Embed Size (px)
Citation preview

American Journal of Medical Genetics 104:331±338 (2001)
Clinical, Genetic, and Biochemical Characterizationof a Leber Hereditary Optic Neuropathy FamilyContaining Both the 11778 and 14484Primary Mutations
Michael D. Brown,1* Jon C. Allen,1 Gregory P. Van Stavern,2 Nancy J. Newman,2,3 andDouglas C. Wallace1
1Center for Molecular Medicine, Emory University School of Medicine, Atlanta, Georgia2Department of Ophthalmology, Emory University School of Medicine, Atlanta, Georgia3Department of Neurology and Department of Neurological Surgery, Emory University School of Medicine, Atlanta,Georgia
Four mitochondrial DNA (mtDNA) muta-tions at nps 3460, 11778, 14484, and 14459account for roughly 90% of cases of Leberhereditary optic neuropathy (LHON) andare designated as ``primary'' LHON muta-tions since they act as major predispositionfactors for LHON. Although each primarymutation can arise independently on differ-ent mtDNA backgrounds during humanevolution, they characteristically do not co-occur in LHON patients. We report here afamily with the simultaneous occurrence ofthe 11778A and 14484C mutations. Neuro-ophthalmological examination of the pro-band, a nine-year-old Caucasian female, re-vealed the bilateral optic atrophy, centralscotomas, and reduced visual acuity typicalof LHON. Her mother had normal appearingoptic discs and is today visually asympto-matic. Analysis of the proband blood mtDNArevealed that she harbored both the 11778A(heteroplasmic, 94% mutant) and the 14484C(homoplasmic mutant) mutation. This geno-type was maintained in proband lympho-blasts and transmitochondrial cybrids. Themother also had both mutations, with the14484C mutation homoplasmic in all celltypes examined. However, only 31% of herblood mtDNAs carried the 11778 mutation,which segregated to essentially 100% wild-
type in lymphoblast and cybrid mtDNA.Complex I-linked respiration and speci®cenzyme activity were consistently lowest inproband lymphoblast and cybrid mitochon-dria compared to those from the mother,11778A patients, 14484C patients, or con-trols, thus demonstrating both a deleterioussynergistic interaction between the 11778Aand 14484C mutations and the magnitude of11778A-associated complex I dysfunction.Remarkably, spontaneous vision recoveryoccurred in the proband, highlighting thecomplexities encountered when associatingmtDNA genotype and complex I functionwith LHON expression. ß 2001 Wiley-Liss, Inc.
KEY WORDS: mitochondrial DNA; muta-tions;Leberhereditaryopticneuropathy; oxidative pho-sphorylation; ophthalmolo-gic disease
INTRODUCTION
Missense mutations in the mitochondrial DNA(mtDNA) can result in Leber hereditary optic neuro-pathy (LHON), which features maternal transmissionof acute or subacute, progressive, and bilateral loss ofcentral vision due to optic nerve dysfunction [Newmanet al., 1991; Harding et al., 1995]. The associationbetween LHON and mtDNA mutations was ®rstidenti®ed in 1988 when a G to A base change atnucleotide pair (np) 11778 was found in nine unrelatedfamilies [Wallace et al., 1988]. Since then, three othermtDNA variants, at nps 3460A, 14484C, and 14459A,have been identi®ed that also cause familial LHON[Howell et al., 1991; Huoponen et al., 1991; Johns et al.,1992; Jun et al., 1994]. Together, these mutations havebeen designated ``primary'' LHON mutations because
Grant sponsor: NIH; Grant numbers: EY11305, NS21328, P30-EY0 6360; Grant sponsor: Research to Prevent Blindness, Inc.
*Correspondence to: Michael D. Brown, Center for MolecularMedicine, Emory University School of Medicine, 420 B DentalBuilding, 1462 Clifton Road, N.E., Atlanta, GA 30322.E-mail: [email protected]
Received 8 June 2001; Accepted 31 July 2001
DOI 10.1002/ajmg.10054
ß 2001 Wiley-Liss, Inc.

they are signi®cant risk factors for blindness. PrimaryLHON mutations generally share the following geneticfeatures: a) they are absent in control populations, b)they occur in multiple LHON families and on differentmtDNA backgrounds, c) they can be homoplasmic orheteroplasmic, and d) they do not typically co-occur inpatients [Brown and Wallace, 1994; Brown et al., 1995;Howell et al., 1995; Howell, 1998]. Although the 14459Amutation can also provoke early onset dystonia [Junet al., 1994; Shoffner et al., 1995], optic atrophy istypically the only clinical manifestation of LHON. Over-all, these mutations account for more than 90% of allreported LHON cases.
