Embed Size (px)
Citation preview

Conformational potential energy surface of BrOONO
Antonija Lesar a,*, Sa�ssa Prebil a, Max M€uuhlh€aauser b, Milan Hodo�ss�ccek a,c
a Department of Physical and Organic Chemistry, Institute Jo�zzef Stefan, Jamova 39, Ljubljana SI-1000, Sloveniab Institut f€uur Physikalische und Theoretische Chemie der Universit€aat Bonn, Wegelerstrasse 12, Bonn 53115, Germany
c Center for Molecular Modeling, National Institute of Chemistry, Hajdrihova 19, Ljubljana SI-1000, Slovenia
Received 14 August 2002; in final form 5 November 2002
Abstract
Cis-perp and trans-perp conformations with respect to N–O and O–O bonds were found to be the only stable ones
on the BrOONO potential energy surface using the CCSD(T)//B3LYP method with the 6-311G* basis set. The energy
for the cis-perp form is 2:0 kcal mol�1 lower than for the trans-perp form while the saddle point connecting the twominima is 9:0 kcal mol�1 above the cis-perp level. A comparison of the relative energetics for stationary points on theBrOONO, ClOONO, and HOONO conformational potential energy surfaces is discussed.
� 2002 Elsevier Science B.V. All rights reserved.
1. Introduction
Already in 1975 Wofsy et al. [1] pointed out that
bromine compounds participate in the destruc-
tion of stratospheric ozone. The first report on
stratospheric significance of bromine nitrate,
BrONO2, was made by Spencer and Rowland [2]
on the basis of UV and IR absorption character-istics measurements and the estimation of the rate
of formation from BrO and NO2. The kinetics
of reaction was studied at room temperature by
discharge-flow-mass spectrometric and flash-pho-
tolysis-UV absorption techniques for low and high
pressure conditions, respectively [3]. The reaction
was found to be pressure dependent and later ki-
netics studies established the negative temperature
dependence of the low-pressure [4] and the high-
pressure [5] rate constant of reaction. While none
of the works [3,4] have been addressed to analyze
the isomer formation, it has nevertheless been
speculated that it will not occur. Gas-phase
structural studies for BrONO2 by the combined
use of electron diffraction intensities and rotational
constants has been provided by Casper et al. [6]. Ina thermal decomposition study of bromine nitrate
by IR spectroscopy, Orlando et al. [7] have con-
sidered the possibility of product formation
other than BrONO2 in a recombination reaction
but they found no obvious evidence of isomer
formation.
The role of BrONO2 as a reservoir for active
bromine in the lower stratosphere is now wellrecognized. The major loss processes of BrONO2in the stratosphere include photolysis at UV
wavelengths [8] during the day and heterogeneous
reactions during the night [9]. The quantum yield
Chemical Physics Letters 368 (2003) 399–407
www.elsevier.com/locate/cplett
* Corresponding author. Fax: +386-1-251-93-85.
E-mail address: [email protected] (A. Lesar).
0009-2614/02/$ - see front matter � 2002 Elsevier Science B.V. All rights reserved.
PII: S0009-2614 (02 )01888-2

measurements of NO3 production in the UV
photolysis of BrONO2 have shown that besides
NO2, NO3 is also an important photolytic product
[10]. Although numerous laboratory studies have
been carried out to elucidate the equivalent reac-
tion of ClO, i.e., ClOþNO2, there still remainsconflicting indirect evidence of the presence of
more than one isomer of chlorine nitrate. On the
other hand, longstanding controversy over the
existence of peroxynitrous acid, HOONO, an iso-
mer of nitric acid, HONO2, was just recently re-
solved with the first direct spectroscopic
observation of HOONO [11] produced in reaction
of NO2 with OH radicals.Thus, we are not aware of any experimental
conclusions that different isomers of BrONO2 may
be formed. Likewise, we do not know of any the-
oretical investigations that have been attributed to
isomeric species of BrONO2, while there exist a
few theoretical studies devoted to the structural,
vibrational and thermochemical parameters of
BrONO2 [12,13]. In this study, we present the first,as far as we know, ab inito calculations on the
BrOONO isomer and its conformational forms by
determining their equilibrium or first-order saddle
point structures, vibrational frequencies, and rel-
ative energies. It is also of interest to compare and
contrast the results with those of ClOONO and
HOONO. The ab initio methods used in the
present investigation are described in Section 2.Section 3 contains the results and discussion, while
the conclusions are summarized in Section 4.
