Embed Size (px)
Citation preview

Clinical Trial Readiness Checklist October 2014
This checklist was developed by the Huntington Society of Canada in partnership with Rx&D, Canada’s Research-Based
Pharmaceutical Companies. While developed to assist the broader research community, this checklist can be amended to reflect
the needs of disease-specific clinical study requirements. It will be updated as required. Page 1
The clinical trial readiness checklist is a tool which can help new and experienced
researchers prepare for upcoming clinical studies. It is designed to be a quick
reference to ensure that you have the basic requirements for conducting clinical
studies, however does not replace the sponsor protocol requirements, Health
Canada/FDA regulations or Research Ethics Board considerations.
HOW IT WORKS:
REQUIRED: The items listed under the REQUIRED category are the basic items needed
for the conduct of any clinical study. Sites should ensure that they have required staff,
space and equipment, training and resources to successfully conduct the study.
RECOMMENDED/OPTIONAL: The items listed under the RECOMMENDED/OPTIONAL
category are protocol and site specific. Not all items will be necessary for you to conduct
a clinical study, so simply check off the ones that you have and make a note to speak to
the sponsor about additional items required for the protocol.
PROCESS OVERVIEW: This section provides a glimpse into the documents, activities and
considerations required in the process of conducting a clinical study.
FUTURE WORK:
This toolkit should be reviewed and updated annually, or at least, as guidance documents,
federal regulations and ethical considerations are updated. This is meant to be a dynamic
document which can be tailored for different clinical trials related to different therapeutic
areas.

Clinical Trial Readiness Checklist October 2014
This checklist was developed by the Huntington Society of Canada in partnership with Rx&D, Canada’s Research-Based
Pharmaceutical Companies. While developed to assist the broader research community, this checklist can be amended to reflect
the needs of disease-specific clinical study requirements. It will be updated as required. Page 2
Staffing Resources
Required Recommended/Optional Principal Investigator, MD (Medical Degree is
protocol dependent)
Optimal to have experience in clinical trials, as s/he ultimately must take personal responsibility for all aspects of study and the delegation of tasks. Optimal to be on-site whenever study subjects are being assessed.
Co-Investigator or Back up Investigator
This individual must understand the protocol, and should be available to fill in for the PI when s/he is away on leave, vacation or becomes ill and cannot continue the PI role.
Study Coordinator
This is usually a clinically trained individual with specific training in the conduct of clinical trials. It is most often a nurse. This person is often involved in all aspects of the study including ethics submission, all pre-study set up (lab draws, ECG, pharmacy , etc.) as well as actual study related activities which may include blood draw, ECGs and some of the rating scales (including cognitive testing), therefore experience is required.
An understanding and appreciation of the unique challenges in dealing with HD is important.
Research Manager
This applies to sites that are likely to have many active studies. This individual will provide project management and oversee the conduct of the research program. Experience in clinical trial design and conduct are an asset. Manager will oversee start-up of projects, negotiation of contracts, ethics committee submissions, protocol compliance, interaction with the monitor, and other activities throughout the course of the study and closeout.
Independent Rater
Some studies require an independent rater blinded for study procedures. This person can be a physician, however this varies according with the protocol and what is being rated.
Research Assistants/Associate
Research assistants/associates assist with the setup of a research visits, may help with lab samples with spinning blood and shipping to labs, as well as data entry. Research assistants/associates may be trained in post-graduate specialized programs focusing on clinical trials but are usually not nurses or other allied health professionals.
