Embed Size (px)
Citation preview

Computational Determination of Fundamental Pathway
and Activation Barriers for Acetohydroxyacid Synthase-
Catalyzed Condensation Reactions of a-Keto Acids
YING XIONG,1,2* JUNJUN LIU,
1,2* GUANG-FU YANG,1CHANG-GUO ZHAN
2
1Key Laboratory of Pesticide and Chemical Biology of the Ministry of Education, College ofChemistry, Central China Normal University, Wuhan 430079, People’s Republic of China2Department of Pharmaceutical Sciences, College of Pharmacy, University of Kentucky,
725 Rose Street, Lexington, Kentucky 40536
Received 11 January 2009; Revised 5 March 2009; Accepted 18 May 2009DOI 10.1002/jcc.21356
Published online 24 June 2009 in Wiley InterScience (www.interscience.wiley.com).
Abstract: Acetohydroxyacid synthase (AHAS) is the first common enzyme in the biosynthetic pathway leading to
the production of various branched-chain amino acids. AHAS is recognized as a promising target for new antituber-
culosis drugs, antibacterial drugs, and herbicides. Extensive first-principles quantum mechanical (QM) and hybrid
quantum mechanical/molecular mechanical (QM/MM) calculations have enabled us, in this study, to uncover the
fundamental reaction pathway, determine the activation barriers, and obtain valuable insights concerning the specific
roles of key amino acid residues for the common steps of AHAS-catalyzed condensation reactions of a-keto acids.
The computational results reveal that the rate-determining step of the AHAS-catalyzed reactions is the second reac-
tion step and that the most important amino acid residues involved in the catalysis include Glu1440, Gln2070,Gly1210, and Gly511 that form favorable hydrogen bonds with the reaction center (consisting of atoms from the sub-
strate and cofactor) during the reaction process. In addition, Glu1440 also accepts a proton from cofactor thiamin
diphosphate (ThDP) through hydrogen bonding during the catalytic reaction. The favorable interactions between the
reaction center and protein environment remarkably stabilize the transition state and, thus, lower the activation bar-
rier for the rate-determining reaction step by �20 kcal/mol. The activation barrier calculated for the rate-determining
step is in good agreement with the experimental activation barrier. The detailed structural and mechanistic insights
should be valuable for rational design of novel, potent AHAS inhibitors that may be used as promising new anti-
tuberculosis drugs, antibacterial drugs, and/or herbicides to overcome drug resistance problem.
q 2009 Wiley Periodicals, Inc. J Comput Chem 31: 1592–1602, 2010
Key words: enzyme; catalytic mechanism; reaction pathway; QM/MM calculation
Introduction
Acetohydroxyacid synthase (AHAS; also known as acetolactate
synthase; EC 2.2.1.6; formerly EC 4.1.3.18) is the first common
enzyme in the biosynthetic pathway leading to the production of
branched-chain amino acids, including valine, leucine, and iso-
leucine, in plants and a wide range of microorganisms (such as
Escherichia coli, Saccharomyces cerevisiae, and Arabidopsisthaliana).1 This essential enzyme catalyzes the condensation of
two a-keto acid molecules, e.g., the condensation of two pyru-
vate molecules to form (S)-a-acetolactate and the condensation
of one pyruvate molecule with one a-ketobutyrate molecule to
form (S)-a-aceto-a-hydroxybutyrate. In addition, AHAS has been
proven to be an efficient catalyst in chiral synthesis and a potent
target in biochemistry engineering.2,3
AHAS is recognized as a promising target for new antituber-
culosis drugs,4 antibacterial drugs,5 and herbicides.6 For exam-
ple, Mycobacterium tuberculosis is the pathogen of tuberculosis
that remains a major threat to the human population. This
human disease is responsible for 2–3 millions deaths per year
worldwide.7 The emergence of drug-resistant and multidrug-
Contract/grant sponsor: Kentucky Science and Engineering Foundation;
contract/grant number: KSEF-925-RDE-008
Contract/grant sponsor: National Natural Science Foundation of China;
contract/grant numbers: 20602014, 20503008, 20432010
*These two authors contributed equally to this work.
Correspondence to: C.-G. Zhan; e-mail: [email protected]; G.-F. Yang;
e-mail: [email protected]
q 2009 Wiley Periodicals, Inc.

resistant tuberculosis has greatly increased the need to identify
new antituberculosis target proteins. It was shown that branched
amino acids auxotrophic strain of Mycobacterium failed to pro-
liferate because of the inability to use amino acids from their
hosts,8 indicating that inhibitors for the branched-chain amino
acid biosynthesis could be used as an antituberculosis agents.9
These observations suggested that AHAS could be a potential
target of new antituberculosis drugs. It was found that the
recombinant AHAS from M. avium, which is another human
tuberculosis pathogen, was efficiently inhibited by sulfonylureas
(SU),10 a herbicide that inhibits plant AHAS.
A number of structurally diverse, potent AHAS inhibitors,
have been reported in literature. Some of the known AHAS
inhibitors, e.g. SU, imidazolinones (IM), triazolopyrimidine sul-
fonanilides (TP), and pyrimidinyloxybenzoates (PB), have been
being used commercially as herbicides.6,11–16 Since their first
introduction in the early 1980s, AHAS inhibitors have been used
worldwide and now constitute the most important herbicide fam-
ily in use due to their unique advantages such as broad-spectrum
weed control at very low application rates, extremely low toxic-
ity to animals, and flexible application timing in a wide variety
of crops. However, constantly extensive use of AHAS-inhibiting
herbicides has resulted in about 95 tolerant weeds worldwide,
and this number has shown to be increasing at an exponential
rate. Most seriously, because the structurally diverse AHAS
inhibitors are competitive with one another with respect to the
binding with the enzyme, tolerance toward a particular herbicide
has resulted in varying degrees of cross-tolerance to the other
chemicals. Therefore, because of the widespread occurrence of
AHAS inhibitor-resistant weed species, the AHAS enzyme has
received considerable attention.
However, the currently known AHAS inhibitors, including all
the existing AHAS-inhibiting herbicides, have been proved to be
‘‘extraneous site inhibitors.’’ That is to say, the binding site of
the known AHAS inhibitors is not the active site of AHAS.
These compounds just bind within the tunnel connecting to the
active site, thus blocking substrate entry into the active site. As
a result, the mutations on amino acid residues in the inhibitor-
binding site resulted in resistance to the drugs/herbicides while
the enzyme activities were kept.17 An ideal strategy to develop
novel AHAS inhibitors for overcoming the drug resistance is to
design novel inhibitors that bind to the catalytic active site of
the enzyme, as amino acid residues in the active site are rather
conservative. With the inhibitor binding to the active site, one
would not need to worry about the resistance due to mutations
on the active site residues, as the mutations on the active site
residues would likely make the enzyme inactive. It is important
for rational design of the novel inhibitors of AHAS to under-
stand the detailed structure and fundamental catalytic mechanism
of the enzyme.
