Embed Size (px)
Citation preview

Biochem. J. (1971) 124, 677-683Printed in Great Britain
Coupling of Glycosaminoglycans to Agarose Beads(Sepharose 4B)
By P.-H. IVERIUSMedicinsk-Kemi8ka Indtitutionen, Upp8ala Univeraitet, 751 22 Uppsala 1, Sweden
(Received 10 June 1971 )
1. Heparin, heparan sulphate, chondroitin sulphate and dermatan sulphatewere covalently attached to beads of agarose activated by cyanogen bromide. Thebond is probably mediated by the amino group of a serine or peptide residue at thereducing end of the polysaccharide chain. 2. The uptake of glycosaminoglycanduring the coupling procedure is about O.9mg/ml of wet gel. However, directanalysis of washed and freeze-dried gels reveals that only about one-third of thisamount is firmly attached to the gel. 3. The use of the gels for polysaccharidaseanalyses is exemplified by a hyaluronidase assay. Further applications, e.g. inter-action studies and preparative purposes, are discussed.
Connective-tissue glycosaminoglycans are nega-tively charged polyelectrolytes with ion-exchangeproperties (Laurent, Wasteson & Obrink, 1969).The presence of charges might account for certainbiological effects, e.g. the anticoagulant property ofheparin and the liberation of lipoprotein lipase intothe blood after heparin injection (Brimacombe &Webber, 1964). Further, it has been stated that thecharged glycosaminoglycans are involved in thefibrillogenesis of collagen (Jackson & Bentley,1968) and the development of atherosclerosis(Muir, 1964). However, on the whole our knowledgein this field is scanty.
Several methods have been used to study theelectrostatic interaction between proteins andpolyelectrolytes. Interactions giving rise to in-soluble precipitates can be detected by assays ofthe supernatants or by turbidity measurements(Cornwell & Kruger, 1961). However, suchtechniques do not reveal the formation of solublecomplexes. Further, the ratio between the proteinand the polysaccharide concentrations determinesthe composition of the complexes, which in turnaffects their solubility (Comwell & Kruger, 1961).Electrophoresis has been used to detect the for-mation of soluble complexes (Pigman, Gramling &Holley, 1961), but the interpretation of suchexperiments has been criticized (Davies, Nichol& Ogston, 1963). Analytical ultracentrifugation(Janado & Nishida, 1967) is an elaborate methodthat suffers from the same drawbacks as electro-phoresis. The interaction between tropocollagenand glycosaminoglyeans has been studied by light-scattering (B. Obrink, unpublished work). Solublecomplexes may be detected by this method, which
avoids the limitations of the dynamic methods ofelectrophoresis and ultracentrifugation. Finally, a
column-chromatographic technique has been em-
ployed by Obrink & Wasteson (1971), who studiedthe interaction between tropocollagen andchondroitin sulphate by using a granulatedgel of tropocollagen cross-linked with glutar-aldehyde.The methods reviewed above have been used to
investigate the interaction between free poly-saccharide chains and other substances. Suchexperiments differ from conditions in vivo, wherethe glycosaminoglycans, except heparin andhyaluronic acid, are immobilized by attachment toa protein core (e.g. see Balazs, 1970).A new approach to interaction studies was intro-
duced by Bettelheim, Laurent & Pertoft (1966),who used granulated gels of chondroitin sulphatecross-linked with epichlorohydrin for ion-exchangechromatography. However, the introduction ofcross-links interferes with the polysaccharidestructure. The covalent coupling of varioussubstances to insoluble polymers that have beenactivated by cyanogen bromide has proved a usefuland mild method for proteins (Ax6n, Porath &Ernback, 1967). Glycosaminoglycans liberatedfrom tissues by proteolysis have a serine or peptideresidue at the reducing end of the polysaccharide-chain and thus possess a free amino group (e.g. see
Balazs, 1970). Therefore it has been possible to use
the cyanogen bromide method for attaching glycos-aminoglycans covalently to agarose beads (Sepha-rose 4B). The present paper describes the prepara-tion and characterization of some glycosamino-glycans and their conjugates with Sepharose 4B.
