Embed Size (px)
Citation preview

holder Ns) that have no BLAST hit at 99% identity forfinished data and 95% identity for light-shotgun datawere considered uncovered. The percentage of eachclone not hit by Celera sequence was calculated bydividing the total length of the uncovered sequence bythe sequence length of the clone. The total number ofnucleotides that have no coverage in the Celera assem-bled contigs was calculated by summing the regions ofno hits for all the clones that covered Celera contigs byless than 90% (95% for finished clones). This cutoffvalue was chosen to eliminate the occasionally lowquality of sequences in the clone sequence data. Thecutoff value of 90% was determined by the amount ofno-hit sequences in 16 light-shotgun clones that arefully contained within three Celera contigs. A highercutoff value (95%) was used for the finished data thanfor the light-shotgun data, because finished clones havebetter sequence quality. The total amount of uncoveredsequence for each light-shotgun clones was calculatedby multiplying the no-hit percentage of the clone by theclone length as determined by sizing on agarose gels(36). For those light-shotgun clones with unreportedinsert sizes, the sequence length, excluding Ns, was used
instead. For finished clones, the amount of uncoveredsequence was calculated by multiplying the no-hit per-cent of the clone by the clone’s length. We created7-kbp subcontig blocks and considered each block to befully present in the draft sequence if it was hit by atleast 500 bp of external sequences. We chose theseparameters conservatively, based on the fact that at 13sequence coverage, the chance of failing to sample a7-kbp region covered by a light-shotgun clone is 1 in106. For the WGS assembly, we identified 1380 blocksthat were hit by less than 500 bp of clone sequence and794 blocks that were completely missed by the clonesequence. The total number of missed blocks is 2174,which represents a total 15.2 Mbp.
34. M. Ashburner et al., Genetics 153, 179 (1999).35. Seven conflicts were identified in this study, six of
which appear to be owing to transposable elements.The remaining represents a 30-kbp insert within aCelera contig that does not match the correspondingclone. This discrepancy is still under investigation.
36. www.sciencemag.org/feature/data/1049666.shl37. S. Altschul et al., Nucleic Acids. Res. 25, 3389 (1997).38. R. A. Hoskins, personal communication.
39. In order to align the Celera sequences unambiguouslyto the external data, all significant HSPs at the param-eters given in (27) were screened to identify “mutuallyunique regions” where the clone and contig sequenceshave a unique, reciprocal match relation.
40. Most negative gaps arise because of inaccuracies inthe distances implied by bundles—the bundle impliesa small amount of overlap between two contigsbecause it is actually short, whereas the reality is thatthere is a small gap at that location. In a very smallnumber of cases, there is an overlap, but it is becausethe distance estimate is too long by 3 standarddeviations, or because there is a small bit of foreignDNA at the tip of a contig because of untrimmedvector or a chimeric read. None of these negativegaps has yet been found to imply incorrect assembly.
41. We wish to thank H. Smith and S. Salzburg for themany collegial exchanges, M. Peterson and his teamfor keeping the machines humming, R. Thompson andhis staff for providing us with an environment con-ducive to such an intense effort, and A. Glodek, C.Kraft, and A. Deslattes Mays, and their staff forgetting the data to us.
R E V I E W
Comparative Genomics of the EukaryotesGerald M. Rubin,1 Mark D. Yandell,3 Jennifer R. Wortman,3 George L. Gabor Miklos,4 Catherine R. Nelson,2
Iswar K. Hariharan,5 Mark E. Fortini,6 Peter W. Li,3 Rolf Apweiler,7 Wolfgang Fleischmann,7 J. Michael Cherry,8
Steven Henikoff,9 Marian P. Skupski,3 Sima Misra,2 Michael Ashburner,7 Ewan Birney,7 Mark S. Boguski,10
Thomas Brody,11 Peter Brokstein,2 Susan E. Celniker,12 Stephen A. Chervitz,13 David Coates,14 Anibal Cravchik,3
Andrei Gabrielian,3 Richard F. Galle,12 William M. Gelbart,15 Reed A. George,12 Lawrence S. B. Goldstein,16
Fangcheng Gong,3 Ping Guan,3 Nomi L. Harris,12 Bruce A. Hay,17 Roger A. Hoskins,12 Jiayin Li,3 Zhenya Li,3
Richard O. Hynes,18 S. J. M. Jones,19 Peter M. Kuehl,20 Bruno Lemaitre,21 J. Troy Littleton,22
Deborah K. Morrison,23 Chris Mungall,12 Patrick H. O’Farrell,24 Oxana K. Pickeral,10 Chris Shue,3
Leslie B. Vosshall,25 Jiong Zhang,10 Qi Zhao,3 Xiangqun H. Zheng,3 Fei Zhong,3 Wenyan Zhong,3 Richard Gibbs,26
J. Craig Venter,3 Mark D. Adams,3 Suzanna Lewis2
A comparative analysis of the genomes of Drosophila melanogaster,Caenorhabditis elegans, and Saccharomyces cerevisiae—and the proteinsthey are predicted to encode—was undertaken in the context of cellular,developmental, and evolutionary processes. The nonredundant proteinsets of flies and worms are similar in size and are only twice that of yeast,but different gene families are expanded in each genome, and the mul-tidomain proteins and signaling pathways of the fly and worm are farmore complex than those of yeast. The fly has orthologs to 177 of the 289human disease genes examined and provides the foundation for rapidanalysis of some of the basic processes involved in human disease.
With the full genomic sequence of three ma-jor model organisms now available, much ofour knowledge about the evolutionary basisof cellular and developmental processes willderive from comparisons between protein do-mains, intracellular networks, and cell-cellinteractions in different phyla. In this paper,we begin a comparison of D. melanogaster,C. elegans, and S. cerevisiae. We first askhow many distinct protein families each ge-nome encodes, how the genes encoding theseprotein families are distributed in each ge-nome, and how many genes are shared amongflies, worms, yeast, and mammals. Next wedescribe the composition and organization ofprotein domains within the proteomes of fly,worm, and yeast and examine the representa-tion in each genome of a subset of genes thathave been directly implicated as causative
agents of human disease. Then we comparesome fundamental cellular and developmen-tal processes: the cell cycle, cell structure,cell adhesion, cell signaling, apoptosis, neu-ronal signaling, and the immune system. Ineach case, we present a summary of what wehave learned from the sequence of the flygenome and how the components that carryout these processes differ in other organisms.We end by presenting some observations onwhat we have learned, the obvious questionsthat remain, and how knowledge of the se-quence of the Drosophila genome will helpus approach new areas of inquiry.
The “Core Proteome”How many distinct protein families are en-coded in the genomes of D. melanogaster, C.elegans, and S. cerevisiae (1), and how do
these genomes compare with that of a simpleprokaryote, Haemophilus influenzae? Wecarried out an “all-against-all” comparison ofprotein sequences encoded by each genomeusing algorithms that aim to differentiateparalogs—highly similar proteins that occurin the same genome—from proteins that areuniquely represented (Table 1). Countingeach set of paralogs as a unit reveals the “coreproteome”: the number of distinct proteinfamilies in each organism. This operationaldefinition does not include posttranslationallymodifed forms of a protein or isoforms aris-ing from alternate splicing.
In Haemophilus, there are 1709 protein cod-ing sequences, 1247 of which have no sequencerelatives within Haemophilus (2). There are 178families that have two or more paralogs, yield-ing a core proteome of 1425. In yeast, there are6241 predicted proteins and a core proteome of4383 proteins. The fly and worm have 13,601and 18,424 (3) predicted protein-coding genes,and their core proteomes consist of 8065 and9453 proteins, respectively. It is remarkable thatDrosophila, a complex metazoan, has a coreproteome only twice the size of that of yeast.Furthermore, despite the large differences be-tween fly and worm in terms of developmentand morphology, they use a core proteome ofsimilar size.
24 MARCH 2000 VOL 287 SCIENCE www.sciencemag.org2204
T H E D R O S O P H I L A G E N O M E
on October 12, 2018
http://science.sciencem
ag.org/D
ownloaded from

Gene DuplicationsMuch of the genomes of flies and wormsconsists of duplicated genes; we next askedhow these paralogs are arranged. The fre-quency of local gene duplications and thenumber of their constituent genes differ wide-ly between fly and worm, although in bothgenomes most paralogs are dispersed. The flygenome contains half the number of localgene duplications relative to C. elegans (4),and these gene clusters are distributed ran-domly along the chromosome arms; in C.elegans there is a concentration of gene du-plications in the recombinogenic segments ofthe autosomal arms (1). In both organisms,approximately 70% of duplicated gene pairsare on the same strand (306 out of 417 for D.melanogaster and 581 out of 826 for C. el-egans). The largest cluster in the fly contains17 genes that code for proteins of unknownfunction; the next largest clusters both consist
of glutathione S-transferase genes, each with10 members. In contrast, 11 of 33 of thelargest clusters in C. elegans consist of genescoding for seven transmembrane domain re-ceptors, most of which are thought to beinvolved in chemosensation. Other than theselocal tandem duplications, genes with similarfunctional assignment in the Gene Ontology(GO) classification (5) do not appear to beclustered in the genome.
We next compared the large duplicated genefamilies in fly, worm, and yeast without regardto genomic location. All of the known andpredicted protein sequences of these three ge-nomes were pooled, and each protein was com-pared to all others in the pool by means of theprogram BLASTP. Among the larger proteinfamilies that are found in worms and flies butnot yeast are several that are associated withmulticellular development, including ho-meobox proteins, cell adhesion molecules, andguanylate cyclases, as well as trypsinlike pep-tidases and esterases. Among the large familiesthat are present only in flies are proteins in-volved in the immune response, such as lectinsand peptidoglycan recognition proteins, trans-membrane proteins of unknown function, andproteins that are probably fly-specific: cuticleproteins, peritrophic membrane proteins, andlarval serum proteins.
Gene SimilaritiesWhat fraction of the proteins encoded bythese three eukaryotes is shared? Compara-tive analysis of the predicted proteins encod-ed by these genomes suggests that nearly30% of the fly genes have putative orthologsin the worm genome. We required that aprotein show significant similarity over atleast 80% of its length to a sequence inanother species to be considered its ortholog(6). We know that this results in an underes-timate, because the length requirement ex-cludes known orthologs, such as homeodo-main proteins, which have little similarityoutside the homeodomain. The number ofsuch fly-worm pairs does not decrease muchas the similarity scores become more strin-gent (Table 2A), which strongly suggests thatwe have indeed identified orthologs, whichmay share molecular function. Nearly 20% ofthe fly proteins have a putative ortholog inboth worm and yeast; these shared proteins
probably perform functions common to alleukaryotic cells.
We also compared the proteins of fly,worm, and yeast to mammalian sequences.Most mammalian sequences are available asshort expressed sequence tags (ESTs), so wedispensed with the requirement for similarityover 80% of the length of the proteins. Table2B presents these data. Half of the fly proteinsequences show similarity to mammalianproteins at a cutoff of E , 10210 (where E isexpectation value), as compared to only 36%of worm proteins. This difference increasesas the criteria become more stringent: 25%versus 15% at E , 10250 and 12% versus 7%at E , 102100. Because many of the compar-isons are with short sequences, it is likely thatmany of these sequence similarities reflectconserved domains within proteins ratherthan orthology. However, it does suggest thatthe Drosophila proteome is more similar tomammalian proteomes than are those ofworm or yeast.
Protein Domains and FamiliesProteins are often mosaic, containing two ormore different identifiable domains, and do-mains can occur in different combinations indifferent proteins. Thus, only a portion of aprotein may be conserved among organisms.We therefore performed a comparative anal-ysis of the protein domains composing thepredicted proteomes from D. melanogaster,C. elegans, and S. cerevisiae using sequencesimilarity searches against the SWISS-PROT/TrEMBL nonredundant protein data-base (7), the BLOCKS database (8), and theInterPro database (9). The 200 most commonfly protein families and domains are listed inTable 3, and the 10 most highly representedfamilies in worm and yeast are shown inTable 4. InterPro analyses plus manual datainspection enabled us to assign 7419 fly pro-teins, 8356 worm proteins, and 3056 yeastproteins to either protein families or domainfamilies. We found 1400 different proteinfamilies or domains in all: 1177 in the fly,1133 in the worm, and 984 in yeast; 744families or domains were common to all threeorganisms.
