D., Hammerschmidt, M. and Chen, J. N.(2007). FoxH1 negatively modulates flk1 gene expression and vascularformation in zebrafish. Dev. Biol. 304, 735-744.
Corti, P., Young, S., Chen, C. Y., Patrick, M. J., Rochon, E. R., Pekkan, K. andRoman, B. L. (2011). Interaction between alk1 and blood flow in thedevelopment of arteriovenous malformations. Development 138, 1573-1582.
David, L., Mallet, C., Mazerbourg, S., Feige, J. J. and Bailly, S. (2007).Identification of BMP9 and BMP10 as functional activators of the orphanactivin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 109, 1953-1961.
David, L., Mallet, C., Keramidas, M., Lamandé, N., Gasc, J. M., Dupuis-Girod,S., Plauchu, H., Feige, J. J. and Bailly, S. (2008). Bone morphogeneticprotein-9 is a circulating vascular quiescence factor. Circ. Res. 102, 914-922.
Dekker, R. J., van Soest, S., Fontijn, R. D., Salamanca, S., de Groot, P. G.,VanBavel, E., Pannekoek, H. and Horrevoets, A. J. (2002). Prolonged fluidshear stress induces a distinct set of endothelial cell genes, most specificallylung Krüppel-like factor (KLF2). Blood 100, 1689-1698.
Dekker, R. J., van Thienen, J. V., Rohlena, J., de Jager, S. C., Elderkamp, Y. W.,Seppen, J., de Vries, C. J., Biessen, E. A., van Berkel, T. J., Pannekoek, H. etal. (2005). Endothelial KLF2 links local arterial shear stress levels to theexpression of vascular tone-regulating genes. Am. J. Pathol. 167, 609-618.
Dekker, R. J., Boon, R. A., Rondaij, M. G., Kragt, A., Volger, O. L., Elderkamp,Y. W., Meijers, J. C., Voorberg, J., Pannekoek, H. and Horrevoets, A. J.(2006). KLF2 provokes a gene expression pattern that establishes functionalquiescent differentiation of the endothelium. Blood 107, 4354-4363.
3411RESEARCH ARTICLEBmp10/Alk1 mediates flow signal
DEVELO
PMENT

3412
Goumans, M. J., Valdimarsdottir, G., Itoh, S., Rosendahl, A., Sideras, P. andten Dijke, P. (2002). Balancing the activation state of the endothelium via twodistinct TGF-beta type I receptors. EMBO J. 21, 1743-1753.
Goumans, M. J., Valdimarsdottir, G., Itoh, S., Lebrin, F., Larsson, J.,Mummery, C., Karlsson, S. and ten Dijke, P. (2003). Activin receptor-likekinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling.Mol. Cell 12, 817-828.
Guttmacher, A. E., Marchuk, D. A. and White, R. I., Jr (1995). Hereditaryhemorrhagic telangiectasia. N. Engl. J. Med. 333, 918-924.
Hay, D. C., Beers, C., Cameron, V., Thomson, L., Flitney, F. W. and Hay, R. T.(2003). Activation of NF-kappaB nuclear transcription factor by flow in humanendothelial cells. Biochim. Biophys. Acta 1642, 33-44.
Johnson, D. W., Berg, J. N., Baldwin, M. A., Gallione, C. J., Marondel, I., Yoon,S. J., Stenzel, T. T., Speer, M., Pericak-Vance, M. A., Diamond, A. et al.(1996). Mutations in the activin receptor-like kinase 1 gene in hereditaryhaemorrhagic telangiectasia type 2. Nat. Genet. 13, 189-195.
Kawakami, K., Takeda, H., Kawakami, N., Kobayashi, M., Matsuda, N. andMishina, M. (2004). A transposon-mediated gene trap approach identifiesdevelopmentally regulated genes in zebrafish. Dev. Cell 7, 133-144.
Khachigian, L. M., Resnick, N., Gimbrone, M. A., Jr and Collins, T. (1995).Nuclear factor-kappa B interacts functionally with the platelet-derived growthfactor B-chain shear-stress response element in vascular endothelial cellsexposed to fluid shear stress. J. Clin. Invest. 96, 1169-1175.
Korzh, S., Pan, X., Garcia-Lecea, M., Winata, C. L., Pan, X., Wohland, T.,Korzh, V. and Gong, Z. (2008). Requirement of vasculogenesis and bloodcirculation in late stages of liver growth in zebrafish. BMC Dev. Biol. 8, 84.
Kwan, K. M., Fujimoto, E., Grabher, C., Mangum, B. D., Hardy, M. E.,Campbell, D. S., Parant, J. M., Yost, H. J., Kanki, J. P. and Chien, C. B. (2007).The Tol2kit: a multisite gateway-based construction kit for Tol2 transposontransgenesis constructs. Dev. Dyn. 236, 3088-3099.
