Embed Size (px)
Citation preview

Complexes of alkali metal cations with trifluoromethyl:
A computational investigation on the structure and stability
of MC–(CF3) (MZLi, Na, K) isomers
Jun Lianga,b,c,*, Haiyang Lia,c,*, Ying Liua,c, Shuang Chenga,c
aDalian Institute of Chemical Physics, Chinese Academy of Science, Dalian 116023, People’s Republic of ChinabDepartment of Physics, Wuhu Teacher College, Anhui Normal University, Wuhu 241008, People’s Republic of China
cAnhui Institute of Optics and Fine Mechanics, Chinese Academy of Sciences, Hefei 230031, People’s Republic of China
Received 27 January 2005; revised 9 March 2005; accepted 10 March 2005
Available online 13 June 2005
Abstract
The stationary points characterizing the potential energy profiles of the complex process of the MC (MZLi, Na, K) and CF3 were
investigated by B3LYP in conjunction with the 6-311CG(2d,2p) basis set. The optimized geometries, chemical bonding and NBO analysis
indicate that the complexes of MC (MZLi, Na, K) and CF3 exist as ion–dipole molecules. The calculated affinity energies of the most stable
isomer of LiC, NaC, and KC complexes exceed 10.1 kJ/mol, and these values suggest that the MC–CF3 (MZLi, Na, K) complexes could be
observed as stable species in gas phase, which supports Fujii’s proposal that LiC ion attachment mass spectrometry can serve as a
conceivable technique to detect and quantify the emissions of the perfluorocarbons.
q 2005 Elsevier B.V. All rights reserved.
Keywords: Gas-phase chemistry; Trifluoromethyl; Alkali metal ion; Ion attachment mass spectrometry
1. Introduction
In 1971, a new method known as alkali metal ion
attachment chemical ionization mass spectrometry was
proposed by Beauchamp [1] and others [2,3]. In this
method, alkali metal ions such as LiC, NaC, and KC form
adduct ions (also referred to as cationized molecules) with
molecular species through association reactions. In general,
these cationized molecules are more stable than radical
molecular ions or protonated molecules in the gas phase.
The unique features of the LiC–MS system have been
extensively used by Fujii and co-workers [4–10] to detect
intermediate free radicals, novel molecular species, and
unfamiliar and interstellar species in various MW discharge
plasmas. Reactions between alkali metal cations and the
perfluoro-compounds (PFCs) have attracted a great deal of
0166-1280/$ - see front matter q 2005 Elsevier B.V. All rights reserved.
doi:10.1016/j.theochem.2005.03.031
* Corresponding authors. Address: Department of Physics, Wuhu
Teacher College, Anhui Normal University, Wuhu 241008, People’s
Republic of China. Tel.: C86 551 559 3204; fax: C86 553 577 1530.
E-mail address: [email protected] (J. Liang).
attention in the last two decades, for these reactions are
involved in a significant number of relevant processes in
atmosphere chemistry [11–15]. CF3 is one of the important
radicals in the stratosphere. In recent years, the CF3 free
radical has been the subject of considerable research
interest. This important species is believed to play a
significant role in the ozone layer depletion in the
atmosphere and a variety of ‘down to earth’ processes
such as semiconductor etching and halocarbon fire suppres-
sion [16–18]. In a recent study, Fujii and Arulmozhiraja [11]
have performed density functional theory (DFT) calcu-
lations to investigate the feasibility of measuring the PFCs
in exhaust from semiconductor industry by LiC ion
attachment mass spectrometry. At the B3LYP/6-311CG(3df) level of theory, their computed LiC affinities range
from 12.3 kcal molK1 for CF4 to 21.1 kcal molK1 for C4F8,
and these values are large enough to suggest that industrial
emissions of these PFCs could be in principle detected and
quantified by LiC ion attachment experiments. Recently, we
have investigated the stationary points characterizing the
potential energy profiles of the complex processes of the
MC (MZH, Li, Na, K) and NF3 by Density Functional
Theory (DFT) using the B3LYP hybrid potential and
Journal of Molecular Structure: THEOCHEM 725 (2005) 151–155
www.elsevier.com/locate/theochem

J. Liang et al. / Journal of Molecular Structure: THEOCHEM 725 (2005) 151–155152
the 6-311Cg (2d, 2p) basis set [19]. It was found that the
combinations of MC (MZLi, Na, K) to the F atoms of NF3
may occur in two distinct ways, leading to the formation of
the monocoordinated isomer and the dicoordinated isomer.
