Embed Size (px)
Citation preview

Clemson University Clemson University
TigerPrints TigerPrints
All Theses Theses
December 2019
Connecting Polymeric Nanomedicine and Systems Biology: An Connecting Polymeric Nanomedicine and Systems Biology: An
Innovative Approach to Glioblastoma Treatment Innovative Approach to Glioblastoma Treatment
Jesse James Westfall Clemson University
Follow this and additional works at: https://tigerprints.clemson.edu/all_theses
Recommended Citation Recommended Citation Westfall, Jesse James, "Connecting Polymeric Nanomedicine and Systems Biology: An Innovative Approach to Glioblastoma Treatment" (2019). All Theses. 3197. https://tigerprints.clemson.edu/all_theses/3197
This Thesis is brought to you for free and open access by the Theses at TigerPrints. It has been accepted for inclusion in All Theses by an authorized administrator of TigerPrints. For more information, please contact [email protected].

CONNECTING POLYMERIC NANOMEDICINE AND SYSTEMS BIOLOGY: AN INNOVATIVE APPROACH TO GLIOBLASTOMA TREATMENT
A Thesis Presented to
the Graduate School of Clemson University
In Partial Fulfillment of the Requirements for the Degree
Master of Science Chemical and Biomolecular Engineering
by Jesse J. Westfall December 2019
Accepted by: Jessica Larsen, PhD, Committee Co-Chair
Marc Birtwistle, PhD, Committee Co-Chair Mark Blenner, PhD F. Alex Feltus, PhD

ii
ABSTRACT
Glioblastoma (GBM) remains a highly lethal brain tumor that continues have overall low survival
rate, with only 5% of patients to five or more years. This thesis proposes a drug- and polymersome-
loaded thermosensitive hydrogel as a therapeutic platform to target and eliminate post-surgical
GBM tumor cells. The experiments presented lay some foundational work in establishing the
feasibility of and optimizing such a platform. Polyethene glycol (PEG)-Polyester pH-responsive
polymersomes were synthesized, optimized and conjugated with peptide ligands to increase cellular
uptake in vitro. A Python workflow was designed, using RNAseq data from the Cancer Genome
Atlas (TCGA) and the Genotype-Tissue Expression project (GTEx) with online proteomic and
binding databases, to find GBM cellular surface targets and the ligands that bind them. A
biomimetic hydrogel, enhanced by chemotaxis signaling molecules, was synthesized as an
alternative tumor eliminating modality. The continuation of this work, merging modern
nanomedicine synthesis techniques and disease-specific data analysis promises to have positive
implications for GBM patient prognosis.

iii
ACKNOWLEDGMENTS
I would first like to thank my advisors, Dr. Jessica Larsen and Dr. Marc Birtwistle. We started our
adventure at Clemson University together and it has been a pleasure to have them both as role
models these last two years. They constantly pushed me to pursue the projects that I was passionate
about, mentoring me at every turn. They have helped me to become a better researcher,
communicator and person.
I would also like to thank my other committee members, Dr. Alex Feltus and Dr. Mark Blenner.
Dr. Feltus has been more than generous with his time and resources and helped me get my project
off the ground. Dr. Blenner has been a supportive role model to me since the first day we spoke.
Thank you to the following people in the Department of Chemical and Biomolecular Engineering
at Clemson University: Nick Gregorich, Dr. Eric Davis, Allison Yaguchi, Maxwell Hilbert, Bipin
Chakravarthy-Paruchuri, Deepraj Sarmah, Charles Wang, Dr. Cemal Erdem, Caitlin Anglin and
my Creative Inquiry team: Chad Eaton, Riley Rapert and Asia Paguntalan. Without their support,
both in and out of the laboratory, I would have never been able to write this text.
Thank you to the thousands of scientists and engineers that have built the foundation on which this
thesis rests, and that have dedicated their lives to the pursuit of helping other people. The results
shown here are in whole or part based upon data generated by the TCGA research network.
Finally, I would like to thank my parents, James and Renae Westfall. They have supported me at
every step of my career. They have given me unconditional love from the moment I drew breath
and have molded me into the man I am today. Better parents simply do not exist.

iv
TABLE OF CONTENTS Page
1 – INTRODUCTION .................................................................................................................. 1 Barriers to GBM Treatment ................................................................................................... 2 Local Delivery by Nanoparticle-Loaded Hydrogels ............................................................. 4 A Systems Biology Approach ............................................................................................... 6 Thesis and Overview ............................................................................................................. 7 2 – POLYMERSOMES FOR LOCAL DELIVERY IN GBM .................................................... 8 Introduction ........................................................................................................................... 8 Methods ................................................................................................................................. 9 Experimental Results and Discussion ................................................................................. 11 Conclusions and Future Outlook ......................................................................................... 17 3 – POLYMERSOME LIGAND ATTACHMENT FOR BIOLOGICAL FUNCTION ............ 20 Introduction ......................................................................................................................... 20 Methods ............................................................................................................................... 21 Experimental Results and Discussion ................................................................................. 23 Conclusions and Future Outlook ......................................................................................... 27 4 – TARGETING GBM USING mRNA Data ........................................................................... 29 Introduction ......................................................................................................................... 29 Methods ............................................................................................................................... 30 Experimental Results and Discussion ................................................................................. 32 Conclusions and Future Outlook ......................................................................................... 35 5 – THE CHEMOTAXIS HYDROGEL .................................................................................... 37 Introduction ......................................................................................................................... 37 Methods ............................................................................................................................... 38

v
TABLE OF CONTENTS (CONTINUED) Page
Experimental Results and Discussion ................................................................................. 40 Conclusions and Future Outlook ......................................................................................... 44 APPENDIX A – PYTHON CODES .......................................................................................... 46 REFERENCES ........................................................................................................................... 50
]

vi
LIST OF FIGURES
Page Figure
1.1 – Kaplan-Meier survival plots for glioblastoma multiforme cases according to year of diagnosis and age .......................................................................................................................... 1 1.2 – Nanoparticle loaded hydrogels ............................................................................................ 5 2.1 – Illustration of a cross section of a polymersome ................................................................. 8 2.2 – Size distribution (diameter) of rehydrated PEGPLA particles .......................................... 13 2.3 – Structure of PEG-based block copolymers ........................................................................ 15 2.4 – DLS measurements for PEGPCL polymersomes – solvent injection ............................... 15 2.5 – DLS measurements for PEGPCL polymersomes – emulsion evaporation method........... 16 3.1 – Illustrative examples of how polymersomes functionalized with targeting ligands bind to cellular surface proteins .............................................................................................................. 20 3.2 – Structure of maleimide-functionalized PEG-based block copolymers .............................. 25 3.3 – Size distribution (diameter) M-PEGPCL polymersomes .................................................. 25 3.4 – Fold change in mean fluorescence (FITC) per HEK293 cell treated with AF488- loaded CTAT-Functionalized and non-functionalized M-PEGPLGA polymersomes ........................... 27 4.1 – Illustrative view of workflow identifying candidate genes ............................................... 33 4.2 – Illustrative view of candidate gene/protein GEPIA verification and ligand selection ...... 34 5.1 – Illustration demonstrating that eukaryotic cells migrate along a gradient toward a higher concentration of chemoattractants .............................................................................................. 38 5.2 – Crosslinking of hydrogel by covalent bonding of maleimide functionalized 4-Arm PEG to sulfhydryl pendant group ............................................................................................................ 40 5.3 – Tissue stiffness for normal tissues ..................................................................................... 42 5.4 – Illustration of transwell invasion assay modified for chemotaxis hydrogel ...................... 43

vii
LIST OF TABLES Page
Table
1.1 – Summary of the Barriers to GBM Treatment ...................................................................... 3 2.1 – PEGPLA Polymersomes in 2%wt/v Mannitol – DLS Measurements............................... 12 2.2 – PEGPLA Polymersomes Before and After Lyophilization – DLS Measurements ........... 14 2.3 – Advantages and Disadvantages of Varying Copolymer, Lyoprotectant and Synthesis Method........................................................................................................................................ 18 3.1 – MPEGM - PEGPCL Polymersomes Before and After conjugation with CysTAT Peptide24 3.2 – M-PEGPLGA Polymersomes Before and After Conjugation with CysTAT Peptide .................................................................................................................................................... 26 5.1 – Chemotaxis Hydrogel Formulation and Young’s Modulus............................................... 41

1
1 – INTRODUCTION
After decades of research, glioblastoma (GBM) remains an aggressive, incurable and lethal brain
tumor with a median survival time of approximately 15 months1. Within two years, many patients
develop recurrent tumors in the periphery of the resection cavity, leading, in nearly every case, to
death2. GBM continues to have a low overall survival rate, with only 5% of patients living to five
or more years3,4. The current “gold standard” treatment for GBM is to surgically remove all
accessible tumor mass followed by a combination of radiotherapy and oral temozolomide (TMZ)
chemotherapy3,5–8. Treatment with TMZ only increases the median survival of patients by an
average of four months, as GBM cells rapidly become resistant to the cytotoxic drug9 (figure 1.1).
Figure 1.1 – Kaplan-Meier survival plots for glioblastoma multiforme cases according to year of diagnosis and age. (A) 20–44 years. (B) 45–64 years. (C) 65–79 years. (D) 80+ years. Note that
2005-2007 data is in the era of TMZ and reflects the slight increase in median survival time. Adapted from10.

2
Barriers to GBM Treatment
Treatment Gap
There is a time gap, generally 2-3 weeks, between surgical resection of the GBM tumor and when
oral TMZ and radiotherapy begins11–13. Despite recent advances in tumor surgery for GBM14,15,
even extensive tumor resections may leave behind invasive tumor cells, as they can evade detection
by radiological imaging2. A small population of tumor cells, free to migrate and proliferate for
weeks in a wound healing environment, could be responsible for seeding a recurrent tumor.
Blood Brain Barrier
The blood brain barrier, (BBB) selectively transports molecules and cells to and from the central
nervous system (CNS), and acts as a barrier to pathogens and harmful chemicals16,17. This barrier,
formed by endothelial cells that form tight junctions, prevents 98% of small molecule drugs from
crossing from plasma of blood into the interstitial or cerebrospinal fluid of the brain18,19. Generally,
only small lipophilic drugs can passively diffuse across the BBB20. This drastically lowers the
number of intravenous and oral therapeutic drug candidates available for treating GBM.
General Toxicity
Chemotherapeutics, when taken orally or intravenously, often have off-target toxicity in sensitive
systems, like the kidneys, lungs and heart21. This puts a limit on the drug dosages that doctors are
able to use to treat GBM tumors. TMZ has a particularly toxic effect on bone marrow and liver
during treatment of GBM22. In some cases, particularly among the elderly, TMZ actually shortens
the survival time of the patient due to adverse effects23. These drugs are not only acutely toxic to
sensitive systems, but can lead to long-term secondary malignancies and sterility24.

3
Temozolomide Resistance
Even, though TMZ is capable of crossing the BBB, at least half of all patients treated with TMZ
do not respond to the drug. Tumors that do respond quickly become resistant to the drug, in as few
as one dose25. The cytotoxic effect of alkylating drugs like TMZ is stopped by O6-methylguanine
DNA-methyltransferase (MGMT), a suicide repair protein25,26. Attempts to target MGMT with a
small molecule drug or inhibitor, to hinder TMZ resistance, have not been successful9.
GBM Tumor Cell Heterogeneity
GBM tumor cells are phenotypically heterogeneous. Within the same tumor, there can be several,
phenotypically diverse sub-populations of tumor cells27–29. Furthermore, there is evidence that
GBM tumor cells are plastic, and can shift phenotypes in response to environmental stimuli,
including surgery and chemotherapy30,31. The heterogeneous and dynamic nature of GBM tumor
cells makes them difficult to treat with a single drug and is the driving force behind combination
drug therapy research for GBM.
Table 1.1 – Summary of Barriers to GBM Treatment
Treatment Gap
• Radiotherapy and TMZ treatment 2-3 weeks after tumor resection.
• Unresected GBM tumor cells can migrate and proliferate, leading to recurrent tumors.
General Toxicity • Oral or intravenous chemotherapeutics lead to toxicity
complications across vital systems in the body • Off-target toxicity limits doses of cytotoxic drugs
Blood Brain Barrier (BBB) • Prevents more than 98% of small molecule drugs from reaching the brain, limiting
TMZ Resistance • GBM tumor cells rapidly become resistant to cytotoxic agents, even after a single dose
Tumor Cell Heterogeneity
• GBM cells are phenotypically diverse, even in the same tumor.
• Tumors are dynamic and plastic, able to change phenotype in response to environment