Many protein families exhibit great dis-parities in abundance, and only the C2H2-type zinc finger proteins and the eukaryotic
1Howard Hughes Medical Institute, 2Department ofMolecular and Cell Biology, Berkeley Drosophila Ge-nome Project, University of California, Berkeley, CA94720, USA. 3Celera Genomics, Rockville, MD, 20850USA. 4GenetixXpress, 78 Pacific Road, Palm Beach,Sydney, Australia 2108. 5Massachusetts General Hos-pital Cancer Center, Building 149, 13th Street,Charlestown, MA 02129 USA. 6Department of Genet-ics, University of Pennsylvania School of Medicine,Philadelphia, PA 19104, USA. 7EMBL-EBI, WellcomeTrust Genome Campus, Hinxton, Cambridge CB101SD, UK. 8Department of Genetics, Stanford Univer-sity, Palo Alto, CA 94305, USA. 9Howard HughesMedical Institute, Fred Hutchinson Cancer ResearchCenter, Seattle, WA 98109, USA. 10National Centerfor Biotechnology Information, National Library ofMedicine, National Institutes of Health, Bethesda, MD20894, USA. 11Neurogenetics Unit, Laboratory ofNeurochemistry, National Institute of NeurologicalDisorders and Stroke, National Institutes of Health,Bethesda, MD 20892, USA. 12Berkeley Drosophila Ge-nome Project, Lawrence Berkeley National Laborato-ry, Berkeley, CA 94720, USA. 13Neomorphic, 2612Eighth Street, Berkeley, CA 94710, USA. 14School ofBiology, University of Leeds, Leeds LS2 9JT, UK. 15De-partment of Molecular and Cellular Biology, HarvardUniversity, 16 Divinity Avenue, Cambridge, MA02138, USA. 16Departments of Cellular and MolecularMedicine and Pharmacology, Howard Hughes MedicalInstitute, University of California–San Diego, La Jolla,CA 92093, USA. 17Division of Biology, California In-stitute of Technology, Pasadena, CA 91125, USA.18Howard Hughes Medical Institute, MassachusettsInstitute of Technology (MIT ), Cambridge, MA 02139,USA. 19Genome Sequence Centre, BC Cancer Re-search Centre, 600 West 10th Avenue, Vancouver, BC,V52 4E6, Canada. 20Molecular and Cell Biology Pro-gram, University of Maryland at Baltimore, Baltimore,MD 21201, USA. 21Centre de Genetique Moleculaire,CNRS, 91198 Gif-sur-Yvette, France. 22Center forLearning and Memory, MIT, 77 Massachusetts Ave-nue, Cambridge, MA 02139, USA. 23Regulation of CellGrowth Laboratory, Division of Basic Sciences, Na-tional Cancer Institute–Frederick Cancer Research andDevelopment Center, National Institutes of Health,Frederick, MD 21702, USA. 24Department of Biochem-istry and Biophysics, University of California, SanFrancisco, CA 94143, USA. 25Center for Neurobiologyand Behavior, Columbia University, New York, NY10032, USA. 26Baylor College of Medicine HumanGenome Sequencing Center, Department of Molecu-lar and Human Genetics, Baylor College of Medicine,Houston, TX 77030, USA.
Table 1. Numbers of distinct gene families versus numbers of predicted genes and their duplicated copiesin H. influenzae, S. cerevisiae, C. elegans, and D. melanogaster. Row one shows the total number of genesin each species. Row two shows the total number of all genes in each genome that appear to have arisenby gene duplication. Row three is the total number of distinct gene families for each genome. Eachproteome was compared to itself using the same parameters as described in (63).
H. influenzae S. cerevisiae C. elegans D. melanogaster
Total no. of predicted genes 1709 6241 18424 13601No. of genes duplicated 284 1858 8971 5536Total no. of distinct families 1425 4383 9453 8065
www.sciencemag.org SCIENCE VOL 287 24 MARCH 2000 2205
T H E D R O S O P H I L A G E N O M E
on October 12, 2018
http://science.sciencem
ag.org/D
ownloaded from
protein kinases are among the top 10 proteinfamilies common to all three organisms.There are 352 zinc finger proteins of theC2H2 type in the fly but only 138 in theworm; whether this reflects greater regulatorycomplexity in the fly is not known. The pro-tein kinases constitute approximately 2% ofeach proteome. Curation of the genomic datarevealed that Drosophila has approximately300 protein kinases and 85 protein phospha-tases, around half of which had previouslybeen identified. In contrast, there are approx-imately 500 kinases and 185 phosphatases inthe worm; the difference is largely due to the
worm-specific expansion of certain familiessuch as the CK1, FER, and KIN-15 families.There are currently approximately 600 ki-nases and 130 phosphatases in humans, and itis expected that these figures will rise to 1100and 300, respectively, when the sequence ofthe human genome is completed (10). Of theproteins uncovered in this analysis, over 70%exhibit sequence similarity outside the kinaseor phosphatase domain to proteins in otherspecies. In the kinase group, approximately75% are serine/threonine kinases, and 25%are tyrosine or dual-specificity kinases. Over90% of the newly discovered kinases are
predicted to phosphorylate serine/threonineresidues; this group includes the first atypicalprotein kinase C isoforms identified in Dro-sophila. In addition, we found counterparts ofthe mammalian kinases CSK, MLK2, ATM,and Peutz-Jeghers syndrome kinase, and ad-ditional members of the Drosophila GSK3B,casein kinase I, SNF1-like, and Pak/STE20-like kinase families. In the fly protein phos-phatase group, approximately 42% are pre-dicted to be serine/threonine phosphatases;48% are tyrosine or dual-specificity phospha-tases. Among the newly discovered phospha-tases, 35% are serine/threonine phosphatases,most of which are related to the protein phos-phatase 2C family, and 65% are tyrosine ordual-specificity phosphatases. The fly andworm both contain close relatives to many ofthe known mammalian lipid kinases andphosphatases; however, no SH2-containinginositol 59 phosphatase SHIP is apparent. Fi-nally, it has been found that the assembly ofkinase signaling complexes in vertebrate cellsis aided by the presence of scaffolding andadaptor molecules, many of which containphosphoprotein binding domains; we found85 such proteins in the fly, including coun-terparts to IRS, VAV, SHC, JIP, and MP1.
Two remarkable findings emerge from thepeptidase data that may reflect different ap-proaches to growth and development in flies,worms, and humans. The pattern and distri-bution of peptidase types are similar betweenthe fly and the worm: there are approximately450 peptidases in the fly and 260 in theworm. The difference is due almost entirelyto the expansion or contraction of a singleclass of trypsin-like (S1) peptidases. C. el-egans has seven of this class and yeast hasone, but the fly has 199. Of these, 163 aresmall proteins of approximately 250 aminoacids containing single trypsin domains; veryfew are mosaic proteins. The remainder haveeither multiple trypsin-like domains or longstretches of amino acids with no readily iden-tifiable motif, usually at the NH2-terminus. Inhumans, trypsin-like peptidases perform di-verse functions in digestion, in the comple-ment cascade, and in several other signalingpathways (11), and flies may have a similarlywide range of uses for these proteins. Theextensively characterized members of thisfamily, which include Snake, Easter, Nudel,and Gastrulation-defective, are all key mem-bers of a regulatory cascade that controlsdorsoventral patterning in the fly (12). Inaddition, flies have only two members of theM10 class of peptidases, which include thematrix metalloproteases, collagenases, andgelatinases that are essential for tissue remod-eling and repair in vertebrates.
The number of identifiable multidomainproteins is similar in the fly and the worm:2130 and 2261, respectively. Yeast has only672 (Table 5). Part of this difference is ac-
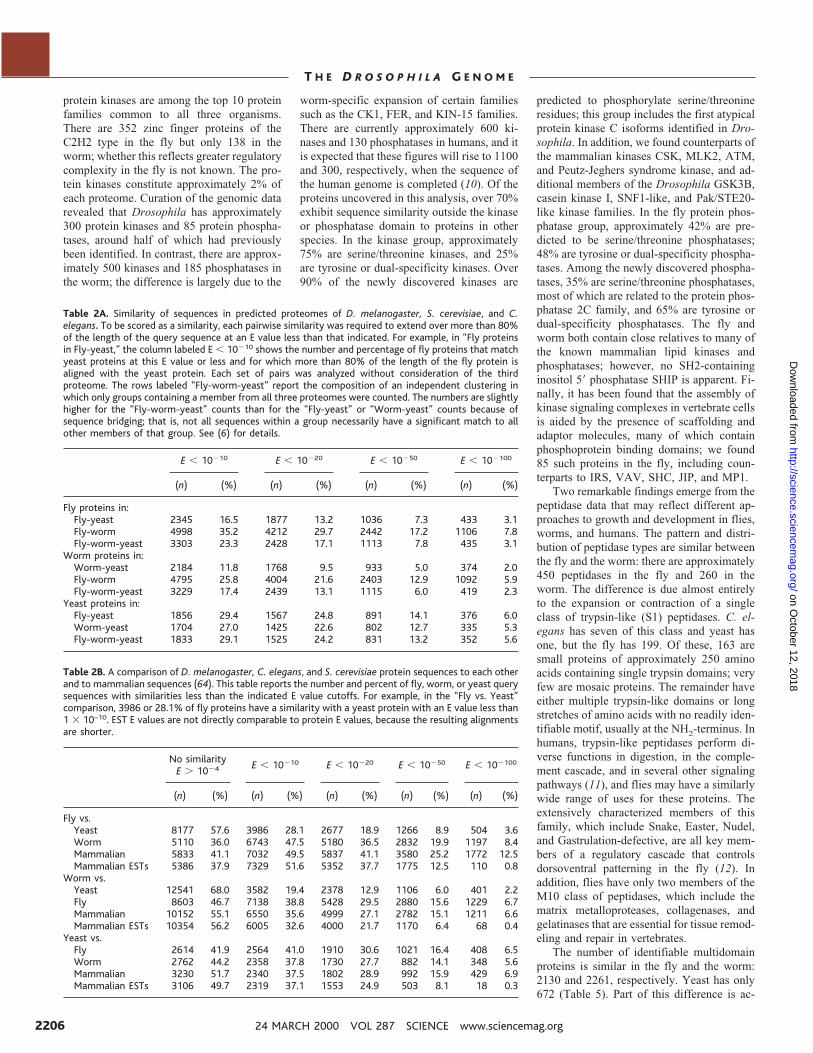
Table 2A. Similarity of sequences in predicted proteomes of D. melanogaster, S. cerevisiae, and C.elegans. To be scored as a similarity, each pairwise similarity was required to extend over more than 80%of the length of the query sequence at an E value less than that indicated. For example, in “Fly proteinsin Fly-yeast,” the column labeled E , 10210 shows the number and percentage of fly proteins that matchyeast proteins at this E value or less and for which more than 80% of the length of the fly protein isaligned with the yeast protein. Each set of pairs was analyzed without consideration of the thirdproteome. The rows labeled “Fly-worm-yeast” report the composition of an independent clustering inwhich only groups containing a member from all three proteomes were counted. The numbers are slightlyhigher for the “Fly-worm-yeast” counts than for the “Fly-yeast” or “Worm-yeast” counts because ofsequence bridging; that is, not all sequences within a group necessarily have a significant match to allother members of that group. See (6) for details.
E , 10210 E , 10220 E , 10250 E , 102100
(n) (%) (n) (%) (n) (%) (n) (%)
Fly proteins in:Fly-yeast 2345 16.5 1877 13.2 1036 7.3 433 3.1Fly-worm 4998 35.2 4212 29.7 2442 17.2 1106 7.8Fly-worm-yeast 3303 23.3 2428 17.1 1113 7.8 435 3.1
Worm proteins in:Worm-yeast 2184 11.8 1768 9.5 933 5.0 374 2.0Fly-worm 4795 25.8 4004 21.6 2403 12.9 1092 5.9Fly-worm-yeast 3229 17.4 2439 13.1 1115 6.0 419 2.3
Yeast proteins in:Fly-yeast 1856 29.4 1567 24.8 891 14.1 376 6.0Worm-yeast 1704 27.0 1425 22.6 802 12.7 335 5.3Fly-worm-yeast 1833 29.1 1525 24.2 831 13.2 352 5.6
Table 2B. A comparison of D. melanogaster, C. elegans, and S. cerevisiae protein sequences to each otherand to mammalian sequences (64). This table reports the number and percent of fly, worm, or yeast querysequences with similarities less than the indicated E value cutoffs. For example, in the “Fly vs. Yeast”comparison, 3986 or 28.1% of fly proteins have a similarity with a yeast protein with an E value less than1 3 10–10. EST E values are not directly comparable to protein E values, because the resulting alignmentsare shorter.