The 3460A, 11778A, 14484C, and 14459A LHONmissense mutations alter the mitochondrial ND1,ND4, and ND6 (both the 14484C and 14459A variants)polypeptides, respectively. Thus, all primary LHONmtDNA mutations modify proteins that are componentsof NADH dehydrogenase or complex I. Complex I isa large, multi-subunit enzyme comprised of sevenmtDNA-encoded (ND1, 2, 3, 4, 4L, 5, and 6) and roughly35 nuclear-encoded polypeptides [Wallace et al., 2001].In oxidative phosphorylation (OXPHOS), electronsenter the mitochondrial electron transport chain fromNADH�H� via complex I. In complex I, the electronstraverse a ¯avin mononucleotide, ®ve to seven iron-sulfur centers, and ultimately reduce ubiquinone toubiquinol. Electrons are transferred to complex III(ubiquinol:cytochrome c oxidoreductase, [EC 1.10.2.2]),then to cytochrome c, next to complex IV (cytochrome coxidoreductase, [EC 1.9.3.1]), and ®nally to atomicoxygen. As electron transfer proceeds through com-plexes I, III, and IV, protons are pumped from thematrix to the inner membrane space creating anelectrochemical gradient (DC).DC is utilized by complexV (ATP synthase, [EC 3.6.1.34]) to condense ADP andinorganic phosphate to ATP. Thus, addition of ADP torespiring mitochondria increases the respiration rate(state III) as the ADP is phosphorylated; mitochondrialrespiration returns to the basal rate (state IV) when theADP is depleted. Addition of an agent that collapses DCuncouples electron transport from ATP synthesis,resulting in an accelerated (uncoupled) respiration rate.
The occurrence of two primary LHON mutations inone family is extremely rare. Here, we describe theclinical, genetic, and biochemical analysis of a patientand her asymptomatic mother, both of whom harbor thenp 11778A and 14484C mutations. Lymphoblasts andtransmitochondrial cybrids (cybrids) derived from theproband and her mother differed only by the presence ofthe 11778A mutation in the proband's cell types, thusproviding a unique opportunity to study both theindividual contributions of each mutation to LHONexpression, as well as genotype-phenotype interactionsamong primary LHON mtDNA variants.
METHODS AND MATERIALS
Clinical Report
A nine-year-old Caucasian girl was ®rst noted to havevisual dif®culties in September 1999. She complain-ed of dif®culty reading the chalkboard at school and
performing her homework, but there was no suddenloss of vision at any time. There were no associatedneurological symptoms, including headache, eye pain,diplopia, weakness, or numbness. There was no pre-ceding or intercurrent systemic illness or exposures.There was no family history of visual loss and thepatient had no siblings. She was initially diagnosed inOctober 1999 with functional visual loss. Computedtomography and magnetic resonance imaging of thebrain, with and without contrast, were normal. Visualevoked responses were abnormal bilaterally, and anelectroretinogram was normal.
Neuro-ophthalmic examination in November 1999revealed best corrected visual acuity of 20/200 O.U.Goldmann visual ®eld testing showed bilateral centralscotomas, right larger than left. Funduscopic exami-nation revealed bilateral optic nerve atrophy withno circumpapillary telangiectasias. The remainder ofthe patient's neuro-ophthalmological and neurologicalexam was unremarkable. Funduscopic examination ofboth parents showed normal appearing optic discs.Complete blood count, vitamin B12 and folate levels,and Lyme and Bartonella titers were normal in thepatient. Follow-up examination three months later wasunchanged. However, follow-up examination in June2000 showed improvement of visual acuity to 20/50 ineach eye with smaller central scotomas. By December2000, visual acuity was 20/40 in each eye and visual®elds showed even further improvement.
DNA Isolation and LHON Mutation Analysis
Genomic DNA was isolated from the buffy coat bloodfraction, lymphoblasts, or cybrids using a Puregene(Gentra Systems) kit. The 3460A, 11778A, and 14484CLHON mutations were analyzed by PCR ampli®cationfollowed by mutation-speci®c restriction endonucleasedigestion [Brown et al., 1995]. For the 11778A muta-tion, the forward primer extended from np 11141 to11158, and the reverse primer from np 11851 to 11868(30 to 50). This mutation creates a novel MaeIIIrestriction site. For the 3460A mutation, the forwardprimer extended from np 3108 to 3127, and the reverseprimer from np 3701 to 3717 (30 to 50). This mutationeliminates a BsaHI site. For the 14484C mutation,the forward primer extended from np 14191 to 14210and the reverse primer from np 14485 to 14510 (30 to 50).The reverse primer had two mismatched cytosines atnucleotide positions 14487 and 14488. These mis-matches create a BstNI site in the presence of the Cto T base change at np 14484. Positive (containingLHON mutation) and negative controls were utiliz-ed for each enzymatic digestion, in part to rule outincomplete digestion, which would confound hetero-plasmy calculations.
Diagnostic restriction endonuclease digestions wereresolved by agarose gel electrophoresis and the DNAfragments were identi®ed using ethidium bromide.Digestion products were visualized using the Eagle EyeII photodocumentation system and heteroplasmy wasquantitated using the ImageQuant software (MolecularDynamics). For proband buffy coat mtDNA exhibiting
332 Brown et al.

heteroplasmy for the np 11778A mutation, we veri®edthe ImageQuant results by cloning. Here, a 404 basepair segment of the mtDNA encompassing the muta-tion was PCR-ampli®ed and cloned into the vectorpCR2.1 and transformed into IVFaF0 cells (TA Cloningkit, Invitrogen, Carlsbad, CA) [Wallace et al., 1997].Sixty white colonies containing inserts were isolatedand the mtDNA PCR-ampli®ed using the same primersutilized to make the cloning insert. The ampli®ed clonemtDNA was then subjected to the restriction digestdiagnostic for the np 11778A mutation. In this way, aheteroplasmic ratio could be obtained by dividingthe number of clones containing the mutant 11778Amutation by the total number of clones examined.Excellent concordance between the ImageQuant andcloning techniques was obtained, with a variance of lessthan 2%.