Recently, six X-ray crystal structures of A. thaliana AHAS in
complex with five different SU and one imidazolinone herbicide
have been reported.18 Like almost all thiamin diphosphate
(ThDP)-dependent enzymes,19 the active site of AHAS20–22 is
formed at the interface between two monomers and, therefore,
the minimum quaternary structure for catalysis is a dimer.
The enzyme-sequestered ThDP (cofactor) plays a central role
in the catalytic mechanism as shown in Figure 1.23 Initially,
ThDP is in its protonated form (R or ES) that then ionizes to the
reactive ylide (INT1); A molecule of pyruvate is attacked by the
ThDP anion to form the lactyl-ThDP (LThDP) intermediate
(INT2) which undergoes decarboxylation.24 In subsequent steps,
the hydroxyethylthiamin diphosphate (HEThDP) enamine/car-
banion (INT3) formed from the decarboxylation attacks the car-
bonyl of a second substrate (pyruvate or 2-ketobutyrate) to form
the product-ThDP adduct. Finally, the product-ThDP adduct dis-
sociates to form ThDP and the free product.25 Direct competi-
tion between alternative substrates for the bound HEThDP deter-
mines the ratio of products formed. In the wild-type enzyme, the
product ratio depends only on the relative amounts of the sub-
strates present and kcat is nearly independent of the amount or
identity of the substrates.25,26 This implies that kcat is determined
Figure 1. Catalytic cycle of AHAS.
1593AHAS-Catalyzed Condensation Reactions
Journal of Computational Chemistry DOI 10.1002/jcc

by the reaction step preceding the product-determining,
carboligation step so that the steps contributing to discrimination
among ‘‘second substrates’’ have little effect on the overall rate
of reaction. The ThDP adducts of LThDP, HEThDP, and the
product acetohydroxyacids (ALThDP and/or AHBThDP) can be
detected quantitatively in the mixture after rapid quenching.25
Determination of the distribution of the key reaction intermedi-
ates by 1H NMR analysis then makes it possible to calculate the
forward unimolecular rate constants for individual microscopic
steps. The forward (net) rate constant for formation of LThDP,
the first detectable step, is overwhelmingly rate determining for
catalysis, whereas the subsequent steps occur with similar, but
much larger rates (e.g., the rates of decarboxylation, carboliga-
tion, and product release are comparable).24,26 Such relative
rates explain why kcat for a given enzyme is essentially the
same for acetohydroxybutyrate (AHB) and acetolactate (AL)
formation, despite the 60-fold preference for 2-ketobutyrate as
substrate.
The present computational study was aimed to understand
the fundamental reaction pathway and energetics for the com-
mon reaction steps concerning the formation of HEThDP enam-
ine/carbanion intermediate before the further reaction with a sec-
ond substrate, particularly for the rate-determining reaction step.
The purpose of this computational study is twofold. One is to
provide fundamental mechanistic insights for a generally inter-
esting type of chemical and biochemical reactions, because the
condensation reactions catalyzed by an enzyme leading to the
eventual formation of branched-chain amino acids, such as
valine, leucine, and isoleucine, are a type of fundamentally inter-
esting chemical/biochemical reactions in biological systems. In
addition, the mechanistic insights obtained from the detailed
computational study can provide a solid, valuable mechanistic
base for rational design of the highly desirable novel AHAS
inhibitors that can tightly bind with the catalytic residues in the
active site. The novel AHAS inhibitors may be used as promis-
ing new antituberculosis drugs, antibacterial drugs, and/or herbi-
cides to overcome the drug resistance. For these purposes, we
have carried out detailed reaction coordinate calculations, for the
first time, on the common steps of AHAS-catalyzed condensa-
tion reactions by using first-principles quantum mechanical
(QM) and hybrid quantum mechanical/molecular mechanical
(QM/MM) methods. The reaction coordinate calculations have
led to valuable mechanistic insights concerning the detailed
reaction pathway and the specific role of some key residues, par-
ticularly for the rate-determining step.
Computational Methods
QM Reaction Coordinate Calculations
Geometries of all molecular structures, including reactants, tran-
sition states, and intermediates, involved in this study were opti-
mized by using density functional theory (DFT) using Becke’s
three-parameter hybrid exchange functional and the Lee-Yang-
Parr correlation functional (B3LYP)27–29 with the 6-311G(d)
basis set. Vibrational frequency calculations were carried out to
ensure that the optimized geometries are indeed associated with
local minima or saddle points on the potential energy surfaces
and to determine the zero-point vibrational energies (ZPVEs)
and thermal corrections to Gibbs free energies at 310 K. Intrin-
sic reaction coordinate (IRC)30,31 calculations were performed at
the B3LYP/6-311G(d) level to confirm the optimized transition
state geometries. The geometries optimized at the B3LYP/
6-311G(d) level were also used to perform single-point energy
calculations with the 6-311G(d), 6-3111G(d,p), and 6-
31111G(d,p) basis sets. All of the QM calculations were per-
formed by using Gaussian03 program.32
Preparation of Initial Structure for QM/MM Calculations
The initial structure used in the QM/MM geometry optimizations
was chosen from the X-ray crystal structure of A. thalianaAHAS in complex with a herbicide molecule (PDB code:
1Z8N). After the herbicide molecule was removed, pyruvate was
docked into the active site of AHAS. The missing hydrogen
atoms were added by using Leap module of Amber8 program.33
Histidine residues were assigned to (neutral) protonation state
HID or HIE based on the local hydrogen bonding network. The
partial atomic charges of substrate pyruvate and cofactors ThDP
and flavin adenine dinucleotide (FAD) required for the structural
preparation using Amber8 program were determined by perform-
ing ab initio QM calculations at the HF/6-31G* level following
by restrained electrostatic potential (RESP)-fitting calculations
using the RESP module of Amber8 program. All other force
field parameters were from the parm99 parameter library.
After the energy minimization of the added atoms using
Amber8 program in the gas phase, the AHAS-pyruvate complex
was solvated in a rectangular box of TIP3P water molecules
with a minimum solute-wall distance of 10 A and neutralized by
addition of counterions (Na1) using the Leap module of Amber8
program. Thus, a system of 95,600 atoms was obtained. To equi-
librate the solvated system, first of all, the protein system was
frozen and the solvent molecules with counterions were allowed
to move during a 3000-step energy minimization process. Sec-
ond, all the atoms were allowed to move during a 5000-step full
minimization process. Then, the water molecules beyond 2.5 A
of the complex were removed and the remained system was
used as the initial structure of QM/MM calculations described
below.