2. Computational methods
Ab initio molecular orbital calculations were
performed using the GAUSSIANAUSSIAN 98 program [14].The geometrical parameters of the BrOONO
conformational forms were obtained using the
second-order M€ooller–Plesset (MP2) method [15],and the DFT approach was obtained using the
Becke three-parameter nonlocal exchange func-
tional [16] with the nonlocal correlation of Lee,
Yang, and Parr (B3LYP) [17,18]. For minima
structures, the single and double excitation cou-pled-cluster method including a perturbation esti-
mate of the effects of connected triple excitations
(CCSD(T)) [19–21] was also used. The standard
triple f plus polarization 6-311G(d) basis set, asimplemented in the computer program [14], was
employed in the optimizations.
The harmonic frequencies were computed from
analytical derivatives at the MP2 and B3LYPlevels for all species, employing the geometries
calculated at these levels of theory, in order to
characterize the nature of the stationary points on
the conformational potential energy surface and to
determine the zero-point vibrational energies
(ZPE).
The anharmonic frequencies were calculated at
the B3LYP level using the 6-311G(d) basis set andthe VSCF [22] method as implemented in the
GAMESS [23] package.
The intrinsic reaction coordinate (IRC), which
follows the minimum-energy path from the first-
order saddle point in both directions, has been
calculated to confirm the connections between the
maxima and the stable forms of BrOONO.
In order to obtain the energy differences withhigh accuracy, we performed single-point calcula-
tions for stationary points on conformational po-
tential energy surface at the CCSD(T) level with
the 6-311G(d) basis set using optimized geometri-
cal parameters obtained with the B3LYP/
6-311G(d) level of theory.
3. Results and discussion
3.1. Geometries and vibrational frequencies
On the conformational potential energy surface
of bromine peroxynitrite, BrOONO, we have
characterized seven stationary points. The geo-
metrical arrangement of nuclei, corresponding tothese points, are shown in Fig. 1. Their structural
parameters, obtained without any symmetry
constraint in the optimizations, are provided in
Table 1. For convenience, the same notation is
used as McGrath and Rowland [24] introduced
for the conformers of HOONO and ClOONO.
They characterized the conformers with respect to
both the sðONO0OÞ and s0ðNO0OBrÞ dihedralangles. The nonplanar cis-perp conformer
(s; s0 ¼� 0; � 90) was found to have the lowest
400 A. Lesar et al. / Chemical Physics Letters 368 (2003) 399–407

energy. The other minimum corresponds to
the trans-perp conformer (s; s0 ¼� 175;� 90). Thelatter conformer results from the rotation of theO0OBr group around the N–O bond. The maxi-
mum energy structures state accompanying this
rotation has the perp–perp conformation (s; s0 ¼� 88; � 85). The examination of the rotation ofthe OBr group around the O0–O bond reveals
four additional stationary points verified to be
maximum energy structures with one imaginary
vibrational frequency. When the OBr unit of thecis-perp conformer is rotated, the corresponding
maxima have cis–cis (s; s0 ¼ 0; 0) and cis–trans
(s; s0 ¼ 0; 180) structures while maxima related tothe trans-perp conformer have trans–cis (s; s0 ¼180; 0) and trans–trans (s; s0 ¼ 180; 180) arrange-ments of the ONO0O and NO0OBr torsional
angles.
From Table 1 it can be seen that for the morestable cis-perp conformational forms of BrOONO,
the MP2 and DFT (B3LYP) methods give sub-
stantially different values for both the bond lengths
and the valence bond angles. The deviation of the
NO0 and OBr bond lengths between the two
methods exceeds 0.4 and 0.2 �AA and is about 20� forthe ONO0 and NO0O bond angles. The geometry
prediction of the more reliable and extensive
CCSD(T) calculations agrees better with that of
the DFT (B3LYP) level of theory. Thus, we must
conclude that the geometry of the cis-perp form is
not adequately described by the MP2 method.
CCSD(T) calculations are also employed for thetrans-perp conformation and all three methods
yield comparable results for the geometrical pre-
diction. These calculations suggest that for our
systems under investigation, containing one third-
row atom, the DFT results are to be preferred over
those resulting from the MP2 approach. The in-
adequacy of MP2 calculations for the cis-perp
conformer may arise from the overemphasizedcontributions from electron correlation or, more
likely, from singlet–triplet instability of the HF
wavefuction as a reference. Likewise, an unusually
long N–O bond, and ONO and NOO bond angles
around 90� at the MP2 level were found in acomputational study on alkali peroxynitrite [27].
The geometrical parameters for the maximum
energy conformeric forms calculated at the MP2and B3LYP levels are quite close to each other.
