Embed Size (px)
Citation preview

UNIVERSITÀ DI MILANO “CENTRO DINO FERRARI”
PER LA DIAGNOSI E LA TERAPIA DELLE MALATTIE NEUROMUSCOLARI E NEURODEGENERATIVE
FONDAZIONE I.R.C.C.S. CA’ GRANDA OSPEDALE MAGGIORE POLICLINICO
FONDAZIONE DI RICOVERO E CURA A CARATTERE SCIENTIFICO DI NATURA PUBBLICA
DIRETTORE PROF. NEREO BRESOLIN
Via F. Sforza, 35 - 20122 Milano – Tel. 02.5503.3809 - Fax 02.5503.3800 E-mail: [email protected] - [email protected] - www.centrodinoferrari.com
COLLABORAZIONI INTERNAZIONALI E
FRONTESPIZI
LAVORI SCIENTIFICI
2016
“ CENTRO DINO FERRARI”
Sezione di Neuroscienze Dipartimento di Fisiopatologia Medico-Chirurgica e dei Trapianti
Università degli Studi di Milano Fondazione I.R.C.C.S. Ca’ Granda - Ospedale Maggiore Policlinico

-2-
UNIVERSITÀ DI MILANO “CENTRO DINO FERRARI”
PER LA DIAGNOSI E LA TERAPIA DELLE MALATTIE NEUROMUSCOLARI E NEURODEGENERATIVE
OSPEDALE MAGGIORE POLICLINICO MANGIAGALLI E REGINA ELENA
FONDAZIONE DI RICOVERO E CURA A CARATTERE SCIENTIFICO DI NATURA PUBBLICA
Istituti di ricerca del “Centro Dino Ferrari”: • IRCCS FONDAZIONE CA' GRANDA OSPEDALE MAGGIORE POLIC LINICO UNIVERSITA’ DEGLI STUDI DI MILANO
• Laboratorio di Biochimica e Genetica • Laboratorio di Cellule Staminali Neurali • Gruppo di Ricerca Disordini del Movimento • Unità Valutativa Alzheimer (U.V.A.) • Centro Sclerosi Multipla • Laboratorio Cellule Staminali • U.O.D. Malattie Neuromuscolari e Rare
• IRCCS ISTITUTO AUXOLOGICO ITALIANO – UNIVERSITA’ DE GLI STUDI DI MILANO
• U.O. Neurologia – Stroke Unit • Laboratorio di Neuroscienze
• IRCCS E. MEDEA
• Laboratorio di Biologia Molecolare, Citogenetica, Analisi Biochimico-Cliniche, Bioinformatica

-3-
Centri Internazionali di Ricerca che collaborano con il “Centro Dino Ferrari”
� Prof. S. Przedborski Motor Neuron Center, Columbia University, Columbia University of New York – USA
� Prof. B. Kaspar - Ohio State University, USA.
� Prof. E. Hedlund - Karolinska Institutet, Stockholm, SWEDEN.
� Prof. S. Di Mauro - Columbia University of New York – USA.
� Prof. L. Stefanis Biomedical Foundation – Academy of Athens, Grecia.
� Dr. Marc Ruepp and Prof. Müehlemann O, University of Bern.
� Prof. H. Moulton Oregon University, USA
� Dr. Enrico Bertini, IRCCS Ospedale Bambino Gesu', Roma 13
� Dr. Franco Pagani, International Centre for Genetic Engineering and Biotechnology (ICGEB), Trieste
� Prof. Luca Imeri, Dipartimento di Fisiologia Umana, Università degli Studi di Milano, Milano
� Dr. Uberto Pozzoli, IRCCS E. Medea Bosisio, Parini, Italy
� Prof. ML Gelmi, Dipartimento di Scienze Farmaceutiche, Università degli Studi di Milano.
� Prof. Cristina Guardia - Laguarta, USA Columbia University New York, N.Y., USA
� Prof. S. Przedborski, , USA Columbia University New York, N.Y., USA
� Prof. D. Re, USA Columbia University New York, N.Y., USA
� Prof. Catarina Quinzii, PhD, Columbia University, New York, N.Y., USA
� Dr. Daniel Claassen il Vanderbilt University Medical Center, Nashville US.
� Prof. Kyproula Christodoulou, The Cyprus Institute of Neurology and Genetics, Cyprus School of Molecular Medicine
� Prof. Bearne Udd - Tampere University, FINLANDIA
� Prof. Martin Rossor - UCL School of Life and Medical Sciences, London, UNITED KINGDOM.
� Prof. Bengt Winblad, Karolinska Institutet, Stockholm, SWEDEN.
� Prof. An Goris, University of Leuven, Belgium
� Dr. Luis Garcia – Institut de Myologie, Université Pierre et Marie Curie, Paris, FRANCE.

-4-
� Prof. Camillo Ricordi, University of Miami (UM), Miami, Florida
� Prof. Giulio Cossu, Institute of Infalmmation and repair, University of Manchester, Manchester, UK
� Prof. Pura Muñoz Cánoves, Department of Experimental and Life Sciences, Pompeu Fabra University, Barcelona, Spain
� Prof. Jacques Tremblay, Centre de recherche, Centre hospitalier de l’Université de Montréal, (CRCHUM), Montréal, Québec, Canada
� Dr. Joao Carlos da Silva Bizario – Muscular Dystrophy Research Center AADM/UNAERP, Ribeirao Preto, SP, BRAZIL.
� Prof. Humberto Cerrel Bazo, Dipartimento Medicina riabilitativa AUSL Piacenza
� Prof. Adolfo Lopez de Munain Arregui, Grupo Nerogenética, Hospital Donostia-Unidad Experimental San Sebastian, Espana
� Prof. Kay Davies, Department of Physiology, Anatomy and Genetics, University of Oxford, Oxford, UK
� Prof. Maurilio Sampaolesi, Stem Cell Research Institute, University Hospital Gasthuisberg, Leuven, Belgium, Human Anatomy Section, University of Pavia, Pavia, Italy, Interuniversity Institute of Myology (IIM), Italy
� Prof. Gillian Butler-Brown and Vincent Mouly, Institut de Myologie, Institut national de la sante´ et de la recherche me´ dicale, and L’Universite´ Pierre et Marie Curie Paris, Paris, France
� Prof. Giuseppe Perale, I.B.I. S/A, Svizzera, Dipartimento di Chimica, Materiali e Ingegneria Chimica "Giulio Natta" Sezione Chimica Fisica Applicata, Politecnico di Milano, Milano
� Prof. Roberto Maggi, Facoltà di Farmacia Università degli Studi di Milano
� Prof. Mario Pellegrino, Dipartimento di Ricerca Traslazionale e delle Nuove Tecnologie in Medicina e Chirurgia, Università di Pisa
� Prof. Daniele Cusi, Università degli Studi di Milano
� Dr. Bernard Brais – McGill University/Montreal Neurological Hospital and Institute, Montreal, Quebec, CANADA
� Dr. Gorka Orive – BTI Biotechnology Institute, Vitoria-Gasteiz, Alava, SPAIN
� Dr. Diana Nordling – Cincinnati Children’s Hospital, Cincinnati, OH, USA
� Prof. Fulvio Mavilio – Institut Genethon, Evry, FRANCE.
� Prof. Guglielmo Foffani, Hospital Nacional Parapléjicos, Toledo, SPAIN.
� Prof. John Rothwell, Sobell Department, UCL Institute of Neurology, London, UNITED KINGDOM.
� Prof. Elena Moro, Department of Psychiatry and Neurology , University Hospital Center of Grenoble, FRANCE.
� Dr. Andre Brunoni, Department of Neurosciences and Behavior, Institute of Psychology, University of Sao Paulo, Sao Paulo, BRAZIL.

-5-
� Prof. Dale J. Lange, M.D. Chair, Department of Neurology Neurologist-in-Chief, Hospital For Special Surgery - New York, USA.
� Prof. Hiroshi Mitsumoto, MD, DSc Head, Eleanor and Lou Gehrig MDA/ALS Research Center, Department of Neurology, Columbia University Medical Center, New York, USA.
� Prof. John E. Landers, - University of Massachusetts Medical School, Worcester, MA, USA.
� Prof. Merit E. Cudkowicz, MD, MSc - Neuromuscular Division Neurology - Massachusetts General Hospital Wang Ambulatory Care Center - Boston, MA, USA
� Prof. Robert H. Brown, Jr., D.Phil., M.D. - Chair, Department of Neurology, UMass Medical School Worcester, USA.
� Prof. Stanley H. Appel MD, Edwards Distinguished Endowed Chair for ALS Director, Methodist Neurological Institute Chair, Department of Neurology - Houston, Texas, USA.
� Prof. Rosa Rademakers - Mayo Clinic Florida, Department of Neuroscience Jacksonville, Florida, USA.
� Prof. Kendall Jensen – Tgen -The Translational Genomics Research Institute, Phoenix, AZ, USA.
� Prof. Matthew Alexander - Children's Hospital Boston - Boston, MA, USA.
� Prof. Christopher Klein- Mayo Clinic - Department of Neurology- Rochester.
� Prof. Joshua Selsby, Iowa State University – Ames - USA.
� Prof. Alexey Belkin, University of Maryland – Baltimore - USA.
� Prof. Toshifumi Yokota- University of Alberta – Edmonton - CANADA.
� Prof. Xiping Cheng- University of Michigan - Ann Arbor - USA.
� Prof. Fred Lublin, Mount Sinai University, New York – USA.
� Prof. Laura Piccio , Washington University, St. Louis – USA.
� Prof. Anne Cross, Washington University, St. Louis – USA
� Prof. Alberto Espay, University of Cincinnati, Cincinnati, USA.
� Prof. Robert Lisak, Wayne State University Medical School, Detroit, MI, USA.
� Prof. Alexis Brice, Sorbonne Universités, Université Pierre et Marie Curie Paris 06, Unité Mixte de Recherche (UMR) S 1127, Institut du Cerveau et de la Moelle Épinière (ICM), Paris France
� Prof. Isabelle Le Ber, Sorbonne Universités, Université Pierre et Marie Curie Paris 06, Unité Mixte de Recherche (UMR) S 1127, Institut du Cerveau et de la Moelle Épinière (ICM), Paris France
� Prof. Bruno Dubois, Institute de la Mémoire et de la Maladie d’Alzheimer (IM2A), Département de Neurologie, Hȏpital de la Pitié-Salpêtrière, AP-HP, Paris, France; INSERM, CNRS, UMR-S975, Institut du Cerveau et de la Moelle Epinière (ICM), Paris, France; Sorbonne, Universités, Université Pierre et marie Curie – paris 6, Paris, France.

-6-
� Prof. Andreas Reif, University of Frankfurt, Germany
� Prof. Albert Ludolph, Dept. Neurology – University of Ulm, Germany.
� Prof. Amman Al-Chalabi, King’s College – London, United Kingdom.
� Prof. Christopher Shaw, King’s College – London , United Kingdom
� Prof. John Powel, King’s College – London , United Kingdom
� P.D. Dr. Marcus Weber, St. Gallen, CH
� Prof. E. I Rugarli – Institute for Genetics CECAD Research Center University of Cologne Joseph-Stelzmann, Köln Germany
� Prof. W Griffith – Institute of Mass Spectrometry College of Medicine Grove Building Swansea University Wales, UK
� Dr. Edward J Hollox – Department of Genetics, University of Leicester, Leicester, UK
� Dr. Nasser M. Al-Daghri, Biochemistry Department College of Science, King Saud University, Kingdom of Saudi Arabia Riyadh 11451(KSA)
� Prof. Prince Mutaib, Biochemistry Department, College of science, King Saud University, Riyadh, KSA
� Dott. Juan Antonio Pineda- Infectious Diseases and Microbiology Clinical Unit. Valme Hospital, Serville, Spain
� Dott. Antonio Caruz – Immunogenetics Unit, Department of Experimenatl Biology, University of Jaen, Spain
� Dott. Manuel Comabella- Hospital Universitari Vall d’Hebron (HUVH). Barcell ona, Spain
� Dott. Matteo Fumagalli – UCL Genetics Institute, Department of Genetics, Evolution and Environment, University College London, London UK
� Prof. Jernej Ule – Department of Molecular Neuroscience, UCL Institute of Neurology London UK
� Translational Core Laboratory, Cincinnati Children’ s Research Foundation, USA
� Prof. Lu Qilong - McColl-Lockwood Laboratory for Muscular Dystroph y Research, Neuromuscular/ALS Center, Carolinas Medical Center, Charlotte, North Carolina, USA.
� Dr. Angelo Monguzzi , Dept. Scienze dei Materiali, Università degli Studi di Milano-Bicocca
� Prof. Anna Spada – Endocrine Unit, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Dept. Of Clinical Science and Community Health, Università degli Studi di Milano
� Dr. Maurilio Bruno – Istituto Ortopedico Galeazzi, IRCCS
� Dr. Francesco Nicassio – Department of Experimental Oncology, European Institute of Oncology, IFOM-IEO Campus
� Prof. Graziella Messina – Dept. Bioscienze, Università degli Studi di Milano
� Prof. Angelo Poletti – Dept. Di Science Farmacologiche e Biomolecolari, Università degli Studi di Milano

-7-
� Prof. Silvio Bicciato, bioinformatics unit, Faculty of Biosciences and Biotechnologies, University of Modena and Reggio Emilia
� Prof. Enrico Tagliafico , clinical Biochemistry, University of Modena and Reggio Emilia
� Prof. Sergio Abrignani, National Institute of Molecular Genetics (INGM), Milan, Italy
� Prof. Silvano Bosari, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico di Milano,
� Prof. Carlo Agostoni, Università degli Studi, IRCCS Ca’ Granda Ospedale Maggiore Policlinico di Milano
� Prof. Lorenza Lazzari, Department of Regenerative Medicine, Fondazione IRCCS Cà Granda Ospedale Maggiore Policlinico di Milano
� Prof. Rosaria Giordano, Department of Regenerative Medicine, Fondazione IRCCS Cà Granda Ospedale Maggiore Policlinico di Milano
� Dr. Massimo Aureli, Università Degli Studi di Milano, Ospedale san Raffaele, Milano
� Prof. Michela Deleidi, Department of Neurodegenerative Diseases, University of Tübingen
� Prof. Cristina Barlassina – Dept. Health Sciences, Università degli Studi di Milano
� Prof. Irene Cettin, Centro di Ricerche Fetali Giorgio Pardi, Università degli Studi di Milano - Polo Universitario Ospedale L. Sacco di Milano
� Prof. Paola Rossi, Dipartimento di Scienze Fisiologiche e Farmacologiche cellulari e molecolari- Sezione di Fisiologia dell’Università di Pavia
� Prof. Umberto Galderisi – Dipartimento di Medicina Sperimentale, Università degli Studi di Milano
� Prof. Rita Maiavacca – UOS Laboratorio di Biochimica, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico
� Dr. Fabio Triulzi - UOC Neuroradiologia - Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico
� Dr. Umberto Giovanella – CNR –ISMAC
� Dr. Anna Villa - Human Genome Lab, Humanitas Clinical and Research Center, Milano
� Prof. Agostino Cortelezzi – UOC Ematologia1 e Centro Trapianti di Midollo, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico di Milano
� Prof. Giuseppe D’Antona, Department of Molecular Medicine, University of Pavia, Pavia, Italy LUSAMMR, Laboratory for Motor Activities in Rare Di seases, Sport Medicine, Centre Voghera, Voghera, Italy
� Prof. Enzo Nisoli, Center for Study and Research on Obesity, Department of Medical Biotechnology and Translational Medicine, University of Milan, Milan, Italy;
� Prof. Dario Parazzoli, Imaging Facility IFOM Foundation – The FIRC Insti tute of Molecular Oncology Foundation, Milan, Italy
� Prof. Stefano Campaner, Center for Genomic Science of IIT@SEMM; Istituto Italiano di Tecnologia (IIT); Milan, Italy
� Prof. Thierry Vanderdriessche – VRIJE Universiteit Brussel

-8-
� Prof. Nadia Grimoldi – Unità di Neurochirurgia, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico di Milano
� Dr. Laura Porretti – Clinical Chemistry and Microbiology Laboratory, Flow Cytometry and Experimental Hepatology Service, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico di Milano
� Prof. Giorgio Roberto Merlo – Dept. Biotecnologie Molecolari Scienze della Salute, Università degli Studi di Torino
� Prof. Luciano Conti – Centro di Biologia Integrata CIBIO, Università degli Studi di Trento
� Prof. Alessandro Quattrone, Director of CiBio, University of Trento
� Prof. Elena Cattaneo, Department of Biosciences and Centre for Stem cell Research, Università degli Studi di Milano
� Prof. Giovanna Cantarella, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico di Milano
� Prof. Mauro Pluderi , Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico di Milano
� Prof. Nadia Grimoldi, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico di Milano
� Prof. Paolo Vezzoni,Unità Operativa di Supporto (UOS) dell’Istituto di Ricerca Genetica e Biomedica (IRGB) del CNR.
� Prof. Marina Bouchè, Unit of Histology, and IIM, Sapienza University, DAHFMO, Rome, Italy
� Prof. Davide Gabellini, Dulbecco Telethon Institute and Division of Regenerative Medicine, San Raffaele Scientific Institute, Milan
� Prof. Franco Rustichelli, Dipartimento di Scienze Cliniche e Odontostomatologiche, Sezione di Biochimica, Biologia e Fisica, Università Politecnica delle Marche, Ancona, Italy
� Prof. Silvia Della Bella, Lab of Clinical and Experimental Immunology, Humanitas Clinical and Research Center, Rozzano (MI), Italy, Department of Medical Biotechnologies and Translational Medicine, University of Milan, Milan, Italy
� Prof. Aldo Pagano, Department of Experimental Medicine,UniversityofGenoa,Genoa,Italy, IRCCS Azienda Ospedaliera Universitaria SanMartino-IST, Genoa, Italy
� Prof. Francesco Meinardi , Università di Milano Bicocca
� Prof. Jose F Rodriguez-Matas-, Department “Giulio Natta”Politecnico di Milano, Italy
� Prof. Jerry Mendell – Nationwide Children’s Hospital, Columbus, USA
DIRETTORE PROF. NEREO BRESOLIN Via F. Sforza, 35 - 20122 Milano – Tel. 02.5503.3809 - Fax 02.5503.3800
E-mail: [email protected] - [email protected] - www.centrodinoferrari.com

1Scientific RepoRts | 6:25960 | DOI: 10.1038/srep25960
www.nature.com/scientificreports
Differential neuronal vulnerability identifies IGF-2 as a protective factor in ALSIlary Allodi1,*, Laura Comley1,*, Susanne Nichterwitz1,*, Monica Nizzardo2, Chiara Simone2, Julio Aguila Benitez1, Ming Cao1, Stefania Corti2,† & Eva Hedlund1,†
The fatal disease amyotrophic lateral sclerosis (ALS) is characterized by the loss of somatic motor neurons leading to muscle wasting and paralysis. However, motor neurons in the oculomotor nucleus, controlling eye movement, are for unknown reasons spared. We found that insulin-like growth factor 2 (IGF-2) was maintained in oculomotor neurons in ALS and thus could play a role in oculomotor resistance in this disease. We also showed that IGF-1 receptor (IGF-1R), which mediates survival pathways upon IGF binding, was highly expressed in oculomotor neurons and on extraocular muscle endplate. The addition of IGF-2 induced Akt phosphorylation, glycogen synthase kinase-3β phosphorylation and β-catenin levels while protecting ALS patient motor neurons. IGF-2 also rescued motor neurons derived from spinal muscular atrophy (SMA) patients from degeneration. Finally, AAV9::IGF-2 delivery to muscles of SOD1G93A ALS mice extended life-span by 10%, while preserving motor neurons and inducing motor axon regeneration. Thus, our studies demonstrate that oculomotor-specific expression can be utilized to identify candidates that protect vulnerable motor neurons from degeneration.
Amyotrophic lateral sclerosis (ALS) is a fatal disease characterized by a progressive loss of somatic motor neu-rons, muscle wasting and paralysis. ALS appears mostly sporadic (sALS), but can be inherited (fALS) due to mutations in e.g. superoxide dismutase 1 (SOD1), TAR DNA-binding protein 43 (TDP-43), Fused in Sarcoma (FUS) and C9ORF72 (chromosome 9 open reading frame 72)1,2. Importantly, data from fALS models indicate that motor neuron intrinsic factors are crucial for initiation and early progression of degeneration3–5. While ALS is characterized by motor neuron loss, certain motor neuron groups are for unknown reasons relatively resistant to degeneration. Among the most resistant are oculomotor neurons6–9, which are located in the brain stem and control eye movement. Consequently, eye-tracking devices are used to enable paralyzed ALS patients to com-municate through computers10. Thus, an investigation of factors intrinsic to oculomotor neurons in health and disease could reveal mechanisms of neuronal resistance and be the basis for future therapeutic strategies to pro-tect vulnerable motor neurons from degeneration. Towards this goal, we and others have previously shown that resistant oculomotor motor neurons display a distinct mRNA and protein signature compared to other vulnerable motor neuron groups11–13. We identified insulin-like growth factor 1 (IGF-1) and 2 (IGF-2) as preferential to ocu-lomotor neurons in the normal rat11. This finding is highly compelling in terms of intrinsic neuronal resistance as IGFs are known motor neuron survival factors14,15. In fact, viral delivery of IGF-1 to motor neurons in the SOD1G93A fALS mouse was neuroprotective and increased the life-span of the mice16. The possible motor neuron protective properties of IGF-2 in ALS has not been previously investigated. IGF-2 can bind to IGF-1 receptors (IGF-1R), IGF-2 receptors (IGF-2R) and insulin receptors. IGF-2R has the highest affinity for IGF-2, but the biological effects of IGF-2 are mediated through IGF-1R and/or insulin receptor, just as for IGF-1. The binding of IGF-2 to IGF-1R, which is a receptor tyrosine kinase, leads to activation of PI3K/Akt and survival pathways or activation of mitogen activated protein kinase (MAPK) pathway and proliferation. Insulin receptor activa-tion leads to proliferation. IGF-2 binding to IGF-2R, which lack the intracellular tyrosine binding domain and
1Department of Neuroscience, Karolinska Institutet, Retzius v. 8, 171 77 Stockholm, Sweden. 2Dino Ferrari Center, Neuroscience Section, Department of Pathophysiology and Transplantation, University of Milan, Neurology Unit, Istituto Di Ricovero e Cura a Carattere Scientifico Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan 20122, Italy. *These authors contributed equally to this work. †These authors jointly supervised this work. Correspondence and requests for materials should be addressed to S.C. (email: [email protected]) or E.H. (email: [email protected])
received: 05 February 2016
accepted: 25 April 2016
Published: 16 May 2016
OPEN

1702
Received November 30, 2015; final revision received April 6, 2016; accepted April 11, 2016.From the Department of Cerebrovascular Disease, IRCCS Foundation Carlo Besta Neurological Institute, Milan, Italy (A.B., G.B.B., E.A.P., N.T.); Stroke
Research Group, Department of Clinical Neurosciences, University of Cambridge, Cambridge, United Kingdom (H.S.M.); Department of Bio-Medical Informatics, University of Pavia, Pavia, Italy (S.Q.); Department of Inherited Cardiovascular Disease, Foundation IRCCS Policlinico San Matteo, Pavia, Italy (E.A., M.G.); Neurology Unit, Department of Neuroscience and Sensory Organs, Maggiore Policlinico Hospital Foundation IRCCS Ca’ Granda, Milan, Italy (S.L., L.C.); Neurology and Stroke Unit, Department of Urgency (G.M., A.C.), Department of Genetics (C.C., G.G.), and Brain MRI 3T Research Center (P.V.), IRCCS Foundation Casimiro Mondino Neurological Institute, Pavia, Italy; Department of Genetics of Neurodegenerative and Metabolic Diseases, IRCCS Foundation C, Besta Neurological Institute, Milan, Italy (F.T., C.G., S.B.); Department of Medical Genetics, Niguarda Ca’ Granda Hospital, Milan, Italy (S.P., L.M.); Department of Genomics for Human Disease Diagnosis and Laboratory of Clinical Molecular Biology, IRCCS San Raffaele hospital, Milan, Italy (P.C., M.F.); University Vita-Salute, Milano, Italy (M.F.); Dino Ferrari Centre, Neuroscience Section, Department of Pathophysiology and Transplantation (DEPT), University of Milan, Milan, Italy (S.C., D.R., G.P.C.); Neurology Unit, Department of Neuroscience and Sensory Organs, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico Milan, Milan, Italy (S.C., D.R., G.P.C.); Department of Molecular Biology, Scientific Institute IRCCS Eugenio Medea, Bosisio Parini, Lecco, Italy (M.T.B.); Center for amyloidosis, Department of medical Thecnologies, IRCCS Foundation San Matteo Policlinico, Pavia, Italy (L.O., G.M.); Vascular Neurology - Spedali Civili, Department of Clinical and Experimental Sciences, University of Brescia, Brescia, Italy (A. Pezzini, A. Padovani); Stroke Unit, Department of Neuroscience and Behaviour Clinical Sciences Circolo Hospital and Macchi Foundation, Varese Hospital Varese, Italy (M.L.D.L., E.P.V., G.B.); Stroke Unit, Department of Neurological Sciences, Sant’Antonio Abate Hospital, Gallarate, Italy (F.M., D.Z.); Stroke Unit, Department of Neurological Sciences, Legnano and Cuggiono Hospital, Legnano, Italy (M.V.C., P.P.); Neurological Unit and Stroke Unit, Department of Neurological Sciences, Ospedale di Desio, Desio, Italy (B.M.M., A.C.); Stroke Unit, Department of Medical and Surgical Sciences, San Gerardo Hospital, Milan Center for Neuroscience, University of Milano-Bicocca, Milano, Italy (S.B., C.F.); Stroke Unit, Department of Neurological Sciences, Azienda Ospedaliera Niguarda Ca Granda, Milan, Italy (C.M., E.A.); Neurological Unit, Department of Neurological Sciences, AO Melegnano, Ospedale di Vizzolo Predabissi, Melegnano, Italy (G.M., F.S.); Stroke Unit, Department of Neurological Sciences, Istituto Clinico Humanitas, Rozzano, Italy (M.C., S.M.); Stroke Unit, Department of Neurology, Neurophysiology and Rehabilitation, San Raffaele Hospital - Milan, Italy (M.S., G.C.); Stroke Unit, Department of Neurological Sciences, Valduce Hospital, Como, Italy (N.C.); Tradate Hospital-Tradate, Italy, Italy (M.G.); UO Neurologia-AO San Paolo, Medical Department, Milan, Italy (D.U.); Stroke Unit, Department of Neurological Sciences, Sant’Anna Hospital, Como, Italy (E.C., L.T., M.A.); Stroke Unit, Department of Neurological Sciences, Istituto Clinico Città Studi, Milan, Italy (B.I., C.S.T.); Stroke Unit, Department of Neurological Sciences, Ospedale di Circolo, Saronno, Italy (L.F., G. Grampa); UO Neurologia-Azienda Ospedaliera di Desio e Vimercate, Department of Neurological Sciences, Vimercate, Italy (M.B.); UO Neurologia AO Fatebenefratelli e Oftalmico, Milano, Department of Medical Sciences, Italy (P.B.); and Department of Neurology, Heidelberg University Hospital, Heidelberg, Germany (C.G.-G.).
*A list of all GENS investigators and monitors are given in online-only Data Supplement.†Deceased.The online-only Data Supplement is available with this article at http://stroke.ahajournals.org/lookup/suppl/doi:10.1161/STROKEAHA.
115.012281/-/DC1.Correspondence to Anna Bersano, MD, PhD, Cerebrovascular Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, via Celoria 11, 20133 Milano,
Italy. E-mail [email protected]
Clinical Pregenetic Screening for Stroke Monogenic DiseasesResults From Lombardia GENS Registry
Anna Bersano, MD, PhD; Hugh Stephen Markus, MD, PhD; Silvana Quaglini, PhD; Eloisa Arbustini, MD, PhD; Silvia Lanfranconi, MD; Giuseppe Micieli, MD; Giorgio B. Boncoraglio, MD;
Franco Taroni, MD; Cinzia Gellera, MD; Silvia Baratta, PhD; Silvana Penco, PhD; Lorena Mosca, PhD; Maurizia Grasso, MD; Paola Carrera, BSc; Maurizio Ferrari, MD;
Cristina Cereda, MD; Gaetano Grieco, PhD; Stefania Corti, MD, PhD; Dario Ronchi, PhD; Maria Teresa Bassi, PhD; Laura Obici, MD; Eugenio A. Parati, MD; Alessando Pezzini, MD, PhD;
Maria Luisa De Lodovici, MD; Elena P. Verrengia, MD; Giorgio Bono, MD, PhD; Francesca Mazucchelli, MD; Davide Zarcone, MD; Maria Vittoria Calloni, MD;
Patrizia Perrone, MD; Bianca Maria Bordo, MD; Antonio Colombo, MD; Alessandro Padovani, MD, PhD; Anna Cavallini, MD; Simone Beretta, MD; Carlo Ferrarese, MD, PhD;
Cristina Motto, MD; Elio Agostoni, MD; Graziella Molini, MD; Francesco Sasanelli, MD; Manuel Corato, MD; Simona Marcheselli, MD; Maria Sessa, MD; Giancarlo Comi, MD, PhD;
Nicoletta Checcarelli, MD; Mario Guidotti, MD; Davide Uccellini, MD; Erminio Capitani; Lucia Tancredi, MD; Marco Arnaboldi, MD; Barbara Incorvaia, MD;
Carlo Sebastiano Tadeo, MD; Laura Fusi, MD; Giampiero Grampa, MD; Giampaolo Merlini, MD, PhD; Nadia Trobia, PhD; Giacomo Pietro Comi, MD, PhD; Massimiliano Braga, MD; Paolo Vitali, MD;
Pierluigi Baron, MD†; Caspar Grond-Ginsbach, PhD; Livia Candelise, MD, PhD; on behalf of Lombardia GENS Group*
© 2016 American Heart Association, Inc.
Stroke is available at http://stroke.ahajournals.org DOI: 10.1161/STROKEAHA.115.012281
Clinical Sciences
by guest on Decem
ber 13, 2016http://stroke.ahajournals.org/
Dow
nloaded from
by guest on Decem
ber 13, 2016http://stroke.ahajournals.org/
Dow
nloaded from
by guest on Decem
ber 13, 2016http://stroke.ahajournals.org/
Dow
nloaded from
by guest on Decem
ber 13, 2016http://stroke.ahajournals.org/
Dow
nloaded from
by guest on Decem
ber 13, 2016http://stroke.ahajournals.org/
Dow
nloaded from
by guest on Decem
ber 13, 2016http://stroke.ahajournals.org/
Dow
nloaded from
by guest on Decem
ber 13, 2016http://stroke.ahajournals.org/
Dow
nloaded from
by guest on Decem
ber 13, 2016http://stroke.ahajournals.org/
Dow
nloaded from

Histological effects of givinostat in boys with Duchennemuscular dystrophy
Paolo Bettica a,*,1, Stefania Petrini b,1, Valentina D’Oria b, Adele D’Amico c, Michela Catteruccia c,Marika Pane d, Serena Sivo d, Francesca Magri e, Simona Brajkovic e, Sonia Messina f,g,
Gian Luca Vita g, Barbara Gatti a, Maurizio Moggio h, Pier Lorenzo Puri i,j, Maurizio Rocchetti k,Giuseppe De Nicolao l, Giuseppe Vita f,g, Giacomo P. Comi e, Enrico Bertini c, Eugenio Mercuri d
a Italfarmaco S.p.A., Milan, Italyb Research Laboratories, Confocal Microscopy Core Facility, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy
c Unit of Neuromuscular and Neurodegenerative Disorders, Laboratory of Molecular Medicine, Department of Neurosciences, Bambino Gesù Children’sHospital, IRCCS, Rome, Italy
d Department of Paediatric Neurology, Catholic University, Rome, Italye Dino Ferrari Centre, Neuroscience Section, Department of Pathophysiology and Transplantation, Neurology Unit, IRCCS Foundation Ca’Granda Ospedale
Maggiore Policlinico, University of Milan, Milan, Italyf Department of Neurosciences, University of Messina, Messina, Italy
g NEMO SUD Clinical Centre for Neuromuscular Disorders, Messina, Italyh Neuromuscular and Rare Disease Unit, Department of Neuroscience, Foundation IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Dino Ferrari Centre,
University of Milan, Milan, Italyi Sanford-Burnham medical Research Institute, Sanford Children’s Health Research Center, La Jolla, California, USA
j IRCCS Fondazione Santa Lucia, Rome, Italyk Independent Consultant, Milan, Italy
l Department of Electrical, Computer and Biomedical Engineering, University of Pavia, Pavia, Italy
Received 16 February 2016; received in revised form 29 June 2016; accepted 7 July 2016
Abstract
Duchenne Muscular Dystrophy (DMD) is caused by mutations in the dystrophin gene leading to dystrophin deficiency, muscle fiberdegeneration and progressive fibrotic replacement of muscles. Givinostat, a histone deacetylase (HDAC) inhibitor, significantly reduced fibrosisand promoted compensatory muscle regeneration in mdx mice. This study was conducted to evaluate whether the beneficial histological effects ofGivinostat could be extended to DMD boys. Twenty ambulant DMD boys aged 7 to <11 years on stable corticosteroid treatment were enrolled inthe study and treated for ≥12 months with Givinostat. A muscle biopsy was collected at the beginning and at the end of treatment to evaluate theamount of muscle and fibrotic tissue. Histological effects were the primary objectives of the study. Treatment with Givinostat significantlyincreased the fraction of muscle tissue in the biopsies and reduced the amount of fibrotic tissue. It also substantially reduced tissue necrosis andfatty replacement. Overall the drug was safe and tolerated. Improvement in functional tests was not observed in this study, but the sample size ofthe study was not sufficient to draw definitive conclusions. This study showed that treatment with Givinostat for more than 1 year significantlycounteracted histological disease progression in ambulant DMD boys aged 7 to 10 years.© 2016 The Authors. Published by Elsevier B.V. This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Keywords: Duchenne muscular dystrophy; Givinostat; Histone deacetylase inhibitor; Histology; Clinical trial
1. Introduction
Duchenne Muscular Dystrophy (DMD) is the most commonmuscular dystrophy in childhood [1]. The disease is caused by
mutations in the dystrophin gene, leading to dystrophindeficiency and subsequent cell membrane instability.This instability determines uncontrolled calcium influx,inflammation, necrosis, and replacement of muscle with fibrotictissue and fat, which leads to severe muscle wasting andweakness.
Pharmacological blockade of the histone deacetylaseactivity, which is constitutively active in DMD muscles [2] by
* Corresponding author. Italfarmaco, Via dei Lavoratori 54, 20092 CiniselloBalsamo, MI, Italy. Fax: +39 02 6443 3554.
E-mail address: [email protected] (P. Bettica).1 Paolo Bettica and Stefania Petrini contributed equally to this article.
http://dx.doi.org/10.1016/j.nmd.2016.07.0020960-8966/© 2016 The Authors. Published by Elsevier B.V. This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Available online at www.sciencedirect.com
Neuromuscular Disorders 26 (2016) 643–649www.elsevier.com/locate/nmd
ScienceDirect

Autophagy in motor neuron disease: Key pathogenetic mechanisms andtherapeutic targets
Maria Sara Cipolat Mis 1, Simona Brajkovic 1, Emanuele Frattini, Alessio Di Fonzo, Stefania Corti ⁎Dino Ferrari Center, Neuroscience Section, Department of Pathophysiology and Transplantation, University of Milan, Neurology Unit, Istituto Di Ricovero e Cura a Carattere Scientifico FoundationCa' Granda Ospedale Maggiore Policlinico, Milan 20122, Italy
a b s t r a c ta r t i c l e i n f o
Article history:Received 12 August 2015Revised 25 January 2016Accepted 29 January 2016Available online 2 February 2016
Autophagy is a lysosome-dependant intracellular degradation process that eliminates long-lived proteins as wellas damaged organelles from the cytoplasm. An increasing body of evidence suggests that dysregulation of thissystem plays a pivotal role in the etiology and/or progression of neurodegenerative diseases including motorneuron disorders. Herein, we review the latest findings that highlight the involvement of autophagy in the path-ogenesis of amyotrophic lateral sclerosis (ALS) and the potential role of this pathway as a target of therapeuticpurposes. Autophagy promotes the removal of toxic, cytoplasmic aggregate-prone pathogenetic proteins, en-hances cell survival, and modulates inflammation. The existence of several drugs targeting this pathway can fa-cilitate the translation of basic research to clinical trials for ALS and other motor neuron diseases.
© 2016 Elsevier Inc. All rights reserved.
Keywords:AuthophagyAmyotrophic lateral sclerosisProtein aggregation
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 842. The autophagic processes in healthy and ALS context . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 853. Accumulation of ALS pathogenetic proteins and autophagy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 864. Autophagy as therapeutic target . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 875. Conclusions. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 88Acknowledgments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 88References. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 88
1. Introduction
There is still no effective treatment for amyotrophic lateral sclerosis(ALS), a fatal, progressive neurodegenerative disease characterized bythe selective loss of upper and lower motor neurons (MNs) (Bucchiaet al., 2015). The degeneration of MNs clinically leads to progressive pa-ralysis and early death due to respiratory failure. About 90% of the ALScases are sporadic, while 10% are familial, inherited with an autosomaldominant/recessive, or X-linked pattern (Marangi and Traynor, 2015).Over 30 causative genes have been identified in familial ALS, howeveronly 10% of the sporadic forms present an involvement of those genes(Chen et al., 2013; Renton et al., 2014; Marangi and Traynor, 2015).
ALS pathology is still not completely understood. Many pathwayshave been suggested to play a role in the disease pathogenesis includingalteration of protein stability, conformation and degradation, distur-bances of RNA metabolism, and axonal transport defects (Bucchiaet al., 2015). In particular, mutated genes that cause ALS, such as super-oxide dismutase 1 (SOD1), TAR DNA-binding protein 43 (TARDBPencoding for TDP-43), and RNA-binding protein FUS, encode proteinsthat can form aggregates (Al-Chalabi et al., 2012). Indeed, the most fre-quent protein cell inclusions detected in the central nervous system(CNS) of apparently sporadic patients are TDP-43 positive (Neumann,2013). Remarkably, these aggregates are detected both in the presenceof mutations in TARDBP gene and in other ALS causative genes such asC9ORF72, SQSTM1, GRN, VCP, UBQLN2, OPTN, and NIPA1 (Marangi andTraynor, 2015).
Mutant aberrant or misfolded proteins, such as TDP-43 and others,aggregates in the cytoplasm, nucleus or extracellular matrix damagingthe cellular organelles and leading to neuronal dysfunction (Walkerand LeVine, 2000). The increased amount of misfolded proteins not
Molecular and Cellular Neuroscience 72 (2016) 84–90
⁎ Corresponding author.E-mail address: [email protected] (S. Corti).
1 These authors equally contributed to this work.
http://dx.doi.org/10.1016/j.mcn.2016.01.0121044-7431/© 2016 Elsevier Inc. All rights reserved.
Contents lists available at ScienceDirect
Molecular and Cellular Neuroscience
j ourna l homepage: www.e lsev ie r .com/ locate /ymcne

Longitudinal follow-up andmuscle MRI pattern of twosiblings with polyglucosanbody myopathy due toglycogenin-1 mutation
Polyglucosan bodies (PBs) are deposits ofamylopectin-like polysaccharides, detectedin muscles of patients affected withglycogenoses-like branching enzyme (GBE)and phosphofructokinase deficiencies.1
Recently, mutations in RBCK1 havebeen associated with a skeletal PBs myop-athy,2 as well as mutations in GYG1,encoding for glycogenyn-1.3 All theseconditions have autosomal recessive inher-itance. Only seven unrelated cases withmutations in GYG1 have been reported,characterised by variable age at onset(from childhood to 7th decade), mainlyproximal weakness, normal creatine
J Neurol Neurosurg Psychiatry July 2016 Vol 87 No 7 797
PostScript
group.bmj.com on December 15, 2016 - Published by http://jnnp.bmj.com/Downloaded from

developed impaired, mainly right-sided,arm abduction. Over the years, shebecame unable to run and could onlyclimb stairs with double support. Her lastexamination, at the age of 64 years,showed scapular winging, positiveGowers’ sign, waddling-type gait, pre-served facial muscles and reduced DTR.At manual testing, muscle power wasmildly impaired at both limb girdles(MRC 5-), but severely compromised atshoulder abduction (MRC 0).
Both sisters had a myopathic EMG,normal CK levels and no pulmonaryabnormalities (normal spirometry andnocturnal oximetry). They underwentechocardiogram, Holter ECG, ECG andcardiac valuation over the previous year,with no evidence of cardiac involvement.
Their skeletal muscle biopsies showed anearly identical vacuolar myopathy (seeonline supplementary figure S1). P.IV-5underwent left and right biceps musclebiopsies, at the age of 36 and 50 years,respectively, which did not show any sig-nificant modifications. PBs were detectedin vacuoles in up to 50% of muscle fibresin P.IV-5 and 40% in P.IV-10. These depos-its were localised in both subsarcolemmaland intracytoplasmic areas, stained faintlywith Gömöri trichrome and showed adiffuse periodic acid-Schiff positivity withoverall resistance to diastase reaction (seeonline supplementary figure S1B–G).Electron microscopy showed that manyvacuoles were filled with mostly homoge-neous or slightly granular filamentousinclusions, consistent with PBs (see onlinesupplementary figure S1H,I), intracyto-plasmic collections of free glycogen, aswell as a few autophagic vacuoles.
Biochemical analysis of enzymesinvolved in glycolysis/glycogen metabol-ism did not reveal any deficiencies (seeonline supplementary table S1). Glycogenspectrum was normal. GBE1 and RBCK1were ruled-out. GYG1 sequencingrevealed that both sisters were homozy-gous for c.143+3G>C mutation.
The sisters underwent shoulder girdle/arm and hip girdle/thigh muscle MRI(figure 1): deltoids and glutaei were themost affected muscles in P.IV-10, all musclesbeing almost substituted in P.IV-5; pseudo-hypertrophy of gracilis, sartorius and rectusfemoris was evident in both patients.
We report two siblings affected with aPBs myopathy due to homozygous c.143+3G>C mutation in GYG1. This mutationaffects the donor splice site in intron 2,causing the skipping of exon 2, and createsa premature stop codon thus causing lackof protein. This variant was detected in 4/7cases of different ethnicities, two
homozygous with disease onset in child-hood/adolescence, and two compound het-erozygous with onset in the fifth–seventhdecade.3 Our findings confirm that thismutation is common in the Italian popula-tion and is associated with adult onset,even though homozygous.Pelvic girdle weakness and atrophy
were reported in all patients with muta-tions in GYG1, half of them presentingalso shoulder girdle involvement.3 Ourcases confirm that early and asymmetricweakness in arm abduction is common.Of note, this asymmetric motor syndromeoverlaps one previously described in amiddle-aged woman with a geneticallyunsolved PBs myopathy.4 Disease progres-sion in our siblings showed variable sever-ity: from mild weakness towheelchair-dependency, the latter pheno-type representing the most severe spec-trum reported so far. Cardiac andrespiratory muscles are spared.3 Musclemorphology shows PBs as the most strik-ing finding.3
Nevertheless, it is unknown how lack ofglycogenin-1 causes PB aggregation inmuscle: these amylase-resistant polysac-charides are also observed in GBE defi-ciency, suggesting an impairment inglycogen synthesis process.Among PB myopathies, muscle MRI
was reported only in one congenital casewith mutations in GBE1 with evidence ofubiquitous fibroadipose replacement.5 Weobserved a correlation between clinicalseverity and degree of fibroadipose substi-tution, with early involvement of deltoidsand glutaei. Upper girdle muscle MRI hasbeen investigated in a few neuromuscularconditions; our siblings’ pattern does notresemble that of other diseases affectingarm abduction, including facioscapulo-humeral muscular dystrophy, in which tra-pezius and serratus anterior are mostlyaffected and are the earliest to beinvolved, with deltoids being spared.6
MRI pattern at lower girdle musclesresembles that observed in late-onsetPompe disease, which, however, showsprominent subscapularis and anterior ser-ratus replacement in the upper girdle.7
In conclusion, our findings demonstratethat a myopathy with PB and early asym-metric impairment of arm abduction isconsistent with mutations in GYG1. Weoutline an unknown variability of clinicalphenotype within the same genetic back-ground. At muscle MRI, early replace-ment of deltoids and glutaei musclesseems to be the hallmark, at least for thec.143+3G>C mutation, an observationto be validated by the analysis of a largercohort with different GYG1 mutations.
Irene Colombo,1 Serena Pagliarani,2
Silvia Testolin,1 Claudia Maria Cinnante,3
Gigliola Fagiolari,1 Patrizia Ciscato,1
Andreina Bordoni,2 Francesco Fortunato,2
Francesca Magri,2 Stefano Carlo Previtali,4
Daniele Velardo,4 Monica Sciacco,1 GiacomoPietro Comi,2 Maurizio Moggio1
1Neuromuscular and Rare Disease Unit, Department ofNeuroscience, Foundation IRCCS Ca’ Granda OspedaleMaggiore Policlinico, University of Milan, Milan, Italy2Department of Pathophysiology and TransplantationNeuroscience Section (DEPT), Neurology Unit, DinoFerrari Centre, Foundation IRCCS Ca’ Granda OspedaleMaggiore Policlinico, University of Milan, Milan, Italy3Unit of Neuroradiology, Department of Neuroscience,Foundation IRCCS Ca’ Granda Ospedale MaggiorePoliclinico, University of Milan, Milan, Italy4Division of Neuroscience and Department ofNeurology, Institute of Experimental Neurology(INSPE), IRCCS San Raffaele Scientific Institute, Milan,Italy
Correspondence to Dr Irene Colombo,Neuromuscular and Rare Disease Unit, Department ofNeuroscience, Foundation IRCCS Ca’ Granda OspedaleMaggiore Policlinico, University of Milan, Via F Sforza35, Milano 20122, Italy; [email protected]
IC and SP contributed equally to this manuscript.
Acknowledgements The authors wish toacknowledge the Italian Association of Myology (AIM),the “Associazione Amici del Centro Dino Ferrari—University of Milan”, and Eurobiobank and theTelethon network of Genetic biobanks [grant numberGTB12001], for providing human biological samples.
Contributors IC was involved in the clinicalevaluation; design of the study; acquisition, analysisand interpretation of the data; and drafting of themanuscript. SP was involved in the design of the study;analysis and interpretation of the data; and drafting ofthe manuscript. ST was involved in the clinicalevaluation; and acquisition, analysis and interpretationof the data. CMC, MS, GPC and MM were involved inthe analysis and interpretation of the data; and revisingof the manuscript. GF and PC were involved in theanalysis and interpretation of the data. AB, FF and DVwere involved in the acquisition of the data. FM wasinvolved in the revising of the manuscript. SCP wasinvolved in the clinical evaluation; acquisition of thedata; and revising of the manuscript.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Foundation IRCCS Ca’ GrandaOspedale Maggiore Policlinico.
Provenance and peer review Not commissioned;externally peer reviewed.
▸ Additional material is published online only. To viewplease visit the journal online (http://dx.doi.org/10.1136/jnnp-2015-310553).
To cite Colombo I, Pagliarani S, Testolin S, et al.J Neurol Neurosurg Psychiatry 2016;87:797–800.
Received 6 February 2015Revised 2 July 2015Accepted 6 July 2015Published Online First 22 July 2015
J Neurol Neurosurg Psychiatry 2016;87:797–800.doi:10.1136/jnnp-2015-310553
J Neurol Neurosurg Psychiatry July 2016 Vol 87 No 7 799
PostScript
group.bmj.com on December 15, 2016 - Published by http://jnnp.bmj.com/Downloaded from

Jean-François Desaphy,PhD
Roberta Carbonara, PhDAdele D’Amico, MDAnna Modoni, MDJulien Roussel, PhDPaola Imbrici, PhDSerena Pagliarani, PhDSabrina Lucchiari, PhDMauro Lo Monaco, MDDiana Conte Camerino,
PhD
Correspondence toDr. Desaphy:[email protected]
Supplemental dataat Neurology.org
Translational approach to address therapyin myotonia permanens due to a newSCN4A mutation
ABSTRACT
Objective: We performed a clinical, functional, and pharmacologic characterization of the novelp.P1158L Nav1.4 mutation identified in a young girl presenting a severe myotonic phenotype.
Methods: Wild-type hNav1.4 channel and P1158L mutant were expressed in tsA201 cells forfunctional and pharmacologic studies using patch-clamp.
Results: The patient shows pronounced myotonia, slowness of movements, and generalized mus-cle hypertrophy. Because of general discomfort with mexiletine, she was given flecainide with sat-isfactory response. In vitro, mutant channels show a slower current decay and a rightward shift ofthe voltage dependence of fast inactivation. The voltage dependence of activation and slow inac-tivation were not altered. Mutant channels were less sensitive to mexiletine, whereas sensitivityto flecainide was not altered. The reduced inhibition of mutant channels by mexiletine was alsoobserved using clinically relevant drug concentrations in a myotonic-like condition.
Conclusions: Clinical phenotype and functional alterations of P1158L support the diagnosis ofmyotonia permanens. Impairment of fast inactivation is consistent with the possible role of thechannel domain III S4-S5 loop in the inactivation gate docking site. The reduced sensitivity ofP1158L to mexiletine may have contributed to the unsatisfactory response of the patient. Thesuccess of flecainide therapy underscores the usefulness of in vitro functional studies to helpin the choice of the best drug for each individual. Neurology® 2016;86:2100–2108
GLOSSARYDM1 5 myotonic dystrophy type 1; IC50 5 half-maximum inhibitory concentration; TB 5 tonic block; UDB 5 use-dependentblock; UDR 5 use-dependent reduction; WT 5 wild-type.
Gain-of-function missense mutations of the skeletal muscle Nav1.4 sodium channel cause myo-tonia or flaccid weakness. Impaired fast-inactivation of hNav1.4 mutants likely induces myoto-nia, while enhancement of activation and impaired slow inactivation contribute to paralyticattacks.1 Various substitutions at the same amino acid (Gly1306) induce various degrees ofchannel alteration, which correlate to the severity of symptoms ranging from the mild myotoniafluctuans (G1306A) to the severe myotonia permanens (G1306E).2 Unusual phenotype is alsoexplained by specific behavior of channel mutant. Carriers of the P1158S mutation showmyotonia in a warm environment and paralytic attacks at cold temperatures.3 Accordingly,heterologously expressed P1158S channels show alteration of activation and slow inactivationat 22°C but not 37°C.4,5
The sodium channel blocker mexiletine is the preferred drug to alleviate myotonia.6 Never-theless, in case of side effects or lack of efficacy, mexiletine can be substituted for by anothersodium channel blocker. We previously demonstrated that flecainide is successful in mexiletine-low responsive patients carrying G1306E, likely due to mutation-specific gating changes.7–9
From the Departments of Biomedical Sciences and Human Oncology (J.-F.D.) and Pharmacy & Drug Sciences (R.C., J.R., P.I., D.C.C.),University of Bari Aldo Moro, Bari; Unit of Neuromuscular and Neurodegenerative Disorders (A.D.), Bambino Gesù Children’s Hospital, Rome;Departments of Geriatrics, Neurosciences, and Orthopedics (A.M., M.L.M.), Institute of Neurology, Catholic University of the Sacred Heart,Rome; Dino Ferrari Centre (S.P., S.L.), Neuroscience Section, Department of Pathophysiology and Transplantation (DEPT), University of Milan;and Neurology Unit (S.P., S.L.), IRCCS Foundation Ca’ Granda, Ospedale Maggiore Policlinico, Milan, Italy.
Go to Neurology.org for full disclosures. Funding information and disclosures deemed relevant by the authors, if any, are provided at the end of the article.The Article Processing Charge was paid by Telethon Italy.
This is an open access article distributed under the terms of the Creative Commons Attribution Licence 4.0 (CC BY), which permits unrestricteduse, distribution, and reproduction in any medium, provided the original work is properly cited.
2100 © 2016 American Academy of Neurology