QM/MM Calculations
QM/MM calculations were performed by using a pseudobond
QM/MM method.34,35 The pseudobond QM/MM method was
initially implemented in revised Gaussian03 and Tinker pro-
grams.34,35 The revised Gaussian03 and Tinker programs can be
used to carry out the QM and MM parts of the QM/MM calcula-
tion iteratively until the full self-consistency is achieved. The
pseudobond QM/MM method uses a seven-valence-electron
atom with an effective core potential35 constructed to replace
the boundary atom of the environment part and to form a pseu-
dobond with the boundary atom of the active part. The main
idea of the pseudobond approach is as follows: one considers
that a large molecule is partitioned into two parts, an active part
and an environment part, by cutting a covalent r-bond Y-X. Y
and X refer to boundary atoms of the environment part and the
1594 Xiong et al. • Vol. 31, No. 8 • Journal of Computational Chemistry
Journal of Computational Chemistry DOI 10.1002/jcc

active part, respectively. Instead of using a hydrogen atom to
cap the free valence of X atom as in the conventional link-atom
approach, a pseudobond Yps-X is formed by replacing the Y atom
with a one-free-valence boundary Y atom (Yps). The Yps atom is
parameterized to make the Yps-X pseudobond mimic the original
Y-X bond with similar bond length and strength, and also to have
similar effects on the rest of the active part.34,35 In the pseudo-
bond approach, the Yps atom and all atoms in the active part form
a well-defined QM subsystem which can be treated by a QM
method. Excluding Y atom, the remaining atoms in the environ-
ment part form the MM subsystem treated by a MM method. The
pseudobond ab initio QM/MM approach has been demonstrated
to be powerful in studies of enzyme reactions.36–38
In this study, we used a newly revised version39 of Gaus-
sian03 and Amber8 programs, instead of the revised Gaussian03
and Tinker programs, to perform the QM/MM calculations,
because the Amber program is capable of parallel computing. In
all of our QM/MM calculations, the QM subsystem included all
atoms of the substrate pyruvate, most atoms of cofactor ThDP,
amino acid residues Gly511 and Ala512 (from one AHAS mole-
cule of the homodimer), and residues Gly1200, Gly1210,Glu1440, and Gln2070 (from the other AHAS molecule of the
homodimer). See below for a figure (Fig. 3) in which the QM
atoms are highlighted. The remaining atoms of the protein
(including Mg21 and all atoms of nonreactive cofactor FAD that
do not directly participate in the reaction) and water molecules
were regarded as MM atoms. All water molecules were included
in the MM subsystem, as none of the water molecules are
expected to be involved in the covalent bond formation/breaking
during the reaction based on the X-ray crystal structure and the
structures modeled in the present study.
The boundary between the QM and MM subsystems was
treated according to the original pseudobond approach.34 The
total energy of the QM/MM system is
ETotal ¼ EMM þ EQM þ EQM=MM (1)
The QM-MM interactions consist of bonding and nonbonding
interactions. The nonbonding interactions between the two sub-
systems include the van der Waals (vdw) and electrostatic inter-
actions calculated through the Lennard-Jones potential and Cou-
lombic term, respectively, in the effective Hamiltonian. The
energy corresponding to the effective Hamiltonian, which is
obtained by QM calculation, is the sum of the QM energy of the
QM subsystem (EQM) and the electrostatic interaction between
the QM and MM subsystems.34 We used the reaction coordinate
driving method37,40 to search for transition states and intermedi-
ates. For example, during the QM/MM reaction coordinate cal-
culations on the second reaction step (rate-determining step) and
also on the first step, the reaction coordinate is represented by
the C2-C internuclear distance, where C2 refers to the carbon
atom at position-2 of ThDP as shown in Figure 1 and C refers
to the carbonyl carbon of pyruvate molecule. It should be noted
that many other geometric parameters should also change while
the C2-C distance changes during the reaction. The changes of
the other geometric parameters are automatic during the QM/
MM reaction coordinate calculations. The same reaction
coordinate approach has been used in many other computational
studies.39,41
Our initial QM/MM reaction coordinate calculations were per-
formed at the HF/3-21G*:Amber level, i.e., the QM calculations
were carried out at the HF/3-21G* level while the MM calcula-
tions were carried out by using the Amber force field imple-
mented in the Amber8 program. The geometry optimizations
were converged to the default criteria of the Gaussian03 (for QM
part) and Amber8 (for MM part) programs. During the QM ge-
ometry optimization process, the pseudobonds were treated with
the well-established effective core potential parameters associated
with 3-21G* basis set.35 In the MM energy minimization process,
only atoms within 20 A of ThDP were allowed to move. No cut-
off for nonbonding interactions was used in the QM/MM calcula-
tions and the MM energy minimizations. The iterative, restrained
geometry optimization/energy minimization processes were
applied repeatedly to different points along the reaction coordi-
nate, resulting in a minimum energy path for the reaction in the
enzymatic environment and its associated potential energy sur-
face. Given that the determined minimum energy path is smooth
and continuous, Hessian matrices for degrees of freedom involv-
ing atoms in the QM subsystem were calculated at stationary
points, leading to determination of the corresponding vibrational
frequencies.41 An energy maximum on the path with one and
only one imaginary frequency is a transition state, whereas an
energy minimum along the path without any imaginary frequency
is characterized as an intermediate. Finally, the geometries opti-
mized at the HF/3-21G*:Amber level along the reaction path
were then used to perform single-point energy calculations at
higher levels, including B3LYP/6-31G*:Amber, B3LYP/
6-311G*:Amber, B3LYP/6-3111G**:Amber, MP2/6-31G*:
Amber, and MP2/6-3111G**:Amber levels.
Most of the QM and QM/MM calculations were performed
in parallel on an IBM X-series Cluster (with 340 nodes and
1,360 processors) at the Center for Computational Sciences, Uni-
versity of Kentucky. Some computations were carried out on a
34-processors IBM x335 Linux cluster and SGI Fuel worksta-
tions in our own laboratory.
Results and Discussion
Fundamental Reaction Pathway and Activation Barriers
from QM Reaction Coordinate Calculations on the Model
Reaction System.