Phlebotomist /ECG Technician
Ability to draw, prepare and ship blood and other biological specimen samples

Clinical Trial Readiness Checklist October 2014
This checklist was developed by the Huntington Society of Canada in partnership with Rx&D, Canada’s Research-Based
Pharmaceutical Companies. While developed to assist the broader research community, this checklist can be amended to reflect
the needs of disease-specific clinical study requirements. It will be updated as required. Page 3
Space and Equipment
Required The equipment needed is protocol specific
and may be on-site or within close proximity
Physical facility to conduct studies including
sufficient space to assess patients, as per protocol
Freezer (-20°C)
Freezer (-70°C)
Fridge (2-8°C) Centrifuge Annual calibration of specific equipment
ECG
Ultrasound or MRI
X-ray if required
Internet (wireless) access IVRS experience EDC experience Experience with electronic patient diaries
Room for coordinator/monitor to work
Secure controlled area with limited access and locked for investigational new drug (IND) or investigational product (IP), with appropriate air temperature and humidity conditions that require daily monitoring and documentation.
Internet access with appropriate security measures.
Ability to provide access to EMR or clinical records for visits and sponsor monitors
Ability to store study related material for the required 25-year period (either on site or via third party)
Clinical Trial Readiness Checklist October 2014
This checklist was developed by the Huntington Society of Canada in partnership with Rx&D, Canada’s Research-Based
Pharmaceutical Companies. While developed to assist the broader research community, this checklist can be amended to reflect
the needs of disease-specific clinical study requirements. It will be updated as required. Page 4
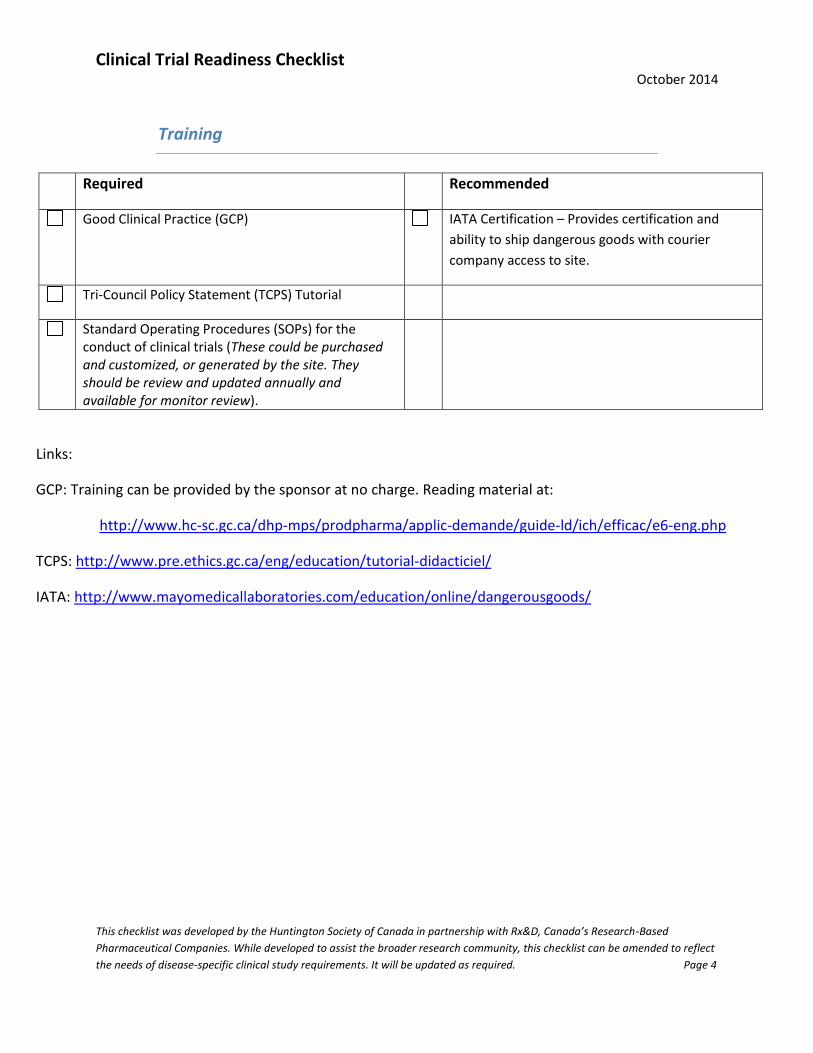
Training
Required Recommended
Good Clinical Practice (GCP) IATA Certification – Provides certification and
ability to ship dangerous goods with courier
company access to site.