No similarityE . 1024 E , 10210 E , 10220 E , 10250 E , 102100
(n) (%) (n) (%) (n) (%) (n) (%) (n) (%)
Fly vs.Yeast 8177 57.6 3986 28.1 2677 18.9 1266 8.9 504 3.6Worm 5110 36.0 6743 47.5 5180 36.5 2832 19.9 1197 8.4Mammalian 5833 41.1 7032 49.5 5837 41.1 3580 25.2 1772 12.5Mammalian ESTs 5386 37.9 7329 51.6 5352 37.7 1775 12.5 110 0.8
Worm vs.Yeast 12541 68.0 3582 19.4 2378 12.9 1106 6.0 401 2.2Fly 8603 46.7 7138 38.8 5428 29.5 2880 15.6 1229 6.7Mammalian 10152 55.1 6550 35.6 4999 27.1 2782 15.1 1211 6.6Mammalian ESTs 10354 56.2 6005 32.6 4000 21.7 1170 6.4 68 0.4
Yeast vs.Fly 2614 41.9 2564 41.0 1910 30.6 1021 16.4 408 6.5Worm 2762 44.2 2358 37.8 1730 27.7 882 14.1 348 5.6Mammalian 3230 51.7 2340 37.5 1802 28.9 992 15.9 429 6.9Mammalian ESTs 3106 49.7 2319 37.1 1553 24.9 503 8.1 18 0.3
24 MARCH 2000 VOL 287 SCIENCE www.sciencemag.org2206
T H E D R O S O P H I L A G E N O M E
on October 12, 2018
http://science.sciencem
ag.org/D
ownloaded from

counted for by proteins with extracellulardomains involved in cell-cell and cell-sub-strate contacts (13), such as the immunoglob-ulin domain–containing proteins, which aremore abundant in flies than in worms (153versus 70) and are nonexistent in yeast. Twoother common extracellular domains occur insimilar numbers in fly and worm: EGF (110versus 109, respectively) and fibronectin typeIII (46 versus 43) but are rare or absent inyeast. Extracellular regions of proteins oftencontain a variety of repeated domains (14),and so these proteins may account for ourfinding that flies have a larger number ofproteins with multiple InterPro domains thaneither worms or yeast (2107 versus 1747 and525, respectively) (Table 6). Some multido-main proteins of the fly are particularly het-erogeneous: Two low-density lipoprotein re-ceptor–related proteins have 75 InterPro do-mains each. Another protein of unknownfunction has 62 InterPro domains; the mostheterogeneous worm and yeast proteins[SWISS-PROT/TrEMBL accession numbers(AC), Q04833 and P32768, respectively]have 61 and 18 InterPro domains, respective-ly. There can be extensive repetition of thesame domain within a protein; for example,an immunoglobulin-like domain is repeated52 times within one protein of unknown func-tion in the fly. The large worm protein UNC-89 contains 48 immunoglobulin-like domains(SWISS-PROT/TrEMBL AC, Q17362). Incontrast, the largest number of repeats in yeast,of a C2H2-type zinc finger domain, occursnine times in the transcription factor TFIIIA(SWISS-PROT/TrEMBL AC, P39933).
The heterotrimeric GTP-binding protein(G protein)–coupled receptors (GPCRs) are alarge protein family in flies, worms, and ver-tebrates whose members are involved in syn-aptic function, hormonal physiology, and theregulation of morphological movements dur-ing gastrulation and germ band extension(15). There are predicted to be at least 700GPCRs in the human genome (16) androughly 1100 GPCRs in C. elegans (17). Wefound approximately 160 GPCR genes in theDrosophila genome, 57 of which appear to beolfactory receptors. Drosophila, C. elegans,and vertebrates each have diverse families ofodorant receptors that, although recognizableas GPCRs, are unrelated by sequence andtherefore apparently evolved independently.The number of odorant receptors in verte-brates ranges from around 100 in zebrafishand catfish to approximately 1000 in themouse; C. elegans also has approximately1000. In the fly, as in zebrafish and mouse,there is a correlation between the number ofodorant receptors and the number of discretesynaptic structures called glomeruli in theolfactory processing centers of the brain (16,18). In the mouse, each glomerulus is dedi-cated to receiving axonal input from neurons
expressing a particular odorant receptor (16).Therefore, the correlation between number ofodorant receptors and number of glomerulimay reflect a conservation in the organiza-tional logic of odor recognition in insect andvertebrate brains. Although the fly odorantreceptors are extremely diverse, there are anumber of subfamilies whose members share50 to 65% sequence identity. The distributionof odorant receptor genes is different amongthese organisms as well. Unlike C. elegans orvertebrate odorant receptors, which are inlarge linked arrays, the fly odorant receptorgenes are distributed as single genes or inarrays of two or three. Vertebrate receptorsare encoded by intronless genes, but both flyand worm receptor genes have multiple in-trons. These distinctions suggest that in addi-tion to differences in the sequences of theodorant receptors of the different organisms,the processes generating the families of re-ceptors may have differed among the lineagesthat gave rise to flies, worms, and vertebrates.
The data suggest conservation of hormonereceptors between flies and vertebrates; nev-ertheless, there is a greater diversity of hor-mone receptors in both C. elegans and verte-brates than in Drosophila. Insects are subjectto complex hormonal regulation, but no ap-parent homologs of vertebrate neuropeptideand hormone precursors were identified.However, many receptors with sequence sim-ilarity to vertebrate receptors for neurokinin,growth hormone secretagogue, leutotropin(follicle-stimulating hormone and luteinizinghormone), thyroid-stimulating hormone, ga-lanin/allatostatin, somatostatin, and vasopres-sin were identified. Other GPCRs include aseventh Drosophila rhodopsin and homologsof adenosine, metabotropic glutamate, g-ami-nobutyric acid (GABA), octopamine, seroto-nin, dopamine, and muscarinic acetylcholinereceptors. In addition, there are GPCRs thatare unique to Drosophila, others with se-quence similarity to C. elegans and humanorphan receptors, and an insect diuretic hor-mone receptor that is closely related to ver-tebrate corticotropin-releasing factor recep-tor. Finally, we found several atypical seven-transmembrane domain receptors, including10 Methuselah (MTH)–like proteins and fourFrizzled (FZ)–like proteins. A mutation inmth increases the fly’s life-span and its resis-tance to various stresses (19); the FZ-likeproteins probably serve as receptors for dif-ferent members of the Wingless/Wnt familyof ligands.
Human Disease GenesStudies in model organisms have providedimportant insights into our understanding ofgenes and pathways that are involved in avariety of human diseases. In order to esti-mate the extent to which different types ofhuman disease genes are found in flies,
worms, and yeast, we compiled a set of 289genes that are mutated, altered, amplified, ordeleted in a diverse set of human diseases andsearched for similar genes in D. melano-gaster, C. elegans, and S. cerevisiae, as de-scribed in the legend to Fig. 1. Of these 289human genes, 177 (61%) appear to have anortholog in Drosophila (Fig. 1). Only pro-teins with similar domain structures wereconsidered to be orthologs; this judgment wasmade by human inspection of the InterProdomain composition of the fly and humanproteins. The importance of human inspec-tion, as well as consideration of publishedinformation, is underscored by the fact thatsome sequences with extremely high similar-ity scores to proteins encoded by fly genes,such as LCK and Myotonic Dystrophy 1,were judged not to be orthologous, but otherswith relatively low scores, such as p53 andRb1, were considered to be orthologs. Weattempted this additional level of analysisonly for the fly proteins, as the lower overalllevel of similarity of worm and yeast proteinsmade these subjective judgments even moredifficult. Some of the human disease genesthat are absent in Drosophila reflect cleardifferences in physiology between the twoorganisms. For instance, none of the hemo-globins, which are mutated in thalassemias,have orthologs in Drosophila. In flies, oxy-gen is delivered directly to tissues via thetracheal system rather than by circulatingerythrocytes. Similarly, several genes re-quired for normal rearrangement of the im-munoglobulin genes do not have Drosophilaorthologs.
Of the cancer genes surveyed, 68% appearto have Drosophila orthologs. In addition topreviously described proteins, these searchesidentified clear protein orthologs for menin(MEN; multiple endocrine neoplasia type 1),Peutz-Jeghers disease (STK11), ataxia telan-giectasia (ATM), multiple exostosis type 2(EXT2), a second bCL2 family member, asecond retinoblastoma family member, and ap53-like protein. Despite its relatively lowsequence similarity to the human genes, theDrosophila gene encoding p53 was consid-ered an ortholog because it shows a con-served organization of functional domains,and its DNA binding domain includes manyof the same amino acids that appear to be hotspots for mutations in human cancer. Com-parison of the fly p53-like protein with thehuman p53, p63, and p73 proteins suggeststhat it may represent a progenitor of thisentire family. In mammalian cells, levels ofp53 protein are tightly regulated in vivo by itsinteraction with the Mdm2 protein, which inturn binds to p19ARF (20). This mode ofregulation, which modulates the activity ofp53 but probably not of p63 or p73 (21), maynot apply to the Drosophila protein, becausewe have not been able to identify orthologs of
www.sciencemag.org SCIENCE VOL 287 24 MARCH 2000 2207
T H E D R O S O P H I L A G E N O M E
on October 12, 2018
http://science.sciencem
ag.org/D
ownloaded from
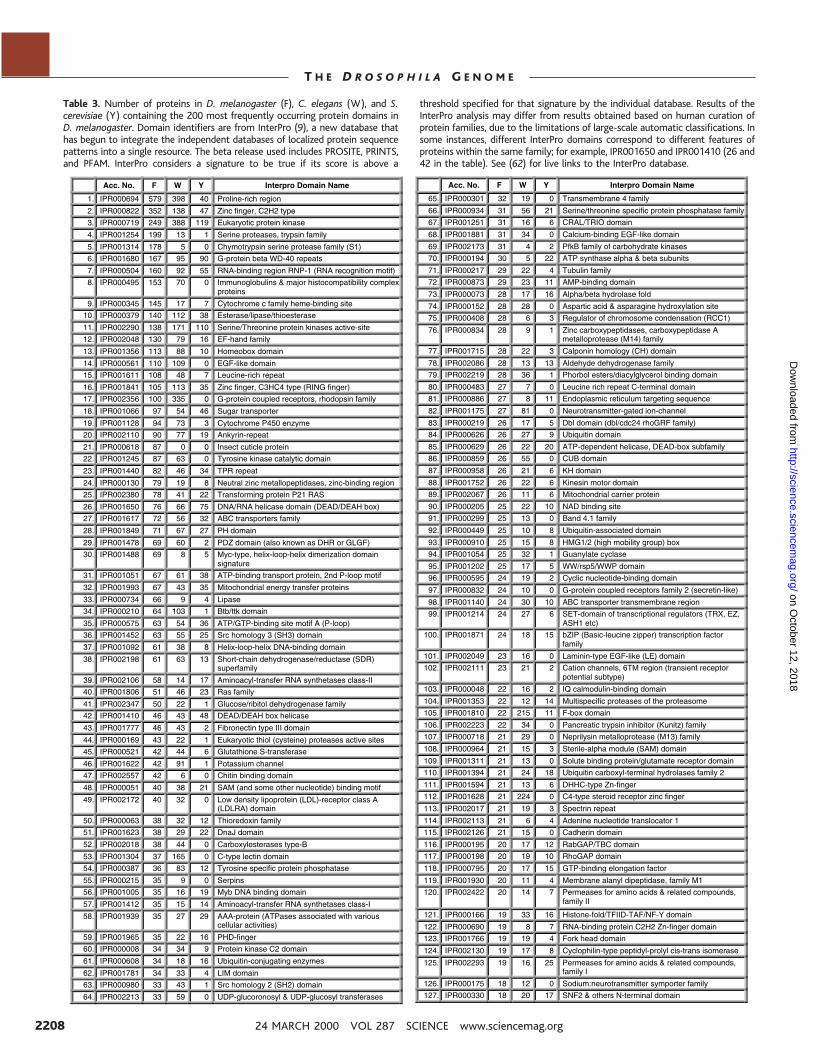
Table 3. Number of proteins in D. melanogaster (F), C. elegans (W), and S.cerevisiae (Y) containing the 200 most frequently occurring protein domains inD. melanogaster. Domain identifiers are from InterPro (9), a new database thathas begun to integrate the independent databases of localized protein sequencepatterns into a single resource. The beta release used includes PROSITE, PRINTS,and PFAM. InterPro considers a signature to be true if its score is above a
threshold specified for that signature by the individual database. Results of theInterPro analysis may differ from results obtained based on human curation ofprotein families, due to the limitations of large-scale automatic classifications. Insome instances, different InterPro domains correspond to different features ofproteins within the same family; for example, IPR001650 and IPR001410 (26 and42 in the table). See (62) for live links to the InterPro database.
