Upload
others
View
1
Download
0
Embed Size (px)
Citation preview
HIGHLIGHTED ARTICLE| INVESTIGATION
Coupling of Human Rhodopsin to a Yeast SignalingPathway Enables Characterization of Mutations
Associated with Retinal DiseaseBenjamin M. Scott,* Steven K. Chen,* Nihar Bhattacharyya,* Abdiwahab Y. Moalim,*
Sergey V. Plotnikov,* Elise Heon,† Sergio G. Peisajovich,* and Belinda S. W. Chang*,‡,§,1
*Department of Cell and Systems Biology, ‡Department of Ecology and Evolutionary Biology, and §Centre for the Analysis ofGenome Evolution and Function, University of Toronto, Ontario M5S 3G5, Canada and †Department of Ophthalmology, Hospital
for Sick Children, Toronto, Ontario M5G 1X8, Canada
ORCID ID: 0000-0002-6525-4429 (B.S.C.)
ABSTRACT G protein-coupled receptors (GPCRs) are crucial sensors of extracellular signals in eukaryotes, with multiple GPCRmutations linked to human diseases. With the growing number of sequenced human genomes, determining the pathogenicity of amutation is challenging, but can be aided by a direct measurement of GPCR-mediated signaling. This is particularly difficult for thevisual pigment rhodopsin—a GPCR activated by light—for which hundreds of mutations have been linked to inherited degenerativeretinal diseases such as retinitis pigmentosa. In this study, we successfully engineered, for the first time, activation by human rhodopsinof the yeast mating pathway, resulting in signaling via a fluorescent reporter. We combine this novel assay for rhodopsin light-dependent activation with studies of subcellular localization, and the upregulation of the unfolded protein response in response tomisfolded rhodopsin protein. We use these assays to characterize a panel of rhodopsin mutations with known molecular phenotypes,finding that rhodopsin maintains a similar molecular phenotype in yeast, with some interesting differences. Furthermore, we compareour assays in yeast with clinical phenotypes from patients with novel disease-linked mutations. We demonstrate that our engineeredyeast strain can be useful in rhodopsin mutant classification, and in helping to determine the molecular mechanisms underlying theirpathogenicity. This approach may also be applied to better understand the clinical relevance of other human GPCR mutations,furthering the use of yeast as a tool for investigating molecular mechanisms relevant to human disease.
KEYWORDS Visual degenerative disease; retinitis pigmentosa; G protein-coupled receptor; disease model; rhodopsin
THEdiversity of biologically relevant signals thatGprotein-coupled receptors (GPCRs) detect make them critical tohow cells sense and respond to their environment. Missensemutationswithin this large family of cell surface receptors cantherefore have serious physiological effects, andmany humandiseases have been linked with missense mutations in GPCRs(Heng et al. 2013). Over 63,000 missense mutations havebeen identified in human GPCRs via large-scale human ge-nome studies (Pándy-Szekeres et al. 2018), but the majorityhave not been studied in detail, so the significance to human
health remains unclear. Missense mutations can disruptGPCR function in many ways, ranging from ligand binding,to protein stability, to changes in downstream signaling andinteractions with negative regulators (Stoy and Gurevich2015). Consequently, disease can arise through many differ-ent molecular phenotypes, and not all mutations are diseasecausing.
To better understand how human missense mutationscontribute to disease, the yeast Saccharomyces cerevisiae hasemerged as a powerful tool for characterizing human pro-tein function due to conserved molecular pathways, rapidgrowth, and ease of genetic manipulation (Laurent et al.2016). These benefits have facilitated yeast models of humandisease (Outeiro and Lindquist 2003; Perocchi et al. 2008)and studies of pathogenic human mutations (Sun et al. 2016;Yang et al. 2017). Synthetic biology approaches, where hu-man protein function and interactions are quantified by
Copyright © 2019 by the Genetics Society of Americadoi: https://doi.org/10.1534/genetics.118.301733Manuscript received June 13, 2018; accepted for publication November 29, 2018;published Early Online December 04, 2018.Supplemental material available at Figshare: https://doi.org/10.25386/genetics.7414109.1Corresponding author: University of Toronto, 25 Harbord St., Toronto, ON M5S3G5, Canada. E-mail: [email protected]
Genetics, Vol. 211, 597–615 February 2019 597
http://orcid.org/0000-0002-6525-4429https://doi.org/10.1534/genetics.118.301733https://doi.org/10.25386/genetics.7414109https://doi.org/10.25386/genetics.7414109mailto:[email protected]
yeast-based gene circuits, have enabled high-throughput ex-periments at an impressive scale (Starita et al. 2015; Sokolinaet al. 2017; Weile et al. 2017; Woodsmith et al. 2017). Withgenetic engineering, human GPCRs can be functionally linkedto the yeast mating pathway, and mating-responsive reportergenes have allowed for detailed studies of GPCR activation(Liu et al. 2016). As human GPCRs retain their natural prefer-ences for ligands and G proteins in yeast (Brown et al. 2000),this application of synthetic biology combines the high-throughput capabilities of yeast-based studies with the abilityto rapidly characterize GPCR function in a cellular context.This has facilitated screens of chemical libraries for novel GPCRligands (Campbell et al. 1999; Horswill et al. 2007), andscreens of mutated GPCRs to characterize specific protein do-mains or to engineer novel function (Erlenbach et al. 2001;Armbruster et al. 2007; Liu et al. 2015).
Despite the power of yeast-based studies of humanGPCRs,only a small proportion of GPCRs have been functionallylinked to the yeast mating pathway, and all have beenligand-activated (Liu et al. 2016). Unlike these GPCRs,rhodopsin exists as a covalent complex between its light-sensitive chromophore 11-cis retinal and the seven-helixtransmembrane opsin apoprotein (Smith 2010). Light ex-posure isomerizes the chromophore, which induces a con-formational change in rhodopsin’s transmembrane helices,activating the associated heterotrimeric G protein, transdu-cin, and triggering the visual transduction cascade that even-tually results in a signal to the brain that light has beenperceived (Smith 2010). Not surprisingly, missense muta-tions in rhodopsin are often associated with retinal diseasesin humans (Athanasiou et al. 2018). Retinitis pigmentosa(RP) is a highly heterogeneous, degenerative retinal disorderthat results in vision impairment and in some cases even-tually blindness, affecting �1 in 4000 people worldwide(Fahim et al. 2000). The heterogeneous nature of this de-generative disease has contributed to the difficulties indeveloping effective prognoses and treatment. Missense mu-tations in rhodopsin have been associated with 20–30% ofautosomal dominant RP and 1% of autosomal recessive RPcases, making rhodopsin one of the most important RP genes(Fahim et al. 2000). Over 150 rhodopsin missense mutationshave been associated with disease (Stenson et al. 2014), andan additional .200 uncharacterized mutations in rhodopsinexons are listed in the Genome Aggregation Database (Leket al. 2016).
Without a tool to rapidly assess rhodopsin function, thisincreasing availability of genetic information has yet to lead toa better understanding of pathogenicity of mutations associ-atedwith RP and other inherited visual diseases. Determiningthe impactofmissensemutationson theability of rhodopsin torespond to light currently relies on in vitro biochemical assaysthat are labor intensive, requiring mammalian cell culture toproduce one mutant protein at a time, followed by immuno-fluorescence or immunoaffinity purification (Sung et al. 1991;Reeves et al. 1996). These technical challenges are com-pounded by the diverse molecular phenotypes of rhodopsin
mutations, ranging from constitutively active, to impropersubcellular localization, to disrupted post-translational mod-ifications (Athanasiou et al. 2018). To efficiently character-ize the wide variety of patient-derived mutations, a rapidmethod that reliably recapitulates light-dependent signalingof rhodopsin is needed.
Here, we use synthetic biology approaches to engineerrhodopsin coupling to the yeast mating pathway, demonstrat-ing for the first time successful rhodopsin light-activatedsignal transduction that can be rapidly quantified using afluorescent reporter gene of mating pathway activation. Wecompared our novel yeast-based assay to more establishedmammalian cell-based methods, using a panel of previouslystudied rhodopsinmutations.We found thatmeasurements ofrhodopsin activation in yeast resemble in vitro results usingrhodopsin purified from mammalian cells, with some excep-tions. We also found that a yeast-based reporter of the un-folded protein response (UPR) produced results consistentwith previous studies in mammalian cells quantifying theeffects of rhodopsin pathogenic mutants on cellular stress.Finally, we used our combined approaches in yeast to inves-tigate recently identified rhodopsin mutations in patientswith retinal disease, and were able to propose pathogenicclassifications that are supported by mammalian cell andclinical data.
Materials and Methods
Yeast strain engineering
The parent yeast strain for all strain engineering was CB008,genotype W303 MATa, far1D, his3, trp1, leu2, ura3 (Supple-mental Material, Table S1 contains all strain genotypes). Allgene knock-outs and knock-ins were conducted using homol-ogous recombination of selectable markers. pFUS1-mCherrywas integrated at the MFA2 locus using plasmid pJW609containing the KanR marker. pFUS1 was defined as the1636 bp immediately upstream of the Fus1 start codon, themCherry sequence used is from Keppler-Ross et al. (2008),and �1 kb homology regions were used. STE2 and SST2were targeted for deletion using TRP1 and HygB selectablemarkers respectively, each with 180 bp of flanking homologyregions identical to the sequences flanking the ORF. The fiveC-terminal amino acids of Gpa1 (KIGII) was replaced with aGpa1-Gat (transducin) chimera, containing the C-terminalamino acids from mammalian Gat (DCGLF), using plasmidpBS600 designed for this study (Figure S1), containing se-lectable marker LEU2, and a sequence homologous to the800 bp 39 to the natural Gpa1 gene. The C. albicans Adhterminator was used downstream of both the pFUS1-mCherryand Gpa1-Gat gene cassettes. Strains were confirmed by PCRand flow cytometry.