Copyright 2016 American Medical Association. All rights reserved.
Association of a Locus in the CAMTA1 Gene With Survivalin Patients With Sporadic Amyotrophic Lateral SclerosisIsabella Fogh, PhD; Kuang Lin, PhD; Cinzia Tiloca, PhD; James Rooney, MD; Cinzia Gellera, PhD; Frank P. Diekstra, MD; Antonia Ratti, PhD;Aleksey Shatunov, PhD; Michael A. van Es, MD, PhD; Petroula Proitsi, PhD; Ashley Jones, PhD; William Sproviero, PhD; Adriano Chiò, MD;Russell Lewis McLaughlin, PhD; Gianni Sorarù, MD, PhD; Lucia Corrado, PhD; Daniel Stahl, PhD; Roberto Del Bo, PhD; Cristina Cereda, PhD;Barbara Castellotti, PhD; Jonathan D. Glass, MD; Steven Newhouse, PhD; Richard Dobson, PhD; Bradley N. Smith, PhD; Simon Topp, MSc;Wouter van Rheenen, MD; Vincent Meininger, MD, PhD; Judith Melki, MD, PhD; Karen E. Morrison, MD; Pamela J. Shaw, MD;P. Nigel Leigh, MD, PhD; Peter M. Andersen, MD, DMSc; Giacomo P. Comi, MD; Nicola Ticozzi, MD; Letizia Mazzini, MD;Sandra D’Alfonso, PhD; Bryan J. Traynor, MD; Philip Van Damme, MD, PhD; Wim Robberecht, MD, PhD; Robert H. Brown, MD, DPhil;John E. Landers, PhD; Orla Hardiman, MD, FRCPI; Cathryn M. Lewis, PhD; Leonard H. van den Berg, MD, PhD; Christopher E. Shaw, MD;Jan H. Veldink, MD, PhD; Vincenzo Silani, MD; Ammar Al-Chalabi, PhD, FRCP; John Powell, PhD
IMPORTANCE Amyotrophic lateral sclerosis (ALS) is a devastating adult-onsetneurodegenerative disorder with a poor prognosis and a median survival of 3 years. However,a significant proportion of patients survive more than 10 years from symptom onset.
OBJECTIVE To identify gene variants influencing survival in ALS.
DESIGN, SETTING, AND PARTICIPANTS This genome-wide association study (GWAS) analyzedsurvival in data sets from several European countries and the United States that werecollected by the Italian Consortium for the Genetics of ALS and the International Consortiumon Amyotrophic Lateral Sclerosis Genetics. The study population included 4256 patients withALS (3125 [73.4%] deceased) with genotype data extended to 7 174 392 variants byimputation analysis. Samples of DNA were collected from January 1, 1993, to December 31,2009, and analyzed from March 1, 2014, to February 28, 2015.
MAIN OUTCOMES AND MEASURES Cox proportional hazards regression under an additivemodel with adjustment for age at onset, sex, and the first 4 principal components of ancestry,followed by meta-analysis, were used to analyze data. Survival distributions for the mostassociated genetic variants were assessed by Kaplan-Meier analysis.
RESULTS Among the 4256 patients included in the analysis (2589 male [60.8%] and 1667female [39.2%]; mean [SD] age at onset, 59 [12] years), the following 2 novel loci weresignificantly associated with ALS survival: at 10q23 (rs139550538; P = 1.87 × 10−9) and inthe CAMTA1 gene at 1p36 (rs2412208, P = 3.53 × 10−8). At locus 10q23, the adjusted hazardratio for patients with the rs139550538 AA or AT genotype was 1.61 (95% CI, 1.38-1.89;P = 1.87 × 10−9), corresponding to an 8-month reduction in survival compared with TTcarriers. For rs2412208 CAMTA1, the adjusted hazard ratio for patients with the GG or GTgenotype was 1.17 (95% CI, 1.11-1.24; P = 3.53 × 10−8), corresponding to a 4-month reductionin survival compared with TT carriers.
CONCLUSIONS AND RELEVANCE This GWAS robustly identified 2 loci at genome-wide levelsof significance that influence survival in patients with ALS. Because ALS is a rare disease andprevention is not feasible, treatment that modifies survival is the most realistic strategy.Therefore, identification of modifier genes that might influence ALS survival could improvethe understanding of the biology of the disease and suggest biological targets forpharmaceutical intervention. In addition, genetic risk scores for survival could be used asan adjunct to clinical trials to account for the genetic contribution to survival.
JAMA Neurol. 2016;73(7):812-820. doi:10.1001/jamaneurol.2016.1114Published online May 31, 2016.
Editorial page 781
Supplemental content atjamaneurology.com
Author Affiliations: Authoraffiliations are listed at the end of thisarticle.
Corresponding Author: IsabellaFogh, PhD, Department of Basic andClinical Neuroscience, Maurice WohlClinical Neuroscience Institute,Institute of Psychiatry, Psychology,and Neuroscience, King’s CollegeLondon, James Black Centre,125 Coldharbour Ln, London SE59NU, England ([email protected]).
Research
Original Investigation
812 (Reprinted) jamaneurology.com
Copyright 2016 American Medical Association. All rights reserved.
Downloaded From: http://jamanetwork.com/ by a Biblioteca IRCCS Ospedale Maggiore - Milano User on 12/15/2016

Copyright 2016 American Medical Association. All rights reserved.
Conclusions
We have identified genetic variants that have a statistically sig-nificant association with survival. The promise of this re-
search is not only to improve our understanding of the biol-ogy of the disease and suggest biological targets forpharmaceutical intervention to extend the survival time of thepatients but also to use genetic risk scores as an adjunct to clini-cal trials to account for the genetic contribution to survival.
ARTICLE INFORMATION
Accepted for Publication: March 17, 2016.
Published Online: May 31, 2016.doi:10.1001/jamaneurol.2016.1114.
Author Affiliations: Department of Basic andClinical Neuroscience, Maurice Wohl ClinicalNeuroscience Institute, Institute of Psychiatry,Psychology, and Neuroscience (IoPPN), King’sCollege London, London, England (Fogh, Lin,Shatunov, Proitsi, Jones, Sproviero, Smith, Topp,C. E. Shaw, Al-Chalabi, Powell); Department ofNeurology and Laboratory of Neuroscience, Istitutodi Ricovero e Cura a Carattere Scientifico (IRCCS)Istituto Auxologico Italiano, Milano, Italy (Tiloca,Ratti, Ticozzi, Silani); Academic Unit of Neurology,Trinity College Dublin, Trinity Biomedical SciencesInstitute, Dublin, Ireland (Rooney); Unit of Geneticsof Neurodegenerative and Metabolic Diseases,Fondazione IRCCS Istituto Neurologico Carlo Besta,Milano, Italy (Gellera, Castellotti); Department ofNeurology and Neurosurgery, Brain Center RudolfMagnus, University Medical Center Utrecht,Utrecht, the Netherlands (Diekstra, van Es, vanRheenen, van den Berg, Veldink); Department ofPathophysiology and Tranplantation, Dino FerrariCenter, Università degli Studi di Milano, Milano, Italy(Ratti, Ticozzi, Silani); Rita Levi MontalciniDepartment of Neuroscience, ALS (AmyotrophicLateral Sclerosis) Centre, University of Torino, Turin,Italy (Chiò); Azienda Ospedaliera Città della Salutee della Scienza, Torino, Italy (Chiò); PopulationGenetics Laboratory, Smurfit Institute of Genetics,Trinity College Dublin, Dublin, Ireland (McLaughlin,Hardiman); Department of Neurosciences,University of Padova, Padua, Italy (Sorarù);Department of Health Sciences, InterdisciplinaryResearch Center of Autoimmune Diseases,A. Avogadro University, Novara, Italy (Corrado,Mazzini, D’Alfonso); Department of Biostatistics,IoPPN, King’s College London, London, England(Stahl); Neurologic Unit, IRCCS Foundation Ca’Granda Ospedale Maggiore Policlinico, Milan, Italy(Del Bo, Comi); Laboratory of ExperimentalNeurobiology, IRCCS C. Mondino National Instituteof Neurology Foundation, Pavia, Italy (Cereda);Department of Neurology, Emory University,Atlanta, Georgia (Glass); National Institute forHealth Research (NIHR) Biomedical ResearchCentre for Mental Health, IoPPN, King’s CollegeLondon, London, England (Newhouse, Dobson);Department of Biostatistics, IoPPN, King’s CollegeLondon, London, England (Newhouse); NIHRBiomedical Research Unit in Dementia, King’sCollege London, London, England (Dobson);Département des Maladies du Système Nerveux,Assistance Publique–Hôpitaux de Paris, Réseau SLA(Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); InstitutNational de la Santé et de la Recherche MedicaleUnité Mixte de Recherché–788 and University ofParis 11, Bicetre Hospital, Paris, France (Melki);School of Clinical and Experimental Medicine,College of Medicine and Dentistry, University ofBirmingham, Birmingham, England (Morrison);
Neurosciences Division, University HospitalsBirmingham National Health Service FoundationTrust, Birmingham, England (Morrison); AcademicNeurology Unit, Department of Neuroscience,Faculty of Medicine, Dentistry and Health,University of Sheffield, Sheffield, England(P. J. Shaw); Section of Neurology, Division ofMedicine, Brighton and Sussex Medical School,Trafford Centre for Biomedical Research, Universityof Sussex, East Sussex, England (Leigh); Institute ofClinical Molecular Biology, Kiel University, Kiel,Germany (Andersen); Department ofPharmacology and Clinical Neuroscience, UmeåUniversity, Umeå, Sweden (Andersen); ALS CenterDepartment of Neurology, Maggiore della CaritàUniversity Hospital, Novara, Italy (Mazzini);Neuromuscular Diseases Research Section,Laboratory of Neurogenetics, National Institute onAging, National Institutes of Health, Bethesda,Maryland (Traynor); Department of Neurosciences,Experimental Neurology, Flanders Instititue forBiotechnology, Vesalius Research Center,Laboratory of Neurobiology, KU Leuven–Universityof Leuven, Leuven, Belgium (Van Damme,Robberecht); Department of Neurology, UniversityHospitals Leuven, Leuven, Belgium (Van Damme);Department of Neurology, University ofMassachusetts Medical School, Worcester (Brown,Landers); IoPPN Genomics and Biomarker Core,Translational Genetics Group, Medical ResearchCouncil Social, Genetic and DevelopmentalPsychiatry Centre, King’s College London, London,England (Lewis); Department of Medical andMolecular Genetics, King’s College London, London,England (Lewis).
Author Contributions: Drs Silani, Al-Chalabi, andPowell are cosenior authors. Drs Fogh and Powellhad full access to all the data in the study and takeresponsibility for the integrity of the data and theaccuracy of the data analysis.Study concept and design: Fogh, Landers,Al-Chalabi, Powell.Acquisition, analysis, or interpretation of data: Allauthors.Drafting of the manuscript: Fogh, Tiloca, Ratti,Al-Chalabi, Powell.Critical revision of the manuscript for importantintellectual content: Lin, Rooney, Gellera, Diekstra,Ratti, Shatunov, van Es, Proitsi, Jones, Sproviero,Chiò, McLaughlin, Sorarù, Corrado, Stahl, Del Bo,Cereda, Castellotti, Glass, Newhouse, Dobson,Smith, Topp, van Rheenen, Meininger, Melki,Morrison, P. J. Shaw, Leigh, Andersen, Comi,Ticozzi, Mazzini, D’Alfonso, Traynor, Van Damme,Robberecht, Brown, Landers, Hardiman, Lewis,van den Berg, C. E. Shaw, Veldink, Silani, Al-Chalabi.Statistical analysis: Fogh, Lin, Rooney, Proitsi, Stahl,Newhouse, Landers, Lewis.Obtained funding: Fogh, Chiò, P. J. Shaw, Ticozzi,D’Alfonso, Van Damme, Robberecht, van den Berg,Al-Chalabi.Administrative, technical, or material support:Gellera, Diekstra, Ratti, van Es, Jones, Del Bo,Castellotti, Smith, Meininger, Morrison, Shaw,Andersen, Ticozzi, Traynor, Van Damme,
Robberecht, Landers, Hardiman, van den Berg,C. E. Shaw, Veldink, Al-Chalabi.Study supervision: Newhouse, Lewis, Silani,Al-Chalabi, Powell.
Conflict of Interest Disclosures: Dr Traynorreports having a patent pending on the clinicaltesting and therapeutic intervention for thehexanucleotide repeat expansion of C9orf72.No other disclosures were reported.
Funding/Support: This study was supported bygrant 905-793 6058 from the Motor NeuroneDisease Association of Great Britain and NorthernIreland and by Emergency Medicine Clinical Trialspilot funding awards from the National Institutes forHealth Research (NIHR) Biomedical Research Centrefor Mental Health at the South London and MaudsleyNational Health Service Foundation Trust andInstitute of Psychiatry, King’s College London(Dr Fogh); by grant NOVALS 2012 from the AgenziaItaliana per la Ricerca sulla Fondazione Italiana diRicerca per la Sclerosi Laterale Amiotrofica(SLA-AriSLA), cofinanced with the contribution of5 × 1000 Healthcare Research support of theMinistry of Health, Ricerca Finalizzata RF-2009-1473856 from the Ministero della Salute andAssociazione Amici Centro Dino Ferrari (Drs Tiloca,Gellera, Ratti, Ticozzi, and Silani); by the HealthResearch Board Clinical Fellowship Programme (DrRooney); by Ricerca Finalizzata RF-2010-2309849from the Ministero della Salute and FP7/2007-2013under grant 259867 from the European Community’sHealth Seventh Framework Programme (Dr Chiò);by AriSLA for the Repeat ALS Project 2013 (DrD’Alfonso); by the Netherlands Organization forHealth Research and Development (Drs Diekstra,van Es [Veni Scheme], van Rheenen, van den Berg[Vici Scheme], and Veldink); by the Thierry LatranFoundation, the Dutch ALS Foundation, and a talentfellowship from the Rudolf Magnus Brain Center (Drvan Es); by the Princess Beatrix Fonds, the PrincessBeatrix Fund Muscle, H. Kersten and M. KerstenFoundation, the Netherlands ALS Foundation, andDr van Dijk and the Adessium Foundation (Dutch,Belgian and Swedish genome-wide association studydata generation); by grant 259867 from theEuropean Community’s Health Seventh FrameworkProgramme; through the E. von Behring Chair forNeuromuscular and Neurodegenerative Disordersand Geneeskundige Stichting Koningin Elisabeth(GSKE) and grant 340429 from the EuropeanResearch Council under the European’s SeventhFramework Programme (Dr Robberecht); by grants259867 and 278611 from the InteruniversityAttraction Poles (IUAP) program P7/16 of the BelgianFederal Science Policy Office and the EuorpeanCommunity’s Seventh Framework Programme(ADAMS project, HEALTH-F4-2009-242257 andFP7/2007-2013); by a clinical investigatorship fromFonds Wetenschappelijk Onderzoek–Vlaanderen(Dr Van Damme); by the Motor Neurone DiseaseAssociation of Great Britain and Northern Ireland, theALS Foundation Netherlands (project MinE), and theEuropean Community’s Health Seventh FrameworkProgramme (Drs Al-Chalabi, Shatunov, Jones, andSproviero); by ZonMW under the framework of
CAMTA1 Gene and Survival in Sporadic Amyotrophic Lateral Sclerosis Original Investigation Research
jamaneurology.com (Reprinted) JAMA Neurology July 2016 Volume 73, Number 7 819
Copyright 2016 American Medical Association. All rights reserved.
Downloaded From: http://jamanetwork.com/ by a Biblioteca IRCCS Ospedale Maggiore - Milano User on 12/15/2016

O R I G I N A L A R T I C L E
iPSC-derived LewisXþCXCR4þb1-integrinþ neural
stem cells improve the amyotrophic lateral sclerosis
phenotype by preserving motor neurons and muscle
innervation in human and rodent modelsMonica Nizzardo1, Monica Bucchia1, Agnese Ramirez1, Elena Trombetta2,Nereo Bresolin1, Giacomo P. Comi1 and Stefania Corti1,*1Dino Ferrari Centre, Neuroscience Section, Department of Pathophysiology and Transplantation (DEPT),University of Milan, Neurology Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy,and 2Flow Cytometry Service, Clinical Chemistry and Microbiology Laboratory Fondazione IRCCS Ca’ GrandaOspedale Maggiore Policlinico, Milan, Italy
*To whom correspondence should be addressed at: Neuroscience Section, Department of Pathophysiology and Transplantation (DEPT), University ofMilan, Neurology Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Via Francesco Sforza 35, 20122 Milan Italy. Tel: þ39 0255033833;Fax: þ39 0250320430; Email: [email protected]
AbstractAmyotrophic lateral sclerosis (ALS) is a fatal incurable neurodegenerative disease characterized by progressive degenerationof motor neurons (MNs), leading to relentless muscle paralysis. In the early stage of the disease, MN loss and consequentmuscle denervation are compensated by axonal sprouting and reinnervation by the remaining MNs, but this mechanism isinsufficient in the long term. Here, we demonstrate that induced pluripotent stem cell-derived neural stem cells (NSCs), inparticular the subpopulation positive for LewisX-CXCR4-b1-integrin, enhance neuronal survival and axonal growth of humanALS-derived MNs co-cultured with toxic ALS astrocytes, acting on both autonomous and non-autonomous ALS disease fea-tures. Transplantation of this NSC fraction into transgenic SOD1G93A ALS mice protects MNs in vivo, promoting their abilityto maintain neuromuscular junction integrity, inducing novel axonal sprouting and reducing macro- and microgliosis. Theseeffects result in a significant increase in survival and an improved neuromuscular phenotype in transplanted SOD1G93Amice. Our findings suggest that effective protection of MN functional innervation can be achieved by modulation of multipledysregulated cellular and molecular pathways in both MNs and glial cells. These pathways must be considered in designingtherapeutic strategies for ALS patients.
IntroductionAmyotrophic lateral sclerosis (ALS) is a fatal incurable neurode-generative disease of unknown aetiology characterized by theselective degeneration of motor neurons (MNs), leading to a
rapidly progressive paralysis and precocious death due to respi-ratory failure, typically within 3–5 years of symptom onset (1,2).Most ALS cases are sporadic (sALS), and only 5–10% of cases arefamilial (fALS) (3). The mechanisms underlying the
Received: March 28, 2016. Revised: May 16, 2016. Accepted: May 19, 2016
VC The Author (2016). Published by Oxford University Press on behalf of the Human Molecular Genetics.All rights reserved. For permissions, please e-mail: [email protected]
1
Human Molecular Genetics, 2016, Vol. 0, No. 0 1–12
doi: 10.1093/hmg/ddw163Advance Access Publication Date: 6 June 2016Original Article
HMG Advance Access published June 19, 2016 at B
iblioteca IRC
CS O
spedale Maggiore - M
ilano on Decem
ber 13, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

O R I G I N A L A R T I C L E
Selective mitochondrial depletion, apoptosis
resistance, and increased mitophagy in human
Charcot-Marie-Tooth 2A motor neuronsFederica Rizzo1, Dario Ronchi1, Sabrina Salani1, Monica Nizzardo1,Francesco Fortunato1, Andreina Bordoni1, Giulia Stuppia1, Roberto Del Bo1,Daniela Piga1, Romana Fato2, Nereo Bresolin1, Giacomo P. Comi1 andStefania Corti1*1Dino Ferrari Centre, Neuroscience Section, Department of Pathophysiology and Transplantation (DEPT),University of Milan, Neurology Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italyand 2Department of Pharmacy and Biotecnology (FaBiT), University of Bologna, Bologna, Italy
*To whom correspondence should be addressed at: Neuroscience Section, Department of Pathophysiology and Transplantation (DEPT), University ofMilan, Neurology Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Via Francesco Sforza 35, 20122 Milan Italy. Tel: þ39 0255033833;Fax: þ39 0250320430; Email: [email protected]
AbstractCharcot-Marie-Tooth 2A (CMT2A) is an inherited peripheral neuropathy caused by mutations in MFN2, which encodes amitochondrial membrane protein involved in mitochondrial network homeostasis. Because MFN2 is expressed ubiquitously,the reason for selective motor neuron (MN) involvement in CMT2A is unclear. To address this question, we generated MNsfrom induced pluripotent stem cells (iPSCs) obtained from the patients with CMT2A as an in vitro disease model. CMT2AiPSC-derived MNs (CMT2A-MNs) exhibited a global reduction in mitochondrial content and altered mitochondrial positioningwithout significant differences in survival and axon elongation. RNA sequencing profiles and protein studies of keycomponents of the apoptotic executioner program (i.e. p53, BAX, caspase 8, cleaved caspase 3, and the anti-apoptotic markerBcl2) demonstrated that CMT2A-MNs are more resistant to apoptosis than wild-type MNs. Exploring the balance betweenmitochondrial biogenesis and the regulation of autophagy-lysosome transcription, we observed an increased autophagic fluxin CMT2A-MNs that was associated with increased expression of PINK1, PARK2, BNIP3, and a splice variant of BECN1 that wasrecently demonstrated to be a trigger for mitochondrial autophagic removal. Taken together, these data suggest that thestriking reduction in mitochondria in MNs expressing mutant MFN2 is not the result of impaired biogenesis, but more likelythe consequence of enhanced mitophagy. Thus, these pathways represent possible novel molecular therapeutic targets forthe development of an effective cure for this disease.
IntroductionCharcot-Marie-Tooth (CMT) type 2A (OMIM 609260, CMT2A� 35%of CMT2) is an inherited sensorimotor axonopathy caused by
missense mutations in the mitofusin 2 (MFN2) gene (1,2) and char-acterized by peripheral motor and sensitive neuron degeneration(3–7). MFN2 is a GTPase dynamin-like protein of the outer
Received: May 25, 2016. Revised: July 21, 2016. Accepted: July 21, 2016
VC The Author 2016. Published by Oxford University Press. All rights reserved. For Permissions, please email: [email protected]
1
Human Molecular Genetics, , Vol. 0, No. 0 1–16
doi: 10.1093/hmg/ddw258Advance Access Publication Date: 9 August 2016Original Article
HMG Advance Access published August 24, 2016 at B
iblioteca IRC
CS O
spedale Maggiore - M
ilano on Decem
ber 13, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

Nature GeNetics VOLUME 48 | NUMBER 9 | SEPTEMBER 2016 1037
l e t t e r s
To identify genetic factors contributing to amyotrophic lateral sclerosis (ALS), we conducted whole-exome analyses of 1,022 index familial ALS (FALS) cases and 7,315 controls. In a new screening strategy, we performed gene-burden analyses trained with established ALS genes and identified a significant association between loss-of-function (LOF) NEK1 variants and FALS risk. Independently, autozygosity mapping for an isolated community in the Netherlands identified a NEK1 p.Arg261His variant as a candidate risk factor. Replication analyses of sporadic ALS (SALS) cases and independent control cohorts confirmed significant disease association for both p.Arg261His (10,589 samples analyzed) and NEK1 LOF variants (3,362 samples analyzed). In total, we observed NEK1 risk variants in nearly 3% of ALS cases. NEK1 has been linked to several cellular functions, including cilia formation, DNA-damage response, microtubule stability, neuronal morphology and axonal polarity. Our results provide new and important insights into ALS etiopathogenesis and genetic etiology.
In recent years, the combination of exome sequencing, segregation analysis and bioinformatic filtering has proven to be an effective strat-egy to rapidly identify new disease genes1. Unfortunately, this method can be difficult to apply to disorders such as ALS, for which late age of onset and low-to-modest variant penetrance make it difficult to obtain large informative multigenerational pedigrees. Owing to high genetic heterogeneity, ALS is also difficult to analyze using filter-ing methods designed to exploit unrelated patient groups2. Recently, we had demonstrated the utility of exome-wide rare variant burden (RVB) analysis as an alternate approach, identifying a replicable association between FALS risk and TUBA4A in a cohort of 363 cases3. In brief, RVB analysis is used to compare the combined frequency of rare variants in each gene in a case–control cohort. Candidate associations are identified by significant differences after multiple-test correction. Since this initial study, we extended our data set to include complete exome sequencing for 1,376 index FALS cases and 13,883 controls. Of these, 1,022 cases and 7,315 controls met all required data, inter-relatedness and ancestral quality control criteria (Supplementary Figs. 1 and 2, and Online Methods).
Successful detection of disease associations through RVB analysis can depend heavily on the appropriate setting of test parameters. As genetic loci often contain many alleles of no or low effect, prior filtering of variants based on minor allele frequency (MAF) and pathogenicity predictors can identify disease signatures otherwise
masked by normal human variability. As appropriate MAF or patho-genicity predictor settings may not be obvious in advance, compre-hensive assessment of all pursuable analysis strategies is desirable but can in turn introduce excessive multiple-test burden. To overcome these limitations, we performed 308 distinct RVB analyses of ten well-established ALS genes using 44 functional and 7 MAF filters (Fig. 1a). All tests included correction for gene coverage and ancestral covariates (Online Methods). In the final cohort, 72 cases and 0 controls harbored known ALS pathogenic mutations in these ten genes (Online Methods). An additional 26 cases harbored a repeat expansion in the C9orf72 gene. Tests differed in their capacity to detect individual known ALS genes (Supplementary Table 1), but we achieved the highest net sensitivity when we restricted analyses to variants with MAF < 0.001 and functional classifications of either non-sense, splice-altering4 or deemed deleterious by functional analysis through hidden Markov models (FATHMM)5. Under these settings, four genes exhibited disease association at exome-wide (Bonferroni-corrected P < 2.5 × 10−6) significance (SOD1, TARDBP, UBQLN2 and FUS), three achieved near exome-wide significance (TUBA4A, TBK1 and VCP), and three displayed modest to marginal disease association (PFN1, VAPB and OPTN) (Fig. 1b). Genes exhibiting the strongest disease associations included those reported as major ALS genes in population-based studies, whereas those exhibiting weaker associa-tions are believed to constitute rarer causes of disease.
Extension of the optimal known ALS gene parameters to all protein- coding genes identified one new gene displaying exome-wide sig-nificant disease association (Fig. 1b). The gene, NEK1 (odds ratio (OR) = 8.2, P = 1.7 × 10−6), encodes the serine/threonine kinase NIMA (never in mitosis gene-A)-related kinase. Retesting NEK1 under alternate analysis parameters identified strong disease associations across most analysis strategies, particularly where we included LOF (nonsense and predicted splice-altering) variants (Supplementary Table 2 and Supplementary Fig. 3). We observed no evidence for systematic genomic inflation (λ = 0.95), confounding related to sample ascertainment (Supplementary Fig. 4) or case–control biases in NEK1 gene coverage (Supplementary Fig. 5). Removal of samples carrying rare variants of known ALS genes did not influence the association (OR = 8.9, P = 7.3 × 10−7).
In an independent line of research, we performed whole-genome sequencing for four ALS patients from an isolated community in the Netherlands (population < 25,000). We observed high inbreeding coefficients for each of the four patients, confirm-ing their high degree of relatedness and supporting a restricted
NEK1 variants confer susceptibility to amyotrophic lateral sclerosis
A full list of authors and affiliations appears at the end of the paper.
Received 25 February; accepted 24 June; published online 25 July 2016; doi:10.1038/ng.3626
npg
© 2
016
Nat
ure
Am
eric
a, In
c. A
ll rig
hts
rese
rved
.np
g©
2016
Nat
ure
Am
eric
a, In
c. A
ll rig
hts
rese
rved
.

Nature GeNetics VOLUME 48 | NUMBER 9 | SEPTEMBER 2016 1041
l e t t e r s
8. Brenner, D. et al. NEK1 mutations in familial amyotrophic lateral sclerosis. Brain 139, e28 (2016).
9. Renton, A.E., Chiò, A. & Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17, 17–23 (2014).
10. Kenna, K.P. et al. Delineating the genetic heterogeneity of ALS using targeted high-throughput sequencing. J. Med. Genet. 50, 776–783 (2013).
11. Lattante, S. et al. Contribution of major amyotrophic lateral sclerosis genes to the etiology of sporadic disease. Neurology 79, 66–72 (2012).
12. Chiò, A. et al. Extensive genetics of ALS: a population-based study in Italy. Neurology 79, 1983–1989 (2012).
13. Thiel, C. et al. NEK1 mutations cause short-rib polydactyly syndrome type majewski. Am. J. Hum. Genet. 88, 106–114 (2011).
14. Shalom, O., Shalva, N., Altschuler, Y. & Motro, B. The mammalian Nek1 kinase is involved in primary cilium formation. FEBS Lett. 582, 1465–1470 (2008).
15. White, M.C. & Quarmby, L.M. The NIMA-family kinase, Nek1 affects the stability of centrosomes and ciliogenesis. BMC Cell Biol. 9, 29 (2008).
16. Lee, J.H. & Gleeson, J.G. The role of primary cilia in neuronal function. Neurobiol. Dis. 38, 167–172 (2010).
17. Lee, L. Riding the wave of ependymal cilia: genetic susceptibility to hydrocephalus in primary ciliary dyskinesia. J. Neurosci. Res. 91, 1117–1132 (2013).
18. Cohen, S., Aizer, A., Shav-Tal, Y., Yanai, A. & Motro, B. Nek7 kinase accelerates microtubule dynamic instability. Biochim. Biophys. Acta 1833, 1104–1113 (2013).
19. Chang, J., Baloh, R.H. & Milbrandt, J. The NIMA-family kinase Nek3 regulates microtubule acetylation in neurons. J. Cell Sci. 122, 2274–2282 (2009).
20. Puls, I. et al. Mutant dynactin in motor neuron disease. Nat. Genet. 33, 455–456 (2003).
21. Ma, X., Peterson, R. & Turnbull, J. Adenylyl cyclase type 3, a marker of primary cilia, is reduced in primary cell culture and in lumbar spinal cord in situ in G93A SOD1 mice. BMC Neurosci. 12, 71 (2011).
22. Chen, Y., Craigen, W.J. & Riley, D.J. Nek1 regulates cell death and mitochondrial membrane permeability through phosphorylation of VDAC1. Cell Cycle 8, 257–267 (2009).
23. Pelegrini, A.L. et al. Nek1 silencing slows down DNA repair and blocks DNA damage-induced cell cycle arrest. Mutagenesis 25, 447–454 (2010).
24. Sama, R.R., Ward, C.L. & Bosco, D.A. Functions of FUS/TLS from DNA repair to stress response: implications for ALS. ASN Neuro 6, 1759091414544472 (2014).
25. Tafuri, F., Ronchi, D., Magri, F., Comi, G.P. & Corti, S. SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front. Cell. Neurosci. 9, 336 (2015).
26. Madabhushi, R., Pan, L. & Tsai, L.H. DNA damage and its links to neurodegeneration. Neuron 83, 266–282 (2014).
27. Coppedè, F. & Migliore, L. DNA damage in neurodegenerative diseases. Mutat. Res. 776, 84–97 (2015).
28. Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell 162, 516–526 (2015).
29. Surpili, M.J., Delben, T.M. & Kobarg, J. Identification of proteins that interact with the central coiled-coil region of the human protein kinase NEK1. Biochemistry 42, 15369–15376 (2003).
kevin P kenna1,40, Perry t c van doormaal2,40, Annelot m dekker2,40, nicola ticozzi3,4,40, Brendan J kenna1, Frank P diekstra2, wouter van Rheenen2, kristel R van eijk2, Ashley R Jones5, Pamela keagle1, Aleksey shatunov5, william sproviero5, Bradley n smith5, michael A van es2, simon d topp5, Aoife kenna1, Jack w miller5, claudia Fallini1, cinzia tiloca3,6, Russell l mclaughlin7, caroline Vance5, claire troakes5, claudia colombrita3,4, gabriele mora8, Andrea calvo9, Federico Verde3,4, safa Al-sarraj5, Andrew king5, daniela calini3, Jacqueline de Belleroche10, Frank Baas11, Anneke J van der kooi12, marianne de Visser12, Anneloor l m A ten Asbroek11, Peter c sapp1, diane mckenna-Yasek1, meraida Polak13, seneshaw Asress13, José luis muñoz-Blanco14, tim m strom15, thomas meitinger16, karen e morrison17, slAgen consortium18, giuseppe lauria19, kelly l williams20, P nigel leigh21, garth A nicholson20,22, Ian P Blair20, claire s leblond23, Patrick A dion23, guy A Rouleau23, Hardev Pall24,25, Pamela J shaw26, martin R turner26, kevin talbot26, Franco taroni27, kevin B Boylan28, marka Van Blitterswijk29, Rosa Rademakers29, Jesús esteban-Pérez30,31, Alberto garcía-Redondo30,31, Phillip Van damme32,33, wim Robberecht32,33, Adriano chio9, cinzia gellera27, carsten drepper34,35, michael sendtner34, Antonia Ratti3,4, Jonathan d glass13, Jesús s mora36, nazli A Basak37, orla Hardiman7, Albert c ludolph38, Peter m Andersen39, Jochen H weishaupt38, Robert H Brown, Jr1, Ammar Al-chalabi5, Vincenzo silani3,4,41, christopher e shaw5,41, leonard H van den Berg2,41, Jan H Veldink2,41& John e landers1,41
1Department of Neurology, University of Massachusetts Medical School, Worcester, Massachusetts, USA. 2Department of Neurology Brain Centre, Brain Centre Rudolf Magnus, University Medical Centre Utrecht, Utrecht, the Netherlands. 3Department of Neurology, IRCCS Istituto Auxologico Italiano, Milan, Italy. 4Department of Pathophysiology and Transplantation, ‘Dino Ferrari’ Center, Università degli Studi di Milano, Milan, Italy. 5Maurice Wohl Clinical Neuroscience Institute, King’s College London, Department of Basic and Clinical Neuroscience, Institute of Psychiatry, Psychology and Neuroscience, London, UK. 6Doctoral School in Molecular Medicine, Department of Sciences and Biomedical Technologies, Università degli Studi di Milano, Milan, Italy. 7Academic Unit of Neurology, Trinity Biomedical Sciences Institute, Trinity College Dublin, Dublin, Ireland. 8Salvatore Maugeri Foundation, IRCSS, Scientific Institute of Milano, Milan, Italy. 9‘Rita Levi Montalcini’ Department of Neuroscience, ALS Centre, University of Torino, Turin, Italy. 10Neurogenetics Group, Division of Brain Sciences, Imperial College London, London, UK. 11Department of Clinical Genetics, Academic Medical Centre, University of Amsterdam, Amsterdam, the Netherlands. 12Department of Neurogenetics and Neurology, Academic Medical Centre, University of Amsterdam, Amsterdam, the Netherlands. 13Department of Neurology, Emory University, Atlanta, Georgia, USA. 14Unidad de ELA, Instituto de Investigación Hospital Gregorio Marañón de Madrid, Madrid, Spain. 15Institute of Human Genetics, Helmholtz Zentrum München–German Research Center for Environmental Health, Neuherberg, Germany. 16Institute of Human Genetics, Technische Universität München, Munich, Germany. 17Faculty of Medicine, University of Southampton, Southampton, UK. 18A list of members and affiliations appears at the end of the paper. 193rd Neurology Unit, Motor Neuron Diseases Center, Fondazione IRCCS Istituto Neurologico ‘Carlo Besta’, Milan, Italy. 20Faculty of Medicine and Health Sciences, Macquarie University, Sydney, New South Wales, Australia. 21Trafford Centre for Medical Research, Brighton and Sussex Medical School, Falmer, UK. 22ANZAC Research Institute, Concord Hospital, University of Sydney, Sydney, New South Wales, Australia. 23Montreal Neurological Institute, Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada. 24Institute of Clinical Studies, College of Medical and Dental Sciences, University of Birmingham, Edgbaston, Birmingham, UK. 25Department of Neurology, Queen Elizabeth Hospital Birmingham, Edgbaston, Birmingham, UK. 26Nuffield Department of Clinical Neurosciences, University of Oxford, Oxford, UK. 27Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico ‘Carlo Besta’, Milan, Italy. 28Department of Neurology, Mayo Clinic Florida, Jacksonville, Florida, USA. 29Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA. 30Unidad de ELA, Instituto de Investigación Hospital 12 de Octubre de Madrid, Madrid, Spain. 31Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) U-723, Madrid, Spain. 32Laboratory of Neurobiology, Department of Neurosciences, KU Leuven and Vesalius Research Centre, VIB, Leuven, Belgium. 33Department of Neurology, University Hospitals, Leuven, Belgium. 34Institute of Clinical Neurobiology, University Hospital Würzburg, Würzburg, Germany. 35Department of Child and Adolescent Psychiatry, University Hospital of Würzburg, Würzburg, Germany. 36ALS Unit/Neurology, Hospital San Rafael, Madrid, Spain. 37NDAL, Department of Molecular Biology and Genetics, Bogazici University, Istanbul, Turkey. 38Neurology Department, Ulm University, Ulm, Germany. 39Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden. 40These authors contributed equally to this work. 41These authors jointly directed this work. Correspondence should be addressed to J.H.V. ([email protected]).
npg
© 2
016
Nat
ure
Am
eric
a, In
c. A
ll rig
hts
rese
rved
.np
g©
2016
Nat
ure
Am
eric
a, In
c. A
ll rig
hts
rese
rved
.

A case report with the peculiar concomitance of2 different genetic syndromesAlberto Lerario, MDa, Irene Colombo, MDb, Donatella Milani, MDc, Lorenzo Peverelli, MDa, Luisa Villaa,Roberto Del Bo, MDd, Monica Sciacco, MDa, Giacomo Pietro Comi, MDd, Susanna Esposito, MDc,∗,Maurizio Moggio, MDa
AbstractRationale: Down syndrome (DS) is the most common chromosome disorder in live born infants, affecting several body systems,but usually sparing skeletal muscles. We present the case of a child with coexistence of DS and dystrophinopathy. Only 1 similar casehas been reported so far.
Patient Concerns: An 8-year-old boy with DS had a history of incidental finding of increased serum creatine kinase levels up to1775U/L (normal values 38–174U/L). He presented no delay in motor development; at the neurological examination, no muscleweakness or fatigability was detected in 2 different evaluations performed over a 6-month period.
Diagnoses: Skeletal muscle biopsy revealed marked dystrophic changes with patchy immunostaining for dystrophin. TheDuchenne muscular dystrophy gene was screened for deletions by multiplex polymerase chain reaction, but no mutations werefound. Sequence analysis of the Duchenne muscular dystrophy gene revealed a splice-site mutation c.1812+1G>A in intron 15 andconfirmed a diagnosis of Becker muscular dystrophy.
Interventions:The patient has started a specific physiotherapy that avoided any deterioration in motor development andmuscularwasting.
Outcomes: A multidisciplinary follow-up was initiated. The genetician that followed the patient for DS was supported by theneurologist, the physiotherapist, the pulmonologist, and the cardiologist.
Lessons: This peculiar “double trouble” case exemplifies the value of careful clinical evaluation and adequate clinical experience toidentify the concomitance of 2 different genetic syndromes in the same patient, and it points out the significance of muscular strengthassessment in DS patients to make the most correct prognosis, and, consequently, to organize the best long-term care.
Abbreviations: BMD = Becker muscular dystrophy, CK = creatine kinase, DMD = Duchenne muscular dystrophy, DS = Downsyndrome.
Keywords: Becker muscular dystrophy, coinheritance, Down syndrome, Duchenne muscular dystrophy
1. Introduction
Down syndrome (DS, OMIM #190685) is 1 of the most commonchromosome abnormalities in humans, occurring in about 1 per
1000 babies born each year.[1] It is caused by triplicate state(trisomy) of all or of a critical portion of chromosome 21, usuallyby nondisjunction.[2] It is characterized mainly by peculiar facialfeatures, physical growth delay, mild to moderate intellectualdisability, high rate of congenital heart defects and hypothyroid-ism, high risk for leukemia and lower, but significant rates ofgastrointestinal malformations, which often require surgerywithin the first days to weeks of life.[3] Other surgical conditionsthat occur at higher rates include Hirschsprung disease,polydactyly, cleft palate, and cataracts.Mutations in Duchenne muscular dystrophy (DMD) gene,
which encodes the protein dystrophin and is located onchromosome X,[4] cause a spectrum of muscle diseases affectingmale individuals and named dystrophinopathies. Two mainforms are recognized, namely Duchenne and Becker musculardystrophies (DMD and BMD, OMIM #310200 and #300376,respectively). DMD is the most common form of inherited muscledisease in childhood, with an estimated incidence of 1 in 3500born males.[5] It presents in early childhood and it is rapidlyprogressive, with affected children being wheelchair-bound by 12years of age. BMD is a generally milder andmore variable form ofdystrophinopathy, with an incidence of 1 in 18,518 malebirths.[5] Indeed, BMD phenotype can range from asymptomaticindividuals with increased serum creatine kinase (CK) levels andmuscle cramps, to more invalidating forms with severe muscleweakness and/or dilated cardiomyopathy. BMD patients usuallyremain ambulatory into their 20s.[6] The Duchenne phenotype is
Editor: Catherine Clelland.
The authors report no conflicts of interest.a Neuromuscular and Rare Disease Unit, Department of Neuroscience,Foundation IRCCS Ca’ Granda Ospedale Maggiore Policlinico, University ofMilan, Milan, b Azienda Ospedaliera di Desio e Vimercate, Neurology Unit, Desio,c Pediatric Highly Intensive Care Unit, Department of Pathophysiology andTransplantation, Università degli Studi di Milano, Fondazione IRCCS Ca’ GrandaOspedale Maggiore Policlinico, d Dino Ferrari Centre, Department ofPathophysiology and Transplantation Neuroscience Section (DEPT), NeurologyUnit, Foundation IRCCS Ca’ Granda Ospedale Maggiore Policlinico, University ofMilan, Milan, Italy.∗Correspondence: Susanna Esposito, Pediatric Highly Intensive Care Unit,
Department of Pathophysiology and Transplantation, Università degli Studi diMilano, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, ViaCommenda 9, 20122 Milan, Italy (e-mail: [email protected]).
Copyright © 2016 the Author(s). Published by Wolters Kluwer Health, Inc. Allrights reserved.This is an open access article distributed under the Creative CommonsAttribution-NoDerivatives License 4.0, which allows for redistribution, commercialand non-commercial, as long as it is passed along unchanged and in whole, withcredit to the author.
Medicine (2016) 95:49(e5567)
Received: 31 August 2016 / Received in final form: 13 November 2016 /Accepted: 15 November 2016
http://dx.doi.org/10.1097/MD.0000000000005567
Clinical Case Report Medicine®
OPEN
1

THE ITALIAN LIMB GIRDLE MUSCULAR DYSTROPHY REGISTRY:RELATIVE FREQUENCY, CLINICAL FEATURES, ANDDIFFERENTIAL DIAGNOSISFRANCESCA MAGRI, MD,1 VINCENZO NIGRO, MD,2,3 CORRADO ANGELINI, MD,4 TIZIANA MONGINI, MD,5
MARINA MORA, MD,6 ISABELLA MORONI, MD,7 ANTONIO TOSCANO, MD,8 MARIA GRAZIA D’ANGELO, MD, PhD,9
GIULIANO TOMELLERI, MD,10 GABRIELE SICILIANO, MD,11 GIULIA RICCI, MD,11 CLAUDIO BRUNO, MD,12
STEFANIA CORTI, MD, PhD,1 OLIMPIA MUSUMECI, MD,8 GIORGIO TASCA, MD, PhD,13 ENZO RICCI, MD, PhD,14
MAURO MONFORTE, MD,14 MONICA SCIACCO, MD,16 CHIARA FIORILLO, MD,17 SANDRA GANDOSSINI, MD,9
CARLO MINETTI, MD,12 LUCIA MORANDI, MD,6 MARCO SAVARESE, PhD,2,3 GIUSEPPINA DI FRUSCIO, PhD,2,3
CLAUDIO SEMPLICINI, MD,15 ELENA PEGORARO, MD,15 ALESSANDRA GOVONI, MD,1 ROBERTA BRUSA, MD,1
ROBERTO DEL BO, PhD,1 DARIO RONCHI, PhD,1 MAURIZIO MOGGIO, MD,16 NEREO BRESOLIN, MD,1 and
GIACOMO PIETRO COMI, MD1
1 Dino Ferrari Centre, Department of Pathophysiology and Transplantation, University of Milan, Neurology Unit, IRCCS FoundationCa’ Granda, Ospedale Maggiore Policlinico, Via Francesco Sforza 35, 20122 Milan, Italy
2 Department of General Pathology, University of Naples, Naples, Italy3 Telethon Institute of Genetics and Medicine, Naples, Italy4 Foundation S. Camillo Hospital, IRCCS, Lido, Venice, Italy5 Department of Neurosciences Rita Levi Montalcini, University of Torino, Torino, Italy6 Neuromuscular Diseases and Neuroimmunology Unit, Fondazione IRCCS Istituto Neurologico C. Besta, Milan, Italy7 Child Neurology Unit, IRCCS Foundation Istituto Neurologico C. Besta, Milan, Italy8 Department of Clinically and Experimental Medicine, University of Messina, Italy9 Neuromuscular Unit–IRCCS E. Medea Bosisio Parini, Bosisio Parini, Italy10 Department of Neurological Sciences, Verona, Italy11 Department of Clinical and Experimental Medicine, University of Pisa, Pisa, Italy12 Center of Myology and Neurodegenerative Diseases, Istituto Giannina Gaslini, Genova13 Don Carlo Gnocchi ONLUS Foundation, Rome, Italy14 Department of Neurology, Policlinico Universitario A. Gemelli, University Cattolica del Sacro Cuore of Rome, Rome, Italy15 Department of Neurosciences, University of Padua, Padua, Italy16 Dino Ferrari Centre, Neuromuscular and Rare Diseases Unit, IRCCS Foundation Ca’ Granda, Ospedale Maggiore Policlinico,
Milan, Italy17 IRCCS Fondazione Stella Maris, Calambrone, Italy
Accepted 13 May 2016
ABSTRACT: Introduction: Limb girdle muscular dystrophies(LGMDs) are characterized by high molecular heterogeneity,clinical overlap, and a paucity of specific biomarkers. Theirmolecular definition is fundamental for prognostic and therapeu-tic purposes. Methods: We created an Italian LGMD registrythat included 370 molecularly defined patients. We revieweddetailed retrospective and prospective data and compared eachLGMD subtype for differential diagnosis purposes. Results:LGMD types 2A and 2B are the most frequent forms in Italy.The ages at disease onset, clinical progression, and cardiacand respiratory involvement can vary greatly between each
LGMD subtype. In a set of extensively studied patients, tar-geted next-generation sequencing (NGS) identified mutations in36.5% of cases. Conclusion: Detailed clinical characterizationcombined with muscle tissue analysis is fundamental to guidedifferential diagnosis and to address molecular tests. NGS isuseful for diagnosing forms without specific biomarkers,although, at least in our study cohort, several LGMD diseasemechanisms remain to be identified.
Muscle Nerve 000:000–000, 2016
Limb girdle muscular dystrophies (LGMDs) areheterogeneous genetic disorders characterized byprogressive muscle impairment, predominantlyinvolving proximal and limb girdle muscles withonset after independent ambulation is achieved,and by histological signs of degeneration andregeneration in muscles.1,2 The clinical spectrumof LGMDs can vary from more severe infantileforms with early loss of independent ambulation tomilder adult-onset and slowly progressive weakness.Cardiac and respiratory involvement can be varia-bly present, and cognitive impairment is usuallyabsent. The estimated incidence of LGMDs innorthern England is 2.27 per 100,000,3 butupdated epidemiological data in Italy are lacking.
Additional supporting information may be found in the online version ofthis article.
Abbreviations: AD, autosomal dominant; AR, autosomal recessive; CK,creatine kinase; ECG, electrocardiogram; EF, ejection fraction; EMG, elec-tromyography; FVC, forced vital capacity; FEV1, forced expiratory volume;IHC, immunohistochemistry; LGMD, limb girdle muscular dystrophy; MFM,motor function measure; MRC, Medical Research Council; 6MWT, 6-minute walking test; NGS, next-generation sequencing; PCR, polymerasechain reaction; RBBB, right bundle branch blocks; SF, shortening fraction;TIGEM, Telethon Institute of Genetic and Medicine; WES, whole exomesequencing; WB, Western blotKey words: differential diagnosis; genotype–phenotype correlations; limbgirdle muscular dystrophy; natural history; next-generation sequencingThis study was supported by grants from Telethon (GUP10006 andGUP11006 to N.G.S.). Telethon Genetic Biobanks Network (GTB07001E)provided the DNA used in this study.
Correspondence to: G.P. Comi; e-mail: [email protected]
VC 2016 Wiley Periodicals, Inc.Published online 00 Month 2016 in Wiley Online Library (wileyonlinelibrary.com). DOI 10.1002/mus.25192
LGMD Italian Registry MUSCLE & NERVE Month 2016 1