The bond lengths differ by less than 0.03 �AA, exceptthat for the perp–perp transition state, where the
NO0 and OBr bond distances differ by 0.06 and 0.4�AA in the two methods, respectively. Also, the dif-ferences in bond angles are small, on the average
less than 2�, with one exception: in the cis–trans
conformer the MP2 and DFT values for the NO0Oangle differ by 4�.The N–O bond length in the NO2 molecule is
1.1944 �AA and the bond angle is predicted to be
134.2� at the B3LYP/6-311G(d) level of treatment,which is in excellent agreement with experimental
values [28] of 1.1934 �AA and 134.1�, respectively.When a comparison is made between NO2 and the
lowest energy structure of BrOO0NO we can see ashrinkage of the N–O bond to 1.133 �AA and an
elongation of the N–O0 bond to 1.684 �AA; the bondangle is distinctly compressed to 110�. Also, theO–Br bond (1.9861 �AA) is elongated compared toits value in the OBr radical, calculated as 1.764 �AAat same level of theory. The NO0O and O0OBr
bond angles are predicted to be 108� and 113�,respectively.The bond lengths for the trans-perp conformer
are changed by less than 0.08 �AA compared to the
Fig. 1. Conformers of bromine peroxynitrite as calculated by
the B3LYP method.
A. Lesar et al. / Chemical Physics Letters 368 (2003) 399–407 401
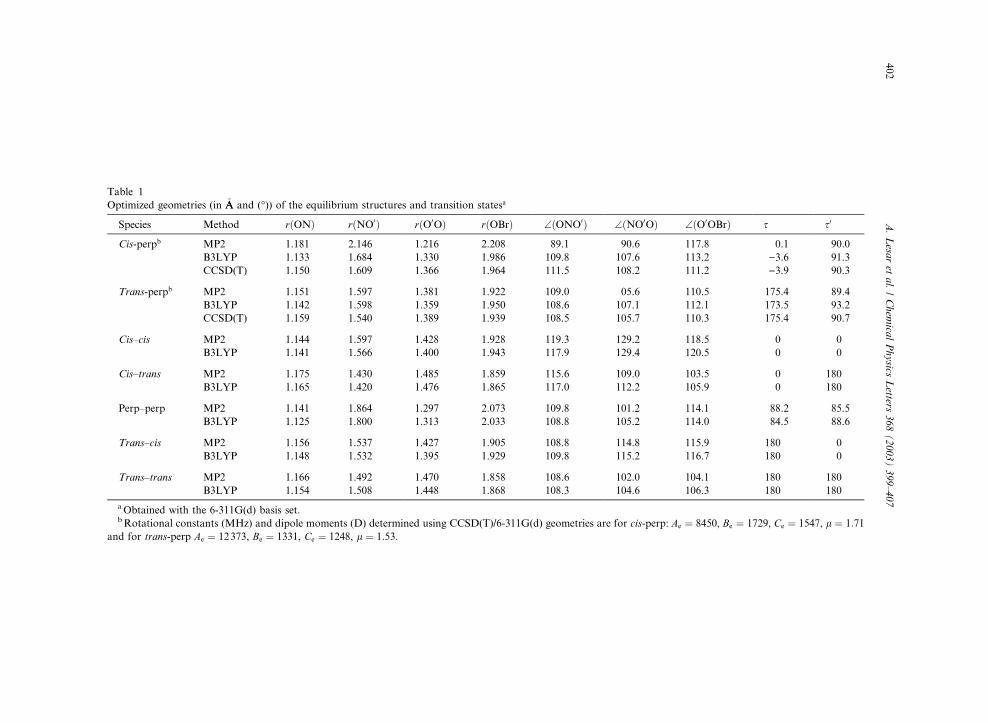
Table 1
Optimized geometries (in �AA and (�)) of the equilibrium structures and transition statesa
Species Method rðONÞ rðNO0Þ rðO0OÞ rðOBrÞ \ðONO0Þ \ðNO0OÞ \ðO0OBrÞ s s0
Cis-perpb MP2 1.181 2.146 1.216 2.208 89.1 90.6 117.8 0.1 90.0
B3LYP 1.133 1.684 1.330 1.986 109.8 107.6 113.2 )3.6 91.3
CCSD(T) 1.150 1.609 1.366 1.964 111.5 108.2 111.2 )3.9 90.3
Trans-perpb MP2 1.151 1.597 1.381 1.922 109.0 05.6 110.5 175.4 89.4
B3LYP 1.142 1.598 1.359 1.950 108.6 107.1 112.1 173.5 93.2
CCSD(T) 1.159 1.540 1.389 1.939 108.5 105.7 110.3 175.4 90.7
Cis–cis MP2 1.144 1.597 1.428 1.928 119.3 129.2 118.5 0 0
B3LYP 1.141 1.566 1.400 1.943 117.9 129.4 120.5 0 0
Cis–trans MP2 1.175 1.430 1.485 1.859 115.6 109.0 103.5 0 180
B3LYP 1.165 1.420 1.476 1.865 117.0 112.2 105.9 0 180
Perp–perp MP2 1.141 1.864 1.297 2.073 109.8 101.2 114.1 88.2 85.5
B3LYP 1.125 1.800 1.313 2.033 108.8 105.2 114.0 84.5 88.6
Trans–cis MP2 1.156 1.537 1.427 1.905 108.8 114.8 115.9 180 0
B3LYP 1.148 1.532 1.395 1.929 109.8 115.2 116.7 180 0
Trans–trans MP2 1.166 1.492 1.470 1.858 108.6 102.0 104.1 180 180
B3LYP 1.154 1.508 1.448 1.868 108.3 104.6 106.3 180 180
aObtained with the 6-311G(d) basis set.bRotational constants (MHz) and dipole moments (D) determined using CCSD(T)/6-311G(d) geometries are for cis-perp: Ae ¼ 8450, Be ¼ 1729, Ce ¼ 1547, l ¼ 1:71
and for trans-perp Ae ¼ 12373, Be ¼ 1331, Ce ¼ 1248, l ¼ 1:53.
