Embed Size (px)
Citation preview

This journal is c The Royal Society of Chemistry 2011 Chem. Commun., 2011, 47, 12527–12529 12527
Cite this: Chem. Commun., 2011, 47, 12527–12529
Indirect 1H NMR characterization of H2@C60 nitroxide derivatives and
their nuclear spin relaxationw
Yongjun Li,aXuegong Lei,
aXia Li,
aRonald G. Lawler,
bYasujiro Murata,
cKoichi Komatsu
d
and Nicholas J. Turro*a
Received 18th August 2011, Accepted 5th October 2011
DOI: 10.1039/c1cc15149e
1H NMR of two H2@C60 nitroxide derivatives has been
characterized indirectly by reducing to their corresponding
hydroxylamines. Nuclear spin relaxation of the endohedral H2
and external protons of the H2@C60 nitroxide and its corresponding
hydroxylamine were measured and analyzed. The observed spectra
are consistent with negligible scalar coupling between the unpaired
electron and the endo-H2. An unexpectedly large bimolecular
relaxivity induced in the hydroxylamine by the corresponding
nitroxide can be explained by rapid hydrogen atom transfer between
the two species.
Nitroxides are stable free radicals that have been widely used
as spin labels in biological systems.1 Recently, research on
dual probe systems in which nitroxide radicals are linked to
fluorophores has attracted intense interest.2 Among them,
fullerene C60 derivatives covalently linked to a nitroxide
radical are of particular interest.3 The interaction between
C60 triplet and the radical makes it a good candidate for
chemically induced dynamic electron polarization (CIDEP)
investigated by time-resolved electron paramagnetic resonance
(EPR) spectroscopy.4,5 C60 nitroxide derivatives are commonly
characterized by mass spectroscopy and/or elemental analysis.3
Due to the paramagnetic nature of the nitroxide radical, it is
usually not possible to obtain detailed structural information by
NMR spectroscopy. It is well-known that paramagnetic nitroxide
radicals can be reduced to their corresponding diamagnetic
hydroxylamines in which NMR information can be obtained.6
However, there have been no reports applying the method
to C60 nitroxide derivatives likely due to the overlapped
NMR peaks.
We report that NMR structural information of C60 nitroxide
derivatives can be easily obtained by in situ reduction to their
corresponding hydroxylamines. We chose hydrazobenzene7
as the reducing reagent because its 1H NMR and the oxidized
product—azobenzene—are in the aromatic region, and do not
interfere with the reduced C60 nitroxide derivatives.
We chose two C60 nitroxide derivatives, 1 and 2, as shown in
Fig. 1 to demonstrate NMR characterization by reducing
them to the hydroxylamines by hydrazobenzene. Two corres-
ponding H2@C60 nitroxide derivatives, H2@1 and H2@2,
have also been synthesized. Measurements on mixtures of
the nitroxide and hydroxylamine exhibit a bimolecular relax-
ivity (R1) contribution to spin relaxation times of the external
and endohedral protons in the hydroxylamine. H2@3 was
used as a control compound.
The synthesis of 1 and 2 has been previously reported.3,8
Analogously, H2@C60 was used as the starting material for the
synthesis of H2@1, H2@2 and H2@3. The 1H NMR spectra of
1 and 2 in CDCl3 are shown in Fig. S1a (ESIw) and Fig. 2a,
respectively. Because of paramagnetic broadening of the nitroxide,
NMR signals corresponding to the protons of the functional
groups are not detected. Note that peaks at 0.9, 1.3 and 2.2 ppm
are probably from impurities from plasticizers or silicone grease as
C60 has a strong tendency to retain these materials.9
The progress of the reduction is readily followed by
monitoring the EPR spectrum of the nitroxide (Fig. S2, ESIw).In the presence of excess hydrazobenzene, CDCl3 solutions of
1 and 2 are completely reduced within 5 minutes. 1H NMR
spectra were taken (32 scans, B5 min) following the addition
and shown in Fig. S1b (ESIw) for reduced 1 and Fig. 2b for
reduced 2. Due to the rather symmetric structure of 1, a singlet
appears at 2.1 ppm from the four equivalent methyl groups,
which is consistent with the 1H NMR spectrum of its
synthetic precursor 3 (Fig. S1c, ESIw). Note that two peaks at
7.5 and 7.9 ppm are from azobenzene—the oxidative product of
hydrazobenzene. For 2, a set of new NMR peaks appear
between 1 and 5 ppm. The assignment of the NMR signals is
Fig. 1 H2@C60 nitroxide derivatives.
