Embed Size (px)
DESCRIPTION
CONFIGURACIÓN ELECTRÓNICA. Debemos recordar que los electrones son fermiones, por lo cual tienen espín semi-entero (+ ½ y -½). Además, tenemos unas “funciones de onda monoelectrónicas” que cumplen las ecuaciones de Hartree-Fock. ECUACIONES DE HARTREE-FOCK. Operador de Fock. - PowerPoint PPT Presentation
Citation preview

iii
iF
iiiiii rrr
CONFIGURACIÓN ELECTRÓNICA
Debemos recordar que los electrones son fermiones, por lo cual tienen espín semi-entero (+½ y -½).
Además, tenemos unas “funciones de onda monoelectrónicas” que cumplen las ecuaciones de Hartree-Fock
ECUACIONES DE HARTREE-FOCK
iF
Operador de Fock
i
i
Energías orbitales
Espín-orbitales moleculares
i
i Función espacial (orbital molecular: OM)
Función de espín

s
ss
0
21
121
121
021
CONFIGURACIÓN ELECTRÓNICA DE CAPA CERRADA (CLOSED SHELL)
Es el caso cuando tenemos n/2 orbitales moleculares ocupados por n electrones, la mitad de ellos con espín +½ y la otra mitad con espín -½
CONFIGURACIÓN ELECTRÓNICA DE CAPA ABIERTA (OPEN SHELL)
Todas las configuraciones que no sean de capa cerrada se clasifican como configuraciones electrónicas de capa abierta.
Energíacreciente
2p
2s
1s
configuración electrónica delátomo de carbono (6 electrones)
Configuración electrónica de capa abierta

MULTIPLICIDAD DE ESPÍN
Si en un sistema tenemos un número p de electrones con espín hacia arriba y un número q de electrones con espín hacia abajo, tendremos que el espín total del sistema es
qpqpS
21
21
21
La multiplicidad de espín del sistema se calcula como
12 Sdadmultiplici
• sistema de capa cerrada: S=0, multiplicidad=1. estamos en presencia de un singulete.
• todos los sistemas de capa cerrada son singulete pero no todos los singulete son de capa cerrada ni todos los estados fundamentales son singulete.
• el estado donde S=1, multiplicidad=3 se denomina triplete (por ejemplo el estado fundamental del carbono).
• el estado donde S=½, 2S+1=2 se denomina doblete (la mayoría de los radicales libres orgánicos son dobletes).
• existen sistemas con multiplicidades mayores (cuadruplete, quintuplete, hexaplete).