4
Local Delivery by Nanoparticle-Loaded Hydrogels
Local drug delivery, in this case, delivering a drug directly into the GBM tumor resection site, has
the potential to overcome several treatment barriers. The drug can be implanted during resection
surgery, eliminating the treatment gap. This would also circumvent complications associated with
the BBB, as the drug would not have to cross from the blood plasma to the CNS. Moreover, as the
drug would not be circulating through the body in the bloodstream, concerns about toxicity in
sensitive systems would be eliminated. The question remains, as to how to deliver the drug to the
tumor cells with a sufficient dosage for long enough to eliminate the tumor mass.
Two drug delivery devices that have been researched for decades are drug-carrying hydrogels and
polymeric nanoparticles. A full review of these systems is beyond the scope of this text. However,
this section will focus on what makes these drug delivery tools attractive for local treatment of
GBM tumors.
Hydrogels
Hydrogels are networks of hydrophilic polymers that remain insoluble due to crosslinking between
the polymer chains. These networks are capable of holding aqueous solutions of drugs and other
therapeutic molecules32. These hydrogels can be synthesized from polymers that can undergo
conformational change when affected by an internal (pH, temperature, redox) or external (enzyme,
magnetic, light) stimulus33,34. Conformational changes can facilitate either the gelation or
degradation of the hydrogel, and when implanted, can be used as a mechanism for releasing a drug
solution into surrounding tissues35,36. A recently popular class of stimuli-responsive hydrogels for
the local treatment of GBM is thermosensitive hydrogels37–40. These solutions remain liquid at room
temperature, but gelate at physiological temperatures, holding a drug solution in place. This is

5
attractive for GBM treatment because a drug-loaded hydrogel can be injected directly into the
tumor resection site during surgery, where the drug can treat the remaining tumor cells.
Polymeric Nanoparticles
Polymeric nanoparticles are nanoscale, highly-dispersed, solid structures, made from synthetic or
organic polymers41. They have a variety of different functional geometries and are capable of
encapsulating hydrophilic and/or hydrophobic drugs41–45. Like hydrogels, when synthesized with
stimuli-responsive polymers, nanoparticles can undergo conformational changes or degradation, in
response to a stimulus, as a mechanism for releasing drugs46–50. Recently, several researchers have
begun to employ drug-carrying nanoparticles to target and treat GBM tumors51–53.
Nanoparticle-Loaded Hydrogels
In 2010, Arai et al. combined nanoparticles a with thermosensitive hydrogel, demonstrating that
the coupling of their properties resulted in prolonged release and therapeutic effects of doxorubicin
in vivo54. Since then, researchers have been combining nanoparticle and hydrogel systems to create
nanoparticle-loaded hydrogels and nanohydrogels (hydrogels synthesized by crosslinking
nanoparticles) 37,40,55,56 (Figure 1.2).
Figure 1.2 – Nanoparticle loaded hydrogels. (a) nanoparticles entrapped in polymer network (b) nanoparticles with physical interactions in polymer matrix (c) nanoparticles as the crosslinkers for
polymer network (d) nanogel made from crosslinked nanoparticles. Adapted from 40.

6
Recently, for treatment in GBM, Bastinach et al. demonstrated that a Lauroyl-gemcitabine
nanoparticle-based hydrogel (GemC12-LNC) lowered the rate of GBM recurrence when surgically
implanted in U87 mouse models. GemC12-LNC was also tolerated in these mouse models for up
to six months, suggesting a very low systemic toxicity38.
Nanoparticle-loaded hydrogels are an attractive platform for the treatment of GBM, as the
combination of stimuli-responsive modalities can facilitate combination therapy and more control
over drug release dynamics.
A Systems Biology Approach
TMZ resistance and GBM tumor cell heterogeneity are not barriers to GBM treatment that can be
overcome by formulating a new nanoparticle or hydrogel. These complications are a consequence
of the still ambiguous disease biology. To solve these problems, a greater understanding of GBMs
underlying biological mechanisms is required. Fortunately, systems biologists have been able to
employ genetic, transcriptomic, proteomic and metabolomic data to classify and quantify GBM
tumor cells in a number of useful ways.
For TMZ resistance, a number of different researchers are analyzing data and building models to
quantify and combat the mechanisms underlying MGMT repair and understand the prognostic
value of MGMT promoter methylation9,26,57,58. To better understand GBM heterogeneity,
researchers are combining new surgical sampling techniques with genomic analysis to track the
evolution dynamics of tumor cell populations31. Other biologists are using single-cell
transcriptomic data and gene-expression based analysis in an effort classify distinct GBM subtypes
for future researchers27,59. In addition, Databases like the Cancer Genome Atlas (TCGA), Cancer
Cell Line Encyclopedia (CCLE) and International Cancer Genome Consortium (ICGC) provide

7
enormous, curated, cancer-specific datasets that help inform the fields of drug discovery and
pharmacology60.
The continuing growth of publicly available biological data and improved analysis methodologies
is crucial to understanding GBM. This analysis can uncover methods to effectively target and
eliminate GBM tumor cells, and is fundamental in informing material, drug and delivery modalities
for future drug delivery systems.
Thesis and Overview
Using stimuli-responsive polymers to create a drug- and nanoparticle-loaded hydrogel, we can
overcome the barriers to improving GBM treatment and improve patient survival. The hydrogel is
to be injected intracranially, filling the cavity of tumor resection, where it releases both a cytotoxic
drug for a short period (<48 hrs), and targeting, drug-loaded polymersomes for a longer period (~40
days). This mode of localized drug delivery erases the complications of crossing the BBB and
systemic toxicity, while simultaneously eliminating the treatment time gap and post-surgical
proliferation of GBM tumor cells. Polymersomes with functionalized surfaces, encapsulating
targeted cancer drugs, facilitate drug delivery and elimination of the diverse array of GBM cell
phenotypes within a patient’s residual tumor.
This text will lay the foundation for the described drug delivery platform for treating GBM.
Chapters 1 focuses on synthesizing and optimizing nanoparticle properties for local treatment of
GBM. Chapter 2 extends the functionality of these particles through the conjugation of surface
ligands, in an effort to increase cellular particle uptake. Chapter 3 introduces a workflow to
elucidate GBM cellular surface targets and the ligands that bind them from transcriptomic datasets
and proteomic databases. Chapter 4 explores a novel tumor elimination modality using biomimetic
hydrogels loaded with chemotaxis signaling molecules.

8
2 – POLYMERSOMES FOR LOCAL DELIVERY IN GBM
Introduction
Amphiphilic block copolymers self-assemble, under aqueous conditions, into a variety of
nanoparticle geometries. However, for polyethylene glycol (PEG) based copolymers, when the
mass of the hydrophilic block is between ~35% ± 10% they assemble into a hollow vesicle known
as a polymersome61. The hydrophobic blocks of the copolymer forms a hydrophobic bilayer
surrounding an aqueous core. This allows the nanoparticle to carry hydrophobic drugs in the
membrane interior and hydrophilic drugs in the core62 (Figure 2.1). Being able to control the
chemical properties and molecular weight of the polymers, polymersomes typically have thicker,
more chemically stable membranes than earlier drug nanoparticles with similar geometries, like
liposomes63.
Figure 2.1 – Illustration of a cross section of a polymersome. The hydrophobic membrane (red) encapsulates hydrophobic molecules while the aqueous core encapsulates water-soluble
compounds. Adapted from 62.
There are a variety of polymers that can undergo conformational change when affected by a
stimulus. When block copolymers are synthesized using these stimuli-responsive polymers, they

9
can assemble into polymersomes that degrade or undergo conformational change in response to
internal (pH, temperature, redox) and external (enzyme, magnetic, light) stimuli by degradation or
conformational change. These mechanisms can be exploited to control drug release from either
aqueous or hydrophobic compartments of the nanoparticles46–49. There has been research
performed using these stimuli-responsive polymersomes for the treatment of GBM53. Using
internal stimuli, Jiang et al. engineered redox-responsive polymersomes to effectively deliver
saporin, a protein toxin therapeutic, in GBM mouse models64. In contrast, Luo et al. used high-
intensity focused ultrasound, an external stimuli, to trigger release and effective delivery of
doxorubicin and perfluorooctyl bromide in a U87 mouse model65.
PH-triggered drug release is attractive mechanism for cancer treatment, due to the acidity of both
the tumor microenvironment and tumor cell endosomes46. Polymersomes assembled from
copolymers with blocks degradable by acid hydrolysis, like polyesters, can be designed to degrade
at different rates in acidic conditions66. It has also been shown that nanoparticles with PEG on their
surface avoid uptake by phagocytic cells as part of the reticuloendothelial system in vivo67. This is
attractive for long-term local therapy of GBM, as microglia are phagocytes that digest particles
within the CNS68.
Polymersomes synthesized from PEG-polyester block copolymers can benefit from both controlled
pH-responsive controllable release and evasion of microglia in the brain. This chapter explores
material and synthesis design choices to for engineering polymersomes optimized for local delivery
to GBM tumor cells.
Methods
Solvent Injection Method for Synthesis of Polymersomes – 1 mg diblock copolymer methoxy
polyethylene glycol polylactide (1kDa-5kDa, Polysciences Inc #24381-1) polyethylene glycol

10
polycaprolactone (2kDA-5kDA) (Aldrich #900648) (PEGPCL) was dissolved in 100 μL miscible
organic solvent dimethyl sulfoxide (VWR BDH Chemicals #BDH1115-1LD) (DMSO), N-methyl-
2-pyrrolidone (VWR Life Science #89500-566) (NMP) or N,N-dimethylformamide (BDH
Chemicals #BDH83634.100) (DMF). In a 4 dram threaded vial, the solution was injected via 0.5
mL syringe (BD #305602) by nano-syringe pump (KD Scientific) into 10 mL continuously stirring
2wt%/v mannitol (BDH VWR Analytical #BDH9248) or inulin (Alfa Aesar #A18425) in water at
a constant rate of 5 μL/min.
Emulsion Evaporation Method for Synthesis of Polymersomes – 1 mg diblock copolymer
methoxy polyethylene glycol polylactide (1kDa-5kDa, Polysciences Inc #24381-1) (PEGPLA) or
methoxy polyethylene glycol polycaprolactone (2kDA-5kDA) (Aldrich #900648) (PEGPCL) was
dissolved in 1 mL immiscible solvent dichloromethane (VWR BDH Chemicals #BDH23373.100E)
(DCM). The solvent solution was added to 9 mL 0.5 wt%/v Pluronic F-68 (Gibco #24040-032) in
water in an 11 dram threaded vial. The mixture was emulsified using a homogenizer (Thermo
Fisher), three cycles of 5 sec on/5 sec off at 50% power. The vial containing the emulsion was
connected to a Rotavapor R-100 (Buchi) via 24/40 vial adapter (Chemglass #CG-1318-40) and
rotated under vacuum until bubbling stopped and solution was clear.
Lyophilization and Characterization of Polymersomes – Polymersome solution was frozen
slowly, first to -20°C for 6 hours, then to -80°C overnight, in a 50 mL conical tube. The solution
was then then lyophilized at 0.04 mbar and -105°C by FreeZone lyophilizer (LABCONCO).
Polymersome diameters and zeta potential were measured by dynamic light scattering (DLS) via
Zetasizer Nano ZS (Malvern).
Loading of Polymersomes – Lyophilized polymersomes were rehydrated in 1 mL aqueous
solution (0.16 mg/mL in water) of Alexa Fluor 488 carboxylic acid tris(triethylammonium) salt

11
(Invitrogen #A33077) (AF488). Unencapsulated AF488 was separated from loaded polymersomes
by centrifugal filter device (Amicon 0.5 mL 100K #UFC5100BK), spinning three times at 14000g
for 10 minutes in a 5424R centrifuge (Eppendorf). Encapsulation efficiency (EE) was determined
with the following equation:
𝐸𝐸𝐸𝐸(%) = 100%− (𝑇𝑇𝑇𝑇𝑇𝑇𝑇𝑇𝑇𝑇 𝑚𝑚𝑇𝑇𝑚𝑚𝑚𝑚 (𝑚𝑚𝑚𝑚) 𝑇𝑇𝑜𝑜 𝐴𝐴𝐴𝐴488 𝑖𝑖𝑖𝑖 𝑜𝑜𝑖𝑖𝑇𝑇𝑇𝑇𝑓𝑓𝑇𝑇𝑇𝑇𝑓𝑓𝑚𝑚
0.16 𝑚𝑚𝑚𝑚 × 100%)
Mass of AF488 in filtrates was calculated using a linear calibration curve (relative fluorescence
units (RFU) vs. AF488 concentration). Fluorescence measurements were made with Synergy H1
microplate reader (BioTek).
Experimental Results and Discussion
The goal of these experiments is to optimize polymersome properties for local delivery in GBM.
The ideal polymersome will have a diameter between 10-200 nm, to prevent rapid clearance by the
renal system and opsonization by phagocytes in the reticuloendothelial system33, be monodisperse,
and be capable of carrying both hydrophobic and/or hydrophilic molecules for delivery. The
polymersomes should also have a significant negative zeta potential, a proxy for nanoparticle
surface charge, and indicator of colloidal stability in solution69. It is also important that the
polymersomes are capable of releasing these molecules in a pH-responsive manner for delivery in
either the acidic GBM tumor microenvironment or acidic intracellular vesicles. Another important
metric to be considered is whether the polymersomes can be lyophilized for long term storage and
rehydrated for future use. These experiments focus on choosing copolymers, synthesis methods
and lyoprotectants to synthesize polymersomes ideal for GBM delivery.

