Embed Size (px)
Citation preview

CONFIDENTIAL
REPORT FOR
SKIN PAIN ASSESSMENT
FOLLOWING APPLICATION OF
THREE TOPICAL FORMULATIONS

CONFIDENTIAL
REPORT FOR
SKIN PAIN ASSESSMENT FOLLOWING APPLICATION OF
THREE TOPICAL FORMULATIONS
HTR STUDY NO. 10-130293-111
SPONSOR NO. P11-001
Final Report
June 14, 2011
FOR
MEDLINE INDUSTRIES, INC.
One Medline Place
Mundelein, IL 60060
BY
HILL TOP RESEARCH
6699 13th
Avenue North
St. Petersburg, FL 33710

HTR Study No. 10-130293-111 Final Report
Sponsor No. P11-001
June 14, 2011
Page 1 of 11
1. SUMMARY
The objective of this study was to assess pain after application of test articles on tape
stripped skin. The study followed a randomized, single-blind, single dose exposure
design. The subjects evaluated a total of five test articles on this study with two or
three test sites per ventral forearm. Skin sensory perception of pain was assessed
through the use of 100 mm anchored visual analog scale (VAS) and a 10-point pain
scale.
The study was divided into a pilot phase and a main phase. A total of 26 subjects
completed the study (6 subjects completed the pilot phase and 20 subjects completed
the main phase).
The test articles used on this study were three currently marketed skin protectant
products and two controls and were identified as follows:
HTR Code Description
A SurePrep® No-Sting Protective Barrier Spray
B MARATHON Liquid Skin Protectant
C 3M™
Cavilon™
No Sting Barrier Film Spray (IO)
D Negative Control – 0.9% Physiological Saline, USP
E Positive Control – 70% Isopropyl Alcohol in water solution
There were no adverse events reported during the course of the study.
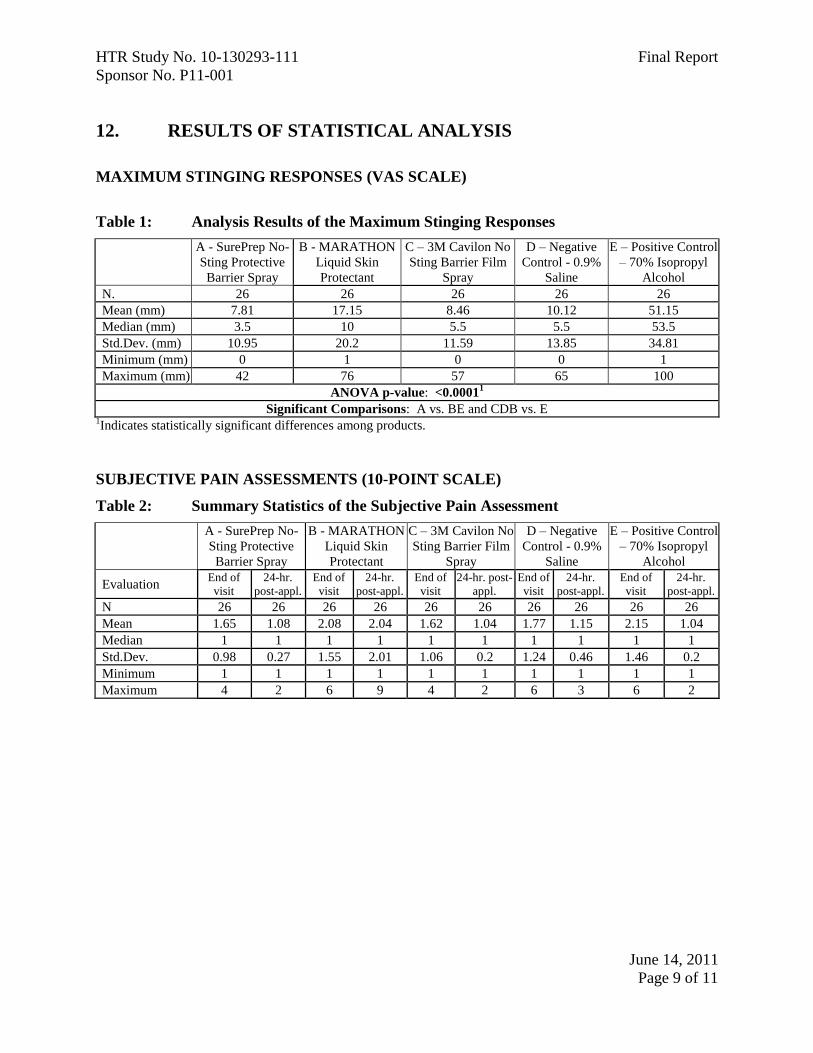
Based upon the results of this study, HTR Code A produced the least pain (mean
score = 7.81 mm), statistically significantly less than HTR Codes B (mean score =
17.15 mm) and E (mean score = 51.15 mm). Analyses of the mean pain scores found
that HTR Codes C (mean score = 8.46 mm) and B produced pain equivalent to the
negative control, HTR Code D (mean score = 10.12 mm), but significantly lower than
the positive control, HTR Code E. Though HTR Code B, MARATHON Liquid Skin
Protectant, received the second highest overall mean pain score, it should be noted
that it was the only test article that required physical touch during the application
procedure due to the nature of the supplied product.
Review of the subjective pain assessment using a 10-point scale found a similar trend
for the test articles at the end of the study visit with the lowest to highest mean pain
scores being HTR Codes C, A, D, B, and E. At the 24-hour follow-up evaluation
HTR Code B produced the highest mean pain score (2.04). It should be noted that for
all test articles at both time points, the mean pain scores were considered at the
threshold pain level or below.

HTR Study No. 10-130293-111 Final Report
Sponsor No. P11-001
June 14, 2011
Page 2 of 11
2. TABLE OF CONTENTS
1. SUMMARY ..................................................................................................................1
2. TABLE OF CONTENTS .............................................................................................2
3. SPONSOR PERSONNEL ............................................................................................3
4. INVESTIGATIVE PERSONNEL ................................................................................3
5. CLINICAL RESEARCH STANDARDS .....................................................................3
6. PROTOCOL .................................................................................................................3
6.1. Study Procedures ..........................................................................................................4
6.2. Protocol Amendments ..................................................................................................5
7. SUBJECTS ...................................................................................................................5
8. STUDY SCHEDULE ...................................................................................................6
9. TEST ARTICLES .........................................................................................................6
10. ADVERSE EVENTS....................................................................................................7
11. METHOD OF STATISTICAL ANALYSIS ................................................................8
11.1. Demographic and Baseline Variable Analysis .............................................................8
11.2. Sensory Assessment Analysis .......................................................................................8
12. RESULTS OF STATISTICAL ANALYSIS ................................................................9
13. CONCLUSIONS ........................................................................................................10
14. SIGNATURE ..............................................................................................................10
15. QUALITY STATEMENT ..........................................................................................11
APPENDICES
APPENDIX I / IRB Approval Letters
APPENDIX II / Protocol and Amendment
APPENDIX III / Statistical Tables
RECORD RETENTION AND PUBLICATION NOTICE

HTR Study No. 10-130293-111 Final Report
Sponsor No. P11-001
June 14, 2011
Page 3 of 11
3. SPONSOR PERSONNEL
Edward Drower
Clinical Project Manager
Medline Industries, Inc.
The study was monitored by Edward Drower on March 27, 2011.
4. INVESTIGATIVE PERSONNEL
Investigator: Micah Humphrey, BS
Technical Director: Jeffrey E. Berg, BS, CCRA
Study Manager: Micah Humphrey, BS
Biostatistician: James P. Bowman, MS
Report Writer: Verda Heinrichs, BA
5. CLINICAL RESEARCH STANDARDS
The clinical investigation, including the informed consent, was reviewed by an Institutional
Review Board in accordance with Title 21 of the Code of Federal Regulations, Parts 50 and 56.
Approval by the Board was obtained on February 3, 2011, prior to initiation of the investigation
(see Appendix I). Revised calendars for the pilot and main studies were approved on February
24, 2011 and Protocol Amendment No. 1 and the revised consent form were approved on March
10, 2011.
This study was conducted according to the principles of Good Clinical Practice, the Standard
Operating Procedures of Hill Top Research, the study protocol and protocol amendment.
6. PROTOCOL
The study protocol was followed (see Appendix II) with the exception of the following
deviations:
During the Day 2 procedures, the 5-minute assessment was completed 6 minutes post-
application and the 15-minute assessment was completed 16 minutes post-application for Site
1 for Subject No. 105.
During the Day 2 procedures, the 15-minute assessment was completed 20 minutes post-
application for Sites 2 and 3 for Subject No. 108.
In the opinion of the Investigator, the deviations did not compromise the integrity of the study.

HTR Study No. 10-130293-111 Final Report
Sponsor No. P11-001
June 14, 2011
Page 4 of 11
6.1. Study Procedures
This was a single center, randomized, single blind, single dose exposure study to assess the skin
sensory perception of pain following test article application on slightly abraded ventral forearm
skin on normal, healthy adult subjects. Skin sensory perception of pain was assessed through the
use of a 100mm, anchored visual analog scale (VAS) and a 10-pt pain scale. The study was
divided into a pilot phase and a main phase. During each phase, subjects were required to make
a total of up to three visits to the test facility over approximately one week. The subjects
evaluated a total of five test articles on this study with two or three test sites per ventral forearm.
The study was divided into a pilot phase and a main phase. The pilot data were reviewed by the
Sponsor and since the results were acceptable the study continued with the main phase. The
sensitivity and magnitude of the control treatment groups was evaluated to assess the validity of
the paradigm. Data was also evaluated to confirm size estimations before the main phase was
initiated.
At Visit 1, potential subjects read a HIPAA form and consent form prior to any study related
procedures being conducted. Study personnel conducted a consent form discussion with the
subject and addressed any questions or concerns related to the study procedures. Written
informed consent was obtained and the subjects received copies of the HIPAA and consent
forms.
Subjects were assigned a screening number and the study inclusion/exclusion criteria were
reviewed and documented. Subject demographics and a brief medical history were documented.
Current medication (prescription and over-the-counter) use over the two weeks prior to the
screening visit were recorded.
Subjects who met all study entrance criteria received an appointment time for Visit 2 (Day 2 –
Dosing) and were dismissed from the test facility. Study Visits 1 and 2 may have been combined
into a single visit if the schedule allowed.
If Visits 1 and 2 were not combined, subjects returned to the test facility at their scheduled
appointment time. Subjects had their study entrance criteria reviewed to confirm ongoing
eligibility and were queried as to the occurrence of any adverse events or changes in their
concomitant medications.
Following confirmation of eligibility, subjects received their final subject number, were
randomized and the following study conduct procedures were performed (Note: tape stripping,
dosing and subject pain assessments proceeded for one test site/test article completely before the
next site/test article was evaluated):
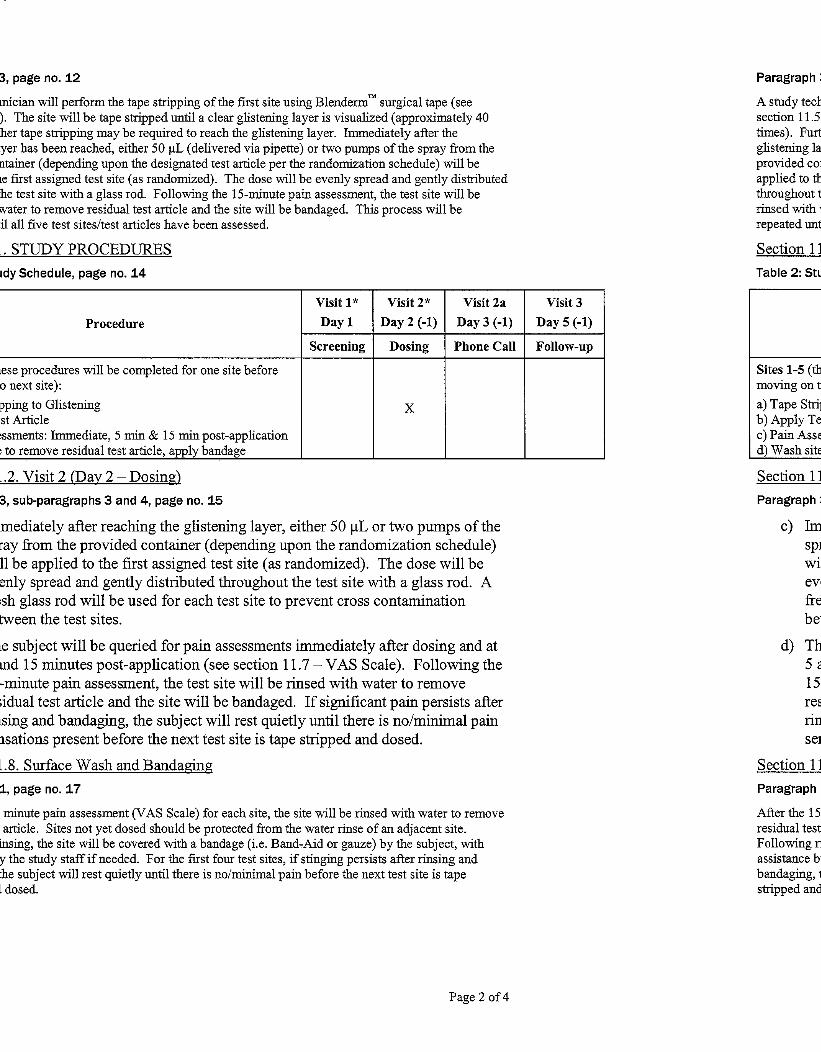
a) Two or three 10 cm2 (2.5 cm x 4 cm) test sites on each ventral forearm (5 sites in total)
were demarcated, with no test site closer than 3 cm from the wrist or antecubital fossa,
and each test site was placed at least 1 cm apart.
b) A study technician performed the tape stripping of the first test site using Blenderm™
surgical tape. The site was tape stripped until a clear glistening layer is visualized
(approximately 40 times). Further tape stripping may have been required to reach the
glistening layer.

HTR Study No. 10-130293-111 Final Report
Sponsor No. P11-001
June 14, 2011
Page 5 of 11
c) Immediately after reaching the glistening layer, either a single use applicator or two
pumps of the spray from the provided container (depending upon the designated test
article per the randomization schedule) was applied to the first assigned test site (as
randomized).
d) The subject was queried for pain assessments immediately after dosing and at 5 and 15
minutes post-application. Following the 15-minute pain assessment, the test site was
bandaged. If significant pain persisted after bandaging, the subject was to rest quietly
until there was no/minimal pain present before the next test site was tape stripped and
dosed.
e) At subsequent intervals, tape stripping, dosing, subject pain assessments and bandaging
was repeated for the remaining test sites/test articles, as described above.
Prior to release from the test facility, the subject was instructed to evaluate their pain at each site
using a 10-point pain scale. They were also instructed to wear protective clothing on the
forearms to prevent sun exposure until all the sites had completely healed. The subjects provided
the test facility with a telephone number for contact for the 24-hour pain evaluation and were
given an appointment time for Visit 3 and dismissed from the test facility.
Subjects were contacted by telephone approximately 24 hours after they were dismissed from the
facility to provide subsequent pain assessments of their sites. The subjects were queried as to the
level of pain at each site based on a 10-point scale.
Subjects returned to the test facility at their scheduled appointment time three days following the
treatment day, where a trained skin grader assessed the abraded skin sites. Subjects were queried
as to the occurrence of any adverse events or changes in their concomitant medications since
their previous visit. Subjects received their study compensation and their participation was
considered complete.
6.2. Protocol Amendments
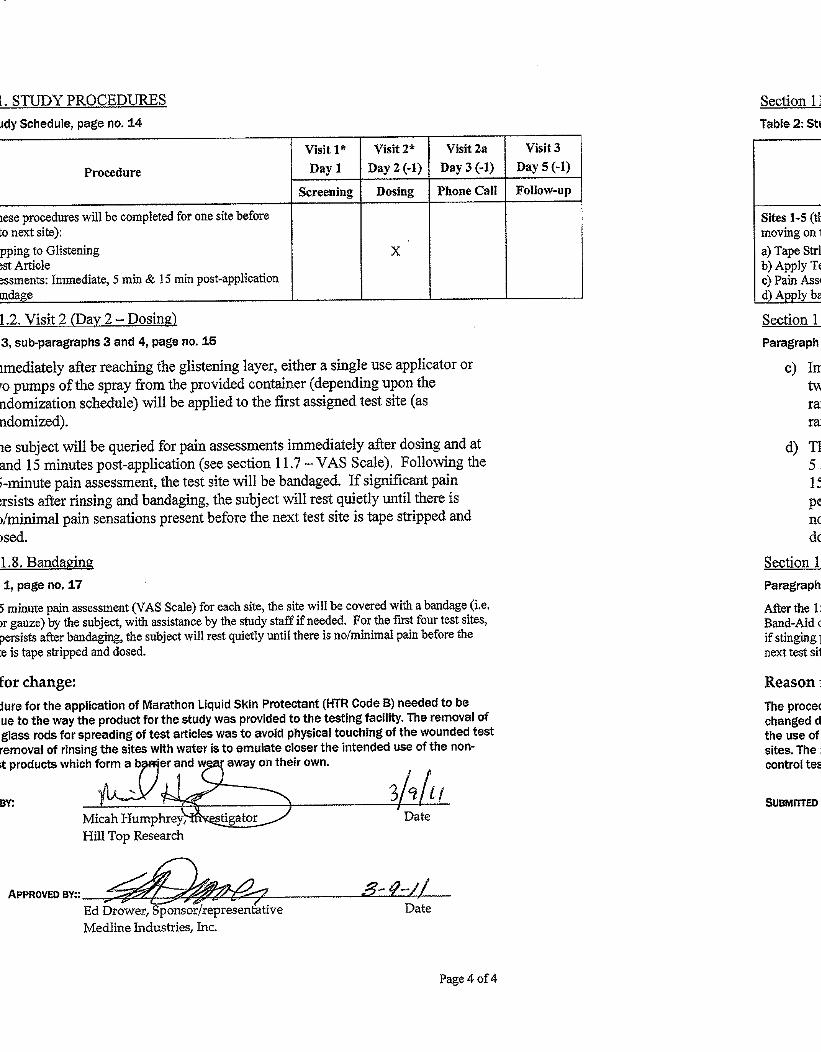
There was one amendment to the protocol (see Appendix II). Protocol Amendment No. 1
modified the test article application procedures (Section 11.0 of the protocol). The application
procedure of Marathon Liquid Skin Protectant (HTR Code B) was changed due to the way the
product was provided to the testing facility by the Sponsor (individual application vials). Use of
glass rods for spreading the test articles was removed to avoid physically touching the wounded
test sites and rinsing of test sites with water was removed to emulate more closely the intended
use of the non-control test products which form a barrier and wear away on their own.
7. SUBJECTS
The study included a pilot phase and a main phase. Subjects were screened and signed the
Consent Form before any study procedures were completed. Only those subjects meeting the
study criteria were enrolled. A total of 27 subjects were enrolled into the study.
One subject, No. 111, withdrew consent and did not complete the study.
A total of 26 subjects completed the study.
HTR Study No. 10-130293-111 Final Report
Sponsor No. P11-001
June 14, 2011
Page 6 of 11
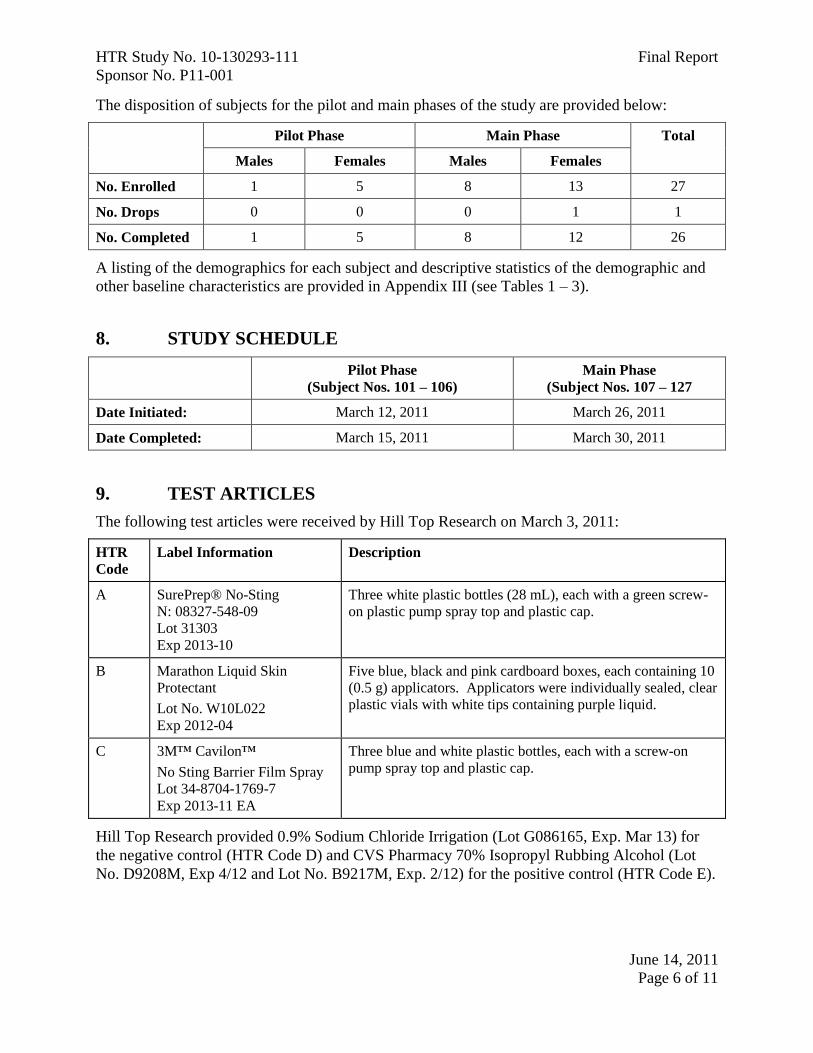
The disposition of subjects for the pilot and main phases of the study are provided below:
Pilot Phase Main Phase Total
Males Females Males Females
No. Enrolled 1 5 8 13 27
No. Drops 0 0 0 1 1
No. Completed 1 5 8 12 26
A listing of the demographics for each subject and descriptive statistics of the demographic and
other baseline characteristics are provided in Appendix III (see Tables 1 – 3).
8. STUDY SCHEDULE
Pilot Phase
(Subject Nos. 101 – 106)
Main Phase
(Subject Nos. 107 – 127
Date Initiated: March 12, 2011 March 26, 2011
Date Completed: March 15, 2011 March 30, 2011
9. TEST ARTICLES
The following test articles were received by Hill Top Research on March 3, 2011:
HTR
Code
Label Information Description
A SurePrep® No-Sting
N: 08327-548-09
Lot 31303
Exp 2013-10
Three white plastic bottles (28 mL), each with a green screw-
on plastic pump spray top and plastic cap.
B Marathon Liquid Skin
Protectant
Lot No. W10L022
Exp 2012-04
Five blue, black and pink cardboard boxes, each containing 10
(0.5 g) applicators. Applicators were individually sealed, clear
plastic vials with white tips containing purple liquid.
C 3M™ Cavilon™
No Sting Barrier Film Spray
Lot 34-8704-1769-7
Exp 2013-11 EA
Three blue and white plastic bottles, each with a screw-on
pump spray top and plastic cap.
Hill Top Research provided 0.9% Sodium Chloride Irrigation (Lot G086165, Exp. Mar 13) for
the negative control (HTR Code D) and CVS Pharmacy 70% Isopropyl Rubbing Alcohol (Lot
No. D9208M, Exp 4/12 and Lot No. B9217M, Exp. 2/12) for the positive control (HTR Code E).
HTR Study No. 10-130293-111 Final Report
Sponsor No. P11-001
June 14, 2011
Page 7 of 11
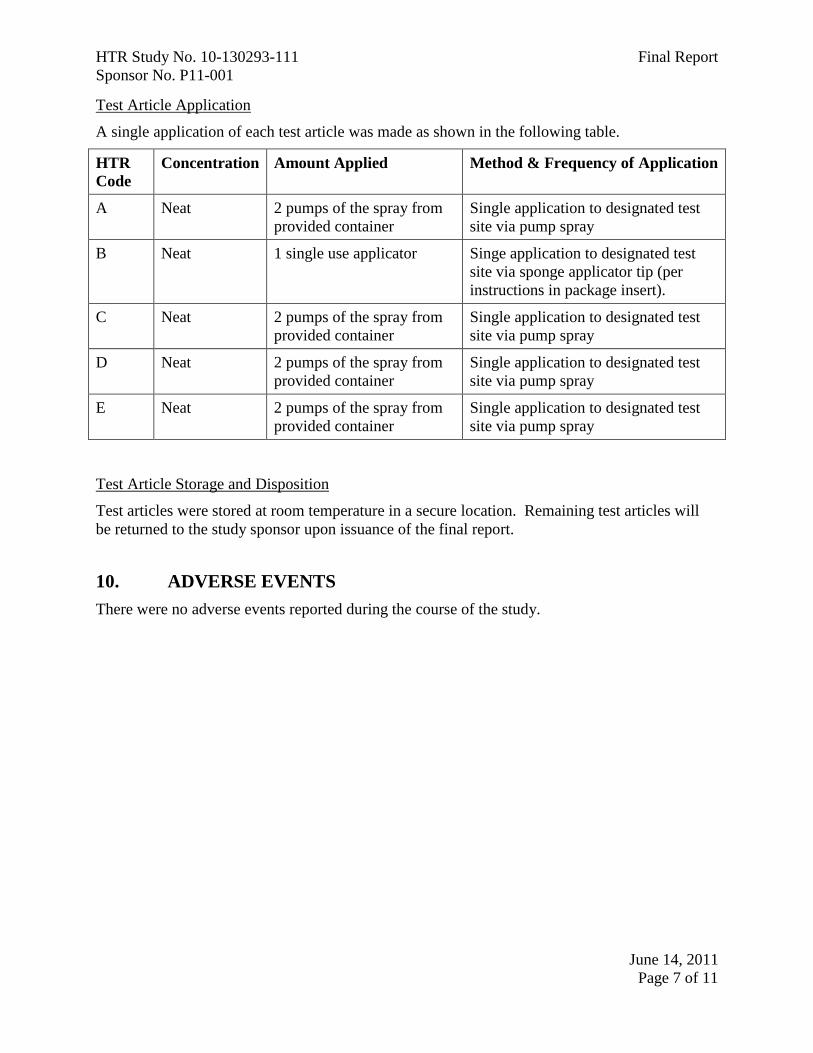
Test Article Application
A single application of each test article was made as shown in the following table.
HTR
Code
Concentration Amount Applied Method & Frequency of Application
A Neat 2 pumps of the spray from
provided container
Single application to designated test
site via pump spray
B Neat 1 single use applicator Singe application to designated test
site via sponge applicator tip (per
instructions in package insert).
C Neat 2 pumps of the spray from
provided container
Single application to designated test
site via pump spray
D Neat 2 pumps of the spray from
provided container
Single application to designated test
site via pump spray
E Neat 2 pumps of the spray from
provided container
Single application to designated test
site via pump spray
Test Article Storage and Disposition
Test articles were stored at room temperature in a secure location. Remaining test articles will
be returned to the study sponsor upon issuance of the final report.
10. ADVERSE EVENTS
There were no adverse events reported during the course of the study.