As depicted in Figure 1, the first three reaction steps are com-
mon for all of concerned condensation reactions of two a-ketoacid molecules no matter whether the second a-keto acid mole-
cule involved in the later reaction step is still pyruvate or not. In
addition, the aforementioned discussion indicates that the rate-
determining step of the condensation reactions is always the
second reaction step. So, our QM reaction coordinate calcula-
tions were focused on the first three reaction steps depicted in
Figure 1. To simplify the QM reaction coordinate calculations,
we used a simplified model system. Specifically, we replaced
the ��P2O7 group of ThDP with ��OH and the protein atoms
were not included in the reaction model. The fundamental reac-
1595AHAS-Catalyzed Condensation Reactions
Journal of Computational Chemistry DOI 10.1002/jcc
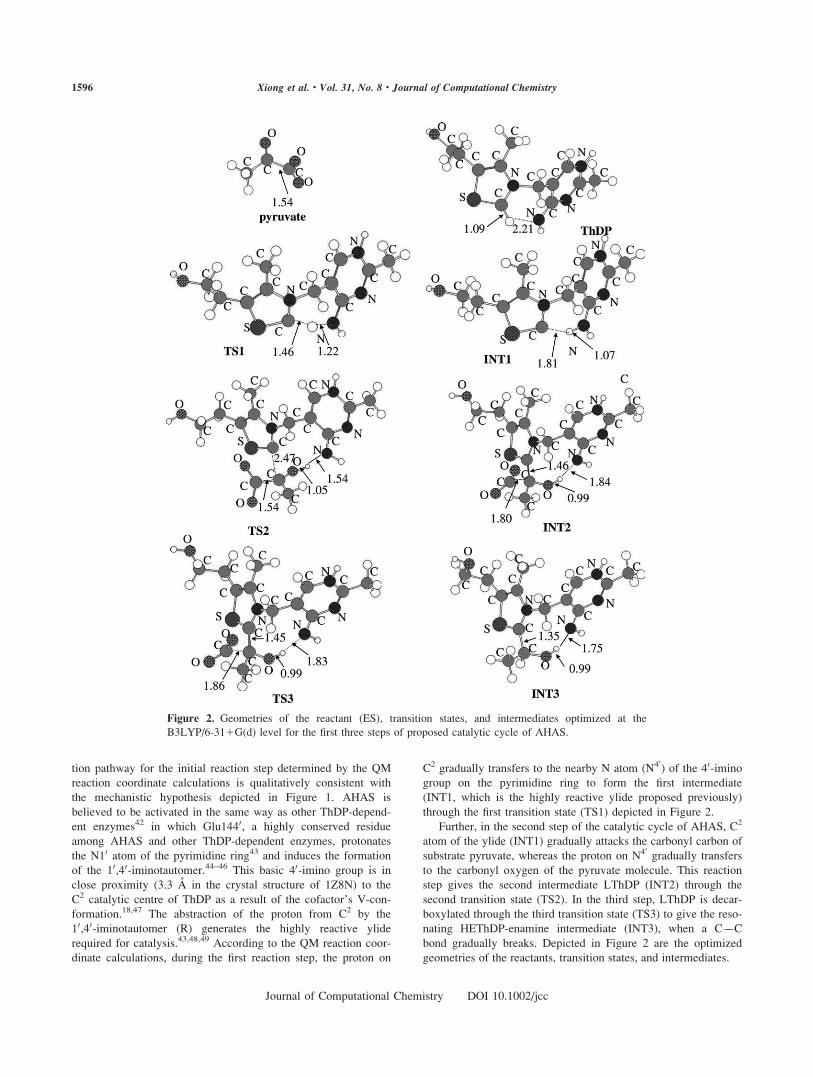
tion pathway for the initial reaction step determined by the QM
reaction coordinate calculations is qualitatively consistent with
the mechanistic hypothesis depicted in Figure 1. AHAS is
believed to be activated in the same way as other ThDP-depend-
ent enzymes42 in which Glu1440, a highly conserved residue
among AHAS and other ThDP-dependent enzymes, protonates
the N10 atom of the pyrimidine ring43 and induces the formation
of the 10,40-iminotautomer.44–46 This basic 40-imino group is in
close proximity (3.3 A in the crystal structure of 1Z8N) to the
C2 catalytic centre of ThDP as a result of the cofactor’s V-con-
formation.18,47 The abstraction of the proton from C2 by the
10,40-iminotautomer (R) generates the highly reactive ylide
required for catalysis.43,48,49 According to the QM reaction coor-
dinate calculations, during the first reaction step, the proton on
C2 gradually transfers to the nearby N atom (N40) of the 40-imino
group on the pyrimidine ring to form the first intermediate
(INT1, which is the highly reactive ylide proposed previously)
through the first transition state (TS1) depicted in Figure 2.
Further, in the second step of the catalytic cycle of AHAS, C2
atom of the ylide (INT1) gradually attacks the carbonyl carbon of
substrate pyruvate, whereas the proton on N40 gradually transfers
to the carbonyl oxygen of the pyruvate molecule. This reaction
step gives the second intermediate LThDP (INT2) through the
second transition state (TS2). In the third step, LThDP is decar-
boxylated through the third transition state (TS3) to give the reso-
nating HEThDP-enamine intermediate (INT3), when a C��C
bond gradually breaks. Depicted in Figure 2 are the optimized
geometries of the reactants, transition states, and intermediates.
Figure 2. Geometries of the reactant (ES), transition states, and intermediates optimized at the
B3LYP/6-311G(d) level for the first three steps of proposed catalytic cycle of AHAS.
1596 Xiong et al. • Vol. 31, No. 8 • Journal of Computational Chemistry
Journal of Computational Chemistry DOI 10.1002/jcc

The energetic results listed in Table 1 show that the highest
activation barrier is associated with the second step, suggesting
the second step is rate determining. This is also qualitatively
consistent with the aforementioned experimental observation.25
However, the activation barrier, �36 kcal/mol calculated at the
B3LYP/6-31111G** level is significantly higher than the acti-
vation barrier (�16 kcal/mol) derived from experimental kinetic
data25 according to the well-known transition state theory in
which k(T) 5 (kBT/h) exp(2DG=/RT). We must account for the
effects of the protein environment on the activation barrier.
Reaction Pathway and Activation Barriers from QM/MM
Calculations
To account for the effects of protein environment on the reaction
pathway and the corresponding activation barrier of the enzy-
matic reaction, we performed QM/MM reaction coordinate
calculations on the first two reaction steps, including the rate-
determining step (i.e. the second step), of the entire enzymatic
reaction system. The QM/MM-optimized geometries of the
enzyme-substrate complex (ES), TS1, INT1, TS2, and INT2 are
depicted in Figure 3.
As seen in the optimized geometries of ES, TS1, and INT1,
during the first reaction step, the proton on C2 of ThDP gradu-
ally transfers to the nearby N atom (N40) of the 40-imino group
on the pyrimidine ring, whereas the proton on N10 atom of the
pyrimidine ring of ThDP gradually transfers to an oxygen atom
of the Glu1440 side chain. So, the first reaction step in the pro-
tein environment is a double-proton transfer process, instead of
the single-proton transfer process found in the aforementioned
model reaction system. The protein environment remarkably
changes the chemical nature of the first reaction step through
coupling with an additional proton transfer from ThDP to the
Glu1440 side chain of the protein. The additional proton transfer
from ThDP to the Glu1440 side chain is expected to significantly
stabilize the TS1 structure and, thus, significantly lower the acti-
vation barrier for this reaction step. The double-proton process
is completed when the first intermediate (INT1) is formed. In
fact, the activation barrier determined for the first reaction step
by the QM/MM calculations at the B3LYP/6-311G*:Amber//HF/
3-21G*:Amber and B3LYP/6-3111G**:Amber//HF/3-21G*:
Amber levels are only 1.90 and 1.94 kcal/mol, respectively. The
QM/MM-calculated activation barrier for this reaction step is
�6 to 7 kcal/mol lower than that calculated for the corresponding
step of the model reaction system.
Because of the different chemical nature of the first reaction
step, the chemical nature of the second reaction step in the protein
environment is also remarkably different from that in the model
reaction system. Without accounting for the protein environment,
the QM reaction coordinate calculations on the aforementioned
model reaction system demonstrated that the proton on N40 gradu-
ally transfers to the carbonyl oxygen of pyruvate, whereas the C2
atom gradually attacks the carbonyl carbon of substrate pyruvate.