12
PEGPLA polymersomes in 2wt%/v mannitol in water – The solvent injection method employed
for synthesizing polymersomes is adapted from Kelly et al70. Table 2.1 shows the size,
polydispersity and zeta potential of the resulting particles.
Table 2.1 – PEGPLA Polymersomes in 2%wt/v Mannitol – DLS Measurements
Diameter (nm) PDI Zeta Potential (mV)
149.00 ± 13.63 0.088 ± 0.022 -30.56 ± 0.91
These polymersomes are in the desired size range, are monodisperse (polydispersity index (PDI) <
0.3)71 and have a large negative zeta potential. Also, the polylactic acid block comprising the
hydrophobic membrane is pH-responsive by acid hydrolysis. These characteristics make these
polymersomes a good candidate for GBM local delivery, assuming they can be loaded with
therapeutic molecules and lyophilized for long term storage.
Lyophilization and loading PEGPLA polymersomes – PEGPLA polymersomes were synthesized
via solvent injection using 2% mannitol and rehydrated by 0.16 mg/mL AF488 dye solution. AF488
is a hydrophilic molecule and is encapsulated in the aqueous core of the polymersomes. Dye
encapsulation efficiency of the polymersomes is shown to be as high as 38.6%, indicating that these
particles are promising carrier for soluble therapeutic molecules. However, DLS measurements
indicate that the rehydrated polymersomes are not monodisperse, likely destabilizing and forming
aggregates (Figure 2.2 (top)). Particles were synthesized in mannitol solutions up to 10%, with
similar results.

13
Figure 2.2 – Size distribution (Diameter) of rehydrated PEGPLA particles (top) in 2%wt/v mannitol in water(bottom) in 2%wt/v inulin in water.
PEGPLA polymersomes were re-synthesized via solvent injection, using 2wt%/v inulin in water as
the lyoprotectant. It has been shown that inulin, a fructose polymer, is effective at lyoprotection of
PEG-based polymersomes72,73. The particles are of similar size and dispersity as the mannitol-
protected particles. When these polymersomes are lyophilized and rehydrated using a 0.16 mg/mL
AF488 dye solution, they maintain their membrane stability and monodisperse nature (Figure 2.2
(bottom)), albeit with a slight increase in diameter. However, these polymersomes show a lower
capacity for soluble molecule encapsulation, having an AF488 encapsulation efficiency of 25.4%
± 2.2%. This is likely due to the larger size of inulin, making it less likely to leave the membrane

14
during rehydration.. Table 2.2 shows the results of DLS measurements on these PEGPLA
polymersomes.
Table 2.2 – PEGPLA Polymersomes Before and After Lyophilization – DLS Measurements
Before lyophilization Rehydrated after Lyophilization Lyoprotectant Diameter (nm) PDI Diameter (nm) PDI 2% mannitol 167.41 ± 25.29 0.108 ± 0.055 100.48 ± 42.01 0.611 ± 0.350 2% inulin 122.14 ± 7.74 0.119 ± 0.023 158.22 ± 6.13 0.175 ± 0.033
Another consideration when choosing a lyoprotectant is the characteristics of the solid product.
Polymersomes lyophilized in mannitol form a powdery substance, easy to store and transfer
between containers. Polymersomes lyophilized in inulin form a slow-flowing, translucent
amorphous, solid, which at room temperature can be difficult to handle and transfer.
While PEGPLA polymersomes are great candidates for pH-responsive delivery of drugs
intravenously, they may be less stable when subjected to extended time frames in the slightly acidic
pH conditions of the GBM tumor microenvironment. In this environment, the polymersome
membrane may prematurely destabilize and release a drug before it is able to reach the GBM tumor
cell. Thus, copolymer blocks made with polycaprolactone (PCL) are attractive as the polymer is
more hydrophobic than PLA. There is a longer carbon chain in each monomer, leading to stronger
hydrophobic interactions in an aqueous environment. There are also less ester groups, sites for acid
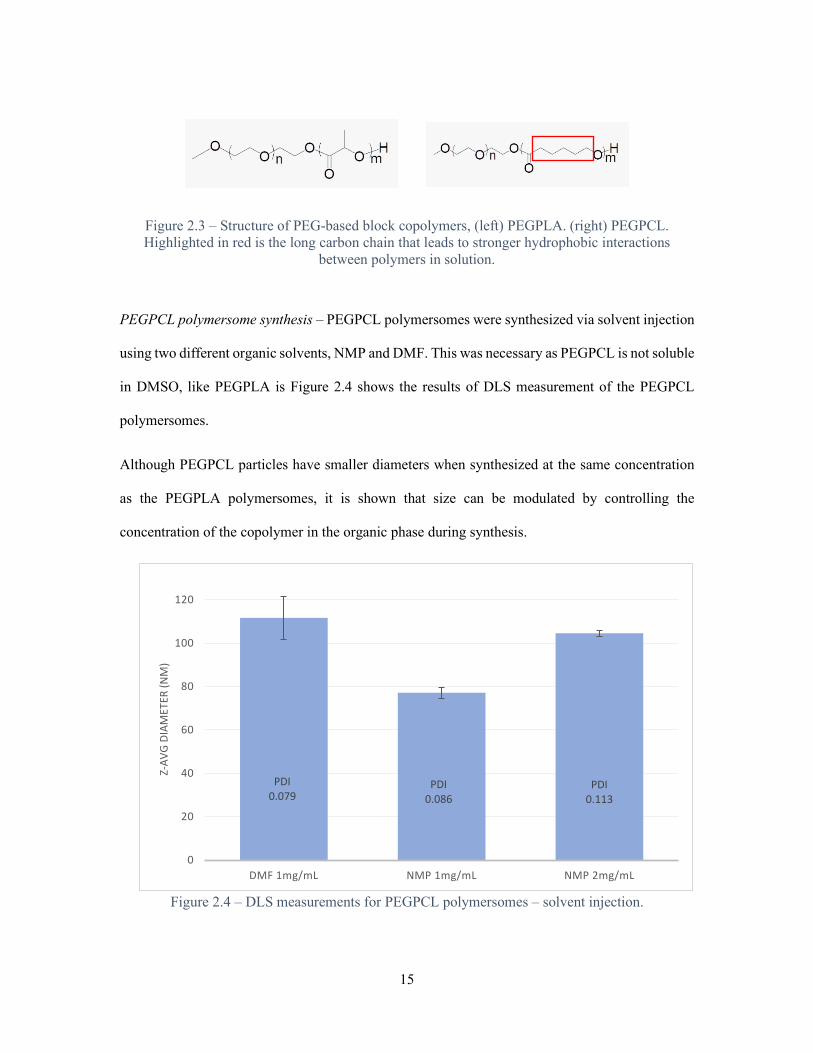
hydrolysis, per block (Figure 2.3). These characteristics should lead to stronger membrane
interactions, and longer stability in the acidic tumor microenvironment, while maintaining a pH-
responsive drug release mechanism.
15
Figure 2.3 – Structure of PEG-based block copolymers, (left) PEGPLA. (right) PEGPCL. Highlighted in red is the long carbon chain that leads to stronger hydrophobic interactions
between polymers in solution.
PEGPCL polymersome synthesis – PEGPCL polymersomes were synthesized via solvent injection
using two different organic solvents, NMP and DMF. This was necessary as PEGPCL is not soluble
in DMSO, like PEGPLA is Figure 2.4 shows the results of DLS measurement of the PEGPCL
polymersomes.
Although PEGPCL particles have smaller diameters when synthesized at the same concentration
as the PEGPLA polymersomes, it is shown that size can be modulated by controlling the
concentration of the copolymer in the organic phase during synthesis.
Figure 2.4 – DLS measurements for PEGPCL polymersomes – solvent injection.
PDI0.079
PDI0.086
PDI0.113
0
20
40
60
80
100
120
DMF 1mg/mL NMP 1mg/mL NMP 2mg/mL
Z-AV
G DI
AMET
ER (N
M)

16
While particles synthesized via solvent injection effectively encapsulate soluble compounds in their
aqueous core, there are GBM therapeutics that have very low solubility in water. There is another
mode of synthesis, single emulsion evaporation (EE) that simultaneously encapsulate hydrophobic
molecules with high efficiency74.
Emulsion evaporation synthesis of PEGPCL polymersomes – PEGPCL polymersomes were
prepared via EE method, adapted from Khalil et al75, with varying concentrations (0.7, 1, 1.3 mg
PEGPCL/1 mL DCM). Figure 2.5 shows the DLS measurements for the PEGPCL polymersomes.
Figure 2.5 – DLS measurements for PEGPCL polymersomes – emulsion evaporation method.
At 1 and 0.7 mg PEGPCL/1 mL DCM and lower concentrations, the polymersomes were desirable
size and monodisperse. At 1.3 mg PEGPCL/1 mL DCM, however, polymersome solutions became
polydisperse. More curious is that there is a negative correlation between polymer concentration
and average diameter. This is in contrast to the positive correlation between polymer concentration
and diameter for solvent injection method discussed earlier. This suggests that polymer
PDI0.299
PDI0.267 PDI
0.426
0
50
100
150
200
0.7 1 1.3
Z-AV
G DI
AMET
ER (N
M)
WT PEG-PCL/VOL DCM (MG/ML)

17
concentration may not be the driving force in the polymersome self-assembly process when using
the EE method. Although literature suggests that size modulation can be achieved through process
design (homogenization, surfactant content, solvents etc.)74,76,77.
Conclusions and Future Outlook
This chapter illustrates the types of material design choices a polymeric nanomedicine researcher
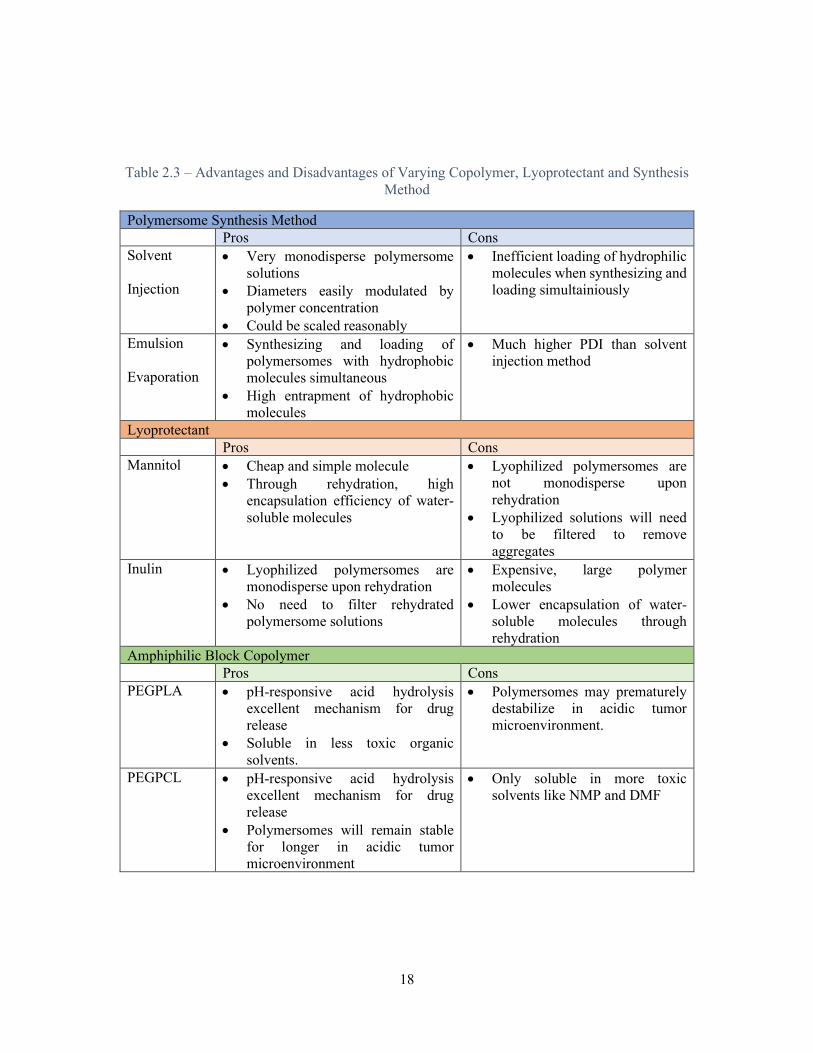
must make when designing drug delivery systems. Table 2.3 summarizes the material, lyoprotectant
and synthesis choices explored in this work.
18
Table 2.3 – Advantages and Disadvantages of Varying Copolymer, Lyoprotectant and Synthesis Method
Polymersome Synthesis Method Pros Cons Solvent
Injection
• Very monodisperse polymersome solutions
• Diameters easily modulated by polymer concentration
• Could be scaled reasonably
• Inefficient loading of hydrophilic molecules when synthesizing and loading simultainiously
Emulsion
Evaporation
• Synthesizing and loading of polymersomes with hydrophobic molecules simultaneous
• High entrapment of hydrophobic molecules
• Much higher PDI than solvent injection method
Lyoprotectant Pros Cons Mannitol • Cheap and simple molecule
• Through rehydration, high encapsulation efficiency of water-soluble molecules
• Lyophilized polymersomes are not monodisperse upon rehydration
• Lyophilized solutions will need to be filtered to remove aggregates
Inulin • Lyophilized polymersomes are monodisperse upon rehydration
• No need to filter rehydrated polymersome solutions
• Expensive, large polymer molecules
• Lower encapsulation of water-soluble molecules through rehydration
Amphiphilic Block Copolymer Pros Cons PEGPLA • pH-responsive acid hydrolysis
excellent mechanism for drug release
• Soluble in less toxic organic solvents.
• Polymersomes may prematurely destabilize in acidic tumor microenvironment.
PEGPCL • pH-responsive acid hydrolysis excellent mechanism for drug release
• Polymersomes will remain stable for longer in acidic tumor microenvironment
• Only soluble in more toxic solvents like NMP and DMF