Lan, Q., Mercurius, K. O. and Davies, P. F. (1994). Stimulation of transcriptionfactors NF kappa B and AP1 in endothelial cells subjected to shear stress.Biochem. Biophys. Res. Commun. 201, 950-956.
Larrivée, B., Prahst, C., Gordon, E., del Toro, R., Mathivet, T., Duarte, A.,Simons, M. and Eichmann, A. (2012). ALK1 signaling inhibits angiogenesis bycooperating with the Notch pathway. Dev. Cell 22, 489-500.
Liu, J., Bressan, M., Hassel, D., Huisken, J., Staudt, D., Kikuchi, K., Poss, K. D.,Mikawa, T. and Stainier, D. Y. (2010). A dual role for ErbB2 signaling in cardiactrabeculation. Development 137, 3867-3875.
Lux, A., Attisano, L. and Marchuk, D. A. (1999). Assignment of transforminggrowth factor β1 and β3 and a third new ligand to the type I receptor ALK-1. J.Biol. Chem. 274, 9984-9992.
Melchionna, R., Porcelli, D., Mangoni, A., Carlini, D., Liuzzo, G., Spinetti, G.,Antonini, A., Capogrossi, M. C. and Napolitano, M. (2005). Laminar shearstress inhibits CXCR4 expression on endothelial cells: functional consequencesfor atherogenesis. FASEB J. 19, 629-631.
Mitchell, D., Pobre, E. G., Mulivor, A. W., Grinberg, A. V., Castonguay, R.,Monnell, T. E., Solban, N., Ucran, J. A., Pearsall, R. S., Underwood, K. W. etal. (2010). ALK1-Fc inhibits multiple mediators of angiogenesis and suppressestumor growth. Mol. Cancer Ther. 9, 379-388.
Moya, I. M., Umans, L., Maas, E., Pereira, P. N., Beets, K., Francis, A., Sents,W., Robertson, E. J., Mummery, C. L., Huylebroeck, D. et al. (2012). Stalkcell phenotype depends on integration of Notch and Smad1/5 signalingcascades. Dev. Cell 22, 501-514.
Neuhaus, H., Rosen, V. and Thies, R. S. (1999). Heart specific expression ofmouse BMP-10 a novel member of the TGF-beta superfamily. Mech. Dev. 80,181-184.
Oh, S. P., Seki, T., Goss, K. A., Imamura, T., Yi, Y., Donahoe, P. K., Li, L.,Miyazono, K., ten Dijke, P., Kim, S. et al. (2000). Activin receptor-like kinase 1modulates transforming growth factor-β 1 signaling in the regulation ofangiogenesis. Proc. Natl. Acad. Sci. USA 97, 2626-2631.
Park, S. O., Lee, Y. J., Seki, T., Hong, K. H., Fliess, N., Jiang, Z., Park, A., Wu, X.,Kaartinen, V., Roman, B. L. et al. (2008). ALK5- and TGFBR2-independent roleof ALK1 in the pathogenesis of hereditary hemorrhagic telangiectasia type 2.Blood 111, 633-642.
Park, J. E., Shao, D., Upton, P. D., Desouza, P., Adcock, I. M., Davies, R. J.,Morrell, N. W., Griffiths, M. J. and Wort, S. J. (2012). BMP-9 inducedendothelial cell tubule formation and inhibition of migration involves Smad1driven endothelin-1 production. PLoS ONE 7, e30075.
Parmar, K. M., Larman, H. B., Dai, G., Zhang, Y., Wang, E. T., Moorthy, S. N.,Kratz, J. R., Lin, Z., Jain, M. K., Gimbrone, M. A., Jr et al. (2006). Integration
of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J. Clin.Invest. 116, 49-58.
Peshkovsky, C., Totong, R. and Yelon, D. (2011). Dependence of cardiactrabeculation on neuregulin signaling and blood flow in zebrafish. Dev. Dyn.240, 446-456.
Ricard, N., Ciais, D., Levet, S., Subileau, M., Mallet, C., Zimmers, T. A., Lee, S.J., Bidart, M., Feige, J. J. and Bailly, S. (2012). BMP9 and BMP10 are critical forpostnatal retinal vascular remodeling. Blood 119, 6162-6171.
Roman, B. L. and Pekkan, K. (2012). Mechanotransduction in embryonicvascular development. Biomech. Model. Mechanobiol. 11, 1149-1168.
Roman, B. L., Pham, V. N., Lawson, N. D., Kulik, M., Childs, S., Lekven, A. C.,Garrity, D. M., Moon, R. T., Fishman, M. C., Lechleider, R. J. et al. (2002).Disruption of acvrl1 increases endothelial cell number in zebrafish cranialvessels. Development 129, 3009-3019.