Stimulated by these findings, we decided to perform DFT
and ab initio calculations on the structure and stability of the
complexes between the alkali metal cation and trifluor-
omethyl radical. From the applied point of view, it is of
interest to investigate whether the alkali metal cation affinity
of CF3 is large enough to suggest the possible experimental
detection and quantification of CF3 by the alkali metal ion
attachment spectrometry. From the fundamental point of
view, the study of the MC–(CF3) (MZLi, Na, K) isomers is
of interest to learn more on the behavior of CF3 as a gaseous
Lewis base.
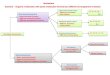
Fig. 1. The optimized geometry of the MC (MZLi, Na, K) and CF3
complexes.
2. Computational details
All calculations were performed using GAUSSIAN 03
program [20]. The Becke’s three parameter hybrid density
functional, B3LYP, which includes a mixture of Hartree–
Fock exchange and DFT exchange correlation has been used
[21,22]. The ligands, as well as complexes, were
optimized first at the B3LYP functional using the 6-311CG(2d,2p) basis set followed by the frequency calculation.
The binding energies (DE) were obtained from the
difference between the total energy of the complex
[E(MCKCF3)] and the sum of the total energies of the
corresponding MC (MZLi, Na, K) ion [E(MC)] and
trifluoromethyl [E(CF3)] using the optimized energies:
DEZ ½EðMC � CF3Þ�K f½EðCF3Þ�C ½EðMCÞ�g. These opti-
mized geometries were characterized by harmonic
vibrational frequency calculations at the same B3LYP/
6-311CG(2d,2p) level. These frequency calculations
also yielded the zero-point energies (ZPE),
which were left unscaled; thermal corrections (at
298.15 K) were needed for the calculation of enthalpies.
Binding enthalpies (MC affinities of ligands)
were then calculated using the following relation: DHZDEC DEZPE CDEthermal CDðPVÞ, where D(PV)ZnRTZK0.593 kcal molK1 at 298.15 K. MC (MZLi, Na,
K) affinities were calculated using the energies of the
optimized structures at B3LYP/6-311CG(2d,2p) level and
corrected by ZPE and thermal corrections, which were
obtained at B3LYP/6-311CG(2d,2p) level. Chemical
bonding analysis was based on the theory of Atoms in
Molecules [23], using the implentation in GAUSSIAN 03 due to
Cioslowski and co-workers [24–28]. In particular, for each
structure we have calculated the B3LYP/6-311CG(2d,2p)
charge density r and the Laplacian of the charge density D2r
at the bond critical points (BCP), intended as the points on
the attractor interaction lines where VrZ0. Atomic charges
were derived using the natural population analysis (NPA)
scheme [29,30].
3. Results and discussion
3.1. Geometries and chemical bonding analysis
The connectivities of the various MC–(CF3) (MZLi, Na,
K) isomers presently located as stationary points on the
B3LYP potential energy surfaces, henceforth indicated as
structures 1–4, are shown in Fig. 1. Their detailed
geometries, the results of chemical bonding analysis and
the NBO charges are collected in Tables 1–3, respectively,
For comparative purposes and also to appreciate the
performance of the various employed theoretical levels,
we have also investigated the uncoordinated ligand CF3.