Rhodopsin mutation selection and patient phenotyping
Rhodopsin mutations were selected from across pheno-typic classes, as reported in a recent comprehensive review
598 B. M. Scott et al.
http://www.yeastgenome.org/locus/S000003693/overviewhttp://www.yeastgenome.org/locus/S000005728/overviewhttp://www.yeastgenome.org/locus/S000002414/overviewhttp://www.yeastgenome.org/locus/S000000523/overviewhttp://www.yeastgenome.org/locus/S000000747/overviewhttp://www.yeastgenome.org/locus/S000005089/overviewhttp://www.yeastgenome.org/locus/S000000532/overviewhttp://www.yeastgenome.org/locus/S000001868/overviewhttp://www.yeastgenome.org/locus/S000004444/overviewhttp://www.yeastgenome.org/locus/S000002414/overviewhttp://www.yeastgenome.org/locus/S000001047/overviewhttps://www.yeastgenome.org/locus/S000001047http://www.yeastgenome.org/locus/S000000523/overviewhttp://www.yeastgenome.org/locus/S000001047/overviewhttp://www.yeastgenome.org/locus/S000001047/overview
(Athanasiou et al. 2018), and with at least one of the follow-ing assays previously published: transducin activation, lo-calization in mammalian cells, or spectroscopy indicatingchromophore binding. The patient cases were selected froman internal database and the phenotype information was col-lected retrospectively. Other than basic demographic and ge-netic information, we collected information about visualacuity (VA), color vision, Goldmann visual fields (GVF), elec-troretinography (ERG), and imaging. Imaging included fun-dus photography (VisucamNM/FA; Carl Zeiss Meditec, Dublin,CA and Optos) and optical coherence tomography (OCT, Cir-rus from Carl Zeiss Meditec). Genetic testing was done usinggene panels based sequencing by CLIA-approved laborato-ries. This study was approved by the Human Research EthicsBoard of the hospital for Sick Children and met the Tenets ofthe declaration of Helsinki.
Cloning and mutagenesis
The human rhodopsin sequence (RefSeq NP_000530.1) wasamplified using Pfu polymerase (Thermo) from plasmid pJETHuRh (Morrow et al. 2017), using primers to insert flankingAarI restriction sites. Following AarI digestion, the rhodopsinsequence was ligated to the yeast centromere plasmidspRS316 and pRS313, which each contained the TDH3 pro-moter (pTDH3; alternatively called pGPD). For mammalianexpression, rhodopsin mutations were introduced into thewild-type bovine rhodopsin sequence in the p1D4 vector forimmunoaffinity purification or the pGFP vector for SK-N-SHimmunofluorescence microscopy (Figure S2). Mutagenesiswas conducted via PCR following the QuikChange site-di-rected mutagenesis protocol (Agilent) and using PfuUltra IIFusion HS DNA Polymerase (Agilent). Mutagenesis primerswere designed with 20 or 21 nucleotides identical to humanor bovine rhodopsin flanking the mutant nucleotide(s).
Yeast plasmid transformation
Yeast strain BS017 or yJW1200 were transformed with indi-vidual plasmids by a standard lithium-acetate method andplated on selective media (SD-URA or SD-HIS, respectively).
Rhodopsin purification
Culturingand transfectionofHEK293Tcellswasperformedaspreviously described (Bhattacharyya et al. 2017). Briefly,p1D4 vector containing a rhodopsin gene was transfectedwith Lipofectamine 2000 (Invitrogen) and cells were har-vested after 48 hours. Rhodopsin was regenerated with11-cis retinal for 2 hours before solubilization in 1% dodecylmaltoside (Anatrace) and immunoaffinity purified with 1D4monoclonal antibody (Molday and MacKenzie 1983). Theultraviolet-visible absorption spectra of the rhodopsin pro-teins were recorded using a Cary 4000 double beam spectro-photometer (Agilent). Pigments were light bleached with aFiber-Lite MI-150 high intensity illuminator (Dolan-Jenner)for 60 sec at 20�. Dark-light difference spectra were calcu-lated by subtracting the light-bleached absorbance spectrafrom the dark spectra.
Yeast light activation assay
Yeast strain BS017 transformed with a human rhodopsinmutant gene in the pRS316 pTDH3 vector was incubatedovernight in SD-URA media in a 30� shaker. The same straintransformed with a plasmid not containing the rhodopsinsequence (Vector) was used as a negative control. Cells werediluted to OD600 0.05 in fresh media containing 5 mM 9-cisretinal (Sigma-Aldrich) and incubated for 2 hours, protectedfrom light, in a 30� shaker; 9-cis retinal is a common alterna-tive to the natural chromophore 11-cis retinal, and givescomparable in vitro results (Opefi et al. 2013). LightSafe50-ml centrifuge tubes (Sigma-Aldrich) were used for 5 mlcultures, and 96-well deep well blocks (VWR) wrapped inaluminum foil for 600 ml cultures. Indicated cultures werethen exposed to light using a Fiber-Lite MI-150 high intensityilluminator (Dolan-Jenner) set to full intensity for 15 min atroom temperature. After 100 ml samples were taken for anal-ysis, an additional 5 mM 9-cis retinal was added to indicatedcultures—to both light exposed and cultures kept in thedark—and placed back in the 30� shaker. Light exposure fol-lowed by retinal addition was conducted every hour for atotal of 6 hours following the first light exposure. Cells werethen treated with the protein synthesis inhibitor cyclohexi-mide, to a final concentration of 10 mg/ml. The mCherryfluorescence of at least 6000 cells was measured for eachsample with a Miltenyi Biotec MACSQuant VYB. The meanmCherry fluorescence was determined using FlowJo. Aftersubtracting the mCherry fluorescence signal of the Vector con-trol, fluorescence values were normalized to the wild-typerhodopsin control used in the same experiment, to allow com-parisons between experiments performed on different days.
Immunofluorescence microscopy
SK-N-SH neuroblastoma (ATCC HTB-11) cells were grownand cultured in full media [DMEM (Life Technologies), 10%FBS (Invitrogen), and 1% Penicillin-Streptomycin (Invi-trogen)] at 37� in 5% CO2 and seeded into 24-well plateswith coverslips (Sarstedt) while under five passages. Oncecells reached �75% confluence, they were transfected with645 ng of pGFP plasmid containing the appropriate bovinerhodopsin gene, using Lipofectamine 2000 (Invitrogen) pro-tocols. After 24 hours, half the wells were incubated withWheat germ Agglutinin (Invitrogen) in HBSS for 10 min at37� to label the plasma membrane. All cells were then rinsedwith PBS and fixed with 2% paraformaldehyde in PBS. Tolabel cells with the endoplasmic reticulum marker antibodyanti-calreticulin (1:400; Abcam), cells were washed andpermeabilized in PBS containing 1% bovine serum albu-min (Sigma) and 0.1% saponin (PBS-BS). Anti-calreticulinwas diluted in PBS-BS and incubated for 1 hours at roomtemperature. After washing with PBS-BS, secondary anti-body (Cy3-conjugated goat anti-rabbit IgG, 1:200; JacksonImmunoresearch) was diluted in PBS-BS and added to thewells for 1 hours. Nuclei were stained with Hoechst (1:1000in PBS, Hoechst type 33258 Invitrogen) for 10 min. Cells
Rhodopsin-Coupled Yeast Signaling 599
http://www.yeastgenome.org/locus/S000003424/overview
were mounted with ProLong Gold Antifade (Thermo), a cov-erslip was applied, and allowed to cure for 24 hours in thedark prior to imaging on Leica TCS SP8 confocal microscope.ImageJ was used to construct Z-stacks, maximum projectionimages, and scale bars.
Yeast microscopy
Yeast strain BS017 expressing human rhodopsin C-terminallytagged with GFP were grown to log phase in selective media,then plated on glass-bottomed dishes (Greiner Bio-One)treated with 1 mg/ml concanavalin A (Sigma-Aldrich). Thecentromere plasmid pRS316 pTDH3 was used, and the GFPsequence used is from Moser et al. (2013). A short aminoacid linker (GGERGS) was introduced between the finalrhodopsin residue and first GFP residue. Images of the cellsadhered to the dishes were acquired using a Leica TCS SP8confocal microscope with a 1003 1.4NA objective and ahybrid detector (Leica). GFP fluorescence was analyzed us-ing a custom Fiji-MATLAB pipeline (File S1 and File S2),which was similar to analyses performed to quantify fluo-rescence peaks in mammalian cells (Cheng et al. 1999). OnFiji using batch mode (Schindelin et al. 2012), yeast cellmaps were generated by first removing high-frequencynoise using ROF Denoise (theta = 25) and preparing forthresholding using Enhance Contrast (0.3 saturation withNormalization). Manual thresholding, filling holes, and fi-nally selecting of cells using the watershed plugin generatedfinal cell maps. Cell membrane maps were defined in MAT-LAB as the seven outer pixels (�0.7 mm) of each cell map.Fluorescent patches at the cell membrane were defined inMATLAB as pixels with fluorescence intensity at least 2.0 SDabove the edge mean. Cells with patchy membrane expres-sion of rhodopsin were defined in MATLAB as cells with atleast 10% of their cell membrane containing membranepatches. Bootstrapped SE were generated in MATLAB byusing the Statistics and Machine Learning Toolbox functions(Mathworks).
UPR activation assay
Yeast strain yJW1200 was generously provided by theWeissman laboratory, University of California, San Francisco.This strain contains a 43 repeat of the unfolded protein re-sponse element upstream of GFP, and the constitutive TEF2promoter upstream of RFP (Jonikas et al. 2009). yJW1200transformed with a human rhodopsin mutant gene in thepRS313 pTDH3 vector was incubated overnight in 3 mlSD-HISmedia in a 30� shaker. The same strain transformedwitha plasmid not containing the rhodopsin sequence (Vector) wasused as a negative control. Cells were diluted to OD600 0.2 in600 ml fresh media and incubated for 4 hours in a 30� shaker.Cells were then treated with the protein synthesis inhibitor cy-cloheximide, to afinal concentration of 10 mg/ml. TheGFP andRFP signal of at least 10,000 cells was measured for eachsample with a Miltenyi Biotec MACSQuant VYB. The meanGFP and RFP fluorescence was determined using FlowJo.Fluorescence values were normalized to the wild-type
rhodopsin control used in the same experiment, to allow com-parisons between experiments performed on different days.
Statistical analyses and graphs
Statistical analyses were performed using Prism (GraphPad),using one-way ANOVA for UPR comparisons to wild-typerhodopsin, and for yeast light anddarkactivation comparisonsto wild-type rhodopsin. Student’s t-test was used to comparethe results of one mutant or condition to one other mutantor condition. Graphs were generated using Prism or Excel(Microsoft).
Data availability statement
Strains and plasmids are available upon request. All supple-mental figures, tables, and files have been uploaded to fig-share. Supplemental material available at Figshare: https://doi.org/10.25386/genetics.7414109.