Scharpfenecker, M., van Dinther, M., Liu, Z., van Bezooijen, R. L., Zhao, Q.,Pukac, L., Löwik, C. W. and ten Dijke, P. (2007). BMP-9 signals via ALK1 andinhibits bFGF-induced endothelial cell proliferation and VEGF-stimulatedangiogenesis. J. Cell Sci. 120, 964-972.
Seemann, P., Brehm, A., König, J., Reissner, C., Stricker, S., Kuss, P., Haupt, J.,Renninger, S., Nickel, J., Sebald, W. et al. (2009). Mutations in GDF5 reveal akey residue mediating BMP inhibition by NOGGIN. PLoS Genet. 5, e1000747.
Sehnert, A. J., Huq, A., Weinstein, B. M., Walker, C., Fishman, M. andStainier, D. Y. (2002). Cardiac troponin T is essential in sarcomere assemblyand cardiac contractility. Nat. Genet. 31, 106-110.
Seki, T., Yun, J. and Oh, S. P. (2003). Arterial endothelium-specific activinreceptor-like kinase 1 expression suggests its role in arterialization andvascular remodeling. Circ. Res. 93, 682-689.
Seki, T., Hong, K. H., Yun, J., Kim, S. J. and Oh, S. P. (2004). Isolation of aregulatory region of activin receptor-like kinase 1 gene sufficient for arterialendothelium-specific expression. Circ. Res. 94, e72-e77.
Star, G. P., Giovinazzo, M. and Langleben, D. (2010). Bone morphogenicprotein-9 stimulates endothelin-1 release from human pulmonarymicrovascular endothelial cells: a potential mechanism for elevated ET-1 levelsin pulmonary arterial hypertension. Microvasc. Res. 80, 349-354.
ten Dijke, P., Yamashita, H., Ichijo, H., Franzén, P., Laiho, M., Miyazono, K.and Heldin, C. H. (1994). Characterization of type I receptors for transforminggrowth factor-beta and activin. Science 264, 101-104.
Topper, J. N., Cai, J., Qiu, Y., Anderson, K. R., Xu, Y. Y., Deeds, J. D., Feeley, R.,Gimeno, C. J., Woolf, E. A., Tayber, O. et al. (1997). Vascular MADs: two novelMAD-related genes selectively inducible by flow in human vascularendothelium. Proc. Natl. Acad. Sci. USA 94, 9314-9319.
Traver, D., Paw, B. H., Poss, K. D., Penberthy, W. T., Lin, S. and Zon, L. I. (2003).Transplantation and in vivo imaging of multilineage engraftment in zebrafishbloodless mutants. Nat. Immunol. 4, 1238-1246.
Urness, L. D., Sorensen, L. K. and Li, D. Y. (2000). Arteriovenous malformationsin mice lacking activin receptor-like kinase-1. Nat. Genet. 26, 328-331.
Villefranc, J. A., Amigo, J. and Lawson, N. D. (2007). Gateway compatiblevectors for analysis of gene function in the zebrafish. Dev. Dyn. 236, 3077-3087.
Wang, D. L., Tang, C. C., Wung, B. S., Chen, H. H., Hung, M. S. and Wang, J. J.(1993). Cyclical strain increases endothelin-1 secretion and gene expression inhuman endothelial cells. Biochem. Biophys. Res. Commun. 195, 1050-1056.
Westerfield, M. (1995). The Zebrafish Book. Eugene, OR: University of OregonPress.
Woods, I. G., Wilson, C., Friedlander, B., Chang, P., Reyes, D. K., Nix, R., Kelly,P. D., Chu, F., Postlethwait, J. H. and Talbot, W. S. (2005). The zebrafish gene map defines ancestral vertebrate chromosomes. Genome Res. 15, 1307-1314.
Xu, P., Liu, J. and Derynck, R. (2012). Post-translational regulation of TGF-βreceptor and Smad signaling. FEBS Lett. 586, 1871-1884.
Yelon, D., Horne, S. A. and Stainier, D. Y. (1999). Restricted expression ofcardiac myosin genes reveals regulated aspects of heart tube assembly inzebrafish. Dev. Biol. 214, 23-37.
Young, K., Conley, B., Romero, D., Tweedie, E., O’Neill, C., Pinz, I., Brogan, L.,Lindner, V., Liaw, L. and Vary, C. P. (2012). BMP9 regulates endoglin-dependent chemokine responses in endothelial cells. Blood 120, 4263-4273.
Zhou, J., Lee, P. L., Tsai, C. S., Lee, C. I., Yang, T. L., Chuang, H. S., Lin, W. W.,Lin, T. E., Lim, S. H., Wei, S. Y. et al. (2012). Force-specific activation ofSmad1/5 regulates vascular endothelial cell cycle progression in response todisturbed flow. Proc. Natl. Acad. Sci. USA 109, 7770-7775.
RESEARCH ARTICLE Development 140 (16)
DEVELO
PMENT





