The addition of MC to the C atom of CF3 leads to the
formation of C3V symmetry ion 1, which was characterized
as a local minimum point for LiC and second-order saddle
points for NaC and KC, respectively, on their respective
energy surfaces. In structure 1, r(Li–C), r(Na–C), r(K–C)
are longer than 2.4 A, which are structurally-telling of ion–
dipole adducts between LiC, NaC, KC and CF3. Accord-
ingly, from Table 2, the BCPs on the attractor interaction
lines corresponding to the MC–C bonds are associated with
charge densities r as small as 0.012, 0.008, and 0.004 e/a.u.3
for LiC, NaC and KC, respectively, and with positive
values of the Laplacian of r, which are typical of
electrostatic interactions. Inspection of Table 3, the NBO
charges on the Li, Na, K are larger than 0.95, but charges on
CF3 less than 0.05. LiC, NaC, KC and CF3 are in the two
different charge units, which indicate weak interactions
between MC (MZLi, Na, K) and CF3. Combining NBO
charge distribution and the above bond length discussion,
MC and CF3 complexes would exist as MC–CF3(MZLi,
Na, K) ion–dipole type complexes. The optimized structure

Table 1
The geometry parameters of the optimized isomers of the MC (MZLi, Na,
K) and CF3 complexes
M Li Na K
Structure1
M–C 2.4994 2.9349 3.5134
C–F 1.3088 1.3119 1.3152
M–C–F 105.25 105.7798 106.3075
F–C–F 113.34 112.8968 112.4411
Structure 2
M–F1 1.828 2.2298 2.6839
C–F 1.2886 1.2975 1.3032
C–F1 1.4248 1.3907 1.3723
F–C–F 115.0422 114.0658 113.4684
F–C–F1 108.5486 109.3388 109.818
M–F1–C 160.1175 176.8916 177.7107
Structure 3
M–F 2.0813 2.4891 2.9515
C–F 1.359 1.3488 1.3419
C–F1 1.2787 1.2877 1.2947
F–C–F 104.76 106.7559 107.9931
F–C–F1 112.945 112.4981 112.1789
Structure 4
M–F 2.4777 2.83199 3.33385
C–F 1.3298 1.3275 1.32600
F–C–F 108.7782 109.5025 110.0746
M–F–C 79.9017 70.5601 38.0466
CF3
C–F 1.3222
F–C–F 111.3562
Bond lengths in angstroms (A), bond angles in degrees (8).
J. Liang et al. / Journal of Molecular Structure: THEOCHEM 725 (2005) 151–155 153
1 and the corresponding bonding analysis reveal also that
the formal attachment of MC to the C atom of CF3 enhances
backdonation from the surrounding fluorines. Thus, at the
B3LYP level of theory, the C–F distances of structure 1 are
shorter than uncoordinated CF3 by ca. 0.013, 0.010 and
0.007 A, respectively, and the F–C–F bond angle are larger
by ca. 1.98, 1.66 and 1.118. Consistently, passing from
uncoordinated CF3 to structure 1, the charge densities of the
C–F bonds slightly increase and corresponding Laplacian
become more negative.
Table 2
B3LYP/6-311C(2d,2p) charge densities r (e/a.u.3) and Laplacian of the charge de
the attractor interaction lines of the MCKCF3 (MZLi, Na, K) complexes 1–4 an
Species Li Na
BCP r V2r BCP r
CF3 C–F 0.29165 K0.3101 C–F 0.
Structure 1 C–F 0.30453 K0.3380 C–F 0.
Li–C 0.01163 C0.0411 Na–C 0.
Structure 2 C–F 0.31860 K0.2551 C–F 0.
C–F1 0.22026 K0.3359 C–F1 0.
Li–F1 0.02901 C0.2450 Na–F1 0.
Structure 3 C–F 0.26630 K0.3770 C–F 0.
C–F1 0.32622 K0.2124 C–F1 0.
Li–F 0.16312 C0.1114 Na–F 0.
Structure 4 C–F 0.28778 K0.3394 C–F 0.
Li–C 0.00587 C0.0405 Na–F 0.
Searching for the complexes arising from the ligation of
MC to the fluorine atoms of CF3 leads to the location of
three distinct structures, namely, the monocoordinated ion 2
of Cs symmetry, the dicoordinated ion 3 of Cs symmetry,
and the tricoordinated ion 4 of C3V symmetry. However, at
the B3LYP/6-311CG(2d,2p) level of theory, only isomers 2
and 3 revealed as true minima on the potential energy
surface, whereas ion 4 revealed second-order saddle points,
unstable with respect to the degenerate bending motion of
the alkali metal atoms. Generally speaking, we first wish to
note that, although the ligation of the alkali metal cations to
the fluorine atom(s) of CF3 leads to sometimes appreciable
structural changes with respect to the uncoordinated ligand,
the BCPs on the attractor interaction lines corresponding to
the M–F (MZLi, Na, K) bonds of structures 2, 3, and 4
are invariably characterized by low values of the
charge densities and positive values of the corresponding
Laplacian, thus suggesting the formation of essentially
electrostatic complexes between MC (MZLi, Na, K) and
CF3. From Table 3, NBO analysis indicates that LiC, NaC,
KC and CF3 are in the two different charge units, and the
positive charges are almost on LiC, NaC, and KC,
respectively, which also indicate that LiC, NaC, KC and
CF3 complexes existing as MC–CF3 (MZLi, Na, K) ion–
dipole type complexes.