24 MARCH 2000 VOL 287 SCIENCE www.sciencemag.org2208
T H E D R O S O P H I L A G E N O M E
on October 12, 2018
http://science.sciencem
ag.org/D
ownloaded from

either Mdm2 or p19ARF in Drosophila. In-terestingly, likely orthologs of the breast can-cer susceptibility genes BRCA1 and BRCA2were not found in Drosophila. In most in-stances, cancer genes that have a Drosophilaortholog also have an ortholog in C. elegans,although the extent of sequence similarity tothe worm gene is lower. In a minority ofinstances, a C. elegans ortholog was clearlyabsent. Cancer genes with orthologs in Dro-sophila and apparently not in C. elegans in-clude p53 and neurofibromatosis type 1 (22),the two genes implicated in tuberous sclerosis(TSC1 and TSC2) (23), and MEN. The twoTSC gene products are thought to bind toeach other and may function in a pathwaythat is conserved between humans and Dro-sophila but is absent in C. elegans and S.cerevisiae. However, the limitations of thistype of analysis are clearly illustrated by ourinability to find a bCL2 ortholog in C. el-egans using these search parameters. The C.elegans ced-9 gene has been shown to func-tion as a bCL2 homolog, and its protein is23% identical to the human protein over itsentire length (24).
Numerous orthologs of neurologicalgenes are also found in the Drosophila ge-
nome. Some, such as Notch (CADASIL syn-drome), the beta amyloid protein precursor-like gene, and Presenilin (Alzheimer’s dis-ease), were already known from previousstudies in the fly. The genome sequencingeffort has uncovered several additional genesthat are likely to be orthologs of human neu-rological genes, such as tau (frontotemporaldementia with Parkinsonism), the Best mac-ular dystrophy gene, neuroserpin (familialencephalopathy), genes for limb girdle mus-cular dystrophy types 2A and 2B, the Fried-reich ataxia gene, the gene for Miller-Diekerlissencephaly, parkin ( juvenile Parkinson’sdisease), and the Tay-Sachs and Stargardt’sdisease genes. Several genes implicated inexpanded polyglutamine repeat diseases, in-cluding Huntington’s and spinal cerebellarataxia 2 (SCA2), are found in the fruit fly.Most human neurological disease genes sur-veyed were also detected in C. elegans, andsome were even found in yeast, although afew examples are apparently present only inDrosophila, such as the Parkin and SCA2orthologs.
Among genes implicated in endocrine dis-eases, those functioning in the insulin path-way are mostly conserved. In contrast, mem-
bers of pathways involving growth hormone,mineralocorticoids, thyroid hormone, and theproteins that regulate body mass in verte-brates, such as those encoding leptin, do notappear to have Drosophila orthologs. Sur-prisingly, a protein that shows significantsequence similarity to the luteinizing hor-mone receptor is present in Drosophila (25).The physiological ligand for this receptor isnot known. A number of genes that have beenimplicated in human renal disorders have or-thologs in Drosophila, despite the differencesbetween human kidneys and insect Mal-pighian tubules. In many instances, thesegene products are involved in fluid and elec-trolyte transport across epithelia. Not surpris-ingly, most disease genes that function inintracellular metabolic pathways appear tohave Drosophila orthologs.
Developmental and Cellular ProcessesDevelopmental strategies in various phyla areovertly very different, from the fixed celllineage of C. elegans to the syncytial embry-ogenic development of the fly, to early em-bryogenesis in amphibians and mammals. Anumber of major processes—cell division,cell shape, signaling pathways, cell-cell and
Table 3 (continued).
www.sciencemag.org SCIENCE VOL 287 24 MARCH 2000 2209
T H E D R O S O P H I L A G E N O M E
on October 12, 2018
http://science.sciencem
ag.org/D
ownloaded from

cell-substrate adhesion, and apoptosis—de-termine the developmental outcomes of thesevery different embryos. Although there aremany more, such as the processes that deter-mine embryonic gradients, cell polarities, andcell movement, here we examine the firstfive, beginning with cell cycle components,and examine what new insights have beengained from the genomic data that affect ourknowledge of the evolution of developmentalprocesses. We then discuss the processes ofneuronal signaling and innate immunity.
Cell cycle. Despite conservation of themechanisms regulating cell cycle progres-sion, many of the functions governing thisprogression are encoded by gene familieswhose individual members are not conservedbetween vertebrates and yeast. For example,the cyclins of S. cerevisiae can be dividedinto a G1 class (Cln1, Cln2, and Cln3) and anS/G2 class (Clb1 through Clb6); it is notpossible to identify orthologs of individualvertebrate cyclins. Consequently, analysis ofthe roles of particular vertebrate cell cyclegenes benefits from a genetic model in whichparallels are more evident. Analysis of theDrosophila genome sequence supports andextends previous suggestions of strong paral-lels between fly and human cell cycle regu-lators. Orthologs of vertebrate cell cycle cy-clins—cyclin A (CycA), CycB, CycB3,CycE, and CycD—have been identified inDrosophila, as have orthologs of cyclins thatappear to have roles in transcription: CycC,CycH, CycK, and CycT. Apparent orthologsof these cyclins can be also be found in C.elegans; however, the level of similarity tothe vertebrate members is invariably substan-tially less. Indeed, BLAST comparisons sug-gest that vertebrate and Drosophila CycA andCycB share more sequence similarity withyeast than with proposed C. elegans or-thologs. Examination of other cell cycle reg-ulators confirms that quite precise compari-sons can be made between vertebrates andflies; parallels with yeast are looser. For ex-ample, like vertebrates, Drosophila uses sev-eral different cyclin-dependent kinases(Cdks) to regulate different aspects of the cellcycle; S. cerevisiae and Schizosaccharomycespombe use only one. Cloning efforts and thegenome sequence revealed Drosophila or-thologs of vertebrate Cdk1 (cdc2) and Cdk2(cdc2c), as well as a single Drosophila Cdk(Cdk4/6) with close similarity to both Cdk4 andCdk6. As in vertebrates, Drosophila has twodistinct kinases that add inhibitory phos-phate to Cdk1, the previously identifiedWee, and a recently recognized homolog ofMyt1, which was initially identified as amembrane-associated inhibitory kinase in Xen-opus (26). C. elegans also has two homologsof these kinases (Wee1.1 and Wee1.3);however, similarity scores do not placethese into distinct Wee1 and Myt1 sub-
types. Each of these genes appears to bepresent in a single copy, a factor that sim-plifies genetic interpretations.
The retinoblastoma gene product pRb is acrucial cell cycle regulator in mammals and isthought to modulate S-phase entry via itsinteractions with the transcriptional regulatorE2F and its dimerization partner (DP). Thisimportant mode of regulation is not found inyeast, but many components of the Rb path-way have been identified and studied in Dro-sophila (27). The sequencing effort uncov-ered a second Rb-related gene in Drosophilaand confirmed the existence of only two E2Ffamily members and a single DP ortholog. C.elegans also has an Rb-related gene, isolatedin a genetic screen for mutations affectingcell fate decisions (28), but it has not beenshown to play a direct role in cell cycle
regulation. Also evident from the sequenceare eight skp-like genes and six cullin-relatedgenes. The Skp and Cullin proteins functionin a complex that mediates the degradation ofspecific target proteins during crucial cellcycle transitions. Further exploration of thegenome sequence should define orthologs tomost vertebrate cell cycle genes and lead togenetic tests of their regulation and function.
Cytoskeleton. A large number of proteinslink events at the cell surface with cytoskel-etal networks and intracellular messengers(13). We found approximately 230 genes (ap-proximately 2% of the predicted genes) thatencode cytoskeletal structural or motor pro-teins; these represent most major familiesfound in other invertebrates and vertebrates(29). The fraction of the Drosophila genomedevoted to cytoskeletal functions appears to
F W Y Cancer + ABL1+ ## ## Acute Myeloid Leukemia-DEK+ ## Adenomat. Polyposis Coli-APC+ AKT2+ Ataxia Telangiectasia-ATM- ## BRCA1- ## ## BRCA2+ Basal Cell Nevus-PTC+ ## B-Cell Lymphoma 2-BCL2- B-Cell Lymphoma 3-BCL3+ Bloom-BLM+ ## Burkitt's Lymphoma-MYC- ## CDKN2C- CSF1R/C-Fms+ Chk2 Protein Kinase- ## PDGFB+ CML-BCR+ Cyclin D1-CCND1+ Cyclin Dep. Kinase 4-CDK4+ EGFR+ ERBB2- ## ETS+ ## E-Cadherin-CDH1+ ## Ewing Sarcoma-FLI-1- ## FGF3- ## ## Fanconi's Anemia A-FANCA- ## ## Fanconi's Anemia C-FANCC- ## ## Fanconi's Anemia G-FANCG+ HNPCC*-MSH2+ HNPCC*-MSH3+ HNPCC*-MSH6+ HNPCC*-MLH1+ HNPCC*-PMS2- KIT- LCK+ ## Lymphoma-MCF2+ ## MADH4- ## MDM2- MET+ ## ## MEN***1+ MEN***2A-RET+ ## Multiple Exostosis 1-EXT1+ ## Multiple Exostosis 2-EXT2- NTRK1+ Neurofibromatosis 1-NF1+ ## Neurofibromatosis 2-NF2+ ## Nijmegen Breakage 1-NBS1+ Nucleoporin-NUP214- ## ## P16-INK4- ## ## P16-INK4A- ## ## P19 ARF+ 1 1 P53+ PTEN+ RAS+ REL+ ## Retinoblastoma-RB1+ STK11+ ## Stem Cell Leukemia-TAL1+ Tuberous Sclerosis 1-TSC1+ ## Tuberous Sclerosis 2-TSC2- ## ## Von Hippel Lindau-VHL- Wilm's Tumor-WT1+ Xeroderma Pigment. A-XPA+ ## Xeroderma Pigment. B-ERCC3+ Xeroderma Pigment. D-XPD+ Xeroderma Pigment. F-XPF+ Xeroderma Pigment. G-XPG
F W Y F W YNeurological+ Adrenoleukodystrophy-ABCD1+ ## Alzheimer-PS1+ ## Alzheimer-APP+ Amyotrophic Lat. Sclero.-SOD1+ Angelman-UBE3A+ Aniridia-PAX6+ ## Best Macular Dystrophy-VMD2+ ## Ceroid-Lipofuscinosis-PPT + Ceroid-Lipofuscinosis-CLN3- ## ## Ceroid-Lipofuscinosis-CLN2- ## ## Charcot-Marie-Tooth 1A-PMP22- ## ## Charcot-Marie-Tooth 1B-MPZ+ Choroideremia-CHM- ## ## Creutzfeldt-Jakob-PRNP+ Deafness, Hereditary-MYO15+ Deafness, X-Linked-TIMM8A+ Diaphanous 1-DIAPH1+ ## Dementia, Multi-Infarct-NOTCH3+ 0 Duchenne MD+-DMD- ## ## Emery-Dreifuss MD+-EMD+ ## Emery-Dreifuss MD+-LMNA+ ## Familial Encephalopathy-PI12+ ## Fragile-X -FRAXA+ Friedreich Ataxia-FRDA+ ## Frontotemporal Dement.-TAU- ## Fukuyama MD+-FCMD+ ## ## Huntington-HD+ Limb Girdle MD+ 2A-CAPN3+ ## Limb Girdle MD+ 2B-YSF- ## ## Limb Girdle MD+ 2E-BSG+ ## Lissencephaly, X-Linked-DCX+ Lowe Oculocerebroren.-OCRL- ## Machado-Joseph-MJD1+ Miller-Dieker Lissen.-PAF- Myotonic Dystrophy-DM1+ Myotubular Myopathy 1-MTM1- ## ## Naito-Oyangi-DRPLA- ## Nemaline Myopathy 2-NEB- ## ## Neuraminidase Defic.-NEU1- ## ## Norrie-NDP- ## ## Ocular Albinism-OA1+ Oculopharyngeal MD+-PABPN1+ Oguchi Type 2-RH KIN- ## Parkinson-SNCA+ Parkinson-PARK2+ Parkinson-UCHL1- ## ## Prog. Myoclonic Epilepsy-CSTB- Retinitis Pigmentosa-RPGR- ## Retinitis Pigmentosa 2-RP2- ## SCA+++ 1-SCA1+ SCA+++ 2-SCA2+ SCA+++ 6-CACNA1A- ## ## SCA+++ 7-SCA7+ ## ## Spinal Muscular Atrophy-SMN1+ Stargardt-ABCA4+ ## Tay-Sachs-HEXA+ Thomsen-CLCN1- ## Usher-USH2A+ Wilson-ATP7B
Cardiovascular+ ## A/V Conduction Defects-CSX+ HDL Deficiency 1-ABCA1+ ## Long Q-T 1-KCNQ1+ Long Q-T 2-KCNH2+ Long Q-T 3-SCN5A+ Fam. Cardiac Myopathy-MYH7
Malformation Syndromes- ## Aarskog-Scott-FGD1+ Achondroplasia-FGFR3+ ## Alagille-JAG1+ Barth-TAZ- ## Beckwith-Wiedemann-CDKN1C- ## ## Cerebral Cavern. Malf.-CCM1
## Chondrodyspl. Punct. 1-ARSE+ ## Cleidocranial Dysplasia-OFC1- Cockayne I-CKN1+ Coffin-Lowry-RPS6KA3+ Diastrophic Dyspl.-SLC26A2+ EEC 3-Ket. P63+ Greig Cephalopolysynd.-GLI3- Hand-Foot-Genital-HOXA13+ Holoprosencephaly 3-SHH+ Holoprosencephaly-SIX3+ ## Holt-Oram-TBX5- ## ## ICF-DNMT3B+ ## Kallman-KAL1- Laterality, X-Linked-ZIC3+ ## Melnick-Fraser-EYA1+ Nail Patella-LMX1B- ## Opitz-MID1+ ## Renal Coloboma-PAX2+ Rieger, Type 1-PITX2- ## ## Rubinstein-Taybi-CREBBP+ ## Saethre-Chotzen-TWIST- ## Septooptic Dysplasia-HESX1+ ## Simpson-Golabi-Behmel-GPC3+ Townes-Brockes-SALL1- ## Treacher-Collins-TCOF1- VMCM-TEK+ Wardenburg-PAX3+ Zellweger-PEX1
Endocrine- ## ## Adrenal Hypoplasia-NR0B1- ## Androgen Receptor-AR- Adrenal Hyperplas. III-CYP21A2+ ## ## Diabetes-INS+ Diabetes-INSR- ## Diabet. Ins. Neurohypop.-AVP- ## Diabet. w/ Hypertens.-PPARG- Dwarfism-GH1- ## ## Dwarfism-GHR- Gonadal Dysgenesis-SRY+ Hyperinsulinism-ABCC8+ ## Hyperinsulinism -KCNJ11- ## Hypothyroidism-TRH+ ## Hypothyroidism-SLC5A5+ ## Leydig Cell Hypoplasia-LHCGR+ ## MODY++ 1-HNF-4A + MODY++ 2-GCK- ## ## MODY++ 3-TCF1- ## MODY++ 4-IPF1- ## ## MODY++ 5-TCF2+ McCune-Albright-GNAS1+ Non-Insulin Dep. Diabet.-PCSK1- ## Obesity-LEP- ## ## Obesity-LEPR- ## Obesity-MC4R- ## ## Obesity-POMC+ ## Pendred-PDS- ## Thyr. Resistance-THRA- ## Thyr. Resistance-THRB- ## ## Thyrotropin Deficiency-TSHB+ ## Vitamin-D Resis. Rickets-VDR
F W Y
F W Y
+
Fig. 1.