Results
Human rhodopsin functionally couples to the yeastmating pathway and signal transduction is dependenton light
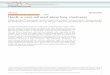
Wehave engineered, for the first time, a vertebrate rhodopsinthat can successfully couple to the yeast mating pathway. Thiswas accomplished by first knocking out the genes encodingFar1, to prevent cell cycle arrest, and the GTPase-activatingprotein Sst2, a negative regulator of the mating pathway(Brown et al. 2000). The endogenous mating pathwayGPCR Ste2 was also knocked out, to prevent unnecessaryinteractions with downstream mating pathway proteins.Next, a chimeric G alpha protein and the fluorescent proteinmCherry under the regulation of the mating-responsiveFUS1 promoter (pFUS1) were inserted into the yeast genome(Figure 1 and Table S1). As rhodopsin is known to interactonly with G alpha proteins containing the same five aminoacid C-terminal sequence [transducin in rod photoreceptorsand Gai1 in engineered systems (Maeda et al. 2014; Sun et al.2015)], only one Gpa1 chimera (Gpa1-Gat) was required forour study. To ensure functional coupling between Gpa1-Gatand human rhodopsin, we first expressed a known constitu-tively active rhodopsin mutant, E113Q M257Y (Han et al.1998). Consistent and high levels of mCherry fluorescencewere observed, regardless of the presence of retinal chromo-phore (Figure S3), indicative of productive rhodopsin expres-sion and the ability to activate the yeast mating pathway.
Wild-type human rhodopsin was then expressed using thesame strain, and, when incubated with retinal, induced theexpression of mCherry only in response to light (Figure 2);5 mM 9-cis retinal in culture media was sufficient to elicit thislight-dependent response—a concentration also used for theheterologous expression of rhodopsin using mammalian cells(Opefi et al. 2013). The 5- to 10-fold increase in mCherryfluorescence in response to GPCR activation was comparableto previous reported activation of the natural mating path-way GPCR Ste2 (Ishii et al. 2008; Kompella et al. 2017).
600 B. M. Scott et al.
http://www.yeastgenome.org/locus/S000000322/overviewhttps://doi.org/10.25386/genetics.7414109https://doi.org/10.25386/genetics.7414109http://www.yeastgenome.org/locus/S000003693/overviewhttp://www.yeastgenome.org/locus/S000004444/overviewhttp://www.yeastgenome.org/locus/S000001868/overviewhttp://www.yeastgenome.org/locus/S000000532/overviewhttp://www.yeastgenome.org/locus/S000001047/overviewhttp://www.yeastgenome.org/locus/S000001047/overviewhttp://www.yeastgenome.org/locus/S000001047/overviewhttp://www.yeastgenome.org/locus/S000001868/overview
Increasing the retinal concentration did not increase activa-tion of the mating pathway, indicating rhodopsin moleculeswere saturated with chromophore (Figure S4). However,mating pathway response was enhanced when retinal wasadded after each hourly light exposure, to account for thelack of retinal recycling enzymes in yeast.
Magnitude of light-activated signal transduction inyeast comparable to assays of rhodopsin expressed inmammalian cells
After establishing light-dependent activation of rhodopsin inyeast, we next sought to compare these new yeast-basedmethods to traditional in vitro methods utilizing protein pu-rified from mammalian cells. Previously characterized rho-dopsin mutations P23H, M39R, and G51A were specificallychosen to establish a gradient of phenotypic severity. P23His the most common RP-associated rhodopsin mutation inNorth America (Dryja et al. 1990; Mendes et al. 2005), andhas been characterized in a number of cell and animal mod-els. P23H rhodopsin consistently displays poor stability(Krebs et al. 2010; Chen et al. 2014), aggregation in the ER(Chiang et al. 2012b), and disrupted transducin activation(Opefi et al. 2013; Chen et al. 2014), leading to severe retinaldegeneration (Cideciyan et al. 1998; Athanasiou et al. 2017;LaVail et al. 2018). The less severe M39R mutation, which isalso associated with RP, has been studied using both bovineand human rhodopsin genes displaying a more severe cyto-solic aggregation phenotype in the human gene background(Davies et al. 2012; Ramon et al. 2014). As M39R rhodopsinis expressed more productively by mammalian cells thanP23H rhodopsin, and a proportion remains able to forma light-responsive complex with retinal, it was selected
as an intermediate RP-associated rhodopsin mutation (Davieset al. 2012; Ramon et al. 2014). G51A is the most commonnonsynonymous rhodopsin mutation in humans (Lek et al.2016), displays a less severe phenotype in vitro and in pa-tients (Cideciyan et al. 1998; Bosch et al. 2003), and may bean asymptomatic variant (Athanasiou et al. 2018). Thesethree rhodopsin mutants were each expressed using themCherry reporter yeast strain. Following exposure to light,the relative mCherry fluorescence was observed as follows:P23H , M39R , G51A = WT (Figure 3A).
Next, we compared rhodopsin activation in yeast to tradi-tional in vitromethods for determining rhodopsin function inresponse to light. The same three mutants were purified fol-lowing heterologous expression in mammalian cells, thenregenerated with retinal. By recording the absorption spectrabefore and after exposure to light, difference spectra showingthe response of rhodopsin to light could be measured. Thismethod has been used extensively to characterize missensemutations suspected to cause inherited retinal disease, as ameasure of the ability of rhodopsin to properly fold and re-spond to light (Sung et al. 1991; Opefi et al. 2013). Therelative response to light displayed a similar range of functionas the yeast-expressed mutants (Figure 3B). This suggestednot only that the function of yeast-expressed rhodopsin wassimilarly impacted by pathogenic mutations, but also that therelative activation of the mating pathway in yeast is compa-rable to the severity of the mutant as measured in vitro usingrhodopsin purified from mammalian cells.
Pathogenic mutations known to disrupt rhodopsinstability or G protein coupling prevent light-activatedsignal transduction in yeast
After establishing yeast as a platform for quantifying light-dependent rhodopsin activation, we investigated a largerpanel of rhodopsin mutations to understand how the widerangeofknown functionalphenotypes translates toa responsein yeast. Effortshavebeenmade to classifymutationsbasedonthese phenotypes, which range from completely inactive toconstitutively active (Figure S5 and Table S2), as discussed indetail in a recent review (Athanasiou et al. 2018). However, asthis new yeast assay examines rhodopsin signaling in an engi-neered cellular system, it was unknown how results wouldcompare to traditional in vitro methods using purified mam-malian cell-expressed rhodopsin. We first focused on patho-genic mutations known to disrupt rhodopsin folding andstability, as we hypothesized these loss-of-function mutationswould be easier to distinguish from wild type. These muta-tions were placed into three groups based on previous char-acterization: mutations intrinsically disrupting rhodopsinstability; mutations indirectly affecting stability by disruptinga post-translational modification (glycosylation) motif; ormutations disrupting G protein coupling, leading to constitu-tive endocytosis (Class 3).
When expressed in yeast and exposed to light, rhodopsinfunction was significantly disrupted by each of the mutationsknown to result inmisfolding or instability, with the exception
Figure 1 Representation of the engineered mating pathway. Rhodopsinactivation was functionally coupled to the expression of a fluorescentreporter protein, mCherry, utilizing the mating-responsive promoterpFUS1. Modifications to the mating pathway included the knockout ofnegative regulator Sst2, the gene encoding Far1, which halts cell growthin the wild-type mating pathway, and the endogenous mating pathwayGPCR Ste2. The chimeric G alpha protein (Gpa1-Gat) contains the fiveC-terminal amino acids of the G alpha subunit of human transducin (Gat).
Rhodopsin-Coupled Yeast Signaling 601
of D190N (Figure 4A). D190N had previously been shown tobe a less severe RP-linked mutation (Fishman et al. 1992;Tsui et al. 2008; Liu et al. 2013), and ERG data from patientsmatches our observation that this missense mutation doesnot completely disrupt rhodopsin function (Sancho-Pelluzet al. 2012). The G89D and L125R mutations also had a re-duced but measurable response to light when expressed inyeast, which fits with previous trends observed (Kaushal andKhorana 1994; Bosch et al. 2003). Interestingly, L125R leadto signaling in the dark as well, equivalent to the mutant’slight-dependent activation, which has not previously beenreported.
Mutations in the N-terminal cap of rhodopsin (V20G,P23H, and Q28H) have been functionally characterized indetail, and are known to poorly activate transducin in re-sponse to light (Opefi et al. 2013). Similarly,mutationsM39R,N55K, G106W, C110Y, G114D, and P171L have each beenshown to disrupt or prevent productive formation of a opsin-retinal complex (Davies et al. 2012; Ramon et al. 2014; Sunget al. 1991, 1993; Hwa et al. 1999; Andrés et al. 2003), whichmatched our observation that light-activated signal transduc-tion was significantly impaired in yeast.
Mutations T4K, N15S, and T17M prevent glycosylation atresidues N2 and N15, resulting in a severe reduction inrhodopsin stability (Kaushal et al. 1994; Opefi et al. 2013).The NXS/T glycosylation consensus sequence is recognizedacross eukaryotes (Lam et al. 2013), and a previous studyindicated that yeast-expressed bovine rhodopsin was glyco-sylated (Mollaaghababa et al. 1996). The reduced stability ofunglycosylated rhodopsin is known to prevent productivetransducin activation in vitro (Opefi et al. 2013), and compa-rable reductions in signaling were observed in our yeast
assay. A general trend of T4K (40% wild-type activity) beingless severe than N15S (4%) and T17M (15%) was observed,similar to previous studies, which indicated glycosylation ismore important on N15 than it is on N2 (Kaushal et al. 1994;Tam and Moritz 2009; Opefi et al. 2013).
Of the rhodopsin mutations we studied with impairedsignaling, R135G is unique as it does not cause misfoldingand it does not prevent the formation of a stable complexwithretinal, when using rhodopsin purified frommammalian cells(Sung et al. 1993). R135G mutates the highly conservedE/DRYmotif, where R135 is the arginine residue in this motifand is crucial for G protein coupling (Acharya and Karnik1996; Rovati et al. 2007). In addition, when heterologouslyexpressed in mammalian cells, R135 mutations cause rho-dopsin to be hyper-phosphorylated, leading to aggregationwith visual arrestin and constitutively undergoing endocyto-sis (Chuang et al. 2004). With two unique molecular mech-anisms contributing to pathogenicity, R135 mutants havebeen placed in their own “Class 3” category (Chuang et al.2004; Athanasiou et al. 2018). The observed absence of sig-naling in yeast was in line with the reported in vitro trans-ducin activation defect using mammalian cell-derived R135Grhodopsin (Min et al. 1993; Acharya and Karnik 1996). How-ever, as our light-activated signal transduction assay couldnot distinguish between disrupted G protein coupling vs. ab-errant endocytosis, this assay alone could not determine themolecular mechanism behind the lack of R135G signaling inyeast.