Tri-Council Policy Statement (TCPS) Tutorial
Standard Operating Procedures (SOPs) for the conduct of clinical trials (These could be purchased and customized, or generated by the site. They should be review and updated annually and available for monitor review).
Links:
GCP: Training can be provided by the sponsor at no charge. Reading material at:
http://www.hc-sc.gc.ca/dhp-mps/prodpharma/applic-demande/guide-ld/ich/efficac/e6-eng.php
TCPS: http://www.pre.ethics.gc.ca/eng/education/tutorial-didacticiel/
IATA: http://www.mayomedicallaboratories.com/education/online/dangerousgoods/
Clinical Trial Readiness Checklist October 2014
This checklist was developed by the Huntington Society of Canada in partnership with Rx&D, Canada’s Research-Based
Pharmaceutical Companies. While developed to assist the broader research community, this checklist can be amended to reflect
the needs of disease-specific clinical study requirements. It will be updated as required. Page 5
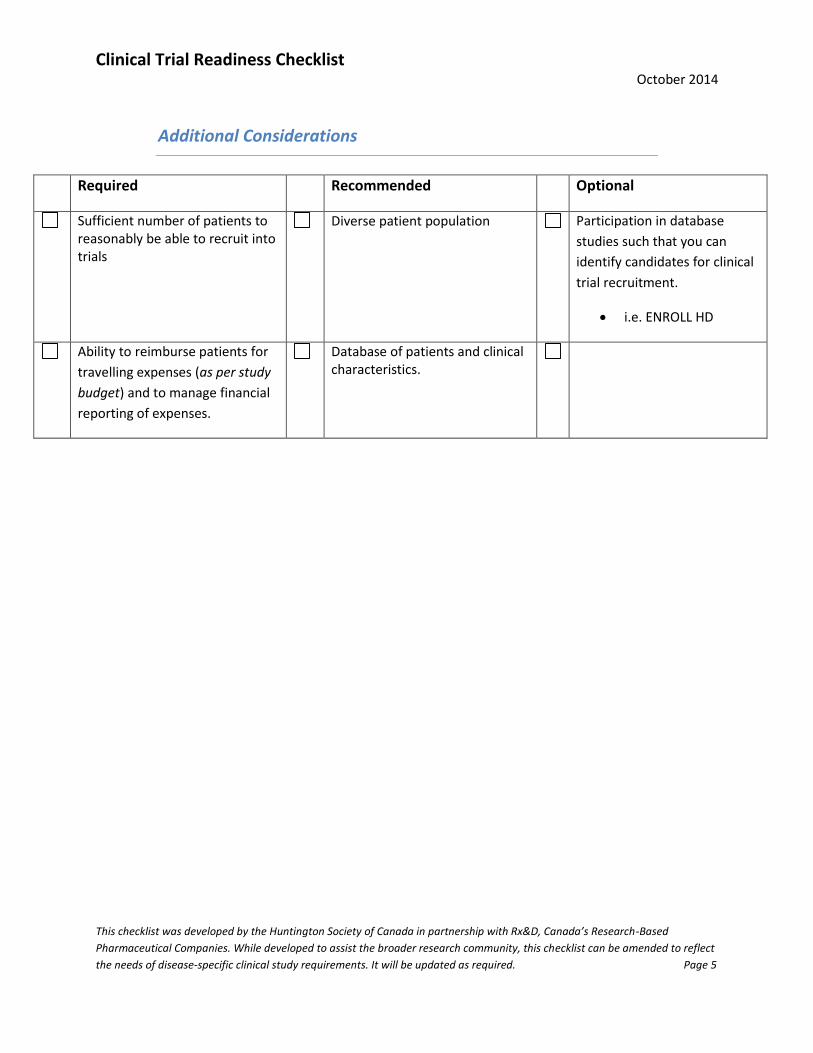
Additional Considerations
Required Recommended Optional
Sufficient number of patients to reasonably be able to recruit into trials
Diverse patient population Participation in database
studies such that you can
identify candidates for clinical
trial recruitment.
i.e. ENROLL HD
Ability to reimburse patients for
travelling expenses (as per study
budget) and to manage financial
reporting of expenses.
