Embed Size (px)
Citation preview

American Journal of Medical Genetics 99:185±189 (2001)
Con®rmation of the Autosomal Recessive Syndromeof Ectopia Lentis and Distinctive CraniofacialAppearance
Randa Haddad,1 Sami Uwaydat,1 Rola Dakroub,1 and Elias I. Traboulsi2*1Department of Ophthalmology, American University of Beirut Medical Center, Beirut, Lebanon2Center for Genetic Eye Diseases, Cole Eye Institute, Cleveland Clinic Division of Ophthalmology, Cleveland, Ohio
We report four members of a LebaneseDruze family with the syndrome of lensdislocation, spontaneous ®ltering blebs,anterior segment abnormalities, and a dis-tinctive facial appearance. The constellationof clinical abnormalities in these patients isnot suggestive of the Marfan syndrome orother connective tissue disorders associatedwith ectopia lentis. We previously describedthis syndrome in another presumably unre-lated and highly inbred Druze family fromthe mountains of Lebanon. We postulatedautosomal recessive inheritance in apseudo-dominant pedigree. A few isolatedreports of similar cases are scattered in theworld literature. We now con®rm that this isa distinct autosomal recessive syndromewhose gene mutation is enriched in theLebanese Druze community.# 2001 Wiley-Liss, Inc.
KEY WORDS: dislocated lens; glaucoma;syndrome; craniofacial dys-morphism; autosomal rec-essive; Lebanon; Druze
INTRODUCTION
Nontraumatic subluxation or dislocation of thecrystalline lens is most commonly observed in Marfansyndrome. It is also a major manifestation of homo-cystinuria, the Weil±Marchesani syndrome, and a fewless common autosomal recessive disorders [Traboulsiand Maumenee, 1998]. Lens dislocation can also occurin the absence of systemic abnormalities in simpleectopia lentis, ectopia lentis et pupillae, aniridia, andrarely in association with anterior segment dysgenesis.We have previously described a family with a rarephenotype of ectopia lentis, progressive scleral thin-
ning with spontaneous ®ltering blebs, peripheraliridocorneal adhesions, glaucoma, and facial abnorm-alities [Shawaf 1995]. We have since examined anotherfamily of the same ethnic Lebanese Druze origin withthe same disorder. The mode of inheritance in thepresent family is clearly autosomal recessive, and thephenotype con®rms our previous observations of aunique and distinct connective tissue disorder.
CLINICAL REPORT
The proband is a 34-year-old woman who presentedwith anterior dislocation of her right cataractous lens.The anterior chamber was ¯at and there was an area ofscleral thinning and bulging superiorly. An intracap-sular cataract extraction was performed through acorneal incision. Her anterior chamber progressivelydeepened but the superior sclera became progressivelythinner, necessitating the performance of two scleralpatch grafts. Her most recent visual acuity in thisaphakic right eye was to count ®ngers at six feet. Herintraocular pressure was 16 mmHg. She counted®ngers at one foot in the left eye in which there was avery thin superior sclera with uveal prolapse under theconjunctiva (Fig. 1), a cataractous subluxated lens withirido-lenticular synechiae, a very shallow anteriorchamber with peripheral irido-corneal adhesions, andareas of iris atrophy. The left fundus could not bevisualized. Ultrasonography did not show any retinaldetachment. The right fundus was normal.
The pedigree is shown in Figure 2. The parents are®rst cousins. The patient's facial features were dis-tinctive (Fig. 3) and resembled those of her threeaffected sisters (Figs. 4±6) but not those of her mother(Fig. 7), deceased father, or unaffected brother (notshown). The nose had an overhanging nasal tip andthere was dental crowding. The mouth was held in aconsistent contour with a pursed appearance. Therewere no other signs of connective tissue disease such aspectus excavatum, lax joints, or ¯at feet. Her skull x-ray was normal. A skeletal survey was obtained whichwas normal and showed no ®ndings suggestive of theMarfan syndrome. Their metacarpal index (the averageof the ratios of the length divided by the midpointwidths of the second, third, fourth, and ®fth metacar-
*Correspondence to: Elias I. Traboulsi, Cole Eye Institute, i32,9500 Euclid Avenue, Cleveland, OH 44195.