402
A.Lesa
ret
al./Chem
icalPhysics
Letters
368(2003)399–407
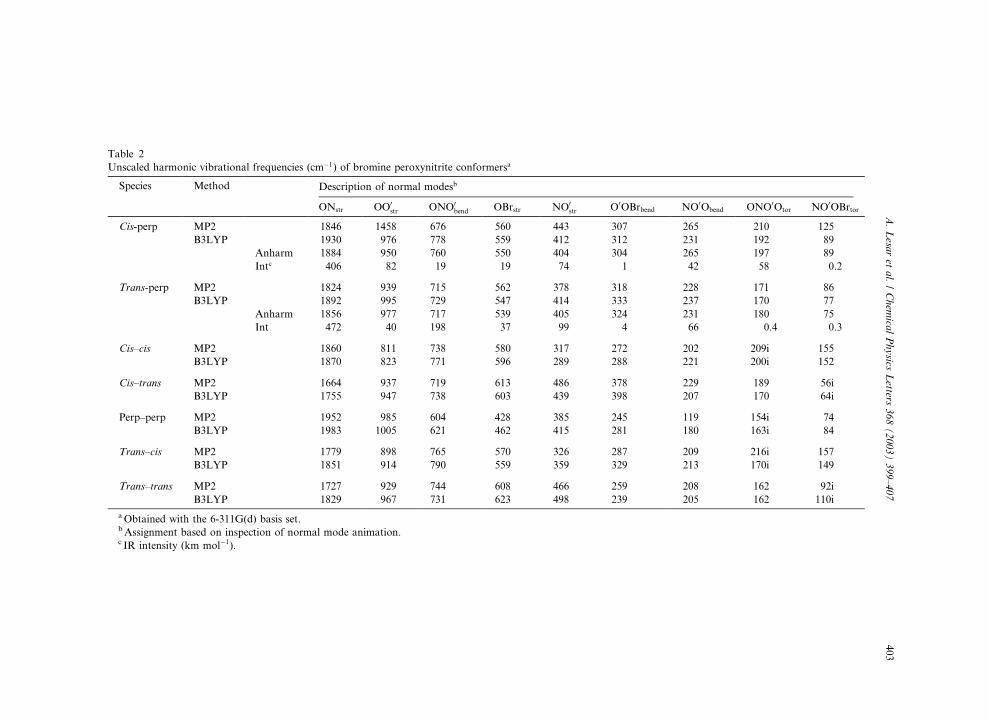
Table 2
Unscaled harmonic vibrational frequencies (cm�1) of bromine peroxynitrite conformersa
Species Method Description of normal modesb
ONstr OO0str ONO0
bend OBrstr NO0str O0OBrbend NO0Obend ONO0Otor NO0OBrtor
Cis-perp MP2 1846 1458 676 560 443 307 265 210 125
B3LYP 1930 976 778 559 412 312 231 192 89
Anharm 1884 950 760 550 404 304 265 197 89
Intc 406 82 19 19 74 1 42 58 0.2
Trans-perp MP2 1824 939 715 562 378 318 228 171 86
B3LYP 1892 995 729 547 414 333 237 170 77
Anharm 1856 977 717 539 405 324 231 180 75
Int 472 40 198 37 99 4 66 0.4 0.3
Cis–cis MP2 1860 811 738 580 317 272 202 209i 155
B3LYP 1870 823 771 596 289 288 221 200i 152
Cis–trans MP2 1664 937 719 613 486 378 229 189 56i
B3LYP 1755 947 738 603 439 398 207 170 64i
Perp–perp MP2 1952 985 604 428 385 245 119 154i 74
B3LYP 1983 1005 621 462 415 281 180 163i 84
Trans–cis MP2 1779 898 765 570 326 287 209 216i 157
B3LYP 1851 914 790 559 359 329 213 170i 149
Trans–trans MP2 1727 929 744 608 466 259 208 162 92i
B3LYP 1829 967 731 623 498 239 205 162 110i
aObtained with the 6-311G(d) basis set.bAssignment based on inspection of normal mode animation.c IR intensity (km mol�1).