Database of patients and clinical characteristics.
Clinical Trial Readiness Checklist October 2014
This checklist was developed by the Huntington Society of Canada in partnership with Rx&D, Canada’s Research-Based
Pharmaceutical Companies. While developed to assist the broader research community, this checklist can be amended to reflect
the needs of disease-specific clinical study requirements. It will be updated as required. Page 1
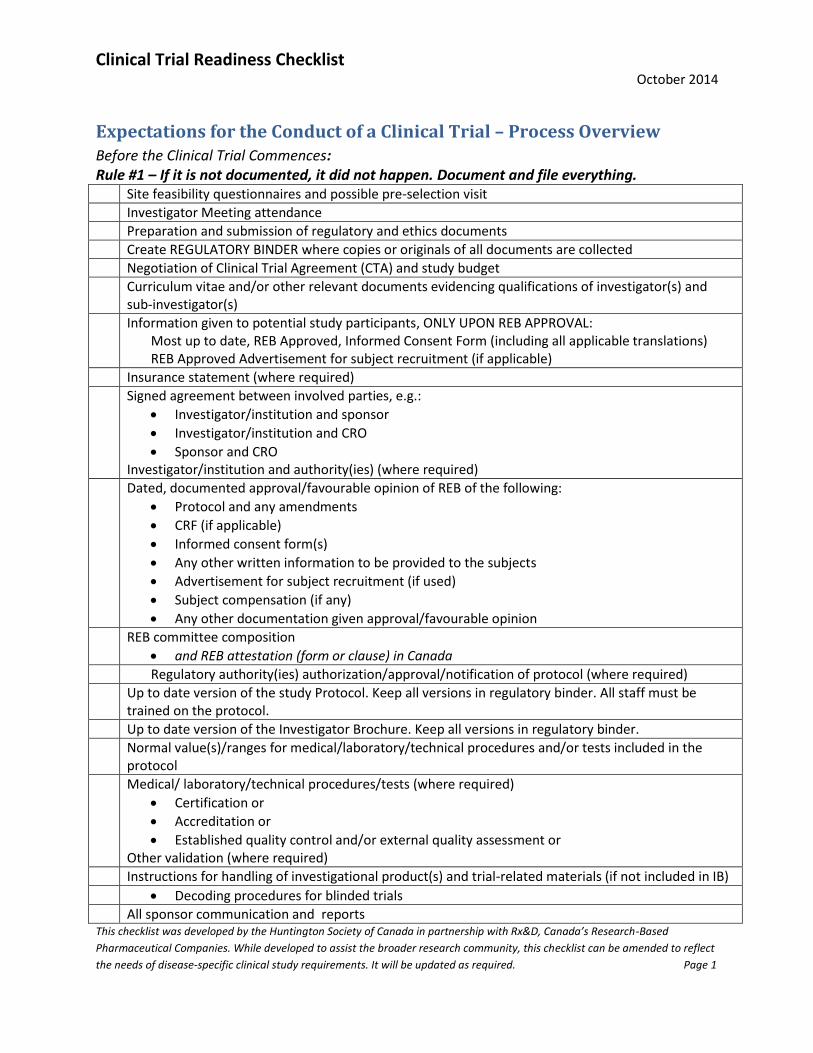
Expectations for the Conduct of a Clinical Trial – Process Overview Before the Clinical Trial Commences: Rule #1 – If it is not documented, it did not happen. Document and file everything. Site feasibility questionnaires and possible pre-selection visit
Investigator Meeting attendance
Preparation and submission of regulatory and ethics documents
Create REGULATORY BINDER where copies or originals of all documents are collected
Negotiation of Clinical Trial Agreement (CTA) and study budget
Curriculum vitae and/or other relevant documents evidencing qualifications of investigator(s) and sub-investigator(s)
Information given to potential study participants, ONLY UPON REB APPROVAL: Most up to date, REB Approved, Informed Consent Form (including all applicable translations) REB Approved Advertisement for subject recruitment (if applicable)
Insurance statement (where required)
Signed agreement between involved parties, e.g.:
Investigator/institution and sponsor
Investigator/institution and CRO
Sponsor and CRO Investigator/institution and authority(ies) (where required)
Dated, documented approval/favourable opinion of REB of the following:
Protocol and any amendments
CRF (if applicable)
Informed consent form(s)
Any other written information to be provided to the subjects
Advertisement for subject recruitment (if used)
Subject compensation (if any)
Any other documentation given approval/favourable opinion
REB committee composition
and REB attestation (form or clause) in Canada
Regulatory authority(ies) authorization/approval/notification of protocol (where required)
Up to date version of the study Protocol. Keep all versions in regulatory binder. All staff must be trained on the protocol.