“Mitochondrial neuropathies”: A survey from the large cohortof the Italian Network
Michelangelo Mancuso a,1,*, Daniele Orsucci a,1, Corrado Angelini b, Enrico Bertini c,Valerio Carelli d, Giacomo Pietro Comi e,f, Antonio Federico g, Carlo Minetti h, Maurizio Moggio i,j,Tiziana Mongini k, Paola Tonin l, Antonio Toscano m, Claudio Bruno n, Elena Caldarazzo Ienco a,
Massimiliano Filosto o, Costanza Lamperti p, Daria Diodato c, Isabella Moroni q,Olimpia Musumeci m, Elena Pegoraro r, Marco Spinazzi r, Naghia Ahmed e,f, Monica Sciacco i,j,
Liliana Vercelli k, Anna Ardissone q, Massimo Zeviani p, Gabriele Siciliano a
a Neurological Clinic, University of Pisa, Pisa, Italyb IRCCS S. Camillo, Venice, Italy
c Unit of Neuromuscular and Neurodegenerative Disorders, Laboratory of Molecular Medicine, Bambino Gesù Hospital IRCCS, Rome, Italyd IRCCS Institute of Neurological Sciences of Bologna, Bellaria Hospital, Bologna, and Neurology Unit, Department of Biomedical and Neuromotor Sciences
(DIBINEM), University of Bologna, Bologna, Italye Dino Ferrari Centre, Neuroscience Section, Department of Pathophysiology and Transplantation (DEPT), University of Milan, Milan, Italy
f Neurology Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italyg Department of Medicine, Surgery and Neurosciences, University Siena and Unit Clinical Neurology and Neurometabolic Diseases, AOUS, Siena, Italy
h Neuropediatric and Muscle Disorders Unit, University of Genoa and G. Gaslini Institute, Genoa, Italyi Neuromuscular Unit, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milano, Italy
j Dino Ferrari Centre, University of Milan, Milan, Italyk Department of Neuroscience, University of Turin, Torino, Italy
l Neurological Clinic, University of Verona, Verona, Italym Department of Clinical and Experimental Medicine, University of Messina, Messina, Italy
n Center of Myology and Neurodegenerative Diseases, G Gaslini Institute, Genova, Italyo Neurological Clinic, University Hospital Spedali Civili, Brescia, Italy
p Unit of Molecular Neurogenetics, The Foundation “Carlo Besta” Institute of Neurology–IRCCS, Milan, Italyq Child Neurology Unit, The Foundation “Carlo Besta” Institute of Neurology–IRCCS, Milan, Italy
r Neurological Clinic, University of Padova, Padova, Italy
Received 24 September 2015; received in revised form 10 February 2016; accepted 15 February 2016
Abstract
Involvement of the peripheral nervous system in mitochondrial disorders has been previously reported. However, the prevalence of peripheralneuropathy in mitochondrial disorders is still unclear. Based on the large database of the “Nation-wide Italian Collaborative Network ofMitochondrial Diseases”, we reviewed the clinical data of 1200 patients, with special regard to peripheral neuropathy (mean age at onset24.3 ± 20.1 years; age at last evaluation 39.8 ± 22.3 years; females 52.7%; childhood onset [before age 16 years] 43.1%). Peripheral neuropathywas present in 143/1156 patients (12.4%), being one of the ten most common signs and symptoms. POLG mutations cause a potentially painful,axonal/mixed, mainly sensory polyneuropathy; TYMP mutations lead to a demyelinating sensory-motor polyneuropathy; SURF1 mutations areassociated with a demyelinating/mixed sensory-motor polyneuropathy. The only mtDNA mutation consistently associated with peripheralneuropathy (although less severely than in the above-considered nuclear genes) was the m.8993T > G (or the rarer T > C) changes, which lead toan axonal, mainly sensory polyneuropathy. In conclusion, peripheral neuropathy is one of the most common features of a mitochondrial disorder,and may negatively impact on the quality of life of these patients. Furthermore, the presence or absence of peripheral neuropathy, as well as itsspecific forms and the association with neuropathic pain (indicative of a POLG-associated disease) can guide the molecular analysis.© 2016 Elsevier B.V. All rights reserved.
Keywords: Disease registry; Mitochondrial myopathies; mtDNA; Neuropathy; Peripheral nerve
* Corresponding author. Neurological Clinic, University of Pisa, Via Roma 67, 56100 Pisa, Italy. Tel.: +39 (0)50 992443; fax: +39 (0)50 992448.E-mail address: [email protected] (M. Mancuso).
1 Both authors contributed equally to this work.
http://dx.doi.org/10.1016/j.nmd.2016.02.0080960-8966/© 2016 Elsevier B.V. All rights reserved.
Available online at www.sciencedirect.com
Neuromuscular Disorders 26 (2016) 272–276www.elsevier.com/locate/nmd
ScienceDirect

RESEARCH ARTICLE
Timed Rise from Floor as a Predictor ofDisease Progression in Duchenne MuscularDystrophy: An Observational StudyElena S. Mazzone1, Giorgia Coratti1, Maria Pia Sormani2, Sonia Messina3, Marika Pane1,Adele D'Amico4, Giulia Colia4, Lavinia Fanelli1, Angela Berardinelli5, Alice Gardani5,Valentina Lanzillotta6, Paola D’Ambrosio7, Roberta Petillo7, Filippo Cavallaro3,Silvia Frosini8, Luca Bello9, Serena Bonfiglio10, Roberto De Sanctis1, Enrica Rolle11,Nicola Forcina1, Francesca Magri12, Gianluca Vita3, Concetta Palermo1, MariaAlice Donati13, Elena Procopio13, Maria Teresa Arnoldi14, Giovanni Baranello14,Tiziana Mongini11, Antonella Pini10, Roberta Battini8, Elena Pegoraro9, Yvan Torrente12,Stefano C. Previtali15, Claudio Bruno6, Luisa Politano7, Giacomo P. Comi12, MariaGrazia D’Angelo16, Enrico Bertini4, Eugenio Mercuri1*
1 Department of Paediatric Neurology, Catholic University, Rome, Italy, 2 Biostatistics Unit, Department ofHealth Sciences, University of Genoa, Genoa, Italy, 3 Department of Neurosciences and Nemo and ClinicalCenter, Psychiatry and Anaesthesiology, University of Messina, Messina, Italy, 4 Unit of Neuromuscular andNeurodegenerative Diseases, Department of Neurosciences, Bambino Gesù Children's Hospital, Rome,Italy, 5 Child Neurology and Psychiatry Unit, “C. Mondino” Foundation, Pavia, Italy, 6 NeuromuscularDisease Unit, G. Gaslini Institute, Genoa, Italy, 7 Cardiomiologia e genetica medica, Dipartimento diMedicina Sperimentale, Seconda Università di Napoli, Napoli, Italy, 8 Department of DevelopmentalNeuroscience, Stella Maris Institute, Pisa,Italy, 9 Department of Neurosciences, University of Padua, Padua,Italy, 10 Child Neurology and Psychiatry Unit, IRCCS Istituto delle Scienze Neurologiche di Bologna,Bologna, Italy, 11 Neuromuscular Center, SG. Battista Hospital, University of Turin, Turin, Italy, 12 DinoFerrari Centre, Neuroscience Section, Department of Pathophysiology and Transplantation (DEPT),University of Milan, Neurology Unit, IRCSS Foudation, Ca’Granda Ospedale Maggiore Policlinico, Milan,Italy, 13 Metabolic and Neuromuscular Unit, Meyer Hospital, Florence, Italy, 14 Developmental NeurologyUnit, Istituto Neurologico “Besta”Milan, Milan, Italy, 15 Neuromuscular repair unit, Inspe and division ofneuroscience, IRCSS San Raffaele Scientific Institute, Milan, Italy, 16 Neuromuscular Disorders Unit,Scientific Institute IRCCS E. Medea, 23842, Bosisio Parini, Lecco, Italy
Abstract
Background
The role of timed items, andmore specifically, of the time to rise from the floor, has been
reported as an early prognostic factor for disease progression and loss of ambulation. The
aim of our study was to investigate the possible effect of the time to rise from the floor test on
the changes observed on the 6MWT over 12 months in a cohort of ambulant Duchenne boys.
Subjects andmethods
A total of 487 12-month data points were collected from 215 ambulant Duchenne boys. The
age ranged between 5.0 and 20.0 years (mean 8.48 ±2.48 DS).
Results
The results of the time to rise from the floor at baseline ranged from 1.2 to 29.4 seconds in
the boys who could perform the test. 49 patients were unable to perform the test at baseline
PLOSONE | DOI:10.1371/journal.pone.0151445 March 16, 2016 1 / 9
OPEN ACCESS
Citation: Mazzone ES, Coratti G, Sormani MP,Messina S, Pane M, D'Amico A, et al. (2016) TimedRise from Floor as a Predictor of DiseaseProgression in Duchenne Muscular Dystrophy: AnObservational Study. PLoS ONE 11(3): e0151445.doi:10.1371/journal.pone.0151445
Editor: Maurilio Sampaolesi, Stem Cell ResearchInstitute, BELGIUM
Received: December 4, 2015
Accepted: February 29, 2016
Published: March 16, 2016
Copyright: © 2016 Mazzone et al. This is an openaccess article distributed under the terms of theCreative Commons Attribution License, which permitsunrestricted use, distribution, and reproduction in anymedium, provided the original author and source arecredited.
Data Availability Statement: All relevant data arewithin the paper and its Supporting Information files.
Funding: This work was supported by Telethon-Uildm (Award number: GUP070009) http://www.telethon.it/, and Parent Project Italy (Award number:not available) http://www.parentproject.it/. Thefunders had no role in study design, data collectionand analysis, decision to publish, or preparation ofthe manuscript.
Competing Interests: The authors have declaredthat no competing interests exist.

Development of Therapeutics for C9ORF72ALS/FTD-Related Disorders
Maria Sara Cipolat Mis1 & Simona Brajkovic1 & Francesco Tafuri1 & Nereo Bresolin1&
Giacomo P. Comi1 & Stefania Corti1
Received: 11 March 2016 /Accepted: 14 June 2016# Springer Science+Business Media New York 2016
Abstract The identification of the hexanucleotide repeat ex-pansion (HRE) GGGGCC (G4C2) in the non-coding regionof the C9ORF72 gene as the most frequent genetic cause ofboth amyotrophic lateral sclerosis (ALS) and frontotemporaldementia (FTD) has opened the path for advances in theknowledge and treatment of these disorders, which remainincurable. Recent evidence suggests that HRE RNA can causegain-of-function neurotoxicity, but haploinsufficiency has al-so been hypothesized. In this review, we describe the recentdevelopments in therapeutic targeting of the pathological ex-pansion of C9ORF72 for ALS, FTD, and other neurodegen-erative disorders. Three approaches are prominent: (1) an an-tisense oligonucleotides/RNA interference strategy; (2) usingsmall compounds to counteract the toxic effects directlyexerted by RNA derived from the repeat transcription (foci),by the translation of dipeptide repeat proteins (DPRs) from therepeated sequence, or by the sequestration of RNA-bindingproteins from the C9ORF72 expansion; and (3) gene therapy,not only for silencing the toxic RNA/protein, but also forrescuing haploinsufficiency caused by the reduced transcrip-tion of the C9ORF72 coding sequence or by the diminishedavailability of RNA-binding proteins that are sequestered byRNA foci. Finally, with the perspective of clinical therapy, we
discuss the most promising progress that has been achieved todate in the field.
Keywords Hexanucleotide repeat expansion .
Haploinsufficiency . Antisense oligonucleotides
Introduction
Amyotrophic lateral sclerosis (ALS) is an incurable and in-variably fatal neurodegenerative condition characterized bythe progressive loss of both upper and lower motor neuronsin the cortex, brainstem, and spinal cord; it clinically results inprogressive paralysis and death within 3–5 years of its onset,often due to respiratory failure [1]. No effective therapy isavailable for this disease. The only drug approved by theFDA and EMA is riluzole, which extends the median lifespanby only 3 months [1]. Thus, the discovery of clinically effec-tive therapies is urgently needed. The vast majority of casesare sporadic (sALS) of unknown origin, while 5–10 % arefamilial (fALS), often with autosomal dominant inheritance[2]. The disease pathogenesis is multilayered, given that sev-eral pathways have been identified as key elements both in theonset and in the progression of the disease. Although severalelements have been investigated as possible targets for treat-ment advancement, the lack of a clear understanding of thecauses of ALS, particularly in cases of sALS, has hamperedthe search for a cure [1]. However, the genetic forms of ALScan offer a solid basis for research since, at least in these cases,the etiopathogenic primum movens is known. The first caus-ative genetic mutations were described in Cu–Zn superoxidedismutase 1 (SOD1) gene in 1993 [3], and due to severalgenome sequencing projects, many of the genes responsiblefor ALS (>30 genes so far) have been described [4]. Theidentification of a hexanucleotide repeat expansion (HRE)
Maria Sara Cipolat Mis and Simona Brajkovic contributed equally to thiswork.
* Stefania [email protected]
1 Dino Ferrari Centre, Neuroscience Section, Department ofPathophysiology and Transplantation (DEPT), University of Milan,Neurology Unit, IRCCS Foundation Ca’Granda Ospedale MaggiorePoliclinico, Via Francesco Sforza 35, 20122 Milan, Italy
Mol NeurobiolDOI 10.1007/s12035-016-9993-0

The genetic basis of undiagnosed musculardystrophies and myopathiesResults from 504 patients
ABSTRACT
Objective: To apply next-generation sequencing (NGS) for the investigation of thegenetic basis of undiagnosed muscular dystrophies and myopathies in a very large cohortof patients.
Methods:We applied an NGS-based platform namedMotorPlex to our diagnostic workflow to testmuscle disease genes with a high sensitivity and specificity for small DNA variants. We analyzed504 undiagnosed patients mostly referred as being affected by limb-girdle muscular dystrophy orcongenital myopathy.
Results:MotorPlex provided a complete molecular diagnosis in 218 cases (43.3%). A further 160patients (31.7%) showed as yet unproven candidate variants. Pathogenic variants were found in47 of 93 genes, and in more than 30% of cases, the phenotype was nonconventional, broadeningthe spectrum of disease presentation in at least 10 genes.
Conclusions: Our large DNA study of patients with undiagnosed myopathy is an example of theongoing revolution in molecular diagnostics, highlighting the advantages in using NGS asa first-tier approach for heterogeneous genetic conditions. Neurology® 2016;87:71–76
GLOSSARYCM 5 congenital myopathy; LGMD 5 limb-girdle muscular dystrophy; MD 5 muscular dystrophy; NGS 5 next-generationsequencing; NMD 5 neuromuscular disorder.
Muscular dystrophies (MDs)1,2 and congenital myopathies (CMs)3 represent the majority ofinherited neuromuscular disorders (NMDs).4,5
Until the development of next-generation sequencing (NGS), routine molecular diagnosis ofNMDs had been based on a gene-by-gene approach.1,6–8 This time-consuming and expensiveapproach9 failed to identify causative variants in more than 40% of cases, detection rates variedwith different genes or conditions, and the size of some large genes, such as TTN or NEB,hampered routine analysis.
Targeted NGS, which focuses only on specific genes of interest,10 has recently been proposedas a cost-effective strategy for the molecular diagnosis of heterogeneous disorders.11
Marco Savarese, PhDGiuseppina Di Fruscio,
PhDAnnalaura Torella, PhDChiara Fiorillo, MDFrancesca Magri, MDMarina Fanin, PhDLucia Ruggiero, MDGiulia Ricci, MDGuja Astrea, PhDLuigia Passamano, MDAlessandra Ruggieri, PhDDario Ronchi, PhDGiorgio Tasca, MDAdele D’Amico, MDSandra Janssens, MDOlimpia Farina, MDMargherita Mutarelli,
PhDVeer Singh Marwah, MScArcomaria Garofalo, MScTeresa Giugliano, PhDSimone Sanpaolo, MDFrancesca Del Vecchio
Blanco, PhDGaia Esposito, BScGiulio Piluso, PhDPaola D’Ambrosio, MDRoberta Petillo, MDOlimpia Musumeci, MDCarmelo Rodolico, MDSonia Messina, MDAnni Evilä, PhDPeter Hackman, PhDMassimiliano Filosto, MDGiuseppe Di Iorio, MDGabriele Siciliano, MDMarina Mora, PhDLorenzo Maggi, MDCarlo Minetti, MDSabrina Sacconi, MDLucio Santoro, MDKathleen Claes, PhDLiliana Vercelli, MDTiziana Mongini, MD
Author list continued on next page
From the Dipartimento di Biochimica Biofisica e Patologia Generale (M.S., G.D.F., A. Torella, A.G., T.G., F.D.V.B., G.E., G.P., V.N.), SecondaUniversità di Napoli; Telethon Institute of Genetics and Medicine (M.S., G.D.F., A. Torella, M. Mutarelli, V.S.M., A.G., T.G., G.E., V.N.),Pozzuoli; U.O.C. Neurologia Pediatrica e Malattie Muscolari (C.F., C.M., C.B.), IRCCS Istituto Giannina Gaslini, Genova; Centro Dino Ferrari,Dipartimento di Fisiopatologia Medico-Chirurgica e dei Trapianti (F.M., D.R., G.P.C.), and Neuromuscular and Rare Disease Unit, Dipartimentodi Neuroscienze (M. Moggio), Università degli Studi di Milano, Fondazione IRCCS Ca’ Granda, Ospedale Maggiore Policlinico, Milan; Dipar-timento di Neuroscienze (M. Fanin, E.P.), Università di Padova; Dipartimento di Neuroscienze e Scienze Riproduttive ed Odontostomatologiche(L.R., L.S.), Università degli Studi di Napoli “Federico II,” Napoli; Dipartimento di Medicina Clinica e Sperimentale (G.R., G.S.), Università degliStudi di Pisa; Medicina Molecolare (G.A., F.M.S.), IRCCS Fondazione Stella Maris, Pisa; Dipartimento di Medicina Sperimentale (L. Passamano,P.D., R.P., L. Politano) and Dipartimento di Scienze Mediche, Chirurgiche, Neurologiche, Metaboliche, e dell’Invecchiamento (O.F., S.S., G.D.I.),Seconda Università di Napoli; Dipartimento di Neuroscienze (A.R., M. Mora, L.M.), Istituto Besta, Milano; Don Carlo Gnocchi ONLUS Foundation(G.T.), Milano; Dipartimento di Neuroscienze (A.D., E.B.), IRCCSOspedale Pediatrico Bambino Gesù, Roma, Italy; Center for Medical Genetics (S.J.,K.C.) and Department of Neurology (J.D.B.), Ghent University Hospital, Belgium; Dipartimento di Neuroscienze (O.M., C.R., S.M., A. Toscano),Università degli Studi di Messina, Italy; Folkhälsan Institute of Genetics (A.E., P.H., B.U.), University of Helsinki, Finland; Section for NeuromuscularDiseases and Neuropathies (M. Filosto), Unit of Clinical Neurology, University Hospital ’Spedali Civili,’ Brescia, Italy; Neuromuscular DiseasesSpecialized Center (S.S.), Archet 1 Hospital, Nice, France; S.S. Malattie Neuromuscolari (L.V., T.M.), Università degli Studi di Torino; Istituto diNeurologia (E.R., E.M.), Università Cattolica del Sacro Cuore, Roma; UOL of Medical Genetics (F.G.), Department of Reproduction and Growth andDepartment of Medical Science, Sant’Anna University Hospital, Ferrara; Department of Life Sciences (R.T.), University of Modena and Reggio Emilia,Modena; and Fondazione Hospital S. Camillo IRCCS (C.A.), Venezia, Italy.
Go to Neurology.org for full disclosures. Funding information and disclosures deemed relevant by the authors, if any, are provided at the end of the article.
© 2016 American Academy of Neurology 71

1Scientific RepoRts | 6:21301 | DOI: 10.1038/srep21301
www.nature.com/scientificreports
Morpholino-mediated SOD1 reduction ameliorates an amyotrophic lateral sclerosis disease phenotypeM. Nizzardo1, C. Simone1, F. Rizzo1, G. Ulzi1, A. Ramirez1, M. Rizzuti1, A. Bordoni1, M. Bucchia1, S. Gatti2, N. Bresolin1, G. P. Comi1 & S. Corti1
Neurotoxicity due to the accumulation of mutant proteins is thought to drive pathogenesis in neurodegenerative diseases. Mutations in superoxide dismutase 1 (SOD1) are linked to familial amyotrophic lateral sclerosis (fALS); these mutations result in progressive motor neuron death through one or more acquired toxicities. Interestingly, SOD1 is not only responsible for fALS but may also play a significant role in sporadic ALS; therefore, SOD1 represents a promising therapeutic target. Here, we report slowed disease progression, improved neuromuscular function, and increased survival in an in vivo ALS model following therapeutic delivery of morpholino oligonucleotides (MOs) designed to reduce the synthesis of human SOD1. Neuropathological analysis demonstrated increased motor neuron and axon numbers and a remarkable reduction in astrogliosis and microgliosis. To test this strategy in a human model, we treated human fALS induced pluripotent stem cell (iPSC)-derived motor neurons with MOs; these cells exhibited increased survival and reduced expression of apoptotic markers. Our data demonstrated the efficacy of MO-mediated therapy in mouse and human ALS models, setting the stage for human clinical trials.
Amyotrophic lateral sclerosis (ALS) is a fatal neurological disease characterized by the degeneration and loss of upper and lower motor neurons (MNs),which leads to paralysis and death within 3–5 years of diagnosis1. Currently, there is no effective treatment for this disease2. The majority of cases of ALS have no clear genetic link-age and are referred to as sporadic (sALS), while 10% of cases are familial (fALS)3. Disease-causing mutations in various genes have been identified3. Mutations in the gene encoding for Cu/Zn superoxide dismutase 1 (SOD1) are relatively frequent4, accounting for 15% of sALS. Mutations in the SOD1 gene are linked to 20% of ALS cases. In these cases, the resulting progressive MN death is likely caused by one or more mutated SOD1-related toxici-ties, as revealed by studies of transgenic rodent models5–8.
Recent evidence supports SOD1 as a toxic factor not only in fALS but also in sALS9. Indeed, changes in oxidation, demetallation, and other types of post-translational modifications are able to induce aberrant con-formations of wild-type (WT) SOD1, which eventually lead to its acquisition of toxic functions comparable to those of fALS-associated SOD1 variants10–12. Due to a series of conformation-specific antibodies, SOD1 has been detected in spinal cord samples from ALS patients and SOD1 rodent models in an altered/abnormal conforma-tion, conventionally referred to as misfolded, which may account for its inherent toxic nature10,13,14. Misfolded “mutant-like” forms of WT SOD1 have also been found in human post-mortem tissue from patients affected by sALS, suggesting a concrete pathogenetic role for these SOD1 variants10,13. This finding, along with reports that a reduction of both WT and mutant SOD1 in astrocytes derived respectively from sALS and fALS patients decreased astrocyte-derived toxicity towards MNs15, provides strong evidence for a pathogenic role of WT SOD1 in sALS.
1Dino Ferrari Centre, Neuroscience Section, Department of Pathophysiology and Transplantation (DEPT), University of Milan, Neurology Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy. 2Centro di Ricerche Chirurgiche Precliniche, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico; Department of Pathophysiology and Transplantation (DEPT), University of Milan, 20122 Milano, Italy. Correspondence and requests for materials should be addressed to M.N. (email: [email protected])
Received: 09 October 2015
accepted: 21 January 2016
Published: 16 February 2016
OPEN

for malignancy in patients with LEMS in whom no pul-monary cancer is discovered.
Daniel B. Simmons, MD1
Thomas M. Duginski, MD1
Jeffrey C. McClean, MD1
Anthony A. Amato, MD2
John H. Sladky, MD1
1Department of Neurology, San Antonio Military MedicalCenter (SAMMC), Fort Sam Houston, Texas, USA2Department of Neurology, Brigham and Women’s Hospitaland Harvard Medical School, Boston, Massachusetts, USA
1. Bobos M, Hytiroglou P, Kostopoulos I, Karkavelas G, PapadimitriouCS. Immunohistochemical distinction between Merkel cell carcinomaand small cell carcinoma of the lung. Am J Dermatopathol 2006;28:99–104.
2. Vernino S. Paraneoplastic disorders affecting the neuromuscularjunction or anterior horn cell. Continuum Lifelong Learning Neurol2009;15:132–146.
3. Eggers SD, Salomao DR, Dinapoli RP, Vernino S. Paraneoplastic andmetastatic neurologic complications of Merkel cell carcinoma. MayoClin Proc 2001;76:327–330.
4. Siau RT, Morris A, Karoo RO. Surgery results in complete cure ofLambert-Eaton myasthenic syndrome in a patient with metastaticMerkel cell carcinoma. J Plast Reconstr Aesthet Surg 2014;67:e162–e164.
Published online 9 October 2015 in Wiley Online Library(wileyonlinelibrary.com). DOI 10.1002/mus.24934
---------------------------------------------------------
ASYMPTOMATIC POMPE DISEASE: CANMUSCLE MAGNETIC RESONANCE IMAGINGFACILITATE DIAGNOSIS?
Glycogen storage disease (GSD) type II, or Pompe dis-ease (PD), is a lysosomal storage disorder caused by acida-1,4-glucosidase deficiency. Its clinical spectrum variesgreatly depending on the age of onset, rate of diseaseprogression, and extent of tissue involvement. Late-onsetPD (LOPD) can be asymptomatic for years, causing diag-nostic delay. Muscle biopsy may be uninformative,because muscle fiber involvement can be patchy.1 Driedblood spot testing (DBS) has been suggested as an initialdiagnostic tool.2 Although we are unsure whetherpatients diagnosed at an asymptomatic stage should betreated with enzyme replacement therapy (ERT) to pre-vent disease progression, there is an increasing need forearly disease markers to monitor closely for overtmyopathy.3
Muscle magnetic resonance imaging (MRI) hasbecome an established and validated non-invasive tech-nique for assessment of selective muscle involvement,mainly in the hereditary myopathies,4–6 using imagingprotocols7 that highlight fatty infiltration using T1-weighted (T1-w) sequences. There is increased interestin short-tau inversion recovery (STIR) and T2 fat-saturation techniques, which both highlight muscle“edema.” These techniques have been used mainly ininflammatory myopathies8 or in selected dystrophies,
such as dysferlinopathy and facioscapulohumeral dystro-phy.9 Here we present the case of an asymptomaticpatient with LOPD who showed a distinctive muscle MRIpattern that helped us make an early diagnosis.
A 16-year-old asymptomatic Italian boy was evaluatedat our center for persistent incidental creatine kinase(CK) elevations (600 IU/L). He exhibited mild, general-ized hyporeflexia. Cardiac and respiratory function testswere normal. DBS was not performed for temporarytechnical problems, so he underwent a quadriceps mus-cle biopsy, which showed normal histochemistry andimmunohistochemistry. He also underwent muscle MRIwith T1-w and STIR sequences on the lower limbs. T1-wsequences did not show any pattern of selective muscularadipose substitution, with the exception of mild involve-ment of the adductor magnus muscles bilaterally. How-ever, both proximal adductor magnus muscles werealtered with an “edema-like” signal on STIR sequences(Fig. 1).
Selective adductor magnus involvement in T1-wsequences is the earliest manifestation of juvenileLOPD.10 However, mildly increased signal intensity onSTIR images has been described in leg muscles in onlya small percentage of LOPD cases. The meaning ofthis is still unknown, although it is probably partiallyrelated to the high glycogen content found in musclebiopsies and described in other GSDs.11,12 The MRIfindings prompted us to test a-glucosidase muscle activ-ity, which proved to be low (9.75 pmol/min/mg, refer-ence range 113 6 41). Molecular analysis of the geneencoding acid a-1,4-glucosidase (compound hetero-zygosity: IVS1-32-13T>G; c.1670T>G) confirmed adiagnosis of PD.
T2 quantification is lacking, and further studies areneeded to confirm this finding on a larger sample todefine its specificity and explain its meaning. However,this case highlights the usefulness of muscle MRI withboth T1-w and T2-STIR sequences in the diagnosis ofLOPD, particularly when clinical and muscle biopsy find-ings are uninformative.
Anna Pichiecchio, MD1
Angela Berardinelli, MD2
Maurizio Moggio, MD, PhD3
Marta Rossi, MD4
Umberto Balottin, MD, PhD4
Giacomo Pietro Comi, MD, PhD3
Stefano Bastianello, MD, PhD1,2,3,4,5
1Neuroradiology Department, C. Mondino National NeurologicalInstitute, Pavia, Italy2Child Neuropsychiatry Unit, Regional Referral Centre forNeuromuscular Disease in Childhood, C. Mondino NationalNeurological Institute, Pavia, Italy3Neuromuscular Unit, Fondazione IRCCS Ca’ GrandaOspedale Maggiore Policlinico, Dino Ferrari Center, Universityof Milan, Milan, Italy4Child Neuropsychiatry Unit, Department of Brain andBehavioral Sciences, University of Pavia, Pavia, Italy5Department of Brain and Behavioral Sciences, University ofPavia, Pavia, Italy
1. Feeney EJ, Austin S, Chien YH, Mandel H, Schoser B, Prater S, et al.The value of muscle biopsies in Pompe disease: identifying lipofuscinVC 2015 Wiley Periodicals, Inc.
326 Letters to the Editor MUSCLE & NERVE February 2016

New Mutations in NEB Gene Discovered by TargetedNext-Generation Sequencing in NemalineMyopathy Italian Patients
Daniela Piga1 & Francesca Magri1 & Dario Ronchi1 & Stefania Corti1 & Denise Cassandrini2 &
Eugenio Mercuri3 & Giorgio Tasca4 & Enrico Bertini5 & Fabiana Fattori5 & Antonio Toscano6 &
Sonia Messina6 & Isabella Moroni7 & Marina Mora7 & Maurizio Moggio8 & Irene Colombo8 &
Teresa Giugliano9,10 & Marika Pane3 & Chiara Fiorillo11 & Adele D’Amico5 & Claudio Bruno11 &
Vincenzo Nigro9,10 & Nereo Bresolin1& Giacomo Pietro Comi1
Received: 20 February 2015 /Accepted: 22 March 2016 /Published online: 22 April 2016# Springer Science+Business Media New York 2016
Abstract Nemaline myopathy represents a group of clinicallyand genetically heterogeneous neuromuscular disorders.Different clinical-genetic entities have been characterized inthe last few years, with implications for diagnostics and geneticcounseling. Fifty percent of nemaline myopathy forms are dueto NEB mutations, but genetic analysis of this large and com-plex gene by Sanger sequencing is time consuming and expen-sive. We selected 10 Italian patients with clinical and biopsyfeatures suggestive for nemaline myopathy and negative forACTA1, TPM2 and TPM3 mutations. We applied a targetednext-generation sequencing strategy designed to analyse NEBcoding regions, the relative full introns and the promoter. Wealso evaluated copy number variations (by CGH array) andtranscriptional changes by RNA Sanger sequencing, wheneverpossible. This combined strategy revealed 11 likely pathogenicvariants in 8 of 10 patients. The molecular diagnosis was fullyachieved in 3 of 8 patients, while only one heterozygous muta-
tion was observed in 5 subjects. This approach revealed to be afast and cost-effective way to analyse the large NEB gene in asmall group of patients and might be promising for the detec-tion of pathological variants of other genes featuring large cod-ing regions and lacking mutational hotspots.
Keywords NEBmutations . Nemaline myopathy .
Next-generation sequencing
Introduction
The term nemaline myopathy (NM) refers to a group of raremuscular disorders that have high clinical and genetic vari-ability. The clinical symptoms of NM include hypotonia, neckflexors and proximal limb muscle involvement, weakness offacial muscles, respiratory insufficiency and feeding prob-
* Giacomo Pietro [email protected]
1 Dino Ferrari Centre, Department of Pathophysiology andTransplantation (DEPT), University of Milan, Neurology Unit,I.R.C.C.S. Foundation Ca’ Granda, Ospedale Maggiore Policlinico,Via F. Sforza 35, 20122 Milan, Italy
2 Molecular Medicine Unit, IRCCS Stella Maris, Pisa, Italy3 Department of Pediatrics, Policlinico Gemelli, Università Cattolica
Sacro Cuore, Roma, Italy4 Don Carlo Gnocchi Onlus Foundation, Milan, Italy5 Department of Laboratories, Unit of Molecular Medicine for
Neuromuscular and Neurodegenerative Disorders, Bambino GesùChildren’s Hospital, Rome, Italy
6 Department of Neurosciences, Psychiatry and Anesthesiology,University of Messina, AOU Policlinico G. Martino, Messina, Italy
7 Pediatric, Neuromuscular Diseases and Neuroimmunology Units,The Foundation “Carlo Besta” IRCCS Neurological Institute,Milan, Italy
8 Dino Ferrari Centre, Neuromuscular Unit, IRCCS Foundation CàGranda Ospedale Maggiore Policlinico, University of Milan,Milan, Italy
9 Department of Biochemistry, Biophysics and General Pathology,Second University of Naples, Naples, Italy
10 Telethon Institute of Genetics and Medicine, Naples, Italy
11 Center of Myology and Neurodegenerative Diseases, GianninaGaslini Institute, Genova, Italy
J Mol Neurosci (2016) 59:351–359DOI 10.1007/s12031-016-0739-2

Mutational analysis of COQ2 in patients with MSA in Italy
Dario Ronchi a,1, Ernesto Di Biase a,1, Giulia Franco a, Valentina Melzi a,Francesca Del Sorbo b, Antonio Elia b, Chiara Barzaghi c, Barbara Garavaglia c,Christian Bergamini d, Romana Fato d, Gabriele Mora e, Roberto Del Bo a,Francesco Fortunato a, Linda Borellini a, Ilaria Trezzi a, Giacomo Monzio Compagnoni a,Edoardo Monfrini a, Emanuele Frattini a, Sara Bonato a, Filippo Cogiamanian f,Gianluca Ardolino f, Alberto Priori g, Nereo Bresolin a, Stefania Corti a,Giacomo Pietro Comi a, Alessio Di Fonzo a,*
a IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Dino Ferrari Center, Neuroscience Section, Department of Pathophysiology andTransplantation, University of Milan, Milan, ItalybNeurology Unit I, Neurological Institute “C. Besta” IRCCS Foundation, Milan, ItalycMolecular Neurogenetics Unit, IRCCS Foundation Istituto Neurologico Carlo Besta, Milano, ItalydDepartment of Pharmacy and Biotecnology (FaBiT), University of Bologna, Bologna, ItalyeDepartment of Neurological Rehabilitation, Fondazione Salvatore Maugeri, IRCCS, Istituto Scientifico di Milano, Milan, ItalyfU.O. Neurofisiopatologia, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, ItalygClinica Neurologica III Università degli Studi di Milano, Dipartimento di Scienze della Salute, Ospedale Santi Paolo e Carlo, Milano, Italy
a r t i c l e i n f o
Article history:Received 10 March 2016Received in revised form 28 April 2016Accepted 30 May 2016Available online 7 June 2016
Keywords:MSACOQ2CoQ10
a b s t r a c t
COQ2 mutations have been implicated in the etiology of multiple system atrophy (MSA) in Japan.However, several genetic screenings have not confirmed the role of its variants in the disease. We per-formed COQ2 sequence analysis in 87 probable MSA. A homozygous change p.A43G was found in anMSA-C patient. Cosegregation analysis and the evaluation of CoQ10 content in muscle and fibroblasts didnot support the pathogenic role of this variant.
� 2016 Elsevier Inc. All rights reserved.
1. Introduction
Multiple system atrophy (MSA) is an adult-onset progressiveneurodegenerative disease characterized by a variable combinationof autonomic dysfunction, cerebellar ataxia, parkinsonian, and py-ramidal signs. Although considered a nongenetic disorder, thereport of familial MSA cases supports the presence of genetic factorsin the etiology of this disease (Hara et al., 2007).
Two Japanese MSA-C families were discovered to carry biallelicmutations in the COQ2 gene, and a heterozygous variant (p.V393A)was reported to increase the risk of sporadic cases (Multiple-System Atrophy Research, 2013). Thereafter most COQ2 mutation
analyses in MSA did not support this association, though rare var-iants in single cases of still unknown significance have been re-ported (Ogaki et al., 2014) (Supplementary Table 1 andSupplementary Fig. 1). COQ2 encodes an enzyme involved in co-enzyme Q10 (CoQ10) biosynthesis and recessive COQ2 mutationswere firstly associated with severe infantile multisystem disordersor nephrotic syndrome featuring primary CoQ10 deficiency (Jakobset al., 2013,; Quinzii et al., 2006).
Here, we report the results of the COQ2molecular screening in acohort of 87 patients with probable MSA.
2. Methods
Clinical diagnosis was performed according to Gilman’sconsensus criteria (Gilman et al., 2008). After written informedconsent, blood samples, muscle, and skin biopsies were collected.
Sequencing analysis, biochemical assays, and CoQ10 dosagewere performed as described in Supplementary Material online.
* Corresponding author at: IRCCS Foundation Ca’ Granda Ospedale MaggiorePoliclinico, Dino Ferrari Center, Neuroscience Section, Department of Pathophysi-ology and Transplantation, University of Milan, Via Francesco Sforza 35, 20122Milan, Italy. Tel.: 00390255033807; fax: 00390255033800.
E-mail address: [email protected] (A. Di Fonzo).1 First coauthorship.
Contents lists available at ScienceDirect
Neurobiology of Aging
journal homepage: www.elsevier .com/locate/neuaging
0197-4580/$ e see front matter � 2016 Elsevier Inc. All rights reserved.http://dx.doi.org/10.1016/j.neurobiolaging.2016.05.022
Neurobiology of Aging 45 (2016) 213.e1e213.e2

ORIGINAL ARTICLE
Anti-sulfatide reactivity in patients with celiac disease
Domenica Saccomannoa, Carolina Tombab, Francesca Magria, Philippe Backelandtc, Leda Roncoronib,d,Luisa Donedad, Maria Teresa Bardellab, Giacomo Pietro Comia, Nereo Bresolina, Dario Conteb and Luca Ellib
aDepartment of Pathophysiology and Transplantation, Neurology Unit, Dino Ferrari Center, University of Milan, Fondazione IRCCS Ca’Granda-Ospedale Maggiore Policlinico, Milan, Italy; bCentre for the Prevention and Diagnosis of Celiac Disease, Gastroenterology andEndoscopy Unit, Fondazione IRCCS Ca’ Granda-Ospedale Maggiore Policlinico, Milan, Italy; cZenTech S. A., Angleur, Belgium; dDepartment ofBiomedical, Surgical and Dental Sciences, University of Milan, Milan, Italy
ABSTRACTObjective: To explore a possible significance of the presence of anti-ganglioside and anti-sulfatide anti-bodies in sera of adult patients with celiac disease (CD) in different clinical scenario.Methods: We selected 22 adult patients with newly diagnosed CD and 20 age–sex matched non-CDcontrols. Patients’ serum was tested – before and after at least 6 months on a gluten-free diet (GFD) –for anti-GM1, GM2, GM3, GD1a, GD1b, GD3, GT1a, GT1b, GQ1b and sulfatide IgM, IgG and IgA auto-antibodies, by means of a dot blot technique and enzyme-linked immunosorbent assay (ELISA).Results: We found the presence of auto-antibodies in untreated patients. In particular, anti-sulfatideIgG antibodies were present in 8 (36%) patients independently of the presence of neurological symp-toms. Anti-sulfatide IgA antibodies were present in 3 (19%) patients. During GFD, anti-sulfatide IgG dis-appeared in all the patients, whereas IgA were observed in 2 patients. Anti-sulfatide, anti-GM1 andanti-GM2 IgM antibodies were also observed in 2 patients on a GFD. All the other auto-antibodieswere absent and no demographic or clinical parameters were associated. Non-CD controls did not pre-sent any auto-antibody.Conclusions: We found anti-sulfatide IgG antibodies in CD patients on a gluten-containing diet.Anti-sulfatide IgA antibodies persisted during GFD together with the occurrence of other IgM auto-antibodies. These data suggest a possible link between gluten and IgG auto-antibodies.
ARTICLE HISTORYReceived 12 July 2016Revised 7 November 2016Accepted 17 November 2016
KEYWORDSAuto-antibodies; celiacdisease; anti-sulfatidereactivity; neurologicaldisorders; gluten-free diet
Introduction
Celiac disease (CD) is a common autoimmune enteropathytriggered by the ingestion of gluten proteins in geneticallysusceptible individuals carrying the HLA type II DQ2 and/orDQ8 haplotypes. Although the small bowel is the main tar-geted organ and localization of the inflammatory damage,CD is considered a systemic disorder as different tissues andorgans, including the central and peripheral nervous systems,are involved. Approximately, 10% of CD patients show neuro-logical symptoms,[1] in particular cerebellar ataxia, peripheralneuropathy (PN) and epilepsy.[2–4]
The pathogenesis of the neurological complications in CDis still unclear; vitamin deficiency could be considered in caseof malabsorption and extensive small-bowel atrophy.However, this mechanism does not justify all the cases, espe-cially those with normal levels of vitamins and without aclear malabsorptive syndrome. It could be supposed that sys-temic inflammation and the consequent increase in cytokinesand auto-antibodies levels would lead to an immune reactionagainst nervous system components. Moreover, the responseof neurological symptoms to a gluten-free diet (GFD) remainscontroversial. From this point of view, if neurological symp-toms are independent of GFD, the presence of a genetic
background facilitating the onset of multiple autoimmunediseases (including neurological autoimmune disorders) withCD may represent a co-factor.[5]
The presence of anti-ganglioside antibodies in the serumof CD patients may explain neurological symptoms in CD, asrecently supposed by different researchers; in fact, IgG anti-bodies against gangliosides have been found in the sera ofadult CD patients with PN.[6]
Gangliosides are glycosphingolipids (GM1, GM2, GM3,GD1a, GD1b, GD3, GT1a, GT1b, GQ1b) which contain sialicacid and are present in large amounts in the nervous system,especially in myelin. They can be antigenic targets in a var-iety of autoimmune neuropathies. The identification of anti-ganglioside antibodies in large cohorts of patients with awide range of acute and chronic peripheral neuropathies andtheir association with particular clinical phenotypes havebeen reported.[7] The possible involvement of anti-sulfatideantibodies in CD remains unknown. Sulfatide is a commonglycolipid in the peripheral nerve myelin and dorsal root gan-glia [8] and may be targeted by IgM auto-antibodies associ-ated with the onset of sensory axonal neuropathy orpredominantly demyelinating sensorimotor neuropathy.[9]
Others authors confirmed the presence of anti-gangliosideIgG antibodies in CD patients but did not find any correlation
CONTACT Luca Elli [email protected] Centre for the Prevention and Diagnosis of Celiac Disease, Gastroenterology and Endoscopy Unit, FondazioneIRCCS Ca’ Granda-Ospedale Maggiore Policlinico, Via Francesco Sforza 35, Milan 20122, Italy� 2016 Informa UK Limited, trading as Taylor & Francis Group
SCANDINAVIAN JOURNAL OF GASTROENTEROLOGY, 2016http://dx.doi.org/10.1080/00365521.2016.1263679

Is Spinal Muscular Atrophy a disease of the motor neurons only: pathogenesis and therapeutic implications?
Chiara Simone1, Agnese Ramirez1, Monica Bucchia1, Paola Rinchetti1, Hardy Rideout2, Dimitra Papadimitriou2,#, Diane B. Re3,#, and Stefania Corti1,#,*
1 Dino Ferrari Centre, NeuroscienceSection, Department of Pathophysiology and Transplantation (DEPT), University of Milan, Neurology Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Via Francesco Sforza 35, 20122, Milan, Italy
2 Division of Basic Neurosciences, Biomedical Research Foundation of the Academy of Athens, BRFAA, Soranou Efesiou 4, 115 27, Athens, Greece
3 Department of Environmental Health Sciences, Columbia University, New York, NY 10032, USA; and Center for Motor Neuron Biology and Disease, Columbia University, New York, NY 10032, USA
Abstract
Spinal Muscular Atrophy (SMA) is a genetic neurological disease that causes infant mortality; no
effective therapies are currently available. SMA is due to homozygous mutations and/or deletions
in the Survival Motor Neuron 1 (SMN1) gene and subsequent reduction of the SMN protein,
leading to the death of motor neurons. However, there is increasing evidence that in addition to
motor neurons, other cell types are contributing to SMA pathology. In this review, we will discuss
the involvement of non-motor neuronal cells, located both inside and outside the central nervous
system, in disease onset and progression. These contribution of non-motor neuronal cells to
disease pathogenesis has important therapeutic implications: in fact, even if SMN restoration in
motor neurons is needed, it has been shown that optimal phenotypic amelioration in animal
models of SMA requires a more widespread SMN correction. It will be crucial to take this
evidence into account before clinical translation of the novel therapeutic approaches that are
currently under development.
Keywords
Spinal Muscular Atrophy; pathogenesis; therapy; central nervous system
1. Introduction
Spinal muscular atrophy (SMA) is the most common genetic neurological disease leading to
infant mortality. It is characterized predominantly by spinal motor neuron loss, muscle
*To whom correspondence should be addressed: Neuroscience Section, Department of Pathophysiology and Transplantation (DEPT), University of Milan, Neurology Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Via Francesco Sforza 35, 20122 Milan Italy. Tel: +39 0255033817; Fax: +39 0250320430; [email protected].#Co-senior authors
HHS Public AccessAuthor manuscriptCell Mol Life Sci. Author manuscript; available in PMC 2016 March 01.
Published in final edited form as:Cell Mol Life Sci. 2016 March ; 73(5): 1003–1020. doi:10.1007/s00018-015-2106-9.
Author M
anuscriptA
uthor Manuscript
Author M
anuscriptA
uthor Manuscript

Nature GeNetics VOLUME 48 | NUMBER 9 | SEPTEMBER 2016 1043
To elucidate the genetic architecture of amyotrophic lateral sclerosis (ALS) and find associated loci, we assembled a custom imputation reference panel from whole-genome-sequenced patients with ALS and matched controls (n = 1,861). Through imputation and mixed-model association analysis in 12,577 cases and 23,475 controls, combined with 2,579 cases and 2,767 controls in an independent replication cohort, we fine-mapped a new risk locus on chromosome 21 and identified C21orf2 as a gene associated with ALS risk. In addition, we identified MOBP and SCFD1 as new associated risk loci. We established evidence of ALS being a complex genetic trait with a polygenic architecture. Furthermore, we estimated the SNP-based heritability at 8.5%, with a distinct and important role for low-frequency variants (frequency 1–10%). This study motivates the interrogation of larger samples with full genome coverage to identify rare causal variants that underpin ALS risk.
ALS is a fatal neurodegenerative disease that affects 1 in 400 people, with death occurring within 3 to 5 years of the onset of symptoms1. Twin-based studies estimate heritability to be around 65%, and 5–10% of patients with ALS have a positive family history1,2. Both of these features are indicative of an important genetic component in ALS etiology. Following initial discovery of a risk-associated C9orf72 locus in ALS genome-wide association studies (GWAS)3–5, identification of a pathogenic hexanucleotide-repeat expansion in this locus revolu-tionized the field of ALS genetics and biology6,7. The majority of ALS heritability, however, remains unexplained, and only two additional risk loci have since been identified robustly3,8.
To discover new genetic risk loci and elucidate the genetic archi-tecture of ALS, we genotyped 7,763 new cases and 4,669 controls and additionally collected genotype data from published GWAS of ALS. In total, we analyzed 14,791 cases and 26,898 controls from 41 cohorts (Supplementary Table 1 and Supplementary Note). We combined these cohorts on the basis of genotyping platform and nationality to form 27 case–control strata. In total, 12,577 cases and 23,475 controls passed quality control (Online Methods and Supplementary Tables 2–5).
For imputation purposes, we obtained high-coverage (~43.7×) whole-genome sequencing data from 1,246 patients with ALS and 615 controls from the Netherlands (Online Methods and Supplementary Fig. 1). After quality control, we constructed a reference panel including
18,741,510 single-nucleotide variants (SNVs). Imputing this cus-tom reference panel into Dutch ALS cases considerably increased the imputation accuracy for low-frequency variants (minor allele frequency (MAF) = 0.5–10%) in comparison to commonly used reference panels from 1000 Genomes Project Phase 1 (ref. 9) and Genome of the Netherlands10 (Fig. 1a). Improvement was also observed when imputing into ALS cases from the UK (Fig. 1b). To benefit from the global diversity of haplotypes, the custom and 1000 Genomes Project panels were combined, which further improved imputation. Given these results, we used the merged reference panel to impute all strata in our study.
In total, we imputed 8,697,640 variants passing quality control into the 27 strata and tested the strata separately for association with ALS risk by logistic regression. We then included the results in an inverse-variance-weighted, fixed-effects meta-analysis, which identi-fied four loci associated at genome-wide significance (P < 5 × 10−8) (Fig. 2a). The previously reported C9orf72 (rs3849943)3–5,8, UNC13A (rs12608932)3,5 and SARM1 (rs35714695)8 loci all reached genome-wide significance, as did a new association for a nonsynonymous variant in C21orf2 (rs75087725, P = 8.7 × 10−11; Supplementary Tables 6–10). This variant was present on only 10 haplotypes in the 1000 Genomes Project reference panel (MAF = 1.3%), whereas it was present on 62 haplotypes in our custom reference panel (MAF = 1.7%). As a result, more strata passed quality control for this variant by pass-ing the allele frequency threshold of 1% (Supplementary Table 11). This result demonstrates the benefit of the merged reference panel with ALS-specific content, which improved imputation and resulted in the identification of a genome-wide significant association.
Linear mixed models (LMMs) can improve power while controlling for sample structure11, which would be particularly important in our study that included a large number of imperfectly balanced strata. Even though LMM analysis for ascertained case–control data poten-tially results in a small loss of power in comparison to meta-analysis11, we judged the advantage of combining all strata while controlling the false positive rate to be more important than this potential loss and therefore jointly analyzed all strata in an LMM to identify additional risk loci. There was no overall inflation of the LMM test statistics in comparison to the meta-analysis test statistics (Supplementary Fig. 2). We observed modest inflation of test statistics in the quantile– quantile plot (λGC = 1.12, λ1,000 = 1.01; Supplementary Fig. 3). LD score regression yielded an intercept of 1.10 (standard error
Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis
A full list of authors and affiliations appears at the end of the paper.
Received 7 January; accepted 20 June; published online 25 July 2016; doi:10.1038/ng.3622
l e t t e r s
npg
© 2
016
Nat
ure
Am
eric
a, In
c. A
ll rig
hts
rese
rved
.np
g©
2016
Nat
ure
Am
eric
a, In
c. A
ll rig
hts
rese
rved
.