Structure 2 is the most stable structure in the four isomers
on their potential surfaces, respectively, and they are global
minima. The optimized structures of structure 2 reveal that
the ligation of MC (MZLi, Na, K) to a single F atom of CF3
induces an appreciable elongation of the involved C–F
bond. Thus, from Table 1, we note that, at the B3LYP level
of theory, the C–F1 bond distance of structure 2 is larger
than the C–F distance of CF3 by ca. 0.10, 0.08, and 0.05 A
for LiCKCF3, NaCKCF3, KCKCF3, respectively. In
addition, the C–F bond distances of structure 2 are
invariably predicted to be shorter than CF3 by ca. 0.033,
0.024, 0.019 A, and the F–C–F bond angle result larger by
ca. 3.70, 2.73 and 2.138 for LiC–CF3, NaC–CF3, KC–CF3,
respectively. From Table 2, all these structural changes
nsities V2r (e/a.u.5), evaluated at the bond critical points (BCP) located on
d CF3 (for connectivities and labeling of the atoms, see Fig. 1)
K
V2r BCP r V2r
29165 K0.3101 C–F 0.29165 K0.3101
30140 K0.3292 C–F 0.29823 K0.3217
00751 C0.0269 K–C 0.00443 C0.0135
31123 K0.2685 C–F 0.30674 K0.2794
24080 K0.3575 C–F1 0.25366 K0.3547
01900 C0.1356 K–F1 0.01335 C0.0700
27261 K0.3559 C–F 0.27719 K0.3420
31880 K0.2360 C–F1 0.31321 K0.2535
11342 C0.0677 K–F 0.00810 C0.0398
28869 K0.3261 C–F 0.28935 K0.3199
00480 C0.0290 K–F 0.00321 C0.0174

Table 3
NBO charges for the equilibrium geometries of the MC (MZ Li, Na, K)
and CF3 complexes
MCKCF3 Structure 1 Structure 2 Structure 3 Structure 4
LiCKCF3
C 0.91761 1.07615 1.07485 1.07461
Li 0.95041 0.99211 0.97360 0.96918
F K0.28934 K0.50149 K0.26339 K0.34793
F K0.28934 K0.28339 K0.39253 K0.34793
F K0.28934 K0.28339 K0.39253 K0.34793
NaCKCF3
C 0.95152 1.06567 1.06352 1.06864
Na 0.95778 0.99439 0.98143 0.97680
F K0.30310 K0.45998 K0.28053 K0.34848
F K0.30310 K0.30004 K0.38221 K0.34848
F K0.30310 K0.30004 K0.38221 K0.34848
KCKCF3
C 0.96340 1.05197 1.05193 1.05756
K 0.98312 0.99578 0.99080 0.98881
F K0.31551 K0.42903 K0.29482 K0.34879
F K0.31551 K0.30936 K0.37396 K0.34879
F K0.31551 K0.30936 K0.37396 K0.34879
NPA atomic charges.
Table 4
The total energies (E), zero-point energies (ZPE) and relative energies (DE)
of the optimized geometries of the MC (MZLi, Na, K) and CF3 complexes
M Li Na K
MCCCF3
E K344.948227 K499.750835 K937.424357
ZPE 0.011833 0.011833 0.011833
DE 0.0 0.0 0.0
Structure 1
E K344.952098 K499.752690 K937.423475
ZPE 0.012781 0.012283 0.012101
DE K10.16 K7.52 2.32
Structure 2
E K344.967558 K499.761318 K937.429266
ZPE 0.012610 0.012232 0.012038
DE K50.75 K27.52 K12.89
Structure 3
E K344.965135 K499.760155 K937.428301
ZPE 0.012821 0.012270 0.012091
DE K44.39 K24.47 K10.35
Structure 4
E K344.954862 K499.755306 K937.426220
ZPE 0.012196 0.012015 0.011905
DE K17.42 K12.26 K4.89
The total energies (E), zero-point energies (ZPE) in hartree. Relative
energies (DE) in kJ/mol.