HTR Study No. 10-130293-111 Final Report
Sponsor No. P11-001
June 14, 2011
Page 8 of 11
11. METHOD OF STATISTICAL ANALYSIS
Safety: Safety analysis was carried out on the data of every subject who received at least one test
article treatment.
Pain Sensory Evaluation: Analysis was carried out on the data of all subjects who completed
the study. The data from withdrawn subjects was not included in the final statistical analyses.
11.1. Demographic and Baseline Variable Analysis
Descriptive statistics of the demographic and other baseline characteristics were summarized and
are presented in Table 1.
11.2. Sensory Assessment Analysis
The source data were the pain scores assigned by the subjects immediately after dosing and at 5
and 15 minutes following topical application of the test articles. The subjects’ responses,
measured in mm, were tabulated for each post-dosing assessment for each test site. Descriptive
statistics (mean, standard deviation, median, minimum, maximum) were provided for the
response to the pain sensory question for each time point post-dosing, and for the maximum
stinging response, for each of the test articles. The data used in the statistical analysis were the
maximum score assigned for each treatment. Data were analyzed using analysis of variance
techniques.
Additionally, pain scores were assigned to each test site by the subjects 24 hours after dosing
utilizing a 1 to 10 scale. These data were tabulated with descriptive statistics.
All statistical tests employed a level of significance of 0.05 and no adjustments were made for
the number of tests being conducted.
HTR Study No. 10-130293-111 Final Report
Sponsor No. P11-001
June 14, 2011
Page 9 of 11
12. RESULTS OF STATISTICAL ANALYSIS
MAXIMUM STINGING RESPONSES (VAS SCALE)
Table 1: Analysis Results of the Maximum Stinging Responses
A - SurePrep No-
Sting Protective
Barrier Spray
B - MARATHON
Liquid Skin
Protectant
C – 3M Cavilon No
Sting Barrier Film
Spray
D – Negative
Control - 0.9%
Saline
E – Positive Control
– 70% Isopropyl
Alcohol
N. 26 26 26 26 26
Mean (mm) 7.81 17.15 8.46 10.12 51.15
Median (mm) 3.5 10 5.5 5.5 53.5
Std.Dev. (mm) 10.95 20.2 11.59 13.85 34.81
Minimum (mm) 0 1 0 0 1
Maximum (mm) 42 76 57 65 100
ANOVA p-value: <0.00011
Significant Comparisons: A vs. BE and CDB vs. E 1Indicates statistically significant differences among products.
SUBJECTIVE PAIN ASSESSMENTS (10-POINT SCALE)
Table 2: Summary Statistics of the Subjective Pain Assessment
A - SurePrep No-
Sting Protective
Barrier Spray
B - MARATHON
Liquid Skin
Protectant
C – 3M Cavilon No
Sting Barrier Film
Spray
D – Negative
Control - 0.9%
Saline
E – Positive Control
– 70% Isopropyl
Alcohol
Evaluation End of
visit
24-hr.
post-appl.
End of
visit
24-hr.
post-appl.
End of
visit
24-hr. post-
appl.
End of
visit
24-hr.
post-appl.
End of
visit
24-hr.
post-appl.
N 26 26 26 26 26 26 26 26 26 26
Mean 1.65 1.08 2.08 2.04 1.62 1.04 1.77 1.15 2.15 1.04
Median 1 1 1 1 1 1 1 1 1 1
Std.Dev. 0.98 0.27 1.55 2.01 1.06 0.2 1.24 0.46 1.46 0.2
Minimum 1 1 1 1 1 1 1 1 1 1
Maximum 4 2 6 9 4 2 6 3 6 2



HTR Study No. 10-130293-111 Final Report
Sponsor No. P11-001
APPENDIX I. IRB APPROVAL LETTERS
Total number of pages including this cover page = 10










HTR Study No. 10-130293-111 Final Report
Sponsor No. P11-001
APPENDIX II. PROTOCOL AND AMENDMENT
Total number of pages including this cover page = 42

CONFIDENTIAL
PROTOCOL FOR
SKIN PAIN ASSESSMENT FOLLOWING APPLICATION OF THREE
TOPICAL FORMULATIONS
HILL TOP RESEARCH STUDY NO. 10-130293-111
SPONSOR PROTOCOL NO. P11-001
FINAL PROTOCOL
JANUARY 28, 2011
FOR
MEDLINE INDUSTRIES, INC
ONE MEDLINE PLACE
MUNDELINE, IL 60060
BY
HILL TOP RESEARCH INCORPORATED
6699 13TH
AVENUE NORTH
ST. PETERSBURG, FL 33710


Sponsor Protocol No. P11-001 Page 3
Final HTR Study No. 10-130293-111
January 28, 2011
2. TABLE OF CONTENTS
1. PROTOCOL SIGNATURE PAGE ..............................................................................2
2. TABLE OF CONTENTS .............................................................................................3
3. SYNOPSIS ...................................................................................................................6
4. CLINICAL INVESTIGATION SITES ........................................................................8
4.1. Site Information ............................................................................................................8
4.2. Sponsor Information .....................................................................................................8
5. INTRODUCTION ........................................................................................................8
5.1. Objective .......................................................................................................................8
5.2. Efficacy Endpoints ........................................................................................................9
5.3. Safety Endpoints ...........................................................................................................9
6. STUDY DESIGN AND DURATION ..........................................................................9
7. ETHICS ........................................................................................................................9
7.1. Institutional Review Board ...........................................................................................9
7.2. Ethical Conduct of the Study ........................................................................................9
7.3. Subject Information and Consent ...............................................................................10
7.4. Authorization to Disclose Protected Health Information ...........................................10
8. STUDY POPULATION .............................................................................................10
8.1. Inclusion Criteria ........................................................................................................10
8.2. Exclusion Criteria .......................................................................................................11
8.3. Additional Study Restrictions .....................................................................................11
9. DOSAGE AND ADMINISTRATION .......................................................................12
9.1. Packaging ....................................................................................................................13
9.2. Storage, Issue and Return of Test Articles .................................................................13
10. SUBJECT NUMBERING AND IDENTIFICATION ................................................13
11. STUDY PROCEDURES ............................................................................................13
11.1. Visit 1 (Day 1 – Screening) ........................................................................................14
11.2. Visit 2 (Day 2 - Dosing) .............................................................................................15
11.3. Visit 2a (Telephone Pain Assessment) .......................................................................16
11.4. Visit 3 (Day 5 – Follow-up) ........................................................................................16
11.5. Tape Stripping ............................................................................................................16

Sponsor Protocol No. P11-001 Page 4
Final HTR Study No. 10-130293-111
January 28, 2011
11.6. Treatment Assignments ..............................................................................................16
11.7. Pain Assessments ........................................................................................................17
11.8. Surface Wash and Bandaging .....................................................................................17
12. STATISTICAL ANALYSIS ......................................................................................18
12.1. Sample Size Calculation .............................................................................................18
12.2. Evaluable Subjects ......................................................................................................18
12.3. Demographic and Baseline Variable Analysis ...........................................................18
12.4. Sensory Assessment Analysis .....................................................................................18
12.5. Adverse Event Analysis ..............................................................................................18
13. ADVERSE EVENTS..................................................................................................19
13.1. Definition of an Adverse Event ..................................................................................19
13.2. Definition of a Serious Adverse Event (SAE) ............................................................19
13.3. Definition of an Unexpected AE ................................................................................20
13.4. Eliciting Reports of Adverse Events ...........................................................................20
13.5. Reporting Adverse Events ..........................................................................................20
13.6. Causality Assessment .................................................................................................21
13.7. Severity Assessment ...................................................................................................21
13.8. Outcome Assessment ..................................................................................................22
13.9. Anticipated Reactions .................................................................................................22
14. PREMATURE DISCONTINUATION AND WITHDRAWAL CRITERIA ............22
14.1. Premature Discontinuation .........................................................................................22
14.2. Criteria for Termination ..............................................................................................23
14.3. Replacement Procedures .............................................................................................23
15. MONITORING ...........................................................................................................23
16. AMENDMENTS /MODIFICATION OF THIS PROTOCOL...................................23
17. SUBJECT CONFIDENTIALITY ..............................................................................23
18. INVESTIGATOR OBLIGATIONS ...........................................................................24
18.1. Investigator Service ....................................................................................................24
18.2. Documentation ............................................................................................................24
18.3. Institutional Review Board (IRB) ...............................................................................24
18.4. Informed Consent .......................................................................................................24
18.5. Performance ................................................................................................................24

Sponsor Protocol No. P11-001 Page 5
Final HTR Study No. 10-130293-111
January 28, 2011
18.6. Use of Investigational Materials .................................................................................25
18.7. Source Documents ......................................................................................................25
18.8. Final Report, Retention and Review of Records ........................................................25
19. PROTOCOL CHANGES AND DEVIATIONS ........................................................25
20. CONFIDENTIALITY ................................................................................................26
21. REFERENCES ...........................................................................................................26
LIST OF TABLES
Table 1: Test Article Identification ...........................................................................................12
Table 2: Study Schedule ...........................................................................................................14

Sponsor Protocol No. P11-001 Page 6
Final HTR Study No. 10-130293-111
January 28, 2011
3. SYNOPSIS
Sponsor: Medline Industries, Inc.
Title of the Study: Skin Pain Assessment Following Application of Three Topical
Formulations
Sponsor Protocol No: P11-001
HTR Study No: 10-130293-111
Objective: The objective of this study is to assess pain after test article application on tape
stripped skin.
Study Design: Randomized, single blind, single dose exposure study to assess the skin sensory
perception of pain following test article application on slightly abraded ventral forearm skin on
normal, healthy adult subjects. The study will be divided into a pilot phase and a main phase.
Number of Subjects: Approximately 6 subjects will be enrolled into the pilot phase of the
study in order to complete with 5 subjects. If the study continues into the main phase,
approximately 22 additional subjects will be enrolled in order to complete with a total of 25
subjects between the two phases.
Main Criteria for Inclusion: Normal, healthy adult male and female subjects, 18 to 65 years
of age (inclusive), with normal skin on their ventral forearms and no history of allergies or
sensitivities to the ingredients in the test articles, adhesives or latex.
Test Products, Dose, and Mode of Administration: Five test articles will be used in this
study (single dose, topically applied to designated randomized, tape-stripped sites on the
ventral forearm):
Formulation A: SurePrep® No-Sting Protective Barrier Spray
Formulation B: MARATHON Liquid Skin Protectant
Formulation C: 3M™
Cavilon™
No Sting Barrier Film Spray (IO)
Formulation D: Negative Control – 0.9% Physiological Saline, USP
Formulation E: Positive Control – 70% Isopropyl Alcohol in water solution
Criteria for Evaluation: Sensory Evaluation: Analysis will be carried out on the data
(subjective pain assessments) of all subjects who complete the study.
Safety: Safety analysis will be carried out on the data of every subject who received at least
one test article treatment. Safety will also be assessed through the collection of adverse events.
Statistical Methods: The subjects‘ pain responses, measured in mm from the visual analogue
scales, will be tabulated for each post-dosing assessment for each test site. Descriptive
statistics (mean, standard deviation, median, minimum, maximum) will be provided for the
response to the pain sensory assessment for each time point post-dosing, and for the maximum
pain response for each of the test articles. The data to be used in the statistical analysis are the
maximum score assigned for each treatment. Data will be analyzed using analysis of variance
techniques.
Additionally, pain scores will be assigned to each test site by the subjects 24 hours after dosing
utilizing a 1 to 10 scale. These data will be tabulated with descriptive statistics.

Sponsor Protocol No. P11-001 Page 7
Final HTR Study No. 10-130293-111
January 28, 2011
Regulatory Compliance: This clinical study, including the Informed Consent, will be
reviewed by an Institutional Review Board in accordance with Title 21 of the Code of Federal
Regulations, Parts 50 and 56. Approval by the Board will be obtained prior to the initiation of
the study. The study will be conducted in accordance with the Principles of Good Clinical
Practice, the Standard Operating Procedures of Hill Top Research and the Sponsor's Protocol
and Protocol Amendment(s).

Sponsor Protocol No. P11-001 Page 8
Final HTR Study No. 10-130293-111
January 28, 2011
4. CLINICAL INVESTIGATION SITES
4.1. Site Information
Subject recruitment will be conducted at a Hill Top Research (HTR) contract recruiting facility
located at 6088 Main & Mill Streets, Miamiville, OH 45147. Subject screening, enrollment, test
article assignments and dermal assessments will be conducted at Hill Top Research, 6699 13th
Avenue North, St. Petersburg, FL 33710.
CLINICAL INVESTIGATOR SITE: Hill Top Research
6699 13th
Avenue North
St. Petersburg, FL 33710
Phone: 727-344-7602
Fax: 727-345-5323
PRINCIPAL INVESTIGATOR: Micah Humphrey, B.Sc.
Technical Director: Jeffrey E. Berg, B.Sc., CCRA
Biostatistician: James P. Bowman, MS
Study Manager: Micah Humphrey, B.Sc.
4.2. Sponsor Information
STUDY SPONSOR: Edward Drower
Clinical Project Manager
Medline Industries, Inc.
One Medline Place
Mundelein, IL 60060
Phone: 847-473-5883
Cell: 224-221-8924
Fax: 866-758-4660
R&D Workroom: 847-643-3849
E-mail: [email protected]
5. INTRODUCTION
The use of wound barrier products has led to increased wound protection and improved overall
healing with minimal side effects.1,2
The polymer composition of these products tends to be
either silicone or cyanoacrylate based. The purpose of this study will evaluate the non-stinging
properties of three of these spray barrier products (Marathon, Sureprep® and Cavilon™).
5.1. Objective
The objective of this study is to assess pain after test article application on tape stripped skin.

Sponsor Protocol No. P11-001 Page 9
Final HTR Study No. 10-130293-111
January 28, 2011
5.2. Efficacy Endpoints
The efficacy endpoints of this study are:
Comparison of subjective pain assessments (VAS Scale at dosing, 10-pt scale at end
of dosing day, and 10-pt scale at 24-hour follow-up phone call) of all subjects who
complete the study.
5.3. Safety Endpoints
The safety endpoint of this study is:
Assessments of safety through the collection of adverse events.
6. STUDY DESIGN AND DURATION
Randomized, single blind, single dose exposure study to assess the skin sensory perception of
pain following test article application on slightly abraded ventral forearm skin on normal, healthy
adult subjects. Skin sensory perception of pain will be assessed through the use of a 100mm,
anchored visual analog scale (VAS) and a 10-pt pain scale. The study will be divided into a pilot
phase and a main phase. During each phase, subjects will be required to make a total of up to
three visits to the test facility over approximately one week. The subjects will evaluate a total of
five test articles on this study with two or three test sites per ventral forearm.
7. ETHICS
7.1. Institutional Review Board
This study will be reviewed by an appropriately constituted Institutional Review Board (IRB) as
outlined in 21 CFR Part 56. The IRB will review the protocol, any amendments, the informed
consent form (ICF), subject instructions, safety information, Investigator‘s curriculum vitae (CV)
and advertisements (if applicable).
A copy of the IRB approval opinion, along with any other documents required under local
regulations or standards must be on file at sponsor headquarters before the first shipment of
study supplies is provided to the study site.
7.2. Ethical Conduct of the Study
This study will be conducted in accordance with 21 Code of Federal Regulations (CFR) Parts 50
and 56).
Medline Industries, Inc. is responsible for the ongoing safety evaluation of the investigational
products and will promptly notify participating Investigators and regulatory authorities of
findings that could adversely affect the safety of subjects, impact the conduct of the study, or
alter the IRB‘s approval to continue the study. Medline Industries, Inc. will promptly report all
adverse reactions related to the study articles that are both serious and unexpected to the
appropriate regulatory authorities and to all Investigators and IRBs currently involved in studies
of this study article.