Nature GeNetics VOLUME 48 | NUMBER 9 | SEPTEMBER 2016 1047
l e t t e r s
wouter van Rheenen1,123, Aleksey shatunov2,123, Annelot m dekker1, Russell l mclaughlin3, Frank P diekstra1, sara l Pulit4, Rick A A van der spek1, Urmo Võsa5, simone de Jong6,7, matthew R Robinson8, Jian Yang8, Isabella Fogh2,9, Perry tc van doormaal1, gijs H P tazelaar1, max koppers1,10, Anna m Blokhuis1,10, william sproviero2, Ashley R Jones2, kevin P kenna11, kristel R van eijk1, oliver Harschnitz1,10, Raymond d schellevis1, william J Brands1, Jelena medic1, Androniki menelaou4, Alice Vajda12,13, nicola ticozzi9,14, kuang lin2, Boris Rogelj15,16, katarina Vrabec17, metka Ravnik-glavac17,18, Blaž koritnik19, Janez Zidar19, lea leonardis19, leja dolenc grošelj19, stéphanie millecamps20, François salachas20–22, Vincent meininger23,24, mamede de carvalho25,26, susana Pinto25,26, Jesus s mora27, Ricardo Rojas-garcía28,29, meraida Polak30,31, siddharthan chandran32,33, shuna colville32, Robert swingler32, karen e morrison34, Pamela J shaw35, John Hardy36, Richard w orrell37, Alan Pittman36,38, katie sidle37, Pietro Fratta39, Andrea malaspina40,41, simon topp2, susanne Petri42, susanne Abdulla43, carsten drepper44, michael sendtner44, thomas meyer45, Roel A ophoff46–48, kim A staats48, martina wiedau-Pazos49, catherine lomen-Hoerth50, Vivianna m Van deerlin51, John Q trojanowski51, lauren elman52, leo mccluskey52, A nazli Basak53, ceren tunca53, Hamid Hamzeiy53, Yesim Parman54, thomas meitinger55, Peter lichtner55, milena Radivojkov-Blagojevic55, christian R Andres56, cindy maurel56, gilbert Bensimon57–59, Bernhard landwehrmeyer60, Alexis Brice61–65, christine A m Payan57,59, safaa saker-delye66, Alexandra dürr67, nicholas w wood68, lukas tittmann69, wolfgang lieb69, Andre Franke70, marcella Rietschel71, sven cichon72–76, markus m nöthen72,73, Philippe Amouyel77, christophe tzourio78, Jean-François dartigues78, Andre g Uitterlinden79,80, Fernando Rivadeneira79,80, karol estrada79, Albert Hofman80,81, charles curtis6,7, Hylke m Blauw1, Anneke J van der kooi82, marianne de Visser82, An goris83, markus weber84, christopher e shaw2, Bradley n smith2, orietta Pansarasa85, cristina cereda85, Roberto del Bo86, giacomo P comi86, sandra d’Alfonso87, cinzia Bertolin88, gianni sorarù88, letizia mazzini89, Viviana Pensato90, cinzia gellera90, cinzia tiloca9, Antonia Ratti9,14, Andrea calvo91,92, cristina moglia91,92, maura Brunetti91,92, simona Arcuti93, Rosa capozzo93, chiara Zecca93, christian lunetta94, silvana Penco95, nilo Riva96, Alessandro Padovani97, massimiliano Filosto97, Bernard muller98, Robbert Jan stuit98, PARAls Registry99, slAlom group99, slAP Registry99, FAls sequencing consortium99, slAgen consortium99, nnIPPs study group99, Ian Blair100, katharine Zhang100, emily P mccann100, Jennifer A Fifita100, garth A nicholson100,101, dominic B Rowe100, Roger Pamphlett102, matthew c kiernan103, Julian grosskreutz104, otto w witte104, thomas Ringer104, tino Prell104, Beatrice stubendorff104, Ingo kurth105, christian A Hübner105, P nigel leigh106, Federico casale91, Adriano chio91,92, ettore Beghi107, elisabetta Pupillo107, Rosanna tortelli93, giancarlo logroscino108,109, John Powell2, Albert c ludolph60, Jochen H weishaupt60, wim Robberecht83,110,111, Philip Van damme83,110,111, lude Franke5, tune H Pers112–116, Robert H Brown11, Jonathan d glass30,31, John e landers11, orla Hardiman12,13, Peter m Andersen60,117, Philippe corcia56,118,119, Patrick Vourc’h56, Vincenzo silani9,14, naomi R wray8, Peter m Visscher8,120, Paul I w de Bakker4,121, michael A van es1, R Jeroen Pasterkamp10, cathryn m lewis6,122, gerome Breen6,7, Ammar Al-chalabi2,124, leonard H van den Berg1,124 & Jan H Veldink1,124
1Department of Neurology, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands. 2Maurice Wohl Clinical Neuroscience Institute, Department of Basic and Clinical Neuroscience, King’s College London, London, UK. 3Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland. 4Department of Medical Genetics, Center for Molecular Medicine, University Medical Center Utrecht, Utrecht, the Netherlands. 5Department of Genetics, University of Groningen, University Medical Centre Groningen, Groningen, the Netherlands. 6MRC Social, Genetic and Developmental Psychiatry Centre, Institute of Psychiatry, Psychology and Neuroscience, King’s College London, London, UK. 7NIHR Biomedical Research Centre for Mental Health, Maudsley Hospital and Institute of Psychiatry, Psychology and Neuroscience, King’s College London, London, UK. 8Queensland Brain Institute, University of Queensland, Brisbane, Queensland, Australia. 9Department of Neurology and Laboratory of Neuroscience, IRCCS Istituto Auxologico Italiano, Milan, Italy. 10Department of Translational Neuroscience, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands. 11Department of Neurology, University of Massachusetts Medical School, Worcester, Massachusetts, USA. 12Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland. 13Department of Neurology, Beaumont Hospital, Dublin, Ireland. 14Department of Pathophysiology and Tranplantation, ‘Dino Ferrari’ Center, Università degli Studi di Milano, Milan, Italy. 15Department of Biotechnology, Jožef Stefan Institute, Ljubljana, Slovenia. 16Biomedical Research Institute BRIS, Ljubljana, Slovenia. 17Department of Molecular Genetics, Institute of Pathology, Faculty of Medicine, University of Ljubljana, Ljubljana, Slovenia. 18Institute of Biochemistry, Faculty of Medicine, University of Ljubljana, Ljubljana, Slovenia. 19Ljubljana ALS Centre, Institute of Clinical Neurophysiology, University Medical Centre Ljubljana, Ljubljana, Slovenia. 20Institut du Cerveau et de la Moelle Epinière, INSERM U1127, CNRS UMR 7225, Sorbonne Universités, UPMC Université Paris 06, UMRS 1127, Paris, France. 21Centre de Référence Maladies Rares SLA Ile de France, Département de Neurologie, Hôpital de la Pitié-Salpêtrière, Paris, France. 22GRC-UPMC SLA et Maladies du Motoneurone, Paris, France. 23Ramsay Generale de Santé, Hôpital Peupliers, Paris, France. 24Réseau SLA Ile de France, Paris, France. 25Institute of Physiology, Institute of Molecular Medicine, Faculty of Medicine, University of Lisbon, Lisbon, Portugal. 26Department of Neurosciences, Hospital de Santa Maria–CHLN, Lisbon, Portugal. 27Department of Neurology, Hospital San Rafael, Madrid, Spain. 28Neurology Department, Hospital de la Santa Creu i Sant Pau de Barcelona, Autonomous University of Barcelona, Barcelona, Spain. 29Centro de Investigación en Red de Enfermedades Raras (CIBERER), Madrid, Spain. 30Department of Neurology, Emory University School of Medicine, Atlanta, Georgia, USA. 31Emory ALS Center, Emory University School of Medicine, Atlanta, Georgia, USA. 32Euan MacDonald Centre for Motor Neuron Disease Research, Edinburgh, UK. 33Centre for Neuroregeneration and Medical Research Council
npg
© 2
016
Nat
ure
Am
eric
a, In
c. A
ll rig
hts
rese
rved
.np
g©
2016
Nat
ure
Am
eric
a, In
c. A
ll rig
hts
rese
rved
.

RESEARCH Open Access
The alliance between genetic biobanks andpatient organisations: the experience of thetelethon network of genetic biobanksChiara Baldo1, Lorena Casareto2, Alessandra Renieri3, Giuseppe Merla4, Barbara Garavaglia5, Stefano Goldwurm6,Elena Pegoraro7, Maurizio Moggio8, Marina Mora9, Luisa Politano10, Luca Sangiorgi11, Raffaella Mazzotti12,Valeria Viotti1, Ilaria Meloni3, Maria Teresa Pellico4, Chiara Barzaghi5, Chiuhui Mary Wang13, Lucia Monaco13
and Mirella Filocamo12*
Abstract
Background: Rare diseases (RDs) are often neglected because they affect a small percentage of the population(6–8 %), which makes research and development of new therapies challenging processes. Easy access to high-qualitysamples and associated clinical data is therefore a key prerequisite for biomedical research. In this context, GeneticBiobanks are critical to developing basic, translational and clinical research on RDs. The Telethon Network of GeneticBiobanks (TNGB) is aware of the importance of biobanking as a service for patients and has started a dialoguewith RD-Patient Organisations via promotion of dedicated meetings and round-tables, as well as by includingtheir representatives on the TNGB Advisory Board. This has enabled the active involvement of POs in draftingbiobank policies and procedures, including those concerning ethical issues. Here, we report on our experiencewith RD-Patient Organisations who have requested the services of existing biobanks belonging to TNGB anddescribe how these relationships were established, formalised and maintained.
Results: The process of patient engagement has proven to be successful both for lay members, who increasedtheir understanding of the complex processes of biobanking, and for professionals, who gained awareness ofthe needs and expectations of the people involved. This collaboration has resulted in a real interest on the partof Patient Organisations in the biobanking service, which has led to 13 written agreements designed to formalise thisprocess. These agreements enabled the centralisation of rare genetic disease biospecimens and their related data,thus making them available to the scientific community.
Conclusions: The TNGB experience has proven to be an example of good practice with regard to patientengagement in biobanking and may serve as a model of collaboration between disease-oriented Biobanksand Patient Organisations. Such collaboration serves to enhance awareness and trust and to encourage thescientific community to address research on RDs.
Keywords: Patient organisations, Rare diseases, Biobanking, Networking, Agreements, Sample and data centralisation,Patient involvement, Research
* Correspondence: [email protected]. Centro di Diagnostica Genetica e Biochimica delle MalattieMetaboliche, Istituto G. Gaslini, Via G. Gaslini 5, 16147 Genoa, ItalyFull list of author information is available at the end of the article
© The Author(s). 2016 Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, andreproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link tothe Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver(http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
Baldo et al. Orphanet Journal of Rare Diseases (2016) 11:142 DOI 10.1186/s13023-016-0527-7

Finally, the TNGB-PO’s agreements demonstrate howactive collaboration between GBs and POs is critical torare disease biobanking.
ConclusionsTo the best of our knowledge, this type of agreement isunique at the national and international level. The set ofrules and tasks of the parties indeed ensures (i) qualityand proper use of the samples, (ii) individuals’ confidenti-ality throughout the entire process and, more importantly,(iii) visibility of and easy access to a specific sample collec-tion for the interested biomedical community.The TNGB experience may therefore serve as a
model of collaboration between disease-orientedBiobanks and Patient Organisations, as it shows howmutual respect and effective collaboration betweenpatients and the scientific community are essential tothe enhancement of awareness and trust, as well as tothe sharing of objectives and efforts to supportresearch on rare diseases.
Additional file
Additional file 1: Agreement template. (PDF 217 kb)
AbbreviationsBBMRI: Biobanking and Biomolecular Resources Research Infrastructure;FP7: Seventh Framework Programme for research and technologicaldevelopment; GB: Genetic Biobank; HUGO: Human Genome Organisation;MTA: Material Transfer Agreement; PO: Patient Organisation; RD: RareDisease; TNGB: Telethon Network of Genetic Biobanks; UNESCO: UnitedNations Educational, Scientific and Cultural Organization
AcknowledgmentsThis work was supported by the Telethon Network of Genetic Biobanks(Project No. GTB12001). We thank the Patient Organisations (Ring 14International, Federazione Italiana Prader Willi, Associazione Mowat Wilson,Associazione Nonsolo15, Gruppo Famiglie Dravet, ASAL-Associazione Sindromedi Alport, AIRETT-Associazione Italiana Rett, Associazione Italiana Sindrome diPoland, ILA-Associazione Italiana Angiodisplasie ed Emangiomi Infantili, LNDFamiglie Italiane, F.O.P. Italia Onlus, Associazione Italiana Sindromi Neurodegen-erative da Accumulo di Ferro, ASSI Gulliver - Sindrome di Sotos) for theireffective engagement. We thank Renza Barbon Galluppi (President ofUNIAMO FIRM till 2015) and Sara Casati (Bioethicist) for their dedicationand active participation within the “Determinazione Rara” project. Finally,the authors would also like to give special thanks to Annamaria Merico, of theTelethon Technology Transfer Office, for her continued support and assistance.This paper is dedicated to the memory of Professor Franca Dagna Bricarelli,a long-time friend and TNGB Coordinator Emeritus until 2014. Francaplayed a pivotal role in developing collaboration with Patient Organisations.Without Franca's knowledge and enthusiasm this work would not have beenpossible.
FundingThis work was partially supported by Telethon Italy, the funding body ofproject no. GTB12001, “Telethon Network of Genetic Biobanks”.
Availability of data and materialsNot applicable.
Authors’ contributionsMF and CBal coordinated the study and drafted the manuscript with theassistance of LC. RM, VV, IM, MTP and CBar were involved in the daily
biobanking operations within the respective agreements. AR, GM, BG, SG, EP,MMog, MMor, LP, LS, CMW and LM helped to draft the manuscript. Allauthors read and approved the final manuscript.
Competing interestsThe authors declare that they have no competing interests.
Consent for publicationNot applicable.
Ethics approval and consent to participateNot applicable.
Author details1S.C. Laboratorio di Genetica Umana, E.O. Ospedali Galliera, Genoa, Italy.2Ufficio Coordinamento Network, c/o U.O.S.D. Centro di Diagnostica Geneticae Biochimica delle Malattie Metaboliche, Istituto G. Gaslini, Genoa, Italy.3Medical Genetics, University of Siena and Genetica Medica, AziendaOspedaliera Universitaria Senese, Siena, Italy. 4Medical Genetics Unit, IRCCSCasa Sollievo della Sofferenza, S. Giovanni Rotondo, FG, Italy. 5U.O.C.Neurogenetica Molecolare, Fondazione I.R.C.C.S. Istituto Neurologico CarloBesta, Milan, Italy. 6Parkinson Institute, ASST Centro Specialistico OrtopedicoTraumatologico G. Pini – CTO, Milan, Italy. 7Università di Padova, AziendaOspedaliera Universitaria, Padova, Italy. 8Neuromuscular and Rare DiseasesUnit, Dino Ferrari Centre, IRCCS Foundation Ca’ Granda Ospedale MaggiorePoliclinico, University of Milan, Milan, Italy. 9Laboratorio di Biologia Cellulare,UO Malattie Neuromuscolari e Neuroimmunologia, Fond. IRCCS IstitutoNeurologico Carlo Besta, Milan, Italy. 10Cardiomiologia e Genetica Medica,Dipartimento di Medicina Sperimentale, Seconda Università di Napoli eAzienda Ospedaliera Universitaria della SUN, Naples, Italy. 11S.S.D. GeneticaMedica e Malattie Rare Ortopediche Istituto Ortopedico Rizzoli, Bologna, Italy.12U.O.S.D. Centro di Diagnostica Genetica e Biochimica delle MalattieMetaboliche, Istituto G. Gaslini, Via G. Gaslini 5, 16147 Genoa, Italy.13Fondazione Telethon, Milan, Italy.
Received: 3 August 2016 Accepted: 18 October 2016
References1. Monaco L, Crimi M, Wang CM. The challenge for a European network
of biobanks for rare diseases taken up by RD-Connect. Pathobiology.2014. doi:10.1159/000358492.
2. Filocamo M, Baldo C, Goldwurm S, Renieri A, Angelini C, Moggio M, Mora M,Merla G, Politano L, Garavaglia B, Casareto L, Bricarelli FD. Telethonnetwork of genetic biobanks staff. Telethon network of geneticbiobanks: a key service for diagnosis and research on rare diseases.Orphanet J Rare Dis. 2013. doi:10.1186/1750-1172-8-129.
3. Thompson R, Johnston L, Taruscio D, Monaco L, Béroud C, Gut IG, Hansson MG,'t Hoen PB, Patrinos GP, Dawkins H, Ensini M, Zatloukal K, Koubi D,Heslop E, Paschall JE, Posada M, Robinson PN, Bushby K, Lochmüller H.RD-Connect: an integrated platform connecting databases, registries,biobanks and clinical bioinformatics for rare disease research. J GenIntern Med. 2014. doi:10.1007/s11606-014-2908-8.
4. Mitchell D, Geissler J, Parry-Jones A, Keulen H, Schmitt D, Vavassori R,Matharoo-Ball B. Biobanking from the patient perspective. Res InvolvemEngagem. 2015. doi:10.1186/s40900-015-0001-z.
5. Graham CE, Molster C, Baynam GS, Bushby K, Hansson M, Mascalzoni D,Kole A, Mora M, Monaco L, Bellgard M, Carpentieri D, Posada M, Riess O,Rubinstein YR, Schaefer F, Taruscio D, Terry SF, Zatloukal K, Knoppers B,Lochmüller H, Dawkins HJS. Current trends in biobanking for rare diseases: areview. J Biorepository Sci Appl Med. 2014;2:49–61. http://dx.doi.org/10.2147/BSAM.S46707.
6. Mora M, Angelini C, Bignami F, Bodin AM, Crimi M, Di Donato JH, Felice A,Jaeger C, Karcagi V, LeCam Y, Lynn S, Meznaric M, Moggio M, Monaco L,Politano L, de la Paz MP, Saker S, Schneiderat P, Ensini M, Garavaglia B,Gurwitz D, Johnson D, Muntoni F, Puymirat J, Reza M, Voit T, Baldo C,Bricarelli FD, Goldwurm S, Merla G, Pegoraro E, Renieri A, Zatloukal K,Filocamo M, Lochmüller H. The EuroBioBank Network: 10 years of hands-onexperience of collaborative, transnational biobanking for rare diseases.Eur J Hum Genet. 2015. doi:10.1038/ejhg.2014.272.
Baldo et al. Orphanet Journal of Rare Diseases (2016) 11:142 Page 7 of 8

Cancer Res Treat. 2016 Mar 25 [Epub ahead of print]
pISSN 1598-2998, eISSN 2005-9256
http://dx.doi.org/10.4143/crt.2015.450
│ http://www.e-crt.org │ 1Copyright ⓒ 2016 by the Korean Cancer AssociationThis is an Open-Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/)
which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Open Access
Coexistence of VHL Disease and CPT2 Deficiency: A Case Report
Case Report
von Hippel-Lindau (VHL) disease is an inherited syndrome manifesting with benign
and malignant tumors. Deficiency of carnitine palmitoyltransferase type II (CPT2) is a
disorder of lipid metabolism that, in the muscle form, manifests with recurrent attacks
of myalgias often associated with myoglobinuria. Rhabdomyolytic episodes may be
complicated by life-threatening events, including acute renal failure (ARF). We report
on a male patient who was tested, at 10 years of age, for VHL disease because of
family history of VHL. He was diagnosed with VHL but without VHL-related manifesta-
tion at the time of diagnosis. During childhood, the patient was hospitalized several
times for diffuse muscular pain, muscle weakness, and dark urine. These recurrent
attacks of rhabdomyolysis were never accompanied by ARF. The patient was found
to be homozygous for the mutation p.S113L of the CPT2 gene. To the best of our
knowledge, this is the first report of the coexistence of VHL disease and CPT2 defi-
ciency in the same individual. Based on findings from animal models, the case illus-
trates that mutations in the VHL gene might protect against renal damage caused by
CPT2 gene mutations.
Key words
von Hippel-Lindau disease,
Carnitine palmitoyltransferase type II,
Neoplasms, Rhabdomyolysis, Acute kidney injury
Introduction
von Hippel-Lindau disease (VHL) is a hereditary, autoso-
mal-dominant, neoplastic disease associated with various
tumor types, including central nervous system (CNS) and
retinal hemangioblastomas, clear cell renal carcinomas,
pheochromocytomas, and pancreatic neuroendocrine
tumors, in addition to pancreatic and renal cysts. The disease
is caused by mutations in the VHL tumor-suppressor gene.
As described by the two-hit hypothesis of Knudson, affected
patients inherit one mutated copy of the VHL gene from an
affected parent through the germline. Later in life, the other
normal copy of the VHL gene undergoes somatic mutation
in susceptible tissues (i.e., the second hit), initiating local
tumorigenesis. The VHL protein is expressed ubiquitously,
and its function is primarily associated with the formation of
a ubiquitin ligase complex with other participating proteins
+ + + + + + + + + + + + + + + + + + + +
+ + + + + + + + + + + + + + + + + + + +
+ + + + + + + + + + + + + + + + + + + +
+ + + + + + + + + + + + + + + + + + + +
+ + + + + + + + + + + + + + + + + + + +
+ + + + + + + + + + + + + + + + + + + +
+ + + + + + + + + + + + + + + + + + + +
+ + + + + + + + + + + + + + + + + + + +
+ + + + + + + + + + + + + + + + + + + +
+ + + + + + + + + + + + + + + + + + + +
+ + + + + + + + + + + + + + + + + + + +
+ + + + + + + + + + + + + + + + + + + +
+ + + + + + + + + + + + + + + + + + + +
Correspondence:
Alfonso Massimiliano Ferrara, MD, PhD
Familial Tumor Unit, Veneto Institute of
Oncology IOV-IRCCS, Via Gattamelata,
68, 35128 Padua, Italy
Tel: 39-0498215503
Fax: 39-0498215502
E-mail: [email protected]
Received November 22, 2015
Accepted March 14, 2016
Alfonso Massimiliano Ferrara, MD, PhD1
Monica Sciacco, MD, PhD2
Stefania Zovato, MD, PhD1
Silvia Rizzati, MD1
Irene Colombo, MD2
Francesca Boaretto, PhD1
Maurizio Moggio, MD, PhD2
Giuseppe Opocher, MD1,3
1Familial Tumor Unit, Veneto Institute of Oncology IOV-IRCCS, Padua, 2Neuromuscular and Rare Disease Unit, Dino Ferrari Centre, IRCCS Foundation Ca' Granda, Ospedale Maggiore Policlinico,University of Milan, Milan, 3Department of Medicine, DIMED University of Padua, Padua, Italy

Rare variants in SQSTM1 and VCP genes and risk of sporadic inclusionbody myositis
Qiang Gang a,b, Conceição Bettencourt a,c,1, Pedro M. Machado a,b,d,1, Stefen Brady b,e,Janice L. Holton b, Alan M. Pittman a,f, Deborah Hughes a, Estelle Healy b,Matthew Parton b, David Hilton-Jones e, Perry B. Shieh g, Merrilee Needhamh,Christina Liang i, Edmar Zanoteli j, Leonardo Valente de Camargo j, Boel De Paepe k,Jan De Bleecker k, Aziz Shaibani l, Michela Ripolonem, Raffaella Violanom,Maurizio Moggiom, Richard J. Barohn n, Mazen M. Dimachkie n, Marina Mora o,Renato Mantegazza o, Simona Zanotti o, Andrew B. Singleton p, Michael G. Hanna a,b,*,Henry Houlden a,b,q,*, The Muscle Study Group and The International IBM GeneticsConsortium
aDepartment of Molecular Neuroscience, Institute of Neurology, University College London, Queen Square, London, UKbMRC Centre for Neuromuscular Diseases, Institute of Neurology, University College London, Queen Square, London, UKcDepartment of Clinical and Experimental Epilepsy, Institute of Neurology, University College London, Queen Square, London, UKdCentre for Rheumatology, Division of Medicine, University College London, London, UKeNuffield Department of Clinical Neurosciences, John Radcliffe Hospital, Oxford, UKfReta Lila Weston Laboratories, UCL Institute of Neurology, London, UKgDepartment of Neurology, University of California, Los Angeles, Los Angeles, CA, USAhWestern Australian Neurosciences Research Institute (WANRI), University of Western Australia and Murdoch University, Fiona Stanley Hospital, Perth,AustraliaiDepartment of Neurology, Royal North Shore Hospital, New South Wales, AustraliajDepartment of Neurology, Medical School of the University of São Paulo (FMUSP), São Paulo, BrazilkDepartment of Neurology and Neuromuscular Reference Centre, Ghent University Hospital, Ghent, BelgiumlNerve and Muscle Center of Texas, Houston, TX, USAmNeuromuscular Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Dino Ferrari Centre, University of Milan, Milan, ItalynThe University of Kansas Medical Centre, Kansas City, KS, USAoNeuromuscular Diseases and Neuroimmunology Unit, Fondazione IRCCS Isitituto Neurologico C. Besta, Milano, Italyp Laboratory of Neurogenetics, National Institute on Aging, National Institute of Health, Bethesda, MD, USAqNeurogenetics Laboratory, Institute of Neurology, University College London, Queen Square, London, UK
a r t i c l e i n f o
Article history:Received 17 March 2016Received in revised form 27 June 2016Accepted 29 July 2016
Keywords:Sporadic inclusion body myositissIBMSQSTM1VCPGenetic risk factor
a b s t r a c t
Genetic factors have been suggested to be involved in the pathogenesis of sporadic inclusion bodymyositis (sIBM). Sequestosome 1 (SQSTM1) and valosin-containing protein (VCP) are 2 key genes asso-ciated with several neurodegenerative disorders but have yet to be thoroughly investigated in sIBM. Acandidate gene analysis was conducted using whole-exome sequencing data from 181 sIBM patients, andwhole-transcriptome expression analysis was performed in patients with genetic variants of interest. Weidentified 6 rare missense variants in the SQSTM1 and VCP in 7 sIBM patients (4.0%). Two variants, theSQSTM1 p.G194R and the VCP p.R159C, were significantly overrepresented in this sIBM cohort comparedwith controls. Five of these variants had been previously reported in patients with degenerative diseases.The messenger RNA levels of major histocompatibility complex genes were upregulated, this elevationbeing more pronounced in SQSTM1 patient group. We report for the first time potentially pathogenicSQSTM1 variants and expand the spectrum of VCP variants in sIBM. These data suggest that defects in
* Corresponding authors at: MRC Centre for Neuromuscular Diseases, Department of Molecular Neuroscience, Institute of Neurology, University College London, QueenSquare, London WC1N 3BG, UK. Tel.: þ44 203 448 4068; fax: þ44 20 3448 4786.
E-mail addresses: [email protected] (M.G. Hanna), [email protected] (H. Houlden).1 Equally contributed to the work.
Contents lists available at ScienceDirect
Neurobiology of Aging
journal homepage: www.elsevier .com/locate/neuaging
0197-4580/� 2016 The Author(s). Published by Elsevier Inc. This is an open access article under the CC BY license (http://creativecommons.org/licenses/by/4.0/).http://dx.doi.org/10.1016/j.neurobiolaging.2016.07.024
Neurobiology of Aging xxx (2016) 1.e1e1.e9

RESEARCH PAPER
LOPED study: looking for an early diagnosis in alate-onset Pompe disease high-risk populationO Musumeci,1 G la Marca,2 M Spada,3 S Mondello,1 C Danesino,4 G P Comi,5
E Pegoraro,6 G Antonini,7 G Marrosu,8 R Liguori,9 L Morandi,10 M Moggio,11
R Massa,12 S Ravaglia,4 A Di Muzio,13 M Filosto,14 P Tonin,15 G Di Iorio,16
S Servidei,17 G Siciliano,18 C Angelini,6,19 T Mongini,20 A Toscano,1 the ItalianGSD II group
For numbered affiliations seeend of article.
Correspondence toProfessor Antonio Toscano,Department of Neurosciences,University of Messina, RegionalReference Center for RareNeuromuscular Disorders, AOUPoliclinico “G Martino”, Via CValeria, Messina 98125, Italy;[email protected]
Received 17 December 2014Revised 28 January 2015Accepted 20 February 2015Published Online First17 March 2015
To cite: Musumeci O,la Marca G, Spada M, et al.J Neurol NeurosurgPsychiatry 2016;87:5–11.
ABSTRACTObjective A multicentre observational study wasaimed to assess the prevalence of late-onset Pompedisease (LOPD) in a large high-risk population, using thedried blood spot (DBS) as a main screening tool.Design/methods 17 Italian neuromuscular centreswere involved in the late-onset Pompe early diagnosis(LOPED) study. Inclusion criteria were: (1) age ≥5 years,(2) persistent hyperCKaemia and (3) muscle weakness atupper and/or lower limbs (limb-girdle muscle weakness,LGMW). Acid α-glucosidase (GAA) activity wasmeasured separately on DBS by fluorometric as well astandem mass spectrometry methods. A DBS retest wasperformed in patients resulted positive at first assay. Forthe final diagnosis, GAA deficiency was confirmed by abiochemical assay in skeletal muscle, whereas genotypewas assessed by GAA molecular analysis.Results In a 14-month period, we studied 1051 cases:30 positive samples (2.9%) were detected by first DBSscreening, whereas, after retesting, 21 samples were stillpositive. Biochemical and molecular genetic studiesfinally confirmed LOPD diagnosis in 17 cases (1.6%).The median time from the onset of symptoms/signs todiagnosis was 5 years. Among those patients, 35%showed presymptomatic hyperCKaemia and 59%showed hyperCKaemia+LGMW, whereas 6% manifestedwith LGMW.Conclusions LOPED study suggests that GAA activityshould be accurately screened by DBS in all patientsreferring for isolated hyperCKaemia and/or LGMW. Atimely diagnosis was performed in five patients withpresymptomatic hyperCKaemia, but two had alreadymanifested with relevant changes on muscle morphologyand MRI. Consequently, enzyme replacement therapywas started in 14/17 patients, including the 2 patientsstill clinically presymptomatic but with a laboratoryevidence of disease progression.
INTRODUCTIONPompe disease (glycogen storage disease type II,GSD II) is a rare autosomal recessive disorder dueto Acid α-glucosidase (GAA) deficiency leading toglycogen accumulation in several tissues, with pre-dilection of skeletal muscle and heart.1 Clinically, itis associated with a range of phenotypes, with vari-able organ involvement, age of onset and severitydegree.2
Late-onset Pompe disease (LOPD) is a slowlyprogressive form with juvenile or adult presenta-tion, characterised by progressive muscle weaknessand, often, respiratory impairment.3
Diagnosis of LOPD is still challenging and oftenquite delayed. This has been hypothesised to bedue to several reasons such as rarity of the disorder,wide clinical spectrum, overlap of signs and symp-toms with other neuromuscular disorders or vari-able diagnostic approach in different countries.4
Recently, Kishnani et al,5 using data from theInternational Pompe Registry, calculated the timeinterval between onset and diagnosis, ‘a diagnosticgap’, in different categories of patients with Pompedisease and still found this delay consistent.Since 2006, enzyme replacement therapy (ERT)
represents the first disease-specific treatment. Inpatients with LOPD, ERTwas less effective in olderjuveniles and adults than in infants, likely becauseof delayed diagnosis.4 6
These considerations suggested development ofmore rapid diagnostic tools, such as the driedblood spot (DBS), to detect GAA activity, whichcould result in an earlier LOPD diagnosis.7–10
This study, based on measuring GAA activity byDBS, reports on the results of a multicentreresearch project looking for the prevalence ofLOPD in a large high-risk population, in order tostart ERT in a timely manner.
PATIENTS AND METHODSThe study was conducted in accordance with GCPand ethical principles deriving from theDeclaration of Helsinki, and from regulations inforce for observational studies.It was approved by the Ethics Committees of the
Coordinator Center (Messina) and participantcentres.The patients, recruited from 17 Italian neuro-
muscular centres, were all admitted for diagnosticpurposes.Inclusion criteria were: (1) age ≥5 years, (2) per-
sistent hyperCKaemia (considered as 1.5-fold theupper normal limit, ie, creatine kinase (CK)>250 UI/L in females and >350 UI/L in males),evidenced at least twice with a minimum interval of1 month, and/or (3) muscle weakness affectingupper and/or lower limbs (limb-girdle muscle weak-ness, LGMW).
Musumeci O, et al. J Neurol Neurosurg Psychiatry 2016;87:5–11. doi:10.1136/jnnp-2014-310164 5
Neuromuscular
group.bmj.com on September 2, 2016 - Published by http://jnnp.bmj.com/Downloaded from

analysis.22 At the end of the retesting, we still found four ‘false’positive cases presenting with presymptomatic hyperCKaemia,no muscle biopsy-specific abnormalities, residual GAA activityin skeletal muscle within normal range and only one mutationidentified by GAA gene sequencing; these results suggest consid-ering them as heterozygous individuals.
The two different methods presented good performances,even if some differences have been revealed. The false-positiverate was 9/1051 (0.85%) for TMS analysis and 11/1051 (1.05%)for fluorometry at first assay, and 4/1051 (0.38%) and 6/1051(0.57%) for retesting, respectively. Even if not appropriate due tosmall numbers of investigated individuals, in this study, TMSidentified all 17 patients with LOPD with a positive predictivevalue (PPV) at first test 0.65, whereas PPV was slightly lower byfluorometry (0.58). Between these two techniques, similar differ-ences in false-positive rate and PPVs have already been reportedin the literature in a larger population.23
In addition, we have here included a revised version of a diag-nostic algorithm (figure 1), where it is indicated that a positiveDBS must be confirmed by biochemical assays on differenttissues and/or by a genetic analysis to complete the diagnosticpanel.4 However, in cases where the molecular analysis, firstrequested after a positive DBS, is not fully confirmatory, it willbe necessary to perform the biochemical analysis to detect GAAactivity levels.
Nowadays, when to start ERT in LOPD is largely debated. In2012, American Association of Neuromuscular &Electrodiagnostic Medicine (AANEM) guidelines recommendedstarting ERT in symptomatic patients and in presymptomaticpatients with proximal muscle weakness, detectable by manualmuscle testing, or with a reduction in respiratory parameters.24
On the other hand, some reports have suggested that ERTshould be started at the earliest onset of symptoms or objectivesigns, to obtain better therapeutic responses.25–27
In fact, it is well known that a progressive muscular derange-ment, caused by enlargement and rupture of glycogen-filledlysosomes as well as autophagic dysfunction, is responsible forirreversible damage of skeletal and respiratory muscles.
Therefore, ERT guidelines have been mainly addressed tosymptomatic patients, without taking into consideration labora-tory or instrumental investigations, such as GAA residual activ-ity, morphological aspects or muscle MRI findings inpresymptomatic patients. ERT was started in 12 patientsshowing LGMW+hyperCKaemia but also in 2/5 presentingwith hyperCKaemia with no clinical symptoms but with clear-cut muscle damage demonstrated by MRI, and altered musclemorphological features.
Finally, we suggest reconsidering the guidelines in order tobetter define possible additional parameters that may influencethe decision for a timely start of ERT. However, only a pro-longed follow-up of early treated cases could confirm theopportunity to treat such patients at the presymptomatic stage.
Therefore, our data suggest that DBS plays a central role inthe diagnostic work up of high-risk patients with presympto-matic hyperCKaemia and/or LGMW of unknown origin whereLOPD is suspected.
Author affiliations1Department of Neurosciences, University of Messina, Messina, Italy2Department of Neurosciences, Psychology, Pharmacology and Child Health,University of Florence, Florence, Italy3Department of Neuroscience, University of Turin, Turin, Italy4University of Pavia, Pavia, Italy5Neuroscience Section, Department of Pathophysiology and Transplantation (DEPT),Neurology Unit, Dino Ferrari Centre, University of Milan, IRCCS Foundation Ca’Granda Ospedale Maggiore Policlinico, Milan, Italy
6Neurological Clinic, University of Padova, Padova, Italy7Department of Neurology, Mental Health and Sensory Organs (NESMOS) Faculty ofMedicine and Psychology University of Rome “Sapienza”, Rome, Italy8ASP 8, Cagliari, Italy9IRCCS Istituto di Scienze Neurologiche and Department of Biomedical andNeuromotor Sciences, University of Bologna, Bologna, Italy10Neuroimmunology and Neuromuscular Diseases Unit, Foundation IRCCSNeurological Institute “Carlo Besta”, Italy11Neuromuscular Unit—Fondazione IRCCS Ca’ Granda Ospedale MaggiorePoliclinico, Milano, Dino Ferrari Centre University of Milan, Milan, Italy12University of Tor Vergata, Roma, Italy13Centro Malattie Neuromuscolari e Centro Studi sull’Invecchiamento (CeSI), Chieti,Italy14University Hospital Spedali Civili, Neurological Clinic, Brescia, Italy15Neurological Clinic, University of Verona, Verona, Italy16University of Napoli, Napoli, Italy17Institute of Neurology, Catholic University, Rome, Italy18Neurological Clinic, University of Pisa, Pisa, Italy19IRCCS S Camillo, Venice, Italy20Department of Neurosciences ‘Rita Levi Montalcini’, University of Turin, Turin, Italy
Collaborators The Italian GSD II group includes: F Montagnese University ofMessina, D Ombrone BSc, Newborn Screening, Clinical Chemistry and PharmacologyLab, Meyer Children’s University Hospital, Firenze, S Pagliardini, MD (University ofTurin), P De Filippi, University of Pavia, D Ronchi, University of Milan, C Semplicini,University of Padova, M Garibaldi, University of Rome, R Piras, University of Cagliari,L Maggi, “C Besta” National Institute of Neurology, V Lucchini, University of Milan,C Terracciano, University of Rome Tor Vergata, A Todeschini University of Brescia, MScarpelli, University of Verona, F Ciccocioppo, Centro Malattie Neuromuscolari eCentro Studi sull’Invecchiamento (CeSI), Chieti, G Primiano, Catholic University,Rome, G Ricci, University of Pisa, L Vercelli, University of Turin, E Barca, University ofMessina.
Contributors All authors provided substantial contributions to the conception ordesign of the work, or the acquisition, analysis or interpretation of data. Allcollaborators collected data and provided and cared for the study patients.
Funding This work has been made possible through unconditional support fromGenzyme-Sanofi.
Competing interests All the authors are active members of the Italian Associationof Myology (AIM) and this project is a part of AIM scientific programmes. AT is amember of the Pompe Global Advisory Board sustained by Genzyme and hasreceived reimbursement for participation in board meetings and invited lectures. CA,TM and MF received reimbursement for participation in board meetings and invitedlectures by Genzyme.
Ethics approval It was approved by the Ethics Committees of the CoordinatorCenter (Messina) and participating centres.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement AT had full access to all of the data in the study andtakes responsibility for the integrity of the data and the accuracy of the dataanalysis.
REFERENCES1 Hers HG. Alpha-glucosidase deficiency in generalized glycogen storage disease
(Pompe’s disease). Biochem J 1963;86:11–16.2 Kishnani P, Hwu W, Mandel H, et al. A retrospective, multinational, multicenter
study on the natural history of infantile-onset Pompe disease. J Pediatr2006;148:671–6.
3 Gaeta M, Barca E, Ruggeri P, et al. Late-onset Pompe disease (LOPD): correlationsbetween respiratory muscles CT and MRI features and pulmonary function. MolGenet Metab 2013;110:290–6.
4 Toscano A, Montagnese F, Musumeci O. Early is better? a new algorithm for earlydiagnosis in late onset Pompe disease (LOPD). Acta Myol 2013;32:78–81.
5 Kishnani PS, Amartino HM, Lindberg C, et al. Timing of diagnosis of patients withPompe disease: data from the Pompe Registry. Am J Med Genet A2013;161:2431–43.
6 Chien YH, Hwu WL, Lee NC. Pompe disease: early diagnosis and early treatmentmake a difference. Pediatr Neonatol 2013;54:219–27.
7 Kallwass H, Carr C, Gerrein J, et al. Rapid diagnosis of late-onset Pompe disease byfluorometric assay of alpha-glucosidase activities in dried blood spots. Mol GenetMetab 2007;90:449–52.
8 la Marca G, Casetta B, Malvagia S, et al. New strategy for the screening oflysosomal storage disorders: the use of the online trapping-and-cleanup liquidchromatography/mass spectrometry. Anal Chem 2009;81:6113–21.
10 Musumeci O, et al. J Neurol Neurosurg Psychiatry 2016;87:5–11. doi:10.1136/jnnp-2014-310164
Neuromuscular
group.bmj.com on September 2, 2016 - Published by http://jnnp.bmj.com/Downloaded from

RESEARCH Open Access
Gene-specific mitochondria dysfunctions inhuman TARDBP and C9ORF72 fibroblastsElisa Onesto1, Claudia Colombrita1,2, Valentina Gumina1,2, Maria Orietta Borghi3,4, Sabrina Dusi5, Alberto Doretti1,2,Gigliola Fagiolari6, Federica Invernizzi5, Maurizio Moggio6, Valeria Tiranti5, Vincenzo Silani1,2 and Antonia Ratti1,2*
Abstract
Dysregulation of RNA metabolism represents an important pathogenetic mechanism in both amyotrophic lateralsclerosis (ALS) and frontotemporal dementia (FTD) due to the involvement of the DNA/RNA-binding proteins TDP-43and FUS and, more recently, of C9ORF72. A potential link between dysregulation of RNA metabolism and mitochondrialdysfunction is recently emerged in TDP-43 disease models. To further investigate the possible relationship between thesetwo pathogenetic mechanisms in ALS/FTD, we studied mitochondria functionality in human mutant TARDBP(p.A382T)and C9ORF72 fibroblasts grown in galactose medium to induce a switch from a glycolytic to an oxidative metabolism. Inthis condition we observed significant changes in mitochondria morphology and ultrastructure in both mutant cellswith a fragmented mitochondria network particularly evident in TARDBP(p.A382T) fibroblasts. From analysis of themitochondrial functionality, a decrease of mitochondria membrane potential with no alterations in oxygenconsumption rate emerged in TARDBP fibroblasts. Conversely, an increased oxygen consumption and mitochondriahyperpolarization were observed in C9ORF72 fibroblasts in association to increased ROS and ATP content. We foundevidence of autophagy/mitophagy in dynamic equilibrium with the biogenesis of novel mitochondria, particularly inmutant C9ORF72 fibroblasts where an increase of mitochondrial DNA content and mass, and of PGC1-α protein wasobserved. Our imaging and biochemical data show that wild-type and mutant TDP-43 proteins do not localize atmitochondria so that the molecular mechanisms responsible for such mitochondria impairment remain to be furtherelucidated. For the first time our findings assess a link between C9ORF72 and mitochondria dysfunction and indicatethat mitochondria functionality is affected in TARDBP and C9ORF72 fibroblasts with gene-specific features in oxidativeconditions. As in neuronal metabolism mitochondria are actively used for ATP production, we speculate that TARDBPand C9ORF72 mutations might trigger cell death by impairing not only RNA metabolism, but also mitochondria activityin ALS/FTD neurons.
Keywords: ALS, FTD, TDP-43, C9ORF72, Fibroblast, Mitochondria dysfunction
IntroductionAbnormal aggregates of TDP-43 protein represent aneuropathological hallmark of the fatal motoneuron dis-ease amyotrophic lateral sclerosis (ALS) and of fronto-temporal dementia (FTD). TDP-43 forms pathologicalcytoplasmic inclusions in affected brain tissues of nearlyall sporadic and familial ALS cases, with the exception ofSOD1-associated forms, and of a subset of patients withFTD (FTD-TDP), representing an important molecular
link between these two apparently different neurodegener-ative disorders [29]. TDP-43 is a DNA/RNA bindingprotein, mainly localized in the nucleus where it is in-volved in regulating splicing, transcription and miRNAbiogenesis, but it has also regulatory functions in thecytoplasm controlling target mRNA stability, transportand translation [26]. The presence of abnormal TDP-43aggregates in the cytoplasm together with the concur-rent depletion of TDP-43 from the nucleus suggest thatTDP-43 may trigger neuronal death by two differentpathogenetic mechanisms including a toxic gain-of-function and/or a loss-of-nuclear function. Independ-ently of the pathogenetic mechanism of TDP-43, RNAmetabolism is strongly impaired at different levels in
* Correspondence: [email protected] of Neurology and Laboratory of Neuroscience, IRCCS IstitutoAuxologico Italiano, Via Zucchi, 18, Cusano Milanino 20095, MI, Italy2Department of Pathophysiology and Transplantation, ‘Dino Ferrari’ Center -Università degli Studi di Milano, Milan, ItalyFull list of author information is available at the end of the article
© 2016 Onesto et al. Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, andreproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link tothe Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver(http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
Onesto et al. Acta Neuropathologica Communications (2016) 4:47 DOI 10.1186/s40478-016-0316-5
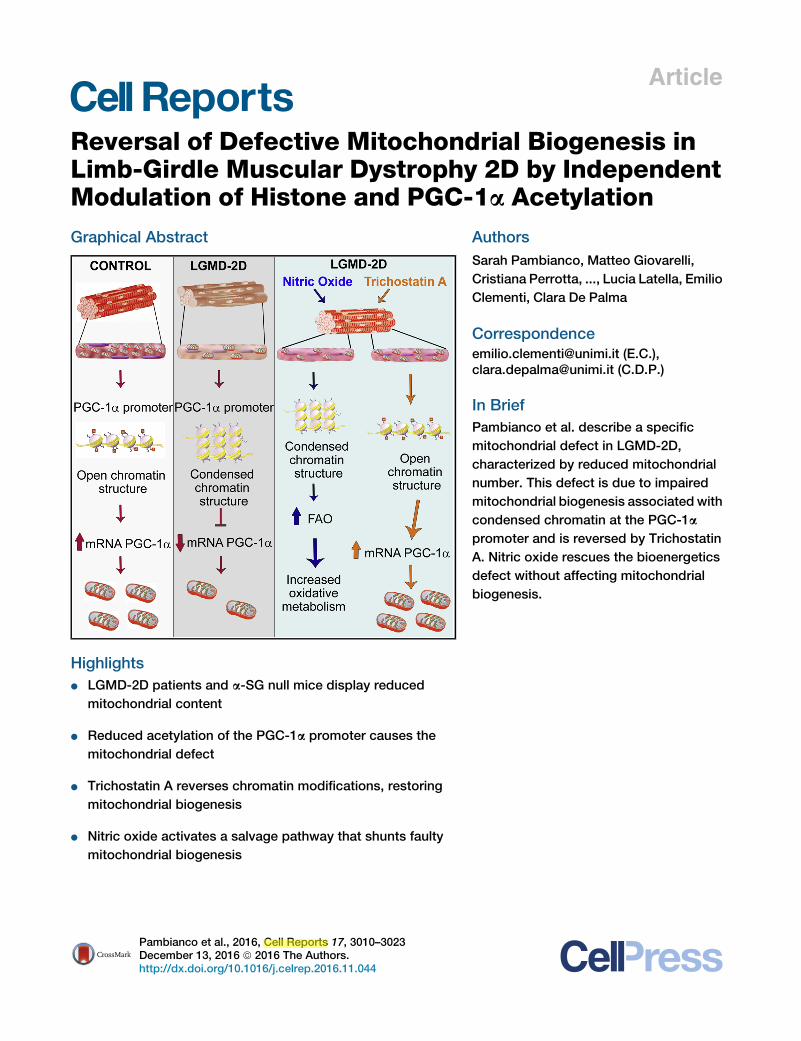
Article
Reversal of Defective Mitochondrial Biogenesis inLimb-Girdle Muscular Dystrophy 2D by IndependentModulation of Histone and PGC-1a Acetylation
Graphical Abstract
Highlights
d LGMD-2D patients and a-SG null mice display reduced
mitochondrial content
d Reduced acetylation of the PGC-1a promoter causes the
mitochondrial defect
d Trichostatin A reverses chromatin modifications, restoring
mitochondrial biogenesis
d Nitric oxide activates a salvage pathway that shunts faulty
mitochondrial biogenesis
Authors
Sarah Pambianco, Matteo Giovarelli,
Cristiana Perrotta, ..., Lucia Latella, Emilio
Clementi, Clara De Palma
[email protected] (E.C.),[email protected] (C.D.P.)
In Brief
Pambianco et al. describe a specific
mitochondrial defect in LGMD-2D,
characterized by reduced mitochondrial
number. This defect is due to impaired
mitochondrial biogenesis associated with
condensed chromatin at the PGC-1a
promoter and is reversed by Trichostatin
A. Nitric oxide rescues the bioenergetics
defect without affecting mitochondrial
biogenesis.
Pambianco et al., 2016, Cell Reports 17, 3010–3023December 13, 2016 ª 2016 The Authors.http://dx.doi.org/10.1016/j.celrep.2016.11.044