MÉTODOS RESTRINGIDOS Y NO- RESTRINGIDOS (RHF, ROHF Y UHF)
RHF: Restricted Hartree-Fock
Dos electrones cuyos espines están apareados ocupan el mismo orbital espacial. Cada orbital espacial está ocupado por dos electrones. Esto implica una restricción.
UHF: Unrestricted Hartree-Fock Se puede levantar la restricción anterior y establecer que, las funciones espaciales sean diferentes para los diferentes espines. Hacemos referencia, entonces, a los métodos no-restringidos.
ROHF: Restricted Open-shell Hartree-Fock
En este método, las partes espaciales de los orbitales doblemente ocupados son las mismas, los electrones apareados tienen la misma función espacial.
Los cálculos restringidos y no-restringidos pueden realizarse en sistemas de capa cerrada o capa abierta ya que son conceptos diferentes. Sin embargo, para los sistemas de capa cerrada suele usarse RHF y para los de capa abierta ROHF y UHF.
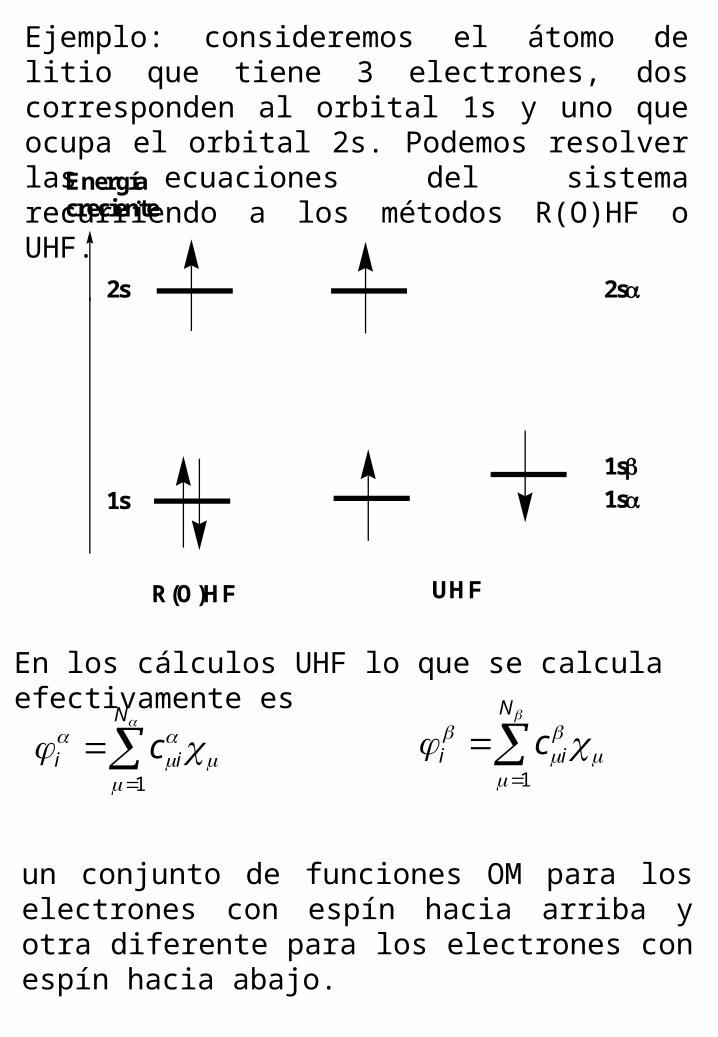
Ejemplo: consideremos el átomo de litio que tiene 3 electrones, dos corresponden al orbital 1s y uno que ocupa el orbital 2s. Podemos resolver las ecuaciones del sistema recurriendo a los métodos R(O)HF o UHF.
Energíacreciente
2s
1s
2s
1s1s
R(O)HF UHF
En los cálculos UHF lo que se calcula efectivamente es
N
ii c1
N
ii c1
un conjunto de funciones OM para los electrones con espín hacia arriba y otra diferente para los electrones con espín hacia abajo.

HFORUHF EE )(
En general (hay excepciones):
• para sistemas no-singulete (como el litio)
Es estos casos los sistemas son mejor representados por cálculos no-restringidos.
• para sistemas singulete, los resultados RHF y UHF suelen ser idénticos y por ello se usa el método RHF ya que requiere menos tiempo de CPU.
Las excepciones implican la presencia de inestabilidades en la función de onda.
CÁLCULO DE ESTABILIDAD SCF
Determina si la función de onda calculada para el sistema molecular es estable o no, es decir, si hay otra función de onda de menor energía que corresponda a una solución diferente de las ecuaciones SCF. Si existe una inestabilidad implica que existe otra función de onda que representa mejor al estado fundamental del sistema en estudio.
Keyword gaussian: stable
stable=opt

Existen diferentes tipos de inestabilidades:
• RHFUHF instability
La función de onda de menor energía es un singulete, pero no de capa cerrada, sino de capa abierta (un birradical, como el ozono.
• RHFUHF instability
Hay un triplete de menor energía que el singulete (como en el oxígeno molecular).
• RHFRHF instability
UHFUHF instability
Hay más de una solución a las ecuaciones SCF para el sistema y el cálculo converge a una solución que no es el mínimo.
CONTAMINACIÓN DE ESPÍN
Es el mayor problema presente en las funciones de onda UHF.
2
S
Hconmuta con
las funciones de onda deben ser funciones propias de
con valor propio S(S+1).
2
S

Mientras que las funciones de onda RHF y ROHF son funciones propias del operador de espín, las funciones UHF no lo son.
Consecuentemente, S(S+1) puede tener un valor muy diferente al teórico.
Si el valor calculado difiere del teórico en más de un 10%, la contaminación de espín es importante y los resultados no son confiables.