Sponsor Protocol No. P11-001 Page 10
Final HTR Study No. 10-130293-111
January 28, 2011
7.3. Subject Information and Consent
Subject consent will be obtained prior to participation in any study conduct as required by the
regulatory guidelines (21 CFR Part 50). Subjects will be given ample opportunity to read the
consent form and have all questions regarding study conduct answered prior to signing the
consent form. Each subject will be provided with a copy of the ICF to retain for his or her
records. The original signed ICF will be retained on file at the study center.
7.4. Authorization to Disclose Protected Health Information
Subjects will be informed of the following information: The purpose of the protected health
information (PHI) being collected, the possibility that the PHI may be re-disclosed, the duration
of the authorization, the right to revoke the authorization, and the right to refuse signature and
limit access to PHI during and following the conduct of the trial. Written authorization to
disclose PHI will be incorporated into the informed consent process and will be obtained prior to
the subject entering the study per HTR standard operating procedures (SOPs). Each subject will
be provided with a signed copy of the authorization and the original will be retained on file at the
study center.
8. STUDY POPULATION
Approximately 6 subjects will be enrolled into the pilot phase of the study in order to complete
with 5 subjects. If the study continues into the main phase, approximately 22 additional subjects
will be enrolled in order to complete with a total of 25 subjects between the two phases of the
study.
All subjects will fulfill the inclusion and exclusion criteria as outlined in Sections 8.1 and 8.2.
8.1. Inclusion Criteria
To participate in the study, each subject must satisfy the following inclusion criteria:
a. Subject has been informed of the nature of the study and has read, understands and
signed an informed consent form and HIPAA statement.
b. Subject is between 18 and 65 years of age, inclusive.
c. Subject is male or female and in reasonably good health.
d. Females of childbearing potential agree to use an adequate method of birth control
during study participation (including oral contraceptives, hormone implant, IUD,
spermicide with barrier method [condom or diaphragm], male sexual partner(s)
surgically sterile or abstinence). Subjects of childbearing potential include those
women who are not postmenopausal (no menses) for at least 1 year or surgically
sterile (hysterectomy or bilateral tubal ligation [both tubes tied] or bilateral
oophorectomy [both ovaries removed]).

Sponsor Protocol No. P11-001 Page 11
Final HTR Study No. 10-130293-111
January 28, 2011
8.2. Exclusion Criteria
A subject will be excluded from participation in the test period for any of the following criteria:
a. Subject has a history of significant skin condition or disorder, such as psoriasis,
atopic dermatitis, eczema, skin cancer, etc.
b. Subject has a current skin condition on the test sites (ventral surface of the forearms),
such as scar tissue, sunburn, tattoos, scratches, bruises, excessive hair or uneven skin
tones that may interfere with the placement of the test sites, their assessments or
could compromise the safety of the subject.
c. Subject has a history of allergy to soaps, lotions, emollients, ointments, creams,
cosmetics, adhesives or latex.
d. Subject has a history of sensitivity/allergy to the ingredients found in the test articles,
isopropyl alcohol or a history of adverse reactions to topical formulations.
e. Subject has a history of keloid formation (abnormal scarring).
f. Subject has heavily pigmented skin that could heal with abnormal darkening at the
test sites.
g. Subject has a medical condition or uses a medication that, in the Investigator‘s
judgment, places the subject at an undue risk.
h. Subject is currently taking, or has been taking on a continuous basis in the last 14
days prior to study start, corticosteroids, nonsteroidal anti-inflammatory drugs,
acetylsalicylic acid (81 mg per day is allowed) or other pain relief medications.
i. Subject is currently taking, or has taken in the last 14 days prior to study start,
anticoagulant medications (e.g. Coumadin).
j. Subject reports intending to start, stop or change dose of any prescription or over-the-
counter (OTC) medication within 48 hours prior to or throughout the study.
Acetaminophen may be taken by the subject, if needed.
k. Subject has used prescription or OTC topical medications on the ventral forearms
within one month prior to study conduct.
l. Subject has a history of alcohol or drug abuse within one year prior to study conduct.
m. Subject is pregnant, lactating or breast-feeding (self report only – urine pregnancy
testing will not be required).
n. Subject has used an investigational drug within the 30 days prior to study start.
Any exceptions to the inclusion/exclusion criteria will be considered on a case-by case basis and
will be documented in the Source Documentation. All exceptions need approval from the Study
Sponsor and Principal Investigator prior to enrollment in the study.
8.3. Additional Study Restrictions a. No exercise with either arm, and no strenuous exercise overall for 24 hours prior to
test article administration and throughout the study.
b. Following the test article applications, subjects will be instructed to wear protective
clothing on the forearms to prevent sun exposure.
Sponsor Protocol No. P11-001 Page 12
Final HTR Study No. 10-130293-111
January 28, 2011
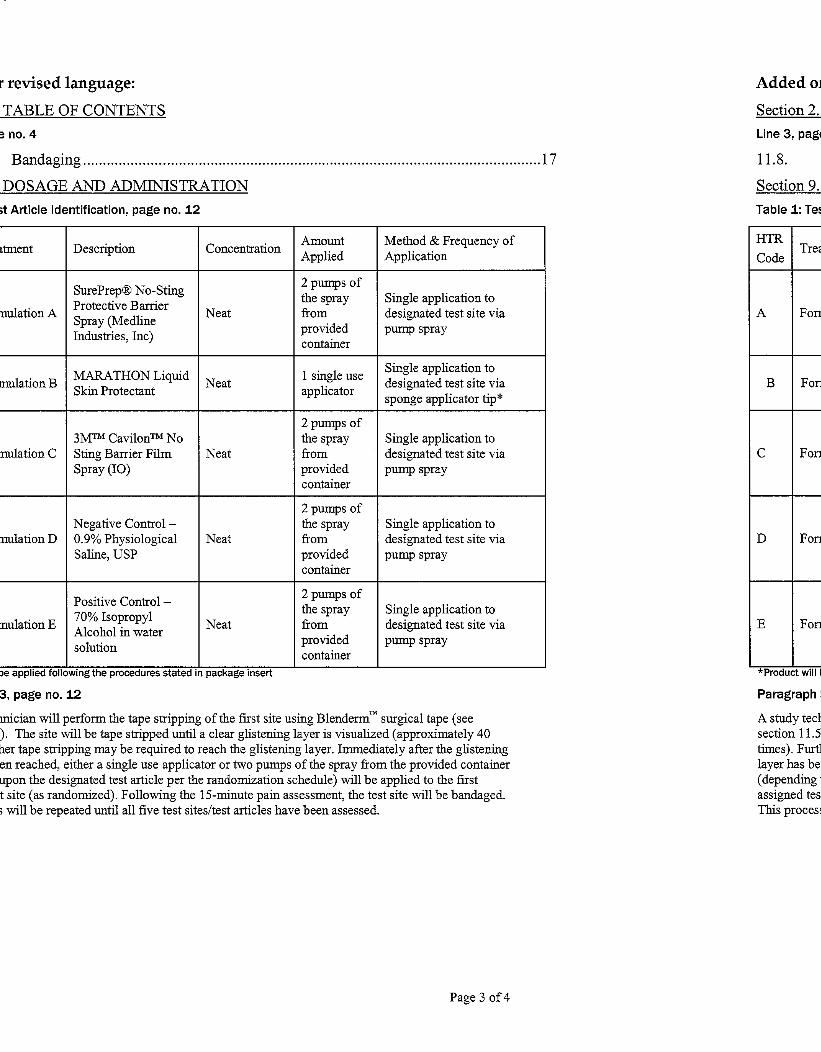
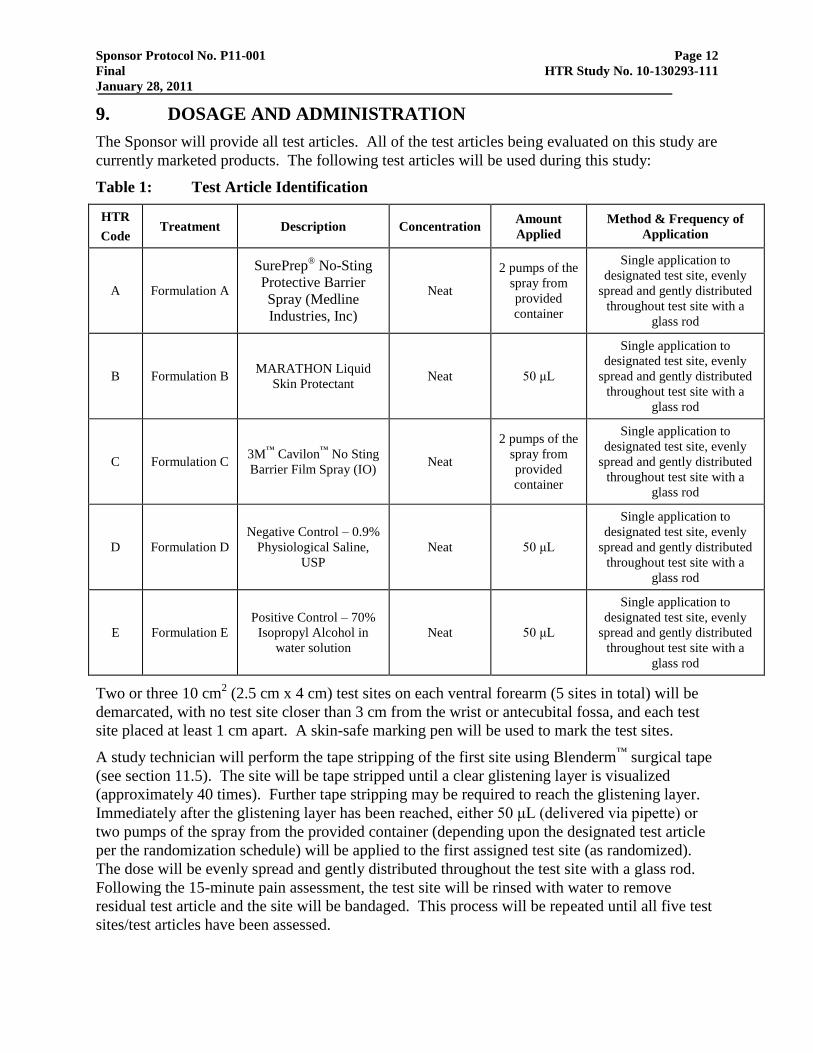
9. DOSAGE AND ADMINISTRATION
The Sponsor will provide all test articles. All of the test articles being evaluated on this study are
currently marketed products. The following test articles will be used during this study:
Table 1: Test Article Identification
HTR
Code Treatment Description Concentration
Amount
Applied
Method & Frequency of
Application
A Formulation A
SurePrep® No-Sting
Protective Barrier
Spray (Medline
Industries, Inc)
Neat
2 pumps of the
spray from
provided
container
Single application to
designated test site, evenly
spread and gently distributed
throughout test site with a
glass rod
B Formulation B MARATHON Liquid
Skin Protectant Neat 50 μL
Single application to
designated test site, evenly
spread and gently distributed
throughout test site with a
glass rod
C Formulation C 3M
™ Cavilon
™ No Sting
Barrier Film Spray (IO) Neat
2 pumps of the
spray from
provided
container
Single application to
designated test site, evenly
spread and gently distributed
throughout test site with a
glass rod
D Formulation D
Negative Control – 0.9%
Physiological Saline,
USP
Neat 50 μL
Single application to
designated test site, evenly
spread and gently distributed
throughout test site with a
glass rod
E Formulation E
Positive Control – 70%
Isopropyl Alcohol in
water solution
Neat 50 μL
Single application to
designated test site, evenly
spread and gently distributed
throughout test site with a
glass rod
Two or three 10 cm2 (2.5 cm x 4 cm) test sites on each ventral forearm (5 sites in total) will be
demarcated, with no test site closer than 3 cm from the wrist or antecubital fossa, and each test
site placed at least 1 cm apart. A skin-safe marking pen will be used to mark the test sites.
A study technician will perform the tape stripping of the first site using Blenderm™
surgical tape
(see section 11.5). The site will be tape stripped until a clear glistening layer is visualized
(approximately 40 times). Further tape stripping may be required to reach the glistening layer.
Immediately after the glistening layer has been reached, either 50 μL (delivered via pipette) or
two pumps of the spray from the provided container (depending upon the designated test article
per the randomization schedule) will be applied to the first assigned test site (as randomized).
The dose will be evenly spread and gently distributed throughout the test site with a glass rod.
Following the 15-minute pain assessment, the test site will be rinsed with water to remove
residual test article and the site will be bandaged. This process will be repeated until all five test
sites/test articles have been assessed.

Sponsor Protocol No. P11-001 Page 13
Final HTR Study No. 10-130293-111
January 28, 2011
A randomization schedule will be generated by Hill Top Research that will identify the number
and location of the five test sites (i.e. 2 or 3 sites on the left or right ventral forearm) and the test
articles will be randomly assigned (HTR Codes A-E) within these five test sites so that each
subject receives each test article one time to one of the five test sites.
9.1. Packaging
The test articles will be provided by the sponsor in plain packaging with labels on each
container. The test article containers will be packaged accordingly so that the subjects will be
blinded with respect to the treatment assignments.
9.2. Storage, Issue and Return of Test Articles
The Sponsor will provide the study site with the study supplies. The study site will administer
the test articles on the appropriate days according to the randomization schedule.
All test articles are to be stored at the study site in a controlled access room at room temperature
15oC-30
oC (59
oF-86
oF).
The Investigator will maintain an accurate record of the receipt of the test articles as shipped by
the Sponsor, including the date and quantity received. The record will also include the test article
code and may include the final weight, as necessary. This inventory record must be available for
inspection at any time.
At the completion of the study, all test articles will be returned to the Sponsor upon issue of the
final study report. The Sponsor will also be notified of any subsequent changes in test article
accountability as they occur.
10. SUBJECT NUMBERING AND IDENTIFICATION
Subjects will be identified by their initials and a unique subject number. Screening numbers (i.e.
01, 02, 03…) will be assigned consecutively by the Investigator or designee. Subjects who
qualify and enter the treatment phase of the study will be assigned a subject number (i.e. 101,
102, 103…).
11. STUDY PROCEDURES
The study will be divided into a pilot phase and a main phase. The Sponsor will review the
results of the pilot phase and if acceptable results occur, the study will continue into the main
phase. The pilot data will be reviewed prior to the continuation of the main phase. The
sensitivity and magnitude of the control treatment groups will be evaluated to assess the validity
of the paradigm. Data will also be evaluated to confirm size estimations before the main phase is
initiated.
The procedures for each study visit for both the pilot and main phase of the study are identical
and are outlined in Table 2 below:
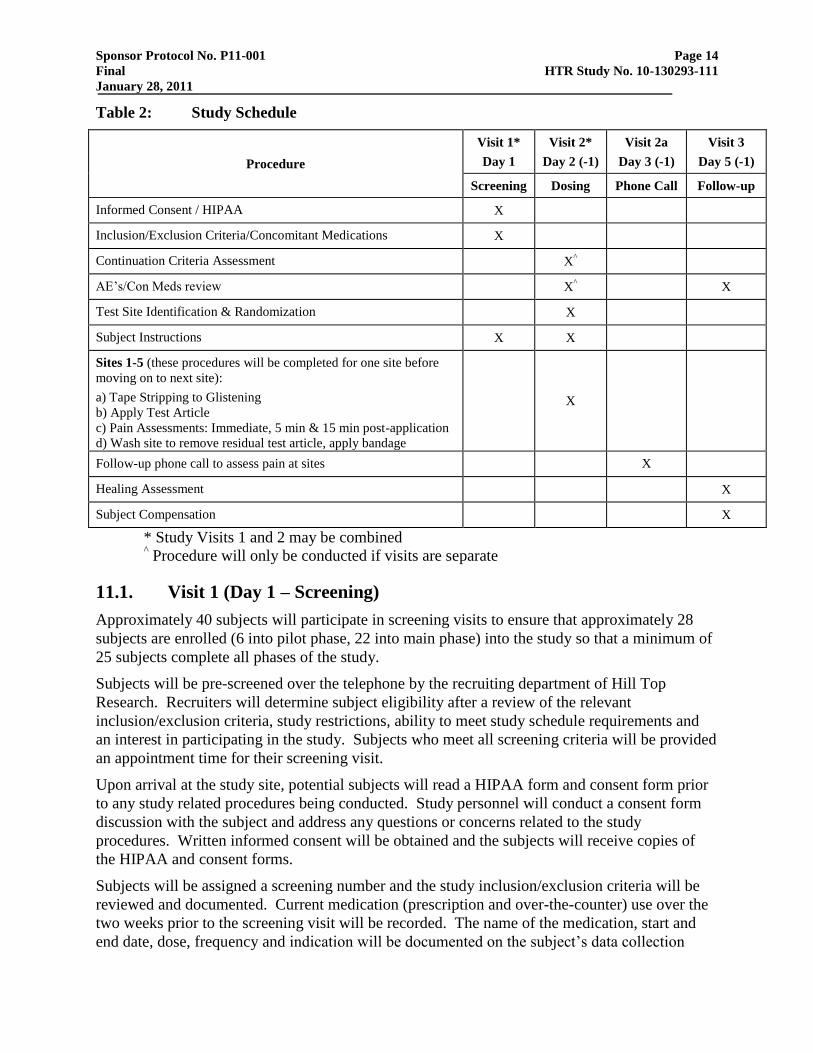
Sponsor Protocol No. P11-001 Page 14
Final HTR Study No. 10-130293-111
January 28, 2011
Table 2: Study Schedule
Procedure
Visit 1*
Day 1
Visit 2*
Day 2 (-1)
Visit 2a
Day 3 (-1)
Visit 3
Day 5 (-1)
Screening Dosing Phone Call Follow-up
Informed Consent / HIPAA X
Inclusion/Exclusion Criteria/Concomitant Medications X
Continuation Criteria Assessment X^
AE‘s/Con Meds review X^ X
Test Site Identification & Randomization X
Subject Instructions X X
Sites 1-5 (these procedures will be completed for one site before
moving on to next site):
a) Tape Stripping to Glistening
b) Apply Test Article
c) Pain Assessments: Immediate, 5 min & 15 min post-application
d) Wash site to remove residual test article, apply bandage
X
Follow-up phone call to assess pain at sites X
Healing Assessment X
Subject Compensation X
* Study Visits 1 and 2 may be combined ^ Procedure will only be conducted if visits are separate
11.1. Visit 1 (Day 1 – Screening)
Approximately 40 subjects will participate in screening visits to ensure that approximately 28
subjects are enrolled (6 into pilot phase, 22 into main phase) into the study so that a minimum of
25 subjects complete all phases of the study.
Subjects will be pre-screened over the telephone by the recruiting department of Hill Top
Research. Recruiters will determine subject eligibility after a review of the relevant
inclusion/exclusion criteria, study restrictions, ability to meet study schedule requirements and
an interest in participating in the study. Subjects who meet all screening criteria will be provided
an appointment time for their screening visit.
Upon arrival at the study site, potential subjects will read a HIPAA form and consent form prior
to any study related procedures being conducted. Study personnel will conduct a consent form
discussion with the subject and address any questions or concerns related to the study
procedures. Written informed consent will be obtained and the subjects will receive copies of
the HIPAA and consent forms.
Subjects will be assigned a screening number and the study inclusion/exclusion criteria will be
reviewed and documented. Current medication (prescription and over-the-counter) use over the
two weeks prior to the screening visit will be recorded. The name of the medication, start and
end date, dose, frequency and indication will be documented on the subject‘s data collection

Sponsor Protocol No. P11-001 Page 15
Final HTR Study No. 10-130293-111
January 28, 2011
form. During the course of the study, if a subject is given any over-the-counter or prescription
medications or if administration of any medication becomes necessary during the study, the
name of the medication, reason for use, dosage information, date and frequency of administration
will also be documented on the subject‘s source documents.
Subjects who meet all study entrance criteria will receive an appointment time for Visit 2 (Day 2
– Dosing) and will be dismissed from the test facility. Study Visits 1 and 2 may be combined
into a single visit if the schedule allows.
11.2. Visit 2 (Day 2 - Dosing)
Study Visits 1 and 2 may be combined into a single visit if the schedule allows.
If Visits 1 and 2 are not combined, subjects will return to the test facility at their scheduled
appointment time. Subjects will have their study entrance criteria reviewed to confirm ongoing
eligibility and will be queried as to the occurrence of any adverse events or changes in their
concomitant medications.
Following confirmation of eligibility, subjects will receive their final subject number, be
randomized and the following study conduct procedures will be performed (Note: tape stripping,
dosing and subject pain assessments will proceed for one test site/test article completely before
the next site/test article is evaluated):
a) Two or three 10 cm2 (2.5 cm x 4 cm) test sites on each ventral forearm (5 sites in total)
will be demarcated, with no test site closer than 3 cm from the wrist or antecubital fossa,
and each test site placed at least 1 cm apart. A skin-safe marking pen will be used to
identify the test sites.
b) A study technician will perform the tape stripping of the first test site using Blenderm™
surgical tape (see section 11.5). The site will be tape stripped until a clear glistening
layer is visualized (approximately 40 times). Further tape stripping may be required to
reach the glistening layer.
c) Immediately after reaching the glistening layer, either 50 μL or two pumps of the spray
from the provided container (depending upon the randomization schedule) will be applied
to the first assigned test site (as randomized). The dose will be evenly spread and gently
distributed throughout the test site with a glass rod. A fresh glass rod will be used for
each test site to prevent cross contamination between the test sites.
d) The subject will be queried for pain assessments immediately after dosing and at 5 and 15
minutes post-application (see section 11.7 – VAS Scale). Following the 15-minute pain
assessment, the test site will be rinsed with water to remove residual test article and the
site will be bandaged. If significant pain persists after rinsing and bandaging, the subject
will rest quietly until there is no/minimal pain sensations present before the next test site
is tape stripped and dosed.
e) At subsequent intervals, tape stripping, dosing, subject pain assessments and
rinsing/bandaging will be repeated for the remaining test sites/test articles, as described
above.