aDepartment of Chemistry, Columbia University, New York,NY 10027, USA. E-mail: [email protected]
bDepartment of Chemistry, Brown University, Providence,Rhode Island 02912, USA
c Institute for Chemical Research, Kyoto University, Kyoto 611-0011,Japan
dDepartment of Environmental and Biological Chemistry,Fukui University of Technology, Gakuen, Fukui 910-8505, Japan
w Electronic supplementary information (ESI) available: 1H NMRspectra of 1, reduction profiles monitored by EPR, and analysis ofcontact shift. See DOI: 10.1039/c1cc15149e
ChemComm Dynamic Article Links
www.rsc.org/chemcomm COMMUNICATION
Dow
nloa
ded
by C
olum
bia
Uni
vers
ity o
n 14
Nov
embe
r 20
11Pu
blis
hed
on 2
6 O
ctob
er 2
011
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C1C
C15
149E
View Online / Journal Homepage / Table of Contents for this issue
12528 Chem. Commun., 2011, 47, 12527–12529 This journal is c The Royal Society of Chemistry 2011
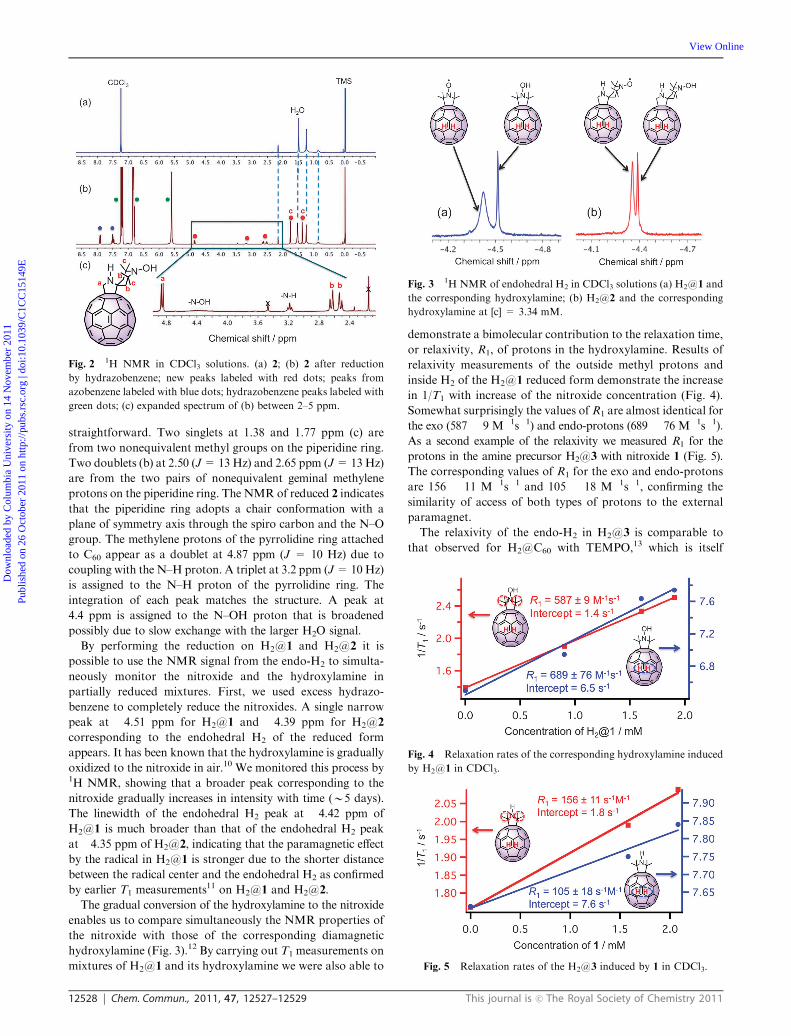
straightforward. Two singlets at 1.38 and 1.77 ppm (c) are
from two nonequivalent methyl groups on the piperidine ring.