Fondazione Ca’ Granda- Ospedale Maggiore Policlinico (Milano, Italy), which
is part of the Telethon Genetic Biobank network.
Oxygen Consumption Measurement
Oxygen consumption was measured using Oroboros O2K oxygraph
(Oroboros Instruments) as described in the Supplemental Experimental
Procedures.
PGC-1a Acetylation Assay
PGC-1a acetylation was detected by immunoprecipitation in TA and dia-
phragm muscles as described (Woldt et al., 2013). In brief, muscle tissues
were homogenized in a buffer containing 50 mM Tris-HCl (pH 7.4), 68 mM su-
crose, 50mMKCl, 10mMEDTA, 0.2%BSA, protease, and phosphatase inhib-
itor mixture. PGC-1a protein was immunoprecipitated from 500 mg of protein
homogenate with Santa Cruz antibody against PGC-1a antibody (Santa Cruz
Biotechnology, sc-13067, 2 mg per sample) followed by western blot analysis
using antibody against acetyl-lysine (1:2,000, Cell Signaling 944, Cell Signaling
Technology) and PGC-1a (Cell Signaling 4259). To demonstrate the specificity
of the Santa Cruz antibody, we have immunoprecipitated muscle lysate with
the Santa Cruz antibody and detected the PGC-1a band with two different an-
tibodies, Cell Signaling 4259 and Millipore, AB3242 (Merck Millipore). The
respective IgG as negative control has been used, as shown in the Figure S7.
Chromatin Immunoprecipitation
ChIP assay was performed as previously described (Albini et al., 2013) withmi-
nor modifications indicated in the Supplemental Experimental Procedures and
using primers specified in Table S2.
Quantitative Real-Time PCR and DNA Quantification
Total RNA and mitochondrial DNA were isolated from human and mouse mus-
cles as described in the Supplemental Experimental Procedures and amplified
using the primers listed in Table S1.
Statistical Analysis
Statistical significance of raw data between the groups in each experiment
was evaluated using unpaired Student’s t test (single comparisons) or one-
way ANOVA followed by Bonferroni or Tukey post-tests (multiple compari-
sons). When data are not normally distributed, the Mann-Whitney test was
used and, when indicated, data belonging from different experiments were
represented and averaged in the same graph. The GraphPad Prism soft-
ware package (Graph Software) was used. The results are expressed as
means ± SEM of the indicated n values. A probability of <5% (p < 0.05)
was considered to be significant.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures,
seven figures, and two tables and can be found with this article online at
http://dx.doi.org/10.1016/j.celrep.2016.11.044.
AUTHORS CONTRIBUTIONS
S.P., M.G., and C.D.P. designed the study, conducted the experiments, and
analyzed and interpreted data. C.P., S.Z., D.C., I.D.R., and C.M. performed
the experiments and participated to the interpretation of the data. P.L.P
and L.L. performed and analyzed ChIP experiments. M.R., R.V., and M.M.
supported the acquisition and analysis of human samples. E.C. participated
in the design of the study and interpretation of data. C.D.P., C.P., D.C.,
M.T.B., P.L.P., L.L., and E.C. wrote the final version of the manuscript. All au-
thors read and approved the final manuscript.
ACKNOWLEDGMENTS
The authors thank Elena Vezzoli and Maura Francolini, at Fondazione Filarete
(Milano, Italy) for the electron microscopy analysis. We also thank The Italian
Association of Myology, Associazione Amici del Centro Dino Ferrari, University
of Milan, Eurobiobank, and Telethon network of Genetic biobanks (grant num-
ber GTB12001) for providing human biological samples. This work was sup-
ported by ‘‘Ministero della Salute Giovani Ricercatori GR-2011-02350544’’
grant to C.D.P.; ‘‘Ricerca corrente 2016’’ and ‘‘Ministero dell’Istruzione, Uni-
versita e Ricerca PRIN2015’’ grants to E.C.; and ‘‘Epigen Project PB.
P01.001.019/Progetto Bandiera Epigenomica IFT’’ grant to L.L.
Received: October 1, 2015
Revised: June 10, 2016
Accepted: November 11, 2016
Published: December 13, 2016
REFERENCES
Albini, S., Coutinho, P., Malecova, B., Giordani, L., Savchenko, A., Forcales,
S.V., and Puri, P.L. (2013). Epigenetic reprogramming of human embryonic
stem cells into skeletal muscle cells and generation of contractile myospheres.
Cell Rep. 3, 661–670.
Ashmore, T., Roberts, L.D., Morash, A.J., Kotwica, A.O., Finnerty, J., West,
J.A., Murfitt, S.A., Fernandez, B.O., Branco, C., Cowburn, A.S., et al. (2015).
Nitrate enhances skeletal muscle fatty acid oxidation via a nitric oxide-
cGMP-PPAR-mediated mechanism. BMC Biol. 13, 110.
Avalos, J.L., Bever, K.M., and Wolberger, C. (2005). Mechanism of sirtuin inhi-
bition by nicotinamide: altering the NAD(+) cosubstrate specificity of a Sir2
enzyme. Mol. Cell 17, 855–868.
Azuara, V., Perry, P., Sauer, S., Spivakov, M., Jørgensen, H.F., John, R.M.,
Gouti, M., Casanova, M., Warnes, G., Merkenschlager, M., and Fisher, A.G.
(2006). Chromatin signatures of pluripotent cell lines. Nat. Cell Biol. 8, 532–538.
Barger, J.L., Kayo, T., Vann, J.M., Arias, E.B., Wang, J., Hacker, T.A., Wang,
Y., Raederstorff, D., Morrow, J.D., Leeuwenburgh, C., et al. (2008). A low
dose of dietary resveratrol partially mimics caloric restriction and retards aging
parameters in mice. PLoS ONE 3, e2264.
Barton, E.R., Morris, L., Kawana, M., Bish, L.T., and Toursel, T. (2005).
Systemic administration of L-arginine benefits mdx skeletal muscle function.
Muscle Nerve 32, 751–760.
Bernstein, B.E., Mikkelsen, T.S., Xie, X., Kamal, M., Huebert, D.J., Cuff, J., Fry,
B., Meissner, A., Wernig, M., Plath, K., et al. (2006). A bivalent chromatin struc-
ture marks key developmental genes in embryonic stem cells. Cell 125,
315–326.
Brunelli, S., Sciorati, C., D’Antona, G., Innocenzi, A., Covarello, D., Galvez,
B.G., Perrotta, C., Monopoli, A., Sanvito, F., Bottinelli, R., et al. (2007). Nitric
oxide release combined with nonsteroidal antiinflammatory activity prevents
muscular dystrophy pathology and enhances stem cell therapy. Proc. Natl.
Acad. Sci. USA 104, 264–269.
Buono, R., Vantaggiato, C., Pisa, V., Azzoni, E., Bassi, M.T., Brunelli, S., Scior-
ati, C., and Clementi, E. (2012). Nitric oxide sustains long-term skeletal muscle
regeneration by regulating fate of satellite cells via signaling pathways
requiring Vangl2 and cyclic GMP. Stem Cells 30, 197–209.
Cacchiarelli, D., Martone, J., Girardi, E., Cesana, M., Incitti, T., Morlando, M.,
Nicoletti, C., Santini, T., Sthandier, O., Barberi, L., et al. (2010). MicroRNAs
involved in molecular circuitries relevant for the Duchenne muscular dystrophy
pathogenesis are controlled by the dystrophin/nNOS pathway. Cell Metab. 12,
341–351.
Chan, M.C., Rowe, G.C., Raghuram, S., Patten, I.S., Farrell, C., and Arany, Z.
(2014). Post-natal induction of PGC-1a protects against severemuscle dystro-
phy independently of utrophin. Skelet. Muscle 4, 2.
Chen, D., and Guarente, L. (2007). SIR2: a potential target for calorie restriction
mimetics. Trends Mol. Med. 13, 64–71.
Chen, Y.W., Zhao, P., Borup, R., and Hoffman, E.P. (2000). Expression
profiling in the muscular dystrophies: identification of novel aspects of molec-
ular pathophysiology. J. Cell Biol. 151, 1321–1336.
Colussi, C., Mozzetta, C., Gurtner, A., Illi, B., Rosati, J., Straino, S., Ragone, G.,
Pescatori, M., Zaccagnini, G., Antonini, A., et al. (2008). HDAC2 blockade by
nitric oxide and histone deacetylase inhibitors reveals a common target in
Cell Reports 17, 3010–3023, December 13, 2016 3021

ORIGINAL COMMUNICATION
A novel clinical tool to classify facioscapulohumeral musculardystrophy phenotypes
Giulia Ricci1,2• Lucia Ruggiero3
• Liliana Vercelli4 • Francesco Sera5•
Ana Nikolic1• Monica Govi1 • Fabiano Mele1
• Jessica Daolio1• Corrado Angelini6 •
Giovanni Antonini7 • Angela Berardinelli8 • Elisabetta Bucci7 • Michelangelo Cao9•
Maria Chiara D’Amico10• Grazia D’Angelo11
• Antonio Di Muzio10•
Massimiliano Filosto12• Lorenzo Maggi13
• Maurizio Moggio14• Tiziana Mongini4 •
Lucia Morandi13• Elena Pegoraro9
• Carmelo Rodolico15• Lucio Santoro3
•
Gabriele Siciliano2• Giuliano Tomelleri16
• Luisa Villa14• Rossella Tupler1,17
Received: 9 January 2016 / Revised: 6 April 2016 / Accepted: 7 April 2016 / Published online: 28 April 2016
� The Author(s) 2016. This article is published with open access at Springerlink.com
Abstract Based on the 7-year experience of the Italian
Clinical Network for FSHD, we revised the FSHD clinical
form to describe, in a harmonized manner, the phenotypic
spectrum observed in FSHD. The new Comprehensive
Clinical Evaluation Form (CCEF) defines various clinical
categories by the combination of different features. The
inter-rater reproducibility of the CCEF was assessed
between two examiners using kappa statistics by evaluating
56 subjects carrying the molecular marker used for FSHD
diagnosis. The CCEF classifies: (1) subjects presenting
facial and scapular girdle muscle weakness typical of
FSHD (category A, subcategories A1–A3), (2) subjects
with muscle weakness limited to scapular girdle or facial
muscles (category B subcategories B1, B2), (3) asymp-
tomatic/healthy subjects (category C, subcategories C1,
C2), (4) subjects with myopathic phenotype presenting
clinical features not consistent with FSHD canonical phe-
notype (D, subcategories D1, D2). The inter-rater relia-
bility study showed an excellent concordance of the final
four CCEF categories with a j equal to 0.90; 95 % CI
(0.71; 0.97). Absolute agreement was observed for cate-
L. Ruggiero and L. Vercelli contributed equally to this work.
Electronic supplementary material The online version of thisarticle (doi:10.1007/s00415-016-8123-2) contains supplementarymaterial, which is available to authorized users.
& Rossella Tupler
1 Department of Life Sciences, University of Modena and
Reggio Emilia, Modena, Italy
2 Department of Clinical and Experimental
Medicine, Neurological Clinic, University of Pisa,
Pisa, Italy
3 Department of Neurosciences, Reproductive and
Odontostomatological Sciences, University Federico II of
Naples, Naples, Italy
4 Department of Neuroscience, Center for Neuromuscular
Diseases, University of Turin, Turin, Italy
5 MRC Centre of Epidemiology for Child Health, UCL
Institute of Child Health, London, UK
6 IRCCS San Camillo, Venice, Italy
7 Department of Neuroscience, Mental Health and Sensory
Organs, S. Andrea Hospital, University of Rome ‘‘Sapienza’’,
Rome, Italy
8 Unit of Child Neurology and Psychiatry, IRCCS ‘‘C.
Mondino’’ Foundation, Pavia, Italy
9 Department of Neurosciences, University of Padua, Padua,
Italy
10 Center for Neuromuscular Disease, CeSI, University ‘‘G.
D’Annunzio’’, Chieti, Italy
11 Department of Neurorehabilitation, IRCCS Institute Eugenio
Medea, Bosisio Parini, Italy
12 Neurology Clinic, ‘‘Spedali Civili’’ Hospital, Brescia, Italy
13 IRCCS Foundation, C. Besta Neurological Institute, Milan,
Italy
14 Neuromuscular Unit, Fondazione IRCCS Ca’ Granda
Ospedale Maggiore Policlinico, Dino Ferrari Center,
University of Milan, Milan, Italy
15 Department of Clinical and Experimental Medicine,
University of Messina, Messina, Italy
16 Department of Neurological, Neuropsychological,
Morphological and Movement Sciences, University of
Verona, Verona, Italy
17 Department of Molecular Cell and Cancer Biology,
University of Massachusetts Medical School, Worcester,
USA
123
J Neurol (2016) 263:1204–1214
DOI 10.1007/s00415-016-8123-2

Multiple Sclerosis Journal
2016, Vol. 22(8) 1007 –1012
DOI: 10.1177/ 1352458515610646
© The Author(s), 2015. Reprints and permissions: http://www.sagepub.co.uk/ journalsPermissions.nav
MULTIPLESCLEROSIS MSJJOURNAL
http://msj.sagepub.com 1007
IntroductionProgranulin (GRN) is a multifunctional protein with important roles in inflammation and tissue repair, and also a neurotrophic factor critical for neuronal survival.1,2 Progranulin has anti-inflammatory prop-erties, but can be cleaved by extracellular proteases, into peptides named granulins, which exert instead pro-inflammatory properties.1
Mutations in GRN, leading to haploinsufficiency, have been associated with autosomal dominant frontotem-poral lobar degeneration (FTLD).3 The role of GRN variability has been assessed in several neurodegener-ative disorders. A contribution of GRN genetic varia-bility has been previously shown in sporadic FTLD and Alzheimer’s disease.4–6 It has been observed that GRN genetic variability influences age of onset and duration of survival in amyotrophic lateral sclerosis (ALS) patients.7 Certain polymorphisms, such as the rs5848, influence the transcription levels of the gene
or the translation of the mRNA,4,6 thus influencing the protein levels and de-regulating the inflammatory activity of progranulin.
Activated microglia and macrophages have been identified as a major source of progranulin in multiple sclerosis (MS) brains.8 Nevertheless, progranulin can be evaluated also in the periphery (in serum or plasma). In FTLD patients carrying mutations caus-ing haploinsufficiency, serum progranulin levels are undetectable, and no progranulin has been observed in brains at autopsy, leading to the hypothesis that this protein is crucial to prevent neurodegeneration.9
Neurodegeneration plays an important role also in MS;10,11 thus progranulin might be involved in the pathogenesis of this disease. Moreover, the involvement of progranulin in inflammation suggests a potential implication in the pathogenic mechanisms of MS.
Progranulin genetic polymorphisms influence progression of disability and relapse recovery in multiple sclerosis
Marco Vercellino, Chiara Fenoglio, Daniela Galimberti, Alessandra Mattioda, Carlotta Chiavazza, Eleonora Binello, Lorenzo Pinessi, Dario Giobbe, Elio Scarpini and Paola Cavalla
AbstractBackground: Progranulin (GRN) is a multifunctional protein involved in inflammation and repair, and also a neurotrophic factor critical for neuronal survival. Progranulin is strongly expressed in multiple sclerosis (MS) brains by macrophages and microglia.Methods: In this study we evaluated GRN genetic variability in 400 MS patients, in correlation with clinical variables such as disease severity and relapse recovery. We also evaluated serum progranulin levels in the different groups of GRN variants carriers.Results: We found that incomplete recovery after a relapse is correlated with an increased frequency of the rs9897526 A allele (odds ratio (OR) 4.367, p = 0.005). A more severe disease course (Multiple Sclerosis Severity Score > 5) is correlated with an increased frequency of the rs9897526 A allele (OR 1.886, p = 0.002) and of the rs5848 T allele (OR 1.580, p = 0.019). Carriers of the variants associated with a more severe disease course (rs9897526 A, rs5848 T) have significantly lower levels of circulating progranulin (80.5 ± 9.1 ng/mL vs. 165.7 ng/mL, p = 0.01).Conclusion: GRN genetic polymorphisms likely influence disease course and relapse recovery in MS.
Keywords: Multiple sclerosis, progranulin, genetic, neuroprotection, inflammation, relapse, disability
Date received: 25 February 2015; revised: 11 August 2015; accepted: 29 August 2015
Correspondence to: Marco Vercellino Neurologia 3 S.C., Department of Neuroscience, City of Health and Science University Hospital of Turin, Corso Bramante 88, 10126 Turin, Italy. [email protected]
Marco Vercellino Dario Giobbe Neurologia 3 S.C., Department of Neuroscience, City of Health and Science University Hospital of Turin, Italy
Chiara Fenoglio Daniela Galimberti Elio Scarpini Department of Pathophysiology and Transplantation, “Dino Ferrari” Center, University of Milan, Fondazione Cà Granda, IRCCS Ospedale Policlinico, Italy
Alessandra Mattioda Carlotta Chiavazza Eleonora Binello Lorenzo Pinessi Paola Cavalla Multiple Sclerosis Center, Department of Neuroscience, City of Health and Science University Hospital of Turin, Italy
610646MSJ0010.1177/1352458515610646Multiple Sclerosis JournalVercellino et al.research-article2015
Original Article

Journal of Alzheimer’s Disease 51 (2016) 277–291DOI 10.3233/JAD-150849IOS Press
277
Genetic Counseling and Testingfor Alzheimer’s Disease and FrontotemporalLobar Degeneration: An Italian ConsensusProtocol
Martina Bocchettaa,b, Anna Megaa, Livia Bernardic, Emilio Di Mariad, Luisa Benussie,Giuliano Binettif , Barbara Borronig, Rosanna Colaoc, Giuseppe Di Fedeh, Silvia Fostinellie,Daniela Galimbertii, Massimo Gennarellij, Roberta Ghidonie, Irene Piacerik, Michela Pievania,Corinna Porteril, Veronica Redaellih, Giacomina Rossih, Silvia Suardih, Claudio Babilonim,Elio Scarpinii, Fabrizio Tagliavinih, Alessandro Padovanig, Benedetta Nacmiask, Sandro Sorbik,Giovanni B. Frisonia,n, SINdem‡, Amalia C. Brunic,∗
‡SINdem Collaborators:
Marco Bozzalio, Lucilla Parnettip, Carlo Ferrareseq, Stefano F. Cappar, Camillo Marras,Carlo Masullot, Innocenzo Rainerou, Vincenzo Silaniv, Giuseppe Sorrentinow, Giuseppe Brunox,Annachiara Cagniny
aLaboratory of Alzheimer’s Neuroimaging and Epidemiology, IRCCS Istituto Centro San Giovanni di DioFatebenefratelli, Brescia, ItalybDepartment of Molecular and Translational Medicine, University of Brescia, Brescia, ItalycCentro Regionale di Neurogenetica, ASP Catanzaro, Lamezia terme (CZ) ItalydDepartment of Health Sciences, University of Genova and Division of Medical Genetics, Galliera Hospital,Genova, ItalyeMolecular Markers Laboratory, IRCCS Istituto Centro San Giovanni di Dio Fatebenefratelli, Brescia, Italyf IRCCS Istituto Centro San Giovanni di Dio Fatebenefratelli, Brescia, ItalygUniversity of Brescia and Centre for Ageing Brain and Neurodegenerative Disorders, Neurology Unit, Brescia,Brescia, ItalyhIRCCS Fondazione Istituto Neurologico Carlo Besta, Milan, ItalyiUniversity of Milan, Fondazione Ca Granda, IRCCS Ospedale Maggiore Policlinico, Milan, ItalyjGenetic Unit, IRCCS Istituto Centro San Giovanni di Dio Fatebenefratelli, Brescia, ItalykDepartment of Neuroscience, Psychology, Drug Research and Child Health, University of Florence,Florence, ItalylBioethics Unit, IRCCS Istituto Centro San Giovanni di Dio Fatebenefratelli, Brescia, ItalymDepartiment of Physiology and Pharmacology, University of Rome “La Sapienza”, Rome, Italy; IRCCS SanRaffaele Pisana of Rome, Italy
∗Correspondence to: Amalia Cecilia Bruni, MD; CentroRegionale di Neurogenetica, Viale Arturo Perugini, 88046Lamezia Terme, Italy; Tel.: +39 0968 208080; Fax: +39 0968208032; E-mail: [email protected].
ISSN 1387-2877/16/$35.00 © 2016 – IOS Press and the authors. All rights reserved

278 M. Bocchetta et al. / ItalianDIAfN Protocol for Genetic Counseling
nMemory Clinic and LANVIE - Laboratory of Neuroimaging of Aging, University Hospitals and University ofGeneva, Geneva, SwitzerlandoNeuroimaging Laboratory, IRCCS Santa Lucia Foundation, Rome, ItalypSection of Neurology, Department of Medicine, Centre for Memory Disturbances, University of Perugia,Perugia, ItalyqSchool of Medicine and Surgery, Milan Center for Neuroscience (NeuroMI), University of Milano Bicocca,Italy; Neurology Unit, San Gerardo Hospital, Monza, ItalyrNeTS Center-Istituto Universitario di Studi Superiori (IUSS), Pavia, ItalysInstitute of Neurology and Center for Neuropsychological Research of the Policlinico Gemelli, CatholicUniversity of Rome, ItalytDepartment of Neuroscience, Institutes of Neurology, Catholic University of the Sacred Heart, Rome, ItalyuNeurology I - Headache Center, Department of Neuroscience “Rita Levi Montalcini, University of Torino,Torino, ItalyvDepartment of Neurology-Stroke Unit and Laboratory of Neuroscience, IRCCS Istituto Auxologico Italiano,“Dino Ferrari” Centre, Department of Pathophysiology and Transplantation, Universita’ degli Studi di Milano,Milan, ItalywUniversity of Naples Parthenope, Naples, ItalyxMemory Clinic, Department of Neurology and Psychiatry, University of Rome “Sapienza", ItalyyDepartment of Neurosciences (DNS), University of Padova, Padua; IRCCS, San Camillo Hospital Foundation,Venice, Italy
Accepted 1 December 2015
Handling Associate Editor: Raquel Sanchez-Valle
Abstract.Background: Genetic testing of familial Alzheimer’s disease (AD) and frontotemporal lobar degeneration (FTLD) is attract-ing interest thanks to innovative primary prevention clinical trials and increased request for information by at-risk individuals.However, ethical, social, and psychological implications are paramount and genetic testing must be supported by structuredgenetic counseling. In Italy, practice parameters and guidelines for genetic counseling in dementia are not available.Objective: To develop a nationally harmonized protocol for genetic counseling and testing of familial AD and FTLD.Method: Activities were carried out in the context of the Italian Dominantly Inherited Alzheimer’s and FrontotemporalNetwork (IT-DIAfN) project, a national network of centers of excellence with expertise in managing patients with familialAD and FTLD. A survey of the literature on genetic counseling protocols and guidelines was conducted. Local protocolsfor genetic counseling were surveyed. Differences and commonalities among protocols were identified and discussed amongproject partners. Consensus was reached following implicit aggregation methods.Results: Consensus was reached on a protocol for patients with clinically diagnosed familial AD or FTLD and a distinctprotocol for their at-risk relatives. Genetic counseling should be provided by a multidisciplinary team including a geneticist,a neurologist/geriatrician, and a psychologist/psychiatrist, according to the following schedule: (i) initial consultation withtailored information on the genetics of the dementias; (ii) clinical, psychological, and cognitive assessment; if deemedappropriate (iii) genetic testing following a structured decision tree for gene mutation search; (iv) genetic testing resultdisclosure; (v) psychological support follow-up.Conclusions: This genetic counseling protocol provides Italian centers with a line of shared practice for dealing with therequests for genetic testing for familial AD and FTLD from patients and at-risk relatives, who may also be eligible participantsfor novel prevention clinical trials.
Keywords: Alzheimer’s disease, frontotemporal degeneration, genetic counseling, genetic testing
INTRODUCTION
Alzheimer’s disease (AD) and frontotemporallobar degeneration (FTLD) are two of the most com-mon forms of dementia. Although the majority of
cases are sporadic (i.e., without a family history),a markedly familial component has been reportedin 60% of early onset (<65 years) AD patients [1]and in 25–50% of FTLD patients [2, 3]. An autoso-mal dominant mode of inheritance is found in about

© 2016 S. Karger AG, Basel1420–8008/16/0414–0172$39.50/0
Original Research Article
Dement Geriatr Cogn Disord 2016;41:172–180
Body Mass Index Predicts Progression of Mild Cognitive Impairment to Dementia Ilaria Cova a Francesca Clerici a, c Laura Maggiore a Simone Pomati a Valentina Cucumo a Roberta Ghiretti a Daniela Galimberti b Elio Scarpini b Claudio Mariani a Barbara Caracciolo c, d
a Center for Research and Treatment on Cognitive Dysfunctions, Institute of Clinical Neurology, Department of Clinical Sciences, ‘Luigi Sacco’ Hospital, University of Milan, and b Department of Neurological Sciences, ‘Dino Ferrari’ Center, University of Milan, IRCCS Fondazione Ospedale Maggiore Policlinico, Milan , Italy; c Aging Research Center, Department of Neurobiology, Health Care Sciences and Society, Karolinska Institutet/Stockholm University, and d Stockholm Gerontology Research Center, Stockholm , Sweden
Key Words Alzheimer’s disease · Body mass index · Dementia · Mild cognitive impairment · Weight loss
Abstract Aims: To examine the relationship between body mass index (BMI) and progression to de-mentia and Alzheimer’s disease (AD) in mild cognitive impairment (MCI). Materials and Methods: Two hundred and twenty-eight MCI subjects (mean age 74.04 ± 6.94 years; 57% female) from a memory clinic were followed for 2.40 ± 1.58 years. Baseline height and weight were used to calculate the BMI. The main outcome was progression to dementia (DSM-IV cri-teria) and AD (NINCDS-ADRDA criteria). Cox proportional hazard models were used to assess the longitudinal association of BMI with dementia and AD, adjusting for a comprehensive set of covariates, including vascular risk factors/diseases and neuroimaging profiles. Results: Out of 228 subjects with MCI, 117 (51.3%) progressed to dementia. Eighty-nine (76%) of the inci-dent dementia cases had AD. In both unadjusted and multi-adjusted models, a higher BMI was associated with a reduced risk of dementia (multi-adjusted HR 0.9; 95% CI 0.8–0.9) and AD (multi-adjusted HR 0.9; 95% CI 0.8–0.9). Being underweight increased the risk of all types of dementia (multi-adjusted HR 2.5; 95% CI 1.2–5.1) but was not specifically associated with
Received: November 11, 2015 Accepted: January 21, 2016 Published online: March 31, 2016
Ilaria Cova Center for Research and Treatment on Cognitive DysfunctionsInstitute of Clinical Neurology, Department of Biomedical and Clinical Sciences ‘Luigi Sacco’ Hospital, University of Milan, Via G.B. Grassi 74, IT–20157 Milan (Italy) E-Mail ilaria.cova @ unimi.it
www.karger.com/dem
DOI: 10.1159/000444216
Ilaria Cova and Francesca Clerici contributed equally to this work. Francesca Clerici passed away on Febru-ary 21, 2015.
Dow
nloa
ded
by:
Ver
lag
S. K
AR
GE
R A
G, B
AS
EL
172.
16.7
.111
- 6
/27/
2016
2:1
4:03
PM

Clinical short communication
Gene promoter methylation and expression of Pin1 differ betweenpatients with frontotemporal dementia and Alzheimer's disease
Evelyn Ferri a,b,⁎,1, Beatrice Arosio a,c,1, Claudio D'Addario d,e, Daniela Galimberti c,f, Cristina Gussago a,Mariangela Pucci d, Martina Casati a,b, Chiara Fenoglio c,f, Carlo Abbate c, Paolo Dionigi Rossi c, Elio Scarpini c,f,Mauro Maccarrone e,g, Daniela Mari a,c
a Geriatric Unit, Department of Medical Sciences and Community Health, University of Milan, Via Pace 9, 20122 Milan, Italyb PhD in Nutritional Sciences University of Milan, Via Festa del Perdono 7, 20122 Milan, Italyc Fondazione Ca' Granda, IRCCS Ospedale Maggiore Policlinico, Via Francesco Sforza 35, 20122 Milan, Italyd Faculty of Bioscience and Technology for Food, Agriculture and Environment, University of Teramo, Piazza Aldo Moro 45, 64100 Teramo, Italye European Center for Brain Research (CERC)/Santa Lucia Foundation, Via del Fosso di Fiorano 64, 00143 Rome, Italyf Department of Pathophysiology and Transplantation, “Dino Ferrari” Center, University of Milan, Via Francesco Sforza 35, 20122 Milan, Italyg Campus Bio-Medico University of Rome, Via Álvaro del Portillo 21, 00128 Rome, Italy
a b s t r a c ta r t i c l e i n f o
Article history:Received 20 October 2015Received in revised form 7 January 2016Accepted 2 February 2016Available online 03 February 2016
Frontotemporal Dementia (FTD) and Alzheimer's Disease (AD) share the accumulation of fibrillar aggregates ofmisfolded proteins. To better understand these neurodegenerative diseases and identify biomarkers in easily ac-cessible cells, we investigated DNA methylation at Pin1 gene promoter and its expression in peripheral bloodmononuclear cells of FTD patients. We found a lower gene expression of Pin1 with a higher DNA methylationin three CpG sites at Pin1 gene promoter analysed in FTD subjects, in contrast to a higher gene expression witha lower methylation in AD subjects and controls. These data suggest an important and distinct involvement ofPin1 in these two types of dementia.
© 2016 Elsevier B.V. All rights reserved.
Keywords:NeurodegenerationFrontotemporal dementiaAlzheimer's diseasePin1MethylationPeripheral marker
1. Introduction
Alzheimer's Disease (AD) and Frontotemporal Dementia (FTD) aretwo of the most common neurodegenerative disorders and the leadingcauses of neurodegenerative dementia [1–3]. These diseases share theaccumulation of fibrillar aggregates of proteins that results from proteinmisfolding [4].
Much alike the late-onset AD is identified by an accumulation of ex-tracellular amyloid beta (Aβ) plaques derived from amyloid precursorprotein (APP), and by intracellular tangles comprised ofhyperphosphorylated tau protein [5, 6].
The hallmark of FTD is the selective atrophy of the frontal and tem-poral cortex, with neuronal loss and gliosis of the superficial layers [7].The pathologic substrate in FTD is an abnormal tau deposition or aggre-gation in neurons and glia [8].
Pin1 is a ubiquitously expressed protein, belonging to the peptidyl-prolyl isomerase family, and that specifically recognizes phosphorylatedPro-directed Ser/Thr peptide sequences [9–12]. Pin1 regulates the con-formation of proteins after their phosphorylation to further control theirfunction [11–15]. Pin1 plays a crucial role in multiple cellular processesand is implicated in pathogenesis of several diseases, including neuro-degenerative disorders [16–22]. Indeed, Pin1 regulates APPmetabolismand facilitates tau dephosphorylation, thus making a subset of proteinsaccessible to kinases and phosphatases [23]. Therefore a potential neu-roprotective function of Pin1 in neurodegenerative diseases has beensuggested [17, 18, 20, 24, 25].
Our group has recently observed a significant increase in Pin1 geneexpression along with a decreased promoter methylation in peripheralblood mononuclear cells (PBMCs) of patients with late-onset AD, com-pared to controls (CT) [26]. Thus, alterations in easily accessiblePBMCsmay be valuable biomarkers in the early diagnosis of AD, and po-tentially of other tauopathies [27].
In this preliminary study, we focused on gene and protein expres-sions of Pin1 in PBMCs of subjects with FTD, aswell as of a larger sampleof patientswith AD and CT compared to our previous study [26].We de-cided to investigate DNA methylation of Pin1 gene promoter in FTD
Journal of the Neurological Sciences 362 (2016) 283–286
⁎ Corresponding author at: Geriatric Unit, Department of Medical Sciences andCommunity Health, University of Milan, Via Pace 9, 20122 Milan, Italy.
E-mail address: [email protected] (E. Ferri).1 These authors contributed equally to this work.
http://dx.doi.org/10.1016/j.jns.2016.02.0040022-510X/© 2016 Elsevier B.V. All rights reserved.
Contents lists available at ScienceDirect
Journal of the Neurological Sciences
j ourna l homepage: www.e lsev ie r .com/ locate / jns

RESEARCH ARTICLE Open Access
Repetitive element hypermethylationin multiple sclerosis patientsK. Y. Neven1,2, M. Piola3, L. Angelici1, F. Cortini4, C. Fenoglio5, D. Galimberti5, A. C. Pesatori1,4, E. Scarpini5
and V. Bollati1,4,6*
Abstract
Background: Multiple sclerosis (MS) is a complex disorder of the central nervous system whose cause iscurrently unknown. Evidence is increasing that DNA methylation alterations could be involved in inflammatoryand neurodegenerative diseases and could contribute to MS pathogenesis. Repetitive elements Alu, LINE-1 andSAT-α, are widely known as estimators of global DNA methylation. We investigated Alu, LINE-1 and SAT-αmethylation levels to evaluate their difference in a case–control setup and their role as a marker of disability.
Results: We obtained blood samples from 51 MS patients and 137 healthy volunteers matched by gender, ageand smoking. Methylation was assessed using bisulfite-PCR-pyrosequencing. For all participants, medical history,physical and neurological examinations and screening laboratory tests were collected. All repetitive elements werehypermethylated in MS patients compared to healthy controls. A lower Expanded Disability Status Scale (EDSS)score was associated with a lower levels of LINE-1 methylation for ‘EDSS = 1.0’ and ‘1.5 ≤ EDSS ≤ 2.5’ compared toan EDSS higher than 3, while Alu was associated with a higher level of methylation in these groups: ‘EDSS = 1.0’and ‘1.5 ≤ EDSS ≤ 2.5’.
Conclusions: MS patients exhibit an hypermethylation in repetitive elements compared to healthy controls. Aluand LINE-1 were associated with degree of EDSS score. Forthcoming studies focusing on epigenetics and themultifactorial pathogenetic mechanism of MS could elucidate these links further.
Keywords: Multiple sclerosis, Hypermethylation, DNA methylation, Repetitive elements, Epigenetics, Expandeddisability status scale
BackgroundMultiple Sclerosis (MS) is a neurodegenerative diseaseinvolving the central nervous system (CNS) in which theinfiltration of focal lymphocytes in myelin causes inflamma-tory lesions leading to axonal damage [1]. MS knows manydifferent disease courses, of which relapsing-remitting is themost common one. This course is typed with attacks ofworsening neurological functioning (relapsing), followed bypartial or complete improvement of the symptoms (remit-ting) [2]. Women are more prone to develop the diseasethan men, and the age of onset of the patient ranges mostly
between 20 and 40 years, with a transition to the progressiveforms at the age of 40 to 50 [3].The causal factors of MS are still poorly understood, but
they are probably heterogeneous [4]. Recent findings sug-gest that the interplay between individual geneticsusceptibility and external, environmental influences modu-late the disease presentation and therapeutic responsiveness[5, 6]. Evidence is increasing that epigenetic mechanismscould be involved in inflammatory and neurodegenerativediseases and some studies have suggested that changes inthese mechanisms could contribute to MS pathogenesis,representing a bridge between genetics and environmentalcausal factors [7]. Epigenetics are stable and heritable pat-terns that modify the phenotype without altering the geno-type. In particular, DNA methylation has been the mostextensively studied epigenetic marker. It involves adding amethyl group to the 5′ cytosine located in a CpG site to form5 methylcytosine (5mC). A recent study from Huynh et al.
* Correspondence: [email protected] of Clinical Sciences and Community Health, EPIGET –Epidemiology, Epigenetics and Toxicology Lab, Università degli Studi diMilano, Milan, Italy4Department of Preventive Medicine, Fondazione IRCCS Ca’ Granda OspedaleMaggiore Policlinico, Epidemiology Unit, Milan, ItalyFull list of author information is available at the end of the article
© 2016 The Author(s). Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, andreproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link tothe Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver(http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
Neven et al. BMC Genetics (2016) 17:84 DOI 10.1186/s12863-016-0395-0

mechanism for MS progression and axonal loss could bevia oxidative stress, caused by a decrease in antioxidantlevels, which might lead to DNA damage [31]. A possiblemode of action is through DNA methyl-transferase(DNMT), which are recruited to sites of DNA damage andhave previously been suggested as being directly involved inDNA damage repair [32]. These DNMTs have been re-ported as important and essential elements in developmentand are responsible for genomic integrity due to theirmethylating capabilities [32, 33]. As Bollati et al. proposed,a LINE-1 hypermethylation could be the consequence ofthis DNMT upregulation [22]. The effect of DNMT overex-pression was observed in brain tissue of mice, where theyassociated this upregulation with an increase in methylationin motor neuron cells [34].Although we found MS patients to have hypermethy-
lated repetitive elements, no distinct methylation differ-ences were observed in different clinical MS groups (i.e.disease activity, phase of MS, days since relapse, year of on-set, multisystem disorder, spinal cord relapse and the pres-ence of oligoclonal bands in CSF). Our data had a limitedamount of MS patients in different clinical groups, whichmight contribute to non-significant results.
ConclusionAs MS has a multifactorial pathology, hypotheses focus-ing solely on environmental or genetic components aremissing key components. However, epigenetics are ableto bridge the gap between these theories and appears tobe promising. In summary, we found that 1) Alu, LINE-1and SAT-α repetitive elements were hypermethylated inMS patients, 2) Alu, LINE-1 and SAT-α are positively cor-related with each other in healthy controls, while onlyLINE-1 and SAT-α are correlated in MS patients and 3)EDSS values were associated with differential methyla-tion in Alu and LINE-1 elements. We suggest that forth-coming investigations should include a higher number ofMS patients to increase statistical power. Future studiesfocusing on epigenetics and both disease course and clin-ical prognostic markers could further elucidate the un-derstanding of the multifactorial pathology of MS.
Additional files
Additional file 1: Figure S1. CpG specific odds ratios were calculatedbetween MS (n = 51) patients and Healthy controls (n = 137 for LINE-1and SAT-α; n = 135 for Alu). Methylation was assessed in 3 CpG sites forAlu and SAT-α and 4 CpG sites in LINE-1. Estimates are presented as oddsratios, adjusted for age, gender and smoking status, and were calculatedusing the multivariate logistic regression analysis. (DOCX 564 kb)
Additional file 2: Table S1. Characteristics of Multiple Sclerosis (MS)patients and healthy controls for each methylation marker. Methylationis subdivided as ‘mean’ (i.e. average of the separate positions) and theindividual positions of the markers. (DOCX 66 kb)
Additional file 3: Table S2. Not significant data is presented as theestimate (ß) and their respective p-value (p). Disease activity is presentedas ‘annualized relapse rate’ (ARR). The presence and amount of oligoclonalbands was measured in cerebrospinal fluid (CSF). (DOCX 18 kb)
Abbreviations5mC, 5-methylcytosine; CSF, cerebrospinal fluid; DNMT, DNA methyl-transferase;EDSS, expanded disability status scale; LINE-1, long interspersed nuclear elements;MS, multiple sclerosis; OR, odds ratio; PCR, polymerase chain reaction; SAT-a,satellite DNA alpha
AcknowledgmentsThis work was supported by Lombardy Region Research Contracts UniMi31557/2010. Prof. Bollati received support from the EU Programme “Ideas”(ERC-2011-StG 282413).
FundingThe authors received no financial support for the research, authorship, and/or publication of this article.
Availability of data and materialsNot applicable for our dataset. Data will not be shared.
Authors’ contributionsKN and CF carried out the epigenetic studies. FC performed clinical markerassessment. KN, MP and VB drafted the manuscript. MP and DG recruitedstudy participants and collected biological samples. LA performed thestatistical analysis. ACP, ES and VB conceived the study, and participated inits design and coordination. All authors read and approved the finalmanuscript.
Competing interestsThe authors declare that they have no competing interests.
Consent for publicationIndividual data is not published.
Ethics and consent to participateThis research was approved by the institutional review board (FondazioneIRCCS Ca’Granda, Ospedale Maggiore Policlinico, Milan. 2009). Informedconsent was received from all the study participants.
Author details1Department of Clinical Sciences and Community Health, EPIGET –Epidemiology, Epigenetics and Toxicology Lab, Università degli Studi diMilano, Milan, Italy. 2Centre for Environmental Sciences, Hasselt University,Diepenbeek, Belgium. 3Neurology Unit, Saronno ASST Valle Olona Hospital,Saronno, Italy. 4Department of Preventive Medicine, Fondazione IRCCS Ca’Granda Ospedale Maggiore Policlinico, Epidemiology Unit, Milan, Italy.5Department of Pathophysiology and Transplantation, Dino Ferrari Centre,Università degli Studi di Milano and Fondazione IRCCS Ca’ Granda OspedaleMaggiore Policlinico, Milan, Italy. 6Department of Clinical Sciences andCommunity Health, Valentina Bollati, Università degli Studi di Milano, Via SanBarnaba 8, 20122 Milan, Italy.
Received: 21 March 2016 Accepted: 10 June 2016
References1. Compston A, Coles A. Multiple Sclerosis. Lancet. 2008;372(9648):1502–17.2. Weinshenker BG. Natural history of multiple sclerosis. Ann Neurol. 1994;36:6–11.3. Milo R, Kahana E. Multiple Sclerosis: geoepidemiology, genetics and the
environment. Autoimmun Rev. 2010;9(5):387–94.4. Steinman L. Multiple Sclerosis: a two-stage disease. Nat Immunol. 2001;2(9):762–4.5. O’Gorman C, Lucas R, Taylor B. Environmental risk factors for multiple
sclerosis: a review with a focus on molecular mechanisms. Int J Mol Sci.2012;13(9):11718–52.
6. Goldenberg MM. Multiple Sclerosis Review. Pharm Ther. 2012;37(3):175–84.7. Huynh JL, Casaccia P. Epigenetic mechanisms in Multiple Sclerosis: implications
for pathogenesis and treatment. Lancet Neurol. 2013;12(2):195–206.
Neven et al. BMC Genetics (2016) 17:84 Page 6 of 7

Short communication
Effect of fingolimod treatment on circulating miR-15b, miR23a andmiR-223 levels in patients with multiple sclerosis
Chiara Fenoglio ⁎, Milena De Riz, Anna M. Pietroboni, Alberto Calvi, Maria Serpente, Sara M.G. Cioffi,Marina Arcaro, Emanuela Oldoni, Elio Scarpini, Daniela GalimbertiDept. of Pathophysiology and Transplantation, “Dino Ferrari” Center, University of Milan, Fondazione Cà Granda, IRCCS Ospedale Maggiore Policlinico, Milan, Italy
a b s t r a c ta r t i c l e i n f o
Article history:Received 14 June 2016Received in revised form 20 July 2016Accepted 30 August 2016
MicroRNAs (miRNAs) have recently found to be dysregulated in serum from multiple sclerosis (MS) patients.Cell free circulating miR-15b, -23a and 223 levels were analyzed by Real Time PCR in a cohort consisting of 30serum samples from Relapsing Remitting MS patients at baseline (T0) and after three, six, nine and twelvemonths (T1, T2, T3, T4) after starting the treatment. A down-regulation of miRNA levels in patients at T0 com-paredwith controlswas present (p b 0.001). MiRNA levels slightly increased at T1 and this trend reached the sta-tistical significance at T2 vs T0 and remains stable at T3 and T4. Our preliminary results suggest that aberrantlevels of circulating miRNAs are recovered in fingolimod treated MS patients. Circulating miRNAs profilingcould thus represent an easy detectable biomarker of disease and response to treatment.
© 2016 Elsevier B.V. All rights reserved.
Keywords:Multiple sclerosisMicroRNAFingolimodBiomarker
1. Introduction
Fingolimod (marketed as Gilenya®) is the first orally available com-pound recently approved in Europe and USA for the treatment of Re-lapsing Remitting Multiple Sclerosis (RRMS), first in class of S1Preceptors modulators. S1P expressed on lymphocyte regulates the nor-mal egress of lymphocytes from lymphoid tissues, whereas S1P recep-tors expressed in the Central Nervous System (CNS) can modulateneurogenesis, neural function and migration. Fingolimod exerts a dou-ble action: immune modulating in the periphery, and protective andreparatory in the CNS (Chun and Hartung, 2010). Fingolimod has dem-onstrated good efficacy in reducing MS clinical activity compared tofirst-line disease modifying treatments (DMTs) and showed a uniquecentral capacity in reducing brain atrophy progression (Cohen et al.,2010). Furthermore, fingolimod has demonstrated the ability to modifythe expression level of several proteins (Chun and Hartung, 2010).MicroRNAs (miRNAs) are small noncoding RNA molecules of ~22–25nucleotides in length which are synthesized by enzymatic cleavage ofRNAprecursor. They act as negative regulators of gene expression inter-fering with mRNA stability and translation (Abdellatif, 2012).
De-regulation of miRNAs have been associated with autoimmunediseases, including MS, and they have been proposed as potential
biomarkers of disease (Fenoglio et al., 2012, 2013). A growing body ofevidence support a modulation of specific miRNA levels by commondrugs used in MS, highlighting a possible role of miRNAs as biomarkersof treatment response (Fenoglio et al., 2012; Muñoz-Culla et al., 2014;De Felice et al., 2014).
Recently, it was demonstrated that natalizumab modified the ex-pression levels of three specific miRNAs (let-7c, miR-125a-5p andmiR-642) after 6-month treatment, while miR-320, miR320b andmiR-629 showed different expression levels in patients who occurredin PML compared with patients who did not develop PML (Muñoz-Culla et al., 2014).
Considering Interferon treatment, a different expression ofmiR-26a-5p in patients at 3 months treatment was shown, keeping stable at6 months treatment (De Felice et al., 2014).
There is instead no information so far about a possible influenceof fingolimod treatment on miRNA levels. Given this premise, we in-vestigated the effect of fingolimod treatment on the expression ofselected cell free circulating miRNAs, miR-15b, miR-23a and miR-223,previously described to be differentially expressed in MS (Fenoglioet al., 2013).
2. Material and methods
2.1. Patients
This study was approved by the Institutional Review Board (IRB) ofthe Fondazione Cà Granda, IRCCS Ospedale Maggiore Policlinico andall subjects provided written informed consent.
Journal of Neuroimmunology 299 (2016) 81–83
⁎ Corresponding author.E-mail addresses: [email protected] (C. Fenoglio), [email protected] (M. De
Riz), [email protected] (A.M. Pietroboni), [email protected] (A. Calvi),[email protected] (M. Serpente), [email protected] (S.M.G. Cioffi),[email protected] (M. Arcaro), [email protected] (E. Oldoni),[email protected] (E. Scarpini), [email protected] (D. Galimberti).
http://dx.doi.org/10.1016/j.jneuroim.2016.08.0170165-5728/© 2016 Elsevier B.V. All rights reserved.
Contents lists available at ScienceDirect
Journal of Neuroimmunology
j ourna l homepage: www.e lsev ie r .com/ locate / jneuro im

original article© The American Society of Gene & Cell Therapy
Duchenne muscular dystrophy is the most common genetic muscular dystrophy. It is caused by mutations in the dystrophin gene, leading to absence of muscular dystrophin and to progressive degeneration of skeletal muscle. We have demonstrated that the exon skipping method safely and efficiently brings to the expression of a functional dystrophin in dystrophic CD133+ cells injected scid/mdx mice. Golden Retriever muscular dystrophic (GRMD) dogs represent the best preclinical model of Duchenne muscular dystrophy, mimicking the human pathology in genotypic and phenotypic aspects. Here, we assess the capacity of intra-arterial delivered autologous engineered canine CD133+ cells of restoring dystrophin expression in Golden Retriever muscular dys-trophy. This is the first demonstration of five-year follow up study, showing initial clinical amelioration followed by stabilization in mild and severe affected Golden Retriever muscular dystrophy dogs. The occurrence of T-cell response in three Golden Retriever muscular dystrophy dogs, consistent with a memory response boosted by the exon skipped-dystrophin protein, suggests an adap-tive immune response against dystrophin.
Received 2 December 2015; accepted 3 August 2016; advance online publication 4 October 2016. doi:10.1038/mt.2016.163
INTRODUCTIONDuchenne muscular dystrophy (DMD), the most common form of muscular dystrophy, is a lethal X-linked recessive disorder caused by a deficiency of dystrophin protein.1–3 In the early phase of the disease; a chronic regenerative process exhausts the self-renewal potential of DMD stem cells (SCs). This condition leads to muscu-lar fibrosis in which most muscle tissue is lost and replaced by con-nective tissue and, consequently, progressive muscle weakness and atrophy arise.4 DMD patients are confined to wheelchair before the age of 12 years and eventually die from heart and respiratory
failure.1,3 No effective treatment exists although novel therapeutic strategies, ranging from new drugs to gene and cell therapy, hold promises for significant advances.5 In particular, different types of SCs have been shown to partially rescue the pathological pheno-type in dystrophic mice.3,6–10 We have previously demonstrated the stem characteristics of circulating human CD133+ cells and their ability to restore dystrophin expression and eventually regenerate the satellite cell pool in dystrophic scid/mdx mice after intramus-cular and intra-arterial delivery.8,11 We have also isolated CD133+ cells from normal and dystrophic muscular biopsies, showing that the intramuscularly injection of muscle-derived CD133+ cells in DMD patient is a safe and feasible procedure.12 In addition, dys-trophic CD133+ cell population derived from skeletal muscle, transduced with a lentivirus carrying antisense oligonucleotides (AONs) able to skip exon 51, can induce the expression of an exon-skipped version of human dystrophin, and participate to muscle regeneration after in vivo transplantation into scid/mdx mice.11 Although these results might have an important impact for DMD therapeutic approach, in order to proceed to a clini-cal trial it is essential to show efficacy in large animal model of muscular dystrophy, mainly in nonsyngeneic transplants. In this context, the dystrophin-deficient dog, the Golden Retriever mus-cular dystrophy (GRMD) dog, fulfills a great importance, because it mimics more closely the human disease than other existing mammalian models of dystrophin deficiency.13 GRMD is caused by a frameshift mutation in intron 6 of the dystrophin gene.14,15 It is a severe form of dystrophy, which displays dystrophic muscle lesions, inflammatory foci, progressive fibrosis, fatty infiltration, early locomotor impairment, and premature death due to respi-ratory or cardiac failure. A wide interindividual variability also figures among the numerous similarities shared by canine and human diseases, even though the walking complications shown by GRMD dogs starting from 8 months of age is a feature only of the canine pathology. Here, we want to assess the long-term efficacy of combined gene and stem cell therapy, represented by
4October2016
1949
1964
Self Immune Response against Restored Dystrophin
Molecular Therapy
10.1038/mt.2016.163
original article
00nov2016
24
11
2December2015
3August2016
© The American Society of Gene & Cell Therapy
The first two authors contributed equally to this work.Correspondence: Yvan Torrente, Stem Cell Laboratory, Department of Pathophysiology and Transplantation, Università degli Studi di Milano, Unit of Neurology, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Centro Dino Ferrari, Via Sforza 35. 20122, Milano. E-mail: [email protected]
Adaptive Immune Response Impairs the Efficacy of Autologous Transplantation of Engineered Stem Cells in Dystrophic DogsClementina Sitzia1, Andrea Farini1, Luciana Jardim2, Paola Razini1, Marzia Belicchi1,3, Letizia Cassinelli1, Chiara Villa1, Silvia Erratico3, Daniele Parolini1, Pamela Bella1, Joao Carlos da Silva Bizario4, Luis Garcia5, Marcelo Dias-Baruffi2, Mirella Meregalli1,3 and Yvan Torrente1,3
1Stem Cell Laboratory, Department of Pathophysiology and Transplantation, Università degli Studi di Milano, Unit of Neurology, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Centro Dino Ferrari, Milano, Italy; 2Faculdade de Ciências Farmacêuticas de Ribeirão Preto, USP, Ribeirão Preto, SP, Brazil; 3Ystem s.r.l., Milan, Italy; 4Muscular Dystrophy Research Center (AADM/UNAERP), School of Medicine, University of Ribeirão Preto, Ribeirão Preto-SP, Brazil; 5Université Versailles St-Quentin, UMR S 1179 Inserm, Montigny-le-Bretonneux, France
Molecular Therapy vol. 24 no. 11, 1949–1964 nov. 2016 1949