However, according to the QM/MM reaction coordinate calcula-
tion, the proton on N40 only forms a hydrogen bond with the
carbonyl oxygen atom of pyruvate (in INT2), whereas the C2
atom gradually attacks the carbonyl carbon of substrate pyruvate.
The proton on N40 does not transfer to the carbonyl oxygen of
pyruvate according to the QM/MM calculations.
In the INT1, TS1, and INT2 geometries depicted in Figure 3,
pyruvate atoms form two hydrogen bonds with amino acid resi-
dues of AHAS. One hydrogen bond exists between the carbonyl
oxygen of pyruvate and the NH2 group of Gln2070 side chain
(through 1HE2 atom on the NH2 group). The other hydrogen bond
exists between the carboxyl oxygen of pyruvate and the backbone
NH of Gly1210. In addition, ThDP atoms also form two hydrogen
bonds with amino acid residues of AHAS. One of the hydrogen
bonds exists between the carbonyl oxygen of Gly511 backbone
and the N40 atom of ThDP (through an H atom on N40). The other
exists between the N10 atom of ThDP and the protonated oxygen
on the side chain of Glu1440. The later is consistent with the
experimental observation reported by Bar-Ilan.42 Altogether, the
four hydrogen bonds between pyruvate-ThDP and the four resi-
dues of AHAS are expected to stabilize the TS2 structure and,
therefore, lower the activation barrier.
Further, a sequence alignment of 24 available AHAS pro-
teins6 from different sources, including plant, fungal, algal, and
bacterial species, reveals that the aforementioned four key resi-
dues forming hydrogen bonds with pyruvate or ThDP are all
highly conserved for AHAS enzymes, suggesting that these four
residues (i.e., Glu144, Gln207, Gly121, and Gly511) may play
an important role in the catalysis for all of the AHAS enzymes.
Our structural results are qualitatively consistent with available
experimental kinetic data in literature. For example, Glu47Gln
and Glu47Ala mutations on E. coli AHAS II (corresponding to
Glu144Gln and Glu144Ala mutations on A. thaliana AHAS II)
decreased the catalytic rate constant (kcat) of the enzyme by
about 10- and 13-fold, respectively.42 This is because the muta-
tions destroy the hydrogen bond between the N10 atom of ThDP
and the protonated oxygen on the side chain of Glu144 (or
Glu47 in E. coli AHAS II). For the same reason, substitution of
ThDP with an analog without N10 or the 40-amino group also
considerably decreased the enzymatic activity.50 These experi-
mental data qualitatively confirm the importance of Glu1440 resi-due in stabilizing the TS2 geometry depicted in Figure 3. Chip-
man and coworkers51 also reported that mutation of a highly
conserved residue, i.e., Gln110 in E. coli AHAS II correspond-
Table 1. Activation Barriers (DG=, in kcal/mol) Obtained from the QM
Calculations on the Model System for the First Three Steps of
AHAS-catalyzed Condensation Reactions.a
Method DG1= b DG2
= b DG3= b
B3LYP/6-311G* 9.27 37.50 0.25
B3LYP/6-3111G** 7.65 37.29 0.29
B3LYP/6-31111G** 8.09 35.83 0.00
Expt.c 16.21
aAll the geometries were optimized at B3LYP/6-311G* level.bDG1
=, DG2=, and DG3
= refer to calculated activation barriers for reaction
steps 1, 2, and 3, respectively, in Figure 1. Included also in DG1=, DG2
=,
and DG3= values are thermal corrections to free energies calculated at
B3LYP/6-311G* level.cThe experimental activation barrier was based on the experimental data
in Ref. 25 and the transition state theory in which k(T) 5 (kBT/h)
exp(2DG=/RT).
1597AHAS-Catalyzed Condensation Reactions
Journal of Computational Chemistry DOI 10.1002/jcc

ing to Gln207 in A. thaliana AHAS II, led to ‘‘a significant loss
of activity,’’ although they did not report specific kinetic param-
eters for the mutations on the residue. Their experimental
observation qualitatively confirms the importance of Gln2070 instabilizing the TS2 geometry depicted in Figure 3. The hydrogen
bond between the N40 atom of ThDP and the backbone oxygen
atom of Gly511 existed in all of the QM-MM-optimized geome-
tries depicted in Figure 3, which is consistent with the observa-
tion by Duggleby et al.52 that such a hydrogen bond existed in
all of the X-ray crystal structures of AHAS.
Depicted in Figure 3(F) are the plots of the relative potential
energies calculated at the HF/3-21G*:Amber, B3LYP/6-31G*,
B3LYP/6-311G*:Amber, B3LYP/6-3111G**:Amber, and
MP2/6-31G*:Amber levels versus the reaction coordinate (the
Figure 3. QM/MM-optimized geometries of ES (A), TS1 (B), INT1 (C), TS2 (D), and INT2 (E) and
the potential energy surface along the reaction coordinate for the rate-determining step (F). In the
optimized geometries, the QM atoms used on the QM/MM calculations are represented by the balls.
1598 Xiong et al. • Vol. 31, No. 8 • Journal of Computational Chemistry
Journal of Computational Chemistry DOI 10.1002/jcc

minimum-energy path). It would be very time consuming to
perform QM/MM calculations on all of the structures along
the reaction path at the MP2/6-3111G**:Amber level. So,
single-point QM/MM energy calculations at the MP2/
6-3111G**:Amber level were only performed on the INT1 and
TS2 geometries used for the QM/MM calculations at the MP2/
6-31G*:Amber level to estimate the activation barrier at the
MP2/6-3111G**:Amber level. The QM/MM-calculated values
of the activation barrier are summarized in Table 2. As one can
see in Table 2, the QM/MM calculations at the HF/3-
21G*:Amber level considerably overestimated the activation bar-
rier for this reaction step. The QM/MM calculations at the
B3LYP/6-31G*:Amber level also significantly overestimated the
activation barrier. Nevertheless, the QM/MM calculations at
more sophisticated levels, including the B3LYP/6-311G*:
Amber, B3LYP/6-3111G**:Amber, MP2/6-31G*:Amber, and
MP2/6-3111G**:Amber levels, all lead to very close results
(15.20 to 15.82 kcal/mol) that are all in good agreement with
the experimental activation barrier of 16.21 kcal/mol.25 We note
that various new density functionals were reported in the past a
few years and that some newly developed functionals could out-
perform B3LYP.53,54 To examine the effect of the choice of the
density functional on the calculated activation barrier, the activa-
tion barrier of the rate-determining step (step 2) was also
calculated by using several other functionals available in the
Gaussian03 program. As shown in Table 2, the activation barrier
calculated with B3LYP is closest to the result calculated with
the MP2 method and the experimental activation barrier of
16.21 kcal/mol25 for this particular QM/MM system.