24 MARCH 2000 VOL 287 SCIENCE www.sciencemag.org2210
T H E D R O S O P H I L A G E N O M E
on October 12, 2018
http://science.sciencem
ag.org/D
ownloaded from

be somewhat smaller than that found in C.elegans (5%) (30); whether this reflects a truebiological difference or a difference in classifi-cation criteria remains to be discovered. Of theDrosophila cytoskeletal genes, 90 encode pro-teins belonging to the kinesin, dynein, or myo-sin motor superfamilies, or accessory or regu-latory proteins known to interact with the motorprotein subunits. Approximately 80 genes en-code actin-binding proteins, including proteinsbelonging to the spectrin/a-actinin/dystrophinsuperfamily of membrane cytoskeletal and ac-tin–cross-linking proteins. Twenty genes en-code proteins that are likely to bind microtu-bules, based on their similarity to microtubule-binding proteins found in other organisms.
Fourteen genes encode members of the actinsuperfamily, 12 encode members of the tubulinsuperfamily, and 5 encode septins. Overall, therepresentation of predicted cytoskeletal proteintypes and families is similar to what has beenfound for C. elegans, although Drosophila hasmany more dyneins, probably because C. el-egans lacks motile cilia and flagella.
Among this collection of cytoskeletal genesare several interesting and in some cases long-sought genes. One gene encodes a protein withstriking homology to proteins of the tau/MAP2/MAP4 family that share a characteristic repeat-ed microtubule-binding domain. Two encodenew tubulins; one appears most closely relatedto a-tubulin, and the other appears most closely
related to b-tubulin, both with approximately50% identity. Neither new tubulin has greatersimilarity to the other, more divergent membersof the tubulin superfamily, such as g-, d-, orε-tubulin (31). Thus, both Drosophila and C.elegans appear to lack d- and ε-tubulin, eventhough d-tubulin is highly conserved betweenChlamydomonas and humans. There are alsothree new members of the central motor domainfamily of kinesins that encode nonmotor pro-teins that regulate microtubule dynamics (32).There are clear homologs of the dystrophincomplex and of dystrobrevin. Finally, the flylacks cytoplasmic intermediate filament pro-teins, other than nuclear lamins, although otherinvertebrates, including C. elegans, appear tohave genes encoding these (33). Drosophilaand C. elegans both also appear to lack a geneencoding kinectin, the proposed receptor forkinesin and cytoplasmic dynein on vesicles andorganelles (34). Flies and worms must thus usedifferent proteins to link microtubule motors tovesicles and organelles.
Cell adhesion. Cell-cell adhesion and cell-substrate adhesion molecules have been crucialto the development of multicellular organismsand the evolution of complex forms of embry-ogenesis (13). The transmembrane extracellularmatrix-cytoskeleton linkage via integrins is an-cient. There are five a and two b integrins inthe fly, two a and one b in C. elegans, and atleast 18 a and eight b in vertebrates. Integrin-associated cytoplasmic proteins (talin, vinculin,a-actinin, paxillin, FAK, p130CAS, and ILK)are encoded by single-copy fly genes, as aretensin and syndecan.
Two genes for type IV collagen subunitsand genes for the three subunits of lamininwere already known in the fly. Analysis ofthe genome revealed no more laminin genesand only one more collagen, which is closestto types XV and XVIII of vertebrates. Acounterpart of this collagen is found in C.elegans, which has on the order of 170 col-lagens. Most important, it appears that thecore components of basement membranes(two type IV collagen subunits, three lamininsubunits, entactin/nidogen, and one perle-can), are all present in flies. This constitutionof basement membranes was clearly estab-lished early in evolution and has been wellconserved in metazoans; remarkably, the flypreserves the linked head-to-head organiza-tion of vertebrate type-IV collagen genes. Incontrast to this conservation, many well-known vertebrate integrin (ECM) ligands areabsent from the fly: fibronectin, vitronectin,elastin, von Willebrand factor, osteopontin,and fibrillar collagens are all missing.
The fly has three classic cadherins, two ofwhich are closely linked, but no protocadherinsof the type found in vertebrates as clusters withcommon cytoplasmic domains (35). Verte-brates have three such clusters encoding over50 protocadherins and close to 20 classical
Renal+ ## Alport-COL4A5+ Bartter-SLC12A1+ ## Congenital Nephrotic-NPHS1+ Dent-CLCN5+ Diabetes Insipidus 2-AQP2+ Gitelman-SLC12A3+ 1º Hyperoxaluria I-AGXT- ## 1º Hypomagnesemia-CLDN16+ ## Hypophosphatasia-ALPL- ## Nephronophthisis 1-NPHP1- ## Polycystic Kidney 1-PKD1+ ## Polycystic Kidney 2-PKD2- ## Pseudohypoaldoster-NR3C2+ Renal Tubul. Acidosis-ATP6B1+ 0 Vitamin D Resis. Rickets-PHEX- ## ## Williams-Beuren-ELN
Hematological+ Chediak-Higashi-CHS1+ Diamond-Blackfan Anem.-RPS19- ## ## Essen.Thrombocythemia-THPO+ G6PD Deficiency-G6PD- ## ## HPLH2-PRF1- ## Hemophilia A-F8C- Hemophilia B-F9
Hered. Spherocytosis-ANK1++ ## Megaloblas. Anemia-SLC19A2+ ## Myeloperoxidase Defic.-MPO- ## ## Osler-Rendu-Weber-ENG- ## ## -Thalassemia-HBA1- ## ## -Thalassemia-HBB- ## ## -Thalassemia-HBD- ## ## -Thalassemia-HBE+ Thrombophilia-PLG- ## Von Willebrand-VWF+ ## Wiskott-Aldrich-WAS
Immune+ Bare Lymphocyte-ABCB3+ Bare Lymphocyte-RFX5- ## ## Bare Lymphocyte-RFX5AP- ## ## Bare Lymphocyte-MHC2TA+ Bruton Agammaglobulin.-BTK- ## Chronic Granulom.-NCF1+ ## Chronic Granulom.-CYBB+ Immunodeficiency-DNA Ligase 1- Immunodeficiency-CD3G- ## ## SCID**-IL2RG- ## ## SCID**-IL7R+ SCID**-JAK3- ## ## SCID**-RAG1- ## ## SCID**-RAG2+ SCID**-ZAP70- ## ## T-Cell Immunodefic.-CD3E- ## X-Linked Lymphoprol.-SH2D1A
Metabolic+ CPT2 Deficiency-CPT2+ 1º Carnitine Defic.-SLC22A5+ ## Citrullinemia, Type I-ASS+ Cystinuria, Type 1-SLC3A1- ## Hypercalcemia-CASR+ Galactokinase-GALK1+ ## Gaucher-GBA- ## ## Hemochromatosis-HFE- ## Lesch-Nyhan-HPRT1+ ## Liddle-SCNN1G+ ## Liddle-SCNN1B+ Menkes-ATP7A+ Niemann-Pick C-NPC1+ SCID**-ADA+ Trimethylaminuria-FMO3+ ## Variegate Porphyria-PPOX+ Wernicke-Korsakoff-TKT
Other+ ## -1-Antitrypsin Deficency-PI- ## ## Alveolar Proteinosis-SFTPB
## Corneal Dystrophy-TGFBI+Cystic Fibrosis-ABCC7+
+ Cystinosis-CTNS+ Darier-White-SERCA- Downreg. in Adenoma-DRA- ## Ehlers-Danlos IV-COL3A1- ## Fam. Mediterr. Fever-MEFV+ Finnish Amyloidosis-GSN+ Glycerol Kinase Defic.-GK+ Hereditary Pancreatitis-PRSS1+ ## Hermansky-Pudlak-HPS+ Hyperexplexia-GLRA2+ Juvenile Glaucoma-GLC1A- Keratoderma-KRT9+ ## Marfan-FBN1- ## ## Mcleod-XK- ## Monilethrix-KRTHB- ## Monilethrix-KRTHB6- ## Osteogenesis Imperf.-COL1A1- ## Spondyloepip. Dysp.-COL2A1- ## ## Vohwinkel-LOR+ ## ## Wolfram-WFS1
F W Y
F W Y
F W Y
F W Y
F W Y
Fig. 1 (continued). Fly (F), worm ( W), and yeast ( Y) genes showing similarity to human diseasegenes. This collection of human disease genes was selected to represent a cross section of humanpathophysiology and is not comprehensive. The selection criteria require that the gene is actuallymutated, altered, amplified, or deleted in a human disease, as opposed to having a functiondeduced from experiments on model organisms or in cell culture. Due to redundancy in gene andprotein sequence databases, a single reference sequence for each gene had to be chosen. Mostreference sequences represent the longest mRNA of several alternatives in GenBank. Authoritativesources in the literature and electronic databases [Online Mendelian Inheritance in Man (OMIM)]were also consulted. In all, 289 protein sequences met these criteria. These were used as queriesto search a database consisting of the sum total of gene products (38,860) found in the completegenomes of fly, worm, and yeast. 12,953 was used as the effective database size (the z parameterin BLAST ). BLASTP searches were conducted as described for full genome searches, except for thez parameter. To control for potential frameshift errors in the Drosophila genome sequence, searchesagainst a six-frame translation of the entire genome (using TBLASTN) were also conducted with thedisease gene sequences using the z parameter above. Only two cases in which matches to genomicsequence were better than to the predicted protein were found, and these were manually correctedto reflect the better TBLASTN scores in the table. Results are scaled according to various levels ofstatistical significance, reflecting a level of confidence in either evolutionary homology or func-tional similarity. White boxes represent BLAST E values .1 3 10–6, indicating no or weaksimilarity; light blue boxes represent E values in the range of 1 3 1026 to 1 3 10240; purple boxesrepresent E values in the range of 1 3 10240 to 1 3 102100; and dark blue boxes represent E values,1 3 102100, indicating the highest degree of sequence conservation. Actual E values can be foundin the Web supplement to this figure (62), where links to OMIM and GenBank may also be found.A plus sign indicates our best estimate that the corresponding Drosophila gene product is thefunctional equivalent of the human protein, based on degree of sequence similarity, InterProdomain composition, and supporting biological evidence, when available. A minus sign indicatesthat we were unable to identify a likely functional equivalent of the human protein.
www.sciencemag.org SCIENCE VOL 287 24 MARCH 2000 2211
T H E D R O S O P H I L A G E N O M E
on October 12, 2018
http://science.sciencem
ag.org/D
ownloaded from

cadherins. The fly has no reelin, an ECM ligandfor CNR-type protocadherins in vertebrates(36). However, there are other fly proteins withcadherin repeats, including the previouslyknown Fat, Dachsous, and Starry night, and anew very large protein related to Fat. C. eleganshas 15 genes containing cadherin repeats; thenumber in humans is now 70 and will undoubt-edly rise (13).