CONJUNTOS DE FUNCIONES DE BASE
b
sssii c
1
s Funciones de base, conjuntos de base (basis sets)
La expansión de una función desconocida (el OM) en términos de un conjunto de funciones conocidas (base) no es una aproximación si la base es completa.
Sin embargo, una base completa implica que debe usarse un número infinito de funciones, lo cual es imposible en cálculos reales.
El éxito de los cálculos radica en la correcta elección de la base. Nos interesa el número de funciones (cuanto más pequeña sea la base peor será la representación) y el tipo de funciones de base ( cuanto mejor sea la función de base menor será el número de funciones requeridas para alcanzar el mismo nivel de precisión) usados.

FUNCIONES DE SLATER Y FUNCIONES GAUSSIANAS
STO: Slater Type Orbitals
GTO: Gaussian Type Orbitals
Las Funciones de Slater tienen la forma
rnmlmln erNYr
1..,, ,,,
N: constante de normalización
Yl,m(): armónicos esféricos
: exponente orbitalLos armónicos esféricos son las funciones propias de los operadores que representan al momento angular.
Las funciones de Slater son muy parecidas a las funciones hidrogenoides, solo que la parte radial es diferente.
,,, ...,, mllnmln YrRr
Las funciones hidrogenoides tienen (n-l-1) nodos radiales, los orbitales STO no tienen nodos radiales. Los nodos radiales se introducen haciendo combinaciones lineales de funciones STO. La dependencia exponencial es adecuada.

• las funciones STO dan buenos resultados ya que se obtienen funciones de onda muy parecidas a las hidrogenoides.
• los cálculos son muy costosos desde el punto de vista computacional, por lo que es casi imposible usar funciones STO.
• normalmente confinadas a los cálculos semiempíricos.
222..,, ,,, rlnmlmln erNYrg
Las Funciones Gaussianas tienen la forma
2
,,,,,rlzlylx
lzlylx ezyNxzyxg
N: constante de normalización
Yl,m(): armónicos esféricos
: exponente orbital que determina la extensión radial (tamaño) de la función.
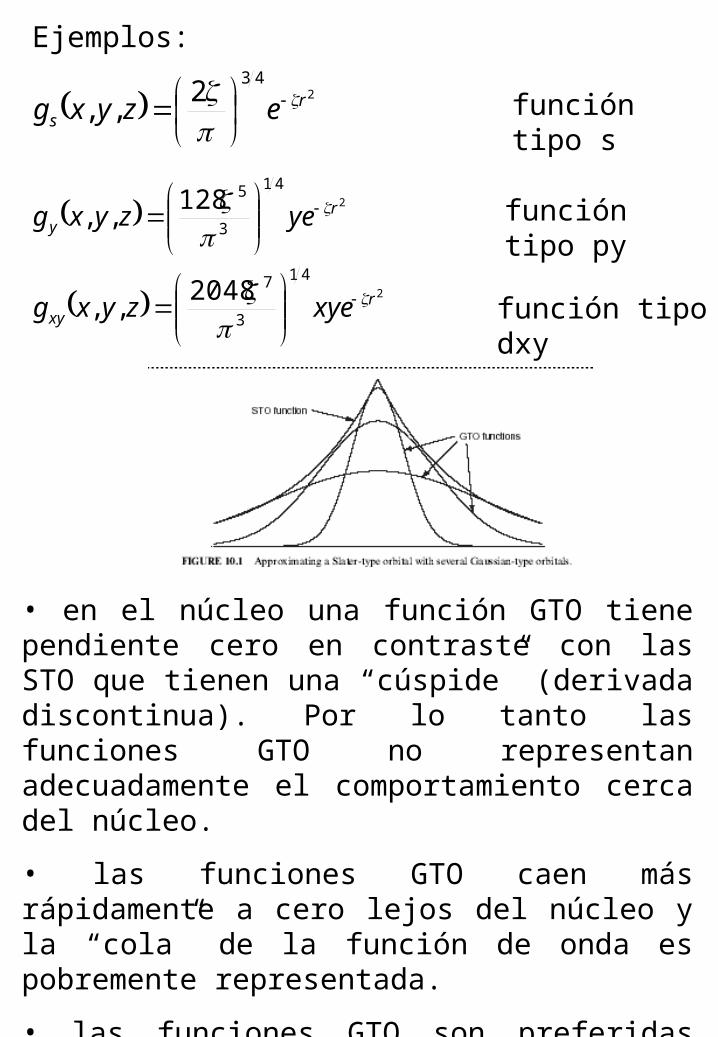
Ejemplos:
243
2,, r
s ezyxg
función tipo s
2
41
3
5128,, r
y yezyxg
función tipo py
2
41
3
72048,, r
xy xyezyxg
función tipo dxy
• en el núcleo una función GTO tiene pendiente cero en contraste con las STO que tienen una “cúspide” (derivada discontinua). Por lo tanto las funciones GTO no representan adecuadamente el comportamiento cerca del núcleo.
• las funciones GTO caen más rápidamente a cero lejos del núcleo y la “cola” de la función de onda es pobremente representada.
• las funciones GTO son preferidas desde el punto de vista de la eficiencia computacional.