Pathogenic mutations that enhance or do not disrupttransducin activation respond similarly in yeast
Based on the wild-type-like activity of G51A we observed inyeast, and the reduced or inactive response of misfolded andunstablemutants, we hypothesized that rhodopsinmutationsthat maintain or increase light-dependent activation in vitromay behave similarly in yeast. Constitutively active rhodop-sin mutants are also associated with disease, causing congen-ital stationary night blindness (CSNB) and RP (Park 2014).We specifically selected a panel of rhodopsin mutationsto characterize in yeast where activity was known to varygreatly, from asymptomatic, to constitutively active, to in-creased signaling in the dark.
Across thisdiversity of function, yeast-expressed rhodopsinagain behaved comparably to rhodopsin purified from mam-malian cells, with both wild-type-like signals and increasedsignaling observed depending on the mutation (Figure 4B).We grouped mutations known to increase downstream trans-ducin activation in vitro, although this increased activity canoccur in the light, dark, or in both states (Park 2014). TheM44T mutation showed a significantly higher response thanwild-type, at over 1.6-fold wild type, which matches in vitrotransducin activation data for M44T (Andrés et al. 2003).T94I trended higher than wild type, and is also believedto cause CSNB due to constitutive activation (119% WT sig-naling in vitro) (Gross et al. 2003), but the increase we ob-served vs. wild type (115% WT signaling in yeast) was not
Figure 2 Light-dependent activation of the mating pathway. Humanrhodopsin was found to activate the mating pathway only in responseto light, requiring the presence of retinal chromophore. Adding retinalafter each hourly light exposure improved the overall response �1.6-fold.Incubating with the same concentration of retinal but keeping the culturein the dark did not result in mating pathway activation. Vector: Yeasttransformed with a plasmid not containing the rhodopsin gene. Datapoints represent results of four individual colonies, each in a 5 ml culture.Error bars represent SD.
602 B. M. Scott et al.
statistically significant. V137M has been reported to activatetransducin 1.25-fold greater than wild type (Andrés et al.2003), but we did not observe an increase in rhodopsin ac-tivity. The V137M mutation is known to have highly variableclinical phenotypes (Ayuso et al. 1996), and it has been sug-gested to be an asymptomatic variant (Rakoczy et al. 2011).
Of the rhodopsin mutations with known increased activitythat we studied, S186W is unique as it is believed to causeautosomal dominant RP due to increased signaling in the dark(Liu et al. 2013). This is a result of reduced thermal stability ofthe inactive dark state, where spontaneous thermal isomeriza-tion of the chromophore leads to signaling in the dark (Liu et al.2013), a phenomenon called “dark noise” (Luo et al. 2011).The increased signaling that we observed in the dark forS186W, equivalent to 30% of activated wild-type rhodopsin,fit with this proposed mechanism of RP pathogenesis. E150Kwas also found to have significantly elevated signaling in thedark, when expressed in yeast. This data is in linewith amousemodel of E150K, which was found to have elevated photore-ceptor signaling in the dark, also believed to be due to reducedthermal stability of the dark state (Zhang et al. 2013). TheE150K mutation is associated with autosomal recessive RP(arRP) and was previously shown to have a 1.3-fold increasedactivation of transducin after light exposure (Zhang et al.2013). An identical value was observed using our yeast-basedassay but was not statistically significant.
To determine if dark state signaling in yeast was retinal-dependent, we investigated signaling without the addition ofretinal for selected mutants (Figure S6). Elevated dark statesignaling appeared to be retinal-dependent for only M44T,L125R, E150K, and S186W. This fits with the proposedmechanism of thermal isomerization of retinal contributingto dark noise for the E150K andS186mutants (Liu et al. 2013;
Zhang et al. 2013), while providing new insight on the M44Tand L125R mutants.
Some rhodopsinmutationsmay cause disease bypreventingthe formation of rhodopsin homodimers (Ploier et al. 2016),but their pathogenicity is debated due to their relativelyhigh frequency in sequenced human genomes (.1:80,000)(Athanasiou et al. 2018). The F45L and V209M mutationswere found to activate the mating pathway at wild-type lev-els when exposed to light, matching published in vitro trans-ducin activation assays (Ploier et al. 2016). F220L was foundto have a 1.5-fold greater response, which was unexpectedlyhigher than the wild-type-like value reported for the F220Cmutation (Ploier et al. 2016). Similarly, although V104I isconsidered asymptomatic, light activation of themating path-way was �1.3-fold greater than wild-type. This mutationdoes not segregate with RP in genetic studies (Macke et al.1993), but transducin activation assays have not previouslybeen performed, so it is unclear how our results in yeast re-late to a potential human phenotype.
Mutations in the C-terminus of rhodopsin disrupt traffick-ing to the rod outer segment (ROS), but do not affect traf-ficking in other mammalian cell types and do not affecttransducin activation in vitro (Sung et al. 1994), thereforewe did not expect their function to differ from wild-typerhodopsin in yeast. Interestingly, V345M significantly af-fected light-activated signaling in yeast, despite the mutationoccurring in the C-terminus, which is not believed to be re-quired for G protein activation. A study of transducin activa-tion using V345M rhodopsin purified from mammalian cellshas not been performed to compare to our yeast results.
There were two examples where light-activated signalingin yeast differed from reported in vitro results using rhodopsinpurified from mammalian cells. The A292E mutation is
Figure 3 Characterization of rhodopsin light-dependent function. (A) Response to light from yeast-expressed rhodopsin mutants, indicating a similarmagnitude of response as the mammalian cell-expressed protein. WT: Wild-type human rhodopsin. Yeast data points represent results of nine individualcolonies, each in a 600 ml culture, minus the mCherry fluorescence of the same strain transformed with empty plasmid control (Vector), and normalizedto wild-type. * P , 0.05 vs. WT or between indicated mutants. (B) Difference spectra of mammalian cell-expressed rhodopsin mutants in response tolight. The peak at 500 nm indicates a light-dependent response.
Rhodopsin-Coupled Yeast Signaling 603
associated with CSNB, and has constitutive activation in vitroin the absence of retinal, but reduced light-dependent trans-ducin activation when retinal is supplied (Gross et al. 2003).In our yeast signaling assay, a 2.1-fold increase in light-activated signaling vs. wild type was observed, greater thanany other mutation we studied. A292E signaling in the darkwith retinal added was not significantly increased, and wasequivalent to not adding retinal. G90D, another constitutivelyactive mutant associated with CSNB (Rao et al. 1994), acti-vated the mating pathway at only 44% of wild-type response.
This result was similar to one transducin activation studyusing G90D (59% of wild type) (Zvyaga et al. 1996), whileothers have shown wild-type-like responses to light but con-stitutive activation in the absence of retinal, similar to A292E(Rao et al. 1994; Gross et al. 2003).
Light-activated signal transduction in yeast correlateswith published assays of rhodopsin function
Of the 33 mutations and controls studied, 23 had previ-ously reported measurements of light-dependent activation of
Figure 4 The light-activated signal transduction of pathogenic rhodopsin mutants in yeast. (A) Missense mutations that intrinsically disrupt rhodopsinstability, or that indirectly affect stability by disrupting a post-translational modification (PTM) motif. Class 3 denotes a unique category of mutations atsite R135, which disrupt G protein coupling and lead to constitutive endocytosis in mammalian cells. (B) Pathogenic and asymptomatic variants thatincrease or do not disrupt rhodopsin activation by light. WT: Wild-type human rhodopsin; Vector: yeast transformed with a plasmid not containing therhodopsin gene; arRP: autosomal recessive RP. Data points represent results of nine individual colonies, each in a 600 ml culture, minus the mCherryfluorescence of Vector, and normalized to wild-type. Error bars represent the 95% CI, * P , 0.05 vs. WT.
604 B. M. Scott et al.
transducin in vitro, or measurements of photoreceptor activ-ity by ERG (Table S2). When plotted together, our yeast-based measurements closely matched this available data onrhodopsin signaling, approaching a 1:1 ratio (Figure S7).This held true for a diverse array of phenotypes, ranging frompathogenic due to inactivation, or pathogenic due to consti-tutive activation, to asymptomatic. A292Ewas found to be anoutlier from this trend. The rate of light-dependent transdu-cin activation has been reported at �80% of wild type, al-though this mutant has constitutive activation in the absenceof retinal in vitro (Gross et al. 2003). As signaling in yeastoccurs in a cellular context, vs. traditional in vitro assays thatuse purified protein, the increase in light-activated signalingwe observed for A292E may have been a combination ofsignaling both with and without retinal bound, or may rep-resent a unique signaling state in yeast. Overall, however, thegeneral trend strongly supports the use of yeast to quantifyrhodopsin-mediated G protein signaling in response to light,as the yeast-based assay was comparable to more laboriousassays for characterizing patient derived mutations.
Subcellular localization of rhodopsin is comparablebetween yeast and mammalian cells
As the majority of disease-linked rhodopsin mutations causethe receptor to misfold (Athanasiou et al. 2018), comparingthe subcellular localization of rhodopsin using mammaliancells is a common technique to study protein trafficking andER retention, and is predictive of pathogenicity (Sung et al.1991; Behnen et al. 2018). To establish phenotypes in mam-malian cells to compare to, we again used the rhodopsinmutations P23H, M39R, and G51A to create a gradient ofphenotypic severity. Rhodopsin mutants were expressed inSK-N-SH neuroblastoma cells with a C-terminal GFP tag thathas previously been shown to not affect rhodopsin stabilityin vitro or in vivo (Moritz et al. 2001)
The P23Hmutation has been characterized in a number ofcell models, consistently showing poor plasma membraneexpression regardless of cell type (Sung et al. 1991; Chianget al. 2012b, 2015). When expressed in SK-N-SH neuro-blastoma cells, immunocytochemistry revealed that P23Hrhodopsin did not localize to the plasma membrane, formingaggregates in the cytosol and colocalized with an ER-specificmarker (Figure 5A and Figure S8). M39R rhodopsin dis-played a nearly wild-type phenotype, colocalizing with aplasma membrane marker but with some evidence of mutantrhodopsin retained within the cell (Figure S8), similar to aprevious study (Davies et al. 2012). G51A displayed robustwild-type-like localization on the plasma membrane, consis-tent with a recent report (Behnen et al. 2018). This compar-ative range of subcellular localization matched what we hadpreviously observed in assays of rhodopsin function for thesethree mutations.