Two doublets (b) at 2.50 (J=13Hz) and 2.65 ppm (J=13Hz)
are from the two pairs of nonequivalent geminal methylene
protons on the piperidine ring. The NMR of reduced 2 indicates
that the piperidine ring adopts a chair conformation with a
plane of symmetry axis through the spiro carbon and the N–O
group. The methylene protons of the pyrrolidine ring attached
to C60 appear as a doublet at 4.87 ppm (J = 10 Hz) due to
coupling with the N–H proton. A triplet at 3.2 ppm (J=10Hz)
is assigned to the N–H proton of the pyrrolidine ring. The
integration of each peak matches the structure. A peak at
4.4 ppm is assigned to the N–OH proton that is broadened
possibly due to slow exchange with the larger H2O signal.
By performing the reduction on H2@1 and H2@2 it is
possible to use the NMR signal from the endo-H2 to simulta-
neously monitor the nitroxide and the hydroxylamine in
partially reduced mixtures. First, we used excess hydrazo-
benzene to completely reduce the nitroxides. A single narrow
peak at �4.51 ppm for H2@1 and �4.39 ppm for H2@2
corresponding to the endohedral H2 of the reduced form
appears. It has been known that the hydroxylamine is gradually
oxidized to the nitroxide in air.10 We monitored this process by1H NMR, showing that a broader peak corresponding to the
nitroxide gradually increases in intensity with time (B5 days).
The linewidth of the endohedral H2 peak at �4.42 ppm of
H2@1 is much broader than that of the endohedral H2 peak
at �4.35 ppm of H2@2, indicating that the paramagnetic effect
by the radical in H2@1 is stronger due to the shorter distance
between the radical center and the endohedral H2 as confirmed
by earlier T1 measurements11 on H2@1 and H2@2.
The gradual conversion of the hydroxylamine to the nitroxide
enables us to compare simultaneously the NMR properties of
the nitroxide with those of the corresponding diamagnetic
hydroxylamine (Fig. 3).12 By carrying out T1 measurements on
mixtures of H2@1 and its hydroxylamine we were also able to
demonstrate a bimolecular contribution to the relaxation time,
or relaxivity, R1, of protons in the hydroxylamine. Results of
relaxivity measurements of the outside methyl protons and
inside H2 of the H2@1 reduced form demonstrate the increase
in 1/T1 with increase of the nitroxide concentration (Fig. 4).
Somewhat surprisingly the values of R1 are almost identical for
the exo (587 � 9 M�1s�1) and endo-protons (689 � 76 M�1s�1).
As a second example of the relaxivity we measured R1 for the
protons in the amine precursor H2@3 with nitroxide 1 (Fig. 5).
The corresponding values of R1 for the exo and endo-protons
are 156 � 11 M�1s�1 and 105 � 18 M�1s�1, confirming the
similarity of access of both types of protons to the external
paramagnet.
The relaxivity of the endo-H2 in H2@3 is comparable to
that observed for H2@C60 with TEMPO,13 which is itself
Fig. 2 1H NMR in CDCl3 solutions. (a) 2; (b) 2 after reduction
by hydrazobenzene; new peaks labeled with red dots; peaks from
azobenzene labeled with blue dots; hydrazobenzene peaks labeled with
green dots; (c) expanded spectrum of (b) between 2–5 ppm.