J. Liang et al. / Journal of Molecular Structure: THEOCHEM 725 (2005) 151–155154
parallel the differences computed in the charge densities of
the C–F bonds of CF3 and structure 2.
The location of the dicoordinated isomer 3 as a true
energy minimum on the potential energy surface provides
the first indication that, with selected electrophiles,
trifluoromethyl may actually behave as a ‘bidentate’
gaseous base. The comparison between the optimized
structure of isomer 3 and uncoordinated CF3 confirms the
expected elongation of the two C–F bonds involved in the
direct interaction with the alkali metal cations. These
structural changes result, however, less pronounced than
structure 2. Consistently, the C–F1 bond lengths result
shorter than CF3 by ca. 0.044, 0.034, 0.028 A for LiC–CF3,
NaC–CF3, KC–CF3, respectively. Once again, compared
with CF3, the charge densities of the C–F bonds of structure
3 are in line with these computed structural changes.
As already pointed out, the tricoordinated structure 4
revealed second-order saddle points on their respective
potential energy surfaces, and their optimized geometries
suggest a rather weak interactions between MC (MZLi,
Na, K) and CF3. Thus, the C–F bond length is appreciably
larger than the same parameter of structures 2 and 3, and the
structural features and charge densities of the CF3 moieties
in the MC–CF3 complexes are only slightly different with
respect to uncoordinated CF3.
3.2. Energies
The relative stabilities of the presently investigated MC–
CF3 (MZLi, Na, K) complexes, obtained at different levels
of theory, are reported in Table 4. The most important
general indication from these calculations is that, irrespec-
tive of the employed basis set and the selected level of
theory, the fluorine-coordinated isomers 2, 3, and 4 are
invariably predicted to be more stable than the carbon-
coordinated isomer 1. In addition, the tricoordinated isomer
4 is invariably predicted as the least stable among the
fluorine-coordinated structures.
One of the aims of the present study was to investigate
the possibility that trifluoromethyl could be detected by MC
(MZLi, Na, K) ion spectrometry. The gas-phase MC cation
affinity (CA) of CF3, namely the minus enthalpy change of
the reaction.
MC CCF3 /MC–ðCF3Þ
From data in Table 4, the affinity energies of various LiC,
NaC, KC complexes are calculated. The CAs of most stable
structure 2 of LiC, NaC, KC complexes are 50.7, 27.5,
12.9 kJ/mol. The affinity energies are 44.4, 24.5, 10.4 kJ/
mol for local minima structure 3, respectively. We see that
the CA energies follow the order LiCONaCOKC in the
four structures 1–4, and the affinity energies of the most
stable isomers exceed 10.1 kJ/mol, which are large enough
to suggest that CF3 could be in principle detected and
quantified by LiC, NaC, KC ion attachment experiments,
and this supports the Fujii’s [8] proposal that LiC ion
attachment mass spectrometry as a conceivable technique to
detect and quantify the PFCs.
Acknowledgements
This work was supported by the National Natural Science
Foundation and the Director Research Grants of Hefei

J. Liang et al. / Journal of Molecular Structure: THEOCHEM 725 (2005) 151–155 155
Institute of Physical Sciences and Anhui Institute of Optics
and Fine Mechanics, Chinese Academy of Science.
References
[1] J.L. Beauchamp, Annu. Rev. Phys. Chem. 22 (1971) 527.
[2] P. Kebarle, Annu. Rev. Phys. Chem. 28 (1977) 445.
[3] T.D. Mark, A.W. Castleman Jr., Adv. At. Mol. Phys. 20 (1984) 65.
[4] T. Fujii, K. Syouji, Phys. Rev. A 46 (1992) 3555.
[5] T. Fujii, K. Syouji, J. Appl. Phys. 74 (1993) 3009.
[6] T. Fujii, K. Syouji, J. Phys. Chem. 97 (1993) 11380.
[7] T. Fujii, H.S. Kim, Chem. Phys. Lett. 268 (1997) 229.