CASE REPORT Open Access
Duchenne muscular dystrophy caused by aframe-shift mutation in the acceptor splicesite of intron 26Mirella Meregalli, Simona Maciotta, Valentina Angeloni and Yvan Torrente*
Abstract
Background: The dystrophin gene is the one of the largest described in human beings and mutations associatedto this gene are responsible for Duchenne or Becker muscular dystrophies.
Case Presentation: Here we describe a nucleotide substitution in the acceptor splice site of intron 26 (c.3604-1G >C) carried by a 6-year-old boy who presented with a history of progressive proximal muscle weakness and elevatedserum creatine kinase levels. RNA analysis showed that the first two nucleotides of the mutated intron 26 (AC) werenot recognized by the splicing machinery and a new splicing site was created within exon 27, generating apremature stop codon and avoiding protein translation.
Conclusions: The evaluation of the pathogenic effect of the mutation by mRNA analysis will be useful in the opticsof an antisense oligonucleotides (AON)-based therapy.
Keywords: DMD, Dystrophin gene, Frame-shift mutation
BackgroundMuscular dystrophies include more than 30 inheriteddisorders causing muscle to weaken and wither away,and Duchenne Muscular Dystrophy (DMD) representsthe most severe form of this family of disorders. It is anX-linked disease, affecting 1 in 5000 male births and iscaused by the absence of the protein dystrophin in skel-etal, cardiac and smooth muscle. Clinical symptomsmanifest at about 3 years of age, and with the progres-sive muscle impairment patients are wheelchair boundby their early teen years and suffer cardiac/respiratoryfailure in their mid- to late twenties [1]. In particular,the incidence of cardiomyopathy in DMD increases withage. Around 25 % of patients have cardiomyopathy at6 years of age and 59 % from 10 years of age, in olderpatients cardiac involvement is ubiquitous, as more than90 % of young men over 18 years of age demonstrateevidence of cardiac dysfunction. No cure is nowadays
available and current treatments include steroid admin-istration and assisted ventilation [2].Dystrophin is a cytoplasmatic protein located at the
membrane of muscle fibres (sarcolemma) representing aconnection between the extracellular matrix and thecytoplasmatic compartment of muscle fibres. For thisreason mechanical stress like contraction during normalexercise impairs dystrophic muscle fibres that degener-ate and, as time goes by, non-contractile fibrotic tissuereplaces muscle tissue [1]. Dystrophin, other than havinga mechanical role, takes part in several signallingpathways spanning from Ca2+ through Nitric Oxide(NO) synthesis and Ras/MAPK pathway, underling evenmore the relevancy of this protein in muscle physiology.In DMD patients absence of dystrophin is caused by
frameshift mutations in the DMD gene that give originto premature stop codons and avoid protein translation.There are also mutations that maintain the open readingframe (ORF) of the DMD gene leading to a milder formof dystrophy called Becker Muscular Dystrophy (BMD).BMD subjects are in fact characterized by low levels offull-length dystrophin or carry in-frame mutations thatallow the generation of shorter but functional forms ofdystrophin. The onset of symptoms in BMD subjects is
* Correspondence: [email protected] of Phatophysiology and Transplantation, Stem Cell Laboratory,Università degli Studi di Milano, Fondazione IRCCS Ca’ Granda OspedaleMaggiore Policlinico di Milano, Centro Dino Ferrari, via F. Sforza 35, 20122Milan, Italy
© 2016 The Author(s). Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, andreproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link tothe Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver(http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
Meregalli et al. BMC Medical Genetics (2016) 17:55 DOI 10.1186/s12881-016-0318-y

ORIGINAL ARTICLE
Exome sequencing identifies variants in two genes encodingthe LIM-proteins NRAP and FHL1 in an Italian patientwith BAG3 myofibrillar myopathy
Francesca D’Avila1,5 • Mirella Meregalli2,4 • Sara Lupoli1,5 • Matteo Barcella1,5 •
Alessandro Orro3 • Francesca De Santis2 • Clementina Sitzia2 • Andrea Farini2 •
Pasqualina D’Ursi3 • Silvia Erratico4 • Riccardo Cristofani6 • Luciano Milanesi3 •
Daniele Braga1,5 • Daniele Cusi1,5 • Angelo Poletti6 • Cristina Barlassina1,5,7 •
Yvan Torrente2,4
Received: 18 May 2016 / Accepted: 9 July 2016
� The Author(s) 2016. This article is published with open access at Springerlink.com
Abstract Myofibrillar myopathies (MFMs) are genetically
heterogeneous dystrophies characterized by the disinte-
gration of Z-disks and myofibrils and are associated with
mutations in genes encoding Z-disk or Z-disk-related pro-
teins. The c.626 C[T (p.P209L) mutation in the BAG3
gene has been described as causative of a subtype of MFM.
We report a sporadic case of a 26-year-old Italian woman,
affected by MFM with axonal neuropathy, cardiomyopa-
thy, rigid spine, who carries the c.626 C[T mutation in
the BAG3 gene. The patient and her non-consanguineous
healthy parents and brother were studied with whole exome
sequencing (WES) to further investigate the genetic basis
of this complex phenotype. In the patient, we found that the
BAG3 mutation is associated with variants in the NRAP
and FHL1 genes that encode muscle-specific, LIM domain
containing proteins. Quantitative real time PCR, immuno-
histochemistry and Western blot analysis of the patient’s
muscular biopsy showed the absence of NRAP expression
and FHL1 accumulation in aggregates in the affected
skeletal muscle tissue. Molecular dynamic analysis of the
mutated FHL1 domain showed a modification in its surface
charge, which could affect its capability to bind its target
proteins. To our knowledge this is the first study reporting,
in a BAG3 MFM, the simultaneous presence of genetic
variants in the BAG3 and FHL1 genes (previously
described as independently associated with MFMs) and
linking the NRAP gene to MFM for the first time.
Keywords Myofibrillar myopathies � Exome sequencing �LIM proteins � BAG3
Introduction
Myofibrillar myopathies (MFMs) are a heterogeneous
group of inherited or sporadic neuromuscular disorders
with clinical and genetic heterogeneity, characterized by
the disintegration of Z disks and myofibrils, followed by
the accumulation of myofibrillar degradation products and
the ectopic accumulation of multiple proteins in the
abnormal fiber regions.
The clinical phenotypes include limb-girdle muscular
dystrophy, distal myopathy, scapuloperoneal syndrome or
rigid spine syndrome. The diagnosis of MFM is based on
clinical findings, electromyography (EMG), nerve con-
duction studies and, most importantly, muscle histology.
D’Avila F. and Meregalli M. are joint first Authors.
& Cristina Barlassina
& Yvan Torrente
1 Department of Health Sciences, Universita degli Studi di
Milano, via A. Rudinı 8, 20100 Milan, Italy
2 Department of Pathophysiology and Transplantation,
Universita degli Studi di Milano, Fondazione IRCCS Ca’
Granda Ospedale Maggiore Policlinico, Centro Dino Ferrari,
via F. Sforza, 35, 20122 Milan, Italy
3 Institute for Biomedical Technologies, CNR, Via Fratelli
Cervi N. 93, Segrate, 20090 Milan, Italy
4 YStem s.r.l., viale Piave 21, 20129 Milan, Italy
5 Filarete Foundation, Viale Ortles, 22, 20100 Milan, Italy
6 Dipartimento di Scienze Farmacologiche e Biomolecolari,
Centro di Eccellenza per lo studio delle malattie
neurodegenerative (CEND), Universita degli studi di Milano,
via Balzaretti, 9, 20100 Milan, Italy
7 Department of Health Sciences, Universita degli Studi di
Milano, c/o Filarete Foundation, Viale Ortles 22/4,
20139 Milan, Italy
123
J Muscle Res Cell Motil
DOI 10.1007/s10974-016-9451-7

RESEARCH ARTICLE
Inositol 1,4,5-trisphosphate (IP3)-dependent Ca2+ signalingmediates delayed myogenesis in Duchenne muscular dystrophyfetal muscleAndrea Farini1, Clementina Sitzia1, Letizia Cassinelli1, Federica Colleoni1, Daniele Parolini1,Umberto Giovanella2, Simona Maciotta1, Augusto Colombo3, Mirella Meregalli1 and Yvan Torrente1,*
ABSTRACTDuchenne muscular dystrophy (DMD) is a progressiveneuromuscular disorder characterized by muscle wasting andpremature death. The defective gene is dystrophin, a structuralprotein, absence of which causes membrane fragility and myofibernecrosis. Several lines of evidence showed that in adult DMD patientsdystrophin is involved in signaling pathways that regulate calciumhomeostasis and differentiation programs. However, secondaryaspects of the disease, such as inflammation and fibrosisdevelopment, might represent a bias in the analysis. Because fetalmuscle is not influenced by gravity and does not suffer frommechanical load and/or inflammation, we investigated 12-week-oldfetal DMD skeletal muscles, highlighting for the first time earlyalterations in signaling pathways mediated by the absence ofdystrophin itself. We found that PLC/IP3/IP3R/Ryr1/Ca2+ signalingis widely active in fetal DMD skeletal muscles and, through thecalcium-dependent PKCα protein, exerts a fundamental regulatoryrole in delaying myogenesis and in myofiber commitment. These dataprovide new insights into the origin of DMD pathology during muscledevelopment.
KEY WORDS: DMD fetus, Dystrophin signaling, Calcium channels,IP3/IP3R pathway, Myosin isoforms
INTRODUCTIONDuchenne muscular dystrophy (DMD) is the most severe form ofmuscular dystrophy. In the early phase of the disease, the skeletalmuscle of DMD patients is characterized by an ongoing process ofdegeneration and regeneration followed by exhaustion of itsregenerative capacity, that leads to infiltration of inflammatorycells, fibrosis and finally to the disruption of the muscle tissuearchitecture (Emery, 2002). Although the dystrophin gene wasdiscovered almost 30 years ago, an effective therapy has not yetbeen found. The membrane fragility, consequent to dystrophinabsence, does not fully explain the divergent expression signature ofboth dystrophic mice (mdx) and DMD patients at different stages ofthe disease versus healthy muscles in terms of muscle wasting,inflammation and fibrosis (Chen and Ende, 2000; Boer et al., 2002;
Porter et al., 2002). Transcriptomic analysis of dystrophic fibersrevealed modification or upregulation of genes involved in calciumhandling, probably related to altered stability of dystrophin-associated proteins (DAPs), in metabolism, extracellular matrixgenes and genes implicated in inflammatory response (Lapidoset al., 2004). It was demonstrated that dystrophin could interact withnitric oxide synthase and syntrophin [regulating other proteinsinvolved in calcium homeostasis (Brenman et al., 1996; Taghli-Lamallem et al., 2014)] and with calmodulin in a calcium-dependent manner (Anderson et al., 1996). These studiessuggested that dystrophin interactions were required for signaltransduction at the membrane but, more importantly, that dystrophinitself modulated different signaling pathways. The inflammation,macrophage infiltration, degenerating and regenerating fibers, andstretch-induced damage are commonly described in DMD assecondary pathology and might interfere with analysis ofdystrophin-related pathways (Boer et al., 2002).
Gharamani et al. partially resolved this issue by silencing thedystrophin gene, through stable RNA interference, in C2C12 cells:they demonstrated a multitude of de-regulated pathways involvingcalcium homeostasis, ion channels, metabolism and musclecontractile unit organization (Ghahramani Seno et al., 2010).However, this in vitro model does not recapitulate the complexarchitecture of mechanically and electrically stimulated muscle.Fetal muscle is not influenced by gravity and does not suffer frommechanical load and/or inflammation; it therefore represents avaluable model in which to elucidate the primary role of dystrophinin regulating not only the structure of membranes but also Ca2+
homeostasis and modulation of signaling/metabolic pathways.Taking into account different studies describing how dystrophinabsence causes an undifferentiated state of myofibers or, at least, analtered or delayed differentiation program (Chen and Ende, 2000),which is evident in mdx embryos (Merrick et al., 2009) and DMDpatients (Pescatori et al., 2007), we also investigated whether itsabsence eventually impaired myogenesis.
Deval et al. suggested that increased calcium release from thesarcoplasmic reticulum (SR) is involved in calcium dysregulation inDMDmyotubes (Deval et al., 2002). The high sensitivity to calciumof dystrophic muscle cells is dependent on two specializedintracellular Ca2+-releasing channels located at the SR membrane:the ryanodine receptor 1 (Ryr1) and inositol 1,4,5-trisphosphate(IP3) receptors (IP3Rs) (Carrasco and Figueroa, 1995; Imbert et al.,1996). Recently, Bellinger et al. identified a structural andfunctional defect in Ryr1 in mdx muscles (Bellinger et al., 2009),and Morel and others suggested that Ryr1 expression could bealtered also in vascular myocytes of mdx mice (Morel et al., 2009).Finally, Andersson et al. hypothesized that leaky Ryr1 channelscould underlie multiple forms of muscular dystrophy linked toReceived 6 May 2015; Accepted 24 December 2015
1Laboratorio di Cellule Staminali, Dipartimento di Fisiopatologia medico-chirurgicae dei Trapianti, Universita degli Studi di Milano, Fondazione IRCCS Ca GrandaOspedale Maggiore Policlinico, Milano, Centro Dino Ferrari, Via Francesco Sforza35, Milan 20122, Centro Dino Ferrari, Italy. 2Consiglio Nazionale delle Ricerche,Istituto per lo Studio delle Macromolecole (CNR-ISMAC), via Bassini 15, Milano20133, Italy. 3Servizio ‘Legge 194’ Dipartimento BDN-Fondazione IRCCS,Policlinico Mangiagalli-Regina Elena, Via Francesco Sforza 35, Milan 20122, Italy.
*Author for correspondence ([email protected])
658
© 2016. Published by The Company of Biologists Ltd | Development (2016) 143, 658-669 doi:10.1242/dev.126193
DEVELO
PM
ENT

RESEARCH ARTICLE
Longitudinal MRI quantification of muscle degeneration inDuchenne muscular dystrophyClaudia Godi1, Alessandro Ambrosi2, Francesca Nicastro3, Stefano C. Previtali4, Corrado Santarosa1,Sara Napolitano5, Antonella Iadanza1, Marina Scarlato4, Maria Grazia Natali Sora4, AndreaTettamanti3, Simonetta Gerevini1, Maria Pia Cicalese5, Clementina Sitzia6, Massimo Venturini7, AndreaFalini1, Roberto Gatti3, Fabio Ciceri8, Giulio Cossu9, Yvan Torrente6 & Letterio S. Politi1,10,11,12
1Neuroradiology Department, Neuroradiology Research Group and CERMAC, San Raffaele Scientific Institute and Vita-Salute San Raffaele
University, Milan, Italy2CUSSB, University Centre for Biomedical Sciences, Vita-Salute San Raffaele University, Milan, Italy3Laboratory of Analysis and Rehabilitation of Motor Function, Division of Neuroscience, San Raffaele Scientific Institute, Milan, Italy4 Division of Neuroscience, Institute of Experimental Neurology (INSpe), San Raffaele Scientific Institute, Milan, Italy5San Raffaele Telethon Institute for Gene Therapy (HSR-TIGET) and Pediatric Immunohematology and Bone Marrow Transplantation Unit, San
Raffaele Scientific Institute, Milan, Italy6Stem Cell Laboratory, Department of Pathophysiology and Transplantation, Universit�a degli Studi di Milano, Fondazione IRCCS C�a Granda
Ospedale Maggiore Policlinico, Centro Dino Ferrari, Milan, Italy7Radiology Department, San Raffaele Scientific Institute and Vita-Salute San Raffaele University, Milan, Italy8Hematology and BMT Unit, San Raffaele Scientific Institute and Vita-Salute San Raffaele University, Milan, Italy9Institute of Inflammation and Repair, University of Manchester, Manchester, United Kingdom10Neuroimaging Research, Division of Hematology/Oncology, Boston Children’s Hospital, Boston, MA, USA11Department of Pediatrics, Harvard Medical School, Boston, MA, USA12University of Massachusetts Memorial Medical Center and University of Massachusetts Medical School, Worcester, MA, USA
Correspondence
Letterio S. Politi, Neuroradiology Department,
Neuroradiology Research Group and
CERMAC, San Raffaele Scientific Institute and
Vita-Salute San Raffaele University, Via
Olgettina 60, 20132 Milan, Italy. Tel: +39
348 3221188; Fax: +39 02 2643 3447;
E-mail: [email protected]
and
Yvan Torrente, Stem Cell Laboratory,
Department of Pathophysiology and
Transplantation, Universit�a degli Studi di
Milano, Fondazione IRCCS C�a Granda
Ospedale Maggiore Policlinico, Centro Dino
Ferrari, via Francesco Sforza 35, 20122
Milan, Italy. Tel: +39 02 55033874; Fax: +39
02 55034723; E-mail: [email protected]
Funding Information
The Associazione Amici Centro Dino Ferrari
and the Italian Ministry of Health (RF-2009-
1547384) supported the publication expenses
of this manuscript.
Received: 11 March 2016; Revised: 22 April
2016; Accepted: 30 April 2016
Annals of Clinical and Translational
Neurology 2016; 3(8): 607–622
doi: 10.1002/acn3.319
Abstract
Objective: The aim of this study was to evaluate the usefulness of magnetic
resonance imaging (MRI) in detecting the progression of Duchenne muscular
dystrophy (DMD) by quantification of fat infiltration (FI) and muscle volume
index (MVI, a residual-to-total muscle volume ratio). Methods: Twenty-six
patients (baseline age: 5–12 years) with genetically proven DMD were longitu-
dinally analyzed with lower limb 3T MRI, force measurements, and functional
tests (Gowers, 10-m time, North Star Ambulatory Assessment, 6-min walking
test). Five age-matched controls were also examined, with a total of 85 MRI
studies. Semiquantitative (scores) and quantitative MRI (qMRI) analyses (signal
intensity ratio – SIR, lower limb MVI, and individual muscle MVI) were carried
out. Permutation and regression analyses according to both age and functional
test-outcomes were calculated. Age-related quantitative reference curves of SIRs
and MVIs were generated. Results: FI was present on glutei and adductor mag-
nus in all patients since the age of 5, with a proximal-to-distal progression and
selective sparing of sartorius and gracilis. Patients’ qMRI measures were signifi-
cantly different from controls’ and among age classes. qMRI were more sensi-
tive than force measurements and functional tests in assessing disease
progression, allowing quantification also after loss of ambulation. Age-related
curves with percentile values were calculated for SIRs and MVIs, to provide a
reference background for future experimental therapy trials. SIRs and MVIs sig-
nificantly correlated with all clinical measures, and could reliably predict func-
tional outcomes and loss of ambulation. Interpretations: qMRI-based indexes
are sensitive measures that can track the progression of DMD and represent a
valuable tool for follow-up and clinical studies.
ª 2016 The Authors. Annals of Clinical and Translational Neurology published by Wiley Periodicals, Inc on behalf of American Neurological Association.
This is an open access article under the terms of the Creative Commons Attribution-NonCommercial-NoDerivs License, which permits use and
distribution in any medium, provided the original work is properly cited, the use is non-commercial and no modifications or adaptations are made.
607

original article© The American Society of Gene & Cell Therapy
Duchenne muscular dystrophy is an inherited fatal genetic disease characterized by mutations in dystrophin gene, causing membrane fragility leading to myofiber necrosis and inflammatory cell recruitment in dystrophic muscles. The resulting environment enriched in proin-flammatory cytokines, like IFN-γ and TNF-α, determines the transformation of myofiber constitutive proteasome into the immunoproteasome, a multisubunit complex involved in the activation of cell-mediate immunity. This event has a fundamental role in producing peptides for antigen presentation by MHC class I, for the immune response and also for cytokine production and T-cell differentiation. Here, we characterized for the first time the presence of T-lymphocytes activated against rever-tant dystrophin epitopes, in the animal model of Duch-enne muscular dystrophy, the mdx mice. Moreover, we specifically blocked i-proteasome subunit LMP7, which was up-regulated in dystrophic skeletal muscles, and we demonstrated the rescue of the dystrophin expression and the amelioration of the dystrophic phenotype. The i-proteasome blocking lowered myofiber MHC class I expression and self-antigen presentation to T cells, thus reducing the specific antidystrophin T cell response, the muscular cell infiltrate, and proinflammatory cytokine production, together with muscle force recovery. We suggest that i-proteasome inhibition should be consid-ered as new promising therapeutic approach for Duch-enne muscular dystrophy pathology.
Received 30 November 2015; accepted 28 July 2016; advance online publication 00 Month 2016. doi:10.1038/mt.2016.162
INTRODUCTIONDuchenne muscular dystrophy (DMD), the most common form of muscular dystrophy, is a genetic disorder caused by mutations in the dystrophin gene.1 Lack of dystrophin causes sarcolemma instability
predisposing to myonecrosis and activation of inflammatory signal-ing cascades. Although inflammation is the pathological hallmark of dystrophic muscular lesion, the mechanisms influencing muscle fiber pathology are still not fully understood.2 The deficiency of dys-trophin causes plasma-membrane instability that leads to a misfold-ing of the multimeric complex of dystrophin glycoprotein complex (DGC) and addresses its components to intracellular proteolysis.3 The ubiquitin-proteasome system is a nonlysosomal, multicatalytic and multisubunit complex involved in the ubiquitin-dependent, selective intracellular degradation of proteins.4 Moreover, protea-some plays a central role in the activation of transcription factors involved in the inflammatory pathways such as NF-κB.5 Many studies demonstrated proteasome participation in muscle fiber degradation that characterized various physiological and patho-logical conditions, therefore suggesting an important role even in DMD.6–8 Interestingly, local intramuscular inhibition of proteasome in murine and canine animal models of DMD leads to dystrophin rescue and DGC proteins expression.7,9–11 The immune cytokine interferon gamma (IFN-γ) can induce the expression of three other catalytically active β-subunits of the 20S proteasome - β1i (PSMB9), β2i (PSMB10), and β5i (PSMB8) to form the induced-proteasome, also referred as immunoproteasome (i-proteasome).6 i-proteasome is involved in the generation of peptides presented by the MHC-I to the antigen receptors of cytotoxic T cells.8 Furthermore, it par-ticipates in several immunological functions such as antigen pre-sentation, cytokine production, T-cell differentiation, and cytotoxic T-cell responses in the periphery. Recently, Foxp3+CD4+ regula-tory T cells (Treg) have been characterized for their capacity to modulate inflammation of acutely and chronically damaged skel-etal muscles.12 Villalta et al. confirmed these evidences showing that Treg depletion from DMD patients and mdx muscles leads to immune cell infiltration and inflammatory response increase, probably due to the inhibition of CD4+-mediated IFN-γ expres-sion.13 Considering the well-known high secretion of inflamma-tory cytokines, i.e., TGF-β and IFN-γ in DMD,13 and the enhanced Treg differentiation and Th1 and Th17 suppression in conditions of
[Q2]
[Q3]
[Q4]
00Month2016
00
00
Immunoproteasome Role in Dystrophic Muscle
Molecular Therapy
10.1038/mt.2016.162
original article
00Month2016
00
00
30November2015
28July2016
© The American Society of Gene & Cell Therapy
The first two authors contributed equally to this work.Correspondence: Yvan Torrente, Dipartimento di Fisiopatologia medico-chirurgica e dei Trapianti, Laboratorio di Cellule Staminali, Università degli Studi di Milano, Fondazione IRCCS Cà Granda Ospedale Maggiore Policlinico, Milano, Centro Dino Ferrari, Via Francesco Sforza 35, 20122 Milan, Italy. E-mail: [email protected]
Therapeutic Potential of Immunoproteasome Inhibition in Duchenne Muscular DystrophyAndrea Farini1, Clementina Sitzia1,2, Barbara Cassani3,4, Letizia Cassinelli1, Rosita Rigoni3,4, Federica Colleoni1, Nicola Fusco5, Stefano Gatti6, Pamela Bella1, Chiara Villa1, Filomena Napolitano7, Rita Maiavacca7, Silvano Bosari6, Anna Villa3,4 and Yvan Torrente1
1Dipartimento di Fisiopatologia medico-chirurgica e dei Trapianti, Laboratorio di Cellule Staminali, Università degli Studi di Milano, Fondazione IRCCS Cà Granda Ospedale Maggiore Policlinico, Milano, Italy; 2Department of Laboratory Medicine, University Milano Bicocca, Desio Hospital, Desio (MB), Italy; 3Milan Unit, Istituto di Ricerca Genetica e Biomedica, Consiglio Nazionale delle Ricerche, Milan, Italy; 4Humanitas Clinical and Research Center, Milan, Italy; 5Department of Pathophysiology and Organ Transplantation, Division of Pathology, Fondazione IRCCS Ca’ Granda, Ospedale Maggiore Policlinico, University of Milan, Milan, Italy; 6Center for Surgical Research, Fondazione IRCCS Ca’ Granda, Ospedale Maggiore Policlinico, Milan, Italy; 7Laboratorio di Chimica Clinica e Microbiologia, Fondazione IRCCS Cà Granda Ospedale Maggiore Policlinico, Milano, Italy
[Q5]
[Q1]
Molecular Therapy 1

38. J. Maier, R. Amin, J. Electrochem. Soc. 155, A339–A344 (2008).39. K. E. Thomas, R. M. Darling, J. Newman, Advances in Lithium-Ion
Batteries (Kluwer Academic/Plenum, 2002).
ACKNOWLEDGMENTS
The x-ray component of this work was supported by the U.S.Department of Energy (DOE), Office of Basic Energy Sciences,Division of Materials Sciences and Engineering (contract DE-AC02-76SF00515). The battery component of this work was supportedby the Ford-Stanford Alliance. The Advanced Light Source issupported by the DOE Office of Basic Energy Sciences undercontract DE-AC02-05CH11231. N.J.S. and D.H.A. acknowledgesupport from the DOE Office of Basic Energy Sciences SBIRprogram under awards DE-SC-0007691 and DE-SC-0009573.
Beam line 5.3.2.1 at the Advanced Light Source was fundedthrough a donation by the King Abdullah University of Science andTechnology. Also supported by a NSF Graduate ResearchFellowship under grant DGE-114747 (Y.L.) and by the GlobalClimate and Energy Project at Stanford University and the DOEOffice of Basic Energy Sciences through the SUNCAT Center forInterface Science and Catalysis (M.Z.B.). N.J.S. and D.H.A. areemployed by Hummingbird Scientific, which designed andmanufactured the microfluidic liquid cell used in theseexperiments. Part of this work was conducted the Stanford NanoShared Facilities and the Stanford Nanofabrication Facility. Wethank J. Nelson Weker, A. Wise, H. W. Shiu, M. Farmand,D. Kilcoyne, S. Fakra, Y. S. Hsieh, and A. Kammers forinsightful discussions and assistance with the experiment.
The raw data for this experiment are available as part of thesupplementary materials.
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/353/6299/566/suppl/DC1Materials and MethodsSupplementary TextFigs. S1 to S19Movies S1 and S2References (40–49)Archive of Image Data
24 November 2015; accepted 8 July 201610.1126/science.aaf4914
NANOMATERIALS
Permanent excimer superstructuresby supramolecular networking ofmetal quantum clustersBeatriz Santiago-Gonzalez,1* Angelo Monguzzi,1*† Jon Mikel Azpiroz,2,3 Mirko Prato,4
Silvia Erratico,5 Marcello Campione,6 Roberto Lorenzi,1 Jacopo Pedrini,1
Carlo Santambrogio,7 Yvan Torrente,5 Filippo De Angelis,2,4
Francesco Meinardi,1† Sergio Brovelli1†
Excimers are evanescent quasi-particles that typically form during collisionalintermolecular interactions and exist exclusively for their excited-state lifetime. Weexploited the distinctive structure of metal quantum clusters to fabricate permanentexcimer-like colloidal superstructures made of ground-state noninteracting gold cores,held together by a network of hydrogen bonds between their capping ligands. Thispreviously unknown aggregation state of matter, studied through spectroscopicexperiments and ab initio calculations, conveys the photophysics of excimers into stablenanoparticles, which overcome the intrinsic limitation of excimers in single-particleapplications—that is, their nearly zero formation probability in ultra-diluted solutions. Invitro experiments demonstrate the suitability of the superstructures as nonresonantintracellular probes and further reveal their ability to scavenge reactive oxygen species,which enhances their potential as anticytotoxic agents for biomedical applications.
Metal quantum clusters are functionalnanoscale materials with potential foruse in sensing (1), bio imaging (2), opto-electronics (3), and nanomedicine (4).With “magic” sizes dictated by the va-
lence of the metal constituents (5) and dimen-sions approaching the Fermi wavelength of theelectron, these few-atom structures bridge thegap between atoms and colloidal nanoparticles.As a result, metal quantum clusters combine amolecule-like electronic structure with quantumconfinement effects that confer them with size-and shape-tunable optical properties, ultralargesurface-to-volume ratios, and unmatched flexibil-ity for tailoring their physical properties throughsurface functionalization. Specifically, ligand-to-metal electron transfer (LM-ET) in metal quan-tum clusters with electron-rich capping agents(6–8) leads to strongly Stokes-shifted emission,which is beneficial for photon management (9)and bio imaging (1) applications. On the otherhand, suppression of LM-ET results in intrinsicluminescence, with energy determined by thequantummechanical combination of single-atom
electronic orbitals (10, 11). Fundamentally, thekey common feature of “intrinsic emitting”metalquantum clusters is their capping with bulkymolecules (12–15) or their encapsulation in supra-molecular vesicles (14, 16, 17), imposing largedistances between the metal cores. The use ofshort ligands, even in the absence of LM-ET,has led instead to a variety of optical behaviors(18–21). This points to a role of intercluster in-teractions in the photophysics of metal quantumclusters and suggests a supramolecular strategyfor tuning their optical properties through con-trolled formation of aggregate species, similarlyto what is achieved with organic chromophores.In molecular physics, aggregate species are
typically divided into two main categories basedon the type of interaction that leads to their for-mation: Molecular dimers arise from ground-state interactions between individual moieties(monomers) (22), whereas excimers are evanes-cent quasi-particles existing exclusively in theexcited state and formed through the aggrega-tion of an excited monomer and a ground-statemonomer. When excimers return to the ground
state, their constituent monomers dissociate (22).As a result, dimers are capable of ground-stateabsorption, whereas excimers exhibit the absorp-tion spectrum of the monomers and long-livedStokes-shifted emission from lower-lying inter-molecular states. Molecular excimers are typi-cally formed through collisional interactionsbetween monomers in concentrated solutions,and their formation probability drops to zeroupon dilution (23). The excimer motif is, there-fore, intrinsically prevented from being used forthe fabrication of stable, self-standing emittersfor single-particle applications.In this work, we overcame this limitation by
demonstrating a previously unknown aggrega-tion state of matter that conveys the photophysicsof excimers into individual particles that can findapplication as nonresonant emitters in cellularimaging and integrated photonic nanotechno-logies. Specifically, we used Aun (gold quantumclusters consisting of n atoms) as building blocksfor fabricating permanent excimer-like colloidalsuperstructures (Au-pXs) held together by a net-work of hydrogen bonds between their cappingligands. As a result of repulsive forces betweenthe metal cores, in the ground state, the net-worked Aun behave as independent chromo-phores, whereas on photoexcitation, they formbimolecular excimers with largely Stokes-shiftedemission (~1 eV). In contrast, encapsulation ofAun in bulky vesicles hinders the excimeric inter-action, resulting in the photophysics of isolatedclusters. Last, we used Au-pXs for in vitro imaging
SCIENCE sciencemag.org 5 AUGUST 2016 • VOL 353 ISSUE 6299 571
1Dipartimento di Scienza dei Materiali, Università degli StudiMilano-Bicocca, Via R. Cozzi 55, 20125 Milano, Italy.2Computational Laboratory for Hybrid and OrganicPhotovoltaics, National Research Council–Institute ofMolecular Science and Technologies (CNR-ISTM), Via Elce diSotto 8, 06123 Perugia, Italy. 3Kimika Fakultatea, EuskalHerriko Unibertsitatea (UPV/EHU), and DonostiaInternational Physics Center, 20080 Donostia, Euskadi,Spain. 4Istituto Italiano di Tecnologia, Via Morego 30, 16163Genova, Italy. 5Dipartimento di Fisiopatologia Medico-Chirurgica e dei Trapianti, Università degli Studi di Milano,Fondazione IRCCS (Istituto di Ricovero e Cura a CarattereScientifico) Cá Granda Ospedale Maggiore Policlinico, CentroDino Ferrari, Via Francesco Sforza 35, 20122 Milano, Italy.6Dipartimento di Scienze dell'Ambiente e del Territorio e diScienze della Terra, Università degli Studi Milano-Bicocca,Piazza della Scienza, 20125 Milano, Italy. 7Dipartimento diBiotecnologie e Bioscienze, Università degli Studi Milano-Bicocca, Piazza della Scienza, 2 20126 Milano, Italy.*These authors contributed equally to this work. †Correspondingauthor. Email: [email protected] (A.M.);[email protected] (F.M.); [email protected] (S.B.)
RESEARCH | REPORTS
on
Aug
ust 4
, 201
6ht
tp://
scie
nce.
scie
ncem
ag.o
rg/
Dow
nloa
ded
from

Tissue-Specific Progenitor and Stem Cells
Impaired Angiogenic Potential of Human PlacentalMesenchymal Stromal Cells in IntrauterineGrowth Restriction
CHIARA MANDO,a,* PAOLA RAZINI,b,* CHIARA NOVIELLI,a GAIA MARIA ANELLI,a MARZIA BELICCHI,b,c
SILVIA ERRATICO,c STEFANIA BANFI,b MIRELLAMEREGALLI,b,c ALESSANDRO TAVELLI,b MARCO BACCARIN,d
ALESSANDRO ROLFO,e SILVIA MOTTA,d YVAN TORRENTE,b,c,f,† IRENE CETINa,g,†
Key Words. Pregnancy x Stem cells x Placenta x Intrauterine growth restriction xMesenchymal stromal cells
ABSTRACT
Human placental mesenchymal stromal cells (pMSCs) have never been investigated in intrauterinegrowth restriction (IUGR). We characterized cells isolated from placental membranes and the basaldisc of six IUGR and five physiological placentas. Cell viability and proliferationwere assessed every 7days during a 6-week culture. Expression of hematopoietic, stem, endothelial, and mesenchymalmarkers was evaluated by flow cytometry. We characterized the multipotency of pMSCs and the ex-pressionof genes involved inmitochondrial content and function. Cell viabilitywashigh in all samples,and proliferation ratewas lower in IUGR comparedwith control cells. All samples presented a startingheterogeneous population, shifting during culture toward homogeneity for mesenchymal markersand occurring earlier in IUGR than in controls. In vitro multipotency of IUGR-derived pMSCs was re-stricted because their capacity for adipocyte differentiation was increased, whereas their ability todifferentiate towardendothelial cell lineagewasdecreased.Mitochondrial content and functionwerehigher in IUGRpMSCs than controls, possibly indicating a shift fromanaerobic to aerobicmetabolism,with the loss of the metabolic characteristics that are typical of undifferentiated multipotent cells.STEM CELLS TRANSLATIONAL MEDICINE 2016;5:451–463
SIGNIFICANCE
This study demonstrates that the loss of endothelial differentiation potential and the increase ofadipogenic ability are likely to play a significant role in the vicious cycle of abnormal placental de-velopment in intrauterine growth restriction (IUGR). This is the first observation of a potential rolefor placentalmesenchymal stromal cells in intrauterine growth restriction, thus leading to newper-spectives for the treatment of IUGR.
INTRODUCTION
Mesenchymal cells are the predominant cellularcomponent in placenta and serve as structuralsupport for trophoblast villi and vascular network[1]. In recent investigations, human placentalmesenchymal stromal cells (pMSCs) have beenisolated according to the meeting criteria pro-posed by the Mesenchymal and Tissue Stem CellCommittee [2] and the International Society forCellular Therapy as cells that adhere to untreatedplastic surfaces and express CD105, CD73, andCD90 but not CD34, CD14, CD19, CD11b,CD79a, or HLA-DR and that differentiate into os-teogenic, adipogenic, and chondrogenic lineagesin vitro [3, 4]. Both fetal (from placental mem-branes and villi) and maternal (from decidua)pMSCs can be isolated from the human placenta,
showing similar phenotypes [5–8]. Indeed, theplacental maternal side comprises the chorionicvilli on one side and maternal decidua on theother. The peripheral region of the placenta onthe maternal side that is in contact with the uter-inewall (called thebasal plate) comprises the cho-rionic villi on one side and maternal deciduabasalis on the other. The significant issue of thepresence of maternal cells in human pMSCs cul-tures was reviewed recently [9].
Despite progress in the characterization ofpMSCs, it remains largely unknown to what de-gree the pMSCs are involved in placenta disease.Investigation of pMSCsmay therefore give impor-tant hints for pregnancy pathologies linked to pla-cental insufficiency, namely intrauterine growthrestriction (IUGR) and preeclampsia (PE) [10].IUGR and PE are leading causes of maternal and
a“L. Sacco” Department ofBiomedical and ClinicalSciences, Center for FetalResearch Giorgio Pardi,Universita degli Studi diMilano, Milan, Italy; bStemCell Laboratory, Departmentof Pathophysiology andTransplantation, FondazioneIRCCS Ca’ Granda OspedaleMaggiore Policlinico, CentroDino Ferrari, Universita degliStudi di Milano, Milan, Italy;cYstem S.R.L., Milan, Italy;dLaboratory of MedicalGenetics, Fondazione IRCCSCa’ Granda OspedaleMaggiore Policlinico, Milan,Italy; eDepartment of SurgicalScience, University of Turin,Turin, Italy, fUNISTEMInterdepartmental Centre forStem Cell Research, Milan,Italy; gDepartment of Motherand Child, Luigi SaccoHospital, Milan, Italy
*Contributed equally.
†Contributed equally.
Correspondence: Irene Cetin,M.D.,Ph.D.,DepartmentofMotherand Child, “L. Sacco” Departmentof Biomedical and ClinicalSciences, School of Medicine,Universita degli Studi di Milano,Luigi Sacco Hospital, via GiovanniBattista Grassi 74, 20157 Milan,Italy. Telephone: 39 0250319804-05; E-Mail: [email protected];or Yvan Torrente, M.D., Ph.D.,Department of Pathophysiologyand Transplantation, Universitadegli Studi di Milano, FondazioneIRCCS Ca’ Granda OspedaleMaggiore Policlinico, viaFrancesco Sforza 35, 20122Milan, Italy. Telephone: 39-0255033874; E-Mail: [email protected]
Received July 15, 2015; acceptedfor publication December 21,2015; published Online First onMarch 8, 2016.
©AlphaMed Press1066-5099/2016/$20.00/0
http://dx.doi.org/10.5966/sctm.2015-0155
STEM CELLS TRANSLATIONAL MEDICINE 2016;5:451–463 www.StemCellsTM.com ©AlphaMed Press 2016
TISSUE-SPECIFIC PROGENITOR AND STEM CELLS

REVIEW
You stole my food! Eating alterations in frontotemporal dementiaMarilena Aielloa, Vincenzo Silanib,c and Raffaella I Rumiatia
aArea of Neuroscience, SISSA, Trieste, Italy; bDepartment of Neurology and Laboratory of Neuroscience, IRCCS Istituto Auxologico Italiano, DinoFerrari Centre, Milan, Italy; cDepartment of Pathophysiology and Transplantation, Università degli Studi di Milano, Milan, Italy
ABSTRACTPatients with different types of dementia may exhibit pathological eating habits, including food fads,hyperphagia, or even ingestion of inanimate objects. Several findings reveal that such eating alterationsare more common in patients with frontotemporal dementia (FTD) than other types of dementia.Moreover, eating alterations may differ between the two variants of the disease, namely the behavioralvariant and semantic dementia (SD). In this review, we summarized evidences regarding four areas:eating and body weight alterations in FTD, the most common assessment methods, anatomicalcorrelates of eating disorders, and finally, proposed underlying mechanisms. An increasing understand-ing of the factors that contribute to eating abnormalities may allow first, a better comprehension of theclinical features of the disease and second, shed light on the mechanism underlying eating behaviors inthe normal population.
ARTICLE HISTORYReceived 8 July 2015Accepted 31 May 2016
KEYWORDSFrontotemporal dementia;eating disorders; bodyweight; hyperorality
Introduction
(He) eats sugar by the spoonful . . . (He) eats four desserts after ameal . . . (She) cooks chicken with mushrooms every day . . . (She)eats raisins soaked in gin 9 times a day . . . (He) eats quickly . . . (She)puts everything in her mouth . . . (He) eats food from others plateswhen he is done with his own . . . (He) shoves food in his mouth.
Quotes from caregivers of frontotemporal dementia patients,adapted from Kertesz (The Banana Lady and Other Stories ofCurious Behavior and Speech, 2006)
Among the many functions that can be affected by dementia,one should also consider eating. The type of impairment can varyfrom one patient to another: One patient may simply forget toeat, another may have trouble using the cutlery when eating,while a third may fail to distinguish food flavors. Furthermore,sometimes dementia can lead to dramatic behavioral changes,as in the cases described by Kertnez in The Banana Lady andOther Stories of Curious Behaviour and Speech (Kertesz, 2006).The “Banana Lady” is a woman who started drinking liters of milkand eating a great quantity of bananas. Moreover, she developedcravings for chocolate and ate sugar by the spoonful. Every dayshe called her husband multiple times just to make sure hebrought home bananas and milk. She stuffed the food into hermouth, pocketing it in her cheeks, without swallowing inbetween bites. In addition, these eating abnormalities wereassociated with profound personality changes. Several yearslater, a postmortem examination confirmed the diagnosis offrontotemporal dementia (FTD). Another patient, Dick, was anobese man who, together with a personality change, developeda series of abnormal eating behaviors: He ate everything in sight,even food left on the table by others; he regularly spent $20 onsnacks and ate them in one sitting, and even hid food under thesofa. The postmortem examination revealed that this patient,too, suffered from a frontotemporal degeneration.
There is good evidence now suggesting that eatingabnormalities are commonly experienced by patients affectedby FTD, more than by patients with other types of dementiasuch as Alzheimer Disease (AD) or vascular dementia (VD)(Ahmed, Irish, et al., 2014; Bathgate, Snowden, Varma,Blackshaw, & Neary, 2001; Bozeat, Gregory, Ralph, & Hodges,2000; Cummings, 1997; Ikeda, Brown, Holland, Fukuhara, &Hodges, 2002; Jenner, Reali, Puopolo, & Silveri, 2006; Liscic,Storandt, Cairns, & Morris, 2007; Mathias & Morphett, 2010;Mendez, Licht, & Shapira, 2008; Miller, Darby, Swartz, Yener, &Mena, 1995; Nagahama, Okina, Suzuki, & Matsuda, 2006).
FTD is a neurodegenerative disease characterized by aprogressive deterioration of the frontal and temporal lobesand comprises three variants: a behavioral variant (bvFTD),semantic dementia (SD), and progressive non-fluent aphasia(PNFA) (Neary et al., 1998; Rascovsky et al., 2011). The bvFTD ischaracterized by progressive behavioral abnormalities, strikingpersonality change, breakdown in social conduct, and execu-tive dysfunction (Gustafson, 1987; Neary et al., 1998), com-monly associated with gray matter atrophy of frontal, insular,and temporal cortices. SD is a disorder primarily of the mean-ing that affects patients’ naming and word comprehension ofseveral types of stimuli (e.g., Snowden, Neary, & Mann, 1996;Warrington, 1975), associated with predominantly left anteriortemporal lobe atrophy involving both gray and white matter(Chao et al., 2007). Finally, PNFA is characterized by hesitant,effortful speech and agrammatism and associated with peri-sylvian atrophy (Gorno-Tempini, Murray, Rankin, Weiner, &Miller, 2004).
The present review is organized as follows. First, we willconsider the literature on bvFTD and SD patients in order toreview the available evidence on eating and body weightalterations in these populations. In doing so, we will describethe diagnostic tools that have been used for evaluating the
CONTACT Marilena Aiello [email protected]
NEUROCASE, 2016http://dx.doi.org/10.1080/13554794.2016.1197952
© 2016 Informa UK Limited, trading as Taylor & Francis Group
Dow
nloa
ded
by [
Mar
ilena
Aie
llo]
at 0
0:03
22
June
201
6

Journal of Alzheimer’s Disease 51 (2016) 277–291DOI 10.3233/JAD-150849IOS Press
277
Genetic Counseling and Testingfor Alzheimer’s Disease and FrontotemporalLobar Degeneration: An Italian ConsensusProtocol
Martina Bocchettaa,b, Anna Megaa, Livia Bernardic, Emilio Di Mariad, Luisa Benussie,Giuliano Binettif , Barbara Borronig, Rosanna Colaoc, Giuseppe Di Fedeh, Silvia Fostinellie,Daniela Galimbertii, Massimo Gennarellij, Roberta Ghidonie, Irene Piacerik, Michela Pievania,Corinna Porteril, Veronica Redaellih, Giacomina Rossih, Silvia Suardih, Claudio Babilonim,Elio Scarpinii, Fabrizio Tagliavinih, Alessandro Padovanig, Benedetta Nacmiask, Sandro Sorbik,Giovanni B. Frisonia,n, SINdem‡, Amalia C. Brunic,∗
‡SINdem Collaborators:
Marco Bozzalio, Lucilla Parnettip, Carlo Ferrareseq, Stefano F. Cappar, Camillo Marras,Carlo Masullot, Innocenzo Rainerou, Vincenzo Silaniv, Giuseppe Sorrentinow, Giuseppe Brunox,Annachiara Cagniny
aLaboratory of Alzheimer’s Neuroimaging and Epidemiology, IRCCS Istituto Centro San Giovanni di DioFatebenefratelli, Brescia, ItalybDepartment of Molecular and Translational Medicine, University of Brescia, Brescia, ItalycCentro Regionale di Neurogenetica, ASP Catanzaro, Lamezia terme (CZ) ItalydDepartment of Health Sciences, University of Genova and Division of Medical Genetics, Galliera Hospital,Genova, ItalyeMolecular Markers Laboratory, IRCCS Istituto Centro San Giovanni di Dio Fatebenefratelli, Brescia, Italyf IRCCS Istituto Centro San Giovanni di Dio Fatebenefratelli, Brescia, ItalygUniversity of Brescia and Centre for Ageing Brain and Neurodegenerative Disorders, Neurology Unit, Brescia,Brescia, ItalyhIRCCS Fondazione Istituto Neurologico Carlo Besta, Milan, ItalyiUniversity of Milan, Fondazione Ca Granda, IRCCS Ospedale Maggiore Policlinico, Milan, ItalyjGenetic Unit, IRCCS Istituto Centro San Giovanni di Dio Fatebenefratelli, Brescia, ItalykDepartment of Neuroscience, Psychology, Drug Research and Child Health, University of Florence,Florence, ItalylBioethics Unit, IRCCS Istituto Centro San Giovanni di Dio Fatebenefratelli, Brescia, ItalymDepartiment of Physiology and Pharmacology, University of Rome “La Sapienza”, Rome, Italy; IRCCS SanRaffaele Pisana of Rome, Italy
∗Correspondence to: Amalia Cecilia Bruni, MD; CentroRegionale di Neurogenetica, Viale Arturo Perugini, 88046Lamezia Terme, Italy; Tel.: +39 0968 208080; Fax: +39 0968208032; E-mail: [email protected].
ISSN 1387-2877/16/$35.00 © 2016 – IOS Press and the authors. All rights reserved