Cell Lines
Lymphoblast cell lines were established by Epstein-Barr Virus (EBV) transformation of leukocytes isolatedfrom whole blood by Ficoll-Hypaque gradients. AllEBV-transformed cell lines were maintained in RPMI1640 medium (Bio-Whitaker, Walkersville, MD) sup-plemented with 15% (vol/vol) heat-inactivated fetalbovine serum (Gibco-BRL life Technologies, GrandIsland, NY). EBV-transformed lymphoblast cell lineshad been maintained in culture for 10 to 20 populationdoublings at the time of mitochondrial isolation.
Transmitochondrial cybrids were prepared by enucl-eating EBV-transformed lymphoblastoid cell lines fromLHON patients and controls and fusing 2� 107 mito-chondria-containing cytoplasts to the 107 WAL2A-r8,cells by electroshock [Trounce et al., 1996]. TheWAL2A-r8 line is a mtDNA-less, HPRT-de®cient cellline derived from lymphoblasts. It is grown in RPMI1640 medium supplemented with 15% heat-inactivatedfetal bovine serum, 4 mg/ml glucose, 50 mg/ml uridine,1 mM pyruvate (GUP media), containing 1 mg of6-thioguanine (6TG). Cybrids were selected in 75 mlof RPMI 1640 medium supplemented with 10% (vol/vol)dialyzed fetal bovine serum (Gibco-BRL Life Technol-ogies), 2 mg/ml glucose, and 1 mg/ml 6TG. Rapid growthof the cybrid cultures was observed 20 to 28 days post-fusion, and cybrid lines were passaged ®ve to 10 popu-lation doublings prior to mitochondrial isolation.
Mitochondrial Isolation, Polarography,and OXPHOS Enzymology
Procedures for mitochondrial isolation, polaro-graphic analysis, and OXPHOS enzymology of sub-mitochondrial particles have been described [Trounceet al., 1996; Jun et al., 1996; Brown et al., 2000; Brownet al., 2001]. Protein concentrations were determinedby the Lowry assay. Lymphoblast and cybrid mitochon-dria were assayed at concentrations ranged from 3.5 to13 mg protein/ml. For respiration analysis of mitochon-dria, two polarographic runs were performed for eachsubstrate [malate plus pyruvate, or malate plusglutamate (site I) and succinate (site II)] involvingtwo additions of ADP (125 nmole for site I and 75 nmole
ADP for site II substrates). The experiment was con-cluded by the addition of the OXPHOS uncoupler 2,4-dinitrophenol. All runs were performed with 250 to500 mg of mitochondrial protein. The respiratory con-trol ratio (RCR) is the mean state III respiration ratedivided by the mean state IV respiration rate. Allmitochondrial isolates included in this study had tohave RCRs of >2 to meet our minimum standard ofmitochondrial integrity. The state III ratio is the meanof the state III respiration rates using site I substratesdivided by the mean of the state III respiration ratesusing site II substrates.
