Embed Size (px)
Citation preview

Constant centrifugal potential approximation for atom-diatom chemical reaction dynamics
Kengo Moribayashi, Shoji Takada, and Hiroki Nakamura Division of Theoretical Studies, Institute for Molecular Science, Myodaiji, Okazaki 444. Japan
(Received 28 September 1993; accepted 8 December 1993)
The constant centrifugal potential approximation (CCPA) is generalized so as to be applicable to the reactions of rotationally excited reactants. The accurate- calculations of reaction probabilities are required only for J< 1 CIil w , Ji , (< * ) where J is the total angular momentum quantum number, ji represents the initial rotational state of a reactant diatomic molecule, pi is the z component of J in the body-fixed frame in the initial arrangement and 1 lnil MAx is the maximum of such I nil’s that give significant contributions to the reaction. The method is applied to the D+Hz(ui=O,ji) reaction with use of the hyperspherical coordinates, and is proved to be useful by comparing the results with those, of the authors’ own accurate calcula- tions. The reaction mechanisms are clarified with respect to the dependence on fii and ji.
I. INTRODUCTION ing the reaction dynamics with much less numerical effort.
Recent progress in the quantum mechanically accurate treatments of atom-diatom chemical reaction dynamics is remarkable,‘-” enabling us to investigate the effects of po- tential energy surface topography on the reaction dynamics without ambiguity and making an interplay among dynam- ics theory, experiment, and quantum chemistry possi- ble.‘6*2@z3 At the same time, however, we have to make more effort to further develop better illuminating approx- imate theories. This is indispensable for challenging the reaction systems involving heavier atoms and polyatomic molecules. As one of such practically useful approxima- tions the constant centrifugal potential approximation (CCPA) (or the energy shift approximation) has been proposed,‘7,‘8,2‘%25 in which the centrifugal potential is re- placed by the constant value at a certain representative position. With use of the accurate results only for J=O, where J is the total angular momentum quantum number, this approximation enables us to estimate integral cross section and rate constant quite accurately and furthermore to analyze the various reaction mechanisms. This can also save a lot of CPU time.
This paper is organized as follows: Section II summa- rizes some basic equations on which the later discussions are based. The equations both in the hyperspherical coor- dinates5 and in the Jacobi coordinates1’24 are provided. Sec- tion III generalizes the CCPA to the case of rotationally excited reactants (j+ 1), being based on the basic S&r& dinger equations in Jacobi coordinates in the body-fixed frame. Section IV explains the actual method of calculation with use of the hyperspherical coordinate approach. The generalized CCPA requires the accurate calculations only at J< I ai] -. Here I nil - is the maximum among these I Cni 1 ‘s which give non-negligible contributions to the reaction. In order to check the validity of the generalized CCPA, however, we have carried out the accurate calcu- lations up to JhlAx at which the cross sections are well converged. Our own method of doing this accurate calcu- lation is explained also in this section. In Sec. V we have applied our method to
The CCPA proposed so far, however, can be applied only to the case of ji=0,17*18 where ji is the initial rota- tional quantum number of a reactant diatomic molecule. This is because the reaction mechanisms for j&O are dif- ferent for J< j, and J>ji. Although there has not been made much work of the quantum mechanically accurate calculations for ji> 1, 14-16 this tendency can be seen from Fig. 1 of Ref. 14 [see also Figs. l(b), l(c), 3(b), and 3(c) of this work]. This comes from the different contributions from the different Szi components, where fii is the z com- ponent of J (or ji) in the body-fixed frame (see Table I of Ref. 1). In the present work we generalize the CCPA to the case of j&l. This is important, because the informa- tion on the dynamics of ji>l is naturally crucial for clar- ifying the overall reaction mechanisms, especially in com- parison with experiment. Together with the power of hyperspherical coordinate approach, this generalized CCPA approximation will be very useful for comprehend-
D+H2( ji,ui=O) -+DH( jf,uf=O) +H, (1) where uk(A=i,f ) represents the vibrational quantum number in the initial (i) or final (f ) channel. The cases of j,+l and 2 are considered at the total energy of E,, =0.6- 1.086 eV. The potential energy surface employed here is the one by Liu-Siegbahn-Truhlar-Horowitz (LSTH) .26p27 Concluding remarks are given in Sec. VI.
II. PRELIMINARIES
For later convenience, wd summarize the basic equa- tions both in the hyperspherical coordinates5 and in the Jacobi coordinates.‘*24 In the actual calculations in the in- ternal region, we use the adiabatically adjusting principal axisshyperspherical (APH) coordinate system composed of six variables, i.e., three Euler angles (c&r) and three hyperspherical coordinates (p,8,~).~ On the other hand, we rely on the equations in the Jacobi coordinates in order to generalize the CCPA approximation.
The total Hamiltonian of triatomic system in the APH coordinates can be written as
4284 J. Chem. Phys. 100 (6), 15 March 1994 0021-9606/94/100(6)/4284/10/$6.00 @ 1994 American Institute of Physics
Downloaded 13 Feb 2001 to 133.30.52.73. Redistribution subject to AIP copyright, see http://ojps.aip.org/jcpo/jcpcpyrts.html

Moribayashi, Takada, and Nakamura: Constant centrifugal potential approximation 4285
H(e4tay :p) = -$ j -g p5 $+H&Q5 :p)
+HJ(w,aY :p) (2)
with
p=/zcz (3)
where mx(X=A, B,C) is the mass of the atom X. Here H,, and HJ represent the J-independent and the J-dependent part of the Hamiltonian at fixed p, respectively, and are explicitly expressed as
Ho(f@:p)=-$ 1
&$,sin28a+- a2
ae sin2 O&G? 1 and
+ V(&#,p) (4)
HJ(&#,a,P,y :p> = fm(~)+mw J(J+l) QP2
+$ i
A(@ fB(@) c(e)- 2 1 J2
z
+ A(e;;pf(e! (J;+J?,
+i a +g We> 3 <S-J+),
where Y(&,A,p) is the potential,
4=7T/2--2x,
1 A(8) =- i+sin 8’
1 B(B) =-
2sh2e’
1 c(e) =- l-sine’
c0se me) ==.
Here Jh is defined as
J,=J,riJ,,
(5)
(6)
(7)
(8)
(9)
(10)
(11)
where J,(J,) is the x(y) component of J. The total wave function is expanded as
\uJ+iwa,m = C P-~+ fj(p)&w4,a,ay :p), n
(12)
where p represents parity and I/?( &p,@,y :p> is the nth eigenfunction of the surface Hamiltonian ( =HO+HJ),
(Ho+Hh,@(&k&y :p> = U~(ph@m+,P,y :p>. (13)
The actual numerical computations are carried out within the framework described above. The scattering ma- trix is obtained by solving the close-coupling scattering equation with respect to p by the R-matrix propagation method.28’2g The details will be described in Sec. IV.
As was done in the previous works,‘7918 in order to obtain the scattering matrix in the asymptotic region we make a transformation from the APH coordinates to the Delves, and finally to the Jacobi coordinates.5
In order to generalize the CCPA approximation, we go back to the Schrijdinger equation in the body-fixed Jacobi coordinate system (R, ,r~ ,e~>. 1924 This is given as
H;;,o+$ 2&l
[J(J+ 1) -2C& -E I y;, ’ /I
= - &, H;,QL+w%A+W’ (14)
where Hfzg is the J-independent part of the Hamiltonian, 2
L?R +ld, ii 3
RL c3R: a r,& A I
2p@2pR:
+ WWa,W, (15)
and Rn(r~) is the mass scaled translational (vibrational) Jacobi coordinates. H z1 oL* I represents the Coriolis cou- pling expressed explicitly as
fi2 H&-+~=-Y d
W-G J(J+l)-&(&*lli?, (16)
where j; are the’ usual lowering ( - ) and raising ( + ) operators corresponding to j,. We should note that fi, is the z, component of j, and J.’ The zL is taken to be along Rk. Eq. (14) will be used in Sec. III in order to introduce the generalized CCPA.
The reaction probability is given by
PJ. ufJ$““iji/2->ji+ 1 ri lf
=& g z lq$~12 If “I
and the corresponding integral cross section and rate con- stant are obtained from
uvfj$‘-ui jiA (18)
and
with kuiji = ,/w/?i, where IA is the orbital angu- lar momentum quantum number in the /z arrangement, Eu,ji represents the initial internal energy of the reactant
J. Chem. Phys., Vol. 100, No. 6, 15 March 1994 Downloaded 13 Feb 2001 to 133.30.52.73. Redistribution subject to AIP copyright, see http://ojps.aip.org/jcpo/jcpcpyrts.html

4286 Moribayashi, Takada, and Nakamura: Constant centrifugal potential approximation
diatomrc molecule, K is the Boltzmann constant, T is tem- perature, and pi is the reduced mass in the initial arrange- ment.
III; CONSTANT CENTRIFUGAL POTENTIAL APPROXIMATION (CCPA) FOR ROTATIONALLY EXCITED REACTANTS
As was demonstrated in Refs. 17 and 18, the CCPA for ji=O was proved to be very useful for estimating integral cross’ section and rate constant for the reactions of rota- tionally unexcited reactants. The accurate information only for J=O is required. This approximation is based on the physical insight that reactive transition occurs in a spatially localized region around the potential ridge. The centrifugal potential [=#J(J+ 1 )/R’] is replaced by a constant value at a certain representative position Rt, and the reaction probability for J>l is approximated as PJ. ~~~~,+4jran(Ea)--P~~~,+,ii=~~[Et~-BtJ(J+l)l
(20) with
(21)
where Et, is the translational energy and n(d’) represents the initial (final) arrangement. This approximation was conflrmed to be effective not only in the RIOSA and the adiabatic-bend approximations,24 but also in the accurate treatment of J=0.‘73’8
In the case of ji> 1, however, approximation (20) can not be directly applied, because there exist nonzero fij channels with different contributions to the reactions. f&. can take the following values, depending on the value of J:
flf= -ji,-ji+l,..., jj-1,ji when J>ji
1 ~~ -J,-J+l,..., J-1,J when J<ji. (22)
Since the number of fii for J< ji is dependent on J and the reaction mechanisms are different for different nls, thus for J< ji and J> ji, we have to carry out the accurate calculations for J<ji in order to generalize the CCPA. That is to say, the generalization of the CCPA should be made on the basis of the accurate results of J= jj for all possible &‘s. ‘In order to do this it is more convenient to employ Eq. (14) in the Jacobi coordinates. This equation with the Coriolis couplings neglected can be rewritten as
If=’ ?- [J(J+l) -2i-k?] -E Y”! JAc+2& I I JJi”i
=
(J(J+l)-jAii+l)) v$;ni
E- $g 1 =o, 03)
where H ~~~ is the Hamiltonian for J= ji with the Coriolis
I y(i) JJi “i
couplings neglected. Since pi ranges over the interval ( - ji,- jj> which is independent of J when J> ji, Eq. (23) suggests that the CCPA can be generalized as follows:
S;$~jn-(E,,) -S;+$f~Wtr) 1’1 I I
~S~~~,~j,..[E,,-BtJ(Jil) ,‘I I I
(24)
or
p J>.ji UfJfltcUiJ.il
(Et,) =P ;;;;~jiA(E,r)
E p J=.ji ,,u.ilAIEtr- BtJ(J+ 1) UfJfA I
with
+Btji(ji+l)l (25)
(26)
The reaction probability P :fjf A, Cu. jiA(Etr) is detined by Eq. ( 17). Approximation (25) is considered to be better than Eq. (24), since the summation and averaging proce- dures are involved in obtaining the probability. In the present work, the actual accurate calculations for J< ji are carried out with use of the hyperspherical coordinate sys- tem as is explained in the next section.
As is mentioned above, the necessity of the accurate calculations for all fii components up to J= ji basically comes from the fact that the number of sli is different for J< ji and J>ji. In some cases, basically at low collision energies, however, not all of the Q components at J= ji contribute significantly to the reactions. In this case, the accurate calculations are required only up to J= 1 nil MAX< ji, where 1 Cni( MAx is the maximum of those 1 fij 1 ‘s which give significant contributions to the dynam- ics. The notations J> ji, J=jj and Btjj( ji+l) in Eqs. (24) and (25) are replaced by J> 1 InilMm, J= Ifhilm, and BtIQiI-(Ifi I i - + 1) , respectively. Naturally, the S-matrix elements for I ail > I &I MAX are assumed to be zero. Unless we can know the relative contributions from each C& Q priori, however, we have to estimate the contributions from all fij and then we can use the accurate results for J= ji in Bqs. (24) and (25).
IV. METHOD OF CALCULATION
In this section our method of accurate calculation of the S matrix for J> 1 is described. As was explained in the previous section, the accurate calculation is necessary for J< ji or I fij I -, even if we employ the CCPA approxi- mation.
J. Chem. Phys., Vol. 100, No. 6, 15 March 1994
Downloaded 13 Feb 2001 to 133.30.52.73. Redistribution subject to AIP copyright, see http://ojps.aip.org/jcpo/jcpcpyrts.html

Moribayashi, Takada, and Nakamura: Constant centrifugal potential approximation 4287
TABLE I. Convergence of the reaction probabilities with respect to n for D+H,(q=O, ji=1,2) +DH(u/=O, j,)+H at J=20 and E,,=l.l eV.
f/-MAX 1 2 3 4 5
(4 ii=1 0 1 2 3 4 5 6 7 8 9
10 11 12
Total (b) /i=2
0 1 2 3 4 5 6 7 8 9
10 11 12
Total
0.6620-02 0.1470-02 0.1710-02 0.1660-01 0.3760-02 0.405 D-02 0.2180-01 0.5160-02 0.4780-02 0.2420-01 0.8360-02 0.6570-02 0.2500-01 0.1570-01 0.1370-01 0.2160-01 0.2150-01 0.2020-01 0.1690-01 0.1960-01 0.1970-01 0.1190-01 0.1420-01 0.1600-01 0.8230-02 0.1470-01 0.1690-01 0.4550-02 0.8260-02 0.1130-01 0.6770-03 0.171 D-02 0.1870-02 0.4380-04 0.2040-03 0.2800-03 0.3460-05 O.lllD-05 0.1180-05 O.l58D+CO O.l15D+OO O.l17D+OO
0.2490-02 0.1440-02 0.1310-02 0.1300-02 0.1300-02 0.9890-02 0.371 D-02 0.3320-02 0.3340-02 0.3320-02 0.1810-01 0.5160-02 0.427 D-02 0.4360-02 0.4340-02 0.2370-01 0.8490-02 0.6090-02 0.6170-02 0.6120-02 0.2750-01 0.1580-01 0.1240-01 0.1220-01 0.1210-01 0.2520-01 0.2180-01 0.1890-01 0.1840-01 0.1830-01 0.1670-01 0.1750-01 0.1710-01 0.1700-01 0.1700-01 0.8660-02 O.lilD-01 0.1190-01 O.l18D-01 0.1180-01 0.5160-02 0.9640-02 0.1050-01 0.1040-01 0.1030-01 0.2700-02 0.5050-02 0.6660-02 0.7260-02 0.7360-02 0.3330-03 0.9300-03 0.9840-03 0.1520-02 0.1530-02 0.1920~04 0.1100-03 0.1240-03 0.9900-04 0.9490-04 0.1650-05 0.8870-06 0.903 D-06 0.8710-06 0.6GOD-05 O.l41D+OO O.lOlD+OO 0.936-D-01 0.9380-01 0.9370-01
0.171 D-02 0.1740-02 0.4090-02 0.4100-02 0.493 D-02 0.495 D-02 0.671 D-02 0.671 D-02 0.1340-01 0.1340-01 0.1950-01 0.1930-01 0.1920-01 0.1910-01 0.1570-01 0.1550-01 0.1630-01 0.1610-01 0.1210-01 0.1240-01 0.303 D-02 0.3030-02 O.l7OD-03 0.1720-03 0.1160-05 0.5060-05 O.l17D+OO O.l17D+OO
The adiabatically adjusting principal axis hyperspher- ical (APH) coordinates5 are used in the actual calculation in the internal reaction zone. The surface eigenfunctions r@’ are expanded as
&w&wty :p) = ; C~~pP~(8,~ :p#&,&@,y), (27)
where Ck are the expansion coefficients,
5&.f(a,fi,,r) = 2J+1
l&2( 1 +Sno) CD &&1&) +
( - 1) J+P+nD Lfi&fk&43, (28)
and DJ M is the Wigner’s D function. Here the basis func- tions @!(0,# :p) are taken to be
cpn, @=” for a=O, r
I $F=’ for ficz)l, (29)
where ~~ are the eigenfunctions of the following eigen- value problem:
where Ho is given by Eq. (4) and a0 is the z component of Jin the APH body-fixed frame [see Bq. (5)]. The notations 0 and fin, should not be confused with fk,(A=i,f ). The reason why we employ this Hamiltonian to construct the basis functions is as follows: Because of the singularity of C( 0) at 8=?r/2 (collinear conformation), the functions {4?=“3 which have nonzero finite amplitudes at 8=r/2 can not be enough as the basis function+ Ln order-to ta-ke _ into account ihis smgularity and to have proper behavior of the wave function there for @l, we have introduced the second term of Ha0 in Eq. (30). Since the basis func- tions {$p=’ 3 properly go to zero at 8=?r/2, we do not need other functions with an,>2 for basis functions. Singu- larities of B(8) and D(e) at 8=0 (triangular conforma- tion) do not cause any trouble, since the eigenfunctions have no significant amplitude at 8 = 0 in the present D + H2 system.
The eigenfunctions (@3 of Ha0 are calculated by the discrete variable representation (DVR) devised by Light and co-workers3op31 in the same way as in Refs. 17 and 18. The surface eigenfunctions and eigenvalues of the total Hamiltonian at fixed p are obtained by diagonalizing the following matrix:
J. Chem. Phys., Vol. 100, No. 6, 15 March 1994 Downloaded 13 Feb 2001 to 133.30.52.73. Redistribution subject to AIP copyright, see http://ojps.aip.org/jcpo/jcpcpyrts.html

4288 Moribayashi, Takada, and Nakamura: Constant centrifugal potential approximation
TABLE II. Cross sections@ 4) for D+H,(u,=O, j,=O) -DH(u,.=O, j/) +H at I&=0.78, 0.93, and 1.086 eV in comparison with the results of Zhang and Miller(ZM) (Ref. 9).
(a) 0.78 eV (b) 0.93 eV (c) 1.086 eV
if Present ZM Present ZM Present ZM
0 0.148&B 0.139Eu O.lOlEo .0.994E- 1 0.6923- 1 0.6753- 1 1 0.354hu 0.377&O 0.276Eo - 0.29530 0.19ohtl 0.202&u 2 0.485&U 0.515&O 0.407E1) 0.463EO 0.29330 0.334EO 3 0.569Eo 0.535hu 0.594Eo 0.597Eo 0.446EO 0.477Eo 4 0.497Eo 0.424hU 0.781BU ~0.66530 0.698&O 0.631Eo 5 0.29830 0.275Eo 0.741hu 0.656EO 0.889hU 0.717lxl 6 0.134EO 0.13ohu 0.485Eo 0.531Eo 0.82230 0.766EO 7 0.545E- 1 0.474E- 1 0.322BJl 0.336EU 0.58OEO 0.681Eo 8. 0.7033-2 O.l14E-1 0.174hu 0.152EXI 0.480&U 0.46OEo 9 0.3963-3 0.591E-3 0.287E- 1 0.357E- 1 0.282lXI 0.23930
10 0.785E--2~ m::~ 0:261E-2 0.554E- 1 0.856E- 1 11 O.l89E-2 0.604E-3 0.733E-2 O.l6OE- 1 12 0.456E-2 0.595E-3
?2 = W f?(p)Gnn&+- (@.yA(B)-tB(~) ,o:W+l)soo.+$
( I a: C(O)-
A(B) +HN GP
2 2 I > cpf’ f126ant
--$ (~:Jc(e) lg%$ml+& (~~IA(e)--B(e)l~>a’><D~~IJ:+J2_I~~,M)
+--f$ (@lo(e) $ l@Ft) ~~~M~J--J+I~~M>,
where in accordance with Eq. (29) fle takes the following value:
I 0 for fi=O, ‘O= 1 for !&l. (32)
As usual, the D functions HoM satisfy the following equa- tions:
J,DJ,,= ,/ f fi (J*Q+~,(JFQ>D.,,,. (33)
The close-coupling equations in the representation of surface adiabatic states {4?> are solved by the diabatic-by sector method and the R-matrix propagation method.28’29 The whole range of p is divided into a large number of small sectors. The basic quantities required to solve the coupled equations are (i) the eigenvalues (Vf( p)} of Eq. ( 13), and (ii) the overlap matrices Q $,$k’ between the eigensurface functions in the adjacent sectors:
Q $;k)E (~~‘k-l’(e,$&?~~ :pk--l) 1 &+k’(@#%~9&Y :pk))
= r;. C~m(pk-l)C,~(pk)q~(k) I I
(34)
with
q”‘k’=(@~(k-l)(e,$ :pkml) I@f’k’(e,+ :Pk)), rs (35)
where pk represents a representative position of p in the kth sector.
(31)
Launay and Dourneuf adopted the wave functions ($F}, which are the eigenfunctions of Ha0 in Eq. (30), directly in the close-coupling method as channel func- tions. lo-r3 Thus they had to employ $? for a,= 0 - 3. On the other hand, we have to calculate #F only for Cn,=O and 1 [see Eq. (29)]. Therefore, the CPU time in our treat- ment can be less.
In the asymptotic region the R matrix propagated in the APH system is transformed into the one in the Delves coordinates and the latter is used to yield the S matrix which satisfies the proper boundary conditions in the Ja- cobi coordinates.5
V. RESULTS AND DISCUSSION
Here we apply our method to
D+Hz(vi=0,jj=1,2)+DH(vf=0,jf)+H (36)
at the total energy Et,,=0.6- 1.086 eV measured from the bottom of the potential in reactant region. The zero point energy of H2 is 0.2683 eV, and the rotational excitation energies of Hz are 0.0147 and 0.0439 eV for ji=l and 2, respectively. In order to check the validity of the presently generalized CCPA approximation, we also carried out ac- curate calculations.
J. Chem. Phys., Vol. 100, No. 6, 15 March 1994 Downloaded 13 Feb 2001 to 133.30.52.73. Redistribution subject to AIP copyright, see http://ojps.aip.org/jcpo/jcpcpyrts.html

Moribayashi, Takada, and Nakamura: Constant centrifugal potential approximation 4289
A. Accurate calculation
In comparison with the results of Launay and Dour- neuf” and those of Zhang and Miller,’ we have tested our quantum mechanically accurate calculations, for J> 1. First, we have investigated the convergence of reaction probability with respect to the number of a, projection of J onto the body-fixed frame z axis [see Eq. (27)] in the APH system. Launay and Dourneuf found in their calcu- lations that the cross sections for H+H, at the total ener- gies of 1.1 and 1.3 eV converged well with aMAx (maxi- mum fi taken into account) =3.” We also tested the convergence with respect to fi in order to save the CPU time and main memory. Table I shows the results for re- action (36) at J= 20 and E,,, = 1.1 eV. From this table we see that while the errors in the case of a-=2 are more than lo%, those in the case of fInMAx=3 are less than 6% except for the case of large j, with very small probabilities. Since the maximum J necessary to get the converged cross sections is roughly equal to 20 at the energies we are in- terested in and the convergence of probability with respect to 0 becomes better at smaller J’s, we can safely take sz -=3 in the present calculation. Next, we have calcu- lated the cross sections for ji=O to compare with those of Zhang and Miller (ZM).9 Table II gives the results for each j, at Et,,= 0.78, 0.93, and 1.086 eV. Table III gives the total cross sections summed over all possible j, as a function of E,, . The discrepancy between the present and the ZM results for individual j, (Table II) amounts to at the most 20% except for the case of large j, with very small probabilities. This discrepancy is acceptable enough for the present purpose, especially if we consider the fact that the two methods are completely different. Moreover, the agreement in Table III is very good and we judge from the above analysis that our code for the accurate calcula- tions works all right.
B. Reaction probability
Here and in the next subsection we demonstrate how the generalized CCPA approximation works. The repre- sentative position Rt of reactive transition is taken to be Rf which is the initial translational Jacobi coordinate of the transition state (saddle point). Figures 1 (a)-1 (c) show the reaction probabilities summed up over all jf for process (36) with j,= 1 as a function of J at Etot=0.6, 0.75, and 1.086 eV. The CCPA (J=O) and CCPA (J= 1)
TABLE III. Cross sections(in 4) for D+H,(u,=O, ji=O)+DH(nf =O,P,jJ +H in comparison with the results of Zhang and Miller(ZM) (Ref. 9).
Total energy (eV) Present
0.6 0.144h-o 0.65 0.741htl 0.7 1.57JzO 0.78 2.54Eo 0.85 3.26hU 0.93 3.9230 1.086 4.81&U
ZM
0.143Eo 0.748EO 1.57hu 2.46Eo 3.16Eo 3.8430 4.68Ed
mean the CCPA approximations based on the accurate result of J=O and J= 1, respectively. The result by Zhao et al. l4 is shown in Fig. 1 (c) just for a comparison. The discrepancy between our results and those by Zhao et al. mainly comes from the fact that we employed the different potential energy surface. We employed LSTH,26927 while they used DMBE.32 In Fig. 1 (a), all three results agree relatively well, namely, even the simplest CCPA( J=O) seems to be applicable. However, in Figs. 1 (b) and 1 (c) the CCPA (J=O) is not working properly. This is because the reaction probabilities for J=O and 1 are quite different at high energies [see Figs. 1 (b) and 1 (c)l.
In order to demonstrate that this big jump comes from different contributions from the different an, components,
”
0 10 20 2
b
FIG. 1. Reaction probabilities for D+H,(u,=O, ji=l)-DH(u/=O, Zj,-) +H as a function of J: (a) E,,=O.6 eV, (b) E,,=O.75 eV, and (c) Etot= 1.086 eV; Present accurate quantum mechanical results (solid line with circles), CCPA(J=O) (dotted line with squares), CCPA (J= 1) (broken line with triangles), and the results by Zhao et al. (dash-dot line with solid rhombuses) (Ref. 14),
J. Chem. Phys., Vol. 100, No. 6, 15 March 1994 Downloaded 13 Feb 2001 to 133.30.52.73. Redistribution subject to AIP copyright, see http://ojps.aip.org/jcpo/jcpcpyrts.html

4290 Moribayashi, Takada, and Nakamura: Constant centrifugal potential approximation
we have estimated the corresponding reaction probabilities for ~i=O and 1 nil = 1 separately, as is shown in Fig. 2. This was carried out by using the transformation between the R matrix in the body-fixed (BF) frame and that in the space-fixed (SF) frame,
@(SF) =cRJW)C-’ ,
where C is a matrix defined by
(37)
&es 6 8.. rA av “A”v JAJV C( j,z,J;n,oo,). (38)
Here T~ represents the vibrational and rotational quantum numbers in the h arrangement collectively, and C( jZJ;fiOn) is a Clebsch-Gordan coefficient. By executing this transformation, we can easily pick up the component of each ai. From Fig. 2, we can see that the components of ] ail = 1 give little contribution to the probability at ener- gies lower than -0.7 eV, but become comparable with the L&=0 component at higher energies. This is the reason why the CCPA(J=O) works all right at E,,=O.6 eV [Fig. l(a)], but fails at E,,,=O.75 and 1.086 eV [Figs. l(b) and l(c)]. This means that 1 Ini]m=O<ji= 1 for Et,,s0.7 eV and I nil MAX= 1 =ji for E,,,ZO.7 eV. In Figs. 1 (b) and l(c) the CCPA(J=O) agree well with the CCPA(J = 1) at JZ 6 and JZ 20, respectively. This comes from the fact that as J increases, the shifted energy in Eq. (25), E,,-- B+J( J+ 1) + Btj,( j i+ 1) , decreases to the region where the contribution of s1,=0 dominates (see Fig. 2).
Figures 3 (a)-3 (c) give the similar results as Fig. 1 for ji=2 at E,,=O.6, 0.75, and 0.95 eV. In this case the CCPA(J=l) works all right up to E,,,=O.75 eV [Fig. 3 (b)], but fails at E,,=O.95 eV [Fig. 3(c)]. The reason for this is the same as before, as is shown in Fig. 4, where the relative contributions from the three components I nil =O, 1, and 2 are shown as a function of total energy. This figure tells that ] nil - may be taken to be 0, 1, and 2 at E,,,SO.55 eV, 0.55 5 E,,,dO.8 eV, and E,,? 0.8 eV, re- spectively. In any case, the above analysis clearly tells that the accurate estimate of reaction probability for J<ji or I&lMAX is necessary for the CCPA approximation to be applicable to the reaction process with a reactant in the
rotationally excited state ji. In Fig. 3 (c) we can also see the agreement between CCPA( J= 1) and CCPA( J=2) at JZ 12 and among three CCPA results at JZ 16. The reason for this is. the same as in the case of ji= 1.
In both cases Of ji= 1 and 2 examined above, the ni=O component dominates the reaction process at low energies. The ni=O component of the total wave function is pro- portional to the spherical harmonics Yi,,o,=c( ei,O), where ei is an angle between the two Jacobi vectors Ri and ri, and thus indicates that the two atoms are apt to concentrate around Gj=O. Roughly speaking, this tells that the reac- tion proceeds mainly along the collinear configuration at low energies, which is in good accordance with the fact that the H3 system has the collinear transition state.
As is seen in Figs. 1 (c) and 3 (c), the CCPA proba- bilities oscillate and deviate from the accurate results ap-
FIG. 2. The IfijI components of the probability for J= 1 for the same reaction as in Fig. 1 as a function of total energy: ai= (solid line with solid circles), and 1 &I = 1 (dotted line with solid squares).
FIG. 3. The same as Fig. 1 for ji=2. (a) E,,,,=O.6 eV, (b) Etot=0.75 eV, and (6) E,,=O.95 eV; CCPA(J=2) (dash-dot line with rhombuses) is added.
J. Chem. Phys., Vol. 100, No. 6, 15 March 1994
Downloaded 13 Feb 2001 to 133.30.52.73. Redistribution subject to AIP copyright, see http://ojps.aip.org/jcpo/jcpcpyrts.html
Moribayashi, Takada, and Nakamura: Constant centrifugal potential approximation
0.6 0.8 1 Enerw(W
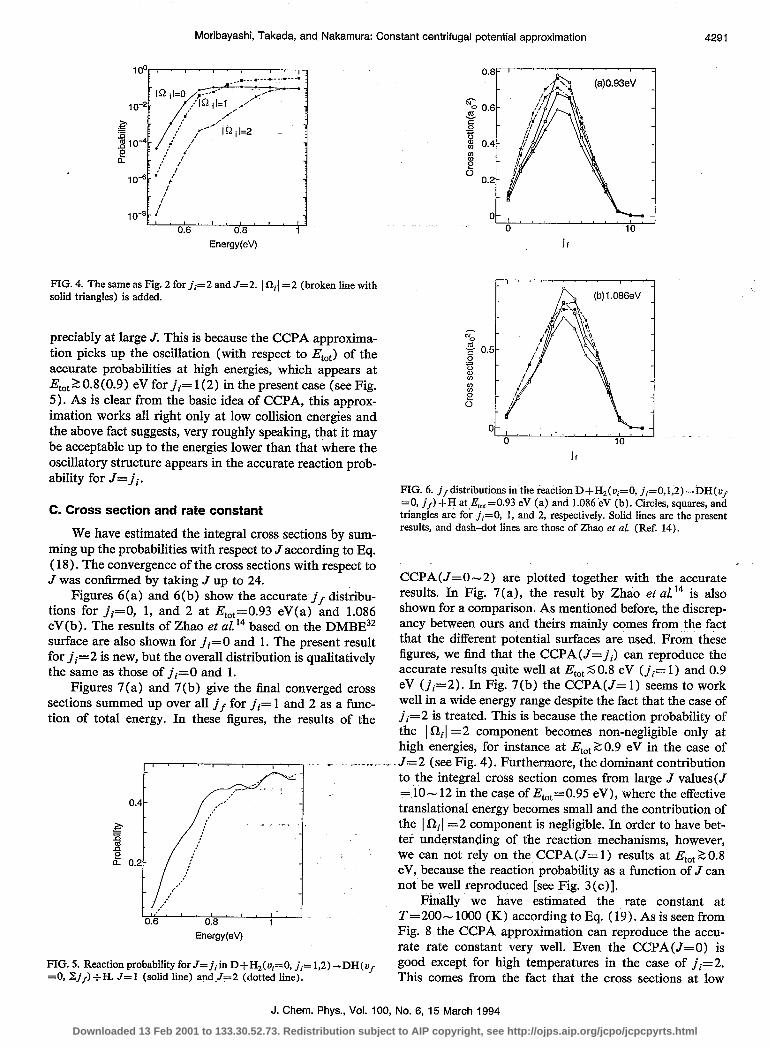
FIG. 4. The same as Fig. 2 for ii= 2 and J= 2. I a,[ = 2 (broken lie with solid triangles) is added.
preciably at large J. This is because the CCPA approxima- tion picks up the oscillation (with respect to E,,) of the accurate probabilities at high energies, which appears at Et,,, 2 0.8 (0.9) eV for ji= l(2) in the present case (see Fig. 5). As is clear from the basic idea of CCPA, this approx- imation works all right only at low collision energies and the above fact suggests, very roughly speaking, that it may be acceptable up to the energies lower than that where the oscillatory structure appears in the accurate reaction prob- ability for J= ji.
C. Cross section and rate constant
We have estimated the integral cross sections by sum- ming up the probabilities with respect to J according to Eq. ( 18). The convergence of the cross sections with respect to J was confirmed by taking J up to 24.
Figures 6(a) and 6(b) show the accurate j, distribu- tions for ji=O, 1, and 2 at Et,,=0.93 eV(a) and 1.086 eV (b) . The results of Zhao et al. l4 based on the DMBE3’ surface are also shown for ji=O and 1. The present result for ji=2 is new, but the overall distribution is qualitatively the same as those of ji=O and 1.
Figures 7(a) and 7(b) give the final converged cross sections summed up over all j, for ji= 1 and 2 as a func- tion of total energy. In these figures, the results of the
Ener9W4
FIG. 5. Reaction probability for J=j,in D+H,(a,=O, ji= 1,2) -DH(a,- ~0, Zjci/) +H. J= 1 (solid line) and~J=2 (dotted line).
0.8
s- - 5
0.6
E ‘G p 0.4 t: 6
0.2
FIG. 6. j,-distributions in’the &action D+H,(u,=O, j,=O,1,2) -DH(u,, =O, jr) +H at &,,=‘0.93 eV (a) and 1.086 eV (b). Circles, squares, and triangles are foi ji=O, 1, and 2, respectively. Solid lines are the present results, and dash-dot lines are those of Zhao et al. (Ref. 14).
CCPA(J=O-2) are plotted together with the accurate results. In Fig. 7(a), the result by Zhao ef al. l4 is also shown for a comparison. As mentioned before, the discrep- ancy between ours and theirs mainly comes from the fact that the different potential surfaces are used. From these figures, we find that the CCPA( J= ji) can reproduce the accurate results quite well at Et,, 5 0.8 eV (ji= 1) and 0.9 eV (ji=2). In Fig. 7(b) the CCPA( J= 1) seems to work well in a wide energy range despite the fact that the case of ji=2 is treated. This is because the reaction probability of the I nil = 2 component becomes non-negligible only at high energies, for instance at Et,,2 0.9 eV in the case of J= 2 (see Fig. 4). Furthermore, the dominant contribution to the integral cross section comes from large J values( J = lo- 12 in the case of E,,,=O.95 eV>, where the effective translational energy becomes small and the contribution of the I &I =2 component is negligible. In order to have bet- ter understanding of the reaction mechanisms, however, we can not rely on the CCPA(J= 1) results at Et,, 2 0.8 eV, bet-ause the reaction probability as a function of J can not be well reproduced [see Fig. 3 (c)l.
Finally we have estimated the rate constant at T=200- 1000 (K) according to Eq. (19). As is seen from Fig. 8 the CCPA approximation can reproduce the accu- rate rate constant very well. Even the CCPA( J=O> is good except for high temperatures in the case of ji=2. This comes from the fact that the cross sections at low
J. Chem. Phys., Vol. 100, No. 6, 15 March 1994
Downloaded 13 Feb 2001 to 133.30.52.73. Redistribution subject to AIP copyright, see http://ojps.aip.org/jcpo/jcpcpyrts.html

4292 Moribayashi, Takada, and Nakamura: Constant centrifugal potential approximation
4-
2-
O- a
FIG. 7. Integral cross sections as a function of total energy: (a) ji= 1 and (b) j~2; Present accurate quantum mechanical results (solid line with circles), CCPA (J=O) (dotted line with squares), CCPA (J=l) (bro- ken line with triangles), CCPA(J=I) (dash-dot line with rhombuses), and the results by Zhao et aZ. (solid rhombuses) (Ref. 14).
energies, where the CCPA is valid, dominate because of the Boltzmann factor.
VI. CONCLUSION
The constant centrifugal potential approximation (CCPA), which has been proposed for the reaction with ji (initial rotational quantum number) =0 and proved to work well for estimating integral cross section and rate constant,“*‘* is generalized so as to be applicable to the case of rotationally excited reactants. This generalized CCPA requires accurate evaluation of the reaction proba- bilities for J< 1 Cnil- \ Ji , (< - ) because the reaction mecha- nisms are different for different L$‘s, thus for J> I Cli( MAx and J< I ail MAx. Here I nil MAx is the maximum of such I &l’s that give non-negligible contributions to the reac- tion. This CCPA (J= I lnil MAx) approximation is demon- strated to be very useful by taking the D+H, reaction as an example. This can save a lot of CPU time, and is still powerful for clarifying the reaction mechanisms. As is ex- pected, the approximation works well at low collision en- ergies, roughly speaking at energies lower than the first maximum of reaction probability. It works very well for reaction rate constant in the region of room temperature. The ni (body-fixed z component of J) dependence and ji dependence of the reaction are also clarified and the neces- sity of accurate calculation for J< I fii I MAx is confirmed. In
FIG. 8. Rate constants as a function of temperature l? (a) j,= 1 and (b) j,=2; Present accurate quantum mechanical results (solid line with cir- cles), CCPA (J=O) (dotted line with squares), CCPA(J=l) (broken line with triangles), and CCPA(J=2) (dash-dot line with rhombuses).
accordance with the collinear transition state, the fii=O component is found to be dominant at low energies.
In the present application of CCPA, we have employed Rf~-(initial translational Jacobi coordinate of transition state) as Rt in Eq. (26). Generally speaking, it is better to take an average,
j&z=; (j+2+$2)) (39)
for heterogeneous reaction systems, as was demonstrated in Ref. 18. The present D +Hz system is quite symmetric, and this dose not give a big difference.‘77’8 An unsolved problem is a choice of Rt in the case of such strongly heterogeneous systems that the conventional transition state (saddle point) is located far from the potential ridge where particle rearrangement occurs. The potential ridge in 3D reaction-is two-dimensional and it is not clear how to define Rt in such cases. Another interesting thing to be further investigated is the applicability of the approxima- tion for estimating differential cross sections, although the accuracy is naturally expected to be worse.
ACKNOWLEDGMENTS
The present work was supported in part by a Grant- in-Aid for Scientific Research on Priority Area “Theory of Chemical Reactions” from the Ministry of Education, Sci-
J. Chem. Phys., Vol. 100, No. 6, 15 March 1994
Downloaded 13 Feb 2001 to 133.30.52.73. Redistribution subject to AIP copyright, see http://ojps.aip.org/jcpo/jcpcpyrts.html

Moribayashi, Takada, and Nakamura: Constant centrifugal potential approximation 4293
ence and Culture of Japan. Numerical calculations were carried out at the computer center of Institute for Molec- ular Science.
‘G. C. Schatz and A. Kuppermann, J. Chem. Phys. 684642 (1976). ‘A. Kuppermann and P. G. Hipes, J. Chem. Phys. 84, 5962 (1986). 3F. Webster and J. C. Light, J. Chem. Phys. 85, 4744 (1986). 4K. Haug, D. W. Schwenke, Y. Shima, D. G. Truhlar, J. Zhang, and D.
J. Kouri, J. Phys. Chem. 98, 6757 (1986). ‘R. T Pack, and G. A. Parker, J. Chem. Phys. 87, 3888 (1987). 6G. C. Schatz, Chem. Phys. Lett. 150, 92 (1988). ‘D. E. Manolopoulos and R. E. Wyatt, Chem. Phys. Lett. 1.59, 123
(1989). *M. Baer, J. Chem. Phys. 90, 3043 (1989). 9J. Z. H. Zhang and W. H. Miller, J. Chem. Phys. 91, 1528 (1989).
“J. M. Launay and M. Lc Doumeuf, Chem. Phys. Lett. 163, 178 (1989). “J. M. Launay and M. Le Doumeuf, Chem. Phys. Lett. 169,473 (1990). 12J. M. Launay and S. B. Padkjax, Chem. Phys. Lett. 181, 95 (1991). 13B Lepetit and J. M. Launay, J. Chem. Phys. 95, 5159 (1991). I’M. Zhao, D. G. Truhlar, D. W. Schwenke, and D. J. Kouri, J. Phys.
Chem. 94, 7074 (1990). “W. J. Keogh, A. I. Boothroyd, P. G. Martin, S. L. Mielke, D. G.
Truhlar, and D. W. Schwenke, Chem. Phys. Lett. 195, 144 (1992). 16D. Neuhauser, R. S. Judson, D. J. Kouri, D. E. Adelman, N. E. Shafer,
D. A. V. Kliner, and R. N. Zare, Science 257, 5 19 (1992).
“S. Takada, A. Ohsaki, and H. Nakamura, J. Chem. Phys. 96, 339 (1992).
“S. Takada K. Tsuda, A. Ohsaki, and H. Nakamura, in Advances in Molecular’ Vibrations and Collision Dynamics: Quantum Reactive Scat- tering, Vol. IIA, edited by J. M. Bowman (JAI, Greenwich, 1993).
19S. E. Branchett, S. B. Padkjsr, and J. M. Launay, Chem. Phys. Lett. 208, 523 (1993).
*OR. E. Continetti, B. A. Balko, and Y. T. Lee, J. Chem. Phys. 93, 5719 (1990).
21W. H. Miller and J. Z. H. Zhang, J. Phys. Chem. 95, 12 (1991). *‘D. A. V. Kliner, K. D. Rinnen, and R. N. Zare. Chem. Phvs. Lett. 166,
107 (1990). 23D E Adelman, N. E. Shafer, D.
&em. Phys. 97, 7323 (1992). A. V. Klmer, and R. N. Zare, J.
24A. Ohsaki, and H. Nakamura, Phys. Rep. 187, 1 (1990). 25J. M. Bowman, Adv. Chem. Phys. 61, 115 (1985). 26P. Siegbahn and B. Liu, J. Chem.Phys. 68, 2457 ( 1978). “D. G. Truhlar and C. J.-Horowitz, J. Chem. Phys. 68, 2466 (1978). **J. C. Light and R. B. Walker, 3. Chem. Phys. 65, 4272 ( 1976). 29E. B. Stechel, R. B. Walker, and J. C. Light, J. Chem. Phys. 69, 3518
(1978). 3oJ. C. Light, I. P. Hamilton, and J. V. Lill, J. Chem. Phys. 82, 1400
(1985). 3’R. M. Whitnell and J. C. Light, J. Chem. Phys. 90, 1774 (1989). 32A. J. C. Varandas, F. B. Brown, C. A. Mead, D. G. Truhlar, and N. C.
Blais, J. Chem. Phys. 86, 6258 (1987).
J. Chem. Phys., Vol. 100, No. 6, 15 March 1994
Downloaded 13 Feb 2001 to 133.30.52.73. Redistribution subject to AIP copyright, see http://ojps.aip.org/jcpo/jcpcpyrts.html





