19
Looking forward, for a local delivery system for the treatment of GBM, PEGPCL polymersomes
are the best nanoparticle on which to build a platform. It has been shown that they can be prepared
by different methods for the loading of both hydrophilic and hydrophobic therapeutic molecules.
These polymersomes can also be loaded and lyophilized, with inulin as a lyoprotectant, for long-
term storage. For local delivery in GBM, PEGPCL polymersome solutions can be used to solubilize
thermosensitive polymers to create polymersome-loaded, temperature-responsive hydrogels. These
hydrogels can be injected into GBM tumor resection sites for extended local treatment of remaining
tumor cells. Furthermore, these polymersomes can be functionalized with different ligands to
enhance biological function, including cell surface targeting and cellular uptake, which will be
explored in the next chapter.

20
3 – POLYMERSOME LIGAND ATTACHMENT FOR BIOLOGICAL FUNCTION
Introduction
Recent research have shown that using ligands to target membrane surface targets can lead to
increased biological function, like initiating receptor-mediated endocytosis and crossing the blood
brain barrier (BBB)78. For example, it was demonstrated that functionalizing nanoparticles with
ApoE, a lipoprotein, will increase the transport of those particles across the blood brain barrier via
interaction with low-density lipoprotein receptors79, which are overexpressed on BBB interfacial
cells17,18. The focus of this thesis, local delivery to GBM, is to bypass the BBB entirely, but we can
use the same concept of active targeting to increase effective drug delivery to GBM cells. In
particular, polymersomes are effective vessels for getting drugs through the membranes of cells
when their surfaces are functionalized with ligands, proteins, antibodies and peptides78 (Figure 3.1).
These functional molecules promote cell adhesion and receptor mediated endocytosis, increasing
the rate at which drugs are internalized by a cancer tumor cell44,51,78,80.
Figure 3.1 – Illustrative examples of how polymersomes functionalized with targeting ligands bind to cellular surface proteins. Adapted from 78.

21
Using active targeting ligands is an attractive modality for the local treatment of GBM. There are
cellular surface proteins, overexpressed in GBM, capable of being targeted (this be explored more
thoroughly in the next chapter). The binding affinities of the ligand to those proteins will facilitate
the preferential delivery of chemotherapeutics to GBM tumor cells.
This chapter will focus on demonstrating that, by functionalizing the surface of polymersomes
discussed in Chapter 2, cellular uptake of these polymersomes can be significantly increased in
vitro. This will provide a platform for synthesizing polymersomes functionalized with active
targeting ligands, and how to quantify their drug delivery efficiency in vitro.
Methods
Embedding Method for Synthesizing Maleimide-Functionalized Polymersomes – 1.5 mg
diblock copolymer methoxy polyethylene glycol polycaprolactone (2kDA-5kDA) (Aldrich
#900648) (PEGPCL) and varying masses (5,10,40 mg) of homobifunctional polyethylene
glycol(3500kDa)-(maleimide)2 (JENKEM Technology USA #A4010-1) (MPEGM) were dissolved
in 100 μL N,N-dimethylformamide (BDH Chemicals #BDH83634.100) (DMF). In a 2 dram
threaded vial, the solvent solution was injected via 0.5 mL syringe (BD #305602) by nano-syringe
pump (KD Scientific) into 1.4 mL continuously stirring Alexa Fluor 488 carboxylic acid
tris(triethylammonium) salt (Invitrogen #A33077) (AF488) (0.114 mg/mL) in water at a constant
rate of 5 μL/min. Unencapsulated AF488 and non-embedded MPEGM were separated from loaded
polymersomes by centrifugal filter device (Amicon 0.5 mL 100K #UFC5100BK), spinning three
times at 14000g for 10 minutes in a 5424R centrifuge (Eppendorf).
Synthesizing Maleimide-Functionalized Polymersomes with M-PEGPLGA/PCL – 1 mg
Poly(ε-caprolactone)-PEG-maleimide (2kDa-5kDa) (Nanosoft Polymers #2667) (M-PEGPCL), or
poly(lactide-co-glycolide)-PEG-maleimide (2kDa-5kDa) (M-PEGPLGA) (Nanosoft Polymers

22
#2794) was dissolved in N,N-dimethylformamide (BDH Chemicals #BDH83634.100) (DMF). In
a 2 dram threaded vial, the solvent solution was injected via 0.5 mL syringe (BD #305602) by nano-
syringe pump (KD Scientific) into 1.4 mL continuously stirring Alexa Fluor 488 carboxylic acid
tris(triethylammonium) salt (Invitrogen #A33077) (AF488) (0.114 mg/mL) in water at a constant
rate of 5 μL/min. Unencapsulated AF488 was separated from loaded polymersomes by centrifugal
filter device (Amicon 0.5 mL 100K #UFC5100BK), spinning three times at 14000g for 10 minutes
in a 5424R centrifuge (Eppendorf).
Encapsulation Efficiency and Polymersome Characterization – AF488 Encapsulation
efficiency (EE) was determined with the following equation:
𝐸𝐸𝐸𝐸(%) = 100%− (𝑇𝑇𝑇𝑇𝑇𝑇𝑇𝑇𝑇𝑇 𝑚𝑚𝑇𝑇𝑚𝑚𝑚𝑚 (𝑚𝑚𝑚𝑚) 𝑇𝑇𝑜𝑜 𝐴𝐴𝐴𝐴488 𝑖𝑖𝑖𝑖 𝑜𝑜𝑖𝑖𝑇𝑇𝑇𝑇𝑓𝑓𝑇𝑇𝑇𝑇𝑓𝑓𝑚𝑚
0.16 𝑚𝑚𝑚𝑚 × 100%)
Mass of AF488 in filtrates was calculated using a linear calibration curve (relative fluorescence
units (RFU) vs. AF488 concentration). Fluorescence measurements were made with Synergy H1
microplate reader (BioTek). Polymersome diameters, polydispersity index (PDI) and zeta potential
were measured via Zetasizer Nano ZS (Malvern).
Conjugation of CysTAT ligand to Polymersomes – 0.25 mg CysTAT(47-57) (Genscript
#RP20343) (amino acid sequence: GRKKRRQRRRPQ) (CTAT) was added to aqueous solution of
0.5 mg loaded, maleimide-functionalized polymersomes in water. Mixture was stirred overnight
(~18 hours). Non-conjugated CTAT was separated from polymersomes by centrifugal filter device
(Amicon 0.5 mL 100K #UFC5100BK), spinning three times at 14000g for 10 minutes in a 5424R
centrifuge (Eppendorf).
HEK293 Cell Culture and Treatment – HEK293 cells (ATCC #CRL-1573, verified by short
tandem repeat profiling) were seeded in a 12-well plate, at 0.1 x 106 cells per well and incubated

23
for 24 hours at 37 °C and 5% CO2 in Delbecco’s modified eagle medium (Gibco #10313-021)
(+10% FBS). At ~50% confluence, the cells were treated with 0.5 mg loaded M-PEGPLGA
polymersomes in Delbecco’s phosphate buffered solution (Alfa Aesar #J61917) (PBS), 0.5 mg non-
loaded M-PEGPLGA polymersomes in PBS and 1 μL (2 mg/mL) AF488 in milli-Q water. Cells
were allowed to incubate for 3.5 hours at 37 °C and 5% CO2.
Flow Cytometry to Determine Polymersome Uptake – HEK293 cells removed from 12-well
plate surface by cell scraper and 1 mL of cell suspension per sample was collected. Flow cytometry
was performed to determine the mean cell FITC-A signal for each sample using a CytoFLEX LX
(Beckman Coulter).
Results and Discussion
This chapter’s series of experiments is designed to show that by conjugating ligands to our
polymersome drug carriers, we can introduce biological function, such as cell surface targeting and
enhanced cellular uptake, beneficial to drug delivery. It this case, transactivator of transcription
(TAT) of HIV will be the ligand and will act as a positively charged, cell-penetrating peptide (CPP)
to increase cellular uptake of polymersomes.
Embedding MPEGM in polymersome membrane – In an effort to locate maleimide functional
groups to the surface of PEGPCL polymersomes, Varying masses of homobifunctional PEG
polymers (MPEGM) were dissolved with 1.5 mg PEGPCL in the organic solvent. Polymersomes
are then synthesized using solvent injection and stirred overnight with 0.25 mg cysteine-terminal
TAT (CTAT). A thiol-maleimide conjugation reaction covalently bonds the CTAT to the PEG
polymer embedded in the polymersome membrane. Table 3.1 shows the polymersome
characteristics, measured by DLS both before and after conjugation with CysTAT peptide.

24
Table 3.1 – MPEGM - PEGPCL Polymersomes Before and After conjugation with CysTAT Peptide
mg MPEGM per 1.5 mg PEGPCL
CTAT Diameter (nm) PDI Zeta potential (mV)
5mg - 92.71 0.118 -31.4 + 88.87 0.081 -20.9
10mg - 92.33 0.103 - + 86.89 0.090 -20.3
40mg - 88.94 0.117 -17.8 + 105.9 0.136 -10.0
It would be expected, that if there was an appreciable amount of positively charged CysTAT peptide
on the surface of the polymersomes, the zeta potential, a proxy for surface charge, would be
positive, or at least have a large positive shift. However, after CysTAT conjugation, the
polymersomes still show a significant negative zeta potential. This underwhelming result could be
a consequence of either the lack of embedded homobifunctional PEG polymers. There may not be
sufficient driving force for much the hydrophilic MPEGM to become embedded in the
polymersome membrane. Instead the MPEGM would be free in solution and subsequently washed
away by the centrifugal device. Without enough embedded MPEGM, there are not enough
maleimides on the surface of the particles for the thiols of the CTAT to bind to.
To eliminate complications derived from incomplete or heterogeneous embedding of MPEGM in
the polymersome membrane, polymersomes were synthesized from PEG-based block copolymers
in which a maleimide functional group was already covalently bonded to the terminal end of the
PEG. For this purpose, maleimide functionalized PEG-polycaprolactone (M-PEGPCL) and PEG-
poly(lactide-co-glycolide) (M-PEGPLGA) were purchased (figure 3.2).

25
Figure 3.2 – Structure of Maleimide-Functionalized PEG-based Block Copolymers, (top) M-PEGPCL. (bottom) M-PEGPLGA.
Using solvent injection method, M-PEGPCL polymersomes are synthesized and characterized
using DLS. These polymersomes have surprisingly small diameters and are quite polydisperse
(figure 3.3 (top)). When a 50:50 blend of M-PEGPCL and PEGPCL is synthesized into
polymersomes by solvent injection method, the resulting nanoparticles are more monodisperse,
though still smaller than their non-functionalized counterparts from the previous chapter (figure 3.3
(bottom)).
Figure 3.3 – Size distribution (diameter) M-PEGPCL polymersomes (top) 100% M-PEGPCL (bottom) 50% M-PEGPCL, 50% PEGPCL.