[8] T. Fujii, Phys. Rev. E 58 (1998) 6495.
[9] T. Fujii, Chem. Phys. Lett. 313 (1999) 733.
[10] P.C. Selvin, T. Fujii, Rev. Sci. Instrum. 72 (2001) 2248.
[11] S. Arulmozhiraja, T. Fujii, J. Phys. Chem. A104 (2000) 9613.
[12] K. Stephan, H. Deutsch, T.D. Mark, J. Chem. Phys. 83 (1985) 5712.
[13] P. Sauvageau, J. Doucet, R. Gilbert, C. Sandorfy, J. Chem. Phys. 61
(1974) 391.
[14] G. Fischer, R.L. Purchase, D.M. Smith, J. Mol. Structure 405 (1997)
159.
[15] A.E. Reed, F. Weinhold, J. Am. Chem. Soc. 108 (1986) 3586.
[16] B.J. Bozlee, J.W. Nibler, J. Chem. Phys. 84 (1986) 3798.
[17] D. Forney, M.E. Jacox, K.K. lrikura, J. Chem. Phys. 101 (1994) 8290.
[18] K. Brudnik, J.T. Jodkowski, E. Ratajczak, J. Mol. Structure 656
(2003) 333.
[19] K.M. Pei, J. Liang, H. Li, J. Mol. Structure 690 (2004) 159.
[20] GAUSSIAN 03 written by M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E.
Scuseria, M.A. Robb, J.R. Cheeseman, V.G. Zakrzewski, J.A.
Montgomery, R.E. Stratmann, J.C. Burant, S. Dapprich, J.M. Millam,
A.D. Daniels, K.N. Kudin, M.C. Strain, O. Farks, J. Tomasi, V.
Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S.
Cliffoird, J. Ochterski, G.A. Petersson, P.Y. Ayala, Q. Cui, K.
Morokuma, D.K. Malick, A.D. Rabuck, K. Raghavachari, J.B.
Foresman, J. Cioslowski, J.V. Ortiz, B.B. Stefanov, G. Liu, A.
Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R.L. Martin, D.J.
Fox, T. Keith, M.A. Al-Laham, C.Y. Peng, A. Nanayakkara, C.
Gonzalez, M. Challacombem, P.M.W. Gill, B.G. Johnson, W. Chen,
M.W. Wong, J.L. Andres, M. Head-Gordon, E.S. Replogle, J.A.
Pople, Gaussian, Inc., Pittsburgh, PA, 2003.
[21] A.D. Becke, J. Chem. Phys. 98 (1993) 5648.
[22] C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 37 (1998) 785.
[23] R.F.W. Bader, Atoms in Molecules: A Quantum Theory, Oxford
University Press, Oxford, 1990.
[24] J. Cioslowski, A. Nanayakkara, M. Challacombe, Chem. Phys. Lett.
203 (1993) 137.
[25] J. Cioslowski, Chem. Phys. Lett. 219 (1994) 151.
[26] J. Cioslowski, P.R. Surjan, J. Mol. Structure (Theochem) 255 (1992)
9.
[27] J. Cioslowski, B.B. Stefanov, Mol. Phys. 84 (1995) 707.
[28] J. Cioslowski, Chem. Phys. Lett. 194 (1992) 73.
[29] A.E. Reed, L.A. Curtiss, F. Weinhold, Chem. Rev. 88 (1988) 899.
[30] A.E. Reed, R.B. Weinstock, F. Weinhold, J. Chem. Phys. 83
(1985) 735.













![Index [application.wiley-vch.de] · alkyl trifluoromethyl (or perfluoroalkyl) sulfoximines 681 alkyl trifluoromethyl sulfoxides [(Alk) S(O)CF 3] applications 491–492 preparation](https://img.pdfslide.net/doc/110x75/6064b9f559fa9f231e4ca32d/index-alkyl-trifluoromethyl-or-perfluoroalkyl-sulfoximines-681-alkyl-trifluoromethyl.jpg)








![Isomers [compatibility mode]](https://img.pdfslide.net/doc/110x75/5590bc1e1a28abbf308b46da/isomers-compatibility-mode-5593e8f124020.jpg)