Received 24 February 2000; Accepted 9 October 2000
Published online 6 February 2001
ß 2001 Wiley-Liss, Inc.

pals) which was calculated at the time of their skeletalsurvey, ranged from 9.12 to 9.74 (normal 5.5±8.0). Anechocardiogram revealed left ventricular hypertrophybut no aortic root dilatation or mitral valve prolapse.
The ocular and systemic ®ndings of her three affectedsisters are summarized in Table I. Homocystinuria wasruled out by a normal serum amino acid quantitationand a negative urine assay in all patients. The mother'socular and systemic examinations were within normallimits. A male sibling died of unknown causes at the ageof three months. A sister died of unknown causes underanesthesia at 15 years of age. She had eye problemssimilar to her sisters'. The father had no ocular orsystemic problems. We reviewed a photographic por-trait that did not show any of the facial features thatwere present in his daughters.
DISCUSSION
Although some of the clinical ®ndings in this familyoverlap with those of Marfan syndrome the autosomalrecessive mode of inheritance, the severity of the oculardisease, and the absence of the characteristic somaticfeatures make the diagnosis of the Marfan syndromeextremely unlikely. In fact, patients do not meet the
revised diagnostic criteria for Marfan syndrome [DePaepe et al., 1996]. Echocardiography in four affectedindividuals failed to reveal aortic root dilatation ormitral valve prolapse, the most commonly encounteredcardiac lesions in the Marfan syndrome. We haveevidence that our patients have long slender ®ngers.The metacarpal index adds scienti®c objectivity to theassessment of elongated or slender long bones in thehands. It increases with age and is elevated not only inthe Marfan syndrome, but in any individual with longslender metacarpals. Furthermore, Walker and Ash-croft [1978] consider the values of 9.6 in males and 10.1in females as the upper limits of normal. If we use theselimits, then the values in our patients would fall withinthe normal range.
We have previously reported a highly inbred familywith clinical ®ndings identical to those in the presentpedigree [Shawaf et al., 1995]. We could not establishany known connections between the two pedigrees. Themembers of this family insisted that they were notrelated to the other one on either side of the family.There were six affected members of both sexes in threeconsecutive generations of the family reported byShawaf et al. [1995]. We postulated an autosomal re-cessive mode of inheritance with pseudodominance inone branch of the pedigree. Three eyes of two patientshad spontaneous ®ltering blebs, whereas all eyes of theaffected individuals had dislocated/subluxated lenses,with severe postoperative scleral thinning. All patientshad distinctive abnormal facial features with a beakednose, long face, and dental crowding, identical to thoseof patients in the present family. These clinical featuresresemble those described in four cases by Kirkham[1970] as mandibulofacial dysostosis with ectopia lentis.None of the patients in that report had a spontaneouslyoccurring ®ltering bleb. The cystic and avascularthinning and elevation of the conjunctiva in three eyesof two patients reported cases by Shawaf et al. [1995],and in one of the present four cases appear to be theresult of progressive spontaneous thinning of the sclera
Fig. 1. Left eye of proband on presentation. The patient is looking down and the superior sclera is thin with uveal tissue (arrow) protruding under theconjunctiva.
Fig. 2. Pedigree is compatible with autosomal recessive inheritance.