Fig. 3 1H NMR of endohedral H2 in CDCl3 solutions (a) H2@1 and
the corresponding hydroxylamine; (b) H2@2 and the corresponding
hydroxylamine at [c] = 3.34 mM.
Fig. 4 Relaxation rates of the corresponding hydroxylamine induced
by H2@1 in CDCl3.
Fig. 5 Relaxation rates of the H2@3 induced by 1 in CDCl3.
Dow
nloa
ded
by C
olum
bia
Uni
vers
ity o
n 14
Nov
embe
r 20
11Pu
blis
hed
on 2
6 O
ctob
er 2
011
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C1C
C15
149E
View Online

This journal is c The Royal Society of Chemistry 2011 Chem. Commun., 2011, 47, 12527–12529 12529
somewhat larger than expected on the basis of the diffusion
coefficients and most likely distance of closest approach of the
radical and fullerene. It is therefore surprising to find that the
relaxivities of both types of protons in the hydroxylamine of
H2@1 are increased relative to H2@3 by nearly a factor of
five, putting them outside the range of values attainable by
reasonable estimates of diffusion coefficients and distances of
approach. We offer as an explanation for the accelerated
relaxivity an additional contribution to the observed relaxation
rate from the limitation of the lifetime of H2 in the hydroxylamine,
in the slow exchange limit,14 due to exchange between the radical
and hydroxylamine by hydrogen atom transfer (HAT)
*RNOH + RNO = *RNO + RNOH
where the star (*) simply indicates molecules containing the
nucleus of interest that would be affected by the HAT process.
This reaction has been shown to occur with other hydroxyl-
amine/nitroxide pairs15 and may be sufficiently fast under some
conditions to produce line broadening in the hydroxylamine.16
Although analogous broadening of the RNOH peaks was
too small to be measured, the effect on T1 may be described in
terms of a classic chemical exchange between two sites with
different T1’s. In this case the T1 of the peak of interest,
RNOH, is ca. 8 times as long as that of the RNO peak,
corresponding to 1/T1 values for the endo-H2 of 6.5 s�1 and
50 s�1, respectively. The corresponding 1/T1 value for the
methyl group of RNOH is 1.4 s�1, and that for the RNO
methyl group is presumably much larger because of the
expected much larger dipolar and scalar interactions between
the methyl protons and unpaired electron in H2@1. This is
consistent with the fact that the corresponding peak is too
broad to be seen by high resolution NMR (Fig. S1a, ESIw).Under these conditions, following the inversion pulse of the
T1 measurement procedure, the RNOH peak exchanges
magnetization with an almost fully relaxed RNO peak. It is
easily shown14 that in this case the recovery of the RNOH
peak is essentially exponential with an effective relaxation rate
given by
1/T1 = 1/T10 + R1,dd [RNO] + kHAT[RNO]
where 1/T10 is the relaxation rate when the concentration
[RNO] is zero; R1,dd, is the intermolecular dipolar contribution
that may be estimated to be the value for H2@3.
Using the above method, the resulting second order rate
constants, kHAT, for the endo and exo-protons of the H2@1
hydroxylamine, obtained by subtracting the corresponding
values of R1 for H2@3 from those for H2@1, are estimated
to be 584 � 78 and 431 � 14 M�1s�1, respectively (Fig. 4 and 5;
error estimate 1 standard deviation). The values are statistically
equal, as expected for protons in the same exchanging species.
The values themselves are near the upper range of values for
such HAT reactions15 and are similar to those obtained when
an intermediate complex has been postulated.16 The presence of
a complex in the present case would also be consistent with the
unusually large dipolar contribution to the relaxivity between
nitroxides and endofullerenes.13 Such complexation is commonly
invoked to explain enhanced relaxivity between paramagnetic
metal ions and diamagnet substrates.17 In the present case,
however, it is not supported by a model calculation13 of the
potential energy between a nitroxide and a C60 molecule in
the gas phase which predicted a purely repulsive interaction as
the molecules approach each other. Further theoretical, and
refined experimental study of both the uni- and bimolecular
relaxation processes involved here would clearly be desirable.