677

P.-H. IVERIUS
EXPERIMENTAL
Materials
Crude heparin (prepared from pig intestinal mucosa;anticoagulant activity, 126 U.S. Pharmacopea units/mg)was purchased from Wilson Laboratories, Chicago, Ill.,U.S.A.Human aortas were obtained from autopsy cases. No
strict selection was made with regard to age or state ofhealth, but severely atheromatous vessels were not used.The vessels were kept frozen until preparation. Bovineaortas were obtained from a slaughterhouse and cooled to400 within lih after death.
Crude papain type II (EC 3.4.4.10), deoxyribonucleaseI (EC 3.1.4.5) from ox pancreas, ribonuclease A type I-A(EC 2.7.7.16) also from ox pancreas, and bovine testicularhyaluronidase type I (EC 3.2.1.35) were purchased fromSigma (London) Chemioal Co. Ltd., London S.W.6, U.K.Bovine testioular hyaluronidase in ampoules containingIOOV.R.U. (viscosity-reducing units) was also suppliedby Dr B. Hogberg, AB Leo, Halsingborg, Sweden (lot15573). Chondroitin sulphate lyase ABC (EC 4.2.99.6) wasobtained from Seikagaku Kogyo Co., Ltd., Tokyo, Japan.Sepharose 4B (lots 4088, 5214 and 5742), Sepharose 6Band Sephadex G-200 were purchased from PharmaciaFine Chemicals AB, Uppsala, Sweden. Cyanogen bromidewas obtained from Schuchardt, Munich, Germany, andethanolamine and acetic acid anhydride were fromE. Merck, A.-G., Darmstadt, Germany.
Analyticat methods
Hydrolysis before hexosamine analysis was performedin sealed glass tubes at 1000C for 14h. After hydrolysis,the samples were evaporated, dissolved in water andapplied to micro-columns (5mm x 35mm) of Dowex5OW (X8; 200-400 mesh; H+ form). The columns were
rinsed with 3ml of water before the hexosamines were
eluted with 1.5ml of 2M-HCI. The samples were analysedfor total hexosamine or for glucosamine/galactosamineratios. The former analysis was performed by a modi-fication of the Elson-Morgan method (Gardell, 1953) withno correction for destruction during hydrolysis. Thelatter analysis was performed by either of two g.l.o.methods (Radhakrishnamurthy, Dalferes & Berenson,1966; Hase & Matsushima, 1969).Uronic acid was determined by the carbazole reaction
as modified by Bitter & Muir (1962), with glucurono-laotone as standard. By comparing the colour yieldsobtained in the presence and in the absence of boraterespectively, the reaotion was also used to identifydifferent glycosaminoglyeans (Bitter & Muir, 1962).Sulphate was determined by the method ofAntonopoulos(1962). The contents of sulphaminohexose in heparin andheparan sulphate were measured by the method ofLagunoff & Warren (1962), with glucosamine as standard.Samples were driedin vacuo at 60°C over P205 to determinetheir dry weight.
Specific optical rotations of glycosaminoglyeans inaqueous solution were measured in a Perkin-Elmer 141polarimeter at 250C and at 589nm (kindly performed byDr L. Ohlsson at the Institute of Organic Chemistry,University of Uppsala). Electrophoresis of polysaccha-
rides was carried out on strips of cellulose acetate in0.1 m-barium acetate (Wessler, 1968) or in 0.1 M-HC1(Wessler, 1971). To improve the separation betweenchondroitin 4-sulphate and chondroitin 6-sulphate, theduration of electrophoresis was increased to 12h. Refer-ence polysaccharides were kindly supplied by Dr E.Wessler of this Institute.Average molecular weights of the polysaccharides were
determined by analytical gel chromatography on columnsof Sephadex G-200 or Sepharose 6B, calibrated with well-characterized fractions ofchondroitin sulphate (Wasteson,1971).