186 Haddad et al.
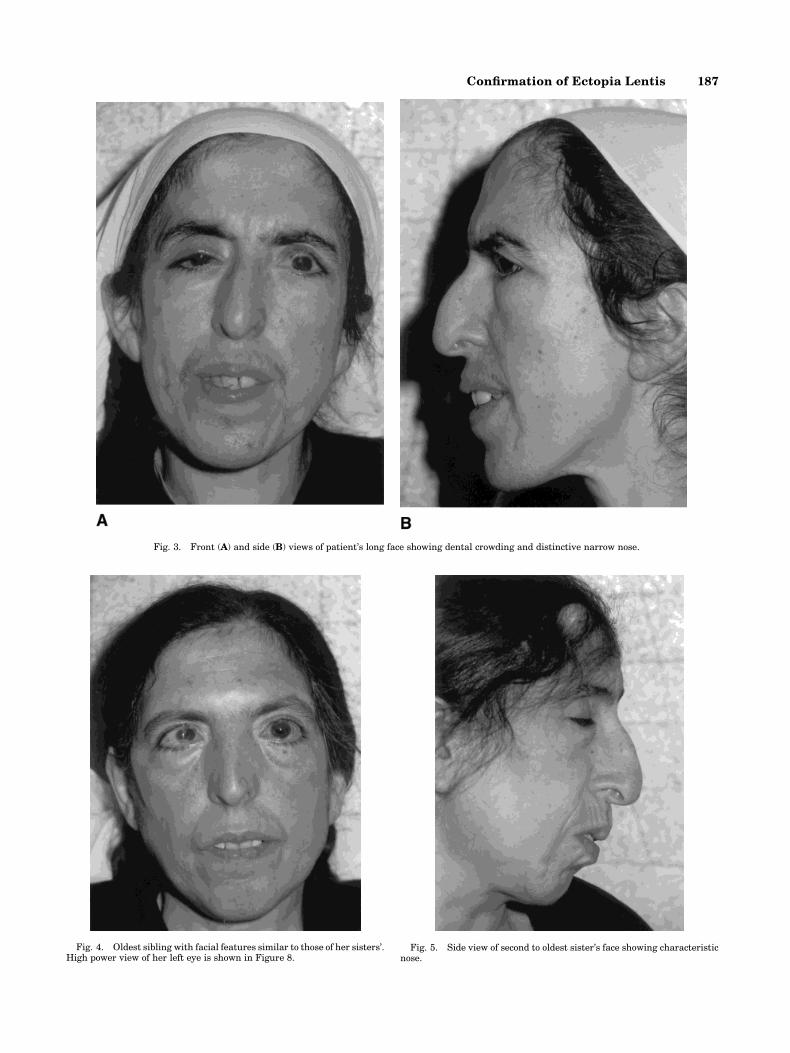
Fig. 3. Front (A) and side (B) views of patient's long face showing dental crowding and distinctive narrow nose.
Fig. 4. Oldest sibling with facial features similar to those of her sisters'.High power view of her left eye is shown in Figure 8.
Fig. 5. Side view of second to oldest sister's face showing characteristicnose.
Con®rmation of Ectopia Lentis 187
close to the limbus. Only four other cases with spon-taneous blebs have been described in the literature.The ®rst patient was reported by Nemet et al. [1973].That patient had Axenfeld±Rieger syndrome andpresented with ocular hypotony secondary to bilateralspontaneous ®ltering blebs. The authors speculatedthat high intraocular pressure caused rupture at thecorneo±scleral junction. Soong et al. [1986] describedtwo patients with Terrien's marginal degenerationwho developed ®ltration blebs without limbal surgery.The blebs occurred after a spontaneous rupture ofDescemet's membrane. Lastly, Pasquale et al. [1991]described a young man with subluxated lenses, andbilateral elevated a vascular and microcystic ®ltrationblebs. Although no comments are made about thepatient's facial features, they were very similar to thoseof patients in this report and in the family described byShawaf et al. [1995].
We believe that the family reported herein, the onedescribed by Shawaf et al. [1995], and some of theisolated cases in the literature [Pasquale et al., 1991]have an autosomal recessive disorder, possibly ofconnective tissue or of a developmetal gene, that isprominently expressed in the face and in the eye. Inaddition to characteristic facial features, the syndrome
manifests itself by lens dislocation and cataract and byscleral thinning and spontaneous ®ltering bleb forma-tion. Chronic apposition of the peripheral iris to thecornea leads to angle closure glaucoma and distortion ofthe iris and angle structure. The long and slender®ngers in the four patients from the present familysuggest that the gene defect may lead to subtle skeletalchanges. The mutation responsible for this disease
Fig. 6. Youngest sister with facial features similar to those of hersisters'.
Fig. 7. Mother of the four affected sisters has no distinctive facialfeatures and normal eyes.
Fig. 8. Superiorly dislocated cataractous lens in oldest affected sibling.The iris is atrophic and distorted superiorly between 10 and 12 o'clock.