A.Lesa
ret
al./Chem
icalPhysics
Letters
368(2003)399–407
403

cis-perp conformer. The main difference is related
to the ONO0O torsional angle being 174� for thetrans-perp conformer. From inspection of the
geometrical parameters of the maximum energy
conformeric forms listed in Table 1, one can see
that major structural changes occur only with re-spect to the two dihedral angles.
To facilitate the experimental identification of
BrOONO, rotational constants and dipole
moments for both minimum energy structures at
the CCSD(T)/6-311G(d) level of calculations are
included in the footnotes of Table 1.
The calculated harmonic frequencies of the two
stable BrOONO conformers are provided in Ta-ble 2 along with those of the corresponding
maximum energy conformeric forms. These fre-
quencies are needed to verify the true nature of
stationary points and to provide the ZPE. The
vibrational frequencies are calculated at the MP2
and B3LYP levels of theory with the 6-311G(d)
basis set. The agreement between the calculated
frequencies at the two levels is satisfactory, exceptfor that of the cis-perp conformer what can be
expected due to the difference in the MP2 geom-
etry. Thus, the MP2 frequencies of the cis-perp
structure are not reliable. While the cis-perp and
trans-perp frequencies in the B3LYP calculations
are very similar, any vibration can be used to
distinguish between them. But the remarkable
difference is related to the computed IR intensitiesof the ONO0 bending vibration and the ONO0O
torsion being substantially more intense for
the trans-perp and cis-perp conformers, respec-
tively. An inspection of normal mode suggests
that the reaction vector corresponding to the
imaginary frequency for the perp–perp transition
can be identified as the ONO0O torsional vibra-
tion rather than the NO0OBr torsion. This can beunderstood from the much heavier bromine mass
than that of oxygen. As expected, the eigenvec-
tors corresponding to the negative eigenvalue of
the force constant matrix for the cis–cis and cis–
trans maximum energy forms are similar, as are
those of the trans–cis and trans–trans maximum
energy, respectively. The former is mainly com-
posed of the ONO0O torsion and twisting while inthe latter eigenvector the twisting mode is less
pronounced.
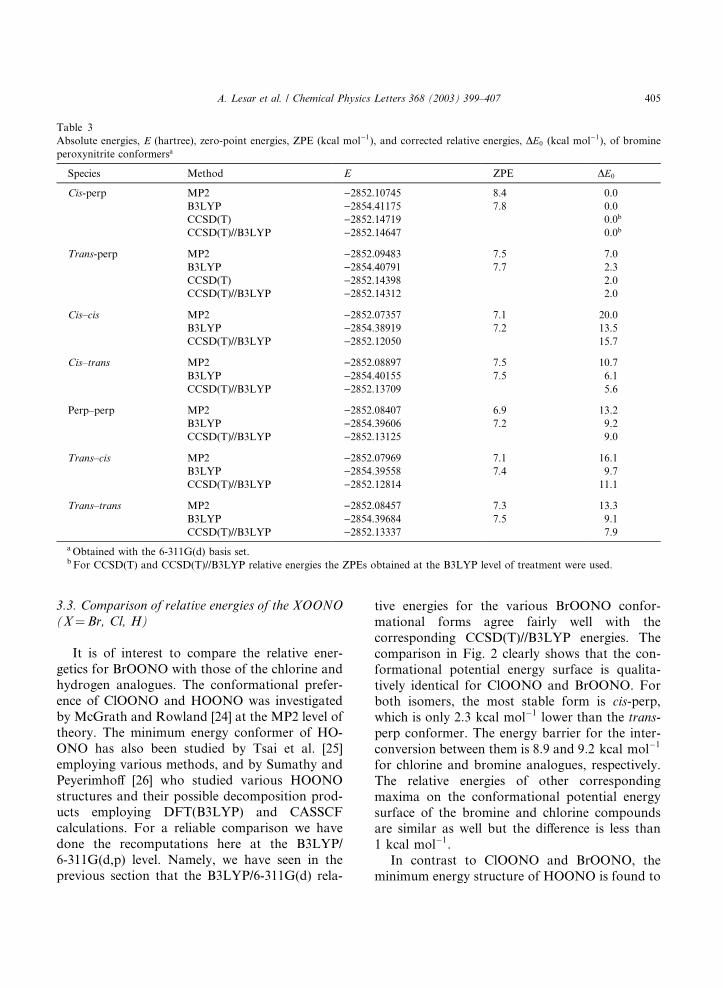
3.2. Relative energetics of BrOONO conformers
The total electronic energies, zero-point ener-
gies (ZPE), and relative energetics with regard to
the cis-perp conformer at the MP2 and B3LYPlevels of treatment, including the ZPE for minima
and maxima, are summarized in Table 3. Also,
results of the more elaborate CCSD(T) calcula-
tions are presented in the table to obtain the most
reliable energy ordering of the structures deter-
mined. For maxima, only CCSD(T) single-point
calculations at the MP2 and B3LYP geometries
were performed. We have seen above that theB3LYP structural parameters for the cis-perp
conformer are more consistent with CCSD(T)
values than MP2 values and thus it is not sur-
prising that the CCSD(T)//MP2 and CCSD(T)//
B3LYP energies differ by 5:4 kcal mol�1. For allother species this difference does not exceed
0:3 kcal mol�1. The CCSD(T)//MP2 energies arenot quoted in the table. Further, the difference inenergy between CCSD(T) and CCSD(T)//B3LYP
is only 0:5 kcal mol�1 for both the cis-perp and thetrans-perp conformers. We believe that the ener-
gies of maximum energy conformeric forms are
reasonably well predicted by the CCSD(T)//
B3LYP level of treatment.