Cell signaling. Components of known sig-naling pathways in the fly and worm havelargely been uncovered by examinations of de-velopmental systems. It is a tribute to the pre-vious genetic analyses done in these organismsthat only a modest number of new componentsof the known signaling pathways were revealedby analysis of the genomic sequence. The corecomponents defined in flies and worms havebeen used in modified and expanded forms invertebrates (37). The predominant pathways—transforming growth factor–b (TGF-b), recep-tor tyrosine kinases, Wingless/Wnt, Notch/lin-12, Toll/IL1, JAK/STAT/cytokine, and Hedge-hog (HH) signaling networks—all have largelyconserved fly and vertebrate components. Theworm, by contrast, does not appear to possessthe HH or Toll/IL1 pathways, nor does it haveall of the components of the Notch/lin-12 net-work (38). Two new proteins of the TGF-bsuperfamily were identified, bringing the totalto seven; all seven are members of the bonemorphogenetic protein (BMP) or b-activin sub-families. We detected no representatives of theother branches of this superfamily, namely theTGF-b, a-inhibin, and Mullerian inhibitingsubstance (MIS) subfamilies. Three new mem-bers of the Wingless/Wnt family were identi-fied, bringing the total to seven. Each of these
proteins has sequence similarity to a differ-ent vertebrate Wnt protein; this ancientfamily clearly underwent much of its ex-pansion before the divergence of the arthro-pod and chordate lineages. There is onlyone member of the Notch and HH families,in contrast to the many members of thesefamilies in vertebrates.
Apoptosis. The core apoptotic machinery ofDrosophila shares many features in commonwith that of mammals. Many apoptosis-induc-ing signals lead to activation of members of thecaspase family of proteases. These proteasesfunction in apoptotic processes as cell deathsignal transducers and death effectors, and innonapoptotic processes in flies and mammals(39). Drosophila contains genes encoding 8caspases, as compared to 4 in the worm and atleast 14 in mammals. Three of the fly caspasescontain long NH2-terminal prodomains of 100to 200 amino acids that are characteristic ofcaspases that function as signal transducers.These prodomains are thought to mediatecaspase recruitment into signaling complexes inwhich activation occurs in response to oli-gomerization. In one pathway described inmammals but not in worms, death signals causethe release of proteins, including cytochrome cand the apoptosis-inducing factor (AIF), frommitochondria (40). The human protein Apaf-1,in conjunction with cytochrome c, activatesCARD domain–containing caspases (41). Dro-sophila has an Apaf-1 counterpart, a CARDdomain–containing caspase, and AIF; Dro-sophila also has counterparts to the caspase-activated DNAse CAD/CPAN/DFF40, its in-hibitor ICAD/DFF45, and the chromatin con-densation factor Acinus (42).
Pro- and anti-apoptotic BCL2 familymembers regulate apoptosis at multiplepoints (43). Drosophila encodes two BCL2family proteins, though more divergent fam-ily members may exist. Fifteen BCL2 familyproteins have been identified in mammalsand two in the worm. In addition, inhibitor ofapoptosis (IAP) family proteins negativelyregulate apoptosis (44). They are defined bythe presence of one or more NH2-terminalrepeats of a BIR domain, a motif that isessential for death inhibition. Drosophila hasfour proteins with this motif, as compared toseven identified thus far in mammals. Thereare several BIR domain–containing proteinsin C. elegans and yeast, but none has beenimplicated in cell death regulation. Reaper(RPR), Wrinkled (W), and Grim are essentialDrosophila cell death activators (45). Or-thologs have not been identified in other or-ganisms, but they are likely to exist becauseRPR, W, and Grim induce apoptosis in ver-tebrate systems and physically interact withapoptosis regulators that include IAPs and theXenopus protein Scythe (46), for which thereis a predicted Drosophila homolog.
Neuronal signaling. The neuronal signalingsystems in flies, worms, and vertebrates revealextensive conservation of some components, aswell as extreme divergence, or the total ab-sence, of others. There is no voltage-activatedsodium channel in the worm (17); flies andvertebrates generate sodium-dependent actionpotentials. The fly genome encodes two pore-forming subunits for sodium channels (Para andNaCP60E), and also four voltage-dependentcalcium channel a subunits, including oneT-type/a1G, one L-type/a1D (Dmca1D), oneN-type/a1A (Dmca1A), and one protein that ismore similar to an outlying C. elegans proteinthan to known vertebrate calcium channels. Ad-ditional fly calcium channel subunits includeone b, one g 2, and three a 2 subunits.
The worm genome encodes over 80 potas-sium channel proteins (17); the fly genome hasonly 30. The extent to which these differentfamily sizes contribute to the establishment ofunique electrical signatures is unknown. The flypotassium channel family includes five Shaker-like genes (Shaker, Shab, Shal, and two Shaws);a large conductance calcium-activated channelgene (slowpoke); a slack subunit relative; threemembers of the eag family (eag, sei, and elk);one small conductance calcium-regulated chan-nel gene; one KCNQ channel gene; and fourcyclic nucleotide–gated channel genes. In ad-dition, there are 50 TWIK members in theworm, but only 11 fly members of the two-pore/TWIK family with four transmembranedomains. There are also three fly members ofthe inward rectifier/two transmembrane family.Finally, neither the fly nor the worm has dis-cernible relatives of a number of mammalianchannel-associated subunits such as minK andmiRP1.
Table 4. The 10 InterPro protein domains occurring in the largest number of different proteins in S.cerevisiae and C. elegans.
Acc. no. InterPro domain nameNo. of
proteins
S. cerevisiaeIPR000719 Eukaryotic protein kinase 119IPR001680 G-protein beta WD-40 repeats 90IPR001650 DNA/RNA helicase domain (DEAD/DEAH box) 75IPR001138 Fungal transcriptional regulatory protein, N-terminus 60IPR001042 TYA transposon protein 57IPR000504 RNA-binding region RNP-1 (RNA recognition motif ) 55IPR001410 DEAD/DEAH box helicase 48IPR000822 Zinc finger, C2H2 type 47IPR001066 Sugar transporter 46IPR001969 Eukaryotic and viral aspartyl proteases active site 42
C. elegansIPR000168 7-Helix G-protein coupled receptor, nematode
(probably olfactory) family545
IPR000694 Proline-rich region 398IPR000719 Eukaryotic protein kinase 388IPR002356 G-protein–coupled receptors, rhodopsin family 335IPR001628 C4-type steroid receptor zinc finger 224IPR001810 F-box domain 215IPR000087 Collagen triple helix repeat 166IPR001304 C-type lectin domain 165IPR002900 Domain of unknown function 142IPR000822 Zinc finger, C2H2 type 138
24 MARCH 2000 VOL 287 SCIENCE www.sciencemag.org2212
T H E D R O S O P H I L A G E N O M E
on October 12, 2018
http://science.sciencem
ag.org/D
ownloaded from

There are also major differences postsynap-tically. C. elegans has approximately 100 mem-bers of a family of ligand-gated ion channels(17); flies have about 50. The worm has 42nicotinic acetylcholine receptor subunits and 37GABA(A)-like receptor subunits; the fly con-tains only 11 nicotinic receptor subunit genesand 12 GABA(A)/glycine-like receptor subunitgenes. In contrast, there are 30 members of theexcitatory glutamate receptor family in the flybut only 10 in the worm. These include sub-types of the AMPA, kainate, NMDA, and deltafamilies. In addition, the fly genome contains alarge number of PDZ-containing genes, ap-proximately a dozen of which encode proteinsthat have high sequence similarity to mamma-lian proteins that interact with specific subsetsof ion channels. We also found a number ofadditional ion channel families, including threevoltage-dependent chloride channels, 14 Trp-like channels, 24 amiloride-sensitive/degenerin-like sodium channels, one ryanodine receptor,one IP3 (inositol 1,4,5-trisphosphate) receptor,eight innexins, and two porins. C. elegans ismissing a nitric oxide synthase gene, copies ofwhich occur in fly and vertebrate genomes.
A large array of proteins mediates specificaspects of synaptic vesicle trafficking and con-tributes to the conversion of electrical signals toneurotransmitter release. These components ofexocytosis and endocytosis are relatively wellconserved with respect to both domain struc-tures and amino acid identities (50 to 90%). Thefly has enzymes for the synthesis of the neuro-transmitters glutamate, dopamine, serotonin,histamine, GABA, acetylcholine, and octopam-ine, and a family of conserved transporters islikely to be involved in loading vesicles withthese neurotransmitters. The conserved vesicu-lar trafficking proteins, with 50 to 80% aminoacid identity, include members of the Munc-18,SCAMP, synaptogyrin, HRS2, tomosyn, cys-teine string protein, exocyst (SEC 5, 6, 7, 8, 10,13, 15, EXO 70, and EXO84), synapsin, rab-philin-3A, RIM, rab-3, CAPS, Mint, Munc-13,NSF, a and g SNAP, DOC-2B, latrophilin,Veli, CASK, VAP-33, Snapin, SV2, and com-plexin families. Generally, there is only onehomolog in Drosophila for every three tofour isoforms in mammals. However, there areeight fly synaptotagmin-like genes, making thisthe largest family of vesicle proteins in Dro-sophila (47). However, there is no homolog ofsynaptophysin, an early candidate for a vesiclefusion pore, which indicates a nonessential rolein exocytosis for this particular protein acrossphyla.
Membrane trafficking also requires inter-actions between compartment-specific vesic-ular and target membrane proteins (v-SNAREs and t-SNAREs, respectively),whose subcellular distribution and combina-torial binding patterns are predicted to defineorganelle identity and targeting specificity(48). The completed fly genome allows us to
address whether there is any correlation be-tween the increased developmental complex-ity of multicellular organisms and a largernumber of SNAREs than that found in uni-cellular organisms. In the fly, we find sixsynaptobrevins, three SNAP-25s, 10 syntax-ins, and four additional t-SNAREs (membrin,BET1, UFE1, and GOS28), and the numberof SNAREs is similar between yeast (49) andDrosophila. Thus, basic subcellular compart-mentalization and membrane trafficking toand between these various compartments hasnot changed dramatically in multicellular ver-sus unicellular organisms. Dynamin, clathrin,the clathrin adapter proteins, amphiphysin,synaptojanin, and a number of additionalgenes that encode proteins with defined en-docytotic motifs are all present.
In contrast to the conservation of the syn-aptic vesicle trafficking machinery, the fewidentified proteins present at mammalian ac-tive zones, namely aczonin, bassoon, and pic-colo, do not have relatives in Drosophila.There are, however, numerous proteins in thefly with combinations of C2 domains, PDZdomains, zinc fingers, and proline-rich do-mains, indicating that the precise proteincomposition of active zones is likely to varyamong metazoans. In addition, Drosophilacontains a neurexin III gene and four neuroli-gin genes that may be part of a neurexin-
neuroligin complex that has been widely pro-posed to provide a synaptic scaffold for link-ing pre- and postsynaptic structures in mam-mals (50). Potential agrin and Musk genes arealso present, though the overall sequencesimilarity is low.