Up to date version of the Investigator Brochure. Keep all versions in regulatory binder.
Normal value(s)/ranges for medical/laboratory/technical procedures and/or tests included in the protocol
Medical/ laboratory/technical procedures/tests (where required)
Certification or
Accreditation or
Established quality control and/or external quality assessment or Other validation (where required)
Instructions for handling of investigational product(s) and trial-related materials (if not included in IB)
Decoding procedures for blinded trials
All sponsor communication and reports
Clinical Trial Readiness Checklist
This checklist was developed by the Huntington Society of Canada in partnership with Rx&D, Canada’s Research-Based
Pharmaceutical Companies. While developed to assist the broader research community, this checklist can be amended to reflect
the needs of disease-specific clinical study requirements. It will be updated as required. Page 2
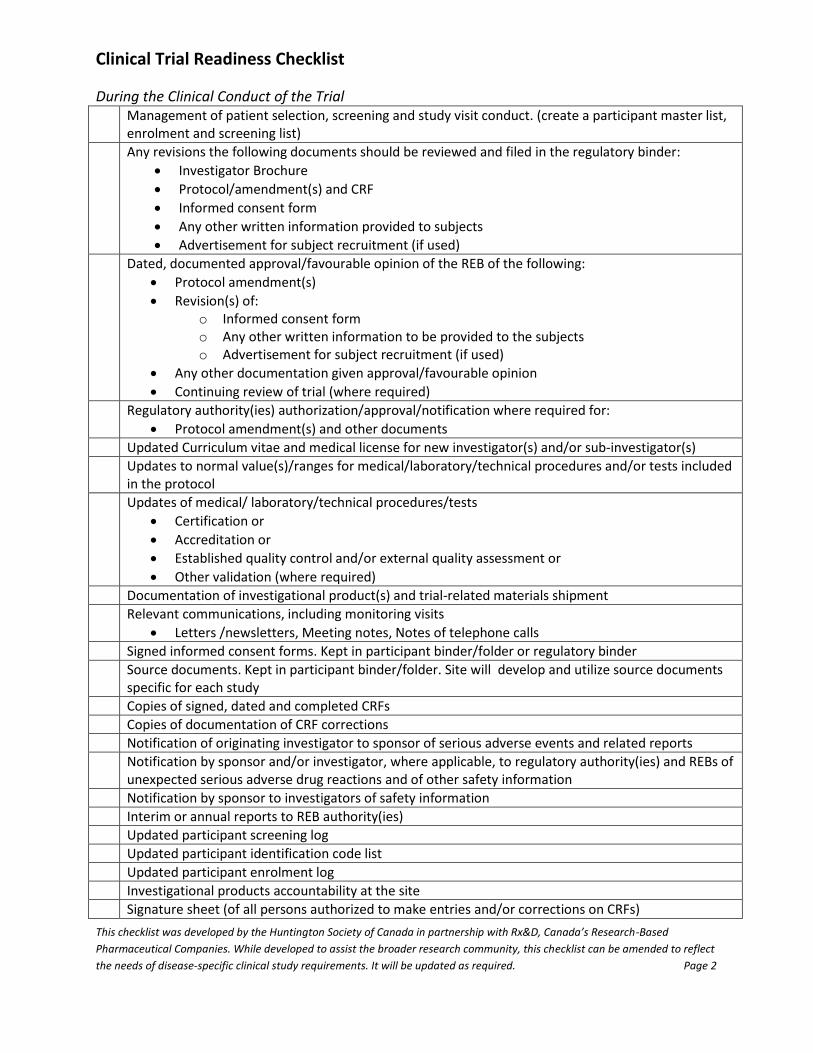
During the Clinical Conduct of the Trial Management of patient selection, screening and study visit conduct. (create a participant master list,
enrolment and screening list)
Any revisions the following documents should be reviewed and filed in the regulatory binder:
Investigator Brochure
Protocol/amendment(s) and CRF
Informed consent form
Any other written information provided to subjects
Advertisement for subject recruitment (if used)
Dated, documented approval/favourable opinion of the REB of the following:
Protocol amendment(s)
Revision(s) of: o Informed consent form o Any other written information to be provided to the subjects o Advertisement for subject recruitment (if used)
Any other documentation given approval/favourable opinion
Continuing review of trial (where required)
Regulatory authority(ies) authorization/approval/notification where required for:
Protocol amendment(s) and other documents
Updated Curriculum vitae and medical license for new investigator(s) and/or sub-investigator(s)
Updates to normal value(s)/ranges for medical/laboratory/technical procedures and/or tests included in the protocol
Updates of medical/ laboratory/technical procedures/tests
Certification or
Accreditation or
Established quality control and/or external quality assessment or
Other validation (where required)
Documentation of investigational product(s) and trial-related materials shipment