OXPHOS enzyme activities were assayed on soni-cated mitochondria, and measured spectrophotometri-cally with a dual-beam UV-visual spectrophotometer(model DW-2000; SLM-Aminco, Urbana, IL). Complex Iactivities were assayed in triplicate using 30 mg ofmitochondrial protein and monitoring the reductionof 10 mM decylubiquinone (DB) at 272 nm by 30 mMNADH. Consistently 90% to 100% of the total complex Iactivity was rotenone sensitive. Citrate synthase (CS)was assayed in duplicate, using 15 mg of mitochondrialprotein.
RESULTS
MtDNA Analysis
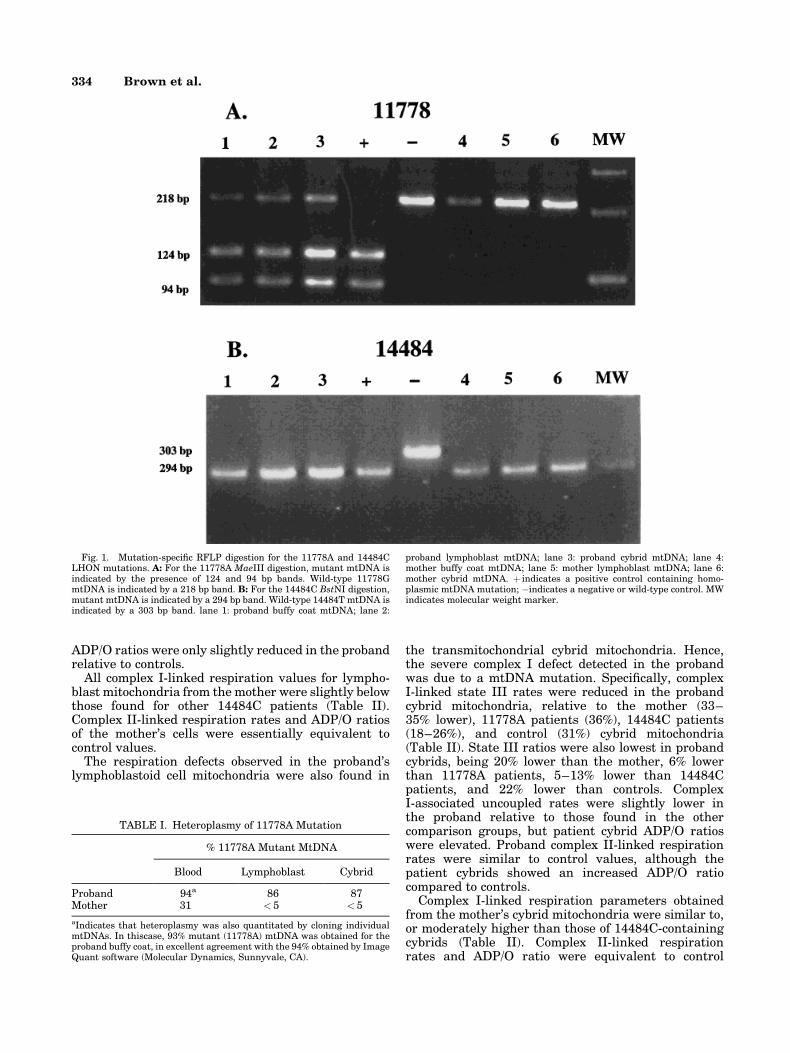
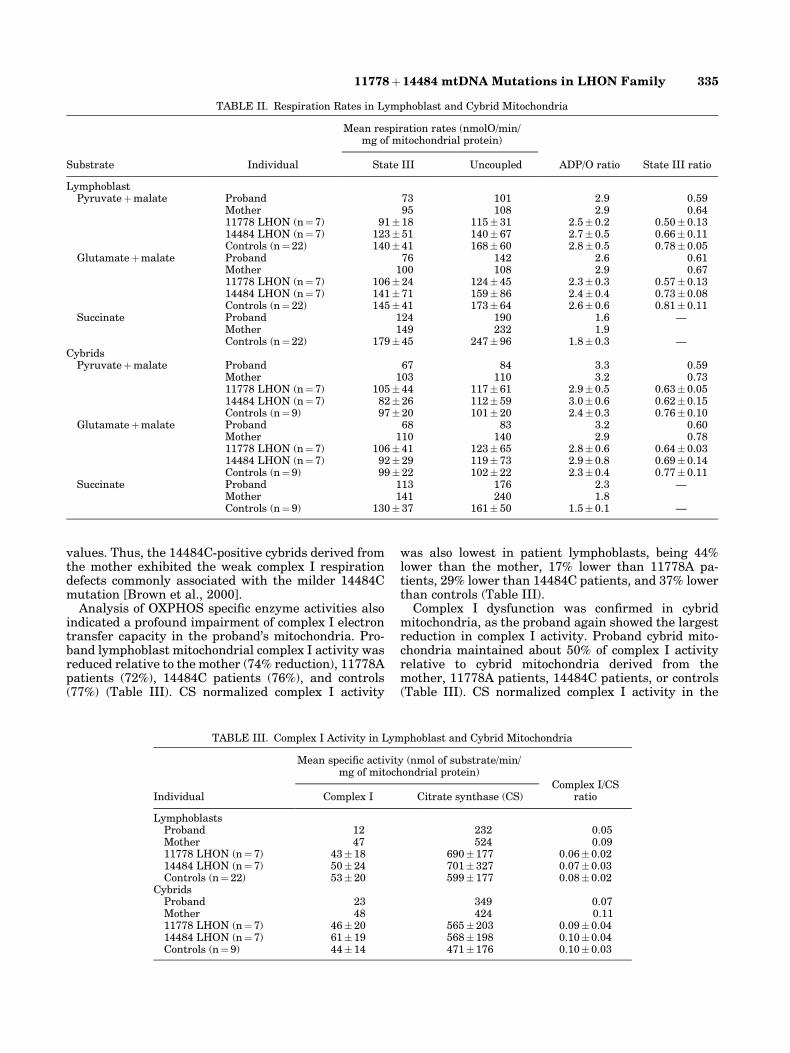
Mutation-speci®c restriction endonuclease digestionof PCR-ampli®ed buffy coat mtDNA indicated the pre-sence of both the 11778A and the 14484C primaryLHON mutations in the proband and her mother. Inboth cases, the 14484C mutation was homoplasmic butthe 11778A mutation was heteroplasmic, with 94% ofthe proband's mtDNA and 31% of the mother's mtDNAbeing mutant (Fig. 1 and Table I). The 11778Amutation remained heteroplasmic in the lymphoblasts(86% mutant) and transmitochondrial cybrids (87%mutant) of the proband, while the mutation segregatedto virtually pure wild-type mtDNA in the mother's celllines, with < 5% of the mtDNAs remaining 11778Amutant (Fig. 1A and Table I). The 14484C mutationwas consistently homoplasmic in all cell types exam-ined for the proband and her mother (Fig. 1B).
OXPHOS Function
Complex I-linked (malate plus pyruvate or gluta-mate), ADP-stimulated (state III), respiration rateswere reduced in proband lymphoblast mitochondriawhen compared with the mother (25% lower), 11778Apatients (20±28%), 14484C patients (41±46%), or con-trols (48%) (Table II). Normalization of state III rateswas accomplished by dividing complex I-linked state IIIrates by complex II (succinate)-linked state III rates.The resulting state III ratios were also decreased in theproband, being 8% lower than the mother, 11% lowerthan 14484C patients, and 24% lower than controls,but were 118±128% of the 11778A patient state IIIratio. Uncoupled rates using complex I substrates wereslightly reduced in the proband, although ADP/O ratioswere not. Succinate-stimulated respiration rates and
11778�14484 mtDNA Mutations in LHON Family 333
ADP/O ratios were only slightly reduced in the probandrelative to controls.