Preparative proceduresProteolysis of tissue. Human or bovine aortas were
freed from adhering tissues, cut into pieces and defattedat 40C with several ohanges of aoetone and ether. Drydefatted material was digested with papain for 24h at6500 in 0.05m-sodium acetate (pH5.5)-0.3M-NaCl-0.01 M-EDTA-0.01 M-cysteine hydrochloride. Papain (1 g)and 150-200g of dry tissue were added to each litre ofbuffer. After 12h the amounts of papain and cysteinehydrochloride were doubled. The final digests werefiltered through glass-wool and then centrifuged toremove insoluble residues.
Other enzymic procedures. Nucleio acids were removedby digestion for 20h at 370C with ribonuclease anddeoxyribonuclease (10mg of each/l) in 0.05M-tris-HClbuffer (pH 7.0)-0.02m-MgC12-0.15 m-NaCl. Digestion withhyaluronidase (Sigma) was performed in 0.1 M-sodiumacetate buffer (pH 5.0)-0.15m-NaCl for 48h at 370C andan enzyme concentration of 0.4 g/l. Hyaluronidase-resistant material was digested for 38h at 370C in 0.05M-tris buffer, pH 8.0, containing chondroitin sulphate lyaseABC (20 units/l) and bovine serum albumin (100 mg/l)(Yamagata, Saito, Habuchi & Suzuki, 1968).
Precipitation of poly8accharide with cetylpyridiniumchloride. Precipitation of polysaccharide with cetyl-pyridinium chloride (Scott, 1960) was performed at theionic strengths of the respective buffers (I 0.3 or less) withthe precipitant present in a threefold excess. The poly-saccharides were converted into their sodium salts bydissolving the cetylpyridinium-polysaccharide precipi-tates in 2m-NaCl at 370C, followed by precipitation with3 vol. of 95% (v/v) ethanol. This procedure was repeatedtwice. Chloride-free polysacoharide precipitates wereobtained by dropwise addition of 4M-sodium acetate to asolution of polysaccharide in 80% (v/v) ethanol. Theprecipitates were washed with acetone and dried under astream of air.
RESULTS
Isolation and characterization of glycosaminoglycanw
Heparin. Crude heparin was repeatedly precipi-tated from 1.2m-sodium chloride by adding cetyl-pyridinium chloride as described by Lindahl,Cifonelli, Lindahl & Roddn (1965). On electro-phoresis in barium acetate most of the materialmigrated as heparin, but a faint spot was also seenin the position ofdermatan sulphate. This impuritywas also detected by g.l.c. of the hexosamines,
-678 1971

MATRIX-BOUND GLYCOSAMINOGLYCANSTable 1. Analysis of polysaccharidefractiomn
679
Percentage values are expressed on a dry-weight basis. The sulphate/disaccharide molar ratios werecalculated by comparing measured sulphate contents with theoretical values for polymers containingrepeating disaccharide units only. Mw is the weight-average molecular weight and S. is the number-averagemolecular weight.