Habiendo decidido el tipo de función (STO/GTO) el factor más importante es el número de funciones a usar.
BASE MÍNIMA: se usan sólo las funciones suficientes para representar todos los electrones del sistema.
H: una función s (1s)
Elementos del segundo período: dos funciones s (1s y 2s) y un conjunto de funciones p (px, py, pz).
Elementos del tercer período: tres funciones s (1s, 2s y 3s), y dos conjuntos de funciones p (2p y 3p).
Ej: STO-3G
BASE DOBLE ZETA (DZ): incluye el doble de funciones de base para todos lo átomos.
H: dos funciones s (1s y 1s’)
Elementos del segundo período: cuatro funciones s (1s, 1s’, 2s y 2s’) y dos funciones p (2p y 2p’).
Elementos del tercer período: seis funciones s y cuatro funciones p.
Estas funciones permiten a los orbitales cambiar de tamaño.

BASE SPLIT VALENCE (DZV): sólo se dobla el número de los orbitales de valencia. Se utiliza una base mínima para los electrones del core (los más internos) y una base DZ para los electrones de valencia.
Ejemplos: 3-21G, 6-31G
BASE TRIPLE ZETA (TZ): estos tipos de base contienen el triple de funciones que una base mínima (6 funciones s y tres funciones p para los elementos del segundo período. Si sólo se realiza para los electrones de valencia tenemos una BASE TRIPLE SPLIT VALENCE (TZV), por ejemplo:6-311G.
BASES EXTENDIDAS: bases cuádruple zeta, quíntuple zeta, etc.
FUNCIONES DE POLARIZACIÓN: permiten a los orbitales cambiar de forma. Corresponden a funciones adicionales con mayor momento angular que las funciones de base originales. Por ej: el C en una base convencional incluye funciones de tipo s y p. la introducción de funciones de polarización corresponde a la introducción de funciones d, f, g, etc.
Ej: 6-31G(d), 6-31G(d,p)

FUNCIONES DIFUSAS: permiten a los orbitales ocupar una región más grande del espacio. Son versiones más grandes de las funciones tipo s y p. Difusas implica que son funciones de base con exponentes menores por lo que se extienden a regiones más lejanas del núcleo.
Son importantes en sistemas donde los electrones están relativamente lejos de los núcleos: moléculas pares de electrones no compartidos, aniones, sistemas excitados, etc.
Ej: 6-31+G(d), 6-31++G(d)
BASES DE ALTO MOMENTO ANGULAR: estas bases agregan múltiples funciones de polarización por átomo a un conjunto de bases TZV.
Ej: 6-31G(2d), 6-311++G(3df,3pd), 6-311+G(3df,2pd,p)
Estas bases se utilizan para describir las interacciones entre electrones en los métodos que incluyen correlación electrónica, no se necesitan en general para los cálculos Hartree-Fock.