All complex I-linked respiration values for lympho-blast mitochondria from the mother were slightly belowthose found for other 14484C patients (Table II).Complex II-linked respiration rates and ADP/O ratiosof the mother's cells were essentially equivalent tocontrol values.
The respiration defects observed in the proband'slymphoblastoid cell mitochondria were also found in
the transmitochondrial cybrid mitochondria. Hence,the severe complex I defect detected in the probandwas due to a mtDNA mutation. Speci®cally, complexI-linked state III rates were reduced in the probandcybrid mitochondria, relative to the mother (33±35% lower), 11778A patients (36%), 14484C patients(18±26%), and control (31%) cybrid mitochondria(Table II). State III ratios were also lowest in probandcybrids, being 20% lower than the mother, 6% lowerthan 11778A patients, 5±13% lower than 14484Cpatients, and 22% lower than controls. ComplexI-associated uncoupled rates were slightly lower inthe proband relative to those found in the othercomparison groups, but patient cybrid ADP/O ratioswere elevated. Proband complex II-linked respirationrates were similar to control values, although thepatient cybrids showed an increased ADP/O ratiocompared to controls.
Complex I-linked respiration parameters obtainedfrom the mother's cybrid mitochondria were similar to,or moderately higher than those of 14484C-containingcybrids (Table II). Complex II-linked respirationrates and ADP/O ratio were equivalent to control
Fig. 1. Mutation-speci®c RFLP digestion for the 11778A and 14484CLHON mutations. A: For the 11778A MaeIII digestion, mutant mtDNA isindicated by the presence of 124 and 94 bp bands. Wild-type 11778GmtDNA is indicated by a 218 bp band. B: For the 14484C BstNI digestion,mutant mtDNA is indicated by a 294 bp band. Wild-type 14484T mtDNA isindicated by a 303 bp band. lane 1: proband buffy coat mtDNA; lane 2:
proband lymphoblast mtDNA; lane 3: proband cybrid mtDNA; lane 4:mother buffy coat mtDNA; lane 5: mother lymphoblast mtDNA; lane 6:mother cybrid mtDNA. � indicates a positive control containing homo-plasmic mtDNA mutation; ÿindicates a negative or wild-type control. MWindicates molecular weight marker.
TABLE I. Heteroplasmy of 11778A Mutation
% 11778A Mutant MtDNA
Blood Lymphoblast Cybrid
Proband 94a 86 87Mother 31 < 5 < 5
aIndicates that heteroplasmy was also quantitated by cloning individualmtDNAs. In thiscase, 93% mutant (11778A) mtDNA was obtained for theproband buffy coat, in excellent agreement with the 94% obtained by ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
334 Brown et al.
values. Thus, the 14484C-positive cybrids derived fromthe mother exhibited the weak complex I respirationdefects commonly associated with the milder 14484Cmutation [Brown et al., 2000].
Analysis of OXPHOS speci®c enzyme activities alsoindicated a profound impairment of complex I electrontransfer capacity in the proband's mitochondria. Pro-band lymphoblast mitochondrial complex I activity wasreduced relative to the mother (74% reduction), 11778Apatients (72%), 14484C patients (76%), and controls(77%) (Table III). CS normalized complex I activity
was also lowest in patient lymphoblasts, being 44%lower than the mother, 17% lower than 11778A pa-tients, 29% lower than 14484C patients, and 37% lowerthan controls (Table III).
Complex I dysfunction was con®rmed in cybridmitochondria, as the proband again showed the largestreduction in complex I activity. Proband cybrid mito-chondria maintained about 50% of complex I activityrelative to cybrid mitochondria derived from themother, 11778A patients, 14484C patients, or controls(Table III). CS normalized complex I activity in the
TABLE II. Respiration Rates in Lymphoblast and Cybrid Mitochondria
Substrate Individual
Mean respiration rates (nmolO/min/mg of mitochondrial protein)
ADP/O ratio State III ratioState III Uncoupled
LymphoblastPyruvate�malate Proband 73 101 2.9 0.59
Mother 95 108 2.9 0.6411778 LHON (n�7) 91�18 115� 31 2.5� 0.2 0.50� 0.1314484 LHON (n�7) 123�51 140� 67 2.7� 0.5 0.66� 0.11Controls (n�22) 140�41 168� 60 2.8� 0.5 0.78� 0.05
Glutamate�malate Proband 76 142 2.6 0.61Mother 100 108 2.9 0.6711778 LHON (n�7) 106�24 124� 45 2.3� 0.3 0.57� 0.1314484 LHON (n�7) 141�71 159� 86 2.4� 0.4 0.73� 0.08Controls (n�22) 145�41 173� 64 2.6� 0.6 0.81� 0.11
Succinate Proband 124 190 1.6 ÐMother 149 232 1.9Controls (n�22) 179�45 247� 96 1.8� 0.3 Ð
CybridsPyruvate�malate Proband 67 84 3.3 0.59
Mother 103 110 3.2 0.7311778 LHON (n�7) 105�44 117� 61 2.9� 0.5 0.63� 0.0514484 LHON (n�7) 82�26 112� 59 3.0� 0.6 0.62� 0.15Controls (n�9) 97�20 101� 20 2.4� 0.3 0.76� 0.10