FractionHeparin (Hep-II)Heparan sulphate(HS-II)
Chondroitinsulphate (CS-I)Dermatan sulphate(DS-III)
Uronic Hexose-acid amine Sulphate(%) (%) (%)
34 20 38
40 34
Sulphamino- Sulphate/ Carbazole (ES30)hexose disaccharide borate/no(%) molar ratio borate ratio [,X]25 Rw28 2.3 1.65 +54 12800 10500
10 13
34 27 20
29 30 21
0.46
1.1
1.1
1.55 +95 58300 40500
2.04 -24 37100 27900
2.83 -62 41500 32600
which were shown to contain about 4% of galactos-amine. The degree of sulphation, as determined byelectrophoresis in 0.1 M-hydrochloric acid, was thesame as that of a reference sample of heparin. Theresults of the analyses are shown in Table 1.Heparan suIphate. Dried human aorta (4.5kg)
was digested with papain. The polysaccharideswere isolated from the digest by precipitation withcetylpyridinium chloride and then subjected tonuclease digestion. Cetylpyridinium chloride pre-cipitation of the nuclease digest yielded 45g ofcrude polygaccharides, which on electrophoresis inbarium acetate showed components having mobili-ties similar to those of chondroitin sulphate,dermatan sulphate and heparan sulphate. Thecrude polysaccharide was further digested withhyaluronidase in 2.5 litres of sodium acetate buffer,precipitated with cetylpyridinium chloride andfinally treated with chondroitin sulphate lyaseABC in 1 litre of tris buffer. The material (5.9g)obtained after a final cetylpyridinium chlorideprecipitation behaved as pure heparan sulphate on
electrophoresis in barium acetate. G.I.c. of theconstituent amino sugars showed that glucosaminewas the only detectable component, confirming thepurity of the preparation. Electrophoresis in0.1 M-hydrochloric acid showed a homogeneous spotwith a mobility about half that of monosulphatedchondroitin sulphate. A sulphate-disaccharidemolar ratio of 0.46 confirmed the result of theelectrophoresis. Additional analytical results are
given in Table 1.Dermatan suphate. The papain digest from
3.6kg of dried bovine aorta was precipitated withcetylpyridinium chloride. The precipitate was
dissolved in 900ml of 2M-sodium chloride andcentrifuged. The clarified solution was mixed with2vol. of 95% (v/v) ethanol. After centrifugation,the supernatant (A) was retained for isolation ofchondroitin sulphate as described below. The
precipitate was subjected to ethanol fractionationin 1 litre of calcium acetate buffer by the procedureofMeyer, Davidson, Linker & Hoffman (1956). Thefraction that was precipitated by raising theethanol concentration from 0% to 25% (v/v) was
converted into the sodium salt. This preparationcontained nucleic acids, as indicated by the u.v.
spectrum, and was therefore digested with nuclease.After the digestion, the polysaccharide was finallyprecipitated with cetylpyridinium chloride, giving4.8g of dried preparation. Electrophoresis inbarium acetate showed a single spot with a mobilityslightly greater than that of the dermatan sulphatestandard. Galactosamine was the only hexosaminedetected by g.l.c. On electrophoresis in 0.1 M-hydrochloric acid, the polysaccharide behaved likemonosulphated standards of chondroitin or
dermatan sulphate. Additional analytical resultsare given in Table 1. The ratio of the extinctionsobtained from the carbazole reaction with andwithout borate was 2.83. Since the ratio fordermatan sulphate from other sources has beenfound to be about 3.6 (Bitter & Muir, 1962; P.-H.Iverius, unpublished work) and that of purechondroitin sulphate 2.0, the value of 2.83 iscompatible with a hybrid structure containing bothL-iduronic acid and D-glucuronic acid (Fransson &Havsmark, 1970).
Chondroitin 8uphate. Supernatant A, obtainedduring the fractionation of glycosaminoglyeansfrom bovine aorta (see above), was mixed withanother 2vol. of 95% (v/v) ethanol. The resultingprecipitate was also subjected to ethanol fraction-ation in 1 litre of calcium acetate buffer by themethod of Meyer et al. (1956). The fractionobtained by raising the ethanol concentration from25 to 40% (v/v) was converted into the chloride-freesodium salt and dried. This fraction, whichweighed 8.9g, behaved as pure chondroitin sulphateon barium acetate electrophoresis. Galactosamine
Vol. 124

P.-H. IVERIUS
was the only hexosamine detected by g.l.c. Theelectrophoretic mobility in O.lM-hydrochloric acidwas the same as that of reference monosulphatedchondroitin sulphate. Electrophoresis for 12h inbarium acetate, which separates chondroitin4-sulphate from chondroitin 6-sulphate, establishedthat chondroitin4-sulphatewas the onlycomponent.The chemical analyses are presented in Table 1.