BASES DE POTENCIAL EFECTIVO DE CORE (ECP): bases utilizadas para los átomos post-tercera fila de la tabla periódica. Estos átomos grandes tienen un gran número de electrones de core que en general no son importantes desde el punto de vista químico y debemos considerar los efectos relativistas. Se usan potenciales efectivos de core o pseudopotenciales para representar los electrones del core y se tratan explícitamente los electrones de valencia.
Ej: LANL2DZ
CONJUNTOS DE BASE CONTRAÍDOS
g
funciones gaussianas=funciones primitivas:
combinación lineal
funciones contraídas:
combinación lineal
orbitales moleculares:

2
,,,,,
rlzlylx
lzlylxp ezyNxzyxg
gaussianas primitivas
pp
p gd
dp: constante para un conjunto de base dado. Los coeficientes de la contracción no cambian durante el curso de un cálculo
: funciones contraídas
p
ppiii gdcc

EJEMPLOS DE BASES:
BASES TIPO POPLE:
STO-nG: n=2-6. Orbitales de tipo Slater que consisten en n primitivas gaussianas. Base mínima. Ej: STO-3G
k-nlmG: Son bases del tipo split valence.
k: número de funciones primitivas usadas para representar los orbitales del core.
Dos valores n,l indican una base DZV, y tres valores n,l,m indican una base TZV. Los números indican cuántas funciones primitivas se usan para representar a los electrones de valencia. Las funciones de polarización se colocan después de la G y las funciones difusas se colocan antes de la G.
Ejemplos:
3-21G: base DZV. 3 primitivas gaussianas para el core. La parte interna de los orbitales de valencia es una contracción de 2 primitivas gaussianas y la parte externa de los orbitales de valencia está representada por 1 gaussiana.

6-31G: Base DZV. Los orbitales del core son una contracción de 6 primitivas gaussianas. La parte interna de los orbitales de valencia es una contracción de 3 primitivas gaussianas y la parte externa de los orbitales de valencia está representada por 1 gaussiana.
6-311G: Base TZV. Core: contracción de 6 primitivas gaussianas. Los orbitales de valencia se dividen en tres funciones representadas por 3, 1 y 1 primitivas.
6-31+G(d): se agregan funciones difusas sp y funciones de polarización d para los átomos pesados.
6-311++G(2df,2pd): se agregan funciones difusas para los átomos pesados y para los hidrógenos. Dos funciones de polarización d y una f para los átomos pesados. Dos funciones de polarización p y una d para los hidrógenos.
BASES DE DUNNING-HUZINAGA:
No tienen la restricción de las bases de Pople que tienen iguales exponentes para las funciones s y p. Son bases más flexibles pero más costosas computacionalmente.
Ej: D95 (base doble zeta)

CORRELATION CONSISTENT BASIS SETS:
Estas bases se utilizan para recuperar la energía de correlación en los electrones de valencia:
cc-pVDZ: correlation consistent polarized Valence Doble Zeta
cc-pVTZ: triple zeta
cc-pVQZ: cuádruple zeta
cc-pV5Z: quíntuple zeta
cc-pV6Z: séxtuple zeta
Las bases pueden ser aumentadas (augmented) por funciones difusas y se agrega el prefijo aug para indicarlo. Ej: aug-cc-pVDZ.

MM
MEE: - ab initio HF
Post-HF CI
MP
CC
MCSCF
- funcionales de la densidad
- métodos semiempíricos
Métodos no
empíricos
Las aproximaciones principales subyacentes en toda la metodología HF son la aproximación de las partículas independientes, la aproximación de los núcleos fijos y el hecho de que cada electrón se trata independientemente del resto, si bien esta influencia es tomada en cuenta por la existencia de un potencial promedio generado por los n-1 electrones restantes.
Estamos despreciando la interacción instantánea entre dos electrones cualesquiera. Esta interacción que no estamos teniendo en cuenta en los cálculos HF es la correlación electrónica.
El movimiento de los electrones está correlacionado.

HFrealncorrelacio EEE Existen diferentes métodos que permiten calcular la energía de correlación.
INTERACCIÓN DE CONFIGURACIONES (CI)
El método HF determina la mejor función de onda representada por un único determinante de Slater.
Una mejor forma de representar al sistema es utilizar una función inicial que contenga más de un determinante de Slater.
En el método CI se desarrolla la función de onda como una combinación lineal de determinantes de Slater.
¿Cómo se seleccionan esos orbitales de Slater? Para ello debemos considerar los conceptos de excitación y configuración electrónica.

simple simpleHF doble doble triple cuádruple
Supongamos: N electrones
M funciones de base (M>N)
Cada uno de los posibles arreglos de los N electrones en los M orbitales recibe el nombre de configuración electrónica.
La solución RHF nos da N/2 OM ocupados y M-N/2 orbitales virtuales o desocupados.
Si se realizan excitaciones (promociones de electrones) se obtienen diferentes determinantes de Slater que pueden incluir excitaciones simples, dobles, triples, cuádruples, etc.