Glutamate�malate Proband 68 83 3.2 0.60Mother 110 140 2.9 0.7811778 LHON (n�7) 106�41 123� 65 2.8� 0.6 0.64� 0.0314484 LHON (n�7) 92�29 119� 73 2.9� 0.8 0.69� 0.14Controls (n�9) 99�22 102� 22 2.3� 0.4 0.77� 0.11
Succinate Proband 113 176 2.3 ÐMother 141 240 1.8Controls (n�9) 130�37 161� 50 1.5� 0.1 Ð
TABLE III. Complex I Activity in Lymphoblast and Cybrid Mitochondria
Individual
Mean speci®c activity (nmol of substrate/min/mg of mitochondrial protein)
Complex I/CSratioComplex I Citrate synthase (CS)
LymphoblastsProband 12 232 0.05Mother 47 524 0.0911778 LHON (n� 7) 43�18 690�177 0.06�0.0214484 LHON (n� 7) 50�24 701�327 0.07�0.03Controls (n� 22) 53�20 599�177 0.08�0.02
CybridsProband 23 349 0.07Mother 48 424 0.1111778 LHON (n� 7) 46�20 565�203 0.09�0.0414484 LHON (n� 7) 61�19 568�198 0.10�0.04Controls (n� 9) 44�14 471�176 0.10�0.03
11778�14484 mtDNA Mutations in LHON Family 335

proband cybrid mitochondria was also 36% lower thanthe mother, 22% lower than 11778A patients, and 30%lower than 14484C patients or controls (Table III).
DISCUSSION
The buffy coat mtDNA from the proband described inthis report was essentially homoplasmic in white bloodcells for both the 11778A and the 14484C primaryLHON mtDNA mutations. Only one other LHON casehas been reported with two primary LHON mutations[Riordan-Eva et al., 1995]. The proband in that studywas a 19-year-old singleton male patient harboring100% of the 11778A and 70% of the 14484C mutations[Riordan-Eva et al., 1995]. He had no affected familymembers, although his mother was also homoplasmicfor the 11778A mutation and heteroplasmic for the14484C mutation [Riordan-Eva, personal communica-tion]. In this patient, vision loss was sequential over atwo month period, such that best corrected visualacuity was hand motion O.D., count ®ngers O.S. Thepatient was followed for 1.5 years, with no signi®cantimprovement [Riordan-Eva, personal communication].Our patient is therefore unique in the literature in thather mtDNA contains two essentially homoplasmic andpathogenic mutations.
The proband reported here is additionally notable inthat she is a female singleton and that the age of onsetwas nine years. Typically, more than 80% of 11778Aand 14484C LHON patients are male and the mean ageof onset associated with both mutations is in the earlyto mid 20s [Newman et al., 1991; Mackey and Howell,1992; Johns et al., 1993; Riordan-Eva et al., 1995].Moreover, the proband experienced spontaneous visualrecovery with improved visual acuity seven monthsafter the initial diagnosis of LHON. Vision recovery israre in 11778A patients, but has been reported inroughly 60% of 14484C patients, with early age ofonset predictive of a high probability of visual recovery[Riordan-Eva et al., 1995]. It is possible that thepresence of both primary mutations precipitated theearly onset of blindness in our proband, but herage and/or gender may have limited the severity ofthe OXPHOS defect and permitted visual recovery.
There is a general correlation between the genotypeand phenotype in this family in that the affecteddaughter carried two deleterious mutations and had apronounced complex I de®cit, while her unaffectedmother had a lower mutational burden associated witha subtle disruption of complex I. However, while theproband's bioenergetic defect may have accounted forthe early age of onset, it is harder to reconcile with thelack of additional clinical signs and the surprisingspontaneous vision improvement. One possible expla-nation is that the blood mtDNA genotype was differentthan that found in ophthalmic tissue, since we do notknow the actual genotypic ratios in the optic nerve/retina of the proband, and hence, the homoplasmymeasured in blood may be irrelevant to disease ex-pression. Nevertheless, pathogenic mtDNA proportionsare most often lower in blood than those foundin affected tissues. For example, Howell et al. [1994]
analyzed postmortem tissue from an 11778A LHONpatient and found that while the patient contained 33%mutant mtDNA in the white blood cells, near homo-plasmic mutant levels were found in the optic nerve,retina, sclera, and muscle tissue [Howell et al., 1994].More recent work suggests that high 11778A mutantproportions in the blood predicts a similarly high mu-tant frequency in the retina and optic nerve [Juvonenet al., 1997; Chinnery et al., 2001]. Thus, given the veryhigh frequency of both the 11778A and the 14484Cmutation in the proband's white blood cells, it is likelythat she had comparable levels in her optic nerve,retina, and other tissues.