Binding of glycosaminoglyeans and ethanolamine toSepharose 4B by covalent linkage
Sepharose 4B was activated by the cyanogenbromide method (Ax6n et al. 1967) as described byKato & Anfinsen (1969). Activated gel (generally75ml) was mixed with polysaccharide (150-160mg,dry wt.) and the volume was made up to 150ml with0.1 M-sodium hydrogen carbonate. The reactionmixture was stirred at 4°C. After 16h, 0.5ml of thesupernatant was withdrawn for analysis andremaining active groups on the gel were eliminatedby stirring for 4h with 7.5 ml of ethanolamine. Thegel was finally transferred to a sintered-glass filterand washed consecutively with 1 litre of water,0.5 litre of 0.5M-sodium chloride and 3 litres ofwater. Parallel to the coupling of the glycosamino-glycan to activated gel, a reference experiment wasperformed in which non-activated gel was substi-tuted for the activated gel. Also gels containingonly ethanolamine were prepared by incubatingactivated gel with ethanolamine for 16h.
Glycosaminoglycan contents of different gel batchesThe glycosaminoglyean contents of the gels were
analysed in three different ways.
One value was based on the transfer of uronicacid from the liquid phase to the gel during thecoupling procedure. The samples withdrawn fromthe supematants, as described above, were diluted20-fold before analysis. The ratio between theextinction in the experiment with activated gel andthe control experiment with non-activated gelrepresents the proportion of the added poly-saccharide that remained unattached. Since theamount of added polysaccharide was known in eachexperiment, the amount bound/vol. of wet gelcould be calculated. Dry-weight contents werecalculated from the wet gel contents by using thevalue 10.5mg of dry gel/ml of wet gel. This valuewas obtained from a dry-weight determinationof the gel heparan sulphate II-Sepharose 4B-Ifraction (HS-II-Seph. 4B-I).Two other values were derived from the sulphate
and hexosamine contents of the freeze-dried gel.The samples for sulphate assays, which variedbetween 3.32 and 10.2mg dry wt. depending on thesulphate contents, were hydrolysed in sealed glasstubes with 2ml of 25% (v/v) formic acid. Thehexosamine determinations were performed onsamples ofabout 10mg dry wt. that were hydrolysedwith 2ml of 4M-hydrochloric acid.The sulphate contents due to the presence of
glycosaminoglycans were obtained by subtractingfrom the total sulphate contents the sulphatecontents of a gel of the same batch, to whichethanolamine had been linked (5.2, 3.2 and4.8,lg/mg for lots no. 4088, 5214 and 5742 respec-tively). Since the sulphate contents of the glyco-saminoglycans were known, the polysaccharidecontents of the gels could easily be calculated.
Table 2. Analy8i8 of gels containing glycosaminoglycansThe amount of polysaccharides taken up by the gels during the coupling procedure was calculated (A) from
uronic acid analyses of supernatants of reaction mixtures (see the text) and also from (B) hexosamine and (C)sulphate analyses of the lyophilized gels. The conversion factor required for each polysaccharide constituentwas computed from Table 1. The nomenclature of the fractions is indicated in Table 1 and the text.