26
When polymersomes were synthesized from M-PEGPLGA using solvent injection method, they
were in the target diameter, PDI and zeta potential ranges established in the previous chapter (See
Table 3.2 below). While PEGPCL is preferable because of its increased stability in the GBM tumor
microenvironment, the following in vitro experiments are performed at neutral pH. As the purpose
of this chapter is to demonstrate an increase in biological function by ligand attachment, the focus
will be on conjugating M-PEGPLGA polymersomes with CTAT in an effort to increase cellular
uptake.
M-PEGPLGA polymersomes were synthesized by solvent injection method then stirred overnight
with 0.25mg of CTAT peptide overnight. Table 3.2 shows diameter, PDI and zeta potential of these
polymersomes both before and after CTAT conjugation.
Table 3.2 – M-PEGPLGA Polymersomes Before and After Conjugation with CTAT Peptide
M-PEGPLGA polymersomes CTAT conjugation
Diameter (nm) PDI Zeta potential (mV)
CysTAT - 90.29 0.133 -33.6 CysTAT + 84.47 0.145 -0.723
A large positive shift, 32.88 mV, is observed after CTAT conjugation, indicating a large number
of covalently bonded CTAT peptide molecules on the surface of the polymersomes.
Observing enhanced cellular uptake by fluorescence flow cytometry – M-PEGPLGA particles are
synthesized by solvent injection with that the aqueous phase of the procedure containing 0.16 mg
AF488 dye. The resulting polymersomes, measured by DLS, had diameters of 87.87 ± 5.96 nm,
PDI of 0.145 ± 0.013, and zeta potentials of -39.2 ± 2.7 mV (n=3). Each batch of polymersomes
was split and half of each sample was stirred with 0.125 mg CysTAT overnight. HEK293 cells
were treated with the loaded polymersomes, both CysTAT- conjugated and unconjugated, for 3.5

27
hours. The cells were analyzed on a flow cytometer to determine the mean fluorescence (FITC) of
each cell population. Normalizing for cell autofluorescence, the mean fluorescence of cells treated
with CysTAT-conjugated increased nearly two-fold over those treated with nonconjugated
polymersomes (Figure 3.4).
Figure 3.4 – Fold change in mean fluorescence (FITC) per HEK293 cell treated with AF488- loaded CTAT-Functionalized and non-functionalized M-PEGPLGA polymersomes
Conclusions and Future Outlook
This chapter illustrates that polymersomes are not just a platform for delivering drug combinations
and extending drug circulation times in patients, but rather they can be functionalized to perform
specific biological functions. In this case, CysTAT ligands increased the rate of cellular uptake of
polymersomes, albeit indiscriminate of cell type. The system is modular, and using similar
bioconjugation techniques, a variety of macromolecules, antibodies, peptides, proteins etc. can be
conjugated to a polymersome surface. In the context of local delivery, particularly in GBM, using
ligands that will increase cellular uptake or adhesion to tumor-specific cell types is an attractive
option.
0
0.5
1
1.5
2
Fold
Cha
nge
Without TAT With TAT

28
Moving forward, the importance of discovering biologically relevant ligands that are specific to the
treatment of specific diseases is paramount, as is optimizing bioconjugation techniques to attach
the ligands to therapeutic polymersomes. This flavor of biology-specific functionalization of
polymersomes could be used to increase drug delivery efficacy for a variety of genetic disorders
and cancers. For GBM, a methodology for finding tumor-specific targets, and the ligands that will
help target them, will be touched on in the next chapter.

29
4 – TARGETTING GBM USING mRNA DATA
Introduction
In previous chapters, it was demonstrated that PEG-polyester polymersomes were potentially
suitable candidates for delivering encapsulated drugs locally to GBM. Furthermore, those
polymersomes can be functionalized with ligands designed to target and improve drug delivery
efficiency to cells. The goal of this chapter is to elucidate GBM surface proteins that, when targeted,
will facilitate an increase in therapeutic efficacy of drug-loaded polymersomes.
The phenotypic heterogeneity of GBM tumor cells confounds the discovery of an effective cellular
surface target. Within a single GBM tumor, there are distinct populations of tumor cells with
varying levels of protein expression27–29,31,81. These populations of tumor cells change phenotypes
in response to stimuli, including immune responses to surgery and chemotherapy82–84. However,
systems biologists have been able to employ genetic, transcriptomic and proteomic data to classify
and quantify heterogeneity of GBM tumor cells in a number of useful ways. Researchers are
combining new surgical sampling techniques with genomic analysis to track the evolution
dynamics of tumor cell populations31. Other biologists are using single-cell transcriptomic data and
gene-expression based analysis in an effort classify distinct GBM subtypes for future
researchers27,59. Furthermore, databases like the Cancer Genome Atlas (TCGA), Cancer Cell Line
Encyclopedia (CCLE) and International Cancer Genome Consortium (ICGC) provide enormous,
curated, cancer-specific datasets that help inform the fields of drug discovery and pharmacology60.
This chapter will focus on using a combination of databases to identify a set of highly expressed
GBM tumor cell surface targets and the ligands that bind to them. These ligands can be conjugated
to drug-carrying polymersomes, creating a combination therapy platform designed to target
multiple GBM cell phenotypes in the same tumor.

30
Methods
Determining High Expression Transcripts in GBM – Feltus TCGA GEM analysis - A gene
expression matrix (GEM), describing fragments per kilobase million (FPKM) of 73599 mRNA
transcripts across 2016 cancerous tumor samples [bladder, thyroid, ovarian, low-grade glioma,
glioblastoma, data from The Cancer Genome Atlas (TCGA)] was built by William Poehlman
(Department of Genetics and Biochemistry, Clemson University), which had been previously
quantile normalized and values converted to log2 values (No outliers detected using Kolmogorov-
Smirnov test)85. From Dr. Alex Feltus (Department of Genetics and Biochemistry, Clemson
University), a key was obtained to convert the knowngene5 UC-Santa Cruz genome database gene
model identifiers to Ensembl gene identifiers, and the transcripts that had no Ensembl ID were
omitted from the GEM. A median FPKM value across the 174 GBM tumor samples was obtained
for each transcript. The transcripts with the 12000 highest median FPKM values were used to
determine genes of high expression in GBM. Westfall TCGA GEM Analysis – RNASeq expression
profiles for 174 GBM tumor samples were downloaded from the TCGA (10/30/2018). The profiles
were merged to form a single GEM. Using python, the data was quantile normalized, and FPKM
values were converted to log2. A median FPKM value across the tumor samples was obtained for
each transcript. The transcripts with the 12000 highest median FPKM values were used to
determine genes of high expression in GBM. (Python code: TCGA_analysis_FELTUS,
TCGA_Analysis_Westfall in Appendix A)
Comparing High Expression GBM Transcripts Against Normal/Low Expression in Normal
Brain Tissue – A GEM, describing FPKM of 56202 mRNA transcripts across 1671 non-diseased
brain tissue samples from the Genotype-Tissue Expression project (GTEx)86, was obtained from
Dr. Alex Feltus (Department of Genetics and Biochemistry, Clemson University), which had been
previously quantile normalized and converted to log2 values. A median FPKM value across the

31
tissue samples was obtained for each transcript. The transcripts with the 5000 highest median
FPKM values were used to determine genes of high expression in healthy brain tissue and were
omitted from the data. The remaining transcripts in the GEM were considered to be of normal or
low expression. The Ensembl gene IDs of the high expression transcripts in the Feltus/Westfall
TCGA GEMs were compared with the Ensembl gene IDs of the normal/low transcripts. Any
overlap in the set identifies a transcript that is highly expressed in GBM and not highly expressed
in non-diseased brain tissue. (Python Code: GTEX_analysis,
TCGA_GTEX_Comparison_FELTUS, TCGA_GTEX_Comparison_Westfall,
Westfall_Feltus_TCGA_COMPARE in Appendix A)
Gene Ontology Annotations to Find Candidate Surface Proteins and Corresponding Binding
Ligands – Ensemble gene IDs of transcripts categorized as highly expressed in GBM and
normal/low expressed in non-diseased brain tissue are uploaded to Uniprot87 where the gene
ontology (GO) annotations are listed for each gene. QuickGO88 is used to search for GO annotations
that identify surface proteins which can be potentially targeted with ligands. Gene IDs for the high
GBM/low non-diseased transcripts are filtered with the surface protein GO annotations to reveal
“candidate genes” as coding for proteins that are potential ligand binding sites for GBM drug
delivery. Candidate genes are cross-checked individually against the Gene Expression Profiling
Interactive Analysis (GEPIA) portal89 to verify that gene has high expression in GBM tumor
samples and normal/low expression in non-diseased brain tissue. To find corresponding binding
ligands, the candidate genes were searched for in the literature-informed online databases Binding
DB90 and RCSB Protein Data Bank91.

32
Experimental Results and Discussion
To find GBM surface proteins for active targeting and the ligands that can be used to target them,
it is necessary to leverage mRNA transcriptome data from the TCGA and GTEx. The workflow to
go from RNAseq data to GBM-specific ligand molecules incorporates a combination of Python
programming and online databases, including GEPIA, Uniprot, QuickGO, RCSB Protein Data
Bank, and Binding DB. The workflow shown in Figure 4.1 illustrates how Python isolates and
compares 73599 mRNA transcripts from TCGA and GTEx to identify genes that are highly
expressed in GBM tumor samples, but not in healthy, non-diseased brain tissues. Using this
method, depending on the source of the TCGA GEM (Feltus or Westfall), there are between 5400
and 7100 transcripts from candidate genes.

33
Figure 4.1 – Illustrative view of workflow identifying candidate genes
Genes identified in this way are uploaded to Uniprot, where groups of genes can be grouped
according to their Gene Ontology (GO) annotations. These annotations identify the many molecular
function, biological process, and cellular component attributes of the proteins these genes encode
for. GO annotations that can identify relevant cellular surface targets can be found using QuickGO.
Candidate genes found to code for the biologically relevant target proteins are then cross-checked
against the GEPIA online database, where expression of individual genes in tumor and healthy
tissue can be visualized based on RNAeq data. This is done as a second check to validate that the
initial python scripts correctly identified transcripts that were indeed abnormally expressed in GBM
tumor samples. These proteins for which candidate genes encode are then run on databases that
Top expressed transcripts from
TCGA GBM dataset
GO Annotations indicating a cellular
surface receptor capable of targetting
Lowest expressed transcripts from GTEX
dataset

34
have ligand binding information based on previous literature, most likely Binding DB or the RCSB
PDB. Figure 4.2 illustrates how gene expression is visualized in by boxplot in GEPIA (axis
representing transcripts per kilobase million (TPM+1)) and the identification of ligands with
binding affinity for the expressed protein.
Figure 4.2 – Illustrative view of candidate gene/protein GEPIA verification and ligand selection. (top) PLAUR (bottom) CXCR4. Boxplots are generated by GEPIA89. Axis is mRNA transcripts per kilobase million (log-2). GBM tumor samples are depicted in red, with healthy brain tissue
samples in gray.
Figure 4.2 also shows two candidate genes/proteins, Urokinase plasminogen activator surface
receptor (PLAUR) and C-X-C chemokine receptor type 4 (CXCR4). PLAUR is located in the
GEPIA RCSB PDB
GEPIA Binding
DB

35
plasma membrane and is shown to have high binding affinity to 2-Amino-5-phenyl-1-pyridin-2-
ylpyrrole-3-carbonitrile according to literature found using BindingDB92. CXCR4 is a
transmembrane protein that has a high binding affinity for (6,6-dimethyl-5,6-dihydroimidazo[2,1-
b][1,3]thiazol-3-yl)methyl N,N'-dicyclohexylimidothiocarbamate, per the literature found in RCSB
PDB93. According to data from GEPIA, transcripts for PLAUR and CXCR4 are found in
significantly higher amount in GBM tumor samples, when compared to healthy non-diseased brain
tissue93. These characteristics (location, binding affinity and mRNA expression) make PLAUR and
CXCR4 good candidates as part of a set of GBM surface targets for targeting with functionalized
polymersomes.
Conclusions and Future Outlook
In this chapter, it has been shown that analysis of large RNAseq datasets can be combined with
extensive data from online transcriptomic and proteomic databases, revealing disease-specific
biologic targets for local drug delivery. This mRNA-to-ligand workflow is great for identifying
GBM targets but can easily be adopted to accommodate other cancers or diseases. It is not
unreasonable to image a workflow like this being adapted for patient- or tumor-derived RNAseq
data and used to develop personalized cancer-targeting drug carriers.
While promising, it is important understand the limitations of this analysis. While there is evidence
that intracellular mRNA levels are correlative with cellular protein levels94–97, there are also
researchers that caution that the correlation is poor and not properly understood98–100. They suggest
that mRNA to protein correlation may be affected by tissue specific factors and epigenetics.
However, as the methods for collecting transcriptomic and proteomic continue to improve, and
abundance of data available increases, it is likely that understanding of the mRNA and protein level
correlation will improve, giving strength to the type of analysis performed in this chapter.

36
Nevertheless, protein level measurements are crucial as additional confirmatory validation step in
future work.
Moving forward, this analysis must be expanded in two ways. First, it is important to find more
candidate genes for GBM. Finding these genes will likely be the result of identifying more GO
annotation combinations indicating a particular protein is a good biological candidate for targeting,
thus expanding the search scope through the highly expressed GBM genes. It is also important to
verify sets of candidate genes are not overexpressed exclusively in the same tumor samples. With
the current dataset of 174 samples, there is likely some overlap of overexpressed genes, but the
goal should be to identify a set of genes that minimizes correlative overexpression of the candidates.
Second, in vitro cellular uptake experiments can be performed on candidates as they are identified.
Cell lines can be transformed to overexpress the candidate protein on the cell surface. Drug or
fluorophore-loaded polymersomes will then be conjugated with the binding ligand associated with
that protein, as in Chapter 3. The expectation is that higher drug/fluorophore payloads will be
internalized by the transformed cells when the loaded polymersomes have been conjugated with
the targeting ligand, quantified by cell death or flow cytometry fluorescence.