We next sought to determine if the same rhodopsin mu-tants expressed in yeast trafficked to the plasmamembrane ina similar manner. The same yeast strain used to functionallycouple human rhodopsin to the mating pathway was used to
express select rhodopsin mutants with GFP fused to theC-terminus. Similar techniques have been used to inves-tigate productive expression of other human GPCRs inyeast, by observing expression on the plasma membraneor the presence of aggregates (O’Malley et al. 2009). Mir-roring our mammalian cell-based observations, P23H andM39R were poorly distributed across the yeast plasmamembrane, with highly localized “patchy” expression,while G51A localized consistently to the membrane (Fig-ure 5A). The peri-nuclear ring observed is similar to thelocalization of other human GPCRs expressed in yeast,indicating the ER membrane (O’Malley et al. 2009; Hashiet al. 2018). Although P23Hwas observed to form aggregatesin the ER of our mammalian cells, this was not observed inyeast; however, M39R did appear to form aggregates in thecytosol of yeast.
We expanded this microscopy-based analysis to additionalrhodopsin mutants expressed in yeast (Figure S9). Imageanalysis software was used to quantify rhodopsin distributionon the plasma membrane of each yeast cell, identifyinglocalized regions or “patches” where rhodopsin appeared toaggregate (Figure 5B). Wild-type rhodopsin was observed tohave a low number of cells displaying a “patchy” phenotype,suggesting even distribution across the plasma membrane.Somemutants associatedwithmisfolding or reduced stability(i.e., N15S, M39R, L125R) exhibited incomplete distributionon the plasma membrane, characterized by a “patchy” phe-notype. Mutations in the C-terminus of rhodopsin (Q344ter,V345M, P347L) disrupt the VXPX motif, which is crucial fortrafficking rhodopsin in photoreceptors (Wang and Deretic2014). However, mutations within this motif do not affectrhodopsin localization in mammalian cells that are not pho-toreceptors (Sung et al. 1991, 1993), and were not found toaffect rhodopsin localization in yeast. In general, these find-ings indicated that rhodopsin maintains its subcellular local-ization when expressed in yeast, which is likely dependent onrhodopsin folding and stability, just as it is with heterologousexpression in mammalian cells.
The yeast unfolded protein response is upregulated bymisfolded rhodopsin mutants
Afterdiscovering that the subcellular localizationof rhodopsinand changes in responses to light were preserved in yeast, weinvestigated additional molecular pathways known to beaffected by pathogenic rhodopsin mutations. Rhodopsin mu-tations P23H and T17M have been shown to activate the UPRin mammalian systems, indicative of severe misfolding (Linet al. 2007; Kunte et al. 2012). P23H has been shown topreferentially activate the IRE1 UPR pathway in mammaliancells (Chiang et al. 2015)—a pathway also present in yeast.Similar to mammalian IRE1, yeast IRE1 serves as a sensor ofmisfolded protein in the ER, which activates the transcriptionfactor HAC1 (Kimata and Kohno 2011). Yeast strains andplasmids have been devised utilizing a HAC1-responsive pro-moter to express reporter genes, which have been used topredict productive expression of other human GPCRs in yeast,
Rhodopsin-Coupled Yeast Signaling 605
http://www.yeastgenome.org/locus/S000001121/overviewhttp://www.yeastgenome.org/locus/S000001863/overviewhttp://www.yeastgenome.org/locus/S000001863/overview
where greater UPR activation was associated with GPCRmisfolding and aggregation (O’Malley et al. 2009). Dueto the conserved pathway, and knowing results established
with other GPCRs, we hypothesized that the severity ofmisfolded rhodopsin could be quantified using a yeast-based sensor of UPR upregulation. The strain designedby Jonikas et al. (2009) has an additional gene cassetteconstitutively expressing RFP, which can be used to correctfor changes in global protein expression (Jonikas et al.2009). We used this strain to study the 33 selected rho-dopsin mutations, plus controls, to quantify their effect onUPR upregulation.
Expressed in the UPR-reporter strain, P23H and T17Mrecapitulated the expected elevated UPR activation in yeast,in addition to several other mutations known to disruptrhodopsin stability (Figure 6). T4K and N15S, which likeT17M disrupt glycosylation, also upregulated the UPR, sug-gesting a crucial stabilizing nature of these posttranslationalmodifications. C110Y prevents the formation of a criticaldisulfide bond in rhodopsin, severely impacting stabilityand function (Hwa et al. 1999), and the observed increasedUPR in yeast matches in silico predictions that this mutant ishighly unstable (Rakoczy et al. 2011). Elsewhere in trans-membrane helix three, the G114D and R135G mutationssimilarly increased the UPR. In mammalian cells, R135G ishyper-phosporylated and aggregates in endosomes (Chuanget al. 2004), but accumulation in the ER or activation of themammalian UPR has not been reported. That R135G acti-vated the yeast UPR suggests that this mutant may be mis-folded in yeast.
The constitutively active mutant A292E showed an upre-gulation in the UPR that has not previously been reported,which may be due to the replacement of a small unchargedresidue with a large negatively charged residue proximal tothe retinal binding pocket. A reduced UPR for mutationsM44T, E150K, and V345M compared to wild type was ob-served, but the physiological relevance is unclear. Interest-ingly, culturing in selective media alone was sufficient toupregulate the UPR, revealed by the difference between thevector control and the untransformed strain grown in richmedium. A log2 GFP/RFP ratio of�1.0 was previously shownto indicate moderate UPR upregulation (Jonikas et al. 2009),which was observed for the Vector control prior to normaliz-ing the data. Expressing wild-type rhodopsin gave a similarvalue, which suggested baseline UPR upregulation was dueto growth in selective media and not to the overexpression ofrhodopsin.
Yeast assays of rhodopsin mutations V81D, A164E, andA164V found in retinal disease patients
Having established a suite of new yeast-based techniques toinvestigate rhodopsin functional and molecular phenotypes,we applied these approaches to study three rhodopsin muta-tions found in degenerative retinal disease patients diagnosedwith RP. One of the rhodopsin mutations, V81D, is a newmutation that has not been previously reported (Case 1),and two of the other mutations are previously reported buthave had little (A164V, Case 2), or no (A164E, Case 3) ex-perimental characterization with respect to those rhodopsin
Figure 5 Representative subcellular localization of rhodopsin mutants (A)Comparative subcellular localization of rhodopsin mutants in SK-N-SHand yeast cells. PM merge: Fluorescence of a plasma membrane marker,merged with GFP-tagged rhodopsin. Images represent maximal projec-tions, with scale bars representing 30 mm and 5 mm respectively. (B)Quantified localization of GFP-tagged rhodopsin mutants on the yeastplasma membrane. Yeast cells with patchy membrane expression of rho-dopsin had at least 10% of their cell membrane containing separatedpatches of rhodopsin. Error bars represent the bootstrapped SE.
606 B. M. Scott et al.
mutations. Missense mutations A164E and A164V have beenpreviously linked to autosomal dominant RP (Fuchs et al.1994; Hwa et al. 1997), but the molecular mechanism un-derlying the disruption of rhodopsin function at this site re-mains to be elucidated. Studies of A164V suggest that helicalpacking may be an issue (Hwa et al. 1997; Stojanovic et al.2003), but the effects of introducing a charged residue at thissite have not yet been investigated. Mutations at the sameresidue in rhodopsin can have highly heterogenous pheno-types (Bosch et al. 2003), so we sought to compare Al64E toA164V in greater detail. We identified a new V81Dmutationin a patient (Case 1)with early-onset autosomal dominant RP(Table S3). This mutation completely removes the V81 codonfrom the rhodopsin DNA sequence, resulting in a deletedamino acid in the central part of the second transmembranedomain (Figure 7A). Such an amino acid deletion could dis-rupt alpha helix formation and stability in the membrane,and is likely to lead to a severe molecular phenotype forV81D rhodopsin.
We characterized V81D, A164E, and A164V using theyeast-based methods for investigating rhodopsin molecularphenotype and function, with experiments conducted in thesamemanner as previously described (Figure 7, B–E). Plasmamembrane localization of all three mutants were poor, with28–40% of yeast cells displaying patchy expression of rho-dopsin. Light-activated signal transduction in yeast was com-pletely abolished for each, similar to the other mutants westudied known to be unstable or misfold. UPR activationwas elevated for all three mutants, with A164E higher thanany other rhodopsin mutant we studied. Together, our re-sults suggest that all three of these mutants have a severephenotype in yeast, based on decreased function, subcellularlocalization, and UPR activation, where we may expect adifference in severity between the A164 mutants based onUPR activation.
Yeast assays comparable to mammalian cell data forrhodopsin mutations V81D, A164E, and A164V
We compared our yeast-based assays of V81D, A164E, andA164V to expression in mammalian cells. A164V colocalizedwith the plasma membrane marker, suggesting a less severephenotype in mammalian cells, which contrasted with theyeast data (Figure 8A). However, A164E and V81D werecompletely retained inside mammalian cells, colocalizingwith the ER marker, indicating they were severely misfolded.Following immunoaffinity purification from mammalian cells,the three RP-associated mutations also showed a range intheir response to light (Figure 8B). Heterologous expressionof V81D produced no functional protein, demonstrating avery severe phenotype. A164E, while not as severe a pheno-type as V81D, expressed poorly and produced limited func-tional protein. A164V expression did produce a high amountof functional protein, consistent with another in vitro study ofthis mutation (Stojanovic et al. 2003). The sum of this cellu-lar and functional data suggested the same functional trendwe first predicted using yeast-based methods, although therewas more evidence of functional A164V protein in mamma-lian cells.
Patient clinical data supports functional trend predictedby yeast and mammalian cells
Next, we looked at patient clinical data. The comparativeseverity in vitro was found to follow the same trend as avail-able patient phenotype information (Table S3). The mostsevere phenotype was exhibited by the patient with theV81D mutation (Case 1). This patient first had symptoms�10 years of age, with difficulty adapting to a dim litenvironment (nyctalopia). This slowly progressed, and at39 years she has moderate visual acuity loss (20/50), mildlyabnormal color vision, constriction of the visual field to thecentral 5�. At age 26 years, electroretinography already
Figure 6 UPR activation in yeast relative to wild-type rhodopsin. GFP expression is a reporter of UPR upregulation, while RFP is expressed constitutivelyto help correct for changes in global protein expression. WT: Wild-type human rhodopsin; Vector: yeast transformed with a plasmid not containing therhodopsin gene; Strain: the yJW1200 strain not transformed with plasmid and grown in rich media; arRP: autosomal recessive retinitis pigmentosa. Datapoints represent results of nine individual colonies, each in a 600 ml culture, normalized to wild-type. Boxes extend from the 25th to 75th percentile, theline across the box represents the median value. Bars represent the min and max recorded values. * P , 0.05 vs. WT.