0
00 ...i
iiT
TTD
DDS
SSCIfull ccccc
El método de interacción de configuraciones completo (full-CI) se basa en expresar la función de onda del sistema como una combinación lineal de todas las configuraciones electrónicas posibles del sistema (determinantes de Slater).
En CI la función de onda se escribe como una combinación lineal de determinantes de Slater y los coeficientes de la expansión se optimizan variacionalmente.
No es posible llevar a cabo cálculos full-CI por lo que debemos recurrir a los
MÉTODOS CI TRUNCADOS
Se consideran algunas de las excitaciones posibles. Por ejemplo podemos incluir las excitaciones simples y las dobles (CISD)
D
DDS
SSCISD ccc 00
También: CISDT, CISDTQ

CONSISTENCIA DE TAMAÑO
Cualquier método CI truncado (diferente de full-CI) conduce a un error denominado de consistencia de tamaño (size consistency).
Por ejemplo, el cálculo CISD de dos moléculas de H2 separadas 100Å no da o mismo que el doble del resultado de un cálculo CISD para una molécula de H2.
Una full-CI es consistente con el tamaño pero todas las formas CI truncadas no lo son (desventaja).
MÉTODOS MULTICONFIGURACIONALES (MCSCF)
MCSCF: Multi-Configuration Self Consistent Field
Los métodos configuracionales pueden considerarse como métodos CI donde no sólo los coeficientes de la combinación lineal de determinantes de Slater se optimizan por el principio variacional, sino que en cada ciclo se optimizan los coeficientes de los orbitales moleculares individuales (en lugar de dejarlos fijos a sus valores HF como en una CI).
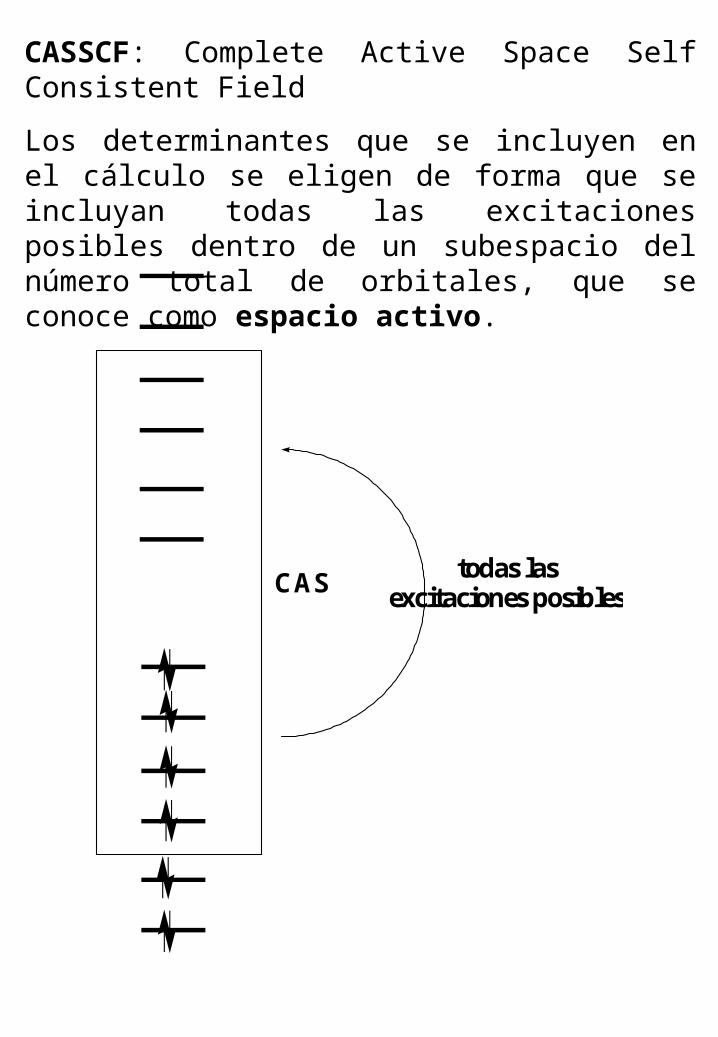
CASSCF: Complete Active Space Self Consistent Field
Los determinantes que se incluyen en el cálculo se eligen de forma que se incluyan todas las excitaciones posibles dentro de un subespacio del número total de orbitales, que se conoce como espacio activo.
CAStodas las
excitaciones posibles