278 M. Bocchetta et al. / ItalianDIAfN Protocol for Genetic Counseling
nMemory Clinic and LANVIE - Laboratory of Neuroimaging of Aging, University Hospitals and University ofGeneva, Geneva, SwitzerlandoNeuroimaging Laboratory, IRCCS Santa Lucia Foundation, Rome, ItalypSection of Neurology, Department of Medicine, Centre for Memory Disturbances, University of Perugia,Perugia, ItalyqSchool of Medicine and Surgery, Milan Center for Neuroscience (NeuroMI), University of Milano Bicocca,Italy; Neurology Unit, San Gerardo Hospital, Monza, ItalyrNeTS Center-Istituto Universitario di Studi Superiori (IUSS), Pavia, ItalysInstitute of Neurology and Center for Neuropsychological Research of the Policlinico Gemelli, CatholicUniversity of Rome, ItalytDepartment of Neuroscience, Institutes of Neurology, Catholic University of the Sacred Heart, Rome, ItalyuNeurology I - Headache Center, Department of Neuroscience “Rita Levi Montalcini, University of Torino,Torino, ItalyvDepartment of Neurology-Stroke Unit and Laboratory of Neuroscience, IRCCS Istituto Auxologico Italiano,“Dino Ferrari” Centre, Department of Pathophysiology and Transplantation, Universita’ degli Studi di Milano,Milan, ItalywUniversity of Naples Parthenope, Naples, ItalyxMemory Clinic, Department of Neurology and Psychiatry, University of Rome “Sapienza", ItalyyDepartment of Neurosciences (DNS), University of Padova, Padua; IRCCS, San Camillo Hospital Foundation,Venice, Italy
Accepted 1 December 2015
Handling Associate Editor: Raquel Sanchez-Valle
Abstract.Background: Genetic testing of familial Alzheimer’s disease (AD) and frontotemporal lobar degeneration (FTLD) is attract-ing interest thanks to innovative primary prevention clinical trials and increased request for information by at-risk individuals.However, ethical, social, and psychological implications are paramount and genetic testing must be supported by structuredgenetic counseling. In Italy, practice parameters and guidelines for genetic counseling in dementia are not available.Objective: To develop a nationally harmonized protocol for genetic counseling and testing of familial AD and FTLD.Method: Activities were carried out in the context of the Italian Dominantly Inherited Alzheimer’s and FrontotemporalNetwork (IT-DIAfN) project, a national network of centers of excellence with expertise in managing patients with familialAD and FTLD. A survey of the literature on genetic counseling protocols and guidelines was conducted. Local protocolsfor genetic counseling were surveyed. Differences and commonalities among protocols were identified and discussed amongproject partners. Consensus was reached following implicit aggregation methods.Results: Consensus was reached on a protocol for patients with clinically diagnosed familial AD or FTLD and a distinctprotocol for their at-risk relatives. Genetic counseling should be provided by a multidisciplinary team including a geneticist,a neurologist/geriatrician, and a psychologist/psychiatrist, according to the following schedule: (i) initial consultation withtailored information on the genetics of the dementias; (ii) clinical, psychological, and cognitive assessment; if deemedappropriate (iii) genetic testing following a structured decision tree for gene mutation search; (iv) genetic testing resultdisclosure; (v) psychological support follow-up.Conclusions: This genetic counseling protocol provides Italian centers with a line of shared practice for dealing with therequests for genetic testing for familial AD and FTLD from patients and at-risk relatives, who may also be eligible participantsfor novel prevention clinical trials.
Keywords: Alzheimer’s disease, frontotemporal degeneration, genetic counseling, genetic testing
INTRODUCTION
Alzheimer’s disease (AD) and frontotemporallobar degeneration (FTLD) are two of the most com-mon forms of dementia. Although the majority of
cases are sporadic (i.e., without a family history),a markedly familial component has been reportedin 60% of early onset (<65 years) AD patients [1]and in 25–50% of FTLD patients [2, 3]. An autoso-mal dominant mode of inheritance is found in about

ORIGINAL ARTICLE
The Italian dementia with Lewy bodies study group(DLB-SINdem): toward a standardization of clinicalprocedures and multicenter cohort studies design
L. Bonanni1 • A. Cagnin2,14 • F. Agosta3 • C. Babiloni4,15 • B. Borroni5 •
M. Bozzali6 • A. C. Bruni7 • M. Filippi3 • D. Galimberti8 • R. Monastero9 •
C. Muscio10 • L. Parnetti11 • D. Perani12 • L. Serra6 • V. Silani13,16 •
P. Tiraboschi10 • A. Padovani5 • On behalf of DLB-SINdem study group
Received: 15 July 2016 / Accepted: 8 September 2016
� Springer-Verlag Italia 2016
Abstract Dementia with Lewy bodies (DLB) causes ele-
vated outlays for the National Health Systems due to high
institutionalization rate and patients’ reduced quality of life
and high mortality. Furthermore, DLB is often misdiag-
nosed as Alzheimer’s disease. These data motivate har-
monized multicenter longitudinal cohort studies to improve
clinical management and therapy monitoring. The Italian
DLB study group of the Italian Neurological Society for
dementia (SINdem) developed and emailed a semi-struc-
tured questionnaire to 572 national dementia centers (from
primary to tertiary) to prepare an Italian large longitudinal
cohort. The questionnaire surveyed: (1) prevalence and
incidence of DLB; (2) clinical assessment; (3) relevance
and availability of diagnostic tools; (4) pharmacological
management of cognitive, motor, and behavioural distur-
bances; (5) causes of hospitalization, with specific focus on
delirium and its treatment. Overall, 135 centers (23.6 %)
contributed to the survey. Overall, 5624 patients with DLB
are currently followed by the 135 centers in a year (2042 of
them are new patients). The percentage of DLB patients
was lower (27 ± 8 %) than that of Alzheimer’s disease and
frontotemporal dementia (56 ± 27 %) patients. The
majority of the centers (91 %) considered the clinical and
neuropsychological assessments as the most relevant
& L. Bonanni
1 Department of Neuroscience Imaging and Clinical Sciences,
University G.d’Annunzio of Chieti-Pescara, Via dei Vestini,
66100 Chieti, Italy
2 Department of Neurosciences, University of Padova, Padua,
Italy
3 Division of Neuroscience, Neuroimaging Research Unit, and
Department of Neurology, Institute of Experimental
Neurology, San Raffaele Scientific Institute, Milan, Italy
4 Department of Physiology and Pharmacology, University of
Rome ‘‘La Sapienza’’, Rome, Italy
5 Neurology Unit, Department of Clinical and Experimental
Sciences, University of Brescia, Piazza Spedali Civili 1,
25100 Brescia, Italy
6 Neuroimaging Laboratory, IRCCS Santa Lucia Foundation,
Rome, Italy
7 Centro Regionale di Neurogenetica ASP CZ,
Lamezia Terme, Italy
8 Neurology Unit, Deptartment of Pathophysiology and
Transplantation, Fondazione Ca’ Granda, IRCCS Ospedale
Policlinico, University of Milan, Milan, Italy
9 Department of Experimental Biomedicine and Clinical
Neuroscience (BioNeC), University of Palermo, Palermo,
Italy
10 Divisione di Neurologia V e Neuropatologia, Fondazione
IRCCS Istituto Neurologico ‘‘Carlo Besta’’, Milan, Italy
11 Clinica Neurologica, Centro Disturbi della Memoria,
Universita di Perugia (I), Perugia, Italy
12 Division of Neuroscience, Nuclear Medicine Unit San
Raffaele Hospital, San Raffaele Scientific Institute, Vita-
Salute San Raffaele University, Milan, Italy
13 Department of Neurology-Stroke Unit and Laboratory of
Neuroscience, IRCCS Istituto Auxologico Italiano,
Milan 20149, Italy
14 IRCCS San Camillo Hospital Foundation, Venice, Italy
15 IRCCS San Raffaele Pisana, Rome, Italy
16 Department of Pathophysiology and Transplantation, ‘‘Dino
Ferrari’’ Center, Universita degli Studi di Milano,
Milan 20122, Italy
123
Neurol Sci
DOI 10.1007/s10072-016-2713-8

Letter to the Editor
Cerebral microbleeds: A new presenting feature ofchromosome 22q11.2 deletion syndrome
Abbreviations:2q11.2DSTopic:22q11.2 deletion syndromeCNSTopic:central nervous systemCMBTopic:cerebral microbleedsArray CGHTopic:array - comparative genomic hybridizationEEGTopic:electroencephalographyCTTopic:computed tomographyMRITopic:magnetic resonance imagingFFETopic:fast-field-echoSWITopic:susceptibility-weighted imagingAPOETopic:apolipoprotein EAPPTopic:amyloid precursor proteinPFA-100Topic:platelet function assayCAATopic:cerebral amyloid angiopathy
Keywords:Cerebral microbleeds22q11.2 deletion syndromePlatelet dysfunction
Dear Editor,22q11.2 deletion syndrome (22q11.2DS) is a contiguous gene
microdeletion syndrome that affects up to 1 in 2000 live births [1]. Thecommonly 22q11.2DS deleted region contains over 35 genes. N70% ofthe patients display congenital heart abnormalities, immune system defi-ciencies, palatal defects, structural renal anomalies and parathyroid hypo-plasia. Structural central nervous system (CNS) abnormalities includespinabifida,microcephalyandreduceddevelopmentof thegreymatter [2].
22q11.2DS patients also show behavioral disturbances, impaired cognitivedevelopment and seizures that can be triggered by cortical dysgenesis orby hypocalcaemia in hypoparathyroidism [3].
We describe a 36-year-old woman, admitted to our neurologicalunit because of episodes of brief loss of consciousness associated withmotor arrest and brief postictal confusion. The patient had experiencedthe first episodes 4 years earlier. The loss of consciousness wassometimes accompanied by tonic-clonic movements. Her past medicalhistory was remarkable for tetralogy of Fallot, surgically treated whenshe was 6 years old. At the age of 30, the patient developed a sustaineddepressed mood, treated with paroxetine for 2 years.
Neurological examination revealed increased muscle stretch reflexes,moderate dysdiadochokinesia and slight difficulty in the tandem walkingtest. Neuropsychological evaluation showed a mild cognitive impairment.EEG showed asymmetric generalised bursts of spike waves and polymor-phic theta activity on the left frontotemporal regions. We investigatedwhether epileptiform activitywas related to hypocalcaemia, but calcaemia(9.2 mg/dL, reference [8].1–10.4), phosphoraemia (3.1 mg/dL, reference[2].8–5.2) and parathyroid hormone level (28.4 ng/L, reference 13.0–64.0) were normal. Brain Magnetic Resonance Imaging (MRI) did not de-tect cortical dysgenesia, or midline development abnormalities. Few focalwhite matter hyperintensities were observed in the frontal lobes. Unex-pectedly, Fast-Field-Echo (FFE) and susceptibility-weighted imaging(SWI) MRI sequences revealed small, round hypointensities in variousareas comprising the cerebellar vermis, left occipital regions, left lentiformnucleus, right frontal lobe and parasagittal portion of the posterior parietalgyrus. These lesions, only detected by FFE and SWI MRI sequences, werestrongly suggestive of tiny accumulations of haemosiderin adjacent to ab-normal fragilemicrovessels named cerebralmicrobleeds (CMBs) [4,5] (Fig.1). As calcium deposits may mimic hemosiderin accumulation [5], weruled out the presence of calcification by CT scan, thus confirming CMBs.
The combination of a conotruncal heart defect with dysmorphicfeatures and short stature (Table 1) was highly suggestive of typical22q11.2DS. Array CGH analysis detected a de novo 2.52-Mb hemizygousdeletion of chromosome 22q11.2, encompassing nucleotides18919942-21440514 (GRCh37/hg 19 release).
To the best of our knowledge, this is the first description of CMBs in22q11.2DS. We investigated the potential risk factors underlying CMBs.
Deep or infratentorial microbleeds usually occur because offibrohyalinosis due to advanced age and atherosclerosis. However, ourpatient was young and did not suffer from hypertension (excluded by24-hour blood pressure monitoring). Another potential factor underly-ing CMBs is cerebral amyloid microangiopathy (CAA) [4,5], that ischaracterized by lobar bleeding. Our patient did not display the typicalCAA features, which include the strictly lobar distribution andhaemorrhagic stroke evidence. We also investigated some of the genesresponsible for hereditable CAA (amyloid-beta fragment of amyloidprecursor protein, APP; cystatin C; transthyretin and gelsolin) andthe APOE haplotype. No mutations were identified; the patient had ane3/e3 genotype, thus she did not carry the alleles predisposing toamyloid deposits in vessel walls (e4) and bleeding (e2).
Journal of the Neurological Sciences 368 (2016) 300–303
http://dx.doi.org/10.1016/j.jns.2016.07.0440022-510X/© 2016 Elsevier B.V. All rights reserved.
Contents lists available at ScienceDirect
Journal of the Neurological Sciences
j ourna l homepage: www.e lsev ie r .com/ locate / jns

Competing interests
The authors declare that they have no competing interests.
Authors' contributions
Study concept and design: BMT, CA. Acquisition of data: CA, BMT, SD,VC, SC, GD. Analysis and interpretation of data: CA, BMT, SD, VC, SJ, GF.Drafting of the manuscript: CA, BMT, SD. All authors revised the manu-script and provided important intellectual content. All authors haveread and approved the final manuscript.
Availability of data and materials
All the data regarding the patient of this case report have beenplaced in the Istituto Auxologico Italiano archives.
Acknowledgments
None.
References
[1] W.L.A. Fung, N.J. Butcher, G. Costain, D.M. Andrade, E. Boot, E.W.C. Chow, et al.,Practical guidelines for managing adults with 22q11.2 deletion syndrome, Genet.Med. 17 (2015) 599–609.
[2] D.M. McDonald-McGinn, K.E. Sullivan, Chromosome 22q11.2 deletion syndrome(DiGeorge syndrome/velocardiofacial syndrome), Medicine (Baltimore) 90 (2011)1–18.
[3] W. González, R.E.D. Bautista, Seizures and EEG findings in an adult patient withDiGeorge syndrome: a case report and review of the literature, Seizure 18 (2009)648–651.
[4] S.M. Greenberg, M.W. Vernooij, C. Cordonnier, A. Viswanathan, R. Al-Shahi Salman,S. Warach, et al., Cerebral microbleeds: a guide to detection and interpretation,Lancet Neurol. (2009) 165–174.
[5] M.A. Ikram, A. van der Lugt, W.J. Niessen, P.J. Koudstaal, G.P. Krestin, A. Hofman,et al., The Rotterdam scan study: design update 2016 and main findings, Eur. J.Epidemiol. 30 (2015) 1299–1315.
[6] S. Lawrence, D.M. McDonald-McGinn, E. Zackai, K.E. Sullivan, Thrombocytopenia in pa-tients with chromosome 22q11.2 deletion syndrome, J. Pediatr. 143 (2003) 277–278.
[7] P.J. Lenting, J.N. Pegon, E. Groot, P.G. De Groot, Regulation of von Willebrand factor-platelet interactions, Thromb. Haemost. (2010) 449–455.
[8] H.P.H. Liang,M.C.Morel-Kopp, J. Curtin,M.Wilson, J. Hewson,W. Chen, et al., Heterozy-gous loss of platelet glycoprotein (GP) Ib-V-IX variably affects platelet function invelocardiofacial syndrome (VCFS) patients, Thromb. Haemost. 98 (2007) 1298–1308.
[9] Z. Chen, J. Wu, C. Yang, P. Fan, L. Balazs, Y. Jiao, et al., DiGeorge syndrome critical re-gion 8 (DGCR8) protein-mediated microRNA biogenesis is essential for vascularsmooth muscle cell development in mice, J. Biol. Chem. 287 (2012) 19018–19028.
[10] C. Rossi, V. De Herdt, N. Dequatre-Ponchelle, H. Hénon, D. Leys, C. Cordonnier,Incidence and predictors of late seizures in intracerebral hemorrhages, Stroke 44(2013) 1723–1725.
M.T. BonatiClinic of Medical Genetics, IRCCS Istituto Auxologico Italiano, Milan, Italy
E-mail address: [email protected]
C. VanelliHematology and Transfusion Medicine, L. Sacco University Hospital andDepartment of Clinical Sciences and Community Health, Università degli
Studi di Milano, Milan, ItalyE-mail address: [email protected]
D. SangalliDepartment of Neurology and Laboratory of Neuroscience, IRCCS Istituto
Auxologico Italiano, Milan, ItalyDepartment of Pathophysiology and Transplantation, “Dino Ferrari” Center,
Università degli Studi di Milano, Milan, ItalyE-mail address: [email protected]
C. SinaDepartment of Neuroradiology, Fondazione IRCCS Ca' Granda, Ospedale
Maggiore Policlinico, Milan, ItalyE-mail address: [email protected]
D. GiardinoLaboratory of Medical Cytogenetics and Molecular Genetics, IRCCS Istituto
Auxologico Italiano, Milan, ItalyE-mail address: [email protected]
J. SassoneDepartment of Medical Sciences, Section of Pharmacology, University of
Ferrara, Ferrara, ItalyE-mail address: [email protected]
F. GirottiDepartment of Neurology and Laboratory of Neuroscience, IRCCS Istituto
Auxologico Italiano, Milan, ItalyE-mail address: [email protected]
V. SilaniDepartment of Neurology and Laboratory of Neuroscience, IRCCS Istituto
Auxologico Italiano, Milan, ItalyDepartment of Pathophysiology and Transplantation, “Dino Ferrari” Center,
Università degli Studi di Milano, Milan, ItalyE-mail address: [email protected]
A. CiammolaDepartment of Neurology and Laboratory of Neuroscience, IRCCS Istituto
Auxologico Italiano, Milan, ItalyCorresponding author at: P.le Brescia 20, 20149 Milan, Italy.
E-mail addresses: [email protected], [email protected]
12 May 2016
Table 2Hematologic parameters.
Hematologic parameters Patient value Normal range
PT 1.06 0.85–1.18PTT 0.99 0.85–1.20Fibrinogen (mg/dL) 220 170–400Platelet count (/μL) 175,000 150,000–450,000Bleeding time (minutes) 6 b7.5Platelet function test (PFA-100) (seconds) Coll/ADP N300 68–121
Coll/epinephrine N300 85–165Cytofluorimetric platelet analysis Reduced P-selectin expressionExpression of membrane glycoprotein GPIIb – IIIa Normal
GP IIIa NormalGP Ib alfa NormalGP IX Normal
Von Willebrand factor (%) 68 70–110Von Willebrand activity (%) 52 60–150Factor XIII (%) 80 70–140
303Letter to the Editor

ORIGINAL COMMUNICATION
Factors predicting survival in ALS: a multicenter Italian study
Andrea Calvo1 • Cristina Moglia1 • Christian Lunetta2,3 • Kalliopi Marinou4 •
Nicola Ticozzi5,6 • Gianluca Drago Ferrante7 • Carlo Scialo8 • Gianni Soraru9 •
Francesca Trojsi10 • Amelia Conte11 • Yuri M. Falzone12 • Rosanna Tortelli13 •
Massimo Russo14 • Adriano Chio1 • Valeria Ada Sansone2,15 • Gabriele Mora4 •
Vincenzo Silani5,6 • Paolo Volanti7 • Claudia Caponnetto8 • Giorgia Querin9 •
Maria Rosaria Monsurro10 • Mario Sabatelli11,16 • Nilo Riva12 • Giancarlo Logroscino13 •
Sonia Messina3,14 • Nicola Fini17 • Jessica Mandrioli17
Received: 24 September 2016 / Accepted: 12 October 2016
� Springer-Verlag Berlin Heidelberg 2016
Abstract The aim of this multicenter, retrospective study
is to investigate the role of clinical characteristics and
therapeutic intervention on ALS prognosis. The study
included patients diagnosed from January 1, 2009 to
December 31, 2013 in 13 Italian referral centers for ALS
located in 10 Italian regions. Caring neurologists collected
a detailed phenotypic profile and follow-up data until death
into an electronic database. One center collected also data
from a population-based registry for ALS. 2648 incident
cases were collected. The median survival time from onset
to death/tracheostomy was 44 months (SE 1.18, CI 42–46).
According to univariate analysis, factors related to survival
from onset to death/tracheostomy were: age at onset,
diagnostic delay, site of onset, phenotype, degree of cer-
tainty at diagnosis according to revised El Escorial criteria
(R-EEC), presence/absence of dementia, BMI at diagnosis,
patients’ provenance. In the multivariate analysis, age at
onset, diagnostic delay, phenotypes but not site of onset,
presence/absence of dementia, BMI, riluzole use, R-EEC
criteria were independent prognostic factors of survival in
ALS. We compared patients from an ALS Registry with
patients from tertiary centers; the latter ones were younger,
& Jessica Mandrioli
1 ‘‘Rita Levi Montalcini’’ Department of Neuroscience, ALS
Center, University of Torino, Turin, Italy
2 NEuroMuscular Omnicentre (NEMO), Serena Onlus
Foundation, Milan, Italy
3 NEMO Sud Clinical Center for Neuromuscular Diseases,
Aurora Onlus Foundation, Messina, Italy
4 Department of Neurorehabilitation ALS Center Scientific
Institute of Milan, Salvatore Maugeri Foundation IRCCS,
Milan, Italy
5 Department of Neurology and Laboratory of Neuroscience,
IRCCS Istituto Auxologico Italiano, Milan, Italy
6 Department of Pathophysiology and Transplantation ‘Dino
Ferrari’ Center, University of Milan, Milan, Italy
7 Neurorehabilitation Unit/ALS Center, Salvatore Maugeri
Foundation, IRCCS, Mistretta, Messina, Italy
8 Department of Neurosciences, Rehabilitation,
Ophthalmology, Genetics Maternal and Child Health
(DINOGMI), University of Genova, IRCCS AOU San
Martino-IST, Genoa, Italy
9 Department of Neurosciences Neuromuscular Center,
University of Padova, Padua, Italy
10 Department of Medical, Surgical, Neurological, Metabolic
and Aging Sciences, MRI Research Center SUN-FISM,
Second University of Naples, Naples, Italy
11 NEuroMuscular Omnicentre (NEMO), Serena Onlus
Foundation: Pol. A. Gemelli Foundation, Rome, Italy
12 Department of Neurology Division of Neuroscience Institute
of Experimental Neurology, San Raffaele Scientific Institute,
Milan, Italy
13 Department of Clinical Research in Neurology, University of
Bari ‘‘A. Moro’’, at Pia Fondazione ‘‘Card. G. Panico’’
Tricase, Lecce, Italy
14 Department of Clinical and Experimental Medicine,
University of Messina and Nemo Sud Clinical Center for
Neuromuscular Diseases, Aurora Foundation, Messina, Italy
15 Department Biomedical Sciences for Health, University of
Milan, Milan, Italy
16 Institute of Neurology, Catholic University of Sacred Heart,
Rome, Italy
17 Department of Neuroscience, S. Agostino-Estense Hospital
and University of Modena and Reggio Emilia, Via Pietro
Giardini n. 1355, 41100 Modena, Italy
123
J Neurol
DOI 10.1007/s00415-016-8313-y

February 2016 | Volume 7 | Article 51
PersPectivepublished: 01 February 2016
doi: 10.3389/fimmu.2016.00005
Frontiers in Immunology | www.frontiersin.org
Edited by: Kenneth Michael Pollard,
The Scripps Research Institute, USA
Reviewed by: Wenxia Song,
University of Maryland, USA David M. Cauvi,
University of California San Diego, USA
*Correspondence:Pier Luigi Meroni
Specialty section: This article was submitted to
B Cell Biology, a section of the journal
Frontiers in Immunology
Received: 29 April 2015Accepted: 08 January 2016
Published: 01 February 2016
Citation: Gerosa M, Poletti B, Pregnolato F, Castellino G, Lafronza A, Silani V,
Riboldi P, Meroni PL and Merrill JT (2016) Antiglutamate Receptor
Antibodies and Cognitive Impairment in Primary Antiphospholipid
Syndrome and Systemic Lupus Erythematosus.
Front. Immunol. 7:5. doi: 10.3389/fimmu.2016.00005
Antiglutamate receptor Antibodies and cognitive impairment in Primary Antiphospholipid syndrome and systemic Lupus erythematosusMaria Gerosa1,2 , Barbara Poletti3 , Francesca Pregnolato4 , Gabriella Castellino5 , Annalisa Lafronza3 , Vincenzo Silani3,6 , Piersandro Riboldi1,5 , Pier Luigi Meroni1,4* and Joan T. Merrill7
1 Department of Clinical Sciences and Community Health, University of Milan, Milan, Italy, 2 Division of Rheumatology, Lupus Clinic, Istituto Ortopedico Gaetano Pini, Milan, Italy, 3 Department of Neurology-Stroke Unit and Laboratory of Neuroscience, IRCCS Istituto Auxologico Italiano, Milan, Italy, 4 Experimental Laboratory of Immunological and Rheumatologic Researches, IRCCS Istituto Auxologico Italiano, Milan, Italy, 5 Allergy, Clinical Immunology and Rheumatology Unit, IRCCS Istituto Auxologico Italiano, Milan, Italy, 6 Department of Pathophysiology and Transplantation, “Dino Ferrari” Center, Università degli Studi di Milano, Milan, Italy, 7 Clinical Pharmacology Research Program, Oklahoma Medical Research Foundation, University of Oklahoma, Oklahoma City, OK, USA
Systemic lupus erythematosus (SLE) and antiphospholipid syndrome have an increased risk to develop cognitive impairment. A possible role for antiphospholipid antibodies (aPL) and antiglutamate receptor (anti-NMDA) antibodies in the pathogenesis of neurological manifes-tations of these two conditions, have been suggested. In particular, the role of anti-NMDA antibodies in the pathogenesis of neuropsychiatric SLE is supported by several experimental studies in animal models and by the finding of a correlation between anti-NMDA positivity in cerebrospinal fluid and neurological manifestations of SLE. However, data from the literature are controversial, as several studies have reported a correlation of these antibodies with mild cognitive impairment in SLE, but more recent studies have not confirmed this finding. The synergism between anti-NMDA and other concomitant autoantibodies, such as aPL, can be hypothesized to play a role in inducing the tissue damage and eventually the functional abnormalities. In line with this hypothesis, we have found a high incidence of at least one impaired cognitive domain in a small cohort of patients with primary APS (PAPS) and SLE. Interestingly, aPL were associated with low scoring for language ability and attention while anti-NMDA titers and mini-mental state examination scoring were inversely correlated. However, when patients were stratified according to the presence/absence of aPL, the cor-relation was confirmed in aPL positive patients only. Should those findings be confirmed, the etiology of the prevalent defects found in PAPS patients as well as the synergism between aPL and anti-NMDA antibodies would need to be explored.
Keywords: systemic lupus erythematosus, antiphospholipid syndrome, mild cognitive impairment, neuropsychological assessment, central nervous system involvement, anti-NMDA/glutamate receptor antibodies
Abbreviations: ANA, antinuclear antibodies; anti-CL, anticardiolipin; anti-dsDNA, antidouble strand DNA; anti-PL, antiphos-pholipid antibodies; anti-β2GPI, anti-β2 glycoprotein I; APS, antiphospholipid syndrome; BBB, blood–brain barrier; CCI, cognitive impairment index; CNS, central nervous system; CSF, cerebrospinal fluid; ELISA, enzyme-linked immunosorbent assay; ENA, extractable nuclear antigen; ICV, intracerebroventricular; Ig, immunoglobulins; LA, lupus anticoagulant; MMSE, mini-mental state examination; NMDA, human glutamate receptor; NPSLE, neuropsychiatric SLE; PAPS, primary APS; SLE, systemic lupus erythematosus.

ARTICLE
Received 10 Aug 2015 | Accepted 7 Mar 2016 | Published 15 Apr 2016
CCNF mutations in amyotrophic lateral sclerosisand frontotemporal dementiaKelly L. Williams1,2,3, Simon Topp4, Shu Yang1,2, Bradley Smith4, Jennifer A. Fifita1,2, Sadaf T. Warraich1, Katharine Y. Zhang1,
Natalie Farrawell5, Caroline Vance4, Xun Hu4, Alessandra Chesi6, Claire S. Leblond7,8, Albert Lee1,9, Stephanie L. Rayner1,
Vinod Sundaramoorthy1,10, Carol Dobson-Stone11,12, Mark P. Molloy1,9, Marka van Blitterswijk13, Dennis W. Dickson13, Ronald C. Petersen14,
Neill R. Graff-Radford15, Bradley F. Boeve14, Melissa E. Murray13, Cyril Pottier13, Emily Don1, Claire Winnick1, Emily P. McCann1,
Alison Hogan1, Hussein Daoud7,8, Annie Levert7,8, Patrick A. Dion7,8, Jun Mitsui16, Hiroyuki Ishiura16, Yuji Takahashi16, Jun Goto16,
Jason Kost17,18, Cinzia Gellera19, Athina Soragia Gkazi4, Jack Miller4, Joanne Stockton20, William S. Brooks11, Karyn Boundy21,
Meraida Polak22, Jose Luis Munoz-Blanco23, Jesus Esteban-Perez24,25, Alberto Rabano26, Orla Hardiman27, Karen E. Morrison20,28,29,
Nicola Ticozzi30,31, Vincenzo Silani30,31, Jacqueline de Belleroche32, Jonathan D. Glass22, John B.J. Kwok11,12, Gilles J. Guillemin1,
Roger S. Chung1, Shoji Tsuji16,33, Robert H. Brown Jr18, Alberto Garcıa-Redondo24,25, Rosa Rademakers13, John E. Landers18, Aaron D. Gitler6,
Guy A. Rouleau7,8, Nicholas J. Cole1,3, Justin J. Yerbury5, Julie D. Atkin1,10, Christopher E. Shaw4, Garth A. Nicholson1,2,3,34 & Ian P. Blair1,2
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are overlapping, fatal neuro-
degenerative disorders in which the molecular and pathogenic basis remains poorly understood.
Ubiquitinated protein aggregates, of which TDP-43 is a major component, are a characteristic
pathological feature of most ALS and FTD patients. Here we use genome-wide linkage analysis in a large
ALS/FTD kindred to identify a novel disease locus on chromosome 16p13.3. Whole-exome sequencing
identified a CCNF missense mutation at this locus. Interrogation of international cohorts identified
additional novel CCNF variants in familial and sporadic ALS and FTD. Enrichment of rare protein-altering
CCNF variants was evident in a large sporadic ALS replication cohort. CCNF encodes cyclin F, a component
of an E3 ubiquitin–protein ligase complex (SCFCyclin F). Expression of mutant CCNF in neuronal cells
caused abnormal ubiquitination and accumulation of ubiquitinated proteins, including TDP-43 and a
SCFCyclin F substrate. This implicates common mechanisms, linked to protein homeostasis, underlying
neuronal degeneration.
DOI: 10.1038/ncomms11253 OPEN
1 Department of Biomedical Sciences, Faculty of Medicine and Health Sciences, Macquarie University, Sydney, New South Wales 2109, Australia. 2 Northcott Neuroscience
Laboratory, ANZAC Research Institute, Sydney, New South Wales 2139, Australia. 3 Sydney Medical School, University of Sydney, Sydney, New South Wales 2006, Australia.4 Medical Research Council Centre for Neurodegeneration Research, Department of Clinical Neuroscience, Institute of Psychiatry, King’s College London, London SE5 8AF, UK.5 Illawarra Health and Medical Research Institute, School of Biological Sciences, University of Wollongong, Wollongong, New South Wales 2522, Australia. 6 Department of
Genetics, Stanford University School of Medicine, Stanford, California 94305, USA. 7 Montreal Neurological Institute and Hospital, Department of Neurology and Neurosurgery,
McGill University, Montreal, Quebec, Canada H3A 2B4. 8 Pathology and Cellular Biology Department, Montreal University, Montreal, QC H3T 1J4 Quebec, Canada. 9 Australian
Proteome Analysis Facility, Macquarie University, Sydney, New South Wales 2109, Australia. 10 Department of Biochemistry, La Trobe University, Melbourne, Victoria 3086,
Australia. 11 Neuroscience Research Australia, Randwick, Sydney, New South Wales 2031, Australia. 12 School of Medical Sciences, University of New South Wales, Kensington,
Sydney, New South Wales 2052, Australia. 13 Department of Neuroscience, Mayo Clinic Florida, Jacksonville, Florida 32224, USA. 14 Department of Neurology, Mayo Clinic
Rochester, Rochester, Minneapolis 55905, USA. 15 Department of Neurology, Mayo Clinic Florida, Jacksonville, Florida, 32224, USA. 16 Medical Genome Center, The University of
Tokyo Hospital, The University of Tokyo, Tokyo 113-8655, Japan. 17 Worcester Polytechnic Institute, Worcester, Massachusetts 01609, USA. 18 Department of Neurology, University
of Massachusetts Medical School, Worcester, Massachusetts 01605, USA. 19 Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto
Neurologico ‘Carlo Besta’, 20133 Milan, Italy. 20 School of Clinical and Experimental Medicine, College of Medical and Dental Sciences, University of Birmingham, Birmingham B15
2TT, UK. 21 The Queen Elizabeth Hospital, Woodville South, South Australia 5011, Australia. 22 Department of Neurology, Emory University, Atlanta, Georgia 30322, USA. 23 Unidad
de ELA, Instituto de Investigacion Hospital Gregorio Maranon de Madrid, Sermas 28007, Spain. 24 Unidad de ELA, Instituto de Investigacion Hospital 12 de Octubre de Madrid,
Sermas 28041, Spain. 25 Centro de Investigacion Biomedica en Red de Enfermedades Raras (CIBERER U-723), Madrid 28029, Spain. 26 Banco de Tejidos, Centro Alzheimer—
Fundacion Reina Sofia, Fundacion CIEN, Madrid 28071, Spain. 27 Academic Unit of Neurology, Trinity Biomedical Sciences Institute, Trinity College Dublin, Dublin 2, Republic of
Ireland. 28 Queen Elizabeth Hospital, University Hospitals Birmingham NHS Foundation Trust, Birmingham B15 2TH, UK. 29 Faculty of Medicine, University of Southampton,
Southampton SO17 1BJ, UK. 30 Department of Neurology and Laboratory of Neuroscience, IRCCS Istituto Auxologico Italiano, 20149 Milan, Italy. 31 Department of Pathophysiology
and Transplantation, ‘Dino Ferrari’ Center—Universita degli Studi di Milano, 20122 Milan, Italy. 32 Neurogenetics Group, Division of Brain Sciences, Imperial College London,
Hammersmith Hospital Campus, London W12 0NN, UK. 33 Department of Neurology, Graduate School of Medicine, The University of Tokyo, Tokyo 113-8655, Japan. 34 Molecular
Medicine Laboratory, Concord Hospital, New South Wales 2139, Australia. Correspondence and requests for materials should be addressed to I.P.B. (email: [email protected]).
NATURE COMMUNICATIONS | 7:11253 | DOI: 10.1038/ncomms11253 | www.nature.com/naturecommunications 1

REPORT
De Novo Mutations in PDE10A CauseChildhood-Onset Chorea with Bilateral Striatal Lesions
Niccolo E. Mencacci,1,2,17 Erik-Jan Kamsteeg,3,17 Kosuke Nakashima,4,17 Lea R’Bibo,1 David S. Lynch,1
Bettina Balint,5,6 Michel A.A.P. Willemsen,7 Matthew E. Adams,8 Sarah Wiethoff,1,9 Kazunori Suzuki,4
Ceri H. Davies,4 Joanne Ng,10,11 Esther Meyer,10 Liana Veneziano,12 Paola Giunti,1 Deborah Hughes,1
F. Lucy Raymond,13 Miryam Carecchio,14,15 Giovanna Zorzi,14 Nardo Nardocci,14 Chiara Barzaghi,15
Barbara Garavaglia,15 Vincenzo Salpietro,1 John Hardy,1,16 Alan M. Pittman,1,16 Henry Houlden,1
Manju A. Kurian,10,11 Haruhide Kimura,4,18 Lisenka E.L.M. Vissers,3,18 Nicholas W. Wood,1,18,*and Kailash P. Bhatia5,18
Chorea is a hyperkinetic movement disorder resulting from dysfunction of striatal medium spiny neurons (MSNs), which form themain
output projections from the basal ganglia. Here, we used whole-exome sequencing to unravel the underlying genetic cause in three
unrelated individuals with a very similar and unique clinical presentation of childhood-onset chorea and characteristic brain MRI
showing symmetrical bilateral striatal lesions. All individuals were identified to carry a de novo heterozygous mutation in PDE10A
(c.898T>C [p.Phe300Leu] in two individuals and c.1000T>C [p.Phe334Leu] in one individual), encoding a phosphodiesterase highly
and selectively present in MSNs. PDE10A contributes to the regulation of the intracellular levels of cyclic adenosine monophosphate
(cAMP) and cyclic guanosine monophosphate (cGMP). Both substitutions affect highly conserved amino acids located in the regulatory
GAF-B domain, which, by binding to cAMP, stimulates the activity of the PDE10A catalytic domain. In silico modeling showed that the
altered residues are located deep in the binding pocket, where they are likely to alter cAMP binding properties. In vitro functional studies
showed that neither substitution affects the basal PDE10A activity, but they severely disrupt the stimulatory effect mediated by cAMP
binding to the GAF-B domain. The identification of PDE10A mutations as a cause of chorea further motivates the study of cAMP
signaling in MSNs and highlights the crucial role of striatal cAMP signaling in the regulation of basal ganglia circuitry. Pharmacological
modulation of this pathway could offer promising etiologically targeted treatments for chorea and other hyperkinetic movement
disorders.
Movement disorders comprise a large clinically and genet-
ically heterogeneous group of disorders, which can be sub-
divided into various clinical entities, including dystonia
and chorea. Although monogenic causes are overall rare,
mutations in greater than >200 genes are known to cause
either an isolated movement disorder or a syndromic form
of movement disorders.1–3 However, in total, mutations in
these genes only explain a small proportion of cases, sug-
gesting that mutations in more genes await discovery.
Chorea is a hyperkinetic movement disorder clinically
characterized by continuous and brief involuntary
movements, which flow from one body part to another
and are unpredictable in terms of timing, speed, and
direction. Chorea is a major feature of several inherited
neurological disorders.4 Functional dysregulation of
striatal GABAergic medium spiny neurons (MSNs), which
form the main output projections from the basal ganglia,
is considered to underlie the pathophysiology of the
choreic movements.5
We have identified three European-descent individuals
affected by a similar childhood-onset movement disorder
predominantly characterized by chorea and bilateral stria-
tal abnormalities on cerebral MRI. The main clinical and
radiological features of the three individuals are presented
in Table 1. In brief, all three individuals presented in child-
hood (age of onset between 5 and 10 years) with a scarcely
progressive movement disorder dominated by chorea.
Developmental milestones were normal, and there were
no other major neurological features, in particular intellec-
tual disability or cognitive decline. Given these clinical
1Department ofMolecular Neuroscience, UCL Institute of Neurology,WC1N 3BG London, UK; 2Department of Neurology and Laboratory of Neuroscience,
IRCCS Istituto Auxologico Italiano, Department of Pathophysiology and Transplantation, Centro Dino Ferrari, Universita degli Studi di Milano, 20149 Mi-
lan, Italy; 3Department of Human Genetics, Donders Centre for Brain, Cognition, and Behavior, Radboud UniversityMedical Center, Geert Grooteplein 10,
6525 GA Nijmegen, the Netherlands; 4CNS Drug Discovery Unit, Pharmaceutical Research Division, Takeda Pharmaceutical Company Limited, 251-8555
Fujisawa, Japan; 5Sobell Department of Motor Neuroscience and Movement Disorders, UCL Institute of Neurology, WC1N 3BG London, UK; 6Department
of Neurology, University Hospital Heidelberg, 69120 Heidelberg, Germany; 7Department of Paediatric Neurology, Donders Centre for Brain, Cognition, and
Behavior, Radboud University Medical Center, Geert Grooteplein 10, 6525 GA Nijmegen, the Netherlands; 8Lysholm Department of Neuroradiology, Na-
tional Hospital for Neurology and Neurosurgery, WC1N 3BG London, UK; 9Center for Neurology and Hertie Institute for Clinical Brain Research, Eberhard
Karls University, 72076 Tubingen, Germany; 10Developmental Neurosciences, UCL Institute of Child Health, WC1N 1EH London, UK; 11Department of
Neurology, Great Ormond Street Hospital, WC1N 3JH London, UK; 12Institute of Translational Pharmacology, National Research Council, 00133 Rome,
Italy; 13Department of Medical Genetics, University of Cambridge, CB2 0XY Cambridge, UK; 14Neuropediatrics Unit, IRCCS Istituto Neurologico Carlo
Besta, 20133 Milan, Italy; 15Molecular Neurogenetics Unit, IRCCS Istituto Neurologico Carlo Besta, 20133 Milan, Italy; 16Reta Lila Weston Institute of
Neurological Studies, UCL Institute of Neurology, WC1N 3BG London, UK17These authors contributed equally to this work18These authors contributed equally to this work
*Correspondence: [email protected]
http://dx.doi.org/10.1016/j.ajhg.2016.02.015.
The American Journal of Human Genetics 98, 763–771, April 7, 2016 763
�2016 The Authors. This is an open access article under the CC BY license (http://creativecommons.org/licenses/by/4.0/).

RESEARCH PAPER
A large-scale multicentre cerebral diffusion tensorimaging study in amyotrophic lateral sclerosisHans-Peter Müller,1 Martin R Turner,2 Julian Grosskreutz,3 Sharon Abrahams,4
Peter Bede,5 Varan Govind,6 Johannes Prudlo,7 Albert C Ludolph,1 Massimo Filippi,8
Jan Kassubek,1 for The Neuroimaging Society in ALS (NiSALS) DTI Study Group
▸ Additional material ispublished online only. To viewplease visit the journal online(http://dx.doi.org/10.1136/jnnp-2015-311952).
For numbered affiliations seeend of article.
Correspondence toProfessor Jan Kassubek,Department of Neurology,University of Ulm, ObererEselsberg 45, Ulm 89081,Germany;[email protected]
Received 6 August 2015Revised 30 October 2015Accepted 9 December 2015Published Online First8 January 2016
▸ http://dx.doi.org/10.1136/jnnp-2015-312882
To cite: Müller H-P,Turner MR, Grosskreutz J,et al. J Neurol NeurosurgPsychiatry 2016;87:570–579.
ABSTRACTObjective Damage to the cerebral tissue structuralconnectivity associated with amyotrophic lateral sclerosis(ALS), which extends beyond the motor pathways, canbe visualised by diffusion tensor imaging (DTI). Theeffective translation of DTI metrics as biomarker requiresits application across multiple MRI scanners and patientcohorts. A multicentre study was undertaken to assessstructural connectivity in ALS within a large sample size.Methods 442 DTI data sets from patients with ALS(N=253) and controls (N=189) were collected for thisretrospective study, from eight international ALS-specialist clinic sites. Equipment and DTI protocols variedacross the centres. Fractional anisotropy (FA) maps ofthe control participants were used to establish correctionmatrices to pool data, and correction algorithms wereapplied to the FA maps of the control and ALS patientgroups.Results Analysis of data pooled from all centres, usingwhole-brain-based statistical analysis of FA maps,confirmed the most significant alterations in thecorticospinal tracts, and captured additional significantwhite matter tract changes in the frontal lobe, brainstemand hippocampal regions of the ALS group thatcoincided with postmortem neuropathological stages.Stratification of the ALS group for disease severity (ALSfunctional rating scale) confirmed these findings.Interpretation This large-scale study overcomes thechallenges associated with processing and analysis ofmultiplatform, multicentre DTI data, and effectivelydemonstrates the anatomical fingerprint patterns ofchanges in a DTI metric that reflect distinct ALS diseasestages. This success paves the way for the use of DTI-based metrics as read-out in natural history, prognosticstratification and multisite disease-modifying studies inALS.
INTRODUCTIONThe use of advanced MRI techniques, in particulardiffusion tensor imaging (DTI) of white mattertracts, has greatly improved the understanding ofthe in vivo cerebral and spinal neuropathology ofthe adult neurodegenerative disorder, amyotrophiclateral sclerosis (ALS).1–5 DTI can quantify theintegrity of large white matter tracts in vivo, usingmetrics such as fractional anisotropy (FA).6 ADTI-based in vivo imaging concept has also beenapplied to the recently introduced neuropatho-logical staging system, indicating that ALS may dis-seminate in regional patterns.7 8 ALS overlaps with
frontotemporal dementia clinically and pathologic-ally. While the majority of patients with ALS donot develop a frank dementia, a large proportionshow cognitive impairments on the same spectrum,and DTI has demonstrated extension of whitematter changes into frontal and temporal lobesaccordingly.9 10
Imaging biomarkers are urgently required forfuture pharmaceutical trials to be used as objectivestudy endpoints. The development of robust prog-nostic and diagnostic markers has become a majorresearch priority in this notoriously heterogeneousdisorder.11 12 Effective biomarkers, particularlythose integrated in therapeutic studies, need to beapplicable across multiple international centresthat, in case of MRI, may vary in their scannerhardware and sequence acquisition parameters.The Neuroimaging Society in ALS (http://www.
nisals.org) was established in 2010, and developeda roadmap for the standardisation and harmonisa-tion of advanced MRI data in ALS.13 The most sen-sitive cerebral pathology found in cross-sectionalMRI studies in patients with ALS has been in thewhite matter tracts,14 in contrast to the characteris-tic hippocampal atrophy that largely definesAlzheimer’s disease. To date, there have been fewlarge-scale multisite DTI studies, and none in ALS.Data analysis of such studies may be hampered bydifferences in scanning protocols,15 16 thus anapproach to pool DTI data acquired with differentprotocols was sought. The objective was to identifyALS-induced alterations in the white matter struc-tural connectivity in a large scale patient cohortfrom multiple sites spread across the world.To this end, a new strategy has been developed
that paves the way to use the large numbers of DTIdata sets from local data bases of different studysites for comprehensive large-scale multicentreimaging studies.
METHODSSubject populations and scanning protocolsFour hundred and forty-two DTI data sets frompatients with ALS (N=253) and control partici-pants (N=189) from eight ALS-specialist clinic sites(tertiary referral centres) were selected ex post factofor this retrospective study (Dublin, Ireland;Edinburgh, UK; Jena, Germany; Miami, USA;Milano, Italy; Oxford, UK; Rostock, Germany;Ulm, Germany). Centre specific details, includingnumber of participants and DTI scanning
570 Müller H-P, et al. J Neurol Neurosurg Psychiatry 2016;87:570–579. doi:10.1136/jnnp-2015-311952
Neurodegeneration
group.bmj.com on January 13, 2017 - Published by http://jnnp.bmj.com/Downloaded from

at two identical scanners39 reporting that the within-session andbetween-session reproducibility was lower than the values forintersubject variability. The initial suggestions on how to correctfor differences in FA maps with different protocols wererecently published.30 40 Common acquisition protocols butinvolving different participants were investigated for groupwiseFA differences between patients and controls on systematicbetween-centre differences.37 During the pooling process, anumber of well-defined parameters that confound the FA resultscould be regressed out. Nevertheless, to some extent, residualcentre-specific FA-influencing factors might be identified. Thatway, matrices for regressing out FA-influencing factors could beapplied in an identical manner to FA-maps of ALS-patients. Thecomparison between the analysis of corrected and the analysisof uncorrected data showed a difference of about 20% in thenumber of significant voxels. However, as in this study the ratiopatients/controls was approximately equal between the centres,the correction effects were less prominent, as it could beexpected had the ratio patients/controls differed more betweenthe centres. Furthermore, this study shows that affectationscould be observed at the multicentre level with high subjectnumbers that would not have been possible to show in single-centre studies with low subject numbers, this methodologicalframework could easily be adapted to further DTI metrics, forexample, radial, axial, or mean diffusivity.
In summary, this study has demonstrated that it is possible tomeaningfully interpret combined DTI data from different MRImanufacturers and software platforms after application ofappropriate compensations for centre-specific differences. Thismight pave the way to repurpose larger numbers of DTI datasets of patients with ALS for more clinically comprehensivelarge-scale multicentric imaging studies.
A limitation of this study compared to other multicentrestudies of neurodegenerative diseases (eg, the PADDINGTONstudy in Huntington’s Disease or the Alzheimer’s DiseaseNeuroimaging Initiative (ADNI)41 42) is the heterogeneousnature of DTI data resource in terms of magnetic field strengthand acquisition sequence parameters used across the centres. Itwould be advantageous in a future prospective multisite studyto try to harmonise as many data acquisition sequence para-meters as possible. A further potential limitation of the currentapproach is the limited number of control data sets per centre,which may or may not be homogeneous over centres, and mightlead to false or overcorrection. However, N=20 (which is thecase in seven out of the eight centres) has been a standardcontrol sample size in single-centre studies.33 43 44 As the ratiopatients/controls did not differ strongly between differentcentres, a systematic centre difference would be averaged out inthe multisite group comparison.
By this multicentre approach involving DTI data sets frommore than 250 patients, it was possible to reveal the in vivopathoanatomy of ALS non-invasively; the alteration pattern wasfound to be in agreement with postmortem neuroanatomicalstudies42 45 so that DTI seems to be prone to expand the poten-tial of other neuroimaging markers such as fluorodeoxyglucose-positron-emission tomography in ALS.46 47 The postmortemcerebral white matter changes in ALS have been long recog-nised45 48 but direct correlation with DTI is still in develop-ment.49 This study represents a framework for mappingALS-specific white matter tract alterations using DTI, and pro-vides encouragement for its extension to other neurodegenera-tive diseases. In other diseases with emerging evidence of aconsistent pathological pattern of spread,50 the white mattertract pathology will be closer to the underlying pathology than
clusters of regional atrophy, and thus the DTI metrics have thepotential to serve as read-outs and biomarkers of disease and itsprogression for future clinical trials.
Author affiliations1Department of Neurology, University of Ulm, Ulm, Germany2Nuffield Department of Clinical Neurosciences, University of Oxford, John RadcliffeHospital, Oxford, UK3Hans-Berger Department of Neurology, Jena University Hospital, Jena, Germany4Human Cognitive Neuroscience, Psychology–PPLS & Euan MacDonald Centre forMND Research & Centre for Cognitive Ageing and Epidemiology, University ofEdinburgh, Edinburgh, UK5Quantitative Neuroimaging Group, Academic Unit of Neurology, Trinity CollegeDublin, Dublin, Ireland6Department of Radiology, University of Miami School of Medicine, Miami, Florida,USA7Department of Neurology, University of Rostock and DZNE, Rostock, Germany8Division of Neuroscience, Neuroimaging Research Unit, Institute of ExperimentalNeurology, San Raffaele Scientific Institute, Vita-Salute San Raffaele University,Milan, Italy
Collaborators NiSALS contributors: Abdulla, Susanne, Department of Neurology,Hannover Medical School, Germany. Agosta, Federica, Neuroimaging Research Unit,Scientific Institute San Raffaele, Milan, Italy. Ajroud-Driss, Senda, Neurology,Northwestern University, USA. Atassi, Nazem, Neurology, Massachusetts GeneralHospital, USA. Bastin, Mark, Brain Imaging Research Centre, Centre for CognitiveAgeing and Epidemiology, University of Edinburgh, Edinburgh, UK. Benatar, Michael,Department of Neurology, University of Miami School of Medicine, USA. Brooks,William, Hoglund Brain Imaging Center, University of Kansas Medical Center, USA.Calvo, Andrea, Rita Levi Montalcini Department of Neuroscience, University ofTorino, Italy. Cardenas-Blanco, Arturo, Plasticity and Neurodegeneration, DZNE,Germany. Chio, Adriano, University of Turin, Torino, Italy. De Carvalho, Mamede,Instituto de Medicina Molecular, Lisbon, Portugal. Dahnke, Robert, Department ofPsychiatry, University Hospital Jena, Jena, Germany. Enzinger, Christian, Departmentof Neurology, Medical University of Graz, Graz, Austria. Ferraro, Pilar Maria,Neuroimaging Research Unit, Scientific Institute San Raffaele, Milan, Italy. Floeter,Mary Kay, SSPU, EMG section, National Institute of Neurological Disorders andStroke, USA. Foerster, Bradley, Radiology, Division of Neuroradiology, University ofMichigan, USA. Gaser, Christian, Department of Psychiatry,Friedrich-Schiller-University of Jena, Jena, Germany. Geraldo, Ana Filipa,Neuroradiology, HSM, Portugal. Gorges, Martin, Department for Neurology,University of Ulm, Ulm, Germany. Grehl, Torsten, Kliniken Bergmannsheil, Bochum,Germany. Groen, Georg, Section Neuropsychology and Functional Imaging,Department of Psychiatry, University of Ulm, Germany. Hardiman, Orla, Departmentof Neurology, Trinity College Dublin, Dublin, Ireland. Hartung, Viktor, Department ofNeurology, University Hospital Jena, Jena, Germany. Jelsone-Swain, Laura,Department of Psychology, University of South Carolina Aiken, USA. Jenkins, Tom,Department of Neurology, Sheffield Institute for Translational Neuroscience, UK.Kalra, Sanjay, Medicine (Neurology), University of Alberta, Canada. Kasper,Elisabeth, University of Rostock, Rostock, Germany. Kitzler, Hagen, Department ofNeuroradiology, Technische Universitaet Dresden, University Hospital, Dresden,Germany. Koritnik, Blaz, Institute of Clinical Neurophysiology, University MedicalCenter Ljubljana, Slovenia. Kuzma–Kozakiewicz, Magdalena, Medical University ofWarsaw, Poland. LaFleur, Karl, Neurology/Psychiatry, University Medical Center—Utrecht, The Netherlands. Lulé, Dorothée, Neurology, University of Ulm, Ulm,Germany. Machts, Judith, Neurology, German Center for NeurodegenerativeDiseases, DZNE, Rostock, Germany. Meoded, Avner, EMG, NINDS, USA. Pettit,Lewis, Human Cognitive Neuroscience, Psychology–PPLS and Euan MacDonaldCentre for MND Research and Centre for Cognitive Ageing and Epidemiology,University of Edinburgh, Edinburgh, UK. Pioro, Erik, Neurology, Cleveland Clinic,USA. Poletti, Barbara, Department of Neurology and Laboratory of Neuroscience,IRCCS Istituto Auxologico Italiano, Italy. Pradat, Pierre-Francois, Department ofNeurology, Pitie-Salpetriere Hospital, France. Prell, Tino, Department of Neurology,University Hospital Jena, Jena, Germany. Proudfoot, Malcolm, Clinical Neuroscience,University of Oxford, UK. Ratti, Elena, Neurology—Neurological Clinical ResearchInstitute (NCRI), Massachusetts General Hospital, USA. Riva, Nilo, Neurology, SanRaffaele Scientific Institute, Italy. Robberecht, Wim, Vlaams Instituut voorBiotechnologie, Leuven, Belgium. Ropele, Stefan, Department of Neurology, MedicalUniversity of Graz, Graz, Austria. Salachas, Francois, Assistance Publique-Hopitauxde Paris, France. Schmidt, Ruben, Neurology, UMC Utrecht, The Netherlands.Schmidt-Wilcke, Kliniken Bergmannsheil, Bochum, Germany. Schuster, Christina,Trinity College Dublin, Ireland. Shaw, Pamela, Sheffield Institute for TranslationalNeuroscience, Sheffield, UK. Sherman, Alex, Neurological Clinical Research Institute,Massachusetts General Hospital, USA. Silani, Vincenzo, Department ofNeurology-Stroke Unit and Department of Neurology and Laboratory ofNeuroscience, IRCCS Istituto Auxologico Italiano, Dino Ferrari Center, Department ofPathophysiology and Transplantation, Universita degli Studi di Milano, Milan, Italy.
Müller H-P, et al. J Neurol Neurosurg Psychiatry 2016;87:570–579. doi:10.1136/jnnp-2015-311952 577
Neurodegeneration
group.bmj.com on January 13, 2017 - Published by http://jnnp.bmj.com/Downloaded from

Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration, 2016; 17: 404–413
RESEARCH ARTICLE
Multicenter validation of CSF neurofilaments as diagnostic biomar-
kers for ALS
PATRICK OECKL1, CLAUDE JARDEL2, FRANCOIS SALACHAS3, FOUDIL LAMARI2,
PETER M. ANDERSEN4, ROBERT BOWSER5,6, MAMEDE DE CARVALHO7,
JULIA COSTA8, PHILIP VAN DAMME9,19, ELIZABETH GRAY10,
JULIAN GROSSKREUTZ11, MARIA HERNANDEZ-BARRAL20, SANNA-
KAISA HERUKKA12, ANDRE HUSS1, ANDREAS JEROMIN5, JANINE KIRBY13,
MAGDALENA KUZMA-KOZAKIEWICZ14, MARIA DEL MAR AMADOR3,
JESUS S. MORA20, CLAUDIA MORELLI15, PETRA MUCKOVA16, SUSANNE PETRI17,
KOEN POESEN18, HEIDRUN RHODE16, ANNA-KARIN RIKARDSSON4,
WIM ROBBERECHT19, ANA I. RODRIGUEZ MAHILLO20, PAMELA SHAW13,
VINCENZO SILANI15,22, PETRA STEINACKER1, MARTIN R. TURNER10,
ERDEM TUZUN21, BERRAK YETIMLER21, ALBERT C. LUDOLPH1 & MARKUS OTTO1
1Department of Neurology, Ulm University Hospital, 89081 Ulm, Germany; 2Department of Metabolic
Biochemistry, Hopitaux universitaires Pitie Salpetriere-Charles Foix, 75651 Paris, France, 3Paris ALS Reference
Center, Neurological Diseases Department, Hopitaux universitaires La Pitie Salpetriere-Charles Foix, 75651 Paris,
France, 4Department of Neurology, University of Umea, 90185 Umea, Sweden, 5Iron Horse Diagnostics, Inc.,
85255 Scottsdale, Arizona, USA, 6Divisions of Neurology and Neurobiology, Barrow Neurological Institute, 85013
Phoenix, Arizona, USA, 7Faculty of Medicine - Instituto de Medicina Molecular, University of Lisbon, 1649-028
Lisbon, Portugal, 8Instituto de Tecnologia Quımica e Biologica Antonio Xavier, Universidade Nova de Lisboa,
2780-157 Oeiras, Portugal, 9University Hospitals Leuven, Department of Neurology, 3000 Leuven, Belgium,10Nuffield Department of Clinical Neurosciences, University of Oxford, OX3 9DU Oxford, UK, 11Department of
Neurology, Jena University Hospital, 07747 Jena, Germany, 12Department of Neurology, University of Eastern
Finland and Kuopio University Hospital, 70211 Kuopio, Finland, 13Department of Neuroscience, Sheffield Institute
for Translational Neuroscience, University of Sheffield, S10 2HQ Sheffield, UK, 14Department of Neurology,
Medical University of Warsaw, 02-097 Warsaw, Poland, 15IRCCS Istituto Auxologico Italiano, Department of
Neurology and Laboratory of Neuroscience, 20149 Milano, Italy, 16Institute of Biochemistry I, Jena University
Hospital, 07743 Jena, Germany, 17Department of Neurology, Hannover Medical School, 30625 Hannover,
Germany, 18Laboratory of molecular neurobiomarker research, University of Leuven and Laboratory Medicine,
University Hospitals of Leuven, 3000 Leuven, Belgium, 19KU Leuven - University of Leuven, Department of
Neurosciences, VIB - Vesalius Research Center, Experimental Neurology - Laboratory of Neurobiology, Leuven,
Belgium, 20ALS Unit, Hospital Carlos III, Madrid, 28029 Madrid, Spain, 21Neuroscience Department, Institute
of Experimental Medical Research, Istanbul University, 34393 Istanbul, Turkey, and 22Department ‘‘Dino Ferrari’’
Center, Department of Pathophysiology and Transplantation, Universita degli Studi di Milano, 20122 Milano, Italy
Abstract
OBJECTIVE: Neurofilaments are leading neurochemical biomarkers for amyotrophic lateral sclerosis (ALS). Here, weinvestigated the effect of preanalytical factors on neurofilament concentrations in cerebrospinal fluid (CSF) in a ‘‘reverse’’round-robin with 15 centers across Europe/U.S. METHODS: Samples from ALS and control patients (5/5 each center,n¼ 150) were analyzed for phosphorylated neurofilament heavy chain (pNfH) and neurofilament light chain (NfL) at twolaboratories. RESULTS: CSF pNfH was increased (p50.05) in ALS in 10 out of 15 centers and NfL in 5 out of 12 centers.The coefficient of variation (CV%) of pNfH measurements between laboratories was 18.7� 19.1%. We calculated adiagnostic cut-off of4568.5 pg/mL for pNfH (sensitivity 78.7%, specificity 93.3%) and41,431pg/mL for NfL (sensitivity
Correspondence: Prof. Dr. med. Markus Otto, Ulm University Hospital, Department of Neurology, Oberer Eselsberg 45, D-89081 Ulm,
Tel: +49-731-500-63010, Fax: +49-731-500-63012, E-Mail: [email protected]
(Received 13 January 2016; revised 4 February 2016; accepted 12 February 2016)
ISSN 2167-8421 print/ISSN 2167-9223 online � 2016 World Federation of Neurology on behalf of the Research Group on Motor Neuron Diseases
DOI: 10.3109/21678421.2016.1167913

CroniconO P E N A C C E S S EC NEUROLOGY
Research Article
The Compound Muscle Action Potential as Neurophysiological Marker for Amyotrophic Lateral Sclerosis
Emanuela Onesti1, Maria Cristina Gori1, Marco Ceccanti1, Giorgio Tartaglia1, Antonio Petrucci2, Vittorio Frasca1, Vincenzo Silani3, and Maurizio Inghilleri1*
1Department of Neurology and Psychiatry, Sapienza University, Rome, Italy2Center of Neuromuscular and Neurological Rare Diseases, S. Camillo-Forlanini Hospital, Rome, Italy3Department of Neurology and Laboratory of Neuroscience, IRCCS Istituto Auxologico Italiano - Department of Pathophysiology and Tranplantation, “Dino Ferrari” Center, University of Milan, Milan, Italy
*Corresponding Author: Maurizio Inghilleri, Department of Neurology and Psychiatry, Viale dell’Università 30 - 00185 Rome, University of Rome “Sapienza”, Italy.
Citation: Maurizio Inghilleri., et al. “The Compound Muscle Action Potential as Neurophysiological Marker for Amyotrophic Lateral Sclerosis”. EC Neurology 3.6 (2016): 509-519.
Received: November 18, 2016; Published: November 26, 2016
Abstract
Objectives: To definite the peripheral nervous involvement in ALS through the repeated use of the compound motor action potential (CMAP) to test the progression of disease, to determine different change of phrenic CMAP and forced vital capacity (FVC) in spinal and bulbar onset, and to establish clinical and neurophysiological features of patients with poor prognosis.
Material & Methods: CMAP from phrenic, ulnar, and medial plantar nerves, Medical Research Council (MRC) score, revised ALS functional rating scale (ALSFRS-R) and FVC were evaluated in 117 ALS patients every three months in one year-period.
Results: Bulbar onset patients had lower FVC but similar amplitude of phrenic CMAP at baseline compared to spinal onset patients. The patients with poor prognosis had lower phrenic CMAP and FVC at baseline. CMAP values, when compared to the rate found in the previous visit, reduced significantly in both poor and good prognosis groups during the entire follow-up period, while the FVC reduced significantly only in the first three months.
Conclusions: CMAP is a reproducible sensitive marker for motor neurons loss and collateral reinnervation in ALS also in a short period of time. The changes in CMAP, MRC, FVC and ALSFRS-R score resulted correlated, but CMAP is the only parameter with the advantage to demonstrate objectively the progression of disease in both patients with poor and good prognosis for the entire period of follow-up. It should be used as clinical outcome of ALS in clinical trials, taking advantage of its objectivity and selectivity for pe-ripheral nervous system study.
Keywords: Amyotrophic Lateral Sclerosis: ALS; Compound Muscle Action Potential: CMAP; Neurophysiological Marker
Introduction
Amyotrophic lateral sclerosis (ALS) is the most frequent adult-onset motor neuron disease characterized by the progressive loss of upper and lower motor neurons (MNs). The death occurs usually on few years after the symptoms onset [1].
Given the lack of biomarkers, diagnosis of ALS is supported by clinical and electrophysiological findings after the exclusion of other

RESEARCH Open Access
Gene-specific mitochondria dysfunctions inhuman TARDBP and C9ORF72 fibroblastsElisa Onesto1, Claudia Colombrita1,2, Valentina Gumina1,2, Maria Orietta Borghi3,4, Sabrina Dusi5, Alberto Doretti1,2,Gigliola Fagiolari6, Federica Invernizzi5, Maurizio Moggio6, Valeria Tiranti5, Vincenzo Silani1,2 and Antonia Ratti1,2*
Abstract
Dysregulation of RNA metabolism represents an important pathogenetic mechanism in both amyotrophic lateralsclerosis (ALS) and frontotemporal dementia (FTD) due to the involvement of the DNA/RNA-binding proteins TDP-43and FUS and, more recently, of C9ORF72. A potential link between dysregulation of RNA metabolism and mitochondrialdysfunction is recently emerged in TDP-43 disease models. To further investigate the possible relationship between thesetwo pathogenetic mechanisms in ALS/FTD, we studied mitochondria functionality in human mutant TARDBP(p.A382T)and C9ORF72 fibroblasts grown in galactose medium to induce a switch from a glycolytic to an oxidative metabolism. Inthis condition we observed significant changes in mitochondria morphology and ultrastructure in both mutant cellswith a fragmented mitochondria network particularly evident in TARDBP(p.A382T) fibroblasts. From analysis of themitochondrial functionality, a decrease of mitochondria membrane potential with no alterations in oxygenconsumption rate emerged in TARDBP fibroblasts. Conversely, an increased oxygen consumption and mitochondriahyperpolarization were observed in C9ORF72 fibroblasts in association to increased ROS and ATP content. We foundevidence of autophagy/mitophagy in dynamic equilibrium with the biogenesis of novel mitochondria, particularly inmutant C9ORF72 fibroblasts where an increase of mitochondrial DNA content and mass, and of PGC1-α protein wasobserved. Our imaging and biochemical data show that wild-type and mutant TDP-43 proteins do not localize atmitochondria so that the molecular mechanisms responsible for such mitochondria impairment remain to be furtherelucidated. For the first time our findings assess a link between C9ORF72 and mitochondria dysfunction and indicatethat mitochondria functionality is affected in TARDBP and C9ORF72 fibroblasts with gene-specific features in oxidativeconditions. As in neuronal metabolism mitochondria are actively used for ATP production, we speculate that TARDBPand C9ORF72 mutations might trigger cell death by impairing not only RNA metabolism, but also mitochondria activityin ALS/FTD neurons.
Keywords: ALS, FTD, TDP-43, C9ORF72, Fibroblast, Mitochondria dysfunction
IntroductionAbnormal aggregates of TDP-43 protein represent aneuropathological hallmark of the fatal motoneuron dis-ease amyotrophic lateral sclerosis (ALS) and of fronto-temporal dementia (FTD). TDP-43 forms pathologicalcytoplasmic inclusions in affected brain tissues of nearlyall sporadic and familial ALS cases, with the exception ofSOD1-associated forms, and of a subset of patients withFTD (FTD-TDP), representing an important molecular
link between these two apparently different neurodegener-ative disorders [29]. TDP-43 is a DNA/RNA bindingprotein, mainly localized in the nucleus where it is in-volved in regulating splicing, transcription and miRNAbiogenesis, but it has also regulatory functions in thecytoplasm controlling target mRNA stability, transportand translation [26]. The presence of abnormal TDP-43aggregates in the cytoplasm together with the concur-rent depletion of TDP-43 from the nucleus suggest thatTDP-43 may trigger neuronal death by two differentpathogenetic mechanisms including a toxic gain-of-function and/or a loss-of-nuclear function. Independ-ently of the pathogenetic mechanism of TDP-43, RNAmetabolism is strongly impaired at different levels in
* Correspondence: [email protected] of Neurology and Laboratory of Neuroscience, IRCCS IstitutoAuxologico Italiano, Via Zucchi, 18, Cusano Milanino 20095, MI, Italy2Department of Pathophysiology and Transplantation, ‘Dino Ferrari’ Center -Università degli Studi di Milano, Milan, ItalyFull list of author information is available at the end of the article
© 2016 Onesto et al. Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, andreproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link tothe Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver(http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
Onesto et al. Acta Neuropathologica Communications (2016) 4:47 DOI 10.1186/s40478-016-0316-5

Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration, 2016; 17: 473–481
RESEARCH ARTICLE
Cognitive assessment in Amyotrophic Lateral Sclerosis by means of
P300-Brain Computer Interface: a preliminary study
BARBARA POLETTI1*, LAURA CARELLI1
*, FEDERICA SOLCA1,
ANNALISA LAFRONZA1, ELISA PEDROLI2, ANDREA FAINI3, STEFANO ZAGO4,
NICOLA TICOZZI1,5, PAOLO MERIGGI6, PIETRO CIPRESSO2, DOROTHEE LULE7,
ALBERT C. LUDOLPH7, GIUSEPPE RIVA2,8 & VINCENZO SILANI1,5
1Department of Neurology and Laboratory of Neuroscience – IRCCS Istituto Auxologico Italiano, Milan, Italy,2Applied Technology for Neuro-Psychology Lab, IRCCS Istituto Auxologico Italiano, Milan, Italy, 3Department of
Cardiovascular, Neural and Metabolic Sciences – IRCCS Istituto Auxologico Italiano, Milan, Italy, 4Department of
Neuroscience and Mental Health, Universita degli Studi di Milano, IRCCS Fondazione Ca’ Granda Ospedale
Maggiore Policlinico, Milan, Italy, 5Department of Pathophysiology and Transplantation, ‘‘Dino Ferrari’’ Center,
Universita degli Studi di Milano, Milan, Italy, 6ICT & Biomedical Technology Integration Unit, Centre for
Innovation and Technology Transfer (CITT), Fondazione Don Carlo Gnocchi Onlus, Milan, Italy, 7Department of
Neurology – University of Ulm, Ulm, Germany, and 8Department of Psychology, Catholic University of Milan,
Milan, Italy
Abstract
Objective: To investigate the use of P300-based Brain Computer Interface (BCI) technology for the administration ofmotor-verbal free cognitive tests in Amyotrophic Lateral Sclerosis (ALS). Methods: We recruited 15 ALS patients and 15age- and education-matched healthy subjects. All participants underwent a BCI-based neuropsychological assessment,together with two standard cognitive screening tools (FAB, MoCA), two psychological questionnaires (BDI, STAI-Y) anda usability questionnaire. For patients, clinical and respiratory examinations were also performed, together with abehavioural assessment (FBI). Results: Correlations were observed between standard cognitive and BCI-based neuropsy-chological assessment, mainly concerning execution times in the ALS group. Moreover, patients provided positive ratesconcerning the BCI perceived usability and subjective experience. Finally, execution times at the BCI-basedneuropsychological assessment were useful to discriminate patients from controls, with patients achieving lower processingspeed than controls regarding executive functions. Conclusions: The developed motor-verbal free neuropsychologicalbattery represents an innovative approach, that could provide relevant information for clinical practice and ethical issues.Its use for cognitive evaluation throughout the course of ALS, currently not available by means of standard assessment,must be addressed in further longitudinal validation studies. Further work will be aimed at refining the developed systemand enlarging the cognitive spectrum investigated.
Keywords: P300 Brain Computer Interface, Amyotrophic Lateral Sclerosis, motor-verbal free cognitive assessment
Introduction
Cognitive and behavioural changes in patients with
Amyotrophic Lateral Sclerosis (ALS) are now
recognized as an integral feature of the disease,
with the most commonly reported alterations
regarding executive functions (1). Besides, the
presence of a spectrum of frontotemporal impair-
ment in ALS has been highlighted (1,2), from
mild-to-moderate cognitive-behavioural changes
to frontotemporal dementia (FTD) (3).
Neuropathological and genetic findings have sup-
ported the link between FTD and ALS (4,5).
Recent studies have also described significant
changes in language (6), memory (7) and social
cognition (8), outlying cognitive impairment in ALS
as an heterogeneous feature. Evidence suggests the
need for some task modifications in order to make
*These authors contributed equally to this work.
Correspondence: Barbara Poletti, PhD, Department of Neurology and Laboratory of Neuroscience, IRCCS Istituto AuxologicoItaliano, Piazzale Brescia, 20 - 20149 - Milan, Italy. Tel: +39 02 619112609. Fax: +39 02 619112038. E-mail:[email protected]
(Received 19 October 2015; revised 9 March 2016; accepted 13 March 2016)
ISSN 2167-8421 print/ISSN 2167-9223 online � 2016 World Federation of Neurology on behalf of the Research Group on Motor Neuron Diseases
DOI: 10.1080/21678421.2016.1181182

Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration, 2016; 17: 489–498
RESEARCH ARTICLE
The validation of the Italian Edinburgh Cognitive and Behavioural
ALS Screen (ECAS)
BARBARA POLETTI1*, FEDERICA SOLCA1
*, LAURA CARELLI1,
FABIANA MADOTTO2, ANNALISA LAFRONZA1, ANDREA FAINI3, ALESSIA MONTI4,
STEFANO ZAGO5, DANIELA CALINI1, CINZIA TILOCA1, ALBERTO DORETTI1,6,
FEDERICO VERDE1,6, ANTONIA RATTI1,6, NICOLA TICOZZI1,6,
SHARON ABRAHAMS7* & VINCENZO SILANI1,6
*
1Department of Neurology and Laboratory of Neuroscience - IRCCS Istituto Auxologico Italiano, Milan, Italy,2Research Centre on Public Health, Department of Medicine and Surgery, University of Milano-Bicocca, Milan,
Italy, 3Department of Cardiovascular, Neural and Metabolic Sciences - IRCCS Istituto Auxologico Italiano, Milan,
Italy, 4Department of Neurorehabilitation Sciences, Casa Cura Policlinico, Milan, Italy, 5Department of
Neuroscience and Mental Health, Universita degli Studi di Milano, IRCCS Fondazione Ca’ Granda Ospedale
Maggiore Policlinico, Milan, Italy, 6Department of Pathophysiology and Transplantation, ‘Dino Ferrari’ Centre,
Universita degli Studi di Milano, Milan, Italy, and 7Euan MacDonald Centre for Motor Neurone Disease
Research, Anne Rowling Regenerate Neurology Clinic, Centre for Cognitive Ageing and Cognitive Epidemiology,
Human Cognitive Neuroscience-PPLS, University of Edinburgh, Edinburgh, UK
Abstract
This study presents the Italian validation of the recently developed Edinburgh Cognitive and Behavioural ALS Screen(ECAS), a short screen for cognitive/behavioural alterations in patients with amyotrophic lateral sclerosis (ALS). Weevaluated the psychometric properties of the ECAS Italian version in terms of reliability and convergent validity for bothcognitive and behavioural features. Furthermore, we investigated the relationship with affective and clinical variables, inaddition to ECAS usability and patients’ insight into cognitive/behaviour changes. Finally, correlations between genetic andcognitive/behavioural data were analysed. We recruited 107 patients with ALS. Normative data were collected on 248healthy subjects. Participants were administered the ECAS and two standard cognitive screening tools (FAB, MoCA), twopsychological questionnaires (BDI, STAI/Y) and an ad hoc usability questionnaire. The FBI was also carried out withcaregivers. Results showed that the ECAS Italian version discriminated well between patients and controls. The mostprevalent deficit occurred in executive functions and fluency. Correlations were observed between the ECAS and standardcognitive screening tools and between the ECAS carer interview and the FBI, supporting its full convergent validity. Inconclusion, the ECAS Italian version provides clinicians with a rapid, feasible and sensitive tool, useful to identify differentprofiles of cognitive-behavioural impairment in ALS.
Key words: Amyotrophic lateral sclerosis (ALS), cognitive assessment, behaviour change, ECAS
Introduction
Over the last 20 years, evidence has been consist-
ently demonstrated of alterations in cognition and
behaviour, particularly in frontal-executive abilities,
in amyotrophic lateral sclerosis (ALS) (1). Recently,
several studies have highlighted a broader cognitive
involvement in ALS (2,3) concerning language (4)
and social cognition (5–7), possibly underestimated
due to the absence of standardized methodologies
used in many studies (8). Some screening assess-
ment tools have been presented for ALS (9,10), even
if not designed to detect heterogeneous cognitive
involvement nor compensating for physical disability.
In order to overcome these limitations, Abrahams
et al. (11) recently developed a rapid cognitive-
behavioural screening tool (Edinburgh Cognitive and
Behavioural ALS Screen – ECAS), specifically
designed for ALS. It includes a range of tests sensitive
Correspondence: B. Poletti, Department of Neurology and Laboratory of Neuroscience, IRCCS Istituto Auxologico Italiano, Piazzale Brescia, 20 - 20149 -
Milan, Italy. Fax: 39 02 619112937. E-mail: [email protected]
*Poletti and Solca are equally contributing authors, as are Abrahams and Silani equally contributing authors.
(Received 7 December 2015; revised 18 March 2016; accepted 28 March 2016)
ISSN 2167-8421 print/ISSN 2167-9223 online � 2016 World Federation of Neurology on behalf of the Research Group on Motor Neuron Diseases
DOI: 10.1080/21678421.2016.1183679

Copyright 2016 American Medical Association. All rights reserved.
Association of a Locus in the CAMTA1 Gene With Survivalin Patients With Sporadic Amyotrophic Lateral SclerosisIsabella Fogh, PhD; Kuang Lin, PhD; Cinzia Tiloca, PhD; James Rooney, MD; Cinzia Gellera, PhD; Frank P. Diekstra, MD; Antonia Ratti, PhD;Aleksey Shatunov, PhD; Michael A. van Es, MD, PhD; Petroula Proitsi, PhD; Ashley Jones, PhD; William Sproviero, PhD; Adriano Chiò, MD;Russell Lewis McLaughlin, PhD; Gianni Sorarù, MD, PhD; Lucia Corrado, PhD; Daniel Stahl, PhD; Roberto Del Bo, PhD; Cristina Cereda, PhD;Barbara Castellotti, PhD; Jonathan D. Glass, MD; Steven Newhouse, PhD; Richard Dobson, PhD; Bradley N. Smith, PhD; Simon Topp, MSc;Wouter van Rheenen, MD; Vincent Meininger, MD, PhD; Judith Melki, MD, PhD; Karen E. Morrison, MD; Pamela J. Shaw, MD;P. Nigel Leigh, MD, PhD; Peter M. Andersen, MD, DMSc; Giacomo P. Comi, MD; Nicola Ticozzi, MD; Letizia Mazzini, MD;Sandra D’Alfonso, PhD; Bryan J. Traynor, MD; Philip Van Damme, MD, PhD; Wim Robberecht, MD, PhD; Robert H. Brown, MD, DPhil;John E. Landers, PhD; Orla Hardiman, MD, FRCPI; Cathryn M. Lewis, PhD; Leonard H. van den Berg, MD, PhD; Christopher E. Shaw, MD;Jan H. Veldink, MD, PhD; Vincenzo Silani, MD; Ammar Al-Chalabi, PhD, FRCP; John Powell, PhD
IMPORTANCE Amyotrophic lateral sclerosis (ALS) is a devastating adult-onsetneurodegenerative disorder with a poor prognosis and a median survival of 3 years. However,a significant proportion of patients survive more than 10 years from symptom onset.
OBJECTIVE To identify gene variants influencing survival in ALS.
DESIGN, SETTING, AND PARTICIPANTS This genome-wide association study (GWAS) analyzedsurvival in data sets from several European countries and the United States that werecollected by the Italian Consortium for the Genetics of ALS and the International Consortiumon Amyotrophic Lateral Sclerosis Genetics. The study population included 4256 patients withALS (3125 [73.4%] deceased) with genotype data extended to 7 174 392 variants byimputation analysis. Samples of DNA were collected from January 1, 1993, to December 31,2009, and analyzed from March 1, 2014, to February 28, 2015.
MAIN OUTCOMES AND MEASURES Cox proportional hazards regression under an additivemodel with adjustment for age at onset, sex, and the first 4 principal components of ancestry,followed by meta-analysis, were used to analyze data. Survival distributions for the mostassociated genetic variants were assessed by Kaplan-Meier analysis.
RESULTS Among the 4256 patients included in the analysis (2589 male [60.8%] and 1667female [39.2%]; mean [SD] age at onset, 59 [12] years), the following 2 novel loci weresignificantly associated with ALS survival: at 10q23 (rs139550538; P = 1.87 × 10−9) and inthe CAMTA1 gene at 1p36 (rs2412208, P = 3.53 × 10−8). At locus 10q23, the adjusted hazardratio for patients with the rs139550538 AA or AT genotype was 1.61 (95% CI, 1.38-1.89;P = 1.87 × 10−9), corresponding to an 8-month reduction in survival compared with TTcarriers. For rs2412208 CAMTA1, the adjusted hazard ratio for patients with the GG or GTgenotype was 1.17 (95% CI, 1.11-1.24; P = 3.53 × 10−8), corresponding to a 4-month reductionin survival compared with TT carriers.
CONCLUSIONS AND RELEVANCE This GWAS robustly identified 2 loci at genome-wide levelsof significance that influence survival in patients with ALS. Because ALS is a rare disease andprevention is not feasible, treatment that modifies survival is the most realistic strategy.Therefore, identification of modifier genes that might influence ALS survival could improvethe understanding of the biology of the disease and suggest biological targets forpharmaceutical intervention. In addition, genetic risk scores for survival could be used asan adjunct to clinical trials to account for the genetic contribution to survival.
JAMA Neurol. 2016;73(7):812-820. doi:10.1001/jamaneurol.2016.1114Published online May 31, 2016.
Editorial page 781
Supplemental content atjamaneurology.com
Author Affiliations: Authoraffiliations are listed at the end of thisarticle.
Corresponding Author: IsabellaFogh, PhD, Department of Basic andClinical Neuroscience, Maurice WohlClinical Neuroscience Institute,Institute of Psychiatry, Psychology,and Neuroscience, King’s CollegeLondon, James Black Centre,125 Coldharbour Ln, London SE59NU, England ([email protected]).
Research
Original Investigation
812 (Reprinted) jamaneurology.com
Copyright 2016 American Medical Association. All rights reserved.
Downloaded From: http://jamanetwork.com/pdfaccess.ashx?url=/data/journals/neur/935408/ by a Bib IRCCS Fond Ist Auxologico Italiano - Milano User on 01/17/2017

Copyright 2016 American Medical Association. All rights reserved.
Conclusions
We have identified genetic variants that have a statistically sig-nificant association with survival. The promise of this re-
search is not only to improve our understanding of the biol-ogy of the disease and suggest biological targets forpharmaceutical intervention to extend the survival time of thepatients but also to use genetic risk scores as an adjunct to clini-cal trials to account for the genetic contribution to survival.
ARTICLE INFORMATION
Accepted for Publication: March 17, 2016.
Published Online: May 31, 2016.doi:10.1001/jamaneurol.2016.1114.
Author Affiliations: Department of Basic andClinical Neuroscience, Maurice Wohl ClinicalNeuroscience Institute, Institute of Psychiatry,Psychology, and Neuroscience (IoPPN), King’sCollege London, London, England (Fogh, Lin,Shatunov, Proitsi, Jones, Sproviero, Smith, Topp,C. E. Shaw, Al-Chalabi, Powell); Department ofNeurology and Laboratory of Neuroscience, Istitutodi Ricovero e Cura a Carattere Scientifico (IRCCS)Istituto Auxologico Italiano, Milano, Italy (Tiloca,Ratti, Ticozzi, Silani); Academic Unit of Neurology,Trinity College Dublin, Trinity Biomedical SciencesInstitute, Dublin, Ireland (Rooney); Unit of Geneticsof Neurodegenerative and Metabolic Diseases,Fondazione IRCCS Istituto Neurologico Carlo Besta,Milano, Italy (Gellera, Castellotti); Department ofNeurology and Neurosurgery, Brain Center RudolfMagnus, University Medical Center Utrecht,Utrecht, the Netherlands (Diekstra, van Es, vanRheenen, van den Berg, Veldink); Department ofPathophysiology and Tranplantation, Dino FerrariCenter, Università degli Studi di Milano, Milano, Italy(Ratti, Ticozzi, Silani); Rita Levi MontalciniDepartment of Neuroscience, ALS (AmyotrophicLateral Sclerosis) Centre, University of Torino, Turin,Italy (Chiò); Azienda Ospedaliera Città della Salutee della Scienza, Torino, Italy (Chiò); PopulationGenetics Laboratory, Smurfit Institute of Genetics,Trinity College Dublin, Dublin, Ireland (McLaughlin,Hardiman); Department of Neurosciences,University of Padova, Padua, Italy (Sorarù);Department of Health Sciences, InterdisciplinaryResearch Center of Autoimmune Diseases,A. Avogadro University, Novara, Italy (Corrado,Mazzini, D’Alfonso); Department of Biostatistics,IoPPN, King’s College London, London, England(Stahl); Neurologic Unit, IRCCS Foundation Ca’Granda Ospedale Maggiore Policlinico, Milan, Italy(Del Bo, Comi); Laboratory of ExperimentalNeurobiology, IRCCS C. Mondino National Instituteof Neurology Foundation, Pavia, Italy (Cereda);Department of Neurology, Emory University,Atlanta, Georgia (Glass); National Institute forHealth Research (NIHR) Biomedical ResearchCentre for Mental Health, IoPPN, King’s CollegeLondon, London, England (Newhouse, Dobson);Department of Biostatistics, IoPPN, King’s CollegeLondon, London, England (Newhouse); NIHRBiomedical Research Unit in Dementia, King’sCollege London, London, England (Dobson);Département des Maladies du Système Nerveux,Assistance Publique–Hôpitaux de Paris, Réseau SLA(Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); InstitutNational de la Santé et de la Recherche MedicaleUnité Mixte de Recherché–788 and University ofParis 11, Bicetre Hospital, Paris, France (Melki);School of Clinical and Experimental Medicine,College of Medicine and Dentistry, University ofBirmingham, Birmingham, England (Morrison);
Neurosciences Division, University HospitalsBirmingham National Health Service FoundationTrust, Birmingham, England (Morrison); AcademicNeurology Unit, Department of Neuroscience,Faculty of Medicine, Dentistry and Health,University of Sheffield, Sheffield, England(P. J. Shaw); Section of Neurology, Division ofMedicine, Brighton and Sussex Medical School,Trafford Centre for Biomedical Research, Universityof Sussex, East Sussex, England (Leigh); Institute ofClinical Molecular Biology, Kiel University, Kiel,Germany (Andersen); Department ofPharmacology and Clinical Neuroscience, UmeåUniversity, Umeå, Sweden (Andersen); ALS CenterDepartment of Neurology, Maggiore della CaritàUniversity Hospital, Novara, Italy (Mazzini);Neuromuscular Diseases Research Section,Laboratory of Neurogenetics, National Institute onAging, National Institutes of Health, Bethesda,Maryland (Traynor); Department of Neurosciences,Experimental Neurology, Flanders Instititue forBiotechnology, Vesalius Research Center,Laboratory of Neurobiology, KU Leuven–Universityof Leuven, Leuven, Belgium (Van Damme,Robberecht); Department of Neurology, UniversityHospitals Leuven, Leuven, Belgium (Van Damme);Department of Neurology, University ofMassachusetts Medical School, Worcester (Brown,Landers); IoPPN Genomics and Biomarker Core,Translational Genetics Group, Medical ResearchCouncil Social, Genetic and DevelopmentalPsychiatry Centre, King’s College London, London,England (Lewis); Department of Medical andMolecular Genetics, King’s College London, London,England (Lewis).
Author Contributions: Drs Silani, Al-Chalabi, andPowell are cosenior authors. Drs Fogh and Powellhad full access to all the data in the study and takeresponsibility for the integrity of the data and theaccuracy of the data analysis.Study concept and design: Fogh, Landers,Al-Chalabi, Powell.Acquisition, analysis, or interpretation of data: Allauthors.Drafting of the manuscript: Fogh, Tiloca, Ratti,Al-Chalabi, Powell.Critical revision of the manuscript for importantintellectual content: Lin, Rooney, Gellera, Diekstra,Ratti, Shatunov, van Es, Proitsi, Jones, Sproviero,Chiò, McLaughlin, Sorarù, Corrado, Stahl, Del Bo,Cereda, Castellotti, Glass, Newhouse, Dobson,Smith, Topp, van Rheenen, Meininger, Melki,Morrison, P. J. Shaw, Leigh, Andersen, Comi,Ticozzi, Mazzini, D’Alfonso, Traynor, Van Damme,Robberecht, Brown, Landers, Hardiman, Lewis,van den Berg, C. E. Shaw, Veldink, Silani, Al-Chalabi.Statistical analysis: Fogh, Lin, Rooney, Proitsi, Stahl,Newhouse, Landers, Lewis.Obtained funding: Fogh, Chiò, P. J. Shaw, Ticozzi,D’Alfonso, Van Damme, Robberecht, van den Berg,Al-Chalabi.Administrative, technical, or material support:Gellera, Diekstra, Ratti, van Es, Jones, Del Bo,Castellotti, Smith, Meininger, Morrison, Shaw,Andersen, Ticozzi, Traynor, Van Damme,
Robberecht, Landers, Hardiman, van den Berg,C. E. Shaw, Veldink, Al-Chalabi.Study supervision: Newhouse, Lewis, Silani,Al-Chalabi, Powell.
Conflict of Interest Disclosures: Dr Traynorreports having a patent pending on the clinicaltesting and therapeutic intervention for thehexanucleotide repeat expansion of C9orf72.No other disclosures were reported.
Funding/Support: This study was supported bygrant 905-793 6058 from the Motor NeuroneDisease Association of Great Britain and NorthernIreland and by Emergency Medicine Clinical Trialspilot funding awards from the National Institutes forHealth Research (NIHR) Biomedical Research Centrefor Mental Health at the South London and MaudsleyNational Health Service Foundation Trust andInstitute of Psychiatry, King’s College London(Dr Fogh); by grant NOVALS 2012 from the AgenziaItaliana per la Ricerca sulla Fondazione Italiana diRicerca per la Sclerosi Laterale Amiotrofica(SLA-AriSLA), cofinanced with the contribution of5 × 1000 Healthcare Research support of theMinistry of Health, Ricerca Finalizzata RF-2009-1473856 from the Ministero della Salute andAssociazione Amici Centro Dino Ferrari (Drs Tiloca,Gellera, Ratti, Ticozzi, and Silani); by the HealthResearch Board Clinical Fellowship Programme (DrRooney); by Ricerca Finalizzata RF-2010-2309849from the Ministero della Salute and FP7/2007-2013under grant 259867 from the European Community’sHealth Seventh Framework Programme (Dr Chiò);by AriSLA for the Repeat ALS Project 2013 (DrD’Alfonso); by the Netherlands Organization forHealth Research and Development (Drs Diekstra,van Es [Veni Scheme], van Rheenen, van den Berg[Vici Scheme], and Veldink); by the Thierry LatranFoundation, the Dutch ALS Foundation, and a talentfellowship from the Rudolf Magnus Brain Center (Drvan Es); by the Princess Beatrix Fonds, the PrincessBeatrix Fund Muscle, H. Kersten and M. KerstenFoundation, the Netherlands ALS Foundation, andDr van Dijk and the Adessium Foundation (Dutch,Belgian and Swedish genome-wide association studydata generation); by grant 259867 from theEuropean Community’s Health Seventh FrameworkProgramme; through the E. von Behring Chair forNeuromuscular and Neurodegenerative Disordersand Geneeskundige Stichting Koningin Elisabeth(GSKE) and grant 340429 from the EuropeanResearch Council under the European’s SeventhFramework Programme (Dr Robberecht); by grants259867 and 278611 from the InteruniversityAttraction Poles (IUAP) program P7/16 of the BelgianFederal Science Policy Office and the EuorpeanCommunity’s Seventh Framework Programme(ADAMS project, HEALTH-F4-2009-242257 andFP7/2007-2013); by a clinical investigatorship fromFonds Wetenschappelijk Onderzoek–Vlaanderen(Dr Van Damme); by the Motor Neurone DiseaseAssociation of Great Britain and Northern Ireland, theALS Foundation Netherlands (project MinE), and theEuropean Community’s Health Seventh FrameworkProgramme (Drs Al-Chalabi, Shatunov, Jones, andSproviero); by ZonMW under the framework of
CAMTA1 Gene and Survival in Sporadic Amyotrophic Lateral Sclerosis Original Investigation Research
jamaneurology.com (Reprinted) JAMA Neurology July 2016 Volume 73, Number 7 819
Copyright 2016 American Medical Association. All rights reserved.
Downloaded From: http://jamanetwork.com/pdfaccess.ashx?url=/data/journals/neur/935408/ by a Bib IRCCS Fond Ist Auxologico Italiano - Milano User on 01/17/2017

LETTER TO THE EDITORS
MRI abnormalities found 1 year prior to symptom onset in a caseof Creutzfeldt–Jakob disease
Federico Verde1,2• Nicola Ticozzi1,2
• Stefano Messina1• Narghes Calcagno2
•
Floriano Girotti1 • Luca Maderna1• Fabio Moda3
• Elisa Scola4• Andrea Falini5 •
Fabrizio Tagliavini3 • Vincenzo Silani1,2
Received: 28 October 2015 / Revised: 4 January 2016 / Accepted: 4 January 2016 / Published online: 12 February 2016
� Springer-Verlag Berlin Heidelberg 2016
Dear Sirs,
Creutzfeldt–Jakob disease (CJD) is a rare disease causing
rapidly progressive dementia, with an annual incidence of
1/1,000,000/year. It is caused by conformational change
and accumulation of the prion protein in the brain, resulting
in spongiform degeneration and producing the typical MRI
appearance of increased DWI signal in cerebral cortex and/
or basal ganglia [1, 2]. We describe a patient in whom
cortical DWI hyperintensity was evident 1 year prior to
symptom onset.
A 65-year-old woman with a history of hypertension and
thyroid disease underwent a neurological evaluation for
chronic tension-type headache in June 2013. Neurological
examination, including mental state evaluation, was nor-
mal. However, brain MRI showed DWI hyperintensity of
the right basal temporo-occipital cortex, with decreased
ADC signal (Fig. 1a, b). A diagnosis of cortical stroke was
made and antiplatelet therapy was recommended; other
conditions that could be suggested by the MRI findings,
namely encephalitis, vasculitis, sarcoidosis, posterior
reversible encephalopathy syndrome, seizures, and
hypoxic-ischemic encephalopathy were excluded as
inconsistent with history and clinical findings [3]. After
12 months in good health, in June 2014 the patient was
diagnosed with invasive ductal breast cancer (stage IIA,
pT1N1M0), which was surgically removed in July. Almost
concomitantly with the diagnosis of cancer, she developed
rapidly progressive mental deterioration, consisting of
forgetfulness, apathy, and difficulties with orientation and
in performing daily activities; removal of the neoplasm did
not influence progression of cognitive decline. She was
therefore admitted to our department in September 2014.
Neurological examination showed markedly reduced motor
and verbal initiative, severe ideomotor apraxia, visual
agnosia with simultanagnosia, and impairment of episodic
memory, attention, and abstract thinking. The remainder of
the examination was normal. Neuropsychological evalua-
tion demonstrated severe multidomain deterioration. Rou-
tine blood tests were unremarkable. Brain MRI showed
extension of the previously documented DWI abnormali-
ties, involving the basal temporo-occipital and parietal
cortex of both hemispheres (Fig. 1c, d). The absence of
contrast enhancement ruled out neoplastic infiltration of the
brain or meninges. Repeated EEG recordings displayed
worsening delta-wave slowing on the right hemisphere.
CSF analysis demonstrated normal protein and cells with-
out intrathecal IgG synthesis, and no autoantibodies (anti-
Hu, -Ri, -Ma1, -Ma2, -CV2, -amphiphysin, -NMDAR, -
VGKC, -thyroglobulin, or -thyroid peroxidase) were
detected in CSF or serum, arguing against inflammatory,
infectious, or paraneoplastic encephalitis. Conversely, CSF
tau protein was markedly elevated (4216 pg/mL, normal
range \400), and 14-3-3 protein was detected. Real-time
quaking-induced conversion (RT-QuIC) assay detected the
& Federico Verde
1 Department of Neurology and Laboratory of Neuroscience,
IRCCS Istituto Auxologico Italiano, Milan, Italy
2 Department of Pathophysiology and Transplantation, ‘‘Dino
Ferrari’’ Centre, University of Milan Medical School, Milan,
Italy
3 Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan,
Italy
4 Department of Neuroradiology, Fondazione IRCCS Ca’
Granda Ospedale Maggiore Policlinico, Milan, Italy
5 Department of Neuroradiology, San Raffaele Scientific
Institute and Vita-Salute San Raffaele University, Milan,
Italy
123
J Neurol (2016) 263:597–599
DOI 10.1007/s00415-016-8022-6

Cell Reports
Article
Reversal of Defective Mitochondrial Biogenesis inLimb-Girdle Muscular Dystrophy 2D by IndependentModulation of Histone and PGC-1a AcetylationSarah Pambianco,1,9 Matteo Giovarelli,1,9 Cristiana Perrotta,1 Silvia Zecchini,2 Davide Cervia,1,3 Ilaria Di Renzo,1
Claudia Moscheni,1 Michela Ripolone,4 Raffaella Violano,4 Maurizio Moggio,4 Maria Teresa Bassi,5 Pier Lorenzo Puri,6,7
Lucia Latella,6,8 Emilio Clementi,2,5,* and Clara De Palma2,10,*1Department of Biomedical and Clinical Sciences ‘‘Luigi Sacco,’’ Universita degli Studi di Milano, 20157 Milano, Italy2Department of Biomedical and Clinical Sciences, Unit of Clinical Pharmacology, University Hospital ‘‘Luigi Sacco’’-ASST Fatebenefratelli
Sacco, National Research Council-Institute of Neuroscience, Universita degli Studi di Milano, 20157 Milano, Italy3Department for Innovation in Biological, Agro-food and Forest systems, Universita degli Studi della Tuscia, 01100 Viterbo, Italy4Neuromuscular Unit, Dino Ferrari Centre, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Universita degli Studi di Milano,
20122 Milano, Italy5IRCCS Eugenio Medea, 23842 Bosisio Parini, Italy6Epigenetics and Regenerative Pharmacology, IRCCS Fondazione Santa Lucia, 00142 Roma, Italy7Sanford Children’s Health Research Center, Sanford Prebys Burnham Medical Discovery Institute, La Jolla, CA 92037, USA8National Research Council-Institute of Translational Pharmacology, 00179 Roma, Italy9Co-first author10Lead Contact
*Correspondence: [email protected] (E.C.), [email protected] (C.D.P.)
http://dx.doi.org/10.1016/j.celrep.2016.11.044
SUMMARY
Mitochondrial dysfunction occurs in many muscledegenerative disorders. Here, we demonstrate thatmitochondrial biogenesis was impaired in limb-girdlemuscular dystrophy (LGMD) 2D patients and miceand was associated with impaired OxPhos capacity.Two distinct approaches that modulated histones orperoxisome proliferator-activated receptor-gammacoactivator 1 a (PGC-1a) acetylation exerted equiva-lent functional effects by targeting different mito-chondrial pathways (mitochondrial biogenesis orfatty acid oxidation[FAO]). The histone deacetylaseinhibitor Trichostatin A (TSA) changed chromatinassembly at the PGC-1a promoter, restored mito-chondrial biogenesis, and enhanced muscle oxida-tive capacity. Conversely, nitric oxide (NO) triggeredpost translation modifications of PGC-1a andinduced FAO, recovering the bioenergetics impair-ment of muscles but shunting the defective mito-chondrial biogenesis. In conclusion, a transcriptionalblockade of mitochondrial biogenesis occurred inLGMD-2D and could be recovered by TSA changingchromatin conformation, or it could be overcome byNO activating a mitochondrial salvage pathway.
INTRODUCTION
Mitochondrial defects accompany several forms of muscular
dystrophies, genetic diseases characterized by progressive
skeletal muscle wasting, local inflammation, and compensatory
regeneration (Barton et al., 2005; Kornegay et al., 2014; Ruegg,
2013). While loss of functional dystrophin protein is the primary
cause of Duchenne muscular dystrophy (DMD), other patho-
genic events have recently been associated with DMD, including
decreased mitochondrial mass (Godin et al., 2012) and reduced
expression of enzymes involved in glycolytic and oxidativemeta-
bolism (Guevel et al., 2011; Percival et al., 2013; Rybalka et al.,
2014). Likewise, in limb-girdle muscular dystrophy 2D (LGMD-
2D), loss of a sarcoglycan (a-SG), an essential component of
the dystrophin-glycoprotein complex, is accompanied with
reduced transcription of nuclear-encoded mitochondrial genes
similar to that observed in DMD (Chen et al., 2000; Pescatori
et al., 2007). This evidence indicates that metabolic dysregula-
tion is a pathogenic event shared by muscular dystrophies in
which deficiency of a component of the dystrophin-associated
complex can impair signaling to the mitochondria.
An important aspect in dystrophic skeletal muscle including
LGMD-2D (Crosbie et al., 2002) is the displacement of the mus-
cle-specific variant of the enzyme neuronal nitric oxide (NO)
synthase (nNOSm), usually localized at the sarcolemma in close
contact with the sarcoglycan-dystroglycan complex, with
reduced generation of NO. NO in skeletal muscle regulates
metabolism and energy expenditure, coupling energy demand
and supply through a variety of actions (De Palma and Clementi,
2012; Stamler and Meissner, 2001). In addition, NO stimulates
mitochondrial biogenesis (Nisoli et al., 2003, 2007), regulates
mitochondrial dynamics (De Palma et al., 2010), and contributes
to muscle physiological growth and repair via several actions
including inhibition of histone deacetylases (HDACs) (Colussi
et al., 2008, 2009; Tidball and Wehling-Henricks, 2014). Normal-
ization of NO production slows disease progression in mouse
3010 Cell Reports 17, 3010–3023, December 13, 2016 ª 2016 The Authors.This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).