Immunity. Multicellular organisms haveelaborate systems to defend against microbialpathogens. Only vertebrates have an acquiredimmune system, but both vertebrates and in-vertebrates share a more primitive innate im-mune system. Innate immunity is based onthe detection of common microbial moleculessuch as lipopolysaccharides and peptidogly-cans by a class of receptors known as patternrecognition receptors (51). We identified alarge family of genes encoding homologs ofreceptors that are involved in microbial rec-ognition in other organisms. These includetwo new homologs of the Drosophila Scav-enger Receptors (dSR-CI), nine members ofthe CD36 family, 11 members of the pepti-doglycan recognition protein (PGRP) family,three Gram-negative binding protein (GNBP)homologs, and several lectins (52).
The recognition of infection by immuno-responsive tissues induces a battery of de-fense genes via Toll/nuclear factor kappa B(NF-kB) pathways in both Drosophila andmammals (53). The Toll receptor was initial-ly discovered as an essential component of
Table 5. Proteins in D. melanogaster, C. elegans, and S. cerevisiae with more than one InterPro domain.These numbers represent the total number of recognizable domains within a single protein, no matterwhether they are multiple copies of the same domain or different domains.
InterPro domains perprotein
D. melanogaster(number of proteins)
C. elegans(number of proteins)
S. cerevisiae(number of proteins)
2 920 1236 4103 388 458 1214 219 182 585 163 98 266 101 72 177 92 53 158 58 27 79 42 25 4
10 22 18 711–15 73 43 616–20 18 17 121–30 22 22 031–50 8 5 051–75 4 5 0
Table 6. Proteins in D. melanogaster, C. elegans, and S. cerevisiae with multiple different InterProdomains. Individual InterPro domains are counted only once per protein, regardless of how many timesthey occur in that protein.
Unique InterProdomains per
protein
D. melanogaster(number of proteins)
C. elegans(number of proteins)
S. cerevisiae(number of proteins)
2 1474 1248 4023 413 335 954 156 114 235 52 38 46 8 9 1
7 or more 4 3 0
www.sciencemag.org SCIENCE VOL 287 24 MARCH 2000 2213
T H E D R O S O P H I L A G E N O M E
on October 12, 2018
http://science.sciencem
ag.org/D
ownloaded from

the pathway that establishes the dorsoventralaxis of the Drosophila embryo. Recent geneticstudies now reveal that Toll signaling pathwaysare key mediators of immune responses to fungiand bacteria in both Drosophila and mice (53).We found seven additional homologs of Tollproteins in Drosophila, all of which are moresimilar to each other than to their mammaliancounterparts. Some of these other Toll proteins,like 18-wheeler, will probably mediate innateimmune responses. In Drosophila, infection byat least some microbes induces a proteolyticcascade that leads to the processing of Spaetzle(SPZ), a cytokine-like protein, which then acti-vates Toll (53). We found two proteins relatedto SPZ with similarities that include most or allof the cysteine residues of SPZ. Given thepresence of multiple Toll-like receptors in Dro-sophila, these new SPZ-like proteins may alsofunction in the immune system. With the ex-ception of the two I-kB kinase homologs andthe three rel proteins (Dorsal, Dif, and Relish),the Drosophila genome appears to contain onlysingle copies of the genes encoding intracellularcomponents of the Toll pathway: Tube, Pelle,and Cactus. How do the different Toll receptorstrigger specific immune responses using thesame intracellular intermediates? One explana-tion is that additional signaling componentsremain unidentified; another explanation iscrosstalk with other signaling pathways. In con-trast, a Toll ortholog has not been identified inC. elegans, although there are some Toll-likereceptors. C. elegans, in addition, does not pos-sess homologs of NF-kB/dorsal transcriptionalactivators that function downstream of Toll.Although it is probable that the worm has re-tained parts of the innate immunity network,there is no clear evidence of an inducible hostdefense system in the worm.
One of the most potent innate immuneresponses in insects is the transcriptional in-duction of genes encoding antimicrobial pep-tides (53). In contrast to Metchnikowin,Drosocin, and Defensin peptides, which areencoded by single genes, the sequence dataindicate that, like the previously identifedcecropin clusters, several antimicrobial pep-tides are encoded by gene families that arelarger than previously suspected. Four genesappear to encode antifungal peptide Droso-mycin isoforms, and two genes each code forthe antibacterial proteins Attacin and Dipteri-cin. These additional genes may generatepeptides with slightly different spectra of an-timicrobial activity or may simply amplifythe antimicrobial response.
Concluding RemarksWhat have we learned about the proteinsencoded by the three sequenced eukaryoticgenomes? Some information emerges readilyfrom the comparison of the fly, worm, andyeast genomes. First, the core proteome sizesof flies and worms are similar and are only
twice the size of that of yeast. This is perhapscounterintuitive, because the fly, a multicel-lular animal with specialized cell types, com-plex development, and a sophisticated ner-vous system, looks more than twice as com-plicated as single-celled yeast. The lesson isthat the complexity apparent in the metazoansis not achieved by sheer number of genes(54). Second, there has been a proliferationof bigger and more complex proteins in thetwo metazoans relative to yeast, including,not surprisingly, more proteins with extracel-lular domains involved in cell-cell and cell-substrate interactions. Finally, the populationof multidomain proteins is somewhat largerand more diverse in the fly than in the worm.There is presently no practical way to quan-tify differences in biological complexity be-tween two organisms, however, so it is notpossible to correlate this increased domainexpansion and diversity in the fly with differ-ences in development and morphology.
The availability of the annotated sequenceof the Drosophila genome enhances the fly’susefulness as an experimental organism. Bygreatly facilitating positional cloning, the ge-nome sequence will increase the efficiency ofgenetic screens that seek to identify genesunderlying many complex processes of cellbiology, development, and behavior. Suchscreens have been the mainstay of Drosoph-ila research and have contributed enormouslyto our knowledge of metazoan biology. Thegenome sequencing effort has revealed anumber of previously unknown counterpartsto human genes involved in cancer and neu-rological disorders; for example, p53, menin,tau, limb girdle muscular dystrophy type 2B,Friedrich ataxia, and parkin. All of these flygenes are present in a single copy in thegenome and can be genetically analyzedwithout uncertainty about redundant copies.More genetic screens are important in orderto uncover interacting network members. Or-thologs of these network members can thenbe sought in the human genome to determineif alterations in any of them predispose hu-mans to the disease in question, an experi-mental paradigm that has already been suc-cessfully executed in several cases. Flies canalso play an important role in exploring waysto rectify disease phenotypes. For example, atleast 10 human neurodegenerative diseasesare caused by expansion of polyglutaminerepeats (55). Human proteins containing ex-panded polyglutamine repeats have been ex-pressed in flies, resulting in the formation ofnuclear inclusions that contain the protein aswell as other shared components (56), just asin humans. It has been shown that directedexpression of the human HSP70 chaperone inthe fly can totally suppress neurodegenera-tion resulting from expression of the humanspinocerebellar ataxia type 3 protein (57).The power and speed of this in vivo system
are unparalleled, and we anticipate the in-creased use of such “humanized” fly models.
Knowing the complete genomic sequencealso allows new experimental approaches tolong-standing problems. For example, itmakes it possible to study networks of genesrather than individual genes or pathways. As-saying the level of transcription of every genein the genome makes it at least theoreticallypossible to monitor the expression of an en-tire network of genes simultaneously. Oneproblem that is approachable this way is thecombinatorial control of gene transcription.The fly genome appears to encode only about700 transcription factors, and mutations inover 170 have already been isolated and char-acterized. The techniques are available tomeasure the changes in expression of everygene in individual cell types as a consequenceof loss or overexpression of each transcrip-tion factor. We can look for common se-quence elements in the promoters of coregu-lated genes and perform chromatin immuno-precipitation to identify the in vivo bindingsites of individual factors. For the first time,we can envision obtaining the data needed tounderstand the behavior of a complex regu-latory network. Of course, collecting thesedata is a massive task, and developing meth-ods to analyze the data is even more daunting.But it is no longer ludicrous to try.
How big is the core proteome of humans?Vertebrates have many gene families withthree or four members: the HOX clusters,calmodulins, Ezrins, Notch receptors, nitricoxide synthases, syndecans, and NF1 tran-scription factor genes are some examples(58). This is evidence for two genome dou-blings during mammalian evolution, super-imposed on which were the amplificationsand contractions over evolutionary time thatuniquely characterize each lineage (59). Thehuman genome, with 80,000 or so genes, islikely to be an amplified version of a very muchsmaller genome, and its core proteome may notbe much larger than that of the fly or worm; thatis, the more complex attributes of a humanbeing are achieved using largely the samemolecular components. The evolution of ad-ditional complex attributes is essentially anorganizational one; a matter of novel interac-tions that derive from the temporal and spa-tial segregation of fairly similar components.
Finally, approximately 30% of the predictedproteins in every organism bear no similarity toproteins in its own proteome or in the pro-teomes of other organisms. In other words,sequence similarity comparisons consistentlyfail to give us information about nearly a thirdof the components that make every organismuniquely itself. What does this mean with re-spect to the evolution and function of theseproteins? Does each genome contain a sub-population of very rapidly evolving genes?One-third of randomly chosen cDNA clones do
24 MARCH 2000 VOL 287 SCIENCE www.sciencemag.org2214
T H E D R O S O P H I L A G E N O M E
on October 12, 2018
http://science.sciencem
ag.org/D
ownloaded from

not cross-hybridize between D. melanogasterand Drosophila virilis (60). Even though theseare distantly related species, they are develop-mentally and morphologically very similar.Crystallographic data will be needed to deter-mine whether these proteins that have divergedin primary sequence have maintained theirthree-dimensional structures or have divergedso far that new folds and domains have formed.
Our first look at the annotated fly genomeprovokes these and other questions. Access tothe genomic sequence will help us design theexperiments needed to answer them. The rel-ative simplicity and manipulability of the flygenome means that we can address some ofthese biological questions much more readilythan in vertebrates. That is, after all, whatmodel organisms are for.
References and Notes1. M. D. Adams et al., Science 287, 2185 (2000); C.
elegans Sequencing Consortium, Science 282, 2012(1998); A. Goffeau et al., Science 274, 546 (1996).
2. R. D. Fleischman et al., Science 269, 496 (1995).3. C. elegans data were taken from A C. Elegans Data-
base (ACEDB) release WS8.4. Local gene duplications were determined by search-
ing for N similar genes within 2N genes on each arm.For example, if three similar genes are found within aregion containing six genes, this counts as one clusterof three genes. Genes were judged to be similar if aBLASTP High Scoring Pair (HSP) with a score of 200 ormore existed between them. Histone gene clusterswere not included. C. elegans data were taken fromACEDB release WS8, containing 18,424 genes.
5. More information about GO is available at http://www.geneontology.org/. The Gene Ontology projectprovides terms for categorizing gene products on thebasis of their molecular function, biological role, andcellular location using controlled vocabularies.
6. Initial results came from an NxN BLASTP analysis per-formed for each fly, worm, and yeast sequence in acombined data set of these completed proteomes. Thedatabases used are as follows: Celera–Berkeley Dro-sophila Genome Project (BDGP), 14,195 predicted pro-tein sequences (1/5/2000); WormPep 18, Sanger Cen-tre, 18,576 protein sequences; and Saccharomyces Ge-nome Database (SGD), 6306 protein sequences (1/7/2000). A version of NCBI-BLAST2 was used with theSEG filter and with the effective search space length ( Yoption) set to 17,973,263. Pairs were formed betweenevery query sequence with a significant BLASTP to oneof the other organisms’ sequences. Significance wasbased on E-value cutoffs and length of match. Thesepairs were then independently grouped using singlelinkage clustering (61). Finally, the number of proteinsfrom each proteome was counted. The requirement for80% alignment of sequences makes this method ofdefining orthology particularly sensitive to errors thatarise from incorrect protein prediction. However, theresults comparing yeast and worm are essentially iden-tical to those previously reported (61), even though theeffective database size was different, the data sets havechanged (Chervitz: yeast 6217 and worm 19,099; thisstudy: yeast 6306, and worm 18,576), and the versionof BLAST used is quite different (Chervitz: WashU BLAST2.0a19MP; this study: NCBI BLAST 2.08).
7. A. Bairoch and R. Apweiler, Nucleic Acids Res. 28, 45(2000).
8. J. G. Henikoff, E. A. Greene, S. Pietrokovski, S. Heni-koff, Nucleic Acids Res. 28, 228 (2000).
9. InterPro (Integrated resource for protein domains andfunctional sites) is a collaborative effort of theSWISS-PROT, TrEMBL, PROSITE, PRINTS, Pfam, andProDom databases to integrate the different patterndatabases into a single resource. The database and adetailed description of the project can be found underhttp://www.ebi.ac.uk/interpro/. PROSITE is described
in K. Hofmann, P. Bucher, L. Falquet, A. Bairoch,Nucleic Acids Res. 27, 215 (1999); PFAM is describedin A. Bateman et al., Nucleic Acids Res.27, 260(1999); and PRINTS is described in T. K. Attwood etal., Nucleic Acids Res. 27, 220 (1999).