Glycosaminoglyean content
FractionHep-II-Seph. 4B-IHep-II-Seph. 4B-IIHep-II-Seph. 4B-IIIHep-II-Seph. 4B-IVHep-II-Seph. 4B-VHep-II-Seph. 4B-VIHS-II-Seph. 4B-ICS-I-Seph. 4B-ICS-I-Seph. 4B-IICS-I-Seph. 4B-IIIDS-III-Seph. 4B-IDS-III-Seph. 4B-II
(mg/ml of wet gel)Lot no. of gel (A)
408840884088408852145742574240884088574240885742
1.211.140.910.710.651.290.880.700.590.900.720.79
(jug/mg of dry gel)
(A) (B) (C)115 39.1 34.6109 41.4 32.886.7 27.8 21.667.6 14.9 12.861.9 30.1 28.1123 43.8 36.283.8 25.9 26.066.7 28.2 21.556.2 25.8 21.185.7 32.0 20.968.6 15.1 15.675.2 26.7 21.5
680 1971

MATRIX-BOUND GLYCOSAMINOGLYCANSThe hexosamine values were all corrected by
subtraction of 0.53,ug/mg. This value was obtainedby analysis of a gel to which ethanolamine only hadbeen linked. Cross-linked dextran, treatea withcyanogen bromide, gives a positive Elson-Morganreaction after hydrolysis (P.-H. Iverius, unpub-lished work). The calculations were then analogousto those based on the sulphate analyses. Allanalyses of the polysaccharide-substituted gels arepresented in Table 2. The polysaccharide contentsof the extensively washed, freeze-dried gels variedfrom about 10 to 40p,g/mg of dry gel (columns Band C). Generally, these values were only one-thirdof the amount of polysaccharide initially trans-ferred from the liquid to the gel phase during thecoupling procedure (columns A).
Inhibition of glycoBaminoglyean binding to gel8 byblocking of reactive groups
Chondroitin sulphate was acetylated essentiallyby the procedure of Fraenkel-Conrat (1957).Chondroitin sulphate (25mg) was dissolved in 2mlof half-saturated sodium acetate and chilled in anice bath. Four portions of acetic acid anhydride(50,1l) were added at 15min intervals. Then,15min after the last addition, the sample wasdialysed against 0.1 M-sodium hydrogen carbonateat 40C. After dialysis, the volume ofthe sample wasadjusted to 5ml.
Activated Sepharose 4B (5ml) was transferred toa 10ml graduated flask. Another 10ml flask wascharged with 5ml of non-activated gel, then 2.4mlof the acetylated chondroitin sulphate solution wasadded to each flask, and the volumes were adjustedto 10ml with O.lM-sodium hydrogen carbonate.The contents of the flasks were stirred for 16h, afterwhich the supernatants were diluted 20-fold andsubjected to uronic acid analysis. In anotherexperiment the activated gel was treated with0.5ml of ethanolamine in lml of O.lM-sodiumhydrogen carbonato before the non-acetylatedchondroitin sulphate was added.
After treatment of the acetylated polysaccharidewith activated gel, the concentration ofpolysaccha-ride in the supernatant was 98% of that obtainedwith a non-activated gel. The corresponding valuerecorded after incubation of non-acetylated poly-saccharide with activated and ethanolamine-treated gel was 99%. In contrast, only 60-70% ofthe polysaccharide remained in the liquid phaseafter the reaction of non-modified chondroitinsulphate with activated gel. It is therefore con-cluded that the covalent binding of glycosamino-glyeans to activated agarose gels could be inhibitedeither by blocking the free amino groups of theglycosaminoglyean by acetylation or by blockingthe active groups in the gel with ethanolamine.
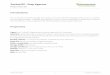
0.2(a)
0 0.1 0.2 0.3Gel added (g)
0.4
0.3
(b)
0.2
0.2
0 5 10 15 20 25Enzyme added (V.R.U.)
Fig. 1. Release of uronic acid by hyaluronidase treatmentof a gel containing chondroitin sulphate. (a) Variousamounts of gel and 15 V.R.U. ofenzyme were incubated ina final volume of 1.25ml for 45 min at 37°C. (b) Enzymewasadded in various amounts to a constant amount of gel(0.5g) and the mixture incubated for 30min at 3700 in afinal volume of 1.25ml as above.