INTERACCIÓN DE CONFIGURACIONES MULTIREFERENCIA (MRCI)
Los métodos CI construyen funciones de onda como combinaciones lineales de determinantes de Slater generados mediante la excitación de electrones a partir de un único determinante de referencia (el determinante de HF).
En los métodos multireferencia, se construye una función de onda CI comenzando con una función multiconfiguracional (MCSCF). Se aplica una CI no a una única configuración de referencia sino a un conjunto de configuraciones de referencia. Es un procedimiento muy costoso.
MÉTODOS PERTURBACIONALES
Método basado en la Teoría de Perturbaciones de Muchos Cuerpos, MBPT (Many-Body Perturbation Theory).

TEORÍA DE PERTURBACIONES
Supongamos que tenemos un sistema con un Hamiltoniano , que es nuestro sistema de interés y cuya E.S. no podemos resolver
H
EH
Supongamos además que tenemos un sistema de referencia para el cual sabemos resolver la E.S.
)0()0()0(0 EH
La idea de la teoría de perturbaciones es que el Hamiltoniano de nuestro sistema puede escribirse de la siguiente manera
10
HHH
0
H sistema sin perturbar
H sistema perturbado
1
H perturbación
entre 0 y 1

Podemos expresar tanto la energía como la función de onda exacta como una serie de potencias del parámetro perturbacional.
...)3(3
)2(2
)1()0(
...)3(3
)2(2
)1()0( EEEEE Introduciendo estas expresiones en la E.S. y agrupando para las diferentes potencias de
)0()0()0(0 EH
)0()1()1()0()0(1)1(0 EEHH
)0()2()1()1()2()0()1(1)2(0 EEEHH
n
iininn EHH
0)()()1(1)(0
El desarrollo anterior rige para cualquier perturbación. Cuando el sistema no-perturbado corresponde a la solución HF (el hamiltoniano no-perturbado corresponde a los operadores de Fock) el método se denomina Teoría de Perturbaciones de Mller-Plesset

TEORÍA DE PERTURBACIONES DE MLLER-PLESSET (MP)
De modo de aplicar la teoría de perturbaciones al cálculo de la energía de correlación, debemos seleccionar el Hamiltoniano no perturbado. La elección más común es tomarlo como una suma de operadores de Fock y esto resulta en el método de MP.
La correlación electrónica se cuenta a partir de MP2.
MP2 típicamente recupera el 80-90% de la energía de correlación y es el método más económico para incluir la correlación electrónica.
Los MP presentan como ventaja sobre la CI que son consistentes respecto al tamaño del sistema.
Sin embargo tienen una desventaja, los MP no son variacionales por lo cual no convergen monotónicamente hacia la energía real del sistema, sino que pueden estar por encima o por debajo de la misma.

COUPLED CLUSTERS (CC)
Los métodos de pares acoplados tienen una filosofía similar a los MP desde el punto de vista de que hay un desarrollo en serie, pero aquí el desarrollo es exponencial
0T
CC e
0
32
!1
...!3
1!2
11
k
kT T
kTTTe
NTTTTT
...321 suma de operadores de cluster
1
T Operador de excitación de una partícula. La aplicación de este operador genera todas las excitaciones simples.
2
T Operador de excitación de dos partícula. La aplicación de este operador genera todas las excitaciones dobles, todas las configuraciones doblemente excitadas.

La idea del método CC es incluir todas las excitaciones posibles de un determinado tipo.
Evidentemente es imposible realizar un tratamiento completo del problema, de modo que debemos truncar la función de onda CC en algún nivel de excitación. Por ejemplo podemos incluir solamente el operador de dobles excitaciones (es el más frecuente)
03
22
22 ...!3
1!2
11
TTTCCD
• consistente con el tamaño
• recupera mucha energía de correlación (comparando CCD vs. MP2)
• muy costoso (mucho más que MP2).