Rhodopsin-Coupled Yeast Signaling 607
documented severe reduction of rod and cone function. Thephenotype of the patient with the A164Vmutation (Case 2) ismilder than that of the patient with the A164E mutation.Although Case 2 had symptoms of nyctalopia since child-hood, the progression of his disease was extremely slow. Atage 64 years his electroretinogram was recordable and onlymildly abnormal. His central visual acuity at 67 years was20/40 and, despite a paracentral scotoma (area of decreasedvision), he maintained a peripheral field. In contrast, the pa-tient with the A164E mutation (Case 3) has a good centralvisual acuity and normal color vision at age 53 years. How-ever, her paracentral scotoma were more severe and pro-gressed to form an annular scotoma at the age of 53 years.Unlike Case 1 (V81D), she also preserved some good periph-eral field of vision at the age of 45 years, and her ERG wasonly moderately abnormal.
The V81D patient is the youngest of the threewith a highlyreduced retinal function, while the A164V patient is the el-dest of the three with the best retinal function (Figure 9).A164E again appeared intermediate to both. These results
show that the overall trend of clinical severity was accuratelypredicted by combining both sets of yeast-derived and mam-malian cell-derived data. The yeast methods provided addi-tional information on UPRupregulation, which also supportedthe difference in severity between A164E and A164V mu-tations, while being less labor intensive than mammalianmicroscopy and expression methods.
Discussion
Yeast provide new methods to investigate rhodopsinstructure and function
In this study, we have engineered the yeast S. cerevisiae tocharacterize both known and novel pathogenic mutations ofthe visual pigment rhodopsin. In comparing our new assaysto traditional mammalian cell-based approaches, we demon-strate that the molecular phenotypes of this light-activatedhuman GPCR are similar in yeast, and that these phenotypesreflect patient clinical data. There are a number of advantagesand differences when compared to traditional techniques,as these yeast-based rhodopsin assays are performed in acellular context, which provides a new perspective on signaltransduction pathways, subcellular localization, and UPRupregulation.
A direct measurement of downstream pathway activationin response to light was achieved by functionally couplingrhodopsin to the yeast mating pathway. This required pro-ductive in vivo activation of a G protein, mimicking the initialstep that occurs in human photoreceptors, even if down-stream signaling differs. By using yeast, many rhodopsin mu-tations could be studied in the same experiment, withmultiple replicates, without the laborious purification stepsthat are traditionally required for in vitro transducin activa-tion assays (Reeves et al. 1996). Not only did yeast-expressedrhodopsin maintain its ability to respond to light, but themagnitude of signal pathway activation was comparable tomany previous studies of mutations expressed in mammaliansystems. We were also able to observe rhodopsin mutationsthat resulted in the activation of dark state rhodopsin, whichhas previously required sensitive spectroscopic assays usingimmunoaffinity purified rhodopsin (Liu et al. 2013), or geneknock-in animal models (Zhang et al. 2013). This includedproviding new data on L125R signaling in the dark, a mutantpoorly expressed in mammalian cells, which has made pre-vious characterization of function challenging (Stojanovicet al. 2003). Mutations at site L125 have been shown to re-duce the thermal stability of rhodopsin (Andrés et al. 2001),which could lead to dark noise through thermal isomeriza-tion of the bound chromophore (Luo et al. 2011). This mech-anism was supported by our finding that L125R dark noisewas retinal-dependent in yeast, which could be a result ofthermal isomerization. Thus, yeast may serve as a platformfor studying difficult-to-express rhodopsin proteins, allowingthe rapid quantification of downstream G protein activationunder various conditions.
Figure 7 Characterization of novel rhodopsin mutants using yeast. (A)Crystal structure of rhodopsin, highlighting residues V81 and A164 in red.The other rhodopsin mutants characterized in this study are highlighted incyan, and the approximate location of the membrane is indicated by thedotted line (1U19.pdb). Quantified phenotype and functional assays ofV81D, A164E, and A164V in yeast (B) Representative subcellular locali-zation, Bar, 5 mm, (C) quantified consistency of rhodopsin localization onthe yeast plasma membrane, (D) light-activated signal transduction, and(E) UPR activation. WT: Wild-type human rhodopsin. The number of bi-ological replicates and error bars are identical to previous figures. * P ,0.05 vs. WT in all panels unless otherwise indicated.
608 B. M. Scott et al.
Mutations known to disrupt rhodopsin folding or stabilitytended to have incomplete localization on the yeast plasmamembrane, similar to mammalian-cell-expressed rhodopsin.Although there was significant heterogeneity between theinvestigated mutants, this observation suggests that thebiochemical properties of rhodopsin were conserved inyeast, despite known differences in glycosylation patterns(Mollaaghababa et al. 1996). We quantified membrane local-ization using a novel automated image analysis procedure,which may be useful for studies of other GPCRs and associ-ated pathogenic mutations when expressed in yeast. How-ever, not all human GPCRs are expressed productively inyeast (O’Malley et al. 2009), so these methods should firstbe validated with the wild-type receptor.
The majority of disease-linked rhodopsin mutations thathave been identified cause the protein to misfold, which can ac-tivate theUPR in the ER, and eventually lead to photoreceptor
cell death (Athanasiou et al. 2018). Modulating the UPRhas been investigated as a potential treatment for RP (Tamet al. 2010; Chiang et al. 2012a; Parfitt et al. 2014), butthe effect on UPR upregulation had only been determinedfor three rhodopsinmutations (Lin et al. 2007; Kunte et al. 2012;Marsili et al. 2015). Determining UPR upregulation by mu-tant rhodopsin has previously required microscopy, immuno-blot, and qPCR methods (Kunte et al. 2012; Marsili et al.2015). We took advantage of an engineered yeast strain thatpossesses a reporter linked to the IRE1UPR pathway, which isthe only one of three UPR pathways that is conserved be-tween mammals and yeast (Kimata and Kohno 2011). Thisyeast-based reporter of UPR activity enables the use of flowcytometry, a more simple and high-throughput method, andoffers the advantage of measuring UPR upregulation directlyin live cells. We found that upregulation of this pathway wasassociated with certain rhodopsin mutants known to misfoldor with reduced stability, which may be predictive of themolecular mechanism contributing to retinal degeneration.
However, yeast do not contain the PERK and ATF6 UPRpathways found in mammalian cells (Kimata and Kohno2011). Thus, although the yeast-based methods provide in-sight into UPR upregulation, these studies would need to becombinedwithmammalian cells to better understand how allUPR pathways may be affected by rhodopsin and other GPCRmutations. This may also explain why nine of the 15 mutantsthat are believed to misfold did not cause an increase UPRactivation in yeast, suggesting significant heterogeneity be-tween rhodopsin mutations. Determining the contribution amissense mutation makes to UPR upregulation is highly rel-evant to pharmacogenomics, as an inability of rhodopsin torespond to light may not necessarily indicate that the patho-genic mutation can be rescued by UPR modulation.
Overall, many known rhodopsin phenotypes were recapit-ulated in yeast, a requirement for any assay of human genefunction seeking to determine the clinical relevance of patientderived missense mutations (Amendola et al. 2016). Thereare also important differences to consider when comparingour yeast-based assays to mammalian cell-based assays. Im-portantly, when a lack of signaling is observed in yeast, it isdifficult to separate rhodopsin mutants that misfold frommutants that fold properly but do not productively activatethe downstream pathway. Although our quantified micros-copy data supported the notion that rhodopsin mutants withinconsistent plasma membrane expression in yeast are alsomutants known to be unstable or misfolded, unique aspectsof yeast cellular machinery or post-translational processingmay influence these mutants. Results from the R135G muta-tion highlight these differences, where this mutant activatedthe UPR in yeast but does not aggregate in the ER of mam-malian cells (Chuang et al. 2004).
The signaling phenotypes of the G90D andA292Emutantsin yeast also differed from the reported constitutive activityin vitro when using purified protein (Gross et al. 2003).That A292E signaled higher and G90D lower than expectedsuggests that constitutive activity in yeast is dependent on
Figure 8 Characterization of novel rhodopsin mutants using mammaliancell expression. (A) Comparative subcellular localization of rhodopsin mu-tants in SK-N-SH cells. PM: Fluorescence of a plasma membrane marker;ER: fluorescence of a ER-specific marker. Bar, 30 mm. (B) Difference spec-tra of mammalian cell-expressed rhodopsin mutants in response to light.The peak at 500 nm indicates a light-dependent response.
Rhodopsin-Coupled Yeast Signaling 609
http://www.yeastgenome.org/locus/S000001121/overview
Figure 9 Clinical assessment of patients with rhodopsin mutations V81D, A164E, and A164V. (A) Goldmann visual fields of the right eye at two timepoints. Normal fields would reach the gray dotted line. The solid blue line outlines the actual field. The hatched areas are scotoma, i.e., areas of loss insensitivity. Darker areas refer to denser scotoma. (B) Structural retinal phenotype of the right eye from cases carrying the A164E and V81D mutations.Optical coherence tomography (OCT) above showing the different retinal layers. Brackets show area of preserved outer retina; A164E . V81D. Unlikefor A164E, the OCT of V81D shows disturbed lamination of the retina with degenerative cysts, reflecting more advanced disease. The retinalphotograph below centered on the posterior pole. Photograph on the right is taken with a wider field camera. ON: Optic nerve. The dotted whiteline indicated the foveal area at the center of the macula. Double white arrow indicates vessel attenuation, while single arrow shows typical pigmentarydeposits (few in these cases). The width of the central visual field corresponds to the area of preserved outer retina on the OCT.
610 B. M. Scott et al.
cellular conditions that would not be revealed in an in vitroassay using purified protein, such as a renewing supply ofboth rhodopsin and G protein. It is interesting to note, how-ever, that A292E has the highest constitutive activity reportedof a CSNB-associated mutation in vitro (Gross et al. 2003),which fits the trend observed in yeast. That light-dependentsignaling in yeast was affected by the V104I, F220L, andV345M mutations was unexpected, which provides interest-ing new data for these previously uncharacterized mutantsthat should be followed up in mammalian cell-based assays.Thus, these yeast-based methods provide complimentary butindependent data to traditional mammalian-cell and in vitrobiochemical techniques, offering a unique perspective on rho-dopsin structure and function in a cellular context.