The functional consequences of two coincident pri-mary LHON mutations was assessed by respirationand enzyme studies using lymphoblasts and trans-mitochondrial cybrids derived from the proband andher mother. The 11778A mutation was maintained athigh levels in the proband lymphoblasts and cybridsbut was lost in the mother's transformed cells, provid-ing the opportunity to study the impact of the 11778A�14484C genotype on complex I function. Two observa-tions can be made. First, the complex I dysfunctionfound in proband mitochondria was greater than eitherthe 11778A or the 14484C mutation imparts indepen-dently, indicating an additive and deleterious interac-tion between these two LHON mutations. This wasmost easily observed in the cybrid data since all con-structs (proband, mother, 11778A patients, 14484Cpatients, and control cybrids) share the same Wal2Anuclear background. From respiration studies, probandcybrid mitochondria state III rates and succinate-normalized state III ratios were 35% and 6% lowerthan 11778A patients, respectively. Curiously, ADP/Oratios were increased in proband cybrid mitochondria,potentially indicating a compensatory mechanism bythe cell. From enzymological analysis, absolute and CS-normalized complex I speci®c activity was diminish-ed by 50% and 22%, respectively, in proband cybridmitochondria relative to 11778A cybrids. Thus, by bothfunctional parameters studied, the 11778A and 14484Cmutations interact synergistically to impart a partial,yet prominent, complex I defect in the proband.
Second, a comparison of complex I function betweenthe 11778A�14484C-containing proband cybrid mito-chondria and the 14484C-containing cybrid mitochon-dria from her mother allowed an assessment of theseverity of the complex I dysfunction associated withthe 11778 mutation [Brown et al., 2000]. Cybridsderived from the proband and her mother shared thesame nucleus and mitochondria. Accordingly, they areisogenic with the exception that the proband cybridmitochondria contained a very high percentage of the11778A mutation, while the mother's cybrid mitochon-dria contained very little of this mutation. The complexI-linked state III respiration rate and state III ratio inproband cybrid mitochondria were 35% and 20% lowerthan those found in the mother's cybrid, respectively.Proband cybrid uncoupled rates were also 20±40%lower than the mother's cybrid, potentially signalinga more generalized respiration defect associated withthe 11778A� 14484C genotype. ADP/O ratios were not
336 Brown et al.

appreciably different between proband and mother.Absolute and CS-normalized complex I speci®c activ-ities were 52% and 36% lower in proband cybrids thanin the mother's cybrids, respectively. Thus, in theWal2A nDNA/14484C mtDNA background, the 11778Amutation results in a �20% reduction in ADP-stimu-lated oxygen consumption and a �36% reduction incomplex I speci®c enzyme activity, indicating the mag-nitude of complex I impairment resulting from thismutation.
Although the 11778A� 14484C genotype results inan unequivocal complex I defect, the pathophysiologicalmechanism by which these mutations result in LHONremains unclear. Both the ND4 (11778A mutation) andthe ND6 (14484C mutation) polypeptides are compo-nents of the hydrophobic, membrane-bound domain ofcomplex I, but their speci®c function and protein-protein interactions are unresolved. If the 11778A and14484C mutations inhibit electron transport, a reduc-tion in ATP production might be expected. However, ifthis were the mechanism for LHON expression, thenthe two mutations should be additive, yet the pheno-type in our 11778A�14484C patient was the same aseach mutation confers independently and the patienthad undergone vision recovery. Alternatively, themutations could have an indirect, yet tissue-speci®c,effect, such as increasing complex I-mediated reactiveoxygen species (ROS) generation [Howell, 1997; Brown,1999]. In this case, the phenotype of the double mutantmight be the same as a single primary LHON mutation,but with an earlier age of onset (as seen in the proband).Such a mechanism does not necessarily require directinteraction between the ND4 and ND6 polypeptides,since ROS production could occur independently byimpairment of complex I at a variety of sites in theenzyme.
In summary, this report documents the ®rst com-prehensive clinical, genetic, and biochemical charac-terization of a patient containing two homoplasmicpathogenic mtDNA mutations. A pronounced complexI defect stands in conceptual contrast to a slightlyatypical LHON presentation and signi®cant visualrecovery observed in the proband, underscoring thecomplexity of genotype-phenotype dynamics in LHON,including the exquisite tissue speci®city associatedwith the most common LHON mutations.
ACKNOWLEDGMENTS
MDB and DCW were recipients of grants from theNational Institutes of Health. Dr. Newman is a reci-pient of a Research to Prevent Blindness Lew R.Wasserman Merit Award.
REFERENCES
Brown MD. 1999. The enigmatic relationship between mitochondrialdysfunction and Leber's hereditary optic neuropathy [editorial; com-ment]. J Neurol Sci 165:1±5.
Brown MD, Wallace DC. 1994. Spectrum of mitochondrial DNA mutationsin Leber's hereditary optic neuropathy. Clin Neurosci 2:138±145.
Brown MD, Torroni A, Reckord CL, Wallace DC. 1995. Phylogeneticanalysis of Leber's hereditary optic neuropathy mitochondrial DNAs
indicates multiple independent occurrences of the common mutations.Hum Mut 6:311±325.
Brown MD, Trounce I, Jun AS, Allen JC, Wallace DC. 2000. Functionalanalysis of lymphoblast and cybrid mitochondria containing the 3460,11778, or 14484 Leber's hereditary optic neuropathy mtDNA mutation.J Biol Chem 275:39831±39836.
Brown MD, Zhadanov S, Allen JC, Hosseini SH, Newman NJ, AtamonovVV, Mikhailovskaya IE, Sukernik RI, Wallace DC. 2001. Novel mtDNAmutations and oxidative phosphorylation dysfunction in RussianLHON families. Hum Genetin press.