188 Haddad et al.

appears to be enriched in the Druze Lebanese popula-tion in which we have found two presumably unrelatedfamilies with identical clinical ®ndings. The rate ofconsanguinity in some of the Lebanese village commu-nities has been estimated at more than 30% [DerKaloustian et al., 1980]. The Druze (single, Durzi) are arelatively small Middle Eastern religious sect charac-terized by an eclectic system of doctrines and by acohesion and loyalty among its members (at timespolitically signi®cant) that have enabled them tomaintain through almost a thousand years of turbulenthistory their close-knit identity and distinctive faith.Propagation of the tenets of this religion began in Cairoin AD 1017, led by Hamzah ibn 'Ali; it is from the nameof Hamzah's subordinate, Muhammad ad-Darazi, thatthe group derives its name. The Druze numbered morethan 2,50,000 in the late 20th century and live mostlyin Lebanon, with smaller communities in Israel andSyria. Druze religious beliefs developed out of Isma'iliteteachings. Various Jewish, Christian, Gnostic, Neopla-tonic, and Iranian elements are combined under adoctrine of strict monotheism.
REFERENCES
De Paepe A, Devereux RB, Dietz HC, Hennekam RC, Pyeritz RE. 1996.Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet62:417±426.
Der Kaloustian VM, Naffah J, Loiselet J. 1980. Genetic diseases inLebanon. Am J Med Genet 7:187±203.
Kirkham TH. 1970. Mandibulofacial dysostosis with ectopia lentis. Am JOphthalmol 70:947±949.
Nemet P, Bracha R, Lazar M. 1973. Spontaneous ®ltering blebs in Axenfeldsyndrome. Am J Ophthalmol 76:590±591.
Pasquale LR, Smith SG, Traboulsi E, Jampel H. 1991. Spontaneous®ltration blebs in a patient with microspherophakia. Am J Ophthalmol112:350±352.
Shawaf S, Noureddin B, Khouri A, Traboulsi E. 1995. A family with asyndrome of ectopia lentis, spontaneous ®ltering blebs, and craniofacialdysmorphism. Ophthalm Genet 16:163±169.
Soong HK, Fitzgerald J, Boruchoff SA, Sugar A, , Mayor RF, , Gabel MG.1986. Corneal hydrops in Terrien's marginal degeneration. Ophthal-mology 93:340±343.
Traboulsi EI, Maumenee IH. 1998. Subluxation of the crystalline lens andassociated systemic disease. In: Traboulsi EI, editor. Genetic diseases ofthe eye. New York: Oxford University Press. p 605±627
Walker TM, Ashcroft MT. 1978. The metacarpal index in Jamaicanchildren and adults. Br J Radiol 51:338±341.
TABLE I. Clinical and Ocular Findings in the Proband's Three Affected Sisters
Patient/age Ocular features Facial features Other
II-5/28
Vision R.E. with �16.00-20/70; Vision L.E. with�17.00�20/50. Wears contact lenses successfully;I.O.P. 11 mmHg in each eye. Bilateral cataractoussubluxated lenses in an inferior direction. Distinctive as in proband.
Normal skull X-ray; Longslender ®ngers; Normal EKGand echocardiogram; Normalamino acid quantitation.
II-2/42
Dislocated lens extraction R.E. at age 30. Now N.L.P.phtisical R.E. Filtering procedure L.E. at age 35.Now C.F. 1'. Shallow Anterior chamber withirido-corneal touch; cataractous superiorlydislocated lens (Fig. 8); C/D�0.9 withdiffuse chorioretinal scars, etiology unclear. Distinctive as in proband. Same as
II-1/44
HM vision in each eye; Bilateral corneal opaci®cation;no view of anterior segment; had undergone surgicalprocedures to both eyes in the past Distinctive as in proband. Same as
Con®rmation of Ectopia Lentis 189










![CASE REPORT / ПРИКАЗ БОЛЕСНИКА Delayed diagnosis of ... · and ectopia lentis (EL) [1]. It has an estimated incidence of 1:50,000–200,000, sufficiently high to consider](https://img.pdfslide.net/doc/110x75/5e452e7fa3e3b7377054df81/case-report-delayed-diagnosis-of-and-ectopia.jpg)


