The activation barrier calculated with the QM/MM method
considering the effects of protein environment is �20 kcal/mol
lower than the corresponding activation barrier calculated with
the QM method neglecting the protein environment. To better
understand how the protein environment contributes to the
�20 kcal/mol decrease of the activation barrier, we also
determined the single-point energy surface of a tailored QM/
MM system in which the MM part includes only the residues
having covalent bonds with the QM atoms. The activation
barrier calculated at the B3LYP/6-3111G**:Amber level for
the tailored QM/MM system is 15.97 kcal/mol, which is only
slightly higher than 15.20 kcal/mol calculated for the complete
QM/MM system. These results suggest that the protein environ-
ment affects the reaction and activation barrier mainly through
maintaining the active site of the enzyme.
To further understand the importance of Glu144 in catalysis,
we also carried out the QM/MM reaction coordinate calculations
on the second reaction step (i.e. the rate-determining reaction
step) associated with the Glu144Ala mutant at the same QM/
MM level of theory used for the native enzyme. The QM/MM-
optimized INT1 and TS2 geometries for the mutant are depicted
in Figure 4. Compared with the corresponding QM/MM-
optimized INT1 and TS2 geometries (in Fig. 3) associated with
the native enzyme, the hydrogen bonding interactions in the
INT1 and TS2 geometries (in Fig. 4) associated with the
Glu144Ala mutant are all similar, except that the hydrogen bond
between residue #144 and ThDP no longer exists in the mutant.
As a result, the activation barrier calculated (at the same MP2/
6-3111G**:Amber//HF/3-21G*:Amber level) for the second
reaction step of the reaction catalyzed by the Glu144Ala mutant
is 17.0 kcal/mol, which is 1.7 kcal/mol higher than the corre-
sponding activation barrier (15.3 kcal/mol in Table 2) calculated
for the second reaction step of the reaction catalyzed by the
native enzyme at the same level of theory. The calculated acti-
vation barrier increase of 1.7 kcal/mol is in good agreement
with the experimental observation that the Glu47Ala mutation
on E. coli AHAS II (corresponding to Glu144Ala mutation on
A. thaliana AHAS II) decreased the catalytic rate constant (kcat)of the enzyme by �13-fold,42 as a �13-fold decrease in kcat cor-responds to a �1.5 kcal/mol increase in activation barrier
according to the conventional transition state theory (CTST).55,56
It should be pointed out that the starting structures used for
the above QM/MM reaction coordinate calculations were all
based on the X-ray crystal structure. Warshel and coworkers57
noted that QM/MM reaction coordinate calculations on an enzy-
matic reaction system using different starting structures could
lead to significantly different activation barrier values. More
recently, Zhang and coworkers38 performed pseudobond QM/
MM reaction coordinate calculations on an enzymatic reaction
by using multiple initial structures, i.e., multiple snapshots of a
molecular dynamics (MD) simulation of the enzyme-substrate
complex. According to the QM/MM calculations by Zhang and
coworkers,38 the fluctuation of the calculated energy barriers is
only 61.1 kcal/mol and the barrier fluctuation has a strong cor-
relation with the values of the geometric parameters associated
Table 2. Activation Barrier (DG2=, in kcal/mol) for Calculated by
Performing QM/MM Calculations on the Second Reaction Step of the
AHAS Catalytic Cycle.
Methoda DG2= (kcal/mol)
QM/MM(HF/3-21G*:Amber) 22.33
QM/MM(B3LYP/6-31G*:Amber) 18.79
QM/MM(B3LYP/6-311G*:Amber) 15.82
QM/MM(B3LYP/6-3111G**:Amber) 15.20
QM/MM(MPWKCIS1K/6-3111G**:Amber) 18.19
QM/MM(BHandHLYP/6-3111G**:Amber) 20.25
QM/MM(B97-2/6-3111G**:Amber) 14.48
QM/MM(mPW1PW91/6-3111G**:Amber) 12.93
QM/MM(B98/6-3111G**:Amber) 14.10
QM/MM(MP2/6-31G*:Amber) 15.33
QM/MM(MP2/6-3111G**:Amber)b 15.31
Expt.c 16.21
aCalculated activation barrier (DG2=) is calculated as the energy change
from INT1 to TS2 on the corresponding minimum-energy path. Thermal
correction to Gibbs free energy at the HF/3-21G* level at T 5 310 K
was included in the calculation of DG2=.
bQM/MM reaction coordinate driving was not carried out at the MP2/6-
3111G**:Amber level because it would be very time-consuming at this
level of theory for this particular enzymatic reaction system. Instead, we
only performed the single-point energy calculations on the local mini-
mum (associated with INT1) and first-order saddle point (associated with
TS2) geometries determined at the MP2/6-31G*:Amber level.cThe experimental activation barrier was based on the experimental data
in Ref. 25 (determined at T 5 310 K) and the classic transition state
theory in which k(T) 5 (kBT/h) exp(2DG=/RT).
1599AHAS-Catalyzed Condensation Reactions
Journal of Computational Chemistry DOI 10.1002/jcc

with the reaction coordinate. When the values of the geometric
parameters associated with the reaction coordinate in a snapshot
are closer to the corresponding average values of the geometric
parameters in the simulated enzyme-substrate complex, the
energy barrier calculated with this snapshot is closer to the aver-
age value of the energy barriers calculated with all of the chosen
snapshots. The pseudobond QM/MM reaction coordinate
approach used in the present study is very similar to that used
by Zhang and coworkers38 Hence, the similar performance and
similar structure-barrier fluctuation correlation, and hopefully the
similarly small energy barrier fluctuation, may be expected for
the present pseudobond QM/MM reaction coordinate calcula-
tions. To examine the possible barrier fluctuation for the current
reaction system, we further carried out an MD simulation on the
INT1 structure for 2 ns and obtained a stable MD trajectory.
The MD simulation was performed in the same way as we did
in our previous computational studies on other protein
systems.58–60 The final snapshot of the MD-simulated INT1
structure was used as the initial structure to repeat the QM/MM
reaction coordinate calculations and calculate the activation
barrier for the second reaction step. The activation barrier calcu-
lated at the MP2/6-3111G**:Amber//HF/3-21G*:Amber level
starting from the MD-simulated INT1 structure is 15.1 kcal/mol,
which is close to the activation barrier of 15.3 kcal/mol calcu-
lated at the same level starting from the X-ray crystal structure.
The close activation barrier values calculated by using different
starting structures suggest that the energetic results calculated in
this study are reliable.
The good agreement between the QM/MM-calculated activa-
tion barrier for the rate-determining step and the corresponding
experimental kinetic data suggests that the reaction pathway
identified in this study is reasonable and that the QM/MM proto-
col used is satisfactory. As the activation barrier calculated by
the QM/MM method accounting for the effects of protein envi-
ronment is �20 kcal/mol lower than the corresponding activa-
tion barrier calculated by the QM method neglecting the protein
environment, the protein environment stabilizes the rate-deter-
mining transition state TS2 more favorably than stabilizing the
intermediate INT1. In light of these new insights, it should be
very interesting to design stable analogs of the substructure of
transition state TS2. The substructure of transition state TS2
may include all atoms of pyruvate and ThDP and there is no
covalent bond between the substructure and the remaining part
of the protein. The stable analogs of the substructure are
expected to be novel, potent AHAS inhibitors that may be used
as promising new antituberculosis drugs, antibacterial drugs,
and/or herbicides to overcome the drug resistance problem.