Chinnery PF, Andrews RM, Turnbull DM, Howell NN. 2001. Leberhereditary optic neuropathy: does heteroplasmy in¯uence the inheri-tance and expression of the G11778A mitochondrial DNA mutation?Am J Med Genet 98:235±243.
Harding AE, Riordan-Eva P, Govan GG. 1995. Mitochondrial DNAdiseases: genotype and phenotype in Leber's hereditary optic neuro-pathy. Muscle Nerve 3:S82±S84.
Howell N. 1997. Leber hereditary optic neuropathy: how do mitochondrialDNA mutations cause degeneration of the optic nerve? J BioenergBiomembr 29:165±173.
Howell N. 1998. Leber hereditary optic neuropathy: respiratory chaindysfunction and degeneration of the optic nerve. Vis Res 38:1495±1504.
Howell N, Bindoff LA, McCullough DA, Kubacka I, Poulton J, Mackey D,Taylor L, Turnbull DM. 1991. Leber hereditary optic neuropathy:identi®cation of the same mitochondrial ND1 mutation in six pedigrees.Am J Hum Genet 49:939±950.
Howell N, Xu M, Halvorson S, Bodis-Wollner I, Sherman J. 1994. Aheteroplasmic LHON family: tissue distribution and transmission ofthe 11778 mutation [letter]. Am J Hum Genet 55:203±206.
Howell N, Kubacka I, Halvorson S, Howell B, McCullough DA, Mackey D.1995. Phylogenetic analysis of the mitochondrial genomes from Leberhereditary optic neuropathy pedigrees. Genetics 140:285±302.
Huoponen K, Vilkki J, Aula P, Nikoskelainen EK, Savontaus ML. 1991. Anew mtDNA mutation associated with Leber hereditary optic neuro-retinopathy. Am J Hum Genet 48:1147±1153.
Johns DR, Neufeld MJ, Park RD. 1992. An ND-6 mitochondrial DNAmutation associated with Leber hereditary optic neuropathy. BiochemBiophys Res Comm 187:1551±1557.
Johns DR, Heher KL, Miller NR, Smith KH. 1993. Leber's hereditary opticneuropathy. Clinical manifestations of the 14484 mutation. ArchOphthal 111:495±498.
Jun AS, Brown MD, Wallace DC. 1994. A mitochondrial DNA mutation atnp 14459 of the ND6 gene associated with maternally inherited Leber'shereditary optic neuropathy and dystonia. Proc Natl Acad Sci USA91:6206±6210.
Jun AS, Trounce IA, Brown MD, Shoffner JM, Wallace DC. 1996. Use oftransmitochondrial cybrids to assign a complex I defect to themitochondrial DNA-encoded NADH dehydrogenase subunit 6 genemutation at nucleotide pair 14459 that causes Leber hereditary opticneuropathy and dystonia. Mol Cell Biol 16:771±777.
Juvonen V, Nikoskelainen E, Lamminen T, Penttinen M, Aula P,Savontaus ML. 1997. Tissue distribution of the ND4/11778 mutationin heteroplasmic lineages with Leber hereditary optic neuropathy.Hum Mut 9:412±417.
Mackey D, Howell N. 1992. A variant of Leber hereditary optic neuropathycharacterized by recovery of vision and by an unusual mitochondrialgenetic etiology. Am J Hum Genet 51:1218±1228.
Newman NJ, Lott MT, Wallace DC. 1991. The clinical characteristics ofpedigrees of Leber's hereditary optic neuropathy with the 11778mutation. Am J Ophthalmol 111:750±762.
Riordan-Eva P, Sanders MD, Govan GG, Sweeney MG, Da Costa J,Harding AE. 1995. The clinical features of Leber's hereditary opticneuropathy de®ned by the presence of a pathogenic mitochondrial DNAmutation. Brain 118:319±337.
Shoffner JM, Brown MD, Stugard C, Jun AS, Pollok S, Haas RH, KaufmanA, Wallace DC. 1995. Leber's hereditary optic neuropathy plus dystoniais caused by a mitochondrial DNA point mutation in a complex Isubunit. Ann Neurol 38:163±169.
Trounce IA, Kim YL, Jun AS, Wallace DC. 1996. Assessment ofmitochondrial oxidative phosphorylation in patient muscle biopsies,lymphoblasts, and transmitochondrial cell lines. Meth Enzymol264:484±509.
11778�14484 mtDNA Mutations in LHON Family 337

Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, ElsasLJ, Nikoskelainen EK. 1988. Mitochondrial DNA mutation asso-ciated with Leber's hereditary optic neuropathy. Science 242:1427±1430.
Wallace DC, Stugard C, Murdock D, Schurr T, Brown MD. 1997. AncientmtDNA sequences in the human nuclear genome: a potential source of
errors in identifying pathogenic mutations. Proc Natl Acad Sci USA94:14900±14905.
Wallace DC, Lott MT, Brown MD, Kerstann K. 2001. Mitochondria andneuro-ophthalmological diseases. In: Scriver CR, Beaudet AL, Sly WS,Valle D, editors. The metabolic and molecular basis of inherited disease,vol. II. New York: McGraw-Hill. p 2425±2509.
338 Brown et al.





