Sponsor Protocol No. P11-001 Page 16
Final HTR Study No. 10-130293-111
January 28, 2011
f) Prior to release from the test facility, the subject will be instructed to evaluate their pain
at each site using a 10-point pain scale (see section 11.7 – 10-point scale). They will also
be instructed to wear protective clothing on the forearms to prevent sun exposure until all
the sites have completely healed. The subject will provide the test facility with a
telephone number for contact for the 24 hour pain evaluation (Visit 2a) and be given an
appointment time for Visit 3 and will be dismissed from the test facility.
11.3. Visit 2a (Telephone Pain Assessment)
Subjects will be contacted by telephone at approximately 24 hours after they are dismissed from
the facility to provide subsequent pain assessments of their sites. The subjects will be queried as
to the level of pain at each site based on a 10-point scale.
11.4. Visit 3 (Day 5 – Follow-up)
Subjects will return to the test facility at their scheduled appointment time three days following
the treatment day, where a trained skin grader will assess the abraded skin sites. Subjects will be
queried as to the occurrence of any adverse events or changes in their concomitant medications
since their previous visit. Subjects will receive their study compensation and their participation
will be considered complete.
11.5. Tape Stripping
Blenderm™
surgical tape (3M™
) will be used for tape stripping. Gloves or finger cots will be
worn by the study staff performing the tape stripping to avoid wound contact. Gloves/finger cots
will be changed between subjects. Skin sites will undergo a tape stripping procedure to create a
superficial wound down to the glistening layer and compromise the skin barrier. The procedure
is outlined below:
Cut strips of Blenderm™
tape to approximately 7.0 x 2.5 cm. Tape strips may be
prepared ahead of time.
Place a tape strip on the marked area of the test site, press it down, rub firmly within
the site marks and remove the strip with a strong and quick stroke. Discard the tape
in the trash.
Repeat stripping using other tape strips, in alternate directions, until a clear glistening
layer can be visualized or after 39 times (40 strips total), whatever comes first. The
number of strippings necessary to reach the glistening layer varies among subjects.
Although not anticipated, further stripping (beyond the 40 strips) may be required to
reach the glistening layer.
11.6. Treatment Assignments
There are a total of two or three sites on each arm for a total of five test sites. Of these, three
sites will be dosed with one of the three test articles (see section 9, Table 1). The remaining two
sites will be dosed with either the negative control (0.9% physiological saline) or the positive
control (70% isopropyl alcohol). Treatment assignments to the test sites will be randomized and
order of application of the test articles to their assigned sites will be randomized. The subjects

Sponsor Protocol No. P11-001 Page 17
Final HTR Study No. 10-130293-111
January 28, 2011
will be blinded with respect to the treatment assignments. The randomization schedule will be
provided by Hill Top Research.
11.7. Pain Assessments
VAS Scale
Immediately after dosing and at approximately 5 and 15 minutes after dose application of each
test article, the subject will be asked the question: ―Do you feel any stinging, pain, or discomfort
sensation at this site?‖. The subject will respond using a 100 mm visual analogue scale
anchored at one end with ‗None‘ (equal to 0 mm) and the other end with ‗Severe‘ (equal to 100
mm). The subject will be instructed to place a single vertical line on the scale that best indicates
the degree of stinging/pain/discomfort. Using calipers, a technician will measure the distance
from zero (i.e. ‗None‘) to the mark on the scale made by the subject, and will record this value
for each post-dosing pain assessment for each test site.
10-point Scale
Prior to leaving the test site following application of the test articles, subjects will be asked the
question: ―How would you rate the pain at site X?‖. The subject will respond for each of their
sites using the 10-pt. scale below:
1 = None, no pain
2 = Threshold pain
3 = Very slight pain
4 = Slight pain
5 = Slight to moderate pain
6 = Moderate pain
7 = Slightly strong pain
8 = Moderately strong pain
9 = Strong pain
10 = Very strong pain
11.8. Surface Wash and Bandaging
After the 15 minute pain assessment (VAS Scale) for each site, the site will be rinsed with water
to remove residual test article. Sites not yet dosed should be protected from the water rinse of an
adjacent site. Following rinsing, the site will be covered with a bandage (i.e. Band-Aid or gauze)
by the subject, with assistance by the study staff if needed. For the first four test sites, if stinging
persists after rinsing and bandaging, the subject will rest quietly until there is no/minimal pain
before the next test site is tape stripped and dosed.
If a site presents with intolerable pain within the 15 minute assessment period, it will be rinsed
with water and the last score carried forward through the remaining assessment times.

Sponsor Protocol No. P11-001 Page 18
Final HTR Study No. 10-130293-111
January 28, 2011
12. STATISTICAL ANALYSIS
12.1. Sample Size Calculation
This is a study to determine the potential for pain sensory response to the application of currently
marketed topical formulations. The sample size is based on clinical judgment and is believed to
be sufficient to satisfy the objectives.
12.2. Evaluable Subjects
Safety: Safety analysis will be carried out on the data of every subject who received at least one
test article treatment.
Pain Sensory Evaluation: Analysis will be carried out on the data of all subjects who completed
the study. The data from withdrawn subjects will not be included in the final statistical analyses.
12.3. Demographic and Baseline Variable Analysis
Descriptive statistics will be provided to summarize the demographic and other baseline
characteristics.
12.4. Sensory Assessment Analysis
The source data are the pain scores assigned by the subjects immediately after dosing and at 5
and 15 minutes following topical application of the test articles (see section 11.6). The subjects‘
responses, measured in mm, will be tabulated for each post-dosing assessment for each test site.
Descriptive statistics (mean, standard deviation, median, minimum, maximum) will be provided
for the response to the pain sensory question for each time point post-dosing, and for the
maximum stinging response, for each of the test articles. The data to be used in the statistical
analysis are the maximum score assigned for each treatment. Data will be analyzed using
analysis of variance techniques.
Additionally, pain scores will be assigned to each test site by the subjects 24 hours after dosing
utilizing a 1 to 10 scale. These data will be tabulated with descriptive statistics.
All statistical tests will employ a level of significance of 0.05 and no adjustments will be made
for the number of tests being conducted.
12.5. Adverse Event Analysis
The frequency of adverse events will be tabulated. A listing of adverse events, including start
and end date/time, severity, relationship to test article, and outcome, will be provided.

Sponsor Protocol No. P11-001 Page 19
Final HTR Study No. 10-130293-111
January 28, 2011
13. ADVERSE EVENTS
13.1. Definition of an Adverse Event
An Adverse Event or Adverse Experience (AE) is any untoward medical occurrence in a subject
administered a test article. It is not necessary that the AE have a causal relationship to treatment
with the test article.
An AE therefore is any unfavorable and unintended sign (for example, a clinically significant
abnormal laboratory finding) symptom, or disease temporally associated with the use of a test
article, whether or not considered related to the test article.
AEs include:
Changes in the general condition of the subject
Subjective symptoms offered by or elicited from the subject
Objective signs observed by the Investigator or study personnel
All concurrent diseases that occur after the start of the trial, including any change in
severity or frequency of pre-existing disease
All clinically relevant laboratory abnormalities or physical findings that occur during the
trial.
Findings not considered clinically significant that are related to the Investigator‘s assessments
are not to be recorded on the AE reporting page. They should instead be recorded on the
relevant source documents.
13.2. Definition of a Serious Adverse Event (SAE)
The Investigator or other study personnel must immediately (within 24 hours) inform Medline
Industries, Inc of all Serious Adverse Events (SAEs) that occur in study subjects.
An SAE is one that:
Results in death
Is an immediate threat to life
Requires inpatient hospitalization, or prolongation of existing hospitalization
Results in persistent or significant disability/incapacity
Is a congenital anomaly or birth defect.
In addition to the above, other important medical events that have not resulted in death, are not
life-threatening, or do not require hospitalization may be considered serious adverse experiences
when, based upon appropriate medical judgment, they are considered to jeopardize the patient or
subject and may require medical or surgical intervention to prevent one of the outcomes listed
above.

Sponsor Protocol No. P11-001 Page 20
Final HTR Study No. 10-130293-111
January 28, 2011
Examples of medical events that might be included as "serious" under this criterion include:
Emergency room treatment or evaluation for signs and symptoms of potentially
serious AEs (e.g., unwitnessed loss of consciousness, allergic bronchospasm
requiring intensive treatment at home or in ER, blood dyscrasias or convulsions that
do not result in hospitalization, anaphylactoid reactions, or the development of drug
dependence or drug abuse). However, emergency room visits that can be defined as
routine outpatient care (e.g., suture removal, treatment for a sprained ankle, earache,
etc.) do not meet this criterion and do not require immediate notification.
Symptomatic overdose if it results in a serious outcome.
Intentional overdose/suicide attempt—with or without any sign or symptom—when
considered to be a threat to life.
Neoplasia (benign or malignant) if judged to be medically serious.
Although not a serious AE in itself, exposure to study article during pregnancy—even if no AE
is produced in the mother—should be reported within 24 hours.
13.3. Definition of an Unexpected AE
An Unexpected Adverse Event or UAE is any adverse experience, the specificity or severity of
which is not consistent with the Investigator‘s Brochure / Manufacturer‘s Safety Information.
13.4. Eliciting Reports of Adverse Events
At each study visit (and during any contact with a subject occurring outside of a defined study
visit, including any contact after study completion), all adverse events reported by the subject or
subject representative or observed or otherwise identified by the Investigator or other study
personnel will be documented.
13.5. Reporting Adverse Events
All AEs must be recorded on the appropriate AE reporting page of the subject‘s source
documents. All AEs must be reported, whether or not they are considered causally related to
study article. For every AE, the Investigator must provide an assessment of the severity,
chronicity, causal relationship to study article, and seriousness of the event; document all actions
taken with regard to study article; and detail any other treatment measures taken for the AE. The
Investigator or other study personnel must immediately (within 24 hours) inform Medline
Industries, Inc. of any AE considered serious or otherwise significant as described in Section
13.2 above. Reporting should not be delayed pending resolution or outcome of an event. If an
outcome for an AE is not available at the time of the initial report, Medline Industries, Inc.
expects follow-up to proceed until such time as an outcome is known.

Sponsor Protocol No. P11-001 Page 21
Final HTR Study No. 10-130293-111
January 28, 2011
Notification may be via telephone or facsimile transmission of a written report. The following
staff at Medline Industries, Inc. should be contacted:
Edward Drower
Clinical Project Manager
Medline Industries, Inc.
One Medline Place
Mundelein, IL 60060
Phone: 847-473-5883
Cell: 224-221-8924
Fax: 866-758-4660
R&D Workroom: 847-643-3849
E-mail: [email protected]
13.6. Causality Assessment
For all AEs, the Investigator will provide an assessment of causal relationship to study article.
The causality assessment must be recorded on the appropriate AE reporting page of the subject‘s
source document. Causal relationship will be classified according to the following criteria:
Unrelated suggests a clearly evident relationship to other etiologies such as
concomitant medications or conditions or subject‘s known clinical state.
Possible suggests that the association of the AE with the study article is unknown.
Other etiologies are also possible.
Probable suggests that a reasonable temporal sequence of the AE with article
administration exists and based upon the Investigator's clinical experience, the
association of the AE with study article seems likely.
Definite suggests that a causal relationship exists between the study article and the
AE, and other conditions (concomitant illness, progression or expression of the
disease state, reaction to concomitant medication) do not appear to explain the AE.
13.7. Severity Assessment
The Investigator will provide an assessment of the severity of each adverse reaction by recording
a severity rating on the appropriate AE reporting page of the subject‘s source document.
(Severity, which is a description of the intensity of manifestation of the AE, is distinct from
seriousness, which implies a patient outcome or AE-required treatment measure associated with
a threat to life or functionality.) Severity will be assessed according to the following scale:
Mild - The AE was an annoyance to the subject, but did not further hinder baseline
functioning; the AE may have been intermittent or continuous.
Moderate - The AE caused the subject to experience some discomfort or some
interference with normal activities, but was not hazardous to health; prescription drug
therapy may have been employed to treat the AE.

Sponsor Protocol No. P11-001 Page 22
Final HTR Study No. 10-130293-111
January 28, 2011
Severe - The AE caused the subject to experience severe discomfort or severely
limited or prevented normal activities and represented a definite hazard to health;
prescription drug therapy and/or hospitalization may have been employed to treat the
AE.
13.8. Outcome Assessment
The Investigator will determine the outcome of the AE. Outcome will be assessed according to
the following scale:
Resolved Without Sequelae - The AE completely healed by end of study (or ongoing
yet unrelated to study, therefore resolved for purposes of study).
Resolved With Sequelae – The AE healed by the end of the study, but aftereffect,
disease or injury is present. Examples would include a stroke that resulted in partial
paralysis; the stroke resolved, but there was residual paralysis or erythema from a
skin reaction that resolved but hyperpigmentation remained.
Ongoing at the End of the Study – The AE had not healed by the end of the study.
This does not mean that the AE was not followed until resolution.
Death
13.9. Anticipated Reactions
During study conduct, the subject will be prompted by the Investigator or designate to report any
new or worsening adverse event.
Subjects may experience some reactions as a result of the test article application, which could
include pain, burning, stinging, tingling, and/or itching or other irritation. In addition, the tape
stripping may cause some minor discomfort (similar to removing a bandage) and dryness,
redness or flaking. These reactions are expected to be confined to the test article application
areas. These responses will not be considered an adverse event except for when the subject
declares it intolerable and requires or requests the test article be removed prior to the 15 minute
test site pain assessment.
It is possible that subjects may have an allergic reaction to the test articles. The occurrence of an
allergic reaction will be considered an adverse event. If any of the test sites become infected,
that will also be considered an adverse event.
14. PREMATURE DISCONTINUATION AND WITHDRAWAL
CRITERIA
14.1. Premature Discontinuation
Every effort will be made to conduct the final evaluation on subjects who discontinue from the
trial prematurely regardless of cause. Final evaluation is defined as completion of the evaluations
scheduled for the final visit.

Sponsor Protocol No. P11-001 Page 23
Final HTR Study No. 10-130293-111
January 28, 2011
14.2. Criteria for Termination
Test article treatment may be terminated for the following reasons:
An Adverse Event that requires study article to be discontinued.
Withdraws consent to continue participation in the study.
Subject is lost to Follow-up
Protocol violation (including lack of compliance)
Other Reasons, such as Administrative Reasons
14.3. Replacement Procedures
Subjects who prematurely drop out of the study will not be replaced.
15. MONITORING
A Sponsor representative may meet with the Investigator and his/her staff prior to the entrance of
the first subject to review the procedures to be followed in conducting the study. After the
enrollment of the first subject, the Investigator will permit the Sponsor to monitor the progress of
the trial on site periodically. The Investigator will make available the source documents as well
as the subjects‘ records and signed consent forms. The Investigator will review the source
documents for any corrected data, and sign the appropriate page(s). Copies of the relevant source
documents will be sent to Hill Top Data Management for data input and analysis. Original
source documents will be maintained by the Investigator.
16. AMENDMENTS /MODIFICATION OF THIS PROTOCOL
Any amendments to the protocol will be proposed to the Investigator in writing by Medline
Industries, Inc. No protocol changes may be implemented before the amended protocol has been
approved by the Institutional Review Board and the signature page, signed by the Investigator
has been received by Medline Industries, Inc.
17. SUBJECT CONFIDENTIALITY
All subject records will be identified only by initials and screening and/or final subject number.
The subject's names are not to be acquired by the Sponsor or their representatives. The
Investigator will keep a master sheet on which the identification information of each subject is
kept.

Sponsor Protocol No. P11-001 Page 24
Final HTR Study No. 10-130293-111
January 28, 2011
18. INVESTIGATOR OBLIGATIONS
18.1. Investigator Service
The Investigator or a designated Sub-Investigator will be readily available by phone at all times
during the study sessions.
18.2. Documentation
The Investigator must provide the following to Medline Industries, Inc. prior to the start of the
study:
Curriculum vitae for the Investigator.
A copy of the original approval for conducting the trial by the Institutional Review
Board as well as amendments and any advertisements. Renewals, with continuance
of the study, must be submitted at least yearly intervals.
A copy of the IRB approved informed consent form.
The Investigator signature page of this protocol signed and dated by the Investigator.
IRB membership roster.
18.3. Institutional Review Board (IRB)
This clinical study, including the Informed Consent, will be reviewed by an Institutional Review
Board in accordance with Title 21 of the Code of Federal Regulations, Parts 50 and 56.
Approval by the Board will be obtained prior to the initiation of the study. Approval of the IRB
prior to the start of the study will be the responsibility of the Investigator. A copy of the
approval letter must be supplied to Medline Industries, Inc. along with a roster of IRB
membership. If an Investigator or any sub-Investigator is on the IRB he must abstain from
voting on this project. During the course of the study, the Investigator shall make timely and
accurate reports to the IRB on the progress of the trial, at intervals not exceeding one year (or as
appropriate), and notify the IRB of SAEs or other significant safety findings as required.
18.4. Informed Consent
The study will be explained to each prospective subject and written informed consent must be
obtained before participation in any study related procedures. Copies of the informed consent
should be given to the subject and placed in the Investigator‘s study files. Each subject who
agrees to participate in the study must sign and date the informed consent.
18.5. Performance
The Investigator must demonstrate reasonable efforts to obtain qualified subjects for the study.

Sponsor Protocol No. P11-001 Page 25
Final HTR Study No. 10-130293-111
January 28, 2011
18.6. Use of Investigational Materials
The Investigator acknowledges that the test articles are investigational and as such must be used
strictly in accordance with the protocol and only under the supervision of the Investigator or sub-
Investigators. Upon receipt of the documentation package, Medline Industries, Inc. will arrange
for the shipping of study supplies. The Investigator must maintain adequate records documenting
the receipt and disposition of all study supplies. Hill Top/Medline Industries, Inc. will supply
forms to record the test articles were received, and document (as appropriate) the lot number of
the test articles, expiration/manufacture date, and a dispensing record to record each subjects use.
This is separate from the dispensing records maintained for each subject in the source
documents. All unused test articles must be returned to Medline Industries, Inc.
18.7. Source Documents
Study data will be captured on appropriately designed source documents. All source document
entries must be made in black ink for duplication purposes. The Investigator or designee must
review all entries for completeness and correctness. When changes or corrections are made on
any source document, a single line must be drawn through the error and initials of the person
making the change and the date of the correction recorded.
18.8. Final Report, Retention and Review of Records
The final report will summarize the method, data and conclusions (when applicable) relative to
the test articles and the subjects. Original source data will be retained by the testing facility for
at least two years. The source data will then be retained on microfilm or another readily
retrievable form indefinitely. A copy of the source documents may be obtained upon request of
the Study Sponsor. Data tables will be incorporated in the report.
The Investigator must maintain all documentation relating to this study. If Medline Industries,
Inc. requests to review any documentation relating to the study, the Investigator must permit
access to such records.
If the Investigator retires, relocates, or for other reasons withdraws from the responsibility of
keeping the study records, custody must be transferred to a person who will accept the
responsibility. Medline Industries, Inc. must be notified in writing of the name and address of
the new custodian.
19. PROTOCOL CHANGES AND DEVIATIONS
For medical, regulatory, or administrative purposes, the Sponsor reserves the right to change the
protocol at any time. The Sponsor also reserves the right to discontinue the study at any time.
If in the interest of safety and/or well being of a particular subject, it is necessary to depart from
the protocol that protocol deviation will be for that subject only. The Investigator must
document this divergence from the protocol for that subject on the appropriate source documents.