As shown in Table 3, the MP2 and B3LYP
energies indicate and the CCSD(T) energy con-firms that the cis-perp is actually the lowest energy
conformer. It lies 2.3 and 2:0 kcal mol�1 at
B3LYP and CCSD(T)//B3LYP levels, respectively,
lower than the trans-perp conformer. The MP2
value of 7:0 kcal mol�1 is significantly higher. It isworth mentioning that the relative energies for
maximum energy conformeric forms obtained at
the B3LYP and CCSD(T)//B3LYP levels are infair agreement with each other.
In the following text we consider the results ob-
tained at the CCSD(T) level for maximum energy
conformeric forms. The energy barrier for rotation
around the N–O0 bond is 9:0 kcal mol�1. The OBrrotation around the peroxide bond in the cis-perp
conformer leads to barriers of 15:7 kcal mol�1 (cis–cis) and 5:6 kcal mol�1 (cis–trans). The corre-sponding barriers for rotation in the trans-perp
conformer were found to be 11:1 kcal mol�1
(trans–cis) and 7:9 kcal mol�1 (trans–trans).
404 A. Lesar et al. / Chemical Physics Letters 368 (2003) 399–407
3.3. Comparison of relative energies of the XOONO
(X¼Br, Cl, H)
It is of interest to compare the relative ener-
getics for BrOONO with those of the chlorine andhydrogen analogues. The conformational prefer-
ence of ClOONO and HOONO was investigated
by McGrath and Rowland [24] at the MP2 level of
theory. The minimum energy conformer of HO-
ONO has also been studied by Tsai et al. [25]
employing various methods, and by Sumathy and
Peyerimhoff [26] who studied various HOONO
structures and their possible decomposition prod-ucts employing DFT(B3LYP) and CASSCF
calculations. For a reliable comparison we have
done the recomputations here at the B3LYP/
6-311G(d,p) level. Namely, we have seen in the
previous section that the B3LYP/6-311G(d) rela-
tive energies for the various BrOONO confor-
mational forms agree fairly well with the
corresponding CCSD(T)//B3LYP energies. The
comparison in Fig. 2 clearly shows that the con-
formational potential energy surface is qualita-
tively identical for ClOONO and BrOONO. Forboth isomers, the most stable form is cis-perp,
which is only 2:3 kcal mol�1 lower than the trans-perp conformer. The energy barrier for the inter-
conversion between them is 8.9 and 9:2 kcal mol�1
for chlorine and bromine analogues, respectively.
The relative energies of other corresponding
maxima on the conformational potential energy
surface of the bromine and chlorine compoundsare similar as well but the difference is less than
1 kcal mol�1.
In contrast to ClOONO and BrOONO, the
minimum energy structure of HOONO is found to
Table 3
Absolute energies, E (hartree), zero-point energies, ZPE (kcal mol�1), and corrected relative energies, DE0 (kcal mol�1), of bromine
peroxynitrite conformersa
Species Method E ZPE DE0
Cis-perp MP2 )2852.10745 8.4 0.0
B3LYP )2854.41175 7.8 0.0
CCSD(T) )2852.14719 0.0b
CCSD(T)//B3LYP )2852.14647 0.0b
Trans-perp MP2 )2852.09483 7.5 7.0
B3LYP )2854.40791 7.7 2.3
CCSD(T) )2852.14398 2.0
CCSD(T)//B3LYP )2852.14312 2.0
Cis–cis MP2 )2852.07357 7.1 20.0
B3LYP )2854.38919 7.2 13.5
CCSD(T)//B3LYP )2852.12050 15.7
Cis–trans MP2 )2852.08897 7.5 10.7
B3LYP )2854.40155 7.5 6.1
CCSD(T)//B3LYP )2852.13709 5.6
Perp–perp MP2 )2852.08407 6.9 13.2
B3LYP )2854.39606 7.2 9.2
CCSD(T)//B3LYP )2852.13125 9.0
Trans–cis MP2 )2852.07969 7.1 16.1
B3LYP )2854.39558 7.4 9.7
CCSD(T)//B3LYP )2852.12814 11.1
Trans–trans MP2 )2852.08457 7.3 13.3
B3LYP )2854.39684 7.5 9.1
CCSD(T)//B3LYP )2852.13337 7.9
aObtained with the 6-311G(d) basis set.b For CCSD(T) and CCSD(T)//B3LYP relative energies the ZPEs obtained at the B3LYP level of treatment were used.