Relevant communications, including monitoring visits
Letters /newsletters, Meeting notes, Notes of telephone calls
Signed informed consent forms. Kept in participant binder/folder or regulatory binder
Source documents. Kept in participant binder/folder. Site will develop and utilize source documents specific for each study
Copies of signed, dated and completed CRFs
Copies of documentation of CRF corrections
Notification of originating investigator to sponsor of serious adverse events and related reports
Notification by sponsor and/or investigator, where applicable, to regulatory authority(ies) and REBs of unexpected serious adverse drug reactions and of other safety information
Notification by sponsor to investigators of safety information
Interim or annual reports to REB authority(ies)
Updated participant screening log
Updated participant identification code list
Updated participant enrolment log
Investigational products accountability at the site
Signature sheet (of all persons authorized to make entries and/or corrections on CRFs)

Clinical Trial Readiness Checklist
This checklist was developed by the Huntington Society of Canada in partnership with Rx&D, Canada’s Research-Based
Pharmaceutical Companies. While developed to assist the broader research community, this checklist can be amended to reflect
the needs of disease-specific clinical study requirements. It will be updated as required. Page 3
Record of retained body fluids/tissue samples (if any)
After Completion or Termination of the Trial Reconcile Investigational product(s) accountability at the site
Documentation of Investigational product destruction
Completed subject identification code list
Final report by investigator to REB where required, and where applicable, to the regulatory authority(ies)
Clinical study report (if applicable)
Measurement of Success a. Reasonable time to start up of study including ethics approvals and contract negotiation. b. Protocol deviations should be held to a minimum. c. Few screening failures, recruited subjects meeting all inclusion/exclusion criteria. d. Reporting to Ethics Committee and sponsor within timelines identified. e. Meeting targets for recruitment. f. Quality data including data entry and source record validation. g. Excellent retention of subjects within study. h. Site protection of subject confidentiality.





