Conclusions
This is the first computational study to uncover the detailed cata-
lytic mechanism for the common steps of AHAS-catalyzed con-
densation reactions of a-keto acids. The first-principles QM and
hybrid QM/MM reaction coordinate calculations have led us to
uncover the fundamental reaction pathway, determine the activa-
tion barriers, and obtain valuable insights concerning the specific
roles of several key amino acid residues for the common steps
of AHAS-catalyzed condensation reactions of a-keto acids. The
QM/MM reaction coordinate calculations have provided valuable
details concerning the reaction process and role of the protein
environment. The most important amino acid residues involved
in the catalysis include Glu1440, Gln2070, Gly1210, and Gly511;
these residues form favorable hydrogen bonds with the reaction
center (consisting of atoms from substrate pyruvate and cofactor
ThDP) during the reaction process to stabilize the transition
states. In addition, Glu1440 also accepts a proton from ThDP
through hydrogen bonding during the catalytic reaction process.
The optimized geometries of the transition states are qualita-
tively consistent with available experimental observations,
including data from site-directed mutagenesis, and provide more
detailed structural and mechanistic information for transition
states and their evolution during the key reaction steps. Com-
pared to the high activation barrier calculated for the model
Figure 4. QM/MM-optimized geometries of INT1 (A) and TS2 (B) for the reaction catalyzed by the
Glu144Ala mutant of A. thaliana AHAS II. The QM atoms used on the QM/MM calculations are
represented by the balls in the figure.
1600 Xiong et al. • Vol. 31, No. 8 • Journal of Computational Chemistry
Journal of Computational Chemistry DOI 10.1002/jcc

reaction system neglecting the protein environment, the favor-
able interactions between the reaction center and protein envi-
ronment remarkably stabilize the transition state and, thus, lower
the activation barrier for the rate-determining reaction step by
�20 kcal/mol. All of the computational results indicate that the
rate-determining step of the AHAS-catalyzed reactions is the
second reaction step and the activation barrier calculated for
the rate-determining step is in good agreement with the experi-
mentally derived activation barrier. The detailed structural and
mechanistic insights should be valuable for rational design of
novel, potent AHAS inhibitors hat may be used as promising
new anti-tuberculosis drugs, antibacterial drugs, and/or herbi-
cides to overcome the drug resistance problem.
Acknowledgment
The authors acknowledge the Center for Computational Sciences
(CCS) at University of Kentucky for supercomputing time on
IBM X-series Cluster with 340 nodes and 1,360 processors.
References
1. Singh, B. K.; Shaner, D. L. Plant Cell 1995, 7, 935.
2. Engel, S.; Vyazmensky, M.; Geresh, S.; Barak, Z.; Chipman, D. M.
Biotechnol Bioeng 2003, 83, 833.
3. Ito, T.; Nakashimada, Y.; Kakizono, T.; Nishio, N. J Biosci Bioeng
2004, 97, 227.
4. Choia, K.-J.; Yub, Y. G.; Hahnc, H. G.; Choid, J.-D.; Yoon, M.-Y.
FEBS Lett 2005, 579, 4903.
5. Boigegrain, R.-A.; Liautard, J.-P.; Kohler, S. Antimicrob Agents
Chemother, 2005, 49, 3922.
6. Duggleby, R. G.; Pang, S. S. J. Biochem Mol Biol 2000, 33, 1.
7. Bloom, B. R.; Murray, C. J. Science 1992, 257, 1055.
8. Guleria, I.; Teitelbaum, R.; McAdam, R. A.; Kalpana, G.; Jacobs,
W. R., Jr.; Bloom, B. R. Nat Med 1996, 2, 334.
9. Grandoni, J. A.; Marta, P. T.; Schloss, J. V. J Antimicrob Chemother
1998, 42, 475.
10. Zohar, Y.; Einav, M.; Chipman, D.M.; Barak, Z. Biochim Biophys
Acta 2003, 1649, 97.
11. Mazur, B. J. Falco, S. C. Annu Rev Plant Physiol Plant Mol Biol
1989, 40, 441.
12. Geier, P. W.; Stahlman, P. W.; Hargett, J. G. Weed Sci 2001, 49, 788.
13. Gerwick, B. C.; Subermanian, V. I.; Loney-Gallant, V. I.; Chander,
D. P. Pestic Sci 1990, 29, 357.
14. Shimizu, T.; Nakayama, I.; Nakao, T.; Nezu, Y.; Abe, H. J. Pestic
Sci 1994, 19, 59.
15. LaRossa, R. A.; Schloss, J. V. J Biol Chem 1984, 259, 8753.
16. Shaner, D. L.; Anderson, P. C.; Stidham, M. A. Plant Physiol 1984,
76, 545.
17. Tranel, P. J.; Wright, T. R. Weed Sci 2002, 50, 700.
18. McCourt, J. A.; Pang, S. S.; King-Scott, J.; Duggleby, R. G.;
Guddat, L. W. Proc Natl Acad Sci USA 2006, 103, 569.
19. Frank, R. A.; Titman, C. M.; Pratap, J. V.; Luisi, B. F.; Perham, R. N.
Science 2004, 306, 872.
20. Pang, S. S.; Duggleby, R. G.; Guddat, L. W. J Mol Biol 2002, 317,
249.
21. Pang, S. S.; Guddat, L. W.; Duggleby, R. G. J Biol Chem 2003,
278, 7639.
22. McCourt, J. A.; Pang, S. S.; Duggleby, R. G.; Guddat, L. W. Bio-
chemistry 2005, 44, 2330.
23. Breslow, R. J Am Chem Soc 1958, 80, 3719.
24. Nemeria, N.; Tittmann, K.; Joseph, E.; Zhou, L.; Vazquez-Coll, M.;
Arjunan, P.; Hubner, G.; Furey, W.; Jordan, F. J Biol Chem 2005,
280, 21473.
25. Tittmann, K.; Golbik, R.; Uhlemann, K.; Khailova, L.; Schneider,
G.; Patel, M.; Jordan, F.; Chipman, D. M.; Duggleby, R. G.; Hubner,
G. Biochemistry 2003, 42, 7885.
26. Tittmann, K.; Vyazmensky, M.; Hubner, G.; Barak, Z. Chipman, D. M.
Proc Natl Acad Sci USA 2005, 102, 553.
27. Becke, A. D. J Chem Phys 1993, 98, 5648.
28. Lee, C.; Yang, W.; Parr, R. G. Phys Rev B 1988, 37, 785.
29. Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J.
J Phys Chem 1994, 98, 11623.