A. Lesar et al. / Chemical Physics Letters 368 (2003) 399–407 405

be the cis–cis conformer, with its cis-perp being
only 0:8 kcal mol�1 higher, followed by the trans-perp with 3:4 kcal mol�1 relative energy. The low-energy cis–cis structure in HOONO is presumablydue to an intramolecular hydrogen bond which is,
of course, not present in the halogen analogues.
The process of interconversion of the cis–cis to the
cis-perp conformer involves the cis-gauche struc-
ture as the maxima. At the B3LYP level this
structure is isoenergetic with the cis-perp con-
former. The energy barrier that corresponds to
rotation around the N–O0 bond amounts to15:1 kcal mol�1, which is significantly higher
compared to the halogen analogues. The rota-
tional barrier calculated in this work is in reason-
able accord with the value of 12:5 kcal mol�1
reported by Sumathy and Peyerimhoff [26]. On the
other hand, all the relative energies of the maxi-
mum energy structures for rotation around the
peroxide bond are significantly lower than thecorresponding barriers in ClOONO and BrO-
ONO.
Let us finally consider the relative stability of
the most stable XOONO isomeric form with the
corresponding XONO2. The BrONO2 is calcu-
lated to be 24.8 and 22:5 kcal mol�1 more stablethan BrOONO at the B3LYP/6-311G(d) and
CCSD(T)/6-311G(d) levels of theory, respectively.
These energy differences are nearly equal to those
found for the analogous chlorine compounds, be-
ing 24.1 and 22:3 kcal mol�1, respectively. At theB3LYP/6-311G(d,p) level of calculations, the
HONO2 is stabilized by 30:0 kcal mol�1, which is
in good agreement with previously reported val-ues: 30:9 kcal mol�1 at the B3LYP/6-311++G(d,p)level [26] and 29:5 kcal mol�1 at the CCSD(T)/CBS level [29].
4. Summary
The conformational potential energy surface ofthe BrOONO isomer of bromine nitrate was in-
vestigated by the MP2, B3LYP and CCSD(T) ab
initio electronic structure methods. The main re-
sults can be summarized as follows.
Two stable conformers of BrOONO were found
differing in their OO0NO torsional angle. The
lower-energy one is a nonplanar cis-perp con-
former, i.e., the Br atom is around 90� out of theplane formed by the other atoms. The other con-
former, trans-perp, having only 2:0 kcal mol�1
higher energy, results from approximately 180�rotation of the O0OBr group around the N–O0
bond. The maximum energy structure accompa-
nying this interconversion has a perp–perp con-
formation and the calculated energy barrier is
9:0 kcal mol�1. Rotation of the O–Br bond in thecis-perp conformer around the O0–O peroxide
bond leads to barriers of 15:7 kcal mol�1 (cis–cis)and 5:6 kcal mol�1 (cis–trans). The correspondingbarriers for rotation of the trans-perp conformer
were found to be 11:1 kcal mol�1 (trans–cis) and7:9 kcal mol�1 (trans–trans).Comparison of structures and relative energies
at the B3LYP/6-311G(d,p) level of treatmentclearly shows that the conformational potential
energy surface for ClOONO is qualitatively
identical to that of BrOONO. In both systems,
the most stable form is cis-perp which is calcu-
lated to be only 2:3 kcal mol�1 lower than thetrans-perp conformer in ClOONO. The energy
barrier for the interconversion between the two
stable structures and the relative energies of othercorresponding maxima on the conformational
potential energy surfaces are similar in BrOONO
Fig. 2. Relative energetics of bromine peroxynitrite (BrOONO)
conformers and comparison with chlorine peroxynitrite (ClO-
ONO) and peroxynitrous acid (HOONO), B3LYP/6-311G(d,p)
level of calculations (c, g, t and p are abbreviation for cis,
gauche, trans and perp, respectively).