Characterizing novel rhodopsin mutations with yeast
Determining mutant function rapidly and accurately has be-come increasingly important with the rise of whole genomesequencing, and the ever-expanding rise in gene mutationswith an unknown impact on human health. Rhodopsin mu-tations linked to inherited retinal disease have been used asexamples of howmany genemutations discovered in patientsare rarely characterized, and that the molecular basis forpathology is poorly understood (Davies 2014; Chiang andGorin 2016). Animal models and traditional in vitro assaysprovide detailed information, but they have not kept pacewith the hundreds (.350) of rhodopsin mutations identifiedto date. This is true of many other genetic diseases, but newmethods of functional characterization are helping to addressthis (Starita et al. 2017), including using yeast-based assays(Sun et al. 2016; Yang et al. 2017).
We investigated the use of yeast to characterize novel andunderstudied pathogenic mutations, and compared to clinicaldata for patients with varying severity of RP. Our yeast-basedapproaches predicted severe phenotypes for the V81D andA164 mutations, which included determinations of their light-activation, subcellular localization, and UPR upregulation. Bycombining yeast and mammalian cell-based assays, the rela-tive severity of these mutations was revealed, as comparedwith clinical phenotypes measuring decline in visual functionin patients. The V81D rhodopsin mutation showed the mostsevere phenotype in our combined assays and the most ex-tensive visual deterioration clinically, in contrast to A164V,which had the mildest phenotype both clinically and experi-mentally, and A164E, which was found to be intermediate.
The difference in severity for the two missense mutationsfound at site 164 highlights the heterogenic nature of RP, andthe importance of characterizing individual disease pheno-types. These results indicate that yeast-based approachescould be useful not only for investigating the molecular basisof retinal disease, but also for better prediction of mutationpathogenicity, to help improve the accuracy of prognoses forpatients associated with specific mutations in rhodopsin. In-tegrating the results of both UPR and light-activation assays(Figure S10) may also help determine the molecular mech-anism of disease, to differentiate between mutations that
cause severe misfolding, vs. disruptions in retinal binding orstability that prevent activation but do not cause cell stress.
Recent successes in gene therapy for inherited retinaldisease means that mutation classification is of utmost im-portance, to determine if such therapy is required (U.S. Foodand Drug Administration 2017). This issue is particularlyimportant to address for degenerative diseases, such asinherited retinal disease, where early intervention is crucial.A method to determine the functional consequences of mu-tations throughout rhodopsin, rapidly and accurately, wouldtherefore be highly beneficial.
The methods presented here could also extend to func-tionally characterizing mutations of many other GPCRs. In-deed, although over 30 human GPCRs have been functionallylinked to the yeast mating pathway, no previous yeast-basedstudy has focused on direct functional characterization ofhuman GPCR mutations. As discussed in a recent review ofGPCR pharmacogenomics, characterizing GPCR mutationscould lead to a better understanding of disease and drugresponses in patients (Hauser et al. 2018). Missense muta-tions that modulate interactions with downstream signalingand regulatory proteins are known to play a role in this, soassays that accurately reflect GPCR function and interactionsin a cellular context, such as the yeast assays presented here,will be key to understanding the impact of GPCR mutationson human health.
Acknowledgments
This study was supported by NSERC Discovery Grants toB.S.W.C. (RGPIN-2015-06279), and S.P (RGPIN-2015-05114),and a Canada Foundation for Innovation Grant to S.P.(#34473).
Literature Cited
Acharya, S., and S. S. Karnik, 1996 Modulation of GDP releasefrom transducin by the conserved Glu134-Arg135 sequence inrhodopsin. J. Biol. Chem. 271: 25406–25411. https://doi.org/10.1074/jbc.271.41.25406
Amendola, L. M., G. P. Jarvik, M. C. Leo, H. M. McLaughlin, Y. Akkariet al., 2016 Performance of ACMG-AMP variant-interpretationguidelines among nine laboratories in the clinical sequencingexploratory research consortium. Am. J. Hum. Genet. 99: 247.https://doi.org/10.1016/j.ajhg.2016.06.001
Andrés, A., A. Kosoy, P. Garriga, and J. Manyosa, 2001 Mutationsat position 125 in transmembrane helix III of rhodopsin affectthe structure and signalling of the receptor. Eur. J. Biochem.268: 5696–5704. https://doi.org/10.1046/j.0014-2956.2001.02509.x
Andrés, A., P. Garriga, and J. Manyosa, 2003 Altered functionalityin rhodopsin point mutants associated with retinitis pigmentosa.Biochem. Biophys. Res. Commun. 303: 294–301. https://doi.org/10.1016/S0006-291X(03)00328-0
Armbruster, B. N., X. Li, M. H. Pausch, S. Herlitze, and B. L. Roth,2007 Evolving the lock to fit the key to create a family of Gprotein-coupled receptors potently activated by an inert ligand.Proc. Natl. Acad. Sci. USA 104: 5163–5168. https://doi.org/10.1073/pnas.0700293104
Rhodopsin-Coupled Yeast Signaling 611
https://doi.org/10.1074/jbc.271.41.25406https://doi.org/10.1074/jbc.271.41.25406https://doi.org/10.1016/j.ajhg.2016.06.001https://doi.org/10.1046/j.0014-2956.2001.02509.xhttps://doi.org/10.1046/j.0014-2956.2001.02509.xhttps://doi.org/10.1016/S0006-291X(03)00328-0https://doi.org/10.1016/S0006-291X(03)00328-0https://doi.org/10.1073/pnas.0700293104https://doi.org/10.1073/pnas.0700293104
Athanasiou, D., M. Aguila, C. A. Opefi, K. South, J. Bellinghamet al., 2017 Rescue of mutant rhodopsin traffic by metformin-induced AMPK activation accelerates photoreceptor de-generation. Hum. Mol. Genet. 26: 305–319. https://doi.org/10.1093/hmg/ddw387
Athanasiou, D., M. Aguila, J. Bellingham, W. Li, C. McCulley et al.,2018 The molecular and cellular basis of rhodopsin retinitispigmentosa reveals potential strategies for therapy. Prog. Retin.Eye Res. 62: 1–23. https://doi.org/10.1016/j.preteyeres.2017.10.002
Ayuso, C., M. J. Trujillo, M. Robledo, C. Ramos, J. Benitez et al.,1996 Novel rhodopsin mutation in an autosomal domi-nant retinitis pigmentosa family: phenotypic variation inboth heterozygote and homozygote Val137Met mutant pa-tients. Hum. Genet. 98: 51–54. https://doi.org/10.1007/s004390050158
Behnen, P., A. Felline, A. Comitato, M. T. Di Salvo, F. Raimondiet al., 2018 A small chaperone improves folding and routingof rhodopsin mutants linked to inherited blindness. iScience 4:1–19. https://doi.org/10.1016/j.isci.2018.05.001
Bhattacharyya, N., B. Darren, R. K. Schott, V. Tropepe, and B. S. W.Chang, 2017 Cone-like rhodopsin expressed in the all-coneretina of the colubrid pine snake as a potential adaptation todiurnality. J. Exp. Biol. 220: 2418–2425. https://doi.org/10.1242/jeb.156430
Bosch, L., E. Ramon, L. J. Del Valle, and P. Garriga, 2003 Structuraland functional role of helices I and II in rhodopsin. A novel in-terplay evidenced by mutations at Gly-51 and Gly-89 in the trans-membrane domain. J. Biol. Chem. 278: 20203–20209. https://doi.org/10.1074/jbc.M301319200
Brown, A. J., S. L. Dyos, M. S. Whiteway, J. H. White, M. A. Watsonet al., 2000 Functional coupling of mammalian receptors tothe yeast mating pathway using novel yeast/mammalian G pro-tein alpha-subunit chimeras. Yeast 16: 11–22. https://doi.org/10.1002/(SICI)1097-0061(20000115)16:1,11::AID-YEA502.3.0.CO;2-K
Campbell, R. M., C. Cartwright, W. Chen, Y. Chen, E. Duzic et al.,1999 Selective A1-adenosine receptor antagonists identifiedusing yeast Saccharomyces cerevisiae functional assays. Bioorg.Med. Chem. Lett. 9: 2413–2418. https://doi.org/10.1016/S0960-894X(99)00398-4
Chen, Y., B. Jastrzebska, P. Cao, J. Zhang, B. Wang et al.,2014 Inherent instability of the retinitis pigmentosa P23Hmutant opsin. J. Biol. Chem. 289: 9288–9303. https://doi.org/10.1074/jbc.M114.551713
Cheng, H., L. S. Song, N. Shirokova, A. Gonzalez, E. G. Lakattaet al., 1999 Amplitude distribution of calcium sparks in confo-cal images: theory and studies with an automatic detectionmethod. Biophys. J. 76: 606–617. https://doi.org/10.1016/S0006-3495(99)77229-2
Chiang, J., and M. B. Gorin, 2016 Challenges confronting preci-sion medicine in the context of inherited retinal disorders. Ex-pert Rev. Precis. Med. Drug Dev. 1: 195–205. https://doi.org/10.1080/23808993.2016.1152159
Chiang, W. C., N. Hiramatsu, C. Messah, H. Kroeger, and J. H. Lin,2012a Selective activation of ATF6 and PERK endoplasmic re-ticulum stress signaling pathways prevent mutant rhodopsinaccumulation. Invest. Ophthalmol. Vis. Sci. 53: 7159–7166.https://doi.org/10.1167/iovs.12-10222
Chiang, W. C., C. Messah, and J. H. Lin, 2012b IRE1 directs pro-teasomal and lysosomal degradation of misfolded rhodopsin.Mol. Biol. Cell 23: 758–770. https://doi.org/10.1091/mbc.e11-08-0663
Chiang, W. C., H. Kroeger, S. Sakami, C. Messah, D. Yasumuraet al., 2015 Robust endoplasmic reticulum-associated degrada-tion of rhodopsin precedes retinal degeneration. Mol. Neurobiol.52: 679–695. https://doi.org/10.1007/s12035-014-8881-8
Chuang, J. Z., C. Vega, W. Jun, and C. H. Sung, 2004 Struc-tural and functional impairment of endocytic pathways byretinitis pigmentosa mutant rhodopsin-arrestin complexes.J. Clin. Invest. 114: 131–140. https://doi.org/10.1172/JCI200421136
Cideciyan, A. V., D. C. Hood, Y. Huang, E. Banin, Z. Y. Li et al.,1998 Disease sequence from mutant rhodopsin allele torod and cone photoreceptor degeneration in man. Proc.Natl. Acad. Sci. USA 95: 7103–7108. https://doi.org/10.1073/pnas.95.12.7103
Davies, W. I., 2014 Challenges using diagnostic next-generationsequencing in the clinical environment for inherited retinaldisorders. Per. Med. 11: 99–111. https://doi.org/10.2217/pme.13.95
Davies, W. I., S. M. Downes, J. K. Fu, M. E. Shanks, R. R. Copleyet al., 2012 Next-generation sequencing in health-care deliv-ery: lessons from the functional analysis of rhodopsin. Genet.Med. 14: 891–899. https://doi.org/10.1038/gim.2012.73
Dryja, T. P., T. L. McGee, L. B. Hahn, G. S. Cowley, J. E. Olssonet al., 1990 Mutations within the rhodopsin gene in pa-tients with autosomal dominant retinitis pigmentosa.N. Engl. J. Med. 323: 1302–1307. https://doi.org/10.1056/NEJM199011083231903
Erlenbach, I., E. Kostenis, C. Schmidt, C. Serradeil-Le Gal, D.Raufaste et al., 2001 Single amino acid substitutions anddeletions that alter the G protein coupling properties of theV2 vasopressin receptor identified in yeast by receptor randommutagenesis. J. Biol. Chem. 276: 29382–29392. https://doi.org/10.1074/jbc.M103203200
Fahim, A. T., S. P. Daiger, and R. G. Weleber, 2000 NonsyndromicRetinitis Pigmentosa Overview. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1417/. Accessed: March 20, 2017.