10. G. D. Plowman, S. Sudarsanam, J. Bingham, D. Whyte,T. Hunter, Proc. Natl. Acad. Sci. U.S.A. 96, 13603,(1999).
11. J. Barrett, N. D. Rawlings, J. F. Wessner, Eds., Hand-book of Proteolytic Enzymes (Academic Press, SanDiego, CA, 1998).
12. C. L. Smith and R. DeLotto, Nature 368, 548 (1994);K. D. Konrad, T. J. Goralski, A. P. Mahowald, J. L.Marsh, Proc. Natl. Acad. Sci. U.S.A. 95, 6819 (1998);E. K. LeMosy, C. C. Hong, C. Hashimoto, Trends CellBiol. 9, 102 (1999).
13. R. O. Hynes, Trends Cell Biol. 9, M33 (1999).14. P. Bork, A. K. Downing, B. Kieffer, I. D. Campbell,
Quart. Rev. Biophys. 29, 119 (1996).15. P. Vernier, B. Cardinaud, O. Valdenaire, H. Philippe,
J.-D. Vincent, Trends Pharmacol. Sci. 16, 375, (1995);J. Colas, J. Launay, J. Vonesch, P. Hickel, L. Maroteaux,Mech. Dev. 87, 77 (1999); M. R. Costa, E. T. Wilson, E.Wieschaus, Cell 76, 1075 (1994).
16. P. Mombaerts, Science 286, 707, (1999).17. C. I. Bargmann, Science 282, 2028 (1998).18. P. J. Clyne et al., Neuron 22, 327 (1999); L. B.
Vosshall, H. Amrein, P. S. Morozov, A. Rzhetsky, R.Axel, Cell 96, 725 (1999); P. P. Laissue et al., J. Comp.Neurol. 405, 543 (1999).
19. Y. J. Lin, L. Seroude, S. Benzer, Science 282, 943(1998).
20. Y. Zhang, Y. Xiong, W. G. Yarbrough, Cell 92, 725(1998).
21. S. N. Jones, A. E. Roe, L. A. Donehower, A. Bradley,Nature 378, 206 (1995).
22. I. The et al., Science 276, 791 (1997).23. N. Ito and G. M. Rubin, Cell 96, 529 (1999).24. M. O. Hengartner and H. R. Horvitz, Cell 76, 665
(1994).25. F. Hauser, H. P. Nothacker, C. J. Grimmelikhuijzen,
J. Biol. Chem. 272, 1002 (1997).26. P. R. Mueller, T. R. Coleman, A. Kumagai, W. G.
Dunphy, Science 270, 86 (1995).27. B. D. Dynlacht, A. Brook, M. Dembski, L. Yenush, N.
Dyson, Proc. Natl. Acad. Sci. U.S.A. 91, 6359 (1994);W. Du, M. Vidal, J.-E. Xie, N. Dyson, Genes Dev. 10,1206 (1996); T. Sawado et al., Biochem. Biophys. Res.Commun. 251, 409 (1998).
28. X. Lu and H. R. Horvitz, Cell 95, 981 (1998).29. T. Kreis and R. Vale, Eds., Guidebook to the Cytoskel-
etal and Motor Proteins (Oxford Univ. Press, Oxford,1999).
30. P. Chang and T. Stearns, Nature Cell Biol. 2, 30(2000).
31. S. K. Dutcher and E. C. Trabuco, Mol. Biol. Cell 9, 1293(1998).
32. A. Desai, S. Verma, T. J. Mitchison, C. E. Walczak, Cell96, 69 (1999).
33. K. Weber, in (29), pp. 291–293.34. J. Kumar, H. Yu, M. P. Sheetz, Science 267, 1834
(1995).35. Q. Wu and T. Maniatis, Cell 97, 779 (1999).36. K. Senzaki, M. Ogawa, T. Yagi, Cell 99, 635 (1999).37. M. P. Belvin and K. V. Anderson, Annu. Rev. Cell Dev.
Biol. 12, 393 (1996); M. Hammerschmidt, A. Brook,A. P. McMahon, Trends Genet. 13, 14 (1997); C. M.Blaumueller and S. Artavanis-Tsakonas, Perspect.Dev. Neurobiol. 4, 325 (1997); T. Hunter, Philos.Trans. R. Soc. London Ser. B 353, 583 (1998); K. M.Cadigan and R. Nusse, Genes Dev. 11, 3286 (1997); J.Capdevila and J. C. Belmonte, Curr. Opin. Genet. Dev.9, 427 (1999); L. Engstrom, E. Noll, N. Perrimon, Curr.Top. Dev. Biol. 35, 229 (1997); B. E. Stronach and N.Perrimon, Oncogene 18, 6172 (1999); P. W. H. Hol-land, J. Garcia-Fernandez, N. A. Williams, A. Sidow,Development (suppl.) (1994), p. 125.
38. G. Ruvkun and O. Hobert, Science 282, 2033 (1998).39. W. C. Earnshaw, L. M. Martins, S. H. Kaufmann, Annu.
Rev. Biochem. 68, 383 (1999); A. Zeuner, A. Eramo, C.Peschle, R. DeMaria, Cell Death Diff. 6, 1075 (1999).
40. X. Liu, C. N. Kim, J. Yang, R. Jemmerson, X. Wang, Cell86, 147 (1996); S. A. Susin et al., Nature 397, 441(1999).
41. P. Li et al., Cell 91, 479 (1997).
42. A. G. Park, Trends Cell Biol. 10, 394 (2000); S. Saharaet al., Nature 401, 168 (1999).
43. A. Gross, J. M. McDonnell, S. J. Korsmeyer, GenesDev.13, 1899 (1999).
44. L. K. Miller, Trends Cell Biol. 9, 323 (1999).45. J. M. Abrams, Trends Cell Biol. 9, 435 (1999).46. K. Thress, W. Henzel, W. Shillinglaw, S. Kornbluth,
EMBO J. 17, 6135 (1998).47. J. T. Littleton, T. L. Serano, G. M. Rubin, B. Ganetzky,
E. R. Chapman, Nature 400, 757 (1999).48. T. Solner et al., Nature 362, 318 (1993).49. R. Jahn and T. C. Sudhof, Annu. Rev. Biochem. 68, 863
(1999).50. K. Ichtchenko et al., Cell 81, 435 (1995).51. R. Medzhitov and C. A. Janeway Jr., Cell 91, 295
(1997).52. A. Pearson, Current Opin. Immunol. 8, 20 (1996);
N. C. Franc et al., Immunity 4, 431 (1996); D. Kang etal., Proc. Natl. Acad. Sci. U.S.A. 95, 10078 (1998);W. J. Lee et al., Proc. Natl. Acad. Sci. U.S.A. 93, 7888(1996).
53. J. A. Hoffmann and J.-M. Reichhart, Trends Cell Biol. 7,309 (1997); K. V. Anderson, Curr. Opin. Immun.12, 13(2000).
54. G. L. G. Miklos, J. Am. Acad. Arts Sci. 127, 197 (1998).55. M. Perutz, Trends Biochem. Sci 24, 58 (1999).56. J. M. Warrick et al., Cell 93, 939 (1998); G. R. Jackson
et al., Neuron 21, 633 (1998).57. J. M. Warrick et al., Nature Genet. 23, 425 (1999).58. J. Spring, FEBS Lett.400, 2 (1997).59. S. Aparicio, Trends Genet. 16, 54 (2000).60. K. J. Schmid and D. Tautz, Proc. Natl. Acad. Sci. USA.
94, 9746 (1997).61. S. A. Chervitz et al., Science 282, 2022 (1998).62. See www.sciencemag.org/feature/data/1049664.shl
for complete protein domain analysis.63. Paralogous gene families ( Table 1) were identified
by running BLASTP. A version of NCBI-BLAST2optimized for the Compaq Alpha architecture wasused with the SEG filter and the effective searchspace length ( Y option) set to 17,973,263. Eachprotein was used as a query against a database ofall other proteins of that organism. A clusteringalgorithm was then used to extract protein familiesfrom these BLASTP results. Each protein sequenceconstitutes a vertex; each HSP between proteinsequences is an arc, weighted by the BLAST Expectvalue. The algorithm identifies protein families byfirst breaking all arcs with an E value greater thansome user-defined value (1 3 10– 6 was used for allof the analyses reported here). The resulting graphis then split into subgraphs that contain at leasttwo-thirds of all possible arcs between vertices.The algorithm is “greedy”; that is, it arbitrarilychooses a starting sequence and adds new se-quences to the subgraph as long as this criterion ismet. An interesting property of this algorithm isthat it inherently respects the multidomain natureof proteins: For example, two multidomain pro-teins may have significant similarity to one anoth-er but share only one or a few domains. In such acase, the two proteins will not be clustered if theunshared domains introduce a large number ofother arcs.
64. An NxN BLASTP analysis was performed for each fly,worm, and yeast sequence in a combined data set ofthese completed proteomes. The databases used areas follows: Celera-BDGP, 14,195 predicted proteinsequences (1/5/2000); WormPep18, Sanger Centre,18,424 protein sequences; and SGD, 6246 proteinsequences (1/7/2000). BLASTP analysis was also per-formed against known mammalian proteins (2/1/2000, GenBank nonredundant amino acid, Human,Mouse, and Rat, 75,236 protein sequences), andTBLASTN analysis was performed against a databaseof mammalian ESTs (2/1/00, GenBank dbEST, Hu-man, Mouse, and Rat). A version of NCBI-BLAST2optimized for the Compaq Alpha architecture wasused with the SEG filter and the effective searchspace length ( Y option) set to 17,973,263.
65. The many participants from academic institutions aregrateful for their various sources of support. Partici-pants from the Berkeley Drosophila Genome Projectare supported by NIH grant P50HG00750 (G.M.R.)and grant P4IHG00739 (W.M.G.).
www.sciencemag.org SCIENCE VOL 287 24 MARCH 2000 2215
T H E D R O S O P H I L A G E N O M E
on October 12, 2018
http://science.sciencem
ag.org/D
ownloaded from

Comparative Genomics of the Eukaryotes
Craig Venter, Mark D. Adams and Suzanna LewisPickeral, Chris Shue, Leslie B. Vosshall, Jiong Zhang, Qi Zhao, Xiangqun H. Zheng, Fei Zhong, Wenyan Zhong, Richard Gibbs, J.Jones, Peter M. Kuehl, Bruno Lemaitre, J. Troy Littleton, Deborah K. Morrison, Chris Mungall, Patrick H. O'Farrell, Oxana K. Fangcheng Gong, Ping Guan, Nomi L. Harris, Bruce A. Hay, Roger A. Hoskins, Jiayin Li, Zhenya Li, Richard O. Hynes, S. J. M.Coates, Anibal Cravchik, Andrei Gabrielian, Richard F. Galle, William M. Gelbart, Reed A. George, Lawrence S. B. Goldstein, Michael Ashburner, Ewan Birney, Mark S. Boguski, Thomas Brody, Peter Brokstein, Susan E. Celniker, Stephen A. Chervitz, DavidE. Fortini, Peter W. Li, Rolf Apweiler, Wolfgang Fleischmann, J. Michael Cherry, Steven Henikoff, Marian P. Skupski, Sima Misra, Gerald M. Rubin, Mark D. Yandell, Jennifer R. Wortman, George L. Gabor , Miklos, Catherine R. Nelson, Iswar K. Hariharan, Mark
DOI: 10.1126/science.287.5461.2204 (5461), 2204-2215.287Science
ARTICLE TOOLS http://science.sciencemag.org/content/287/5461/2204
REFERENCES
http://science.sciencemag.org/content/287/5461/2204#BIBLThis article cites 76 articles, 22 of which you can access for free
PERMISSIONS http://www.sciencemag.org/help/reprints-and-permissions
Terms of ServiceUse of this article is subject to the
registered trademark of AAAS. is aScienceAmerican Association for the Advancement of Science. No claim to original U.S. Government Works. The title
Science, 1200 New York Avenue NW, Washington, DC 20005. 2017 © The Authors, some rights reserved; exclusive licensee (print ISSN 0036-8075; online ISSN 1095-9203) is published by the American Association for the Advancement ofScience
on October 12, 2018
http://science.sciencem
ag.org/D
ownloaded from





