30. Gonzalez, C.; Schlegel, H. B. J Chem Phys 1989, 90, 2154.
31. Gonzalez, C.; Schlegel, H. B. J Phys Chem 1990, 94, 5523.
32. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb,
M. A.; Cheeseman, J. R.; Montgomery, J. A., Jr.; Vreven, T.; Kudin,
K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Bar-
one, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson,
G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.;
Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.;
Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross,
J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.;
Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.;
Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg,
J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.;
Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Fores-
man, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cio-
slowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.;
Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M.
A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.;
Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A.
Gaussian 03, Revision B. 05, Gaussian: Pittsburgh, PA, 2003.
33. Case, D. A.; Darden, T. A.; Cheatham, T. E., III; Simmerling, C. L.;
Wang, J.; Duke, R. E.; Luo, R.; Merz, K. M.; Wang, B.; Pearlman,
D. A.; Crowley, M.; Brozell, S.; Tsui, V.; Gohlke, H.; Mongan, J.;
Hornak, V.; Cui, G.; Beroza, P.; Schafmeister, C.; Caldwell, J. W.;
Ross, W. S.; Kollman, P. A. AMBER 8; University of California:
San Francisco, 2004.
34. Zhang, Y.; Lee, T.; Yang, W. J Chem Phys 1999, 110, 46.
35. Zhang, Y. J Chem Phys 2005, 122, 024114.
36. Corminboeuf, C.; Hu, P.; Tuckerman, M. E.; Zhang, Y. J Am Chem
Soc 2006, 128, 4530.
37. Zhang, Y.; Liu, H.; Yang, W. J Chem Phys 2000, 112, 3483.
38. Hu, P.; Zhang, Y. J Am Chem Soc 2006, 128, 1272.
39. Zheng, F.; Yang, W.; Ko, M.-C.; Liu, J.; Cho, H.; Gao, D.; Tong, M.;
Tai, H.-H.; Woods, J. H.; Zhan, C.-G. J Am Chem Soc 2008, 130, 12148.
40. Williams, I. H.; Maggiora, G. M. Theochem 1982, 6, 365.
41. Zhang, Y.; Liu, H.; Yang, W. Ab Initio QM/MM and Free Energy
Calculations of Enzyme Reactions. In Computational Methods for
Macromolecular Modeling - Challenges and Applications; Schlick, T.;
Gan, H. H., Eds.; Springer-Verlag: New York, 2002; pp. 332–354.
42. Bar-Ilan, A.; Balan, V.; Tittmann, K.; Golbik, R.; Vyazmensky, M.;
Hubner, G.; Barak, Z.; Chipman, D.M. Biochemistry 2001, 40,
11946.
43. Kern, D.; Kern, G.; Neef, H.; Tittmann, K.; Killenberg-Jabs, M.;
Wickner, C.; Schneider, G.; Hubner, G. Science 1997, 275, 67.
44. Jordan, F.; Zhang, Z.; Sergienko, E. Bioorg Chem 2002, 30, 188.
45. Jordan, F. Nat Prod Rep 2003, 20, 184.
46. Nemeria, N.; Baykal, A.; Joseph, E.; Zhang, S.; Yan, Y.; Furey, W.;
Jordan, F. Biochemistry 2004, 43, 6565.
47. Dyda, F.; Furey, W.; Swaminathan, S.; Sax, M.; Farrenkopf, B.;
Jordan, F. Biochemistry 1993, 32, 6165.
1601AHAS-Catalyzed Condensation Reactions
Journal of Computational Chemistry DOI 10.1002/jcc

48. Jordan, F.; Mariam, Y. F. J Am Chem Soc 1978, 100, 2534.
49. Lie, M. A.; Celik, L.; Jørgensen, K. A.; Schiøtt, B. Biochemistry
2005, 44, 14792.
50. Golbik, R.; Neef, H.; Hubner, G.; Konig, S.; Seliger, B.; Meshalkina,
L.; Kochetov, G. A.; Schellenberger, A. Bioorg Chem 1991, 19, 10.
51. Engel, S.; Vyazmensky, M.; Vinogradov, M.; Berkovich, D.;
Bar-Ilan, A.; Qimron, U.; Rosiansky, Y.; Barak, Z.; Chipman, D. M.
J Biol Chem 2004, 279, 24803.
52. Duggleby, R. G.; McCourt, J. A.; Guddat, L. W. Plant Physiol
Biochem 2008, 46, 309.
53. Zhao, Y.; Gonzlez-Garca, N.; Truhlar, D. G. J Phys Chem A 2005,
109, 2012.
54. Sousa, S. F.; Fernandes, P. A.; Ramos, M. J. J Phys Chem A, 2007,
111, 10439.
55. Alvarez-Idaboy, J. R.; Galano, A.; Bravo-Perez, G.; Ruiz, M. E.
J Am Chem Soc 2001, 123, 8387.
56. Pan, Y; Gao, D.; Zhan, C.-G. J Am Chem Soc 2008, 130, 5140.
57. Klahn, M.; Braun-Sand, S.; Rosta, E.; Warshel, A. J Phys Chem B
2005, 109, 15645.
58. Pan, Y.; Gao, D.; Yang, W.; Cho, H.; Yang, G.; Tai, H.-H.; Zhan,
C.-G. Proc Natl Acad Sci USA 2005, 102, 16656.
59. Xiong, Y.; Lu, H.; Zhan, C.-G. J Comput Chem 2008, 29, 1259.
60. Xiong, Y.; Lu, H.; Li, Y.; Yang, G.; Zhan, C.-G. Biophys J 2006,
91, 1858.
1602 Xiong et al. • Vol. 31, No. 8 • Journal of Computational Chemistry
Journal of Computational Chemistry DOI 10.1002/jcc



![Prostaglandin H Synthase-catalyzed Metabolism and DNA ...[CANCER RESEARCH 47, 4007-4014, August 1, 1987] Prostaglandin H Synthase-catalyzed Metabolism and DNA Binding of 2-Naphthylamine](https://img.pdfslide.net/doc/110x75/6125125eba335f0b336d21dc/prostaglandin-h-synthase-catalyzed-metabolism-and-dna-cancer-research-47-4007-4014.jpg)



![SYNF ORM - Thieme...Highly Enantioselective Synthesis of 3,4-Dihydropyrans through a Phosphine-Catalyzed [4+2] Annulation of Allenones and β,γ-Unsaturated α-Keto Esters C o n T](https://img.pdfslide.net/doc/110x75/5f74fef2290f8207a9090d52/synf-orm-thieme-highly-enantioselective-synthesis-of-34-dihydropyrans-through.jpg)
![Visible Light-Induced Salan-Copper(II)-Catalyzed ... · salan-Zr complex[10] can catalyze the a-hydroxylation of b-keto methyl esters with excellent yields and enantioselectivities](https://img.pdfslide.net/doc/110x75/5f7409768c693b1104744c12/visible-light-induced-salan-copperii-catalyzed-salan-zr-complex10-can-catalyze.jpg)




