37
5 – THE CHEMOTAXIS HYDROGEL
Introduction
In an earlier chapter, the concepts of drug-loaded and nanoparticle-loaded hydrogels were
introduced. Extensive research optimizing these systems, employing innovative materials and
informed by modern understanding of disease biology, has been performed over the last
decade37,38,40,54,56,101. However, the ultimate goal for these systems has remained the same: to
efficiently deliver therapeutics to a location/cell/tumor in the effort to treat or eliminate the disease.
However, modern technologies are allowing researchers to ponder quite the opposite: bringing the
diseased cells to the therapeutic location for elimination.
There are a few researchers currently creating and optimizing hydrogels that are great candidates
for cell migration: PEG based hydrogels, crosslinked by enzyme degradable peptides (EDP). These
PEG-EDP hydrogels mimic the extracellular matrix of the human body and are capable of
encapsulating cells and providing an environment for the cells to migrate. When the cell’s receptor
contacts the EDPs, mobility pathways in the cell are activated and the cell excretes the appropriate
degrading enzyme and migrates through the gel102–106.
This chapter focuses on taking the concepts of the PEG-EDP hydrogel and framing them as a tool
for local treatment of cancer. The hydrogels can act as a “black hole”, allowing invasive tumor
cells, particularly those left after tumor resection, to invade instead of migrating to the surrounding
tissue. The direction of the cell migration, that is toward the hydrogel, can be manipulated using a
chemoattractant gradient107,108 (Figure 5.1).

38
Figure 5.1 – Illustration demonstrating that eukaryotic cells migrate along a gradient toward a higher concentration of chemoattractants. Adapted from107.
Chemotaxis is the process by which that mammalian cells migrate toward the higher concentration
of chemical signals, which will in this case, be originating from the hydrogel.
Methods
Synthesis of Chemotaxis Hydrogel – 50 mg maleimide-functionalized 4-arm polyethylene glycol
(20KDa) (JENKEM Technology USA # A7029-1) (4armPEG) was dissolved in 0.5 mL milli-Q
water, varying concentrations of enzyme degradable peptide (Genscript #UI630DK260-1) (amino
acid sequence: CIPESLRAGC) (EDP) (20,40,100 mg/mL) was dissolved in 0.5 mL of milli-Q
water and added to 4armPEG solution and vortexed for 1 second. Mixture was quickly pipetted
into 12-well plate and allowed ~2 hours to gelate.
Determination of Hydrogel Stiffness – Stiffness of hydrogel was determined using Poroelastic
relaxation indentation (PRI). A custom-built apparatus for performing PRI was provided by Dr.
Eric Davis and operated by Nick Gregorich (Department of Chemical and Biomolecular
Engineering, Clemson University). A high-resolution linear actuator (M-230, physike intrumente)
was connected to a S-beam load cell (Futek LSB200, FUTEK Advanced Sensor Technology, Inc)
with a rigid glass indenter of radius of curvature R = 5.187mm. The actuator was mounted to a

39
high-performance linear stage with 46mm of travel range (M433, Newport). The indenter was
lowered by the actuator at a speed of 3 μm/s until contact with the hydrogel was reached. Upon
contact the indenter velocity changed to 10 μm/s until a load of 29.4mN was reached. The indenter
was held, fixed for 5 seconds, and released from the sample. A LabVIEW custom program, written
by Jaime Idarraga-Mora (Department of Chemical and Biomolecular Engineering, Clemson
University), was used to acquire load and indenter height data as a function of time from the S-
beam load cell during the experiments. Hooke’s law was used to calculate Young’s modulus from
collected data109.
Transwell Invasion Assay – Cell Culture – MDA-MB-231 cells and U-87 MG cells (ATCC
#HTB-26, ATCC #HTB-14, both verified by short tandem repeat profiling) were seeded in separate
T-75 flasks, at 2.0 x 106 cells per flask, and incubated for 36-48 hours at 37 °C and 5% CO2, in full
media Delbecco’s modified eagle medium (Gibco #10313-021) (DMEM +10% FBS +2mM L-
glutamine). Hydrogel inserts – 1 mL Chemotaxis hydrogel was synthesized and split into two
separate aliquots. 1 μg Doxorubicin-HCL (Fisher #BP2516-10) (DOX) was added to one of the
aliquots. For each cell line, 75 μL hydrogel was added to a 6.5 mm Transwell permeable support
with 80 μm pore (Costar #3422), and 75 μL hydrogel+DOX was added to another insert. Insert
hydrogels were allowed 2 hours to gelate. Each insert hydrogel was soaked overnight in starving
media (DMEM with no additives), or until hydrogel was visibly saturated. Cell migration – At
~70% confluence, MDAMB231 and U87 cells were lifted from T-75 flask with 0.25% Trypsin-
EDTA (Gibco #25200056), centrifuged, aspirated, then resuspended in starving media (DMEM
with no additives) to a concentration of 1.0 x 106 cells/mL. For each cell line, 100 μL (1.0 x 105
cells) were placed on top of a hydrogel inset, a hydrogel+DOX insert and a blank insert (directly
on membrane). Inserts were placed in 24-well plate containing 550 μL assay media (DMEM +0.5%
FBS +2mM L-glutamine +2ng/mL EGF). Plates, containing cells and inserts are incubated for 48

40
hours at 37 °C and 5% CO2. Plate wells were inspected under Revolve microscope (Echo
Laboratories) at 10x magnification to determine, qualitatively, whether cells had migrated through
the membrane and adhered to well surface.
Experimental Results and Discussion
The goal of the set of experiments in this chapter is to formulate a hydrogel from PEG and an
enzyme degradable peptide (EDP) that is degradable by one or more matrix metalloprotease. Also,
the hydrogel should have a stiffness similar to the tissue type it will be implanted into, which for
the purposes of treating GBM, is brain tissue. Furthermore, the hydrogel should facilitate cell
migration through degradation of the EDP, similar to the way a cell moves through the ECM.
Hydrogel formulation and gelation – This experiment was designed and performed by Chad Eaton
(Department of Bioengineering, Clemson University). A peptide sequence, CIPESLRAGC,
degradable by MMP-2105 (linker), was purchased and used to crosslink 4-Arm PEG functionalized
with terminal maleimides (4ARMPEG). Figure 5.2 illustrates the covalent bonding reaction
between the maleimide groups and the sulfhydryl pendant groups of the terminal cysteines of the
EDP.
Figure 5.2 – Crosslinking of hydrogel by covalent bonding of maleimide functionalized 4-Arm PEG to sulfhydryl pendant group
50 mg 4ARMPEG in 0.5 mL Milli-Q water is combined quickly with 20 mg linker in 0.5 mL Milli-
Q Water. The transparent hydrogel forms and gelates completely in ~2 hours.

41
Determining hydrogel stiffness – This experiment was designed and performed by Chad Eaton and
Riley Rapert (Department of Bioengineering, Clemson University) with assistance and equipment
provided by Nick Gregorich and Dr. Eric Davis (Department of Chemical and Biomolecular
Engineering, Clemson University). Now that a hydrogel can be formulated, it is important to
determine whether the hydrogel is a stiffness that is close to biological tissue, and whether that
stiffness can be modulated by formulation.
A set of hydrogels was synthesized by keeping the amount of 4ARMPEG constant and varying the
amount of linker. 50 mg 4ARMPEG in 50 mL Milli-Q water was combined with varying masses
of linker in 50 mL Milli-Q water and allowed to gelate for 2 hours (Table 5.1). Poroelastic
Relaxation Indentation (PRI) analysis was performed to determine Young’s modulus, a measure of
mechanical stiffness109,110. The following equation, a derivation of Hooke’s Law, was used:
𝐸𝐸 =(1 − 𝑣𝑣2)
𝜋𝜋∆(𝑃𝑃ℎ)∆𝑓𝑓
Where P is the load, h is the probe depth and r is the radius of contact of the spherical probe. An
assumption is made that the substance is isotropic, meaning that the stiffness will be the same in
the hydrogel regardless of direction. Thus, ν is assumed to be 0.5. Table 5.1 shows the Young’s
modulus of each formulation.
Table 5.1 – Chemotaxis Hydrogel Formulation and Young’s Modulus
Hydrogel 4ARMPEG (mg/mL H2O)
Linker (mg/mL H2O) Young’s modulus (kPa)
1 100 20 1.21 2 100 40 1.86 3 100 100 2.01

42
There is a positive correlation between the amount of linker used and the stiffness of the hydrogel.
Figure 5.3 shows the typical Young’s modulus for a variety of tissue types, according to their
Collagen-1 content111. GBM tumor varies from 0.5 - 2 kPa. This indicates that the hydrogels
formulated are similar in stiffness to GBM tumor. Also, it is shown that stiffness of the hydrogel
can be modulated by varying the amount of cross-linker available during gelation.
Figure 5.3 – Tissue stiffness for normal tissues. Highlighted in blue is a range of stiffnesses for GBM tumors. Adapted from111
Cell invasion assay – This experiment was designed and performed by Asia Paguntalan
(Department of Genetics and Biochemistry, Clemson University). The goal of this experiment is to
observe, and quantify, movement of cells into or through the hydrogel toward a chemotaxis signal.

43
A common way to quantify movement of cells toward chemoattractants is a transwell membrane
invasion assay. The assay was modified such that thin chemotaxis hydrogels would be formed
across the inserts above the membrane (Figure 5.4). 75 μL hydrogel (same formulation as hydrogel
#2 in table 5.1) was added and gelated on top of the membrane in the insert, then soaked in DMEM
with no additives. The receiver well was filled with DMEM +0.5% FBS +2mM L-glutamine
+2ng/mL Epidermal growth factor (EGF). The EGF is a chemoattractant for the invading
MDAMB231 breast cancer cells and the U87 glioblastoma cells. It is expected that the EGF would
diffuse through the hydrogel, creating a gradient, that would signal the cells to migrate toward the
receiver well through the hydrogel by degrading the EDP linker. Cells, in DMEM (with no
additives) were deposited on top of the hydrogel and incubated for 48 hours at 37 °C and 5% CO2.
Figure 5.4 – Illustration of transwell invasion assay modified for chemotaxis hydrogel
Unfortunately, in every trial, both U87 and MDAMB231 cells, microscope inspection, failed to
migrate through the hydrogel and membrane and into the receiver well. Upon further inspection,

44
the cells seemed to have remained above the hydrogel entirely. It may be unrealistic to assume
that in 48 hours, EGF would diffuse across the hydrogel and the cells would invade and
transverse through the hydrogel, which is approximately 2mm thick after soaking in media. The
assay can be modified such that the thickness of the gelated hydrogel in the insert is closer to that
of a thin film. Another modification to this experiment is to embed the cells in the hydrogel as it
gelates. Specifically, instead of dissolving 50mg of 4ARMPEG in 0.5 mL of Milli-Q water,
dissolve it in 0.5 mL of cell suspension in media. This approach would lower the time for
diffusing EGF to reach the cells, as they would already be dispersed in the hydrogel.
Conclusions and Future Outlook
This chapter is an illustrative example of innovative materials design, generated by eschewing the
traditional “drug-loaded-hydrogel” delivery concept. These experiments are preliminary work
towards a goal of making a modular platform of disease-fighting chemotaxis hydrogels. The EDP
crosslinker chosen for this work can be interchanged with dozens of other EDPs, each specific to
mobility enzymes secreted by cells of varying tumor type. RNAseq analysis, described in the
previous chapter, can even reveal which chemotaxis signal receptors are expressed in a specific
tumor type, in turn, informing which chemoattractants would be most effective for the system.
Furthermore, this analysis shows that the chemotaxis hydrogel stiffness can be modulated through
formulation, allowing these hydrogels to be implanted in a variety of tissue types.
Moving forward, the most important focus will be observing and quantifying cell movement
through the hydrogel. Likely, the best way to accomplish this is thorough automated, 3D live-cell
imaging, where cells can be tracked in real-time. Hydrogels can be synthesized, using a cell
suspension in lieu of Milli-Q water, embedding the cells in the hydrogel, in multi-well plates. An
automated imaging system like the Cytation 5 and Biospa (BioTek) can be used to image the cells

45
in the hydrogels over several days, with the imaging software tracking selected cells. Furthermore,
chemotaxis signals can be injected at the edge of the well and allowed to diffuse, creating a gradient
across the hydrogel. This will show, qualitatively, whether migration direction of cells in the
hydrogel can be controlled by chemotaxis signaling.

46
APPENDIX A – PYTHON CODES
All Jupyter notebooks, containing these codes, are located on Birtwistle Lab Microsoft OneDrive (Folder: GBM Westfall) Dependent Files: All files are located on Birtwistle Lab Microsoft OneDrive (Folder: GBM Westfal) TCGAV1.txt TCGAIDtoESNID.txt GTEXV1.txt DATA (contains .txt files TCGA used for TCGA_analysis_Westfall) TCGA_analysis_FELTUS import pandas as pd df = pd.read_table("TCGAV1.txt", index_col=0) df.index.name = 'TCGA_ID' GBM={} for i in range(2016): if df.iloc[:,i].name.startswith('GBM'): GBM[df.iloc[:,i].name]=df.iloc[:,i] GBMData=pd.DataFrame(data=GBM) GBMData['Median'] = GBMData.median(axis=1) ensids = pd.read_table("TCGAIDtoENSID.txt", sep=' ', index_col=0) GBMData = GBMData.join(ensids, how='outer') GBMData.sort_values(by='Median', ascending=False, inplace=True) with_ensid = GBMData[GBMData['Ensembl_Gene_ID'].notnull()] largest_median_with_ensid = with_ensid[['Median','Ensembl_Gene_ID']][:12000] largest_median_with_ensid['Ensembl_Gene_ID'].to_csv(r'C:\Users\jjwes\Documents\Python Workspace\GBM\topTCGA.txt',index=0) largest_median_with_ensid.to_csv(r'C:\Users\jjwes\Documents\Python Workspace\GBM\TCGA_high.txt') TCGA_analysis_Westfall

47
import pandas as pd import numpy as np import glob files = glob.glob('**/*.FPKM.txt', recursive=True) base = {} base_frame = pd.DataFrame(base) for i in range(len(files)): base_frame[files[i]] = pd.read_table(files[i],index_col=0, names=['FPKM'])['FPKM'] FPKM=base_frame rank_mean = FPKM.stack().groupby(FPKM.rank(method='first').stack().astype(int)).mean() FPKM_Qnormal=FPKM.rank(method='min').stack().astype(int).map(rank_mean).unstack() FPKM_Qnormal_Nan = FPKM_Qnormal.replace(0, np.nan) Log2_Qnormal_Nan = np.log2(FPKM_Qnormal_Nan) Log2_Qnormal=Log2_Qnormal_Nan.replace(np.nan,0) Log2_Qnormal.index.name = 'Ensembl_Gene_ID' Log2_Qnormal['Median'] = Log2_Qnormal.median(axis=1) Log2_Qnormal.sort_values(by='Median', ascending=False, inplace=True) Log2_Qnormal.index=Log2_Qnormal.index.str[:15] largest_median_TCGA_WESTFALL = Log2_Qnormal[['Median']][:12000] largest_median_TCGA_WESTFALL.to_csv(r'C:\Users\jjwes\Documents\Python Workspace\GBM\TCGA_high_WESTFALL.txt') GTEX_analysis import pandas as pd GTEXData = pd.read_table("GTEXV1.txt") GTEXData['Median'] = GTEXData.median(axis=1) GTEXData.sort_values(by='Median', ascending=True, inplace=True)

48
GTEXData.index=GTEXData.index.str[:15] GTEX_smallest_median = GTEXData['Median'][:50000] GTEX_smallest_median.to_csv(r'C:\Users\jjwes\Documents\Python Workspace\GBM\GTEX_low.txt') TCGA_GTEX_Comparison_FELTUS import pandas as pd import numpy as np TCGA_high = pd.read_table('TCGA_high.txt', sep=',',index_col=0) GTEX_low = pd.read_table('GTEX_low.txt',sep=',',names=['Ensembl_Gene_ID','Median']) p=0 for i in range(12000): if GTEX_low['Ensembl_Gene_ID'].str.contains(TCGA_high['Ensembl_Gene_ID'].iloc[i]).any(): print (TCGA_high['Ensembl_Gene_ID'].iloc[i]) p=p+1 p TCGA_GTEX_Comparison_Westfall import pandas as pd import numpy as np TCGA_high_WESTFALL = pd.read_table('TCGA_high_WESTFALL.txt', sep=',',index_col=0) GTEX_low = pd.read_table('GTEX_low.txt',sep=',',names=['Ensembl_Gene_ID','Median']) p=0 for i in range(12000): if GTEX_low['Ensembl_Gene_ID'].str.contains(TCGA_high_WESTFALL.index[i]).any(): print (TCGA_high_WESTFALL.index[i]) p=p+1 p Westfall_Feltus_TCGA_COMPARE TCGA_high_F = pd.read_table('TCGA_high.txt', sep=',',index_col=0) TCGA_high_W = pd.read_table('TCGA_high_WESTFALL.txt', sep=',',index_col=0)

49
p=0 for i in range(12000): if TCGA_high_W.index.str.contains(TCGA_high_F['Ensembl_Gene_ID'].iloc[i]).any(): print (TCGA_high_F['Ensembl_Gene_ID'].iloc[i]) p=p+1 p

50
REFERENCES
1. Koshy, M. et al. Improved survival time trends for glioblastoma using the SEER 17
population-based registries. J. Neurooncol. 107, 207–12 (2012).
2. Berens, M. E. & Giese, A. “...those left behind.” Biology and Oncology of Invasive
Glioma Cells. Neoplasia 1, 208–219 (1999).
3. Yang, L.-J., Zhou, C.-F. & Lin, Z.-X. Temozolomide and Radiotherapy for Newly
Diagnosed Glioblastoma Multiforme: A Systematic Review. Cancer Invest. 32, 31–36
(2014).
4. Smoll, N. R., Schaller, K. & Gautschi, O. P. Long-term survival of patients with
glioblastoma multiforme (GBM). J. Clin. Neurosci. 20, 670–675 (2013).
5. Hottinger, A. F., Abdullah, K. G. & Stupp, R. Current Standards of Care in Glioblastoma
Therapy. in Glioblastoma 73–80 (Codon Publications, 2016). doi:10.1016/B978-0-323-
47660-7.00006-9
6. Darlix, A. et al. Prolonged administration of adjuvant temozolomide improves survival in
adult patients with glioblastoma. Anticancer Res. 33, 3467–74 (2013).
7. Gallego, O. Nonsurgical treatment of recurrent glioblastoma. Curr. Oncol. 22, e273-81
(2015).
8. Johnson, D. R. & O’Neill, B. P. Glioblastoma survival in the United States before and
during the temozolomide era. J. Neurooncol. 107, 359–364 (2012).
9. Tiek, D. M. et al. Alterations in Cell Motility, Proliferation, and Metabolism in Novel
Models of Acquired Temozolomide Resistant Glioblastoma. Sci. Rep. 8, (2018).

51
10. Darefsky, A. S., King, J. T. & Dubrow, R. Adult glioblastoma multiforme survival in the
temozolomide era: A population-based analysis of Surveillance, Epidemiology, and End
Results registries. Cancer 118, 2163–2172 (2012).
11. Wolbers, J. G. Novel strategies in glioblastoma surgery aim at safe, supra-maximum
resection in conjunction with local therapies. Chin. J. Cancer 33, 8–15 (2014).
12. Disease State. Available at: http://gliadel.com/hcp/disease-state.php. (Accessed: 16th
August 2019)
13. VandenDriessche, T. & Chuah, M. K. Glioblastoma: bridging the gap with gene therapy.
Lancet. Oncol. 14, 789–90 (2013).
14. Hu, S. et al. Real-Time Imaging of Brain Tumor for Image-Guided Surgery. Adv. Healthc.
Mater. 7, e1800066 (2018).
15. Lara-Velazquez, M. et al. Advances in Brain Tumor Surgery for Glioblastoma in Adults.
Brain Sci. 7, (2017).
16. von Roemeling, C., Jiang, W., Chan, C. K., Weissman, I. L. & Kim, B. Y. S. Breaking
Down the Barriers to Precision Cancer Nanomedicine. Trends in Biotechnology 35, 159–
171 (2017).
17. Dobbing, J. The Blood‐Brain Barrier. Developmental Medicine & Child Neurology 3,
311–314 (1961).
18. Abbott, N. J., Rönnbäck, L. & Hansson, E. Astrocyte-endothelial interactions at the blood-
brain barrier. Nature Reviews Neuroscience 7, 41–53 (2006).
19. Pardridge, W. M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow

52
Metab. 32, 1959–1972 (2012).
20. Wolak, D. J. & Thorne, R. G. Diffusion of macromolecules in the brain: Implications for
drug delivery. Mol. Pharm. 10, 1492–1504 (2013).
21. Peer, D. et al. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol.
2, 751–760 (2007).
22. Niewald, M. et al. Toxicity after radiochemotherapy for glioblastoma using temozolomide
- a retrospective evaluation. Radiat. Oncol. 6, 141 (2011).
23. Saito, K. et al. Toxicity and outcome of radiotherapy with concomitant and adjuvant
temozolomide in elderly patients with glioblastoma: a retrospective study. Neurol. Med.
Chir. (Tokyo). 54, 272–9 (2014).
24. Hanna, N. et al. Long-Term Toxicity of Chemotherapy. (2003).
25. Zhang, J., F.G. Stevens, M. & D. Bradshaw, T. Temozolomide: Mechanisms of Action,
Repair and Resistance. Curr. Mol. Pharmacol. 5, 102–114 (2011).
26. D’Alessandris, Q. G., Montano, N., Larocca, L. M., Maira, G. & Pallini, R. Prognostic
impact of MGMT promoter methylation in glioblastoma - A systematic review. J. Cancer
Sci. Ther. 6, 136–141 (2014).
27. Patel, A. P. et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary
glioblastoma. Science (80-. ). 344, 1396–1401 (2014).
28. Qazi, M. A. et al. Intratumoral heterogeneity: pathways to treatment resistance and relapse
in human glioblastoma. Annals of oncology : official journal of the European Society for
Medical Oncology 28, 1448–1456 (2017).

53
29. Eskilsson, E. et al. EGFR heterogeneity and implications for therapeutic intervention in
glioblastoma. Neuro. Oncol. 20, 743–752 (2018).
30. Iwadate, Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncology
Letters 11, 1615–1620 (2016).
31. Sottoriva, A. et al. Intratumor heterogeneity in human glioblastoma reflects cancer
evolutionary dynamics. Proc Natl Acad Sci U S A 110, 4009–4014 (2013).
32. Peppas, N. A., Bures, P., Leobandung, W. & Ichikawa, H. Hydrogels in pharmaceutical
formulations. Eur. J. Pharm. Biopharm. 50, 27–46 (2000).
33. Wagner, A. M., Spencer, D. S. & Peppas, N. A. Advanced architectures in the design of
responsive polymers for cancer nanomedicine. J. Appl. Polym. Sci. 135, 46154 (2018).
34. Cheng, W., Gu, L., Ren, W. & Liu, Y. Stimuli-responsive polymers for anti-cancer drug
delivery. Mater. Sci. Eng. C 45, 600–608 (2015).
35. Wolinsky, J. B., Colson, Y. L. & Grinstaff, M. W. Local drug delivery strategies for
cancer treatment: Gels, nanoparticles, polymeric films, rods, and wafers. Journal of
Controlled Release 159, 14–26 (2012).
36. Koetting, M. C., Peters, J. T., Steichen, S. D. & Peppas, N. A. Stimulus-responsive
hydrogels: Theory, modern advances, and applications. Mater. Sci. Eng. R Reports 93, 1–
49 (2015).
37. Bastiancich, C., Danhier, P., Préat, V. & Danhier, F. Anticancer drug-loaded hydrogels as
drug delivery systems for the local treatment of glioblastoma. Journal of Controlled
Release 243, 29–42 (2016).

54
38. Bastiancich, C. et al. Injectable nanomedicine hydrogel for local chemotherapy of
glioblastoma after surgical resection. J. Control. Release 264, 45–54 (2017).
39. Huang, H., Qi, X., Chen, Y. & Wu, Z. Thermo-sensitive hydrogels for delivering
biotherapeutic molecules: A review. Saudi Pharm. J. (2019).
doi:10.1016/j.jsps.2019.08.001
40. Basso, J. et al. Hydrogel-Based Drug Delivery Nanosystems for the Treatment of Brain
Tumors. Gels 4, 62 (2018).
41. Couvreur, P. Nanoparticles in drug delivery: Past, present and future. Advanced Drug
Delivery Reviews 65, 21–23 (2013).
42. Batrakova, E. V., Bronich, T. K., Vetro, J. A. & Kabanov, A. V. Polymer micelles as drug
carriers. Dep. Pharm. Sci. 57–93 (2006). doi:10.1142/9781860949074_0005
43. Cho, H. K., Cheong, I. W., Lee, J. M. & Kim, J. H. Polymeric nanoparticles, micelles and
polymersomes from amphiphilic block copolymer. Korean Journal of Chemical
Engineering 27, 731–740 (2010).
44. Yu, X. et al. Design of Nanoparticle-Based Carriers for Targeted Drug Delivery. Journal
of Nanomaterials 2016, (2016).
45. Letchford, K. & Burt, H. A review of the formation and classification of amphiphilic
block copolymer nanoparticulate structures: micelles, nanospheres, nanocapsules and
polymersomes. Eur. J. Pharm. Biopharm. 65, 259–269 (2007).
46. Meng, F., Zhong, Z. & Feijen, J. Stimuli-responsive polymersomes for programmed drug
delivery. Biomacromolecules 10, 197–209 (2009).

55
47. Kamaly, N., Yameen, B., Wu, J. & Farokhzad, O. C. Degradable controlled-release
polymers and polymeric nanoparticles: Mechanisms of controlling drug release. Chem.
Rev. 116, 2602–2663 (2016).
48. Shin, T. H. & Cheon, J. Synergism of nanomaterials with physical stimuli for biology and
medicine. Accounts of Chemical Research 50, 567–572 (2017).
49. Felber, A. E., Dufresne, M. H. & Leroux, J. C. PH-sensitive vesicles, polymeric micelles,
and nanospheres prepared with polycarboxylates. Advanced Drug Delivery Reviews 64,
979–992 (2012).
50. Jang, W. S. et al. Enzymatically triggered rupture of polymersomes. Soft Matter 12, 1014–
1020 (2016).
51. Steichen, S. D., Caldorera-Moore, M. & Peppas, N. A. A review of current nanoparticle
and targeting moieties for the delivery of cancer therapeutics. European Journal of
Pharmaceutical Sciences 48, 416–427 (2013).
52. Jiang, Y., Zhang, J., Meng, F. & Zhong, Z. Apolipoprotein e Peptide-Directed Chimeric
Polymersomes Mediate an Ultrahigh-Efficiency Targeted Protein Therapy for
Glioblastoma. ACS Nano 12, 11070–11079 (2018).
53. Raucher, D., Dragojevic, S. & Ryu, J. Macromolecular drug carriers for targeted
glioblastoma therapy: Preclinical studies, challenges, and future perspectives. Frontiers in
Oncology 8, 624 (2018).
54. Arai, T. et al. Novel local drug delivery system using thermoreversible gel in combination
with polymeric microspheres or liposomes. Anticancer Res. 30, 1057–1064 (2010).
55. Jin, S. et al. Redox/pH stimuli-responsive biodegradable PEGylated P(MAA/BACy)

56
nanohydrogels for controlled releasing of anticancer drugs. Colloids Surfaces A
Physicochem. Eng. Asp. 484, 47–55 (2015).
56. Pitorre, M. et al. Recent advances in nanocarrier-loaded gels: Which drug delivery
technologies against which diseases? Journal of Controlled Release 266, 140–155 (2017).
57. Binabaj, M. M. et al. The prognostic value of MGMT promoter methylation in
glioblastoma: A meta-analysis of clinical trials. J. Cell. Physiol. 233, 378–386 (2018).
58. Zhang, K., Wang, X. Q., Zhou, B. & Zhang, L. The prognostic value of MGMT promoter
methylation in Glioblastoma multiforme: A meta-analysis. Familial Cancer 12, 449–458
(2013).
59. Verhaak, R. G. W. et al. Integrated Genomic Analysis Identifies Clinically Relevant
Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR,
and NF1. Cancer Cell 17, 98–110 (2010).
60. Yang, Y. et al. Databases and web tools for cancer genomics study. Genomics, Proteomics
and Bioinformatics 13, 46–50 (2015).
61. Discher, D. E. & Ahmed, F. Polymersomes. Annu. Rev. Biomed. Eng. 8, 323–41 (2006).
62. Lee, J. S. & Feijen, J. Polymersomes for drug delivery: Design, formation and
characterization. Journal of Controlled Release 161, 473–483 (2012).
63. Rideau, E., Dimova, R., Schwille, P., Wurm, F. R. & Landfester, K. Liposomes and
polymersomes: a comparative review towards cell mimicking. Chem. Soc. Rev. 47, 8572–
8610 (2018).
64. Jiang, Y., Yang, W., Zhang, J., Meng, F. & Zhong, Z. Protein Toxin Chaperoned by LRP-

57
1-Targeted Virus-Mimicking Vesicles Induces High-Efficiency Glioblastoma Therapy In
Vivo. Adv. Mater. 30, 1800316 (2018).
65. Luo, Z. et al. On-Demand Drug Release from Dual-Targeting Small Nanoparticles
Triggered by High-Intensity Focused Ultrasound Enhanced Glioblastoma-Targeting
Therapy. ACS Appl. Mater. Interfaces 9, 31612–31625 (2017).
66. Ahmed, F. & Discher, D. E. Self-porating polymersomes of PEG-PLA and PEG-PCL:
Hydrolysis-triggered controlled release vesicles. J. Control. Release 96, 37–53 (2004).
67. Sanchez, L., Yi, Y. & Yu, Y. Effect of partial PEGylation on particle uptake by
macrophages. Nanoscale 9, 288–297 (2017).
68. Galloway, D. A., Phillips, A. E. M., Owen, D. R. J. & Moore, C. S. Phagocytosis in the
Brain: Homeostasis and Disease. Front. Immunol. 10, 790 (2019).
69. Tutorials, E. The Zeta Potential. Building 1–4 (1999).
70. Kelly, J. M., Gross, A. L., Martin, D. R. & Byrne, M. E. Polyethylene glycol-b-poly(lactic
acid) polymersomes as vehicles for enzyme replacement therapy. Nanomedicine 12,
2591–2606 (2017).
71. Bhattacharjee, S. DLS and zeta potential - What they are and what they are not? Journal
of Controlled Release 235, 337–351 (2016).
72. Kelly, J. M., Pearce, E. E., Martin, D. R. & Byrne, M. E. Lyoprotectants modify and
stabilize self-assembly of polymersomes. Polymer (Guildf). 87, 316–322 (2016).
73. Ayen, W. Y. & Kumar, N. A systematic study on lyophilization process of polymersomes
for long-term storage using doxorubicin-loaded (PEG) 3 -PLA nanopolymersomes. Eur. J.

58
Pharm. Sci. 46, 405–414 (2012).
74. Li, M., Rouaud, O. & Poncelet, D. Microencapsulation by solvent evaporation: State of
the art for process engineering approaches. International Journal of Pharmaceutics 363,
26–39 (2008).
75. Khalil, N. M. et al. Pharmacokinetics of curcumin-loaded PLGA and PLGA-PEG blend
nanoparticles after oral administration in rats. Colloids Surfaces B Biointerfaces 101, 353–
360 (2013).
76. Lee, Y., Chang, J. B., Kim, H. K. & Park, T. G. Stability studies of biodegradable
polymersomes prepared by emulsion solvent evaporation method. Macromol. Res. 14,
359–364 (2006).
77. Iqbal, M., Zafar, N., Fessi, H. & Elaissari, A. Double emulsion solvent evaporation
techniques used for drug encapsulation. International Journal of Pharmaceutics 496, 173–
190 (2015).
78. Bazak, R., Houri, M., El Achy, S., Kamel, S. & Refaat, T. Cancer active targeting by
nanoparticles: a comprehensive review of literature. J. Cancer Res. Clin. Oncol. 141, 769–
784 (2015).
79. Neves, A. R., Queiroz, J. F., Lima, S. A. C. & Reis, S. Apo E-Functionalization of Solid
Lipid Nanoparticles Enhances Brain Drug Delivery: Uptake Mechanism and Transport
Pathways. Bioconjug. Chem. 28, 995–1004 (2017).
80. Liu, Y., Xu, C. F., Iqbal, S., Yang, X. Z. & Wang, J. Responsive Nanocarriers as an
Emerging Platform for Cascaded Delivery of Nucleic Acids to Cancer. Adv. Drug Deliv.
Rev. 115, 98–114 (2017).

59
81. De Vleeschouwer, S. Glioblastoma. (Codon Publications, 2017).
doi:http://dx.doi.org/10.15586/codon.glioblastoma.2017 Edited
82. Iwadate, Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol. Lett.
11, 1615–1620 (2016).
83. Wang, Q. et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes
Associates with Immunological Changes in the Microenvironment. Cancer Cell 32, 42-
56.e6 (2017).
84. Ceccarelli, M. et al. Molecular Profiling Reveals Biologically Discrete Subsets and
Pathways of Progression in Diffuse Glioma. Cell 164, 550–563 (2016).
85. Roche, K. E., Weinstein, M., Dunwoodie, L. J., Poehlman, W. L. & Feltus, F. A. Sorting
Five Human Tumor Types Reveals Specific Biomarkers and Background Classification
Genes. Sci. Rep. 8, 8180 (2018).
86. Carithers, L. J. et al. A Novel Approach to High-Quality Postmortem Tissue Procurement:
The GTEx Project. Biopreserv. Biobank. 13, 311–319 (2015).
87. Bateman, A. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 47,
D506–D515 (2019).
88. Binns, D. et al. QuickGO: A web-based tool for Gene Ontology searching. Bioinformatics
25, 3045–3046 (2009).
89. Tang, Z. et al. GEPIA: A web server for cancer and normal gene expression profiling and
interactive analyses. Nucleic Acids Res. 45, W98–W102 (2017).
90. Gilson, M. K. et al. BindingDB in 2015: A public database for medicinal chemistry,

60
computational chemistry and systems pharmacology. Nucleic Acids Res. 44, D1045–
D1053 (2016).
91. Berman, H. M. et al. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr.
58, 899–907 (2002).
92. Omar, M. A. et al. Synthesis and docking studies of novel antitumor benzimidazoles.
Bioorganic Med. Chem. 20, 6989–7001 (2012).
93. Wu, B. et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic
peptide antagonists. Science 330, 1066–1071 (2010).
94. Liu, Y., Beyer, A. & Aebersold, R. On the Dependency of Cellular Protein Levels on
mRNA Abundance. Cell 165, 535–550 (2016).
95. Greenbaum, D., Colangelo, C., Williams, K. & Gerstein, M. Comparing protein
abundance and mRNA expression levels on a genomic scale. Genome Biol. 4, 117 (2003).
96. Edfors, F. et al. Gene‐specific correlation of RNA and protein levels in human cells and
tissues . Mol. Syst. Biol. 12, 883 (2016).
97. Vogel, C. & Marcotte, E. M. Insights into the regulation of protein abundance from
proteomic and transcriptomic analyses. Nat. Rev. Genet. 13, 227–232 (2012).
98. Nancy Kendrick. A gene’s mRNA level does not usually predict its protein leve. Kendrick
labs.Inc (2014).
99. Perl, K. et al. Reduced changes in protein compared to mRNA levels across non-
proliferating tissues. BMC Genomics 18, 305 (2017).
100. Pascal, L. E. et al. Correlation of mRNA and protein levels: Cell type-specific gene

61
expression of cluster designation antigens in the prostate. BMC Genomics 9, 246 (2008).
101. Shen, M., Xu, Y. Y., Sun, Y., Han, B. S. & Duan, Y. R. Preparation of a Thermosensitive
Gel Composed of a mPEG-PLGA-PLL-cRGD Nanodrug Delivery System for Pancreatic
Tumor Therapy. ACS Appl. Mater. Interfaces 7, 20530–20537 (2015).
102. Kyburz, K. A. & Anseth, K. S. Synthetic Mimics of the Extracellular Matrix: How Simple
is Complex Enough? Ann. Biomed. Eng. 43, 489–500 (2015).
103. Tibbitt, M. W. & Anseth, K. S. Dynamic microenvironments: The fourth dimension.
Science Translational Medicine 4, (2012).
104. Singh, S. P. et al. A synthetic modular approach for modeling the role of the 3D
microenvironment in tumor progression. Sci. Rep. 5, (2015).
105. Jansen, L. E., Negrón-Piñeiro, L. J., Galarza, S. & Peyton, S. R. Control of thiol-
maleimide reaction kinetics in PEG hydrogel networks. Acta Biomater. 70, 120–128
(2018).
106. Deforest, C. A., Polizzotti, B. D. & Anseth, K. S. Sequential click reactions for
synthesizing and patterning three-dimensional cell microenvironments. Nat. Mater. 8,
659–664 (2009).
107. Wang, Y., Chen, C. L. & Iijima, M. Signaling mechanisms for chemotaxis. Development
Growth and Differentiation 53, 495–502 (2011).
108. Jin, T. Gradient sensing during chemotaxis. Curr. Opin. Cell Biol. 25, 532–537 (2013).
109. Sakai, M. & Shimizu, S. Indentation rheometry for glass-forming materials. J. Non. Cryst.
Solids 282, 236–247 (2001).

62
110. Nadermann, N. K., Davis, E. M., Page, K. A., Stafford, C. M. & Chan, E. P. Using
Indentation to Quantify Transport Properties of Nanophase-Segregated Polymer Thin
Films. Adv. Mater. 27, 4924–4930 (2015).
111. Pfeifer, C. R., Alvey, C. M., Irianto, J. & Discher, D. E. Genome variation across cancers
scales with tissue stiffness - An invasion-mutation mechanism and implications for
immune cell infiltration. Current Opinion in Systems Biology 2, 103–114 (2017).





