Methodfor the assay of poly8accharidameA gel containing chondroitin sulphate (fraction
CS-I-Seph. 4B-I) was washed with O.lM-sodiumacetate-sodium chloride buffer, pH5.0. In oneexperiment various amounts (0-0.5g) of packed gelwere weighed into small test tubes. The tubes wereplaced in an ice bath and 15 V.R.U. ofhyaluronidase(Leo) was added to each. Then the total volume ofthe contents was adjusted to 1.25ml. Immediatelyafter the contents had been mixed, the tubes weretransferred to a water bath at 370C. During theincubation the tubes were agitated at 5min inter-vals. After 45min the samples were chilled in anice bath and centrifuged at 4°C, then the super-natants were analysed for uronic acid. In anotherexperiment constant amounts of gel (0.5g) wereweighed into a series of tubes. Various amounts of
Vol. 124 681

682 P.-H. IVERIUS 1971hyaluronidase (0-25 V.R.U.) were added and thevolumes adjusted to 1.25ml. The experimentalprocedure was otherwise identical with thatdescribed above, except that the incubation timeat 370C was 30 min.The first experiment (Fig. la) shows that when
the enzyme concentration in the assay is keptconstant the activity observed depends on theamount of gel added. The second experiment(Fig. lb) demonstrates how the enzyme activityobserved varies with the enzyme concentration fora constant amount of gel. The graph seems to beapproximately linear at the lower enzyme con-centrations and should therefore be useful as acalibration curve in an enzyme assay. At higherenzyme activities, however, the graph is non-linear, presumably because the matrix-boundglycosaminoglycan substrate had been used up,since approx. 80% of the polysaccharide wasliberated from the gel after incubation with25 V.R.U. of enzyme for 30min.
DISCUSSION
Cyanogen bromide activation provides a methodfor the coupling of compounds bearing free aminogroups to a gel matrix (Ax6n et al. 1967). In theglycosaminoglycans the free amino groups of serineor peptide residues are available (see e.g. Balazs,1970). The hexosamine residues also contain aminogroups, but in the native state they are eitheracetylated or, in heparin and heparan sulphate,sulphated. The sulphamino groups are acid-labileand might be a potential source of free aminogroups (Brimacome & Webber, 1964). It thusseems reasonable to assume that covalent bondsare formed between the amino groups of the glyco-saminoglycans and the cyanogen bromide-activatedagarose. As acetylation primarily affects free aminogroups (Fraenkel-Conrat, 1957), this assumption isfurther supported by the finding that acetylatedchondroitin sulphate did not react with activatedgel.The glycosaminoglyean contents of the gels
(Table 2) calculated from hexosamine (column B)and sulphate (column C) analyses are in fair agree-ment, although the values based on hexosaminedeterminations are generally somewhat higher. Itis difficult to decide which analytical technique isbetter, since a relatively large blank correction hadto be made in both methods. The values calculatedfrom the transfer of glycosaminoglyean to the gelduring the coupling procedure (columns A) are,however, about three times as great. The mostprobable explanation for this discrepancy is thatmany of the polysaccharide molecules are boundto 'brittle' parts of the gel beads and are thus
easily lost during the extensive washing that followsthe coupling procedure.
The. matrix-bound glycosaminoglyeans wereoriginally produced to provide a simple tool forstudying the interactions between glycosamino-glyeans and other substances such as, for example,human serum lipoproteins (P.-H. Iverius, unpub-lished work). The gels may also be used for prepara-tive work. Lipoprotein lipase from bovine milk hasbeen purified considerably by affinity chromato-graphy on a heparin-containing gel (Olivecrona,Egelrud, Iverius & Lindahl, 1971). A thirdapplication, the assay of polysaccharidase activity,is described above.
This work was supported by grants from the SwedishMedical Research Council (B71-13X-4-07C), the SwedishCancer Society (53-B70-04XB), the Gustav V 80-arsfond,the Ostermans fond, the Svenska Sallskapet f6r MedicinskForskning, the Stiftelsen Lars Hiertas Minne and theUniversity of Uppsala. The author is also indebted toMr E. Lindberg for invaluable help with the preparations.The skilful technical assistance of Miss K. Lilja andMiss A. Sjostrom is gratefully acknowledged.