Fishman, G. A., K. Vandenburgh, E. M. Stone, L. D. Gilbert, K. R.Alexander et al., 1992 Ocular findings associated with rhodop-sin gene codon 267 and codon 190 mutations in dominant ret-initis pigmentosa. Arch. Ophthalmol. 110: 1582–1588. https://doi.org/10.1001/archopht.1992.01080230082026
Fuchs, S., H. Kranich, M. J. Denton, E. Zrenner, S. S. Bhattacharyaet al., 1994 Three novel rhodopsin mutations (C110F, L131P,A164V) in patients with autosomal dominant retinitis pigmen-tosa. Hum. Mol. Genet. 3: 1203. https://doi.org/10.1093/hmg/3.7.1203
Gross, A. K., V. R. Rao, and D. D. Oprian, 2003 Characterizationof rhodopsin congenital night blindness mutant T94I. Biochem-istry 42: 2009–2015. https://doi.org/10.1021/bi020613j
Han, M., S. O. Smith, and T. P. Sakmar, 1998 Constitutive activa-tion of opsin by mutation of methionine 257 on transmembranehelix 6. Biochemistry 37: 8253–8261. https://doi.org/10.1021/bi980147r
Hashi, H., Y. Nakamura, J. Ishii, and A. Kondo, 2018 Modifyingexpression modes of human neurotensin receptor type 1 al-ters sensing capabilities for agonists in yeast signaling bio-sensor. Biotechnol. J. 13: e1700522. https://doi.org/10.1002/biot.201700522
Hauser, A. S., S. Chavali, I. Masuho, L. J. Jahn, K. A. Martemyanovet al., 2018 Pharmacogenomics of GPCR drug targets. Cell172: 41–54.e19. https://doi.org/10.1016/j.cell.2017.11.033
Heng, B. C., D. Aubel, and M. Fussenegger, 2013 An overviewof the diverse roles of G-protein coupled receptors (GPCRs)in the pathophysiology of various human diseases. Biotechnol.Adv. 31: 1676–1694. https://doi.org/10.1016/j.biotechadv.2013.08.017
Horswill, J. G., U. Bali, S. Shaaban, J. F. Keily, P. Jeevaratnamet al., 2007 PSNCBAM-1, a novel allosteric antagonist atcannabinoid CB1 receptors with hypophagic effects in rats.Br. J. Pharmacol. 152: 805–814. https://doi.org/10.1038/sj.bjp.0707347
612 B. M. Scott et al.
https://doi.org/10.1093/hmg/ddw387https://doi.org/10.1093/hmg/ddw387https://doi.org/10.1016/j.preteyeres.2017.10.002https://doi.org/10.1016/j.preteyeres.2017.10.002https://doi.org/10.1007/s004390050158https://doi.org/10.1007/s004390050158https://doi.org/10.1016/j.isci.2018.05.001https://doi.org/10.1242/jeb.156430https://doi.org/10.1242/jeb.156430https://doi.org/10.1074/jbc.M301319200https://doi.org/10.1074/jbc.M301319200https://doi.org/10.1002/(SICI)1097-0061(20000115)16:13.0.CO;2-Khttps://doi.org/10.1002/(SICI)1097-0061(20000115)16:13.0.CO;2-Khttps://doi.org/10.1002/(SICI)1097-0061(20000115)16:13.0.CO;2-Khttps://doi.org/10.1002/(SICI)1097-0061(20000115)16:13.0.CO;2-Khttps://doi.org/10.1016/S0960-894X(99)00398-4https://doi.org/10.1016/S0960-894X(99)00398-4https://doi.org/10.1074/jbc.M114.551713https://doi.org/10.1074/jbc.M114.551713https://doi.org/10.1016/S0006-3495(99)77229-2https://doi.org/10.1016/S0006-3495(99)77229-2https://doi.org/10.1080/23808993.2016.1152159https://doi.org/10.1080/23808993.2016.1152159https://doi.org/10.1167/iovs.12-10222https://doi.org/10.1091/mbc.e11-08-0663https://doi.org/10.1091/mbc.e11-08-0663https://doi.org/10.1007/s12035-014-8881-8https://doi.org/10.1172/JCI200421136https://doi.org/10.1172/JCI200421136https://doi.org/10.1073/pnas.95.12.7103https://doi.org/10.1073/pnas.95.12.7103https://doi.org/10.2217/pme.13.95https://doi.org/10.2217/pme.13.95https://doi.org/10.1038/gim.2012.73https://doi.org/10.1056/NEJM199011083231903https://doi.org/10.1056/NEJM199011083231903https://doi.org/10.1074/jbc.M103203200https://doi.org/10.1074/jbc.M103203200https://www.ncbi.nlm.nih.gov/books/NBK1417/https://www.ncbi.nlm.nih.gov/books/NBK1417/https://doi.org/10.1001/archopht.1992.01080230082026https://doi.org/10.1001/archopht.1992.01080230082026https://doi.org/10.1093/hmg/3.7.1203https://doi.org/10.1093/hmg/3.7.1203https://doi.org/10.1021/bi020613jhttps://doi.org/10.1021/bi980147rhttps://doi.org/10.1021/bi980147rhttps://doi.org/10.1002/biot.201700522https://doi.org/10.1002/biot.201700522https://doi.org/10.1016/j.cell.2017.11.033https://doi.org/10.1016/j.biotechadv.2013.08.017https://doi.org/10.1016/j.biotechadv.2013.08.017https://doi.org/10.1038/sj.bjp.0707347https://doi.org/10.1038/sj.bjp.0707347
Hwa, J., P. Garriga, X. Liu, and H. G. Khorana, 1997 Structureand function in rhodopsin: packing of the helices in thetransmembrane domain and folding to a tertiary structurein the intradiscal domain are coupled. Proc. Natl. Acad.Sci. USA 94: 10571–10576. https://doi.org/10.1073/pnas.94.20.10571
Hwa, J., P. J. Reeves, J. Klein-Seetharaman, F. Davidson, and H. G.Khorana, 1999 Structure and function in rhodopsin: furtherelucidation of the role of the intradiscal cysteines, Cys-110,-185, and -187, in rhodopsin folding and function. Proc. Natl.Acad. Sci. USA 96: 1932–1935. https://doi.org/10.1073/pnas.96.5.1932
Ishii, J., T. Tanaka, S. Matsumura, K. Tatematsu, S. Kuroda et al.,2008 Yeast-based fluorescence reporter assay of G protein-coupled receptor signalling for flow cytometric screening:FAR1-disruption recovers loss of episomal plasmid caused bysignalling in yeast. J. Biochem. 143: 667–674. https://doi.org/10.1093/jb/mvn018
Jonikas, M. C., S. R. Collins, V. Denic, E. Oh, E. M. Quan et al.,2009 Comprehensive characterization of genes required forprotein folding in the endoplasmic reticulum. Science 323:1693–1697. https://doi.org/10.1126/science.1167983
Kaushal, S., and H. G. Khorana, 1994 Structure and functionin rhodopsin. 7. Point mutations associated with autosomaldominant retinitis pigmentosa. Biochemistry 33: 6121–6128.https://doi.org/10.1021/bi00186a011
Kaushal, S., K. D. Ridge, and H. G. Khorana, 1994 Structure andfunction in rhodopsin: the role of asparagine-linked glycosyla-tion. Proc. Natl. Acad. Sci. USA 91: 4024–4028. https://doi.org/10.1073/pnas.91.9.4024
Keppler-Ross, S., C. Noffz, and N. Dean, 2008 A new purple fluo-rescent color marker for genetic studies in Saccharomyces cere-visiae and Candida albicans. Genetics 179: 705–710. https://doi.org/10.1534/genetics.108.087080
Kimata, Y., and K. Kohno, 2011 Endoplasmic reticulum stress-sensing mechanisms in yeast and mammalian cells. Curr.Opin. Cell Biol. 23: 135–142. https://doi.org/10.1016/j.ceb.2010.10.008
Kompella, P. S., A. M. Moses, and S. G. Peisajovich, 2017 Introductionof premature stop codons as an evolutionary strategy to res-cue signaling network function. ACS Synth. Biol. 6: 446–454.https://doi.org/10.1021/acssynbio.6b00142
Krebs, M. P., D. C. Holden, P. Joshi, C. L. Clark, III, A. H. Lee et al.,2010 Molecular mechanisms of rhodopsin retinitis pigmentosaand the efficacy of pharmacological rescue. J. Mol. Biol. 395:1063–1078. https://doi.org/10.1016/j.jmb.2009.11.015
Kunte, M. M., S. Choudhury, J. F. Manheim, V. M. Shinde, M. Miuraet al., 2012 ER stress is involved in T17M rhodopsin-inducedretinal degeneration. Invest. Ophthalmol. Vis. Sci. 53: 3792–3800. https://doi.org/10.1167/iovs.11-9235
Lam, P. V., R. Goldman, K. Karagiannis, T. Narsule, V. Simonyanet al., 2013 Structure-based comparative analysis and predic-tion of N-linked glycosylation sites in evolutionarily distant eu-karyotes. Genomics