406 A. Lesar et al. / Chemical Physics Letters 368 (2003) 399–407

and ClOONO as well, the difference being less
than 1 kcal mol�1. On the other hand, the mini-
mum energy structure of HOONO is the cis–cis
conformer, next is the cis-perp conformer being
only 0:8 kcal mol�1 higher, followed by the trans-perp conformer with 3:4 kcal mol�1 higher rela-tive energy. The lowest structure might be favored
by some intramolecular hydrogen bonding. The
process of interconversion of the cis–cis to the cis-
perp conformer involves the cis-gauche structure
proven to be the first-order saddle point. At the
B3LYP level of treatment this structure is isoen-
ergetic with the cis-perp conformer. The relative
energy of maximum energy structures for rotationaround the N–O0 bond is higher while those for
rotation around the O0–O bond are significantly
lower than the corresponding energies of ClO-
ONO and BrOONO.
Acknowledgements
This work was funded by the Ministry of Ed-
ucation, Science and Sport of Slovenia, Grant No.
P-544 and partly by the NATO collaborative
linkage Grant EST.CLG.977083. The authors
thank Prof. S.D. Peyerimhoff for valuable discus-
sion and critical reading of the manuscript.
References
[1] S.C. Wofsy, M.B. McElory, Y.L. Young, Geophys. Res.
Lett. 2 (1975) 215.
[2] J.E. Spencer, F.S. Rowland, J. Phys. Chem. 82 (1978)
7.
[3] S.P. Sander, G.W. Ray, R.T. Watson, J. Chem. Phys. 85
(1981) 199.
[4] F. Danis, F. Caralp, J. Masanet, R. Lesclaux, Chem. Phys.
Lett. 167 (1990) 450.
[5] R.P. Thorn, E.P. Daykin, P.H. Wine, Int. J. Chem. Kin. 25
(1993) 521.
[6] B. Casper, P. Lambotte, R. Minkowitz, H. Oberhammer, J.
Phys. Chem. 97 (1993) 9992.
[7] J.J. Orlando, G.S. Tyndall, J. Phys. Chem. 100 (1996)
19398.
[8] J.B. Burkholder, A.R. Ravishankara, S. Solomon, J.
Geophys. Res. 100 (1995) 16793.
[9] D.R. Hanson, A.R. Ravishankara, Geophys. Res. Lett. 22
(1995) 385.
[10] M.H. Harwood, J.B. Burkholder, A.R. Ravishankara, J.
Phys. Chem. A 102 (1998) 1309.
[11] S.A. Nizkorodov, P.O. Wennberg, J. Phys. Chem. A 106
(2002) 855.
[12] S. Parthiban, T.J. Lee, J. Chem. Phys. 109 (1998) 525.
[13] S. Parthiban, T.J. Lee, J. Chem. Phys. 113 (2000) 145.
[14] M.J. Frisch et al., GAUSSIANAUSSIAN 98, Revision A.7, Gaussian,
Inc, Pittsburgh, PA, 1998.
[15] W.J. Hehre, L. Radom, P.v.R. Schleyer, A.J. Pople, Ab
initio Molecular Orbital Theory, Wiley-Interscience, New
York, 1986.
[16] A.D. Becke, J. Chem. Phys. 98 (1993) 5648.
[17] C. Lee, W. Yang, W. Parr, Phys. Rev. B. 37 (1988) 785.
[18] B. Miehlich, A. Savin, H. Stoll, H. Preuss, Chem. Phys.
Lett. 157 (1989) 200.
[19] R.J. Bartlett, G.D. Purvis, Int. J. Quant. Chem. 14 (1978)
516.
[20] R.J. Bartlett, G.D. Purvis, J. Chem. Phys. 76 (1982) 1910.
[21] J.A. Pople, M.H. Gordon, K. Raghavachari, J. Chem.
Phys. 87 (1987) 5968.
[22] G.M. Chaban, J.O. Jung, R.B. Gerber, J. Chem. Phys. 111
(1999) 1823.
[23] M.W. Schmidt et al., J. Comput. Chem. 14 (1993) 1347.
[24] M.P. McGrath, F.S. Rowland, J. Phys. Chem. 98 (1994)
1061.
[25] H.-H. Tsai, T.P. Hamilton, J.-H.M. Tsai, M.v.d. Woerd,
J.G. Harrison, M.J. Jablonsky, J.S. Beckman, W.H.
Koppenol, J. Phys. Chem. 100 (1996) 15087.
[26] R. Sumathi, S.D. Peyerimhoff, J. Chem. Phys. 107 (1997)
1872.
[27] H.-H. Tsai, T.P. Hamilton, J.-H.M. Tsai, J.G. Harrison,
J.S. Beckman, J. Phys. Chem. 100 (1996) 6942.
[28] G. Herzberg, Molecular Spectra and Molecular Structure
III, Electronic Structure of Polyatomic Molecules, Van
Nostrand, New York, 1966.
[29] D.A. Dixon, D. Feller, C.-G. Zhan, J. Phys. Chem. A 106
(2002) 3191.
A. Lesar et al. / Chemical Physics Letters 368 (2003) 399–407 407