REFERENCES
Antonopoulos, C. A. (1962). Acta chem. ascnd. 16, 1521.Ax6n, R., Porath, J. & Ernback, S. (1967). Nature, Lond.,
214, 1302.Balazs, E. A. (1970). Chemistry and Molecular Biology of
the Intercellular Matrix, vol. 2, pp. 703-960. Londonand New York: Academic Press.
Bettelheim, F. A., Laurent, T. C. & Pertoft, H. (1966).Carbohyd. Res. 2, 391.
Bitter, T. & Muir, H. M. (1962). Analyt. Biochem. 4,330.
Brimacombe, J. S. & Webber, J. M. (1964). Mucopoly-saccharides, pp. 92-146. Amsterdam: ElsevierPublishing Co.
Cornwell, D. G. & Kruger, F. A. (1961). J. Lipid Res. 2,110.
Davies, M., Nichol, L. W. & Ogston, A. G. (1963). Biochim.biophys. Acta, 75, 436.
Fraenkel-Conrat, H. (1957). In Methods in Enzymology,vol. 4, p. 251. Ed. by Colowick, S. P. & Kaplan, N. 0.New York: Academio Press Inc.
Fransson, L. A. & Havsmark, B. (1970). J. biol. Chem.245, 4770.
Gardell, S. (1953). Acta chem. scand. 7, 207.Hase, S. & Matsushima, Y. (1969). J. Biochem., Tokyo,
66, 57.Jackson, D. S. & Bentley, J. P. (1968). In Treatise on
Collagen, vol. 2A, p. 189. Ed. by Gould, B. S. Londonand New York: Academic Press.
Janado, M. & Nishida, T. (1967). Agric. biol. Chem.,Tokyo, 31, 802.
Kato, I. & Anfinsen, C. B. (1969). J. biol. Chem. 244,5849.
Lagunoff, D. & Warren, G. (1962). Archs Biochem.Biophys. 99, 396.

Vol. 124 MATRIX-BOUND GLYCOSAMINOGLYCANS 683Laurent, T. C., Wasteson, A. & Obrink, B. (1969). InAging of Connective and Skeletal Ti88ue, p. 65. Ed.by Engel, A. & Larsson, T. Stockholm: NordiskaBokhandelns Forlag.
Lindahl, U., Cifonelli, J. A., Lindahl, B. & Roden, L.(1965). J. biol. Chem. 240, 2817.
Meyer, K., Davidson, E., Linker, A. & Hoffman, P. (1956).Biochim. biophy8. Acta, 21, 506.
Muir, H. (1964). In Biological A8pects of OcclusiveVascular Disease, p. 60. Ed. by Chalmers, D. G. &Gresham, G. A. Cambridge University Press.
Obrink, B. & Wasteson, A. (1971). Biochem. J. 121,227.
Olivecrona, T., Egelrud, T., Iverius, P.-H. & Lindahl, U.(1971). Biochem. biophy8. Re8. Commun. 43, 524.
Pigman, W., Gramling, E. & Holley, H. L. (1961).Biochim. biophy8. Acta, 46, 100.
Radhakrishnamurthy, B., Dalferes, E. R. & Berenson,G. S. (1966). Analyt. Biochem. 17, 545.
Scott, J. E. (1960). In Method8 of Biochemical Analy8i8,vol. 8, p. 145. Ed. by Glick, D. New York: Inter-science Publishers Inc.
Wasteson, A. (1971). J. Chromat. 59, 87.Wessler, E. (1968). Analyt. Biochem. 26, 439.Wessler, E. (1971). Analyt. Biochem. 41, 67.Yamagata, T., Saito, H., Habuchi, 0. & Suzuki, S. (1968).
J. biol. Chem. 243, 1523.