Embed Size (px)
Citation preview

Review
1052
Construction of Linear Polymers, Dendrimers,Networks, and Other Polymeric Architecturesby Copper-Catalyzed Azide-AlkyneCycloaddition ‘‘Click’’ Chemistry
Jeremiah A. Johnson, M. G. Finn, Jeffrey T. Koberstein,Nicholas J. Turro*
The enrichment of materials synthesis with the diverse chemical building blocks and func-tional groups of small molecule organic chemistry has been greatly accelerated in recent yearsby the introduction of the copper-catalyzed azide-alkyne cycloaddition (CuAAC). The efficiencyand modular nature of this unique reaction enablesmaterials chemists to prepare novel functionalmaterials of unprecedented complexity. This reviewsummarizes the application of CuAAC to the field ofmaterials construction, defined here as the prep-aration of materials with architectural integritydependent upon the triazole linkage. Recent examples,including linear polymers, dendrimers, polymer net-works, polymeric nanoparticles, and other polymericarchitectures, are described.
J. A. Johnson,Department of Chemistry, Columbia University, 3000 Broadway,MC3157, New York, New York 10027, USAM. G. FinnDepartment of Chemistry and The Skaggs Institute for ChemicalBiology, The Scripps Research Institute, 10550 North Torrey PinesRoad, La Jolla, California 92037, USAJ. T. KobersteinDepartment of Chemical Engineering, Columbia University, 500W. 120th Street, New York, New York 10027, USAN. J. TurroDepartment of Chemistry and Chemical Engineering, ColumbiaUniversity, 3000 Broadway, MC3157, New York, New York 10027Department of Chemistry and The Skaggs Institute for ChemicalBiology, The Scripps Research Institute, 10550 North Torrey PinesRoad, La Jolla, California 92037, USAE-mail: [email protected]
Macromol. Rapid Commun. 2008, 29, 1052–1072
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
Introduction
Every chemistry student learns the second law of
thermodynamics: the entropy of the universe is always
observed to increase. A glance at the recent explosion of
papers describing the use of click chemistry may leave one
with a similar sense of increasing disorder. However, as a
referee pointed out to us, the terms of thermodynamics
provide a more apt analogy: the field of click chemistry is
an open system in constant exchange with its environ-
ment (other disciplines). As in living systems, entropymay
also decrease locally when certain forces intersect, and
click chemistry certainly has helped to give life to the
realization of long-held goals in materials science, as
we discuss in several examples below. The timeline of the
development and evolution of this enabling philosophy
and technology is instructive.
DOI: 10.1002/marc.200800208

Construction of Linear Polymers, Dendrimers, Networks, and . . .
Jeremiah A. Johnson received a B.S. in biomedical engineering with a secondmajor in chemistry fromWashington University inSt. Louis in 2004 where he studied novel fluoropolymer-based antifouling coatings in the laboratory of Professor Karen L.Wooley. He is currently a fourth year graduate student at Columbia University working in the laboratories of Professor NicholasJ. Turro and Professor Jeffrey T. Koberstein. He has spent his graduate school summers working at The Scripps ResearchInstitute in the laboratory of Professor M. G. Finn. In his graduate work, Jeremiah has developed a general synthesis of ozone-and photo-degradable polymeric materials of well-defined structure, and has studied both the copper-catalyzed andstrain-promoted ‘‘click’’ crosslinking of these materials, the latter in collaboration with Professor Carolyn R. Bertozzi. He alsocontributed to the development of ‘‘universal ligands’’ for the synthesis of functionalized nanoparticles. Jeremiah was therecipient of an American Chemical Society Division of Organic Chemistry graduate fellowship in 2007 and recently received the2008 Hammett award from Columbia University’s Department of Chemistry.M.G. Finn received his bachelor’s degree in chemistry from the California Institute of Technology in 1980, and his Ph.D. from theMassachusetts Institute of Technology in 1986. His doctoral research with Professor K. Barry Sharpless focused on themechanism of the titanium-tartrate catalyzed asymmetric epoxidation reaction. After an NIH postdoctoral fellowship withProfessor James P. Collman at Stanford University, he joined the faculty of the University of Virginia in 1988. There he developedorganometallic reagents for organic synthesis, until a sabbatical at The Scripps Research Institute exposed him to opportunitiesat the intersections of chemistry and biology. He moved to Scripps in 1999, where he helped establish ‘‘click chemistry’’ as aconcept and implement it in the laboratory for purposes of bioconjugation, synthesis, and materials science. He has alsodeveloped virus-like capsid particles as practical scaffolds for the polyvalent display of functional molecules. With thesestructures, his laboratory now pursues targets in cancer biology, immunology, and enzyme evolution.Jeffrey T. Koberstein received a Bachelor’s degree in Chemical Engineering from the University of Wisconsin, and a Doctorate inChemical Engineering from the University of Massachusetts in 1979. After a year of postdoctoral research at the Centre deRecherches sur les Macromolecules in Strasbourg, France, he joined the Chemical Engineering faculty at Princeton University.From 1986 to 1999 he was a member of the Polymer Program within the Department of Chemical Engineering and Institute ofMaterials Science at the University of Connecticut, where he was Associate Professor (1986–89) and Professor (1989–1999) ofChemical Engineering. From2000 to 2005 hewas Professor and Chair of Chemical Engineering at ColumbiaUniversity. In 2003 hewas appointed as the Percy K. and Vida L. W. Hudson Professor of Chemical Engineering. Recently, Professor Koberstein chairedthe Materials Engineering and Science Division of the American Institute of Chemical Engineers and received the Stine awardfrom that division in 2006. In 2007, he was elected fellow of AIChE. He is currently co-director of the NSF-IGERT program inMultiscale Phenomena in Polymer Materials. Current research interests include: DNA and carbohydrate microarray technology;ultrathin ceramic and nanoparticle composite films for gas separation; reorganization,modification, and patterning of functionalpolymer surfaces; surface modification of nanoparticles; and smart polymer surfaces for biomaterial applications.Nicholas J. Turro has been teaching at Columbia University since 1964 and is currently the Wm. P. Schweitzer Professor ofChemistry and Professor of Chemical Engineering and Applied Chemistry, as well as Professor of Earth and EnvironmentalEngineering. He received his B.A. from Wesleyan University in Connecticut in 1960 and his Ph.D. in 1963 from the CaliforniaInstitute of Technology under George S. Hammond. Dr. Turro had a postdoctoral position at Harvard University from 1963 to1964. He has written over 800 scientific publications and 2 textbooks on molecular photochemistry. His recent awards includethe 2005 Theodor Forster Award from the German Chemical Society and the 2007 NicholsMedal by the New York Section of theAmerican Chemical Society. He is a member of the National Academy of Sciences and the American Academy of Arts andSciences.
Sharpless and coworkers introduced the click chemistry
concept in 2001, explicitly attempting to bring the
chemical sensibility of polymer chemistry to the world
of organic chemists.[1] Of course all chemists prize
reactions of high efficiency, and the click reactions
introduced in that original paper were mostly already
known. Sharpless and coworkers focused their attention,
however, on the enabling conviction that all branches of
chemical science could benefit from the same abiding
focus on function, and the same acceptance of a relatively
limited but extraordinarily powerful palette of bond-
forming reactions that has always characterized the field
of polymer and materials synthesis. The subsequent few
years brought a resounding affirmation of that expecta-
tion, with an explosion of papers worldwide applying the
Macromol. Rapid Commun. 2008, 29, 1052–1072
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
tenets of click chemistry to a range of applications which
could arguably not be approached by any other means.
‘‘Click chemistry’’ focuses on the extraordinary power of
a very few reactions which form desired bonds under
diverse reaction conditions, with highly diverse building
blocks, in high yields and with no (or conveniently
separated) byproducts.[2] For materials chemists, an
appreciation of the value of such processes is decidedly
not new. The reactions that modern click chemistry
has created, and one process in particular, have thereby
brought new capabilities to a field better prepared
than any other to take advantage of them. That process
is the popular copper-catalyzed azide-alkyne cyclo-
addition (CuAAC), which is the subject of this review
(Scheme 1).
www.mrc-journal.de 1053

J. A. Johnson, M. G. Finn, J. T. Koberstein, N. J. Turro
Scheme 1. Copper-catalyzed azide-alkyne cycloaddition (CuAAC).Throughout this review, triazole units will be shown in blue.
Scheme 2. Materials construction versus functionalization. Func-
1054
The CuAAC reaction was first reported in 2002 by the
laboratories of Meldal and Sharpless, in the latter case as a
direct result of an appreciation of the potential for such a
reaction in the context of the click chemistry concept.[3,4]
The mechanistic and procedural details of this reaction are
largely beyond the scope of this review and we direct the
reader to several references from the primary[5–8] and
review[9] literature on these topics. Recent reports of new
classes of accelerating copper-binding ligands,[10] the
isolation of a copper(I)triazolide click intermediate,[11]
and a novel way to remove all traces of copper from click
reaction mixtures are particularly relevant.[12] We will
focus on the application of CuAAC to the construction of
interesting materials in the rest of this review. A number
of excellent review articles in the past year have
summarized the use of CuAAC in polymer and materials
synthesis,[13–23] but new reports appear quite frequently in
this fast-moving field. We therefore endeavor to bring the
reader the most recent advances available, but also to set
them in context with older examples when appropriate.
tionalization route a) involves decoration of a material by attach-ment of a modifying moiety. Route b) involves alteration of thematerial without the incorporation of new agents (for exampletert-butyl ester conversion to carboxylic acid by hydrolysis). Use ofCuAAC for the materials construction step is the focus of thisreview although it has also proven powerful for materials func-tionalization via route a.Materials Modification versus Construction
There are two fundamentally different ways to use any
reaction inmaterials synthesis: to constructmaterials or to
modify them (Scheme 2). Modification (‘‘decoration’’ or
‘‘functionalization’’), pertains to the attachment or
removal of functionality to or from an already existing
material thereby imbuing it with new properties. Scheme
2 illustrates two potential means by which materials can
be modified - route a in which new modifier building
blocks are added to the material, and route b in which one
or more existing blocks are chemically altered. Perhaps the
most important requirements of route a modification
reactions are chemoselectivity (to ensure functionalization
at the desired site) and high yield (to ensure complete
functionalization), challenges that the CuAAC reaction is
well equipped tomeet. The vast majority of applications of
CuAAC reactions to materials synthesis therefore involve
the modification of polymers, surfaces, dendrimers,
nanoparticles, viruses, networks, and other structures
via route a. Furthermore, thanks to the orthogonal
reactivity of CuAAC, route b schemes can be used in
conjunction with CuAAC functionalization to yield che-
mically diverse macromolecular structures [for example,
constructing and modifying materials derived from
poly(tert-butyl acrylate) (PtBA) enables the orthogonal
Macromol. Rapid Commun. 2008, 29, 1052–1072
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
route b conversion of hydrophobic PtBA to hydrophilic
poly(acrylic acid) (PAA) via acid hydrolysis].
Material construction pertains to building an architec-
ture and the reactions thus employed provide the mortar
with which the building blocks of macroscopic architec-
tures are connected (Scheme 2). For materials synthesis
applications, high yields and chemoselectivity are neces-
sary, and the concept ofmodularity is crucial. Furthermore,
the more easily the required functional groups can be
introduced into potential building blocks, the more
structurally and chemically diverse the material that
can be synthesized. Because azides and alkynes are readily
attached to molecular scaffolds via a variety of simple
techniques, and are stable toward a wide variety of
solvents and reaction conditions, CuAAC allows the user to
bring almost any necessary building block to the problem
at hand without worrying about the connection reaction.
We focus here on the efforts of our laboratories and
others to employ the CuAAC reaction for the construction
of materials, but references are also provided for a number
of recent materials modification applications which utilize
DOI: 10.1002/marc.200800208

Construction of Linear Polymers, Dendrimers, Networks, and . . .
Scheme 3. Representative polymeric architectures in order ofincreasing complexity. Red and blue regions correspond to thefundamental material building blockswhichmay be comprised ofpolymers themselves or small molecules. Materials constructionconcerns the efficient connection of these building blocks, achallenge uniquely met by CuAAC. Synthesis of brush (graft)polymers and star polymers is viewed as a modification in thisreview because both situations involved attachment of polymersto a polymer backbone or multifunctional core, respectively.
CuAAC. In many cases, a reaction can be seen as both a
modification and a construction reaction. For example, the
attachment of alkyne end-functionalized polymers to
azide side-chain derivatized polymers to yield graft
copolymers can be considered both the modification of
the azide derivatized polymer and the construction of a
graft copolymer. Due to space limitations, such cases are
considered modification reactions and only limited
examples are given.
A triazole-containingmaterialwill be defined as resulting
from CuAAC ‘‘construction’’ if at least one triazole within
the structure can be traced in every direction along the
structure to another triazole. For the graft copolymer
example given above, every triazole in the structure is the
result of a modification reaction because it can be traced
directly to the end of the alkyne end-functionalized
polymer. Even within this subset of CuAAC construction
our review is not comprehensive, but instead summarizes a
number of interesting applications of CuAAC to materials
synthesis starting from the construction of linear polymers
and progressing to complicated cross-linked networks of
nanocrystals and nanotubes (Scheme 3). And since the
interactions of click chemistry with materials science are
only beginning, we also look forward to the many new and
exciting architectures and materials that will be made
accessible by CuAAC in the years to come.
Scheme 4. Schematic of linear polymer construction using CuAAC.Such materials have been prepared by addition type polymeri-zation of AB (i) or AA and BB (ii) type monomers, or by iterativeCuAAC/end-group transformation reactions (iii).
Linear ‘‘Click’’ Polymers and Oligomers(From Small Molecule Precursors)
If materials construction is viewed as progressing from
simpler to more complex structures (Scheme 3), then it
should come as no surprise that CuAACwas first applied to
Macromol. Rapid Commun. 2008, 29, 1052–1072
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
materials synthesis in the construction of linear polymers
and oligomers and has since been employed in a variety of
unique ways for the synthesis of such materials
(Scheme 4). The earliest example, from 2004, was reported
by Finn and coworkers as a control reaction in their
discovery of metal adhesives. The authors showed that a
bis-azide and bis-alkyne formed a soluble linear polymer
thus confirming their hypothesis that CuAAC could be
used for addition polymerizations (Scheme 5, 24).[24]
Further details of this work, along with a recent article
by Ravelo and coworkers describing linear CuAAC poly-
mers incorporated into networks (Scheme 5, 25),[25] will be
discussed later in the section on network synthesis.
In 2005, Angelo and coworkers reported using an
iterative diazo-transer/CuAAC process to construct pepti-
domimetic oligomers that possessed a ‘‘zigzag’’ secondary
structure resulting from the large dipole moment of the
triazole CuAAC product (Scheme 5, 26).[26] In this work, the
triazoles of 26 were viewed as replacements for the
traditional amide peptide bond, while the R groups
mimicked amino-acid side chains. The authors suggested
that this strategy could lead to peptide mimics with
enhanced stability in in vivo applications. Around the
same time, Reek and coworkers reported the construction
of novel conjugated polymers derived from A,A and B,B
diazide and dialkyne fluorene monomers (Scheme 5,
27).[27] Introduction of bipyridine-type functionalities into
these polymers opened a route to novel metal-coordinated
conjugated polymers.
Meudtner and Hecht recently reported an example of a
triazole polymer which possessed secondary structure and
metal-binding pyridine units. Step-growth polymerization
of 2,6-diethynylpyridines with a bifunctional aromatic
azide yielded linear polymers with 2,6-bis(1,2,3-triazol-
4-yl)pyridine units on the backbone which are known to
adopt an anti–anti conformation thus giving rise to helical
polymers (Scheme 5, 28).[28] Circular dichroism spectro-
scopy confirmed the existence of chiral helices and when a
www.mrc-journal.de 1055

J. A. Johnson, M. G. Finn, J. T. Koberstein, N. J. Turro
Scheme 5. Representative structures of linear polymers prepared by CuAAC connection of small molecule building blocks. Triazole unitsmade by CuAAC events are shown in blue. Throughout this review, the numbers listed with structures refer to the reference fromwhich thestructure was taken.
1056
variety of metal salts (Zn2þ, Fe2þ, Eu3þ) were added to
dilute solutions of 28 cross-linking occurred yielding
supramolecular metallogels which have potential as novel
magnetic and/or emissive materials.
In 2006, an interesting one-pot synthesis of triazole
oligomerswas reportedbyAucagneandLeigh (Scheme6).[29]
The authors utilized a trimethylsilyl-protected acetylene
(TMS-alkyne) - azide A,B type monomer which was reacted
withanalkyneunderCuAACconditions.Uponcompletionof
this reaction, a silver(I) salt was added along with a second
azide to the same reactionmixture facilitating deprotection
of the TMS-alkyne and subsequent CuAAC with the newly
addedazide. Thismethodologyhas thepotential to beuseful
in a range of applications including the one-pot synthesis of
ABC block copolymers.
Burgess and coworkers later reported an iterative
construction of oligomers derived from the CuAAC
connection of triethylene glycol (TEG) subunits (Scheme
5, 30).[30] By utilizing TEG fragments possessing an alkyne
on one end and a tosylate on the other, the authors were
able to repeatedly perform CuAAC followed by nucleo-
philic displacement of the tosylate with sodium azide to
grow oligomers which then could be end-functionalized
with any desired moiety via a final CuAAC step (R and R0 in
30 could be varied). The water-solubility of these materials
makes them attractive linkers for bioconjugation applica-
tions. Finally, Qing and coworkers prepared addition
polymers by CuAAC which possessed perfluoro-cyclobutyl
(PFCB) groups along the main chain (Scheme 5, 31).[31] The
Scheme 6. Method presented by Leigh et al. for the one-potconstruction of trifunctional oligomers via CuAAC and Ag(I)-catalyzed deprotection of a terminal alkyne.
Macromol. Rapid Commun. 2008, 29, 1052–1072
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
resulting polymers were thermally stable up to �400 8Cand were soluble in organic solvents making them
attractive candidates for low dielectric and other high
performance coating applications.
Linear ‘‘Click’’ Polymers and Oligomers(from Polymeric Precursors)
All of the aforementioned examples utilize CuAAC to
couple small molecules together to construct polymers or
oligomers. Because azides and alkynes have such a narrow
spectrum of reactivity, they can also be incorporated into
polymerization initiators to make end-functionalized
macromonomers. The use of such building blocks to yield
high molecular weight block copolymers has been
described by several laboratories. We also include the
formation of circular polymers, derived from end-
functionalized linear macromolecules, in this section.
In 2005, Opsteen and van Hest reported the construc-
tion of a variety of polystyrene (PS), poly(methyl
methacrylate) (PMMA), and poly(ethylene glycol) (PEG)
block copolymers by a click chemistry strategy.[32] Atom
transfer radical polymerization (ATRP) of the selected
monomers followed by end-group transformation of the
terminal bromine to an azide using sodium azide yielded
polymers which were coupled with other ATRP-derived
polymers grown from an alkyne initiator. The same
authors recently reported an extension of this technique
using triisopropylsilyl-alkyne (TIPS-alkyne) ATRP initiators
to prepare a,v-heterobifunctional polymers possessing
TIPS-alkyne on one end and azides on the other.[33] CuAAC
followed by deprotection and subsequent CuAAC with
another azide-terminated polymer yielded ABC triblock
copolymers (Scheme 7, 33). Notably, the authors mention
that ATRP using a TMS-alkyne initiator resulted in a
significant degree of TMS loss during the ATRP process. The
TIPS group, on the other hand, was found to be completely
stable during ATRP. It would perhaps be interesting to see
if these polymers could be used in conjunction with
DOI: 10.1002/marc.200800208

Construction of Linear Polymers, Dendrimers, Networks, and . . .
Scheme 7. Representative structures of linear polymers prepared by CuAAC connection of polymeric building blocks. Triazole units, theproduct of CuAAC are shown in blue. The corresponding numbers refer to the reference from which the structure was taken.
Scheme 8. Synthesis of a double-stranded DNA catenane byBrown and coworkers.[38] CuAAC circularization of a single DNAstrand (shown in red) followed by annealing of the complemen-tary strand (shown in blue) and CuAAC gave the desired structure(triazoles represented by blue pentagons).
Scheme 9. Schematic of dendrimer modification versus construc-tion. In this reviewwe only describe dendrimers constructed usingCuAAC. These dendrimers possess triazoles at branching pointswithin the dendrimer structure rather than only at the surface.
Leigh’s method described above (CuAAC followed by Ag(I)
deprotection and CuAAC again) to prepare ABC triblocks in
a one-pot procedure.
Matyjaszewski and coworkers reported the synthesis of
an a,v-heterobifunctional-PS which was prepared by ATRP
from an alkyne initiator followed by nucleophilic substitu-
tion of the bromine end group with sodium azide.[34]
Polymerizationof thesemacromonomers (MACs)byCuAAC
gave high molecular polymer products as well as a small
amount of cyclic material (Scheme 7, 34). The authors
mentioned an attempt to perform a one-pot end-group
exchange/CuAAC coupling without success.
The next year, Laurent and Grayson reported the
synthesis of macrocyclic polymers (Scheme 7, 35) derived
from the same polymers reported by Matyjaszewski and
coworkers above, but under different CuAAC conditions
(i.e., higher dilution to favor the cyclization reaction).[35] In
2007, Tunca and coworkers elegantly achieved a one-pot
click construction of an ABC triblock polymer by utilizing
tandem CuAAC and Diels-Alder reactions to yield
PMMA-PS-PEG triblocks (Scheme 7, 36).[36]
Another example of a CuAAC constructed linear
polymer was reported in 2007 by Wang and coworkers.[37]
The authors coupled alkyne-telechelic PEGs with
2,2-bis(azidomethyl)propane-1,3-diol by CuAAC to yield
higher molecular weight PEG polymers with pendant
functionalities (R groups, Scheme 7, 37) including an
alendronate-PEG for potential bone-targeted drug delivery.
The high yield of CuAAC was demonstrated by the
production of hydrogels if no monofunctional PEG was
added to the polymerization mixture (indicating extre-
mely high molecular weight).
Brown and coworkers have recently described the
fascinatingandpotentiallyveryuseful synthesisofacircular
DNA catenane.[38] The authors prepared a single-stranded
DNAwith a 50-alkyne and a 30-azide. CuAAC coupling under
dilute conditions yielded a macrocyclic DNA. When the
complementary DNA strand, also possessing alkyne and
azide termini, was allowed to anneal to the closed DNA loop
and treated with Cu(I), a double-stranded DNA catenane
resulted,connectedwithtwostable triazoleunits (Scheme8).
Macromol. Rapid Commun. 2008, 29, 1052–1072
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
Network/Branched Architectures
Dendrimers
Anumberofgroupshave recentlyutilizedCuAAC inelegant
ways to modify the surfaces of discrete molecular objects
like dendrimers.[39–41] In this review we only mention
the use of CuAAC to construct dendritic architectures
(Scheme 9) and direct the reader to the aforementioned
reviews to find a wealth of examples of CuAAC for
dendrimer modification.
www.mrc-journal.de 1057

J. A. Johnson, M. G. Finn, J. T. Koberstein, N. J. Turro
1058
The synthesis of dendritic molecules requires extremely
efficient reactions for each iterative step to ensure
quantitative conversion of the geometrically increasing
number of functionalities as the dendrimer grows. For this
purpose, chemists in the field of dendrimer synthesis had
been utilizing click-type reactions for years[42–44] but itwas
Fokin,Hawker, and coworkerswhofirst brought theCuAAC
reaction to the field in 2004.[45] Soon thereafter, theWooley
laboratory reported a complementary divergent synthesis
of triazole dendrimers.[46] Hawker and coworkers then
highlighted the chemoselectivity and efficiency of the
CuAAC reaction in this context by making multivalent,
bifunctional dendrimers that onewould be hard-pressed to
prepare so easily by conventional means (Scheme 10).[47]
The final structure featured 16 surface-active mannose
groups and two fluorescent coumarin units derived from
two dendrons, each made with triazole linkages and then
Scheme 10. Hawker’s multivalent, bifunctional dendrimer bearing pr
Macromol. Rapid Commun. 2008, 29, 1052–1072
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
subsequently joined by a CuAAC reaction. The resulting
dendrimer was shown to be 240 times more potent than
monomericmannose ina standardhemagluttinationassay
thereby providing a nice demonstration of the polyvalent
effect of highly functionalized macromolecular scaffolds.
CuAAC has repeatedly been shown to be an efficient
way to functionalize solid supports.[48–54] For example,
Santoyo-Gonzalez and coworkers convergently prepared
glyco-dendrimers with a silica core and demonstrated
their effectiveness as materials for affinity chromatogra-
phy separation of proteins (Scheme 11).[55]
A particularly interesting combination of CuAAC and
ATRP for the synthesis of polymeric dendrimers was
introduced by Monteiro and coworkers.[56] In an approach
similar to thatofOpsteen[33] andMatyjaszewski,[34]ATRPof
styrene using a bifunctional initiator provided azido-
telechelic PS. CuAAC capping of each end of this polymer
otein-binding mannose units and fluorescent coumarin moieties.
DOI: 10.1002/marc.200800208

Construction of Linear Polymers, Dendrimers, Networks, and . . .
Scheme 11. Triazole (shaded blue) glyco-dendrimers on solid SiO2 supports were shown to be effective stationary phases for affinitychromatography.
with tripropargylamine provided tetra-alkyne terminated
PS, which was then addressed with an azido-terminated
PAA. The result was a polymeric dendrimer possessing a
hydrophobic PS corewith ahydrophilic PAA shell. The same
authors recently expanded this approach by using degrad-
able branching units to yield higher generation polymeric
dendrimers with a cleavable periphery (Scheme 12). These
amphiphilic dendrimers self-assembled into micelles in
aqueous environments, and degradation was achieved
via basic hydrolysis of ester groups built into the structure
at the branching points.[57] This work provides a nice
example of the compatibility of ATRP and CuAAC by
offering a straightforward route to complicated polymeric
dendrimers.
Dendrimer synthesis typically involves iterative
activation-functionalization steps requiring two synthetic
steps per generation. Recently, Malkoch and coworkers
reported a beautifully streamlined scheme for the
Scheme 12. Schematic representation of polymeric dendrimersprepared by Monteiro and coworkers. Three triazoles fromCuAAC, represented by blue circles, are present at each branchingpoint. Connection of the triazole units to the polymers by esterlinkages enabled hydrolytic degradation of the dendrimer struc-ture.
Macromol. Rapid Commun. 2008, 29, 1052–1072
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
preparation of 2,2-bis(methylol)propionic acid (bis-MPA)-
triazole and benzyl ether (Frechet type)-triazole dendri-
mers.[58] Because neither azides nor alkynes require
protection during etherification or esterification, and
because azide-alkyne ligation can take place in the
presence of the unprotected nucleophiles and electrophiles
required for etherification or esterification, every step in
the synthetic sequence was used to grow the polymer.
Fourth generation dendrimers possessing 24 surface
hydroxyls were prepared in only four steps, thereby
greatly reducing the time required tomake such structures
and increasing their commercial viability (Scheme 13).
Astruc and coworkers have pioneered the use of the
metal binding properties of triazole dendrimers in the field
of catalysis. Recently, they reported the catalysis of a
number of carbon-carbon bond forming reactions using
triazole dendrimer-bound palladium metal.[59] These third
generation dendrimers, with 81 surface ferrocene units,
were prepared by iterative hydrosilylation-CuAAC reac-
tions and represent novel nanoreactors (Scheme 14).[60]
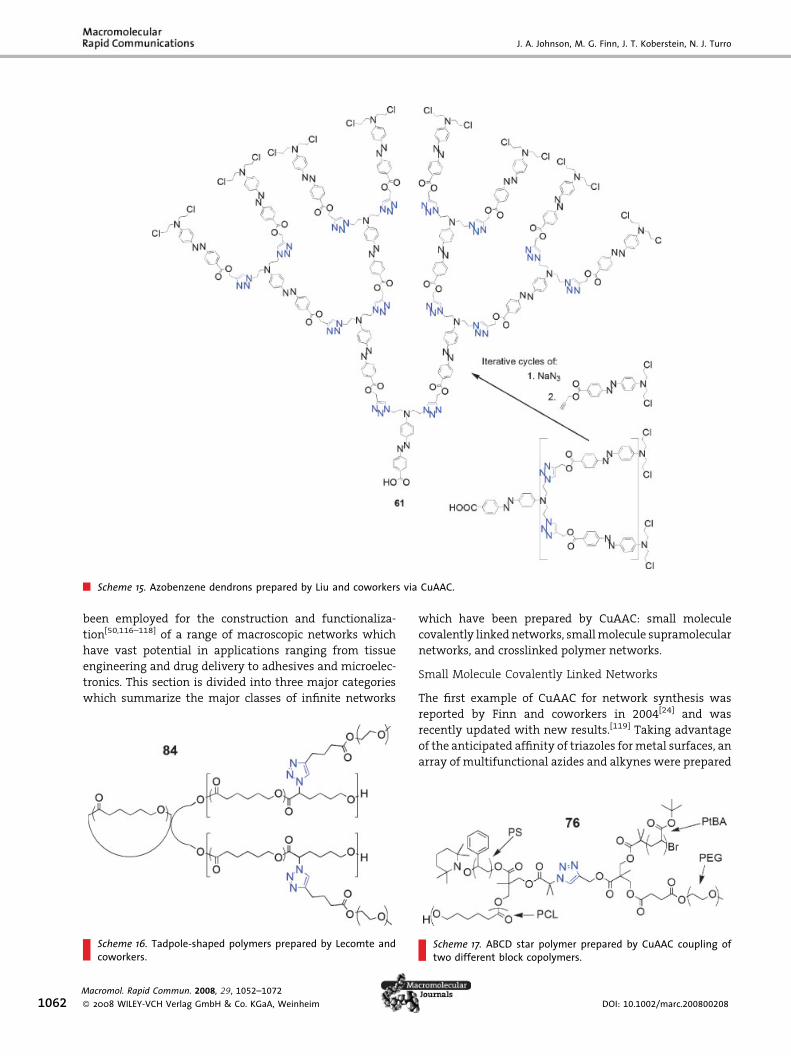
During the preparation of this manuscript, Liu and
coworkers reported a novel synthesis of triazole dendrons
which possessed azobenzene chromophores at each
branching unit.[61] Structures with as many as 15
azobenzene moieties were prepared by iterative CuAAC/
SN2 reactions (Scheme 15), taking advantage of the special
anchimeric assistance reactivity of b-haloamine electro-
philes with azide.[62] It was shown that, despite the steric
hindrance within the dendrimer structure, all of the
azobenzene groups could quantitatively be converted
between cis and trans isomers reversibly using UV and
visible light, respectively. The authors mention the
potential of these dendrimers as fast, nanoscale, photo-
active switches.
www.mrc-journal.de 1059

J. A. Johnson, M. G. Finn, J. T. Koberstein, N. J. Turro
Scheme 13. Triazole/benzyl-ether dendrimer prepared by Malkoch and coworkers in four steps.
1060
Other Discreet Polymeric Structures (Hyperbranched,Brush, and Star Polymers).
Hyperbranched polymers possess properties similar to
dendrimers and are typically easier tomake at the expense
of being polydisperse. The only example of soluble
hyperbranched polymer synthesis performed with the
aid of the CuAAC reaction to have appeared so far is found
in a 2007 report by Sumerlin and coworkers.[63] Here, the
click reaction was used to conveniently combine the
sensitive acryloyl and trithiocarbonate groups into an
initiator for reversible addition-fragmentation chain
transfer (RAFT) polymerization. This initiator then pro-
vided hyperbranched triazole-based polymers upon reac-
tion with styrene or isopropylacrylamide under standard
RAFT conditions.
In recent years CuAAC has been utilized by a number of
groups for modular ligation of linear polymers with other
molecules to give novel graft/brush polymers,[64–74] star
Macromol. Rapid Commun. 2008, 29, 1052–1072
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
polymers,[75–83] and other architectures.[84,85] Since these
can be considered asmodification rather than construction
applications of CuAAC (see Scheme 2), we only highlight a
few interesting examples in this review, with apologies to
those omitted because of space considerations. First,
Lecomte and coworkers have presented novel tadpole-
shaped (Scheme 16)[84] and eight-shaped[85] polymers
using a cyclic tin(IV) dialkoxide initiator for controlled
ring-opening polymerization of e-caprolactones (CL)
including an a-chlorinated monomer. Conversion of the
chlorine groups to azides and subsequent CuAAC with
alkyne-PEG yielded complicated structures with poten-
tially novel properties including unexplored modes of
self-assembly.
Tunca and coworkers have recently presented an elegant
CuAAC-dependent synthesis of star polymers possessing
four chemically different arms (ABCD stars, Scheme 17).[76]
The authors utilized two novel hetero-trifunctional
molecules, the first of which possessed a nitroxide for
DOI: 10.1002/marc.200800208

Construction of Linear Polymers, Dendrimers, Networks, and . . .
Scheme 14. Ferrocene-terminated triazole dendrimers used byAstruc and coworkers to bind palladium. The resulting metallo-dendrimers were useful nanoreactors for the catalysis of a num-ber of carbon-carbon bond forming reactions.
nitroxide-mediated radical polymerization (NMRP), an
alcohol for tin(IV)-mediated ring-opening polymerization,
and a bromine for later conversion to an azide for CuAAC.
The second molecule contained an alkyne for CuAAC, an
ATRP initiator, and an alcohol. Ring-opening polymeriza-
tion of CL from the hydroxyl group of the first molecule,
NMRP of styrene from the nitroxide group, and conversion
of the bromine to an azide yielded a PS-PCL diblock
possessing a single azide at the center. ATRP of methyl
acrylate or tert-BA, and carbodiimide coupling of PEG to the
second molecule yielded both PtBA-PEG and PMMA-PEG
diblock copolymers possessing a single alkyne at the center.
CuAAC proved to be an efficient means to couple the
PS-PCL-N3 block to the PtBA-PEG-alkyne or PMMA-
PEG-alkyne block to yield ABCD star polymers with
potentially novel properties.
Nanoscale Objects Constructed by CuAAC
A number of groups have recently utilized CuAAC for the
decoration of biological,[86–95] polymeric,[96–98] inor-
ganic,[99–106] and carbon[107] nanoscale objects, but only a
few examples exist wherein CuAAC was employed for the
direct constructionof suchmaterials.Wooleyandcoworkers
have demonstrated the synthesis of both shell[108] and
core[109] crosslinked polymeric nanoparticles derived from
amphiphilic PS–PAA diblock copolymers possessing
alkynes on a fraction of either the hydrophilic PAA block
Macromol. Rapid Commun. 2008, 29, 1052–1072
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
or the hydrophobic PS block (for shell and core crosslinking,
respectively). These polymers formed micelles in aqueous
solution and were subsequently CuAAC crosslinked
with bifunctional azides and multifunctional dendritic
azides to form polymeric nanoparticles whichwere further
functionalized with fluorescent moieties (Scheme 18). Liu
and coworkers recently pursued a similar approach by
preparing core crosslinked nanoparticles comprised of
poly(N,N-dimethylacrylamide) (PDMA) and poly(N- isopro-
pylacrylamide-co-azidopropylacrylamide).[110]
Caruso and coworkers reported a fascinating synthesis
of ultrathin microcapsules by a unique layer-by-layer
(LBL)/CuAAC approach.[111] The authors started by making
use of electrostatic and hydrogen bonding interactions
between PAA and silica to deposit a thin layer of PAA,
which had a fraction of its carboxylic acid units converted
to azides (PAA-N3), onto the silica particle. A layer of PAA
displaying alkynes (PAA-alkyne) was then coated and
coupled to the previous layer via CuAAC. This process was
repeated for the desired number of iterations yielding
covalently crosslinked PAA of controllable thickness on the
silica surface (Scheme 19). Fluorescent rhodaminemoieties
were attached to the remaining alkynes for imaging of the
particles. Degradation of the silica interior with hydro-
fluoric acid produced covalently linked capsules which
were shown to swell and deswell reversibly with changing
pH.
Hennink and coworkers recently described the synthesis
of biodegradable microcapsules using CuAAC between
azide- and alkyne-modified dextrans (Scheme 20).[112] The
clickable functional groups were linked to dextran
precursors via hydrolyzable carbonates whichwere shown
to break down under physiological conditions to release
their components at controlled rates. These materials
show promise in drug delivery applications where
controlled delivery of a bioactivemoiety is often necessary.
CuAAC for the Synthesis of Macroscopic ‘‘Infinite’’[113]
Networks
Much of our ownwork on thematerials applications of the
CuAAC reaction has been related to the synthesis of infinite
networks (materials of macroscopic dimensions) from
small molecule and/or polymeric precursors. Such materi-
als require efficient reactions for their preparation as there
is a necessary critical conversion percentage of functional
groups (depending on the branching functionality)
which must be achieved before an infinite network (or
gelation) is reached.[114,115] In this vein, CuAAC provides
a route to diversely functional crosslinked networks for
the same reasons that make it so useful throughout
materials synthesis: ease of functional group introduction,
chemoselectivity, andhighyield. In recent years, CuAAChas
www.mrc-journal.de 1061
J. A. Johnson, M. G. Finn, J. T. Koberstein, N. J. Turro
Scheme 15. Azobenzene dendrons prepared by Liu and coworkers via CuAAC.
1062
been employed for the construction and functionaliza-
tion[50,116–118] of a range of macroscopic networks which
have vast potential in applications ranging from tissue
engineering and drug delivery to adhesives and microelec-
tronics. This section is divided into three major categories
which summarize the major classes of infinite networks
Scheme 16. Tadpole-shaped polymers prepared by Lecomte andcoworkers.
Macromol. Rapid Commun. 2008, 29, 1052–1072
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
which have been prepared by CuAAC: small molecule
covalently linkednetworks, smallmolecule supramolecular
networks, and crosslinked polymer networks.
Small Molecule Covalently Linked Networks
The first example of CuAAC for network synthesis was
reported by Finn and coworkers in 2004[24] and was
recently updated with new results.[119] Taking advantage
of the anticipated affinity of triazoles formetal surfaces, an
array of multifunctional azides and alkynes were prepared
Scheme 17. ABCD star polymer prepared by CuAAC coupling oftwo different block copolymers.
DOI: 10.1002/marc.200800208

Construction of Linear Polymers, Dendrimers, Networks, and . . .
Scheme 18. Molecular structure of shell-CuAAC-crosslinked poly-meric nanoparticles prepared by Wooley and coworkers. Bothlinear and dendritic azide linkers (R) were used for the cross-linking. However, only the smallest dendrimer bearing four azidespossessed the necessary valency and solubility to yield stablenanostructures.
Scheme 20. CuAAC-constructed dextran microcapsules withdegradable carbonate linkages.
which were polymerized between copper, brass, alumi-
num, and zinc plates yielding adhesives (Scheme 21). The
addition of copper catalyst was not required for adhesive
formation on metallic copper and brass plates, indicating
that a sufficient amount of Cu(I) is present on the surface to
catalyze the CuAAC reaction. The addition of copper salts
did accelerate the reaction, however, as did performing the
crosslinking at elevated temperatures (although the final
adhesive strength did not depend strongly on tempera-
ture). In the cases where no catalyst was added, the final
crosslinked products contained between 2 and 5 wt.-% of
copper indicating that the triazole products efficiently
leach copper ions from the surface. This study identified a
tetrafunctional alkyne and trifunctional azide combina-
tion that yielded adhesives which were twice as strong as
commercially available metal adhesives in peel tests.
Several crosslinked polymers analogous to the adhesive
Scheme 19. Polymeric microcapsules prepared using LBL assemblyof alkyne- and azide-functionalized PAA on silica particles fol-lowed by CuAAC crosslinking.
Macromol. Rapid Commun. 2008, 29, 1052–1072
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
materials were made in bulk and characterized by
standard techniques. Interestingly, the glass transition
temperatures, of thesematerials were found to be asmuch
as 80 8C higher than the curing temperatures employed
indicating a high degree of crosslinking even in the
diffusion-restricted glassy state.[120] Potential contributing
factors include the high mobility of catalytic Cu(I) centers
through a developing matrix of triazoles, the great
efficiency of the CuAAC coupling reaction, and the great
amount of energy that it releases (approximately
50 kcal �mol�1). Recently, Katritzky et al. reported a similar
study wherein a library of multifunctional azides and
alkynes were polymerized in solution and characterized by
standard techniques.[121]
Finn and coworkers also utilized two of their previously
studied adhesive molecules, tripropargyl amine with
1,6-diazidohexane, in a solution phase CuAAC reaction
to yield a highly crosslinked polymer which was observed
to reversibly swell and deswell when treated with
trifluoroacetic acid due to the amine functionalities within
the material (Scheme 22).[122] The authors suggest that the
polarity, stability, and p-stacking properties of the triazole
products can potentially be exploited for a range of
applications, a notion that has been abundantly confirmed
by reports that have appeared in the past few years.[2]
Small Molecule Supramolecular Networks
The first example of small molecule supramolecular
hydrogelators prepared by CuAAC was presented by
Kim and coworkers in 2003.[123] The authors appended a
Scheme 21. Schematic structure of crosslinked network derivedfrom triazides and trialkynes which were CuAAC crosslinkedbetween two copper plates.
www.mrc-journal.de 1063

J. A. Johnson, M. G. Finn, J. T. Koberstein, N. J. Turro
Scheme 22. Acid-swellable CuAAC-crosslinked network reportedby Finn and coworkers.
1064
variety of groups to the C5 position of 20-deoxyuridines via
the click reaction. They then showed that these molecules
formed lamellar or fibrous hydrogen-bonded gels in water
depending on the nature of the C5 substituent. Gallardo
Scheme 23. CuAAC stabilized organogels reported by Finn and coworthermoreversible gels with greater mechanical stability.
Macromol. Rapid Commun. 2008, 29, 1052–1072
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
and coworkers have utilized CuAAC to prepare
chiral[124,125] and nonlinear diaryl[126] triazole compounds
which were shown to possess liquid crystalline properties
thus warranting their categorization as supramolecular
infinite networks. The authors note the simplicity of
CuAAC for preparing these compounds and indicate they
are screening a library of triazole structures in a search for
novel properties.
The CuAAC process was used to ‘‘stabilize’’ organogels
by Finn and coworkers in 2006.[127] The authors reasoned
that the introduction of azides and alkynes onto the
termini of the alkyl chains of the undecylamide of
trans-1,2-diaminocyclohexane, a molecule known to self-
assemble into supramolecular networks via hydrogen
bonding, would not disrupt gelation but perhaps provide a
way to strengthen the gel products (Scheme 23). Indeed,
self-assembly followed by CuAAC provided a simple and
novel means to alter the properties (glass transition
temperature and rigidity) of these materials without
sacrificing the thermoreversibility of gelation which is
unique to supramolecular gels.
An interesting example of CuAAC for the synthesis of
metal-organic frameworks (MOFs) was recently reported
by Ferey and coworkers.[128] This work explored the
isoreticular principle—the concept that the size of pores
kers. CuAAC crosslinking of preformed supramolecular gels yielded
DOI: 10.1002/marc.200800208

Construction of Linear Polymers, Dendrimers, Networks, and . . .
Scheme 24. Supramolecular networks prepared by interaction between TPP-TS and tetra-functional CDs. Different structures (2-D networkvs. linear) were observed depending on which CD derivative (R) was employed for the crosslinking. CuAAC was utilized to construct theself-assembling building blocks by attachment of CDs to a zinc porphyrin.
in MOFs can be tuned solely by altering the length of a
rigid organic linker connecting the inorganic metal
portions. The authors exploited the synthetic convenience
provided by the CuAAC to test the principle by installing
small flexible units at the ends of rigid organic core
molecules. The resulting materials lacked permanent
microporosity as a result of contraction facilitated by
the small flexible organic components, thereby confirming
that the isoreticular principle is limited to completely rigid
organic linkers.
Ding and coworkers recently described an example of a
different type of supramolecular small molecule network
enabled by CuAAC.[129] The authors prepared meso-
tetraaryl zinc(II) porphyrins decorated with anion-binding
b-cyclodextrin (b-CD) or permethyl-b-CD units by CuAAC
attachment of the tetra-alkyne porphyrin to CD azides.
Addition of tetraphenylporphyrin tetrasulfonate (TPP-TS)
to these compounds resulted in either networks with
tetrafunctional branching points (as observed for the
permethylated CD derivative, Scheme 24) or one-
Macromol. Rapid Commun. 2008, 29, 1052–1072
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
dimensional nanorods (as observed for the standard CD
derivative, Scheme 24). The difference in network structure
was proposed to depend on the orientations of the
cup-shaped CDs: either ‘‘expansive’’ (all four cups facing
outward) or ‘‘contractive’’ (only two cups facing outward).
The expansive state was observed for the permethyl
derivative, allowing each sulfonate group of TPP-TS to be
bound by a CD host, resulting in a tetrafunctional
branched network. In contrast, the standard CD derivative
was found to be locked in the contracted state, resulting in
the formation of linear networks via binding of only two
TPP-TS moieties in a trans fashion.
Crosslinked Polymer Networks
CuAAC has proven useful for the synthesis of a range of
crosslinked polymeric materials. In 2006, Ossipov and
Hilborn presented the first example of CuAAC for the
synthesis of polymeric hydrogels.[130] By modifying
poly(vinyl alcohol) (PVA) with a fraction of either azide
or alkyne functionalities, the authors showed that
www.mrc-journal.de 1065

J. A. Johnson, M. G. Finn, J. T. Koberstein, N. J. Turro
Scheme 25. Representative examples of hydrogels crosslinked by CuAAC. Triazole CuAAC products which are used for network constructionare shown in blue. Gel 135 possesses triazoles for both construction and modification. Gel 136 is a supramolecular network which resultsfrom hydrophobic/hydrophilic interactions at various temperatures.
1066
hydrogel materials with tunable mechanical properties
could be prepared by CuAAC crosslinking with either
bifunctional crossinkers bearing the antagonist azide or
alkyne functionality, or by crosslinking of the azide-PVA
with alkyne-PVA (Scheme 25, 130).
Soonafter thiswork,Hawker and coworkers reported the
first application of CuAAC to the synthesis of end-linked
polymeric ‘‘model network’’ hydrogels. Bifunctional alky-
ne-PEGs were crosslinked with tetrafunctional azide-PEGs
(Scheme25,131),[131] toproducematerialswithmechanical
strengths about ten times greater than conventional
photo-crosslinked PEG materials. Any remaining azide
and alkyne groups could be functionalized by CuAAC
post-gelation to ‘‘decorate’’ the gel with imaging moieties.
Importantly, gels crosslinked by CuAAC in the presence of
additives such as a radical scavenger, carbon black, and
TiO2 still possessed excellent mechanical properties. In
contrast, photocrosslinking under these conditions would
be inefficient due to adsorption of light and quenching of
excited state radicals by the additives. Lastly, PEGs
containing ester linkages were shown to yield hydro-
lytically degradable materials whereas ether linked gels
were completely stable to changes in pH.
A recent article by Lamanna and coworkers describes
the CuAAC synthesis of novel hyaluronan hydrogels from
Macromol. Rapid Commun. 2008, 29, 1052–1072
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
azide and alkyne-modified polymers (Scheme 25, 132).[132]
Crosslinking in the presence of the drug molecules
benzidamine and doxorubicin yielded loaded gels which
released the drugs at rates dependent on the degree of
crosslinking. Furthermore, CuAAC crosslinking in the
presence of yeast cells provided porous materials with
homogeneously distributed live cells. This report contra-
dicts the conventional wisdom that the copper AAC
catalyst is toxic to in vivo applications of the reaction.
While this expectation is certainly reasonable and is
undoubtedly true in some circumstances, it has yet to be
conclusively tested, especially in the presence of
Cu-binding ligands. Furthermore, one can certainly expect
many cells to survive exposure to Cu-containing media,
since copper is a common element and cells possess
effective means to regulate Cu trafficking. Thus, work by
Lamanna, and other reports in nonmaterials areas,[133–135]
suggest that CuAAC in the presence of live cells is a
promising approach to biocompatible scaffolds for tissue
engineering, among other biomaterials applications.
Recently, Lecomte and coworkers utilized ring-opening
polymerization of an a-chloro caprolactone followed by
conversion of the halides to azides to obtain azide-
functionalized PCL. These materials were then subjected
to CuAAC crosslinking in one pot with a PEG dialkyne and
DOI: 10.1002/marc.200800208

Construction of Linear Polymers, Dendrimers, Networks, and . . .
Scheme 26. Photodegradable PtBA model networks reported by Turro and coworkers.
N,N-dimethyl propargylamine to yield amphiphilic mate-
rials with grafted amine functionalities (Scheme 25,
136).[136] The authors demonstrated the pH-dependent
swelling of the final materials as well as the pH-dependent
release of a model dye cargo. This work provides another
example of the tailoring of physical properties of networks
such as hydrogels by the use of alternately functionalized
click components (alkynylamines in this case) in the
CuAAC step.
A very recent publication by Baker and coworkers
described an interesting synthesis of biodegradable
propargyl glycolide polymers (Scheme 25, 137).[137] By
CuAAC grafting of different ratios of decyl and PEG chains
to the glycolide backbone, polymer brushes that displayed
lower critical solution temperature (LCST) behavior were
produced. LCST occurs when polymers form a supramo-
lecular crosslinked gel driven by the expulsion of water as
temperature is lowered. The cloud point of these materials
was found to vary between 25 and 70 8C in direct
proportion to the percentage of PEG chains (from 55 to
90%, respectively) employed, and the authors suggest that
expanding these boundaries may be possible. Such
materials provide novel, biodegradable, and thermore-
sponsive hydrogels which may be useful for a range of
biomedical applications.
Anseth and coworkers have reported the synthesis of
PEG-peptide model hydrogels derived from tetra-
azido-terminated PEG star polymers coupled with linear
alkyne-telechelic peptides.[138] The peptide domains fea-
tured photo-reactive alloxycarboxyl (Alloc) groups which
were coupled to fluorescent, cysteine-containing peptides
via transparency-based photolithography to yield intern-
ally patterned materials and gradient-functionalized
hydrogel networks. The latter are of biological importance,
as cell migration can often be controlled using chemical
gradients. Although we do not describe it in this review,
our laboratories are pursuing an alternate pathway to
model networks having chemical and mechanical gradi-
ents by photolithography of materials possessing selec-
tively photodegradable units.[83]
Macromol. Rapid Commun. 2008, 29, 1052–1072
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
The above examples all describe hydrogel networks,
however, a number of groups have reported examples of
other polymeric crosslinked networks prepared from click
chemistry. Turro and coworkers have utilized CuAAC to
prepare novel degradable end-linked PtBA model net-
works.[139] ATRP polymerization of PtBA from a bifunc-
tional initiator containing an internal double bond, and
subsequent end-group transformation with sodium azide
yielded ozone-cleavable a,v-azido-PtBA macromonomers.
CuAAC crosslinking with tri- and tetra-functional alkynes
yielded end-linked model networks that possessed pore
sizes related to the molecular weight of the starting
macromonomer. Ozonolysis of thesematerials yielded star
polymer products of defined molecular weight, as deter-
minedbySECanalysis, dependingonwhichcrosslinkerwas
used. The same authors later reported an extension of this
work wherein the ozonizable double bond was replaced
with a photocleavable nitrobenzyloxycarbonyl linkage to
yield photodegradable materials of defined structure.[83] In
this article, they also demonstrated the first tandem
CuAAC/ATRP reaction for the assembly of clickable four-
armed structures. In this case, the resulting tetra-
azido-terminated photodegradable star polymers were
crosslinked with a bifunctional alkyne to give photode-
gradablematerials in a route complementary to that based
on linear polymers described above (Scheme 26).
In 2007, a paper by Yagci and coworkers highlighted the
ability of CuAAC to introduce orthogonal functionality for
post-polymerization processing in a convenient fashion, in
this case for the production of thermally curable PS.[140]
First, a PS-co-poly(chloromethylstyrene) random copoly-
mer was prepared by NMRP. The p-chloromethyl groups
were exchanged with azides by nucleophilic substitution
which was followed by CuAAC to attach alkyne-
functionalized benzooxazine moieties. Benzooxazines are
known to polymerize under thermal conditions and the
resulting crosslinked networks (Scheme 27) obtained upon
heating of the benzooxazine-functionalized PS showed
strongermechanical properties than networks prepared by
typical copolymerization with divinyl monomers.
www.mrc-journal.de 1067

J. A. Johnson, M. G. Finn, J. T. Koberstein, N. J. Turro
Scheme 28. Model liquid crystal networks prepared by CuAACcrosslinking of azido-telechelic poly(cyclooctene) derivatives andtripropargylamine.
Scheme 27. Thermally cured PS networks presented by Yagci andcoworkers.
1068
Ravelo and coworkers recently utilized diazide and
dialkyne organogelators to prepare novel organogels.[25] In
this case, however, the CuAAC reaction was performed
prior to gelation yielding linear polymers which were then
shown to form thermoreversible gels by hydrogen bonding
in dimethylsulfoxide (DMSO) and other DMSO-organic
solvent mixtures. Interestingly, two unexpected require-
ments of gel formation were the presence of an SO2 group
on the backbone of the polymers and the presence of some
residual copper from the CuAAC reaction. The presumed
interaction of Cu2þ with the triazole products of CuAAC
modification is thought to facilitate gelation, allowing
these materials to be considered as ‘‘metallogels’’.
Grubbs and coworkers recently reported the synthesis of
liquid crystal gel networks which were shown to be
reversible electro-optic switcheswhich converted between
a ‘‘scattering polydomain state’’ and a ‘‘transmissive
monodomain state’’ in the presence of an electric field.[141]
These gels were prepared by CuAAC crosslinking of
azido-telechelic poly(cyclooctene) polymers which pos-
sessed a mesogenic cyano-biphenyl group on each repeat
unit (Scheme 28). As the authors note, the modularity of
the synthetic methodmakes it possible to study the effects
of polymer chain length and structure on the electro-
optical properties of liquid crystals.
Crosslinked Polymer Surfaces and Thin Films
Thin polymer films are important in a range of applica-
tions including sensors, electronics, displays, and gas
separation. An obvious approach would be covalent LBL
construction, but relatively few examples exist due to a
lack of orthogonal, high-yielding reactions capable of
producing such materials. As a result, most LBL surfaces
rely on noncovalent interactions such as hydrogen
bonding or electrostatic attraction between polymers,
and so their mechanical properties and chemical diversity
may be limited for certain applications. Recently, CuAAC
has provided a unique route to covalently linked thin films
and has effectively opened the field of LBL synthesis to
nearly any polymeric precursors.
Macromol. Rapid Commun. 2008, 29, 1052–1072
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
The first example of CuAAC for LBL construction of thin
films was reported by Caruso and coworkers in 2006.[142]
The authors utilized PAA-N3 and PAA-alkyne to iteratively
CuAAC-couple PAA layers onto a variety of macroscopi-
cally flat surfaces. UV-Vis and FTIR measurements con-
firmed the increase in film thickness with each subsequent
CuAAC reaction as well as the high yield of the CuAAC
process. This methodology was later used for the synthesis
of ultrathin spherical capsules[111] as mentioned above
(Scheme 19).
Polyethylene (PE) is well known to be resistant to LBL
modification, but Chance and coworkers recently incorpo-
rated CuAAC into such a scheme.[143] Oxidation of PE with
CrO3/H2SO4 and functionalization of the resulting car-
boxylic acids with alkynes provided a substrate that could
be addressed with water-soluble azide and alkyne-
functionalized poly(alkylacrylamides) (PAM) in an LBL
fashion. The resulting PAM-coated PE materials were
further derivatized with small molecules in a final CuAAC
step. As is frequently the case, the CuAAC chemistry here
was performed in water, a reaction medium of increasing
interest in materials processing because of its perceived
environmental benefits.
Hawker and coworkers recently reported LBL film
synthesis using dendritic alkynes and azides
(Scheme 29).[144] The authors hypothesized that the
monodispersity and high degree of functionality of
dendrimers should provide for stable films with greater
control over the thickness growth with each layer. Indeed,
DOI: 10.1002/marc.200800208

Construction of Linear Polymers, Dendrimers, Networks, and . . .
Scheme 29. LBL assembly of dendrimers on silicon wafers using iterative CuAAC/wash steps with alternating azide and alkynesurface-functionalized dendrimers.
by ellipsometrymeasurements the authors confirmed that
the film thickness increase with each layer was dependent
on the generation of dendrimer used and was a constant
for each new layer. Interestingly, the fifth generation
bis-MPA dendrimers give a drastic increase in thickness
due to the ‘‘dendritic’’ effect, in which steric hindrance
causes the structure to avoid compression and remain
spherical. As expected, linear polymeric azide and alkyne
analogs gave much larger and more variable increases in
thickness for each LBL step.
Figure 1. Construction of covalently linked gold nanorod chains byCuAAC. Image taken from ref. [145].
Crosslinked Structures Derived from NanosizedBuilding Blocks
Covalent crosslinking of nanoscale objects is a blossoming
field which promises to give rise to novel materials
possessing potentially unprecedented properties. Rao and
coworkers have recently studied the structural diversity of
CuAAC covalently crosslinked nanorods, nanoparticles,
and nanotubes.[145] An interesting example was provided
by their observation that thioazide- and thioalkyne-
capped gold nanorods formed chain-like structures
(Figure 1) whereas spherical gold particles formed an
organized network. The authors suggested that the rods
form chain structures because the thiol linkers prefer to
Macromol. Rapid Commun. 2008, 29, 1052–1072
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
bind to the ends of the gold rods where the (1,1,1) face of
gold is available. These results hint at the diversity of
phenomena to be uncovered in the exciting field of
covalent nanostructure assembly. These investigators
have also provided exciting TEM images of CuAAC
crosslinked CdSe nanocrystals showing the formation of
periodic arrays as well as the marriage of alkyne-
www.mrc-journal.de 1069

J. A. Johnson, M. G. Finn, J. T. Koberstein, N. J. Turro
1070
functionalized gold nanoparticles with azide-
functionalized carbon nanotubes to yield novel nanoscopic
hybrid structures.
Conclusion
The CuAAC reaction has proven effective for a broad range
of materials construction applications. From small mole-
cules to linear polymers to branched polymers to
nanoscale and macroscopic materials, the CuAAC process
has been successfully applied to building blocks of every
scale and chemical functionality. Furthermore, structure–
property relationships of many complicated materials (for
example the electro-optic properties of liquid crystals as a
function of polymer chain length[141]) can now be studied
in great depth, as the synthesis of the required polymeric
precursors is made reliable by CuAAC. Lastly, the process is
user friendly: many first-time attempts are successful,
with off-the-shelf and inexpensive catalyst components.
These features place even greater emphasis on the
function of the materials that can be envisioned, rather
than on the potential problems of their construction. We
are excited to see the continued use of CuAAC for an
increasingly creative and diverse array of materials
applications.
Received: April 5, 2008; Accepted: April 25, 2008; DOI: 10.1002/marc.200800208
Keywords: alkyne; azide; click; CuAAC; dendrimers; functionali-zation of polymers; hydrogels; organogels
[1] H. C. Kolb, M. G. Finn, K. B. Sharpless, Angew. Chem., Int. Ed.2001, 40, 2004.
[2] C. J. Hawker, V. V. Fokin, M. G. Finn, K. B. Sharpless, Aust. J.Chem. 2007, 60, 381.
[3] V. V. Rostovtsev, L. G. Green, V. V. Fokin, K. B. Sharpless,Angew. Chem., Int. Ed. 2002, 41, 2596.
[4] C. W. Tornoe, C. Christensen, M. Meldal, J. Org. Chem. 2002,67, 3057.
[5] W. G. Lewis, F. G. Magallon, V. V. Fokin, M. G. Finn, J. Am.Chem. Soc. 2004, 126, 9152.
[6] V. O. Rodionov, V. V. Fokin, M. G. Finn, Angew. Chem., Int. Ed.2005, 44, 2210.
[7] V. O. Rodionov, S. I. Presolski, D. D. Diaz, V. V. Fokin, M. G.Finn, J. Am. Chem. Soc. 2007, 129, 12705.
[8] S. Punna, J. Kuzelka, Q. Wang, M. G. Finn, Angew. Chem., Int.Ed. 2005, 44, 2215.
[9] P. Wu, V. V. Fokin, Aldrichim. Acta 2007, 40, 7.[10] V. O. Rodionov, S. I. Presolski, S. Gardinier, Y.-H. Lim, M. G.
Finn, J. Am. Chem. Soc. 2007, 129, 12696.[11] C. Nolte, P. Mayer, B. F. Straub, Angew. Chem., Int. Ed. 2007,
46, 2101.[12] J. E. Macdonald, J. A. Kelly, J. G. C. Veinot, Langmuir 2007, 23,
9543.
Macromol. Rapid Commun. 2008, 29, 1052–1072
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
[13] I. Aprahamian, O. S. Miljanic, W. R. Dichtel, K. Isoda, T.Yasuda, T. Kato, J. F. Stoddart, Bull. Chem. Soc. Jpn. 2007,80, 1856.
[14] W. H. Binder, R. Sachsenhofer, Macromol. Rapid Commun.2007, 28, 15.
[15] A. Dondoni, Chem. Asian J. 2007, 2, 700.[16] R. A. Evans, Aust. J. Chem. 2007, 60, 384.[17] D. Fournier, R. Hoogenboom, U. S. Schubert, Chem. Soc. Rev.
2007, 36, 1369.[18] G. W. Goodall, W. Hayes, Chem. Soc. Rev. 2006, 35, 280.[19] J.-F. Lutz, Angew. Chem., Int. Ed. 2007, 46, 1018.[20] J. E. Moses, A. D. Moorhouse, Chem. Soc. Rev. 2007, 36, 1249.[21] H. Nandivada, X. Jiang, J. Lahann,Adv. Mater. 2007, 19, 2197.[22] B. Voit, New J. Chem. 2007, 31, 1139.[23] J.-F. Lutz, H. Schlaad, Polymer 2008, 49, 817.[24] D. D. Diaz, S. Punna, P. Holzer, A. K. McPherson, K. B. Sharp-
less, V. V. Fokin, M. G. Finn, J. Polym. Sci., Part A: Polym.Chem. 2004, 42, 4392.
[25] D. D. Diaz, J. J. Marrero Tellado, D. G. Velazquez, A. G. Ravelo,Tetrahedron Lett. 2008, 49, 1340.
[26] N. G. Angelo, P. S. Arora, J. Am. Chem. Soc. 2005, 127, 17134.[27] D. J. V. C. van Steenis, O. R. P. David, G. P. F. van Strijdonck,
J. H. van Maarseveen, J. N. H. Reek, Chem. Commun. 2005,4333.
[28] R. M. Meudtner, S. Hecht, Macromol. Rapid Commun. 2008,29, 347.
[29] V. Aucagne, D. A. Leigh, Org. Lett. 2006, 8, 4505.[30] G. Lu, S. Lam, K. Burgess, Chem. Commun. 2006, 1652.[31] Y. Zhu, Y. Huang, W.-D. Meng, H. Li, F.-L. Qing, Polymer 2006,
47, 6272.[32] J. A. Opsteen, J. C. M. van Hest, Chem. Commun. 2005, 57.[33] J. A. Opsteen, J. C. M. van Hest, J. Polym. Sci., Part A: Polym.
Chem. 2007, 45, 2913.[34] N. V. Tsarevsky, B. S. Sumerlin, K. Matyjaszewski, Macro-
molecules 2005, 38, 3558.[35] B. A. Laurent, S. M. Grayson, J. Am. Chem. Soc. 2006, 128,
4238.[36] H. Durmaz, A. Dag, O. Altintas, T. Erdogan, G. Hizal, U. Tunca,
Macromolecules 2007, 40, 191.[37] X.-M. Liu, A. Thakur, D. Wang, Biomacromolecules 2007, 8,
2653.[38] R. Kumar, A. El-Sagheer, J. Tumpane, P. Lincoln, L. M.
Wilhelmsson, T. Brown, J. Am. Chem. Soc. 2007, 129, 6859.[39] C. J. Arnusch, H. Branderhorst, B. de Kruijff, R. M. J. Liskamp,
E. Breukink, R. J. Pieters, Biochemistry 2007, 46, 13437.[40] M. Ortega-Munoz, J. Morales-Sanfrutos, F. Perez-Balderas, F.
Hernandez-Mateo, D. Giron-Gonzalez Ma, N. Sevillano-Tripero, R. Salto-Gonzalez, F. Santoyo-Gonzalez, Org. Biomol.Chem. 2007, 5, 2291.
[41] M. Touaibia, A. Wellens, T. C. Shiao, Q. Wang, S. Sirois, J.Bouckaert, R. Roy, Chem. Med. Chem. 2007, 2, 1190.
[42] M. J. Frechet Jean, Proc. Natl. Acad. Sci. USA 2002, 99, 4782.[43] D. A. Tomalia, A. M. Naylor, W. A. Goddard, III, Angewandte
Chemie 1990, 102, 119.[44] C. J. Hawker, J. M. J. Frechet, J. Am. Chem. Soc. 1990, 112,
7638.[45] P. Wu, A. K. Feldman, A. K. Nugent, C. J. Hawker, A. Scheel, B.
Voit, J. Pyun, J. M. J. Frechet, K. B. Sharpless, V. V. Fokin,Angew. Chem., Int. Ed. 2004, 43, 3928.
[46] M. J. Joralemon, R. K. O’Reilly, J. B. Matson, A. K. Nugent, C. J.Hawker, K. L. Wooley, Macromolecules 2005, 38, 5436.
[47] P. Wu, M. Malkoch, J. N. Hunt, R. Vestberg, E. Kaltgrad, M. G.Finn, V. V. Fokin, K. B. Sharpless, C. J. Hawker, Chem. Com-mun. 2005, 5775.
DOI: 10.1002/marc.200800208

Construction of Linear Polymers, Dendrimers, Networks, and . . .
[48] C. Bouillon, A. Meyer, S. Vidal, A. Jochum, Y. Chevolot, J.-P.Cloarec, J.-P. Praly, J.-J. Vasseur, F.Morvan, J. Org. Chem. 2006,71, 4700.
[49] G. Chen, L. Tao, G. Mantovani, J. Geng, D. Nystroem, D. M.Haddleton, Macromolecules 2007, 40, 7513.
[50] G. Chen, L. Tao, G. Mantovani, V. Ladmiral, D. P. Burt, J. V.Macpherson, D. M. Haddleton, Soft Matter 2007, 3, 732.
[51] Z. Guo, A. Lei, X. Liang, Q. Xu, Chem. Commun. 2006, 4512.[52] S. Punna, E. Kaltgrad, M. G. Finn, Bioconjugate Chem. 2005,
16, 1536.[53] M. Slater, M. Snauko, F. Svec, J. M. J. Frechet, Anal. Chem.
2006, 78, 4969.[54] K. Zeitler, I. Mager, Adv. Synth. Catal. 2007, 349, 1851.[55] M. Ortega-Munoz, J. Lopez-Jaramillo, F. Hernandez-Mateo, F.
Santoyo-Gonzalez, Adv. Synth. Catal. 2006, 348, 2410.[56] M. R. Whittaker, C. N. Urbani, M. J. Monteiro, J. Am. Chem.
Soc. 2006, 128, 11360.[57] C. N. Urbani, C. A. Bell, D. Lonsdale, M. R. Whittaker, M. J.
Monteiro, Macromolecules 2008, 41, 76.[58] P. Antoni, D. Nystroem, C. J. Hawker, A. Hult, M. Malkoch,
Chem. Commun. 2007, 2249.[59] A. K. Diallo, C. Ornelas, L. Salmon, J. R. Aranzaes, D. Astruc,
Angew. Chem., Int. Ed. 2007, 46, 8644.[60] C. Ornelas, J. Ruiz Aranzaes, E. Cloutet, S. Alves, D. Astruc,
Angew. Chem., Int. Ed. 2007, 46, 872.[61] X. Shen, H. Liu, Y. Li, S. Liu, Macromolecules 2008, 41, 2421.[62] A. Converso, K. Burow, A. Marzinzik, K. B. Sharpless, M. G.
Finn, J. Org. Chem. 2001, 66, 4386.[63] A. P. Vogt, S. R. Gondi, B. S. Sumerlin, Aust. J. Chem. 2007, 60,
396.[64] M. Malkoch, R. J. Thibault, E. Drockenmuller, M. Messersch-
midt, B. Voit, T. P. Russell, C. J. Hawker, J. Am. Chem. Soc.2005, 127, 14942.
[65] B. Parrish, R. B. Breitenkamp, T. Emrick, J. Am. Chem. Soc.2005, 127, 7404.
[66] R. Riva, S. Schmeits, F. Stoffelbach, C. Jerome, R. Jerome, P.Lecomte, Chem. Commun. 2005, 5334.
[67] A. P. Vogt, B. S. Sumerlin, Macromolecules 2006, 39, 5286.[68] H. Gao, K. Matyjaszewski, J. Am. Chem. Soc. 2007, 129, 6633.[69] X. Jiang, M. C. Lok, W. E. Hennink, Bioconjugate Chem. 2007,
18, 2077.[70] F. Morvan, A. Meyer, A. Jochum, C. Sabin, Y. Chevolot, A.
Imberty, J.-P. Praly, J.-J. Vasseur, E. Souteyrand, S. Vidal,Bioconjugate Chem. 2007, 18, 1637.
[71] D. Quemener, M. Le Hellaye, C. Bissett, T. P. Davis, C. Bar-ner-Kowollik, M. H. Stenzel, J. Polym. Sci., Part A: Polym.Chem. 2007, 46, 155.
[72] R. Riva, S. Schmeits, C. Jerome, R. Jerome, P. Lecomte,Macro-molecules 2007, 40, 796.
[73] N. V. Tsarevsky, S. A. Bencherif, K. Matyjaszewski, Macro-molecules 2007, 40, 4439.
[74] Z. Li, Q. Zeng, G. Yu, Z. Li, C. Ye, Y. Liu, J. Qin,Macromol. RapidCommun. 2008, 29, 136.
[75] O. Altintas, G. Hizal, U. Tunca, J. Polym. Sci., Part A: Polym.Chem. 2006, 44, 5699.
[76] O. Altintas, G. Hizal, U. Tunca, J. Polym. Sci., Part A: Polym.Chem. 2008, 46, 1218.
[77] O. Altintas, B. Yankul, G. Hizal, U. Tunca, J. Polym. Sci., Part A:Polym. Chem. 2006, 44, 6458.
[78] O. Altintas, B. Yankul, G. Hizal, U. Tunca, J. Polym. Sci., Part A:Polym. Chem. 2007, 45, 3588.
[79] G. Deng, D. Ma, Z. Xu, Eur. Polym. J. 2007, 43, 1179.[80] H. Gao, K. Matyjaszewski, Macromolecules 2006, 39, 4960.[81] J. Luo, Y. Chen, X. X. Zhu, Synlett 2007, 2201.
Macromol. Rapid Commun. 2008, 29, 1052–1072
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
[82] J. Zhu, X. Zhu, E. T. Kang, K. G. Neoh, Polymer 2007, 48, 6992.[83] J. A. Johnson, M. G. Finn, J. T. Koberstein, N. J. Turro, Macro-
molecules 2007, 40, 3589.[84] H. Li, R. Riva, R. Jerome, P. Lecomte,Macromolecules 2007, 40,
824.[85] H. Li, R. Riva, R. Kricheldorf Hans, R. Jerome, P. Lecomte,
Chemistry 2008, 14, 358.[86] S. Sen Gupta, K. S. Raja, E. Kaltgrad, E. Strable, M. G. Finn,
Chem. Commun. 2005, 4315.[87] Q. Zeng, T. Li, B. Cash, S. Li, F. Xie, Q. Wang, Chem. Commun.
2007, 1453.[88] G. Destito, R. Yeh, C. S. Rae, M. G. Finn, M. Manchester, Chem.
Biol. 2007, 14, 1152.[89] E. Kaltgrad, S. Sen Gupta, S. Punna, C.-Y. Huang, A. Chang,
C.-H. Wong, M. G. Finn, O. Blixt, ChemBioChem 2007, 8, 1455.[90] P.-C. Lin, S.-H. Ueng, M.-C. Tseng, J.-L. Ko, K.-T. Huang, S.-C. Yu,
A. K. Adak, Y.-J. Chen, C.-C. Lin, Angew. Chem., Int. Ed. 2006,45, 4286.
[91] J. Geng, G. Mantovani, L. Tao, J. Nicolas, G. Chen, R. Wallis,D. A. Mitchell, B. R. G. Johnson, S. D. Evans, D. M. Haddleton,J. Am. Chem. Soc. 2007, 129, 15156.
[92] D. E. Prasuhn, Jr., P. Singh, E. Strable, S. Brown, M. Manche-ster, M. G. Finn, J. Am. Chem. Soc. 2008, 130, 1328.
[93] P. M. E. Gramlich, C. T. Wirges, J. Gierlich, T. Carell, Org. Lett.2008, 10, 249.
[94] M. Fischler, U. Simon, H. Nir, Y. Eichen, G. A. Burley, J.Gierlich, P. M. E. Gramlich, T. Carell, Small 2007, 3, 1049.
[95] M. Fischler, A. Sologubenko, J. Mayer, G. Clever, G. Burley, J.Gierlich, T. Carell, U. Simon, Chem. Commun. 2008, 169.
[96] R. K. O’Reilly, M. J. Joralemon, C. J. Hawker, K. L. Wooley,Chem. Eur. J. 2006, 12, 6776.
[97] R. K. O’Reilly, M. J. Joralemon, C. J. Hawker, K. L. Wooley,J. Polym. Sci., Part A: Polym. Chem. 2006, 44, 5203.
[98] B. Li, A. L. Martin, E. R. Gillies, Chem. Commun. 2007, 5217.[99] D. A. Fleming, C. J. Thode, M. E. Williams, Chem. Mater. 2006,
18, 2327.[100] R. Ranjan,W. J. Brittain,Macromol. Rapid Commun. 2007, 28,
2084.[101] M. A. White, J. A. Johnson, J. T. Koberstein, N. J. Turro, J. Am.
Chem. Soc. 2006, 128, 11356.[102] G.-L. Huang, T.-C. Liu, M.-X. Liu, X.-Y. Mei, Anal. Biochem.
2005, 340, 52.[103] P.-C. Lin, S.-H. Ueng, S.-C. Yu, M.-D. Jan, A. K. Adak, C.-C. Yu,
C.-C. Lin, Org. Lett. 2007, 9, 2131.[104] L. Polito, D. Monti, E. Caneva, E. Delnevo, G. Russo, D.
Prosperi, Chem. Commun. 2008, 621.[105] A. Gole, C. J. Murphy, Langmuir 2008, 24, 266.[106] K. Patel, S. Angelos, W. R. Dichtel, A. Coskun, Y.-W. Yang, J. I.
Zink, J. F. Stoddart, J. Am. Chem. Soc. 2008, 130, 2382.[107] H. Li, F. Cheng, A.M. Duft, A. Adronov, J. Am. Chem. Soc. 2005,
127, 14518.[108] M. J. Joralemon, R. K. O’Reilly, C. J. Hawker, K. L. Wooley,
J. Am. Chem. Soc. 2005, 127, 16892.[109] R. K. O’Reilly, M. J. Joralemon, C. J. Hawker, K. L. Wooley,New
J. Chem. 2007, 31, 718.[110] X. Jiang, J. Zhang, Y. Zhou, J. Xu, S. Liu, J. Polym. Sci., Part A:
Polym. Chem. 2008, 46, 860.[111] G. K. Such, E. Tjipto, A. Postma, A. P. R. Johnston, F. Caruso,
Nano Lett. 2007, 7, 1706.[112] B. G. De Geest, W. Van Camp, F. E. Du Prez, S. C. De Smedt, J.
Demeester, W. E. Hennink, Chem. Commun. 2008, 190.[113] P. J. Flory, J. Am. Chem. Soc. 1941, 63, 3083.[114] P. J. Flory, J. Am. Chem. Soc. 1941, 63, 3091.[115] P. J. Flory, J. Am. Chem. Soc. 1941, 63, 3096.
www.mrc-journal.de 1071

J. A. Johnson, M. G. Finn, J. T. Koberstein, N. J. Turro
1072
[116] D. D. Evanoff, Jr., S. E. Hayes, Y. Ying, G. H. Shim, J. R.Lawrence, J. B. Carroll, R. D. Roeder, J. M. Houchins, C. F.Huebner, S. H. Foulger, Adv. Mater. 2007, 19, 3507.
[117] H. Nandivada, H.-Y. Chen, L. Bondarenko, J. Lahann, Angew.Chem., Int. Ed. 2006, 45, 3360.
[118] Q. Shi, X. Chen, T. Lu, X. Jing, Biomaterials 2008, 29, 1118.[119] Y. Liu, D. D. Diaz, A. A. Accurso, K. B. Sharpless, V. V. Fokin,
M. G. Finn, J. Polym. Sci., Part A: Polym. Chem. 2007, 45, 5182.[120] N. Le Baut, D. D. Diaz, S. Punna, M. G. Finn, H. R. Brown,
Polymer 2007, 48, 239.[121] A. R. Katritzky, N. K. Meher, S. Hanci, R. Gyanda, S. R. Tala, S.
Mathai, R. S. Duran, S. Bernard, F. Sabri, S. K. Singh, J.Doskocz, D. A. Ciaramitaro, J. Polym. Sci., Part A: Polym.Chem. 2007, 46, 238.
[122] C. Li, M. G. Finn, J. Polym. Sci., Part A: Polym. Chem. 2006, 44,5513.
[123] S. M. Park, Y. S. Lee, B. H. Kim, Chem. Commun. 2003, 2912.[124] G. Conte, F. Ely, H. Gallardo, Liq. Cryst. 2005, 32, 1213.[125] H. Gallardo, F. Ely, A. Bortoluzzi, G. Conte, Liq. Cryst. 2005, 32,
667.[126] G. Conte, R. Cristiano, F. Ely, H. Gallardo, Synth. Commun.
2006, 36, 951.[127] D. D. Diaz, K. Rajagopal, E. Strable, J. Schneider, M. G. Finn,
J. Am. Chem. Soc. 2006, 128, 6056.[128] T. Devic, O. David,M. Valls, J. Marrot, F. Couty, G. Ferey, J. Am.
Chem. Soc. 2007, 129, 12614.[129] Y. Liu, C.-F. Ke, H.-Y. Zhang, J. Cui, F. Ding, J. Am. Chem. Soc.
2008, 130, 600.[130] D. A. Ossipov, J. Hilborn, Macromolecules 2006, 39, 1709.[131] M. Malkoch, R. Vestberg, N. Gupta, L. Mespouille, P. Dubois,
A. F. Mason, J. L. Hedrick, Q. Liao, C. W. Frank, K. Kingsbury,C. J. Hawker, Chem. Commun. 2006, 2774.
Macromol. Rapid Commun. 2008, 29, 1052–1072
� 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
[132] V. Crescenzi, L. Cornelio, C. Di Meo, S. Nardecchia, R.Lamanna, Biomacromolecules 2007, 8, 1844.
[133] M. Sawa, T.-L. Hsu, T. Itoh, M. Sugiyama, S. R. Hanson,P. K. Vogt, C.-H. Wong, Proc. Natl. Acad. Sci. USA 2006,103, 12371.
[134] A. E. Speers, G. C. Adam, B. F. Cravatt, J. Am. Chem. Soc. 2003,125, 4686.
[135] S. I. van Kasteren, H. B. Kramer, H. H. Jensen, S. J. Campbell, J.Kirkpatrick, N. J. Oldham, D. C. Anthony, B. G. Davis, Nature2007, 446, 1105.
[136] J. Zednik, R. Riva, P. Lussis, C. Jerome, R. Jerome, P. Lecomte,Polymer 2008, 49, 697.
[137] X. Jiang, E. B. Vogel, M. R. Smith, III, G. L. Baker, Macromol-ecules 2008, 41, 1937.
[138] B. D. Polizzotti, B. D. Fairbanks, K. S. Anseth, Biomacromo-lecules 2008, 9, 1084.
[139] J. A. Johnson, D. R. Lewis, D. D. Diaz, M. G. Finn, J. T.Koberstein, N. J. Turro, J. Am. Chem. Soc. 2006, 128,6564.
[140] M. Ergin, B. Kiskan, B. Gacal, Y. Yagci, Macromolecules 2007,40, 4724.
[141] Y. Xia, R. Verduzco, R. H. Grubbs, J. A. Kornfield, J. Am. Chem.Soc. 2008, 130, 1735.
[142] G. K. Such, J. F. Quinn, A. Quinn, E. Tjipto, F. Caruso, J. Am.Chem. Soc. 2006, 128, 9318.
[143] D. E. Bergbreiter, B. S. Chance, Macromolecules 2007, 40,5337.
[144] R. Vestberg, M. Malkoch, M. Kade, P. Wu, V. V. Fokin, K. B.Sharpless, E. Drockenmuller, C. J. Hawker, J. Polym. Sci., PartA: Polym. Chem. 2007, 45, 2835.
[145] R. Voggu, P. Suguna, S. Chandrasekaran, C. N. R. Rao, Chem.Phys. Lett. 2007, 443, 118.
DOI: 10.1002/marc.200800208





