In summary, we have demonstrated that 1H NMR structural
information of C60 nitroxide derivatives are obtained indirectly
by reduction in situ to the corresponding hydroxylamine by
hydrazobenzene. The quantitative reduction reaction enables us
to identify and assign all NMR peaks. Due to simplicity and
convenience, the present method would be very useful for
characterization of newly synthesized C60 nitroxide derivatives.
We have also demonstrated the use of the endo-H2 signal from
the H2@fullerene to simultaneously monitor and study the
relaxation of mixtures of the nitroxides and hydroxylamines.
The authors thank the National Science Foundation for its
generous support through Grant CHE 07-17518.
Notes and references
1 P. P. Borbat, A. J. Costa-Filho, K. A. Earle, J. K. Moscicki andJ. H. Freed, Science, 2001, 291, 266.
2 J. P. Blinco, K. E. Fairfull-Smith, B. J. Morrow and S. E. Bottle,Aust. J. Chem., 2011, 64, 373.
3 F. Arena, F. Bullo, F. Conti, C. Corvaja, M. Maggini, M. Pratoand G. Scorrano, J. Am. Chem. Soc., 1997, 119, 789.
4 C. Corvaja, M. Maggini, M. Prato, G. Scorrano and M. Venzin,J. Am. Chem. Soc., 1995, 117, 8857.
5 E. Sartori, A. Toffoletti, C. Corvaja and L. Garlaschelli, J. Phys.Chem. A, 2001, 105, 10776.
6 T. D. Lee and J. F. W. Keana, J. Org. Chem., 1975, 40, 3145.7 A. D. Malievskii and A. B. Shapiro, Kinet. Catal., 2005, 46, 472.8 M. Mazzoni, L. Franco, A. Ferrarini, C. Corvaja, G. Zordan,G. Scorrano and M. Maggini, Liq. Cryst., 2002, 29, 203.
9 J. Nossal, R. K. Saini, L. B. Alemany, M. Meier and W. E. Billups,Eur. J. Org. Chem., 2001, 4167.
10 A. A. Bobko, I. A. Kirilyuk, I. A. Grigor’ev, J. L. Zweier andV. V. Khramtsov, Free Radical Biol. Med., 2007, 42, 404.
11 Y. Li, X. Lei, R. G. Lawler, Y. Murata, K. Komatsu andN. J. Turro, J. Phys. Chem. Lett., 2010, 1, 2135.
12 Preliminary analysis (ESIw) of the shifts and widths of the endo H2
peaks in Fig. 3 indicates that there is no detectable contactinteraction between the protons and unpaired electron and a negligiblecontribution of scalar relaxation to the nitroxide linewidths. Theobserved chemical shift difference between the endo-H2 in the nitroxideand hydroxylamine must therefore arise primarily from differences inshielding of the nuclei in the slightly different electronic environments.
13 E. Sartori, M. Ruzzi, N. J. Turro, K. Komatsu, Y. Murata,R. G. Lawler and A. L. Buchachenko, J. Am. Chem. Soc., 2008,130, 2221.
14 J. Schotland and J. S. Leigh, J. Magn. Reson., 1983, 51, 48.15 A. Wu, E. A. Mader, A. Datta, D. A. Hrovat, W. T. Borden and
J. M. Mayer, J. Am. Chem. Soc., 2009, 131, 11985.16 R. W. Kreilick and S. I. Weissman, J. Am. Chem. Soc., 1966,
88, 2645.17 L. Banci, I. Bertini and C. Luchinat, Nuclear and Electronic
Relaxation, VCH, Weinheim, Germany, 1991.
Dow
nloa
ded
by C
olum
bia
Uni
vers
ity o
n 14
Nov
embe
r 20
11Pu
blis
hed
on 2
6 O
ctob
er 2
011
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/C1C
C15
149E
View Online





