Sponsor Protocol No. P11-001 Page 26
Final HTR Study No. 10-130293-111
January 28, 2011
20. CONFIDENTIALITY
The Protocol and source documents provided to Hill Top Research for review by the
Investigator, the staff, and applicable Institutional Review Board(s) are confidential. It is
understood that this information provided to Hill Top Research by Medline Industries, Inc. in
connection with this study, will not be disclosed to others without written authorization from
Medline Industries, Inc.
21. REFERENCES
1. Marathon Brochure
2. Sureprep Brochure

Sponsor Protocol No. P11-001 Page 27
Final HTR Study No. 10-130293-111
January 28, 2011
MarathonTM
Brochure
Sponsor Protocol No. P11-001 Page 28
Final HTR Study No. 10-130293-111
January 28, 2011
Sponsor Protocol No. P11-001 Page 29
Final HTR Study No. 10-130293-111
January 28, 2011
Sponsor Protocol No. P11-001 Page 30
Final HTR Study No. 10-130293-111
January 28, 2011

Sponsor Protocol No. P11-001 Page 31
Final HTR Study No. 10-130293-111
January 28, 2011

Sponsor Protocol No. P11-001 Page 32
Final HTR Study No. 10-130293-111
January 28, 2011

Sponsor Protocol No. P11-001 Page 33
Final HTR Study No. 10-130293-111
January 28, 2011

Sponsor Protocol No. P11-001 Page 34
Final HTR Study No. 10-130293-111
January 28, 2011
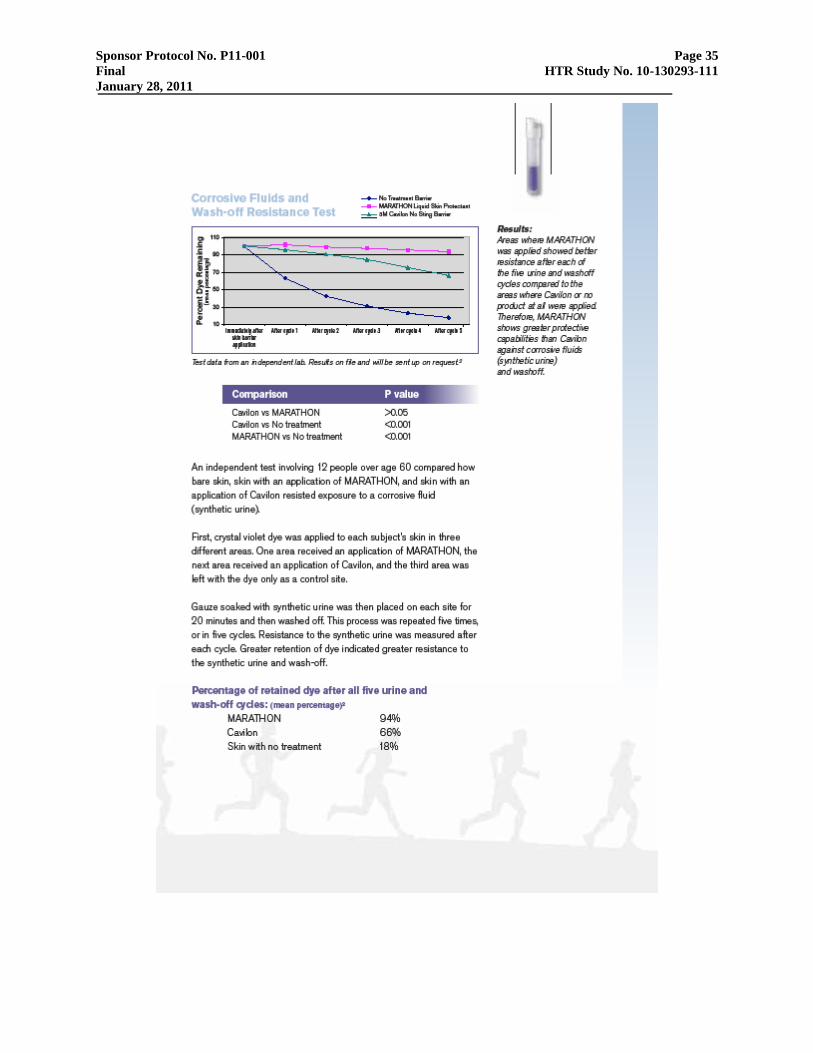
Sponsor Protocol No. P11-001 Page 35
Final HTR Study No. 10-130293-111
January 28, 2011

Sponsor Protocol No. P11-001 Page 36
Final HTR Study No. 10-130293-111
January 28, 2011

Sponsor Protocol No. P11-001 Page 37
Final HTR Study No. 10-130293-111
January 28, 2011
Sureprep Brochure

HTR Study No. 10-130293-111 Final Report
Sponsor No. P11-001
APPENDIX III. STATISTICAL TABLES
Total number of pages including this cover page = 30
Table 1 Tabulation of Subject Demographics
Table 2 Frequency Distribution of Gender and Fitzpatrick Skin Type, by Population
Table 3 Summary Statistics – Age, by Population
Table 4 Overall Pain Assessment, with Maximum Stinging Response
Table 5 Summary Statistics – Overall Pain Assessment
Table 6 Summary Statistics – Maximum Stinging Response
Table 7 Analysis of Variance of Maximum Stinging Response
Table 8 Subjective Pain Assessment
Table 9 Summary Statistics – Subjective Pain Assessment
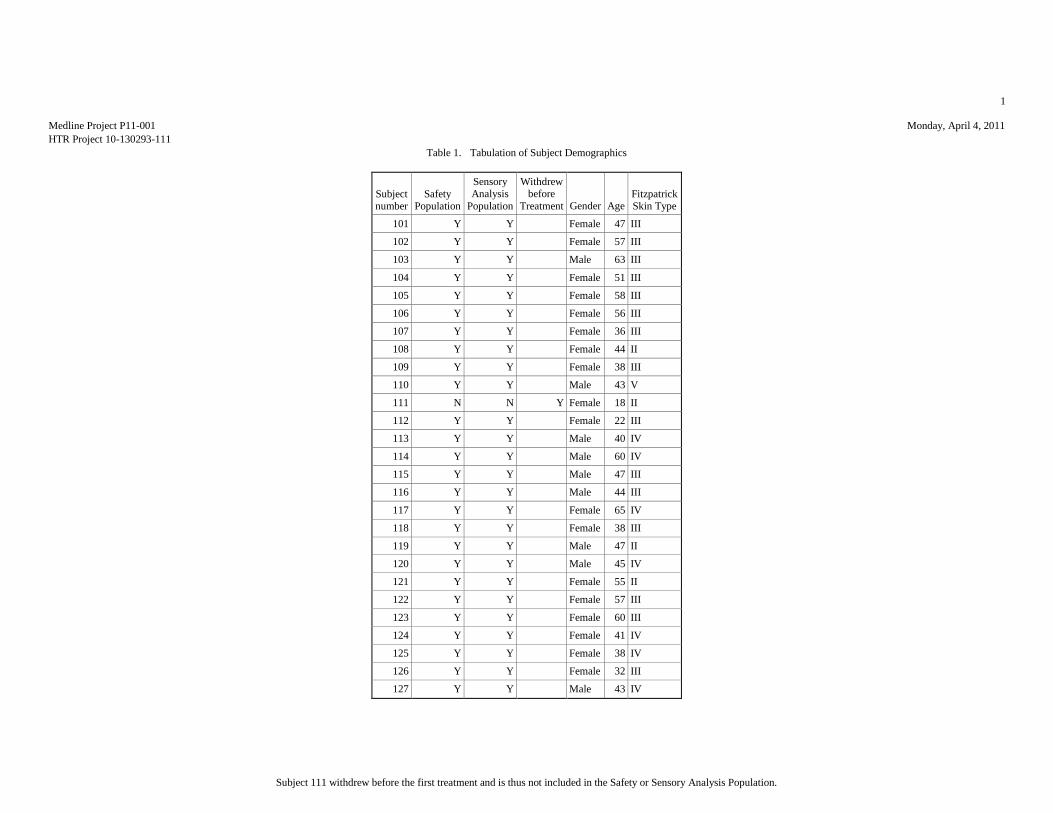
Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 1. Tabulation of Subject Demographics
Subject 111 withdrew before the first treatment and is thus not included in the Safety or Sensory Analysis Population.
1
Subject number
Safety Population
Sensory Analysis
Population
Withdrew before
Treatment Gender Age Fitzpatrick Skin Type
101 Y Y Female 47 III
102 Y Y Female 57 III
103 Y Y Male 63 III
104 Y Y Female 51 III
105 Y Y Female 58 III
106 Y Y Female 56 III
107 Y Y Female 36 III
108 Y Y Female 44 II
109 Y Y Female 38 III
110 Y Y Male 43 V
111 N N Y Female 18 II
112 Y Y Female 22 III
113 Y Y Male 40 IV
114 Y Y Male 60 IV
115 Y Y Male 47 III
116 Y Y Male 44 III
117 Y Y Female 65 IV
118 Y Y Female 38 III
119 Y Y Male 47 II
120 Y Y Male 45 IV
121 Y Y Female 55 II
122 Y Y Female 57 III
123 Y Y Female 60 III
124 Y Y Female 41 IV
125 Y Y Female 38 IV
126 Y Y Female 32 III
127 Y Y Male 43 IV
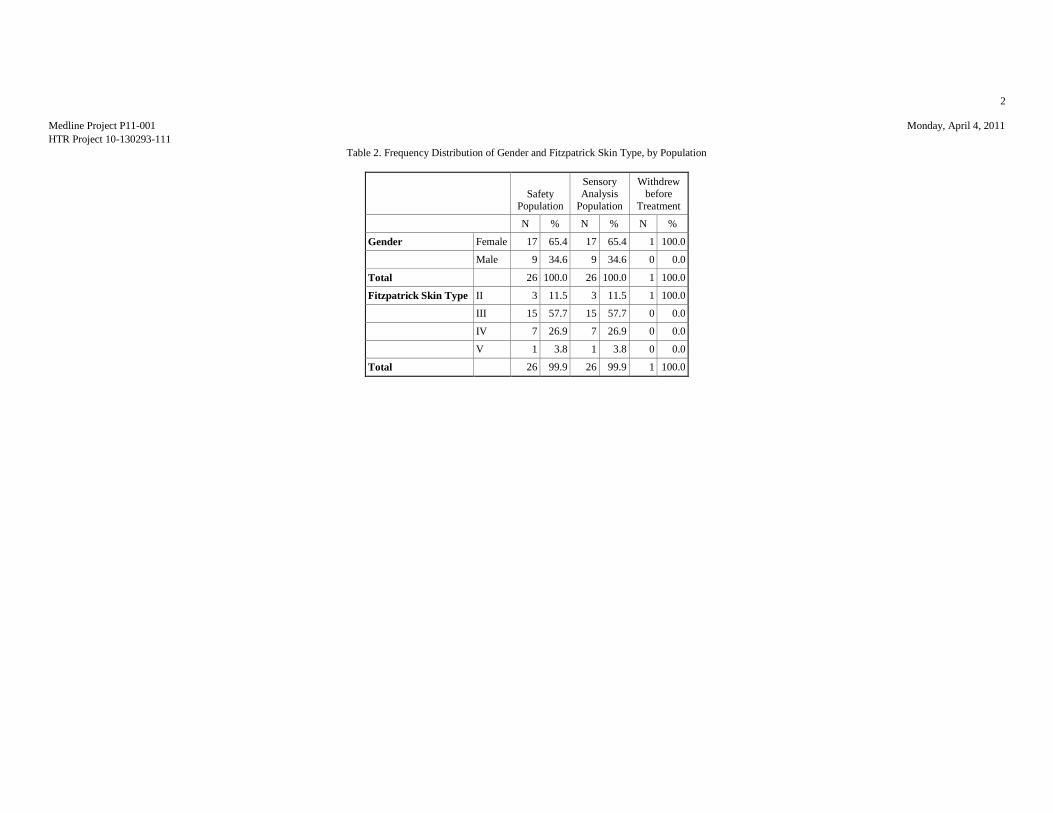
Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 2. Frequency Distribution of Gender and Fitzpatrick Skin Type, by Population
2
Safety
Population
Sensory Analysis
Population
Withdrew before
Treatment
N % N % N %
Gender Female 17 65.4 17 65.4 1 100.0
Male 9 34.6 9 34.6 0 0.0
Total 26 100.0 26 100.0 1 100.0
Fitzpatrick Skin Type II 3 11.5 3 11.5 1 100.0
III 15 57.7 15 57.7 0 0.0
IV 7 26.9 7 26.9 0 0.0
V 1 3.8 1 3.8 0 0.0
Total 26 99.9 26 99.9 1 100.0
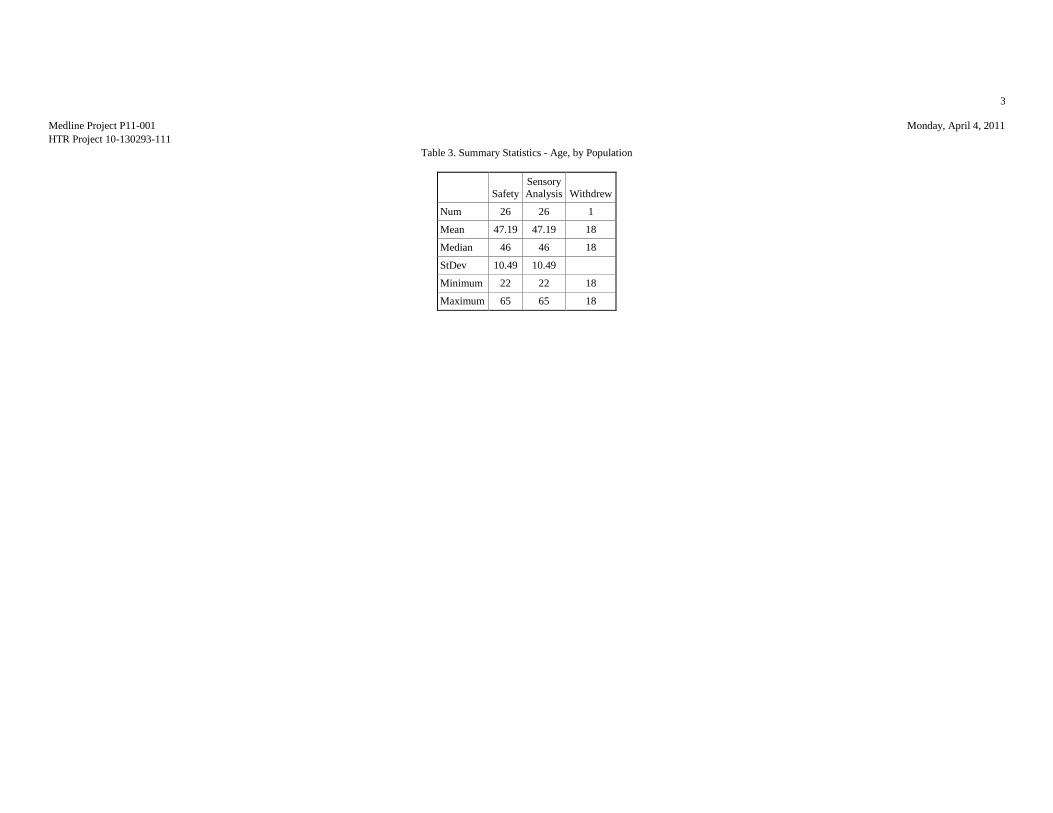
Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 3. Summary Statistics - Age, by Population
3
Safety
Sensory Analysis Withdrew
Num 26 26 1
Mean 47.19 47.19 18
Median 46 46 18
StDev 10.49 10.49
Minimum 22 22 18
Maximum 65 65 18
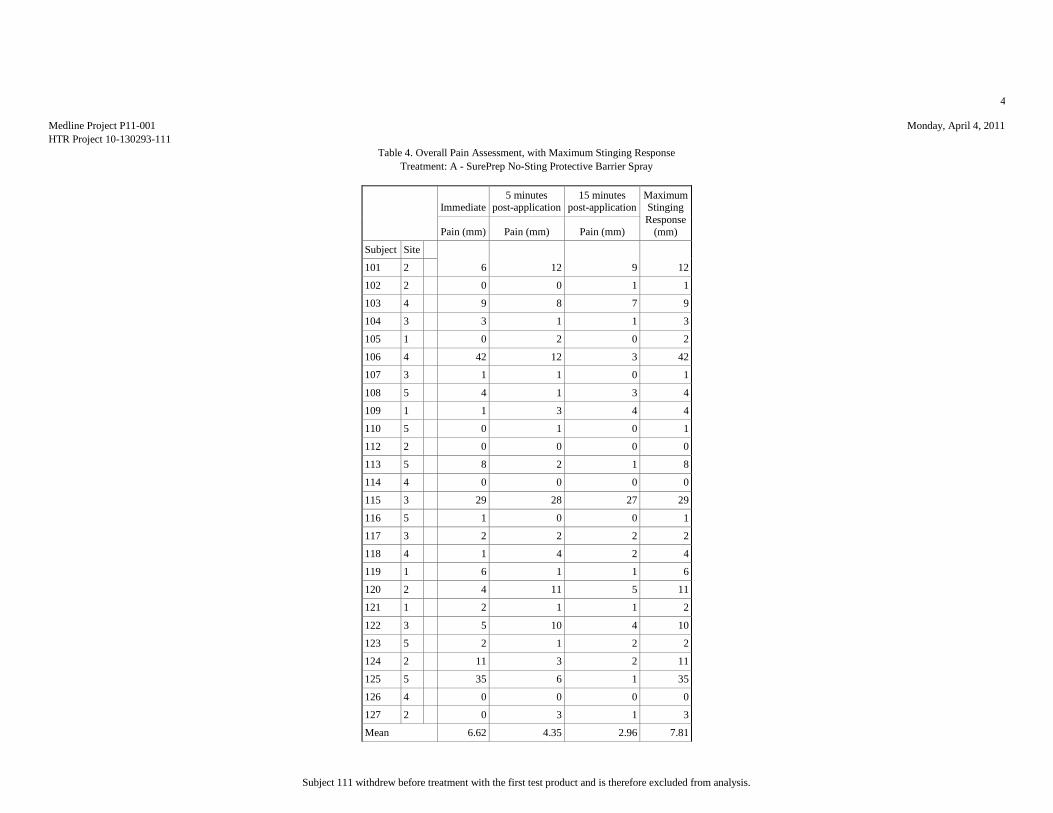
Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 4. Overall Pain Assessment, with Maximum Stinging Response
Treatment: A - SurePrep No-Sting Protective Barrier Spray
Subject 111 withdrew before treatment with the first test product and is therefore excluded from analysis.
4
Immediate
5 minutes post-application
15 minutes post-application
Maximum Stinging
Response
(mm) Pain (mm) Pain (mm) Pain (mm)
Subject Site
6 12 9 12 101 2
102 2 0 0 1 1
103 4 9 8 7 9
104 3 3 1 1 3
105 1 0 2 0 2
106 4 42 12 3 42
107 3 1 1 0 1
108 5 4 1 3 4
109 1 1 3 4 4
110 5 0 1 0 1
112 2 0 0 0 0
113 5 8 2 1 8
114 4 0 0 0 0
115 3 29 28 27 29
116 5 1 0 0 1
117 3 2 2 2 2
118 4 1 4 2 4
119 1 6 1 1 6
120 2 4 11 5 11
121 1 2 1 1 2
122 3 5 10 4 10
123 5 2 1 2 2
124 2 11 3 2 11
125 5 35 6 1 35
126 4 0 0 0 0
127 2 0 3 1 3
Mean 6.62 4.35 2.96 7.81
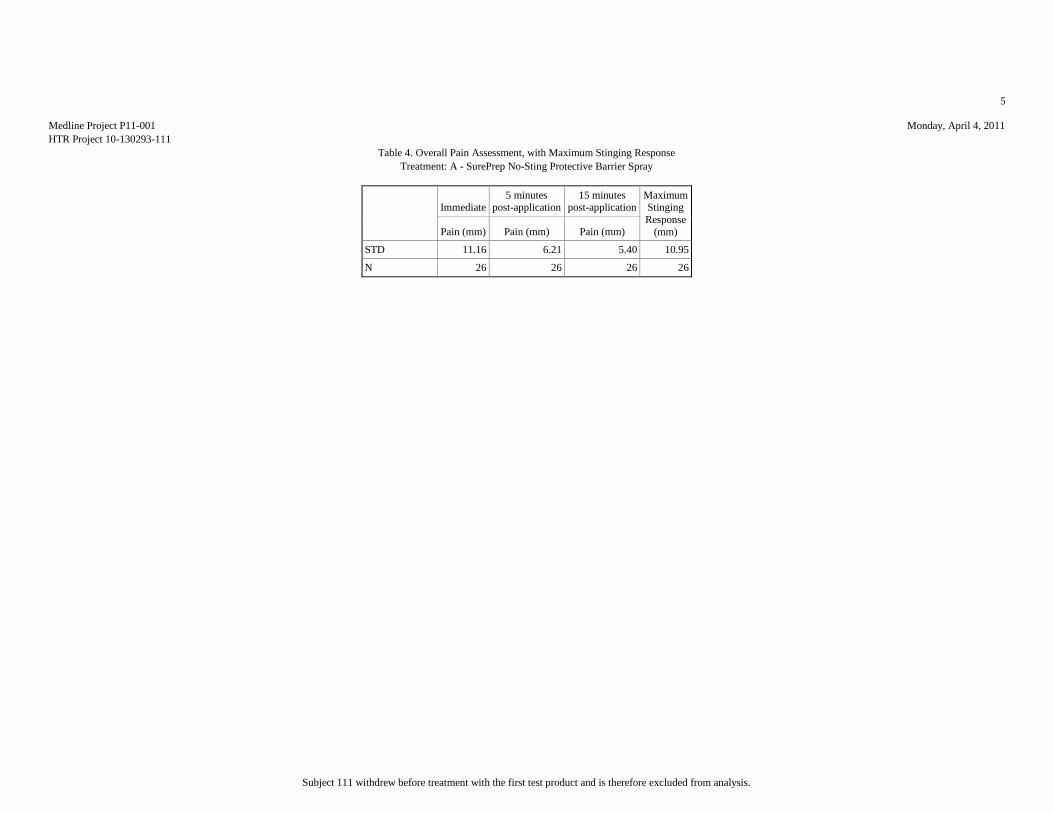
Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 4. Overall Pain Assessment, with Maximum Stinging Response
Treatment: A - SurePrep No-Sting Protective Barrier Spray
Subject 111 withdrew before treatment with the first test product and is therefore excluded from analysis.
5
Immediate
5 minutes post-application
15 minutes post-application
Maximum Stinging
Response
(mm) Pain (mm) Pain (mm) Pain (mm)
STD 11.16 6.21 5.40 10.95
N 26 26 26 26
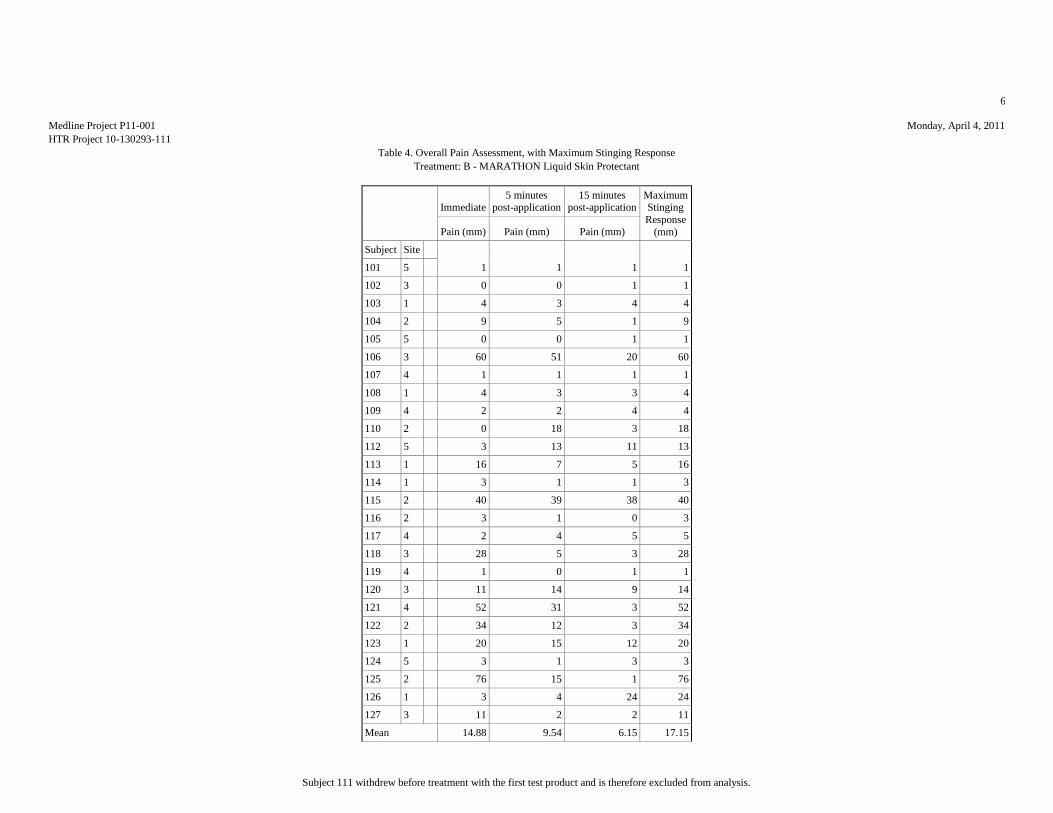
Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 4. Overall Pain Assessment, with Maximum Stinging Response
Treatment: B - MARATHON Liquid Skin Protectant
Subject 111 withdrew before treatment with the first test product and is therefore excluded from analysis.
6
Immediate
5 minutes post-application
15 minutes post-application
Maximum Stinging
Response
(mm) Pain (mm) Pain (mm) Pain (mm)
Subject Site
1 1 1 1 101 5
102 3 0 0 1 1
103 1 4 3 4 4
104 2 9 5 1 9
105 5 0 0 1 1
106 3 60 51 20 60
107 4 1 1 1 1
108 1 4 3 3 4
109 4 2 2 4 4
110 2 0 18 3 18
112 5 3 13 11 13
113 1 16 7 5 16
114 1 3 1 1 3
115 2 40 39 38 40
116 2 3 1 0 3
117 4 2 4 5 5
118 3 28 5 3 28
119 4 1 0 1 1
120 3 11 14 9 14
121 4 52 31 3 52
122 2 34 12 3 34
123 1 20 15 12 20
124 5 3 1 3 3
125 2 76 15 1 76
126 1 3 4 24 24
127 3 11 2 2 11
Mean 14.88 9.54 6.15 17.15
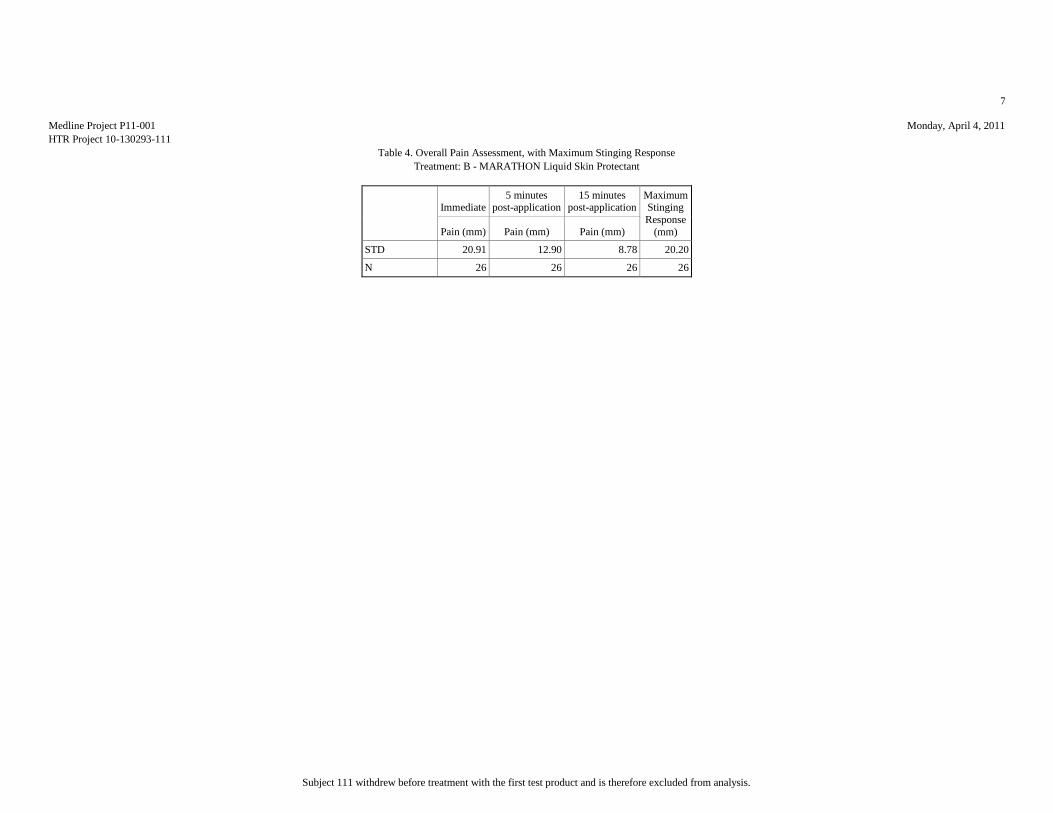
Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 4. Overall Pain Assessment, with Maximum Stinging Response
Treatment: B - MARATHON Liquid Skin Protectant
Subject 111 withdrew before treatment with the first test product and is therefore excluded from analysis.
7
Immediate
5 minutes post-application
15 minutes post-application
Maximum Stinging
Response
(mm) Pain (mm) Pain (mm) Pain (mm)
STD 20.91 12.90 8.78 20.20
N 26 26 26 26
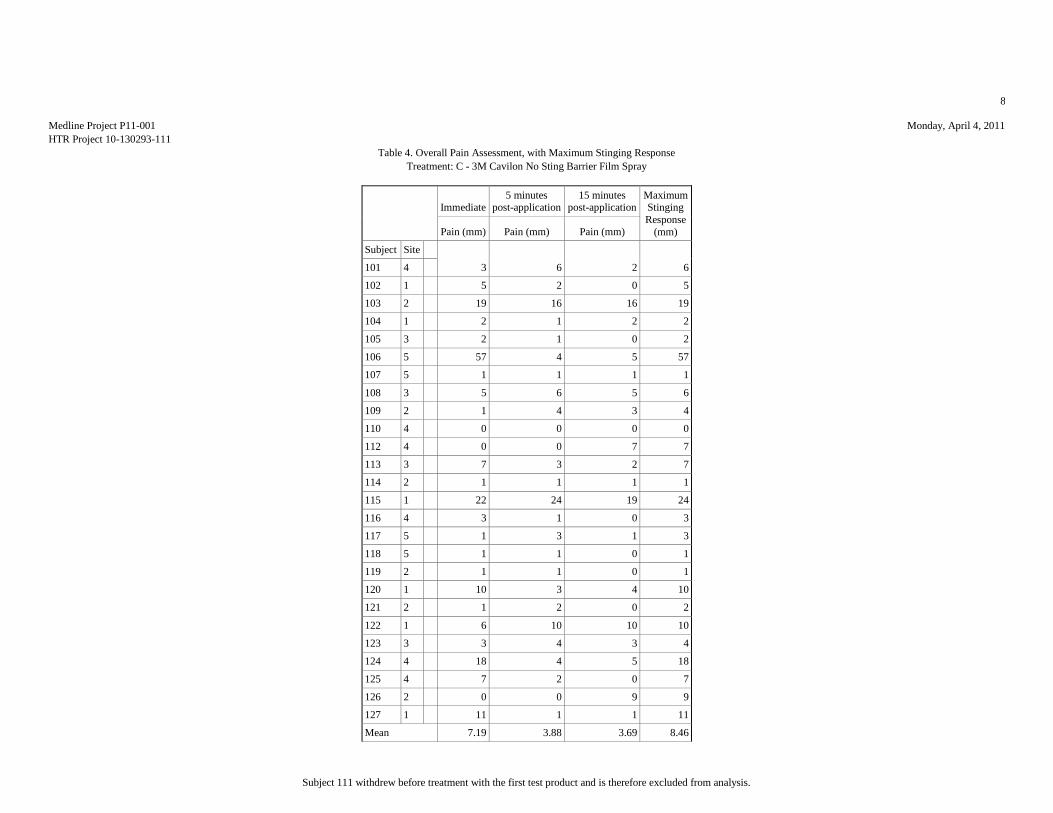
Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 4. Overall Pain Assessment, with Maximum Stinging Response
Treatment: C - 3M Cavilon No Sting Barrier Film Spray
Subject 111 withdrew before treatment with the first test product and is therefore excluded from analysis.
8
Immediate
5 minutes post-application
15 minutes post-application
Maximum Stinging
Response
(mm) Pain (mm) Pain (mm) Pain (mm)
Subject Site
3 6 2 6 101 4
102 1 5 2 0 5
103 2 19 16 16 19
104 1 2 1 2 2
105 3 2 1 0 2
106 5 57 4 5 57
107 5 1 1 1 1
108 3 5 6 5 6
109 2 1 4 3 4
110 4 0 0 0 0
112 4 0 0 7 7
113 3 7 3 2 7
114 2 1 1 1 1
115 1 22 24 19 24
116 4 3 1 0 3
117 5 1 3 1 3
118 5 1 1 0 1
119 2 1 1 0 1
120 1 10 3 4 10
121 2 1 2 0 2
122 1 6 10 10 10
123 3 3 4 3 4
124 4 18 4 5 18
125 4 7 2 0 7
126 2 0 0 9 9
127 1 11 1 1 11
Mean 7.19 3.88 3.69 8.46
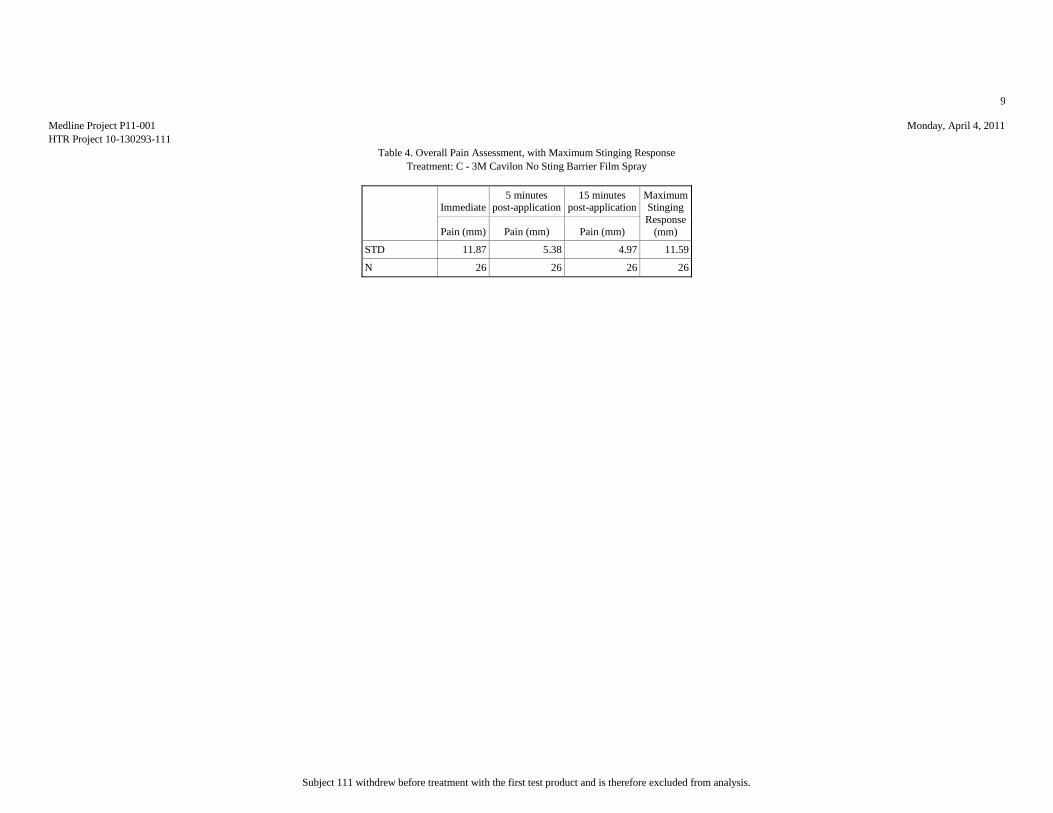
Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 4. Overall Pain Assessment, with Maximum Stinging Response
Treatment: C - 3M Cavilon No Sting Barrier Film Spray
Subject 111 withdrew before treatment with the first test product and is therefore excluded from analysis.
9
Immediate
5 minutes post-application
15 minutes post-application
Maximum Stinging
Response
(mm) Pain (mm) Pain (mm) Pain (mm)
STD 11.87 5.38 4.97 11.59
N 26 26 26 26
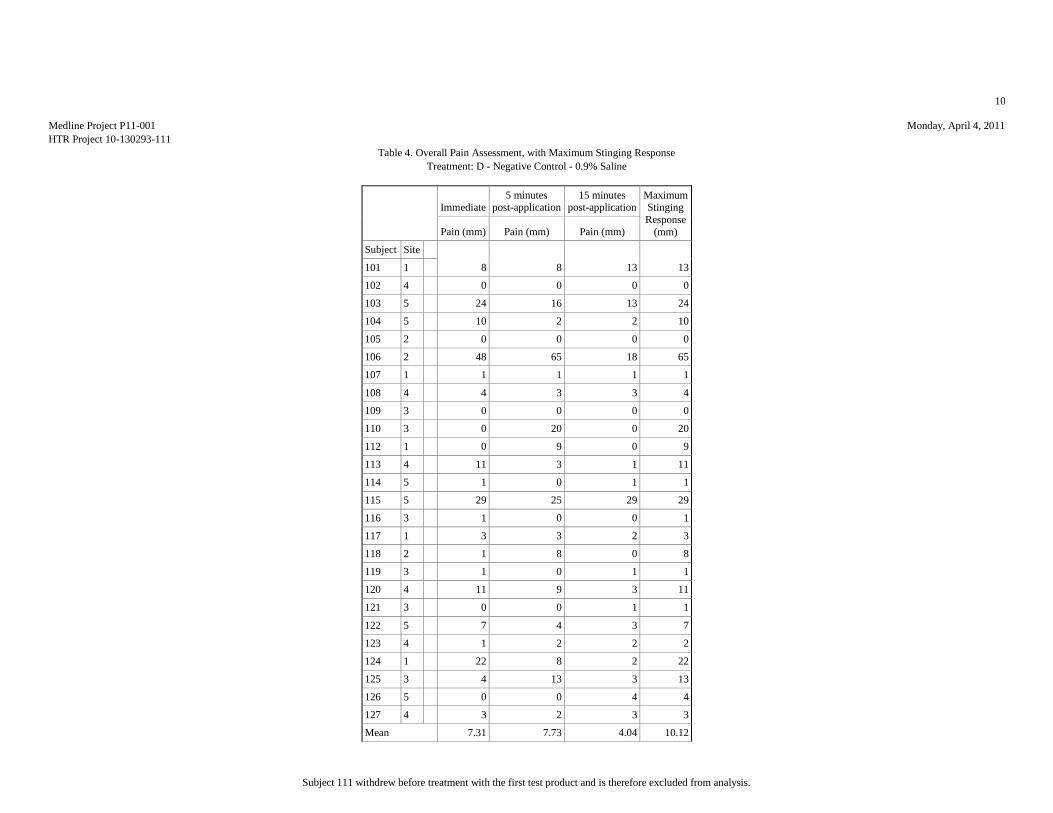
Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 4. Overall Pain Assessment, with Maximum Stinging Response
Treatment: D - Negative Control - 0.9% Saline
Subject 111 withdrew before treatment with the first test product and is therefore excluded from analysis.
10
Immediate
5 minutes post-application
15 minutes post-application
Maximum Stinging
Response
(mm) Pain (mm) Pain (mm) Pain (mm)
Subject Site
8 8 13 13 101 1
102 4 0 0 0 0
103 5 24 16 13 24
104 5 10 2 2 10
105 2 0 0 0 0
106 2 48 65 18 65
107 1 1 1 1 1
108 4 4 3 3 4
109 3 0 0 0 0
110 3 0 20 0 20
112 1 0 9 0 9
113 4 11 3 1 11
114 5 1 0 1 1
115 5 29 25 29 29
116 3 1 0 0 1
117 1 3 3 2 3
118 2 1 8 0 8
119 3 1 0 1 1
120 4 11 9 3 11
121 3 0 0 1 1
122 5 7 4 3 7
123 4 1 2 2 2
124 1 22 8 2 22
125 3 4 13 3 13
126 5 0 0 4 4
127 4 3 2 3 3
Mean 7.31 7.73 4.04 10.12
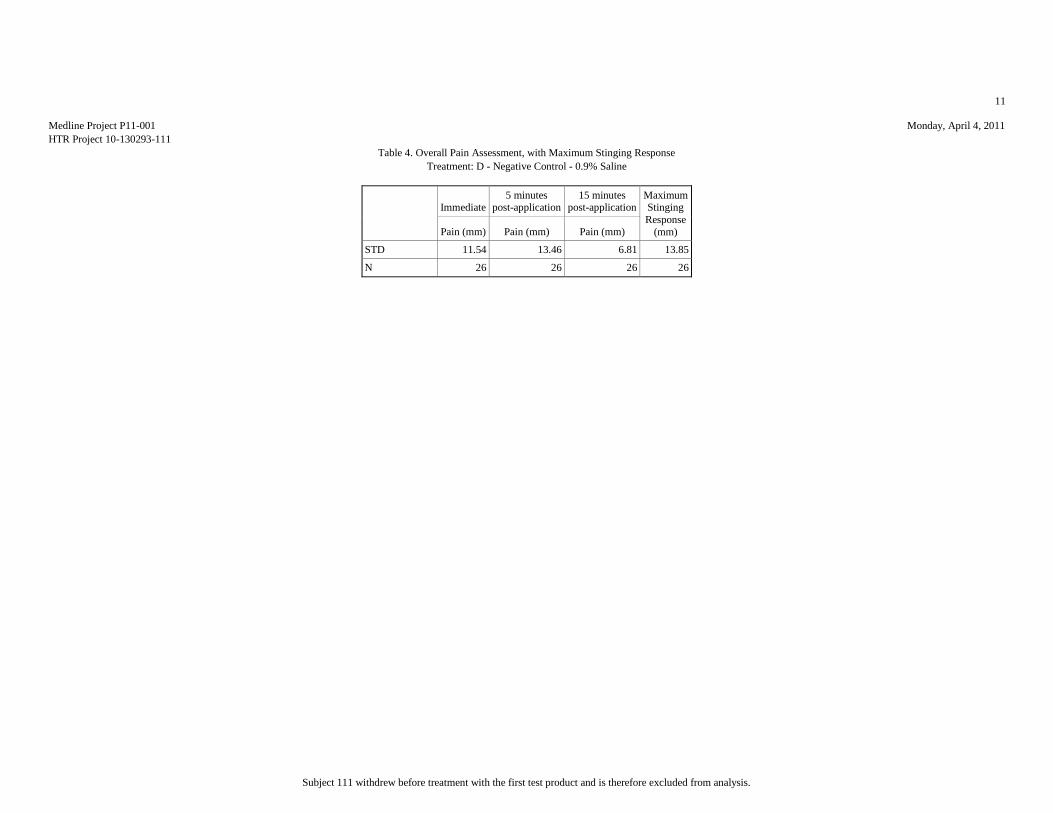
Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 4. Overall Pain Assessment, with Maximum Stinging Response
Treatment: D - Negative Control - 0.9% Saline
Subject 111 withdrew before treatment with the first test product and is therefore excluded from analysis.
11
Immediate
5 minutes post-application
15 minutes post-application
Maximum Stinging
Response
(mm) Pain (mm) Pain (mm) Pain (mm)
STD 11.54 13.46 6.81 13.85
N 26 26 26 26
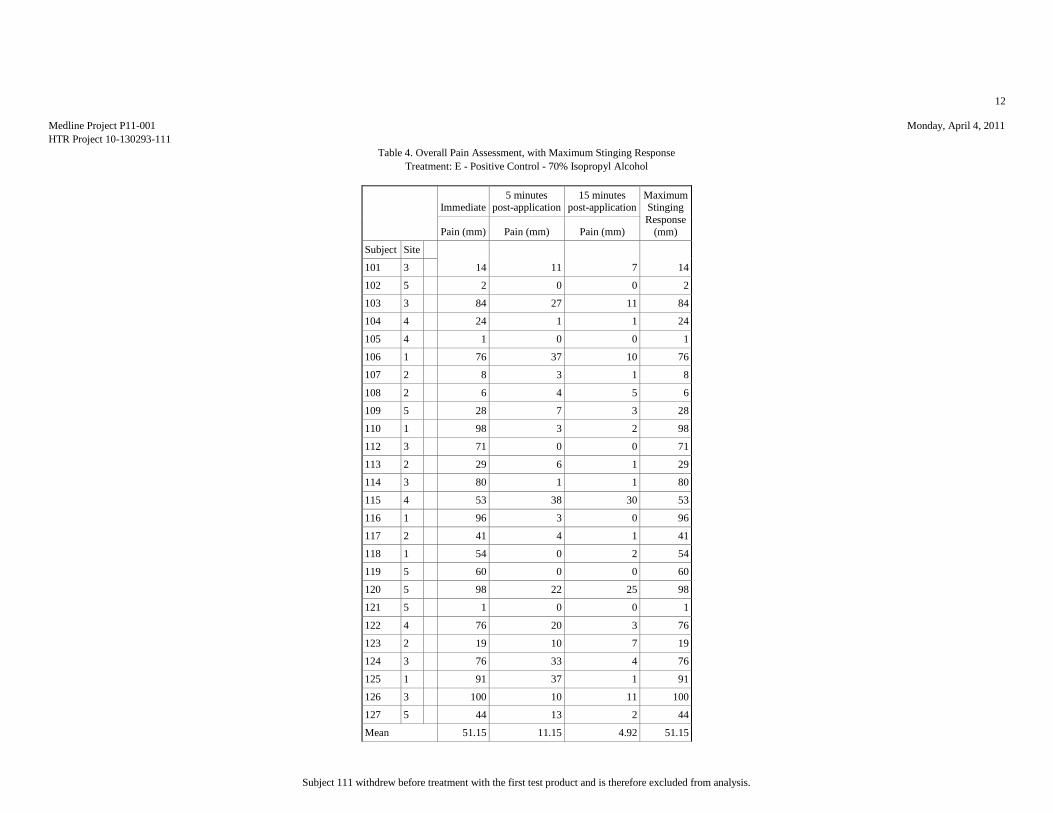
Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 4. Overall Pain Assessment, with Maximum Stinging Response
Treatment: E - Positive Control - 70% Isopropyl Alcohol
Subject 111 withdrew before treatment with the first test product and is therefore excluded from analysis.
12
Immediate
5 minutes post-application
15 minutes post-application
Maximum Stinging
Response
(mm) Pain (mm) Pain (mm) Pain (mm)
Subject Site
14 11 7 14 101 3
102 5 2 0 0 2
103 3 84 27 11 84
104 4 24 1 1 24
105 4 1 0 0 1
106 1 76 37 10 76
107 2 8 3 1 8
108 2 6 4 5 6
109 5 28 7 3 28
110 1 98 3 2 98
112 3 71 0 0 71
113 2 29 6 1 29
114 3 80 1 1 80
115 4 53 38 30 53
116 1 96 3 0 96
117 2 41 4 1 41
118 1 54 0 2 54
119 5 60 0 0 60
120 5 98 22 25 98
121 5 1 0 0 1
122 4 76 20 3 76
123 2 19 10 7 19
124 3 76 33 4 76
125 1 91 37 1 91
126 3 100 10 11 100
127 5 44 13 2 44
Mean 51.15 11.15 4.92 51.15
Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 4. Overall Pain Assessment, with Maximum Stinging Response
Treatment: E - Positive Control - 70% Isopropyl Alcohol
Subject 111 withdrew before treatment with the first test product and is therefore excluded from analysis.
13
Immediate
5 minutes post-application
15 minutes post-application
Maximum Stinging
Response
(mm) Pain (mm) Pain (mm) Pain (mm)
STD 34.81 13.09 7.51 34.81
N 26 26 26 26
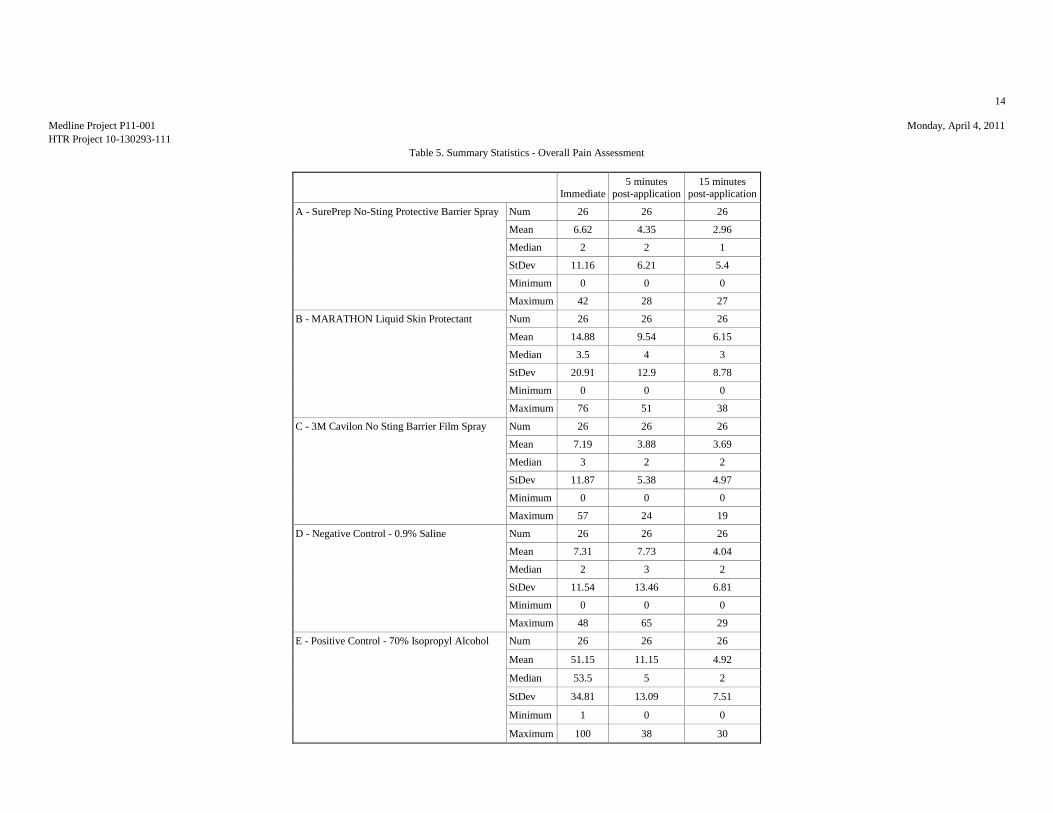
Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 5. Summary Statistics - Overall Pain Assessment
14
Immediate
5 minutes post-application
15 minutes post-application
A - SurePrep No-Sting Protective Barrier Spray Num 26 26 26
Mean 6.62 4.35 2.96
Median 2 2 1
StDev 11.16 6.21 5.4
Minimum 0 0 0
Maximum 42 28 27
B - MARATHON Liquid Skin Protectant Num 26 26 26
Mean 14.88 9.54 6.15
Median 3.5 4 3
StDev 20.91 12.9 8.78
Minimum 0 0 0
Maximum 76 51 38
C - 3M Cavilon No Sting Barrier Film Spray Num 26 26 26
Mean 7.19 3.88 3.69
Median 3 2 2
StDev 11.87 5.38 4.97
Minimum 0 0 0
Maximum 57 24 19
D - Negative Control - 0.9% Saline Num 26 26 26
Mean 7.31 7.73 4.04
Median 2 3 2
StDev 11.54 13.46 6.81
Minimum 0 0 0
Maximum 48 65 29
E - Positive Control - 70% Isopropyl Alcohol Num 26 26 26
Mean 51.15 11.15 4.92
Median 53.5 5 2
StDev 34.81 13.09 7.51
Minimum 1 0 0
Maximum 100 38 30
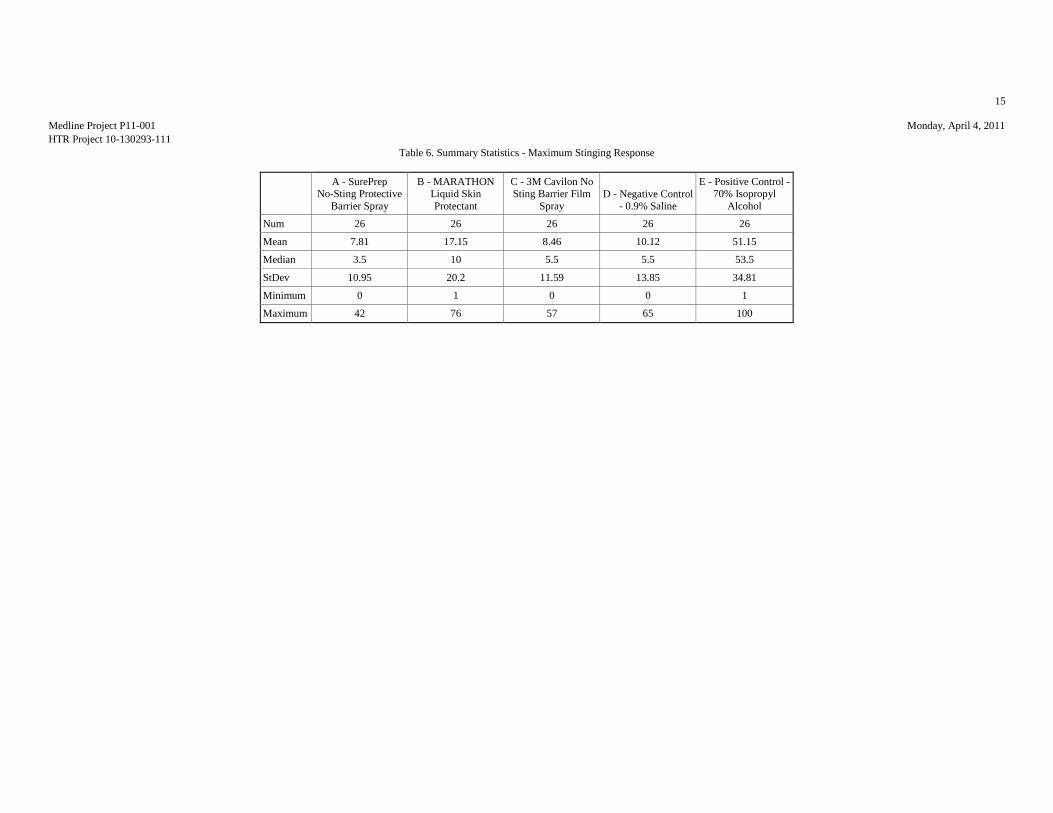
Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 6. Summary Statistics - Maximum Stinging Response
15
A - SurePrep
No-Sting Protective
Barrier Spray
B - MARATHON Liquid Skin
Protectant
C - 3M Cavilon No Sting Barrier Film
Spray D - Negative Control
- 0.9% Saline
E - Positive Control - 70% Isopropyl
Alcohol
Num 26 26 26 26 26
Mean 7.81 17.15 8.46 10.12 51.15
Median 3.5 10 5.5 5.5 53.5
StDev 10.95 20.2 11.59 13.85 34.81
Minimum 0 1 0 0 1
Maximum 42 76 57 65 100

Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 7. Analysis of Variance of Maximum Stinging Response
The GLM Procedure
16
Class Level Information
Class Levels Values
sbj1n 27 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127
trt 5 A B C D E
Number of Observations Read 135
Number of Observations Used 130
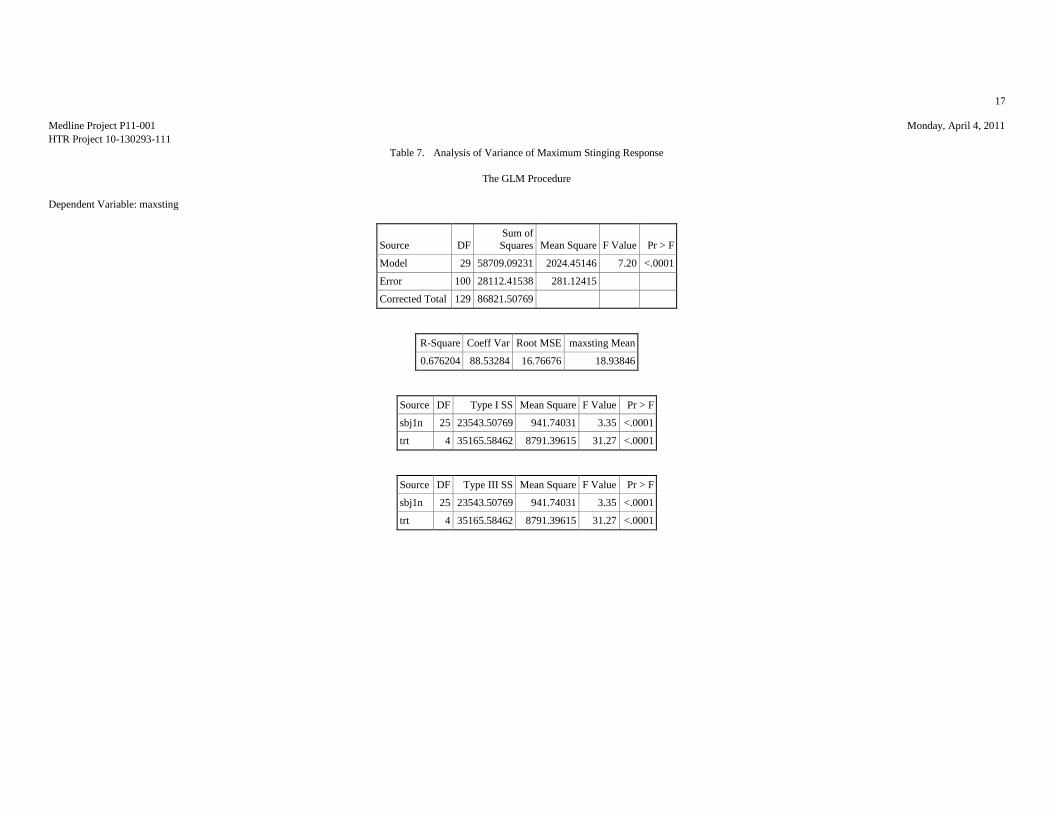
Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 7. Analysis of Variance of Maximum Stinging Response
The GLM Procedure
Dependent Variable: maxsting
17
Source DF Sum of
Squares Mean Square F Value Pr > F
Model 29 58709.09231 2024.45146 7.20 <.0001
Error 100 28112.41538 281.12415
Corrected Total 129 86821.50769
R-Square Coeff Var Root MSE maxsting Mean
0.676204 88.53284 16.76676 18.93846
Source DF Type I SS Mean Square F Value Pr > F
sbj1n 25 23543.50769 941.74031 3.35 <.0001
trt 4 35165.58462 8791.39615 31.27 <.0001
Source DF Type III SS Mean Square F Value Pr > F
sbj1n 25 23543.50769 941.74031 3.35 <.0001
trt 4 35165.58462 8791.39615 31.27 <.0001

Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 7. Analysis of Variance of Maximum Stinging Response
The GLM Procedure
t Tests (LSD) for maxsting
18
Note: This test controls the Type I comparisonwise error rate, not the experimentwise error rate.
Alpha 0.05
Error Degrees of Freedom 100
Error Mean Square 281.1242
Critical Value of t 1.98397
Least Significant Difference 9.226
Means with the same letter are not significantly
different.
t Grouping Mean N trt
A 51.154 26 E
B 17.154 26 B
B
C B 10.115 26 D
C B
C B 8.462 26 C
C
C 7.808 26 A
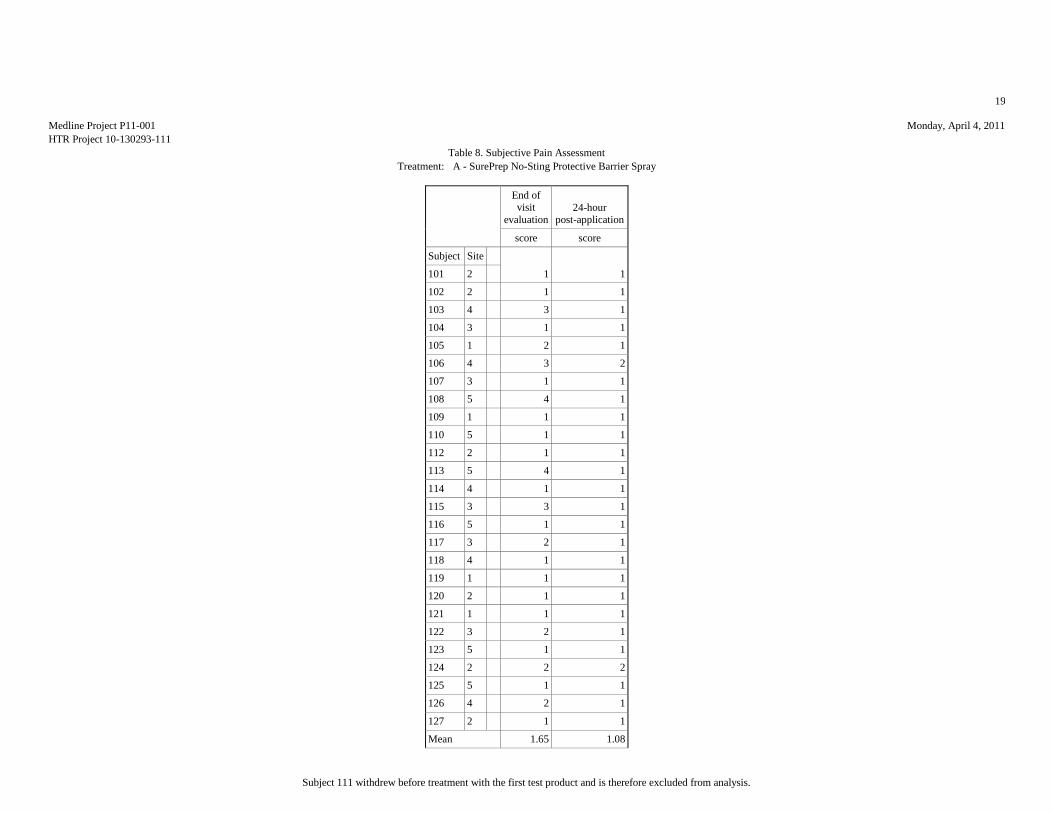
Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 8. Subjective Pain Assessment
Treatment: A - SurePrep No-Sting Protective Barrier Spray
Subject 111 withdrew before treatment with the first test product and is therefore excluded from analysis.
19
End of visit
evaluation 24-hour
post-application
score score
Subject Site
1 1 101 2
102 2 1 1
103 4 3 1
104 3 1 1
105 1 2 1
106 4 3 2
107 3 1 1
108 5 4 1
109 1 1 1
110 5 1 1
112 2 1 1
113 5 4 1
114 4 1 1
115 3 3 1
116 5 1 1
117 3 2 1
118 4 1 1
119 1 1 1
120 2 1 1
121 1 1 1
122 3 2 1
123 5 1 1
124 2 2 2
125 5 1 1
126 4 2 1
127 2 1 1
Mean 1.65 1.08

Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 8. Subjective Pain Assessment
Treatment: A - SurePrep No-Sting Protective Barrier Spray
Subject 111 withdrew before treatment with the first test product and is therefore excluded from analysis.
20
End of visit
evaluation 24-hour
post-application
score score
STD 0.98 0.27
N 26 26
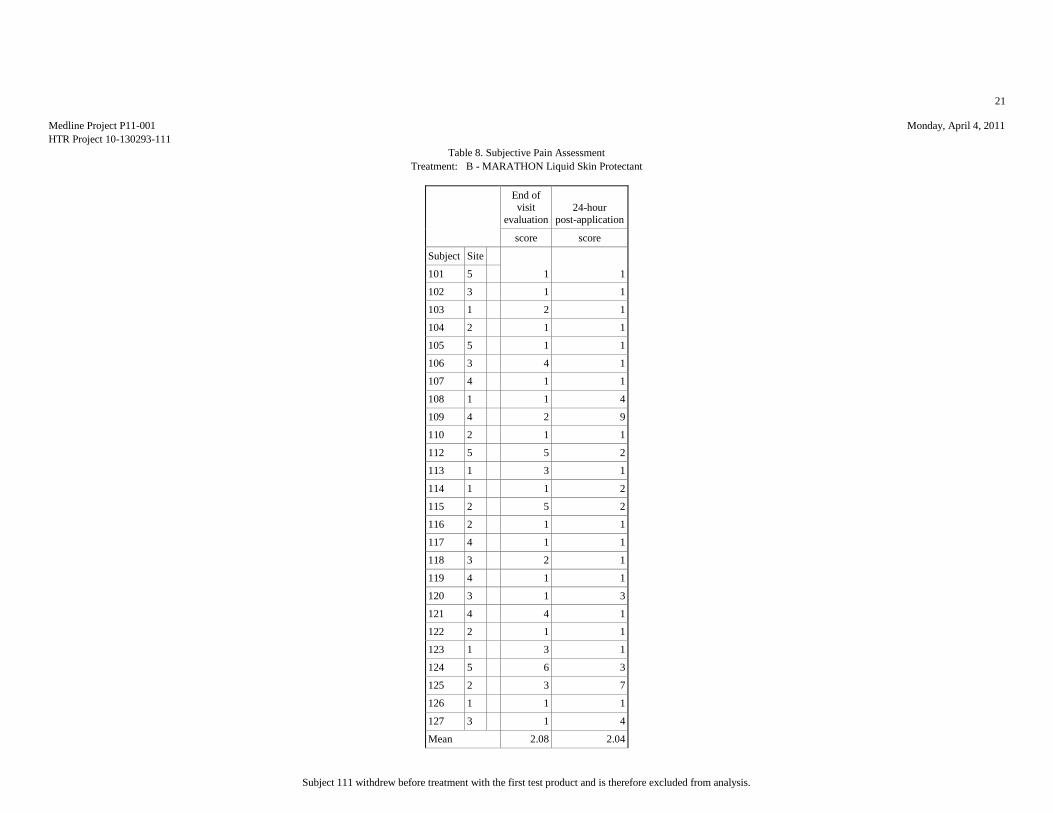
Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 8. Subjective Pain Assessment
Treatment: B - MARATHON Liquid Skin Protectant
Subject 111 withdrew before treatment with the first test product and is therefore excluded from analysis.
21
End of visit
evaluation 24-hour
post-application
score score
Subject Site
1 1 101 5
102 3 1 1
103 1 2 1
104 2 1 1
105 5 1 1
106 3 4 1
107 4 1 1
108 1 1 4
109 4 2 9
110 2 1 1
112 5 5 2
113 1 3 1
114 1 1 2
115 2 5 2
116 2 1 1
117 4 1 1
118 3 2 1
119 4 1 1
120 3 1 3
121 4 4 1
122 2 1 1
123 1 3 1
124 5 6 3
125 2 3 7
126 1 1 1
127 3 1 4
Mean 2.08 2.04

Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 8. Subjective Pain Assessment
Treatment: B - MARATHON Liquid Skin Protectant
Subject 111 withdrew before treatment with the first test product and is therefore excluded from analysis.
22
End of visit
evaluation 24-hour
post-application
score score
STD 1.55 2.01
N 26 26
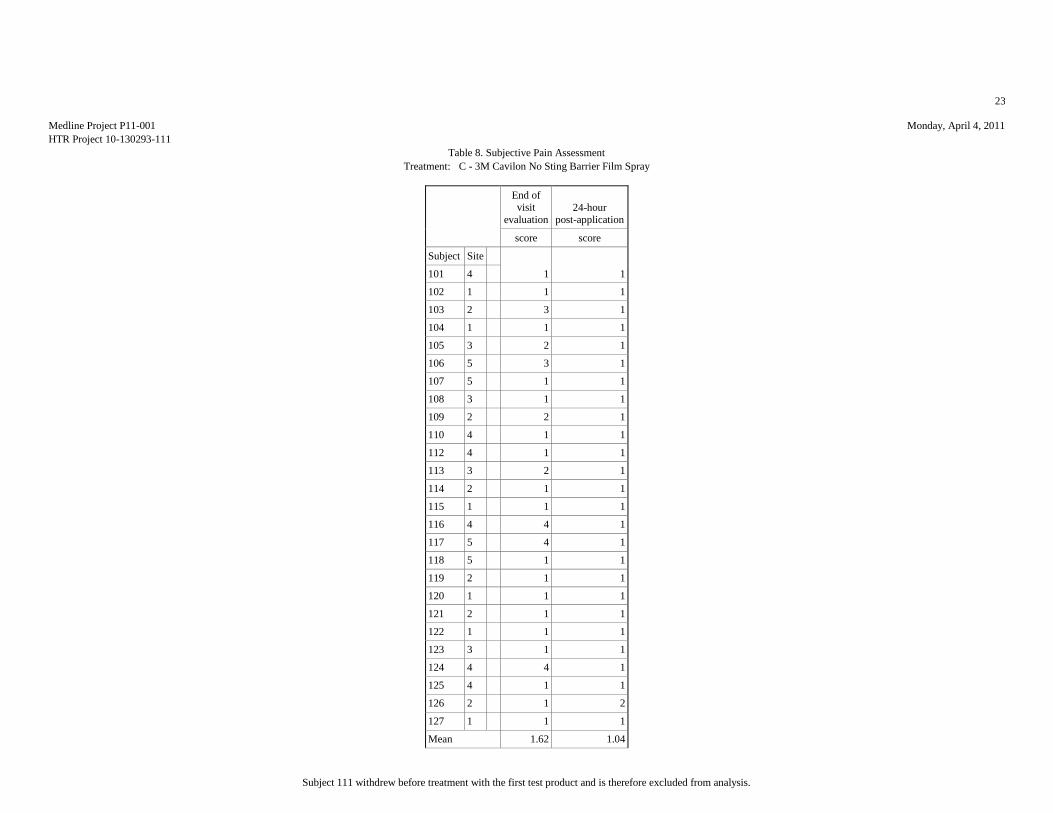
Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 8. Subjective Pain Assessment
Treatment: C - 3M Cavilon No Sting Barrier Film Spray
Subject 111 withdrew before treatment with the first test product and is therefore excluded from analysis.
23
End of visit
evaluation 24-hour
post-application
score score
Subject Site
1 1 101 4
102 1 1 1
103 2 3 1
104 1 1 1
105 3 2 1
106 5 3 1
107 5 1 1
108 3 1 1
109 2 2 1
110 4 1 1
112 4 1 1
113 3 2 1
114 2 1 1
115 1 1 1
116 4 4 1
117 5 4 1
118 5 1 1
119 2 1 1
120 1 1 1
121 2 1 1
122 1 1 1
123 3 1 1
124 4 4 1
125 4 1 1
126 2 1 2
127 1 1 1
Mean 1.62 1.04

Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 8. Subjective Pain Assessment
Treatment: C - 3M Cavilon No Sting Barrier Film Spray
Subject 111 withdrew before treatment with the first test product and is therefore excluded from analysis.
24
End of visit
evaluation 24-hour
post-application
score score
STD 1.06 0.20
N 26 26
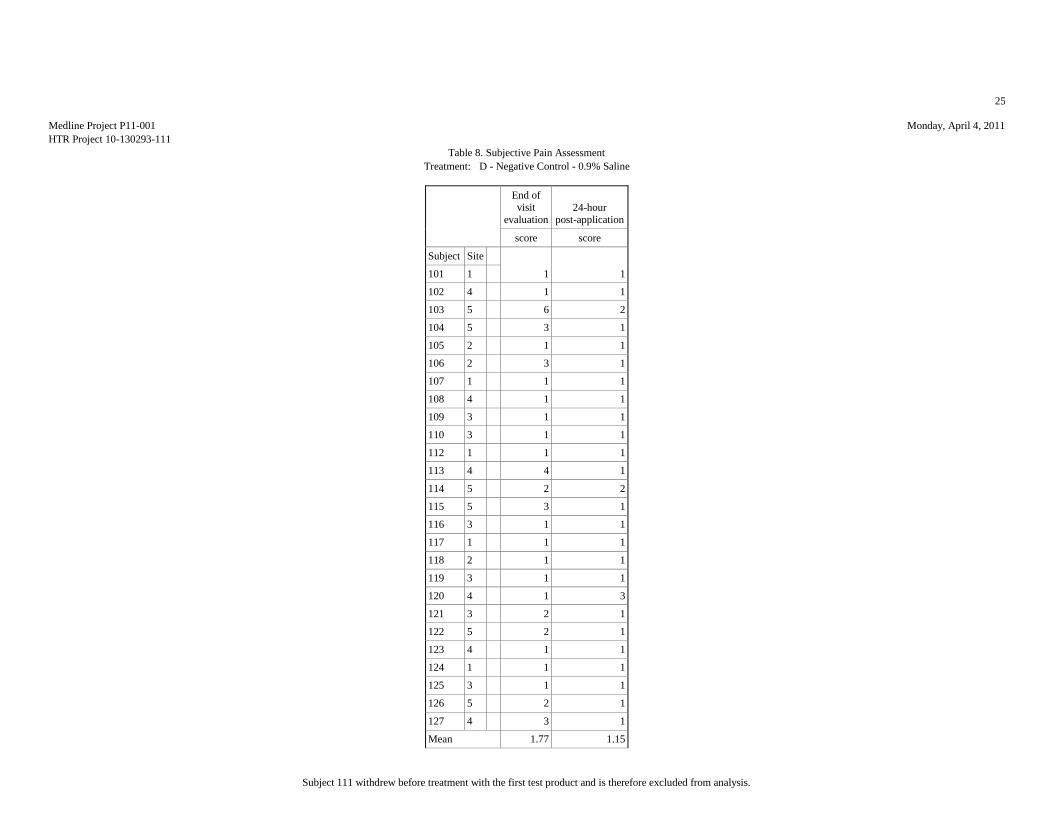
Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 8. Subjective Pain Assessment
Treatment: D - Negative Control - 0.9% Saline
Subject 111 withdrew before treatment with the first test product and is therefore excluded from analysis.
25
End of visit
evaluation 24-hour
post-application
score score
Subject Site
1 1 101 1
102 4 1 1
103 5 6 2
104 5 3 1
105 2 1 1
106 2 3 1
107 1 1 1
108 4 1 1
109 3 1 1
110 3 1 1
112 1 1 1
113 4 4 1
114 5 2 2
115 5 3 1
116 3 1 1
117 1 1 1
118 2 1 1
119 3 1 1
120 4 1 3
121 3 2 1
122 5 2 1
123 4 1 1
124 1 1 1
125 3 1 1
126 5 2 1
127 4 3 1
Mean 1.77 1.15

Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 8. Subjective Pain Assessment
Treatment: D - Negative Control - 0.9% Saline
Subject 111 withdrew before treatment with the first test product and is therefore excluded from analysis.
26
End of visit
evaluation 24-hour
post-application
score score
STD 1.24 0.46
N 26 26
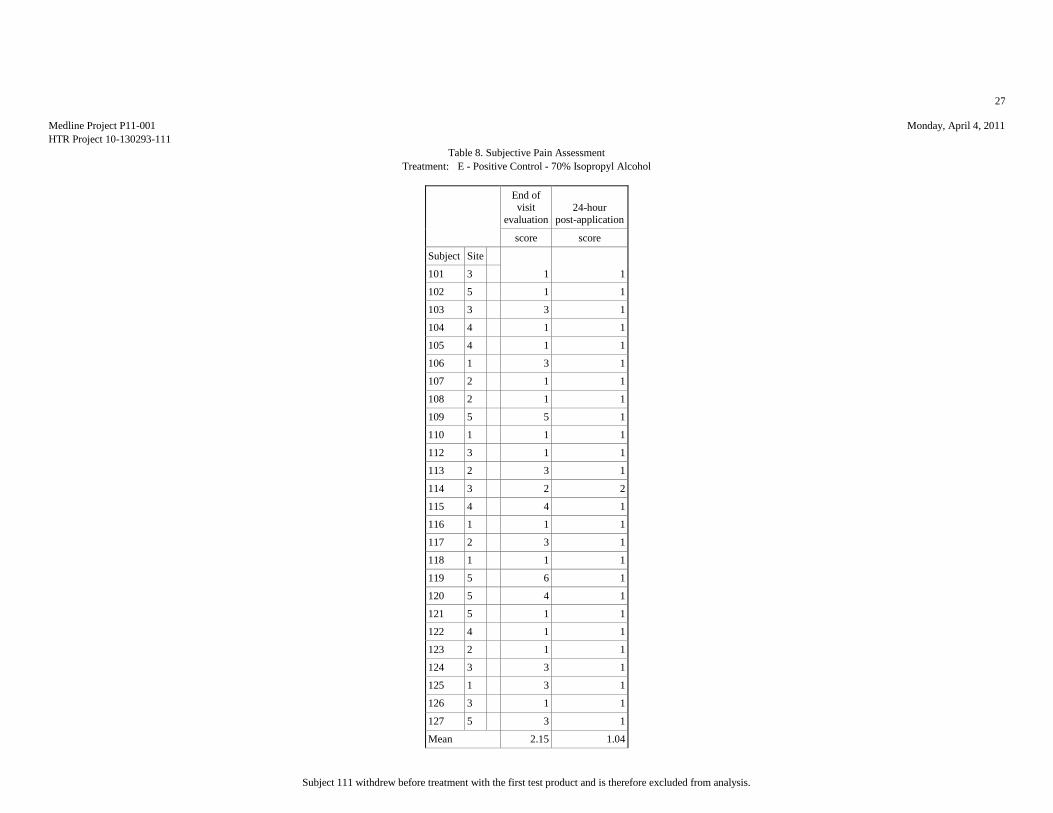
Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 8. Subjective Pain Assessment
Treatment: E - Positive Control - 70% Isopropyl Alcohol
Subject 111 withdrew before treatment with the first test product and is therefore excluded from analysis.
27
End of visit
evaluation 24-hour
post-application
score score
Subject Site
1 1 101 3
102 5 1 1
103 3 3 1
104 4 1 1
105 4 1 1
106 1 3 1
107 2 1 1
108 2 1 1
109 5 5 1
110 1 1 1
112 3 1 1
113 2 3 1
114 3 2 2
115 4 4 1
116 1 1 1
117 2 3 1
118 1 1 1
119 5 6 1
120 5 4 1
121 5 1 1
122 4 1 1
123 2 1 1
124 3 3 1
125 1 3 1
126 3 1 1
127 5 3 1
Mean 2.15 1.04
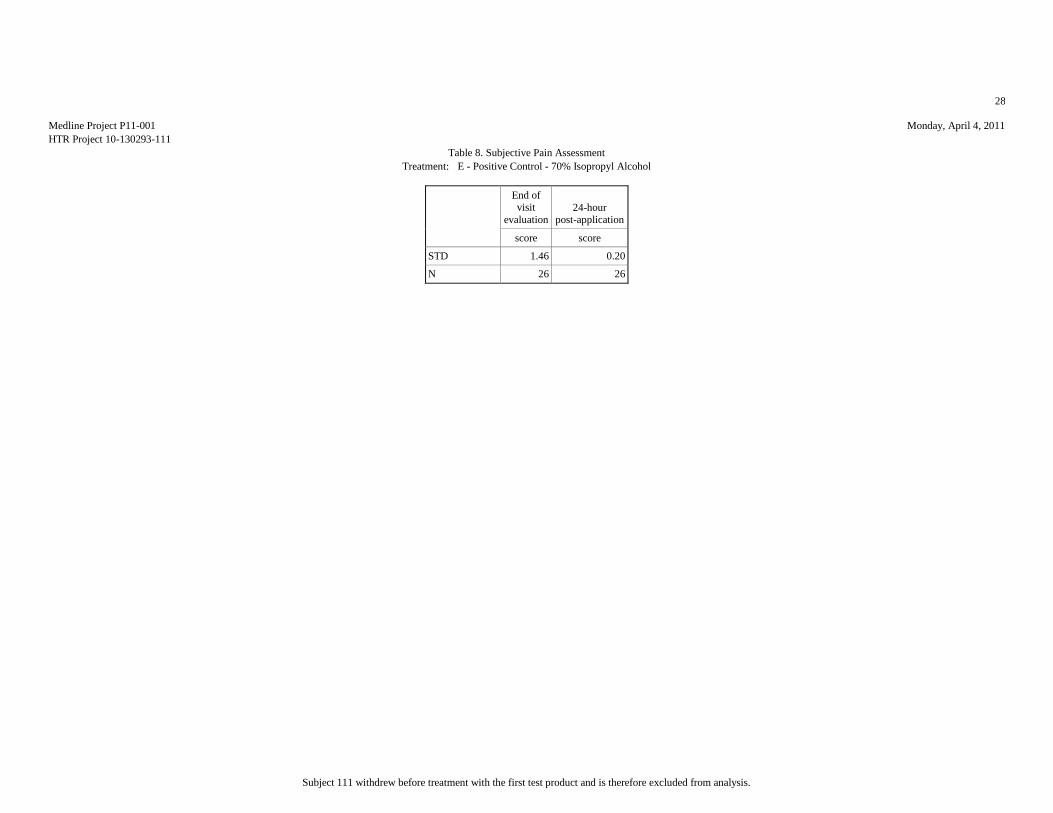
Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 8. Subjective Pain Assessment
Treatment: E - Positive Control - 70% Isopropyl Alcohol
Subject 111 withdrew before treatment with the first test product and is therefore excluded from analysis.
28
End of visit
evaluation 24-hour
post-application
score score
STD 1.46 0.20
N 26 26
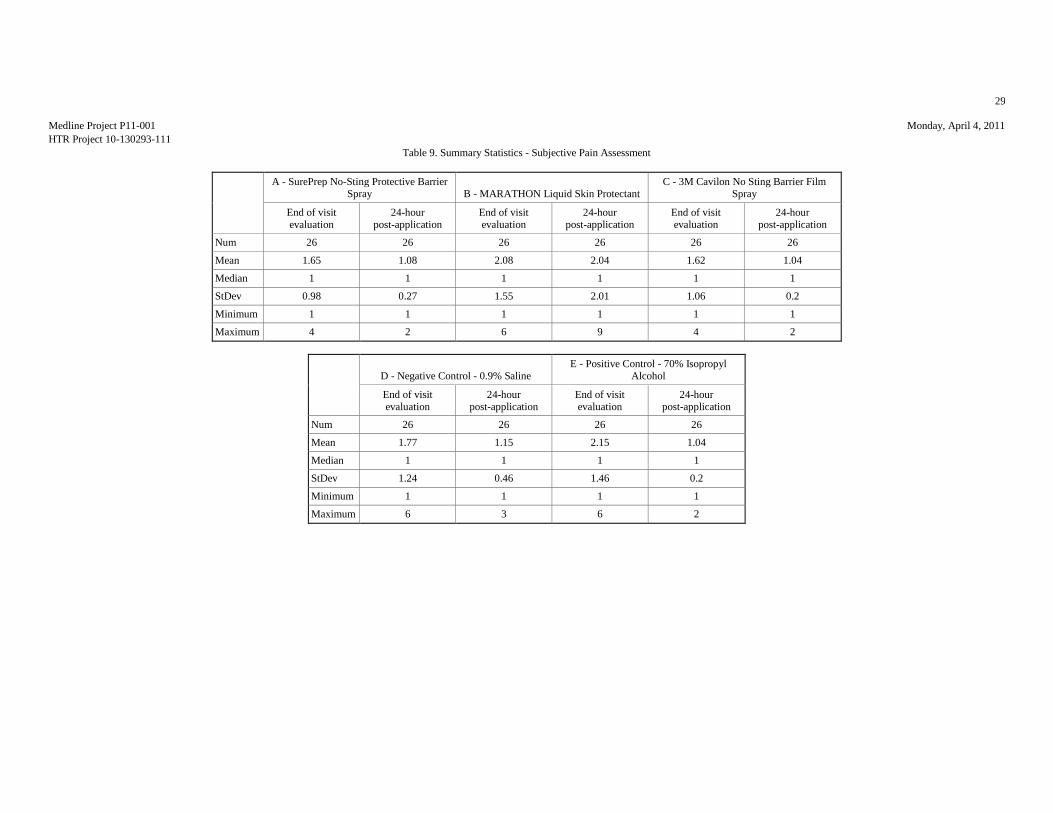
Medline Project P11-001 Monday, April 4, 2011
HTR Project 10-130293-111
Table 9. Summary Statistics - Subjective Pain Assessment
29
A - SurePrep No-Sting Protective Barrier Spray B - MARATHON Liquid Skin Protectant
C - 3M Cavilon No Sting Barrier Film Spray
End of visit evaluation
24-hour post-application
End of visit evaluation
24-hour post-application
End of visit evaluation
24-hour post-application
Num 26 26 26 26 26 26
Mean 1.65 1.08 2.08 2.04 1.62 1.04
Median 1 1 1 1 1 1
StDev 0.98 0.27 1.55 2.01 1.06 0.2
Minimum 1 1 1 1 1 1
Maximum 4 2 6 9 4 2
D - Negative Control - 0.9% Saline
E - Positive Control - 70% Isopropyl Alcohol
End of visit evaluation
24-hour post-application
End of visit evaluation
24-hour post-application
Num 26 26 26 26
Mean 1.77 1.15 2.15 1.04
Median 1 1 1 1
StDev 1.24 0.46 1.46 0.2
Minimum 1 1 1 1
Maximum 6 3 6 2

HTR Study No. 10-130293-111 Final Report
Sponsor No. P11-001
RECORD RETENTION AND PUBLICATION NOTICE
Hill Top Research submits this report with the understanding that the Sponsor may use the study
report for its own purposes. The Sponsor agrees, however, not to use the name Hill Top
Research, or any derivation thereof, in advertising or marketing without the prior written
consent of Hill Top.
All study documents and a copy of the final report will be on file in the Hill Top Research
Records Center at Main and Mill Streets, Miamiville, Ohio 45147, for a period of not less than
two years, unless indicated otherwise on the study protocol or financial contract. A permanent
record will be retained in the form of microfilm or in another readily retrievable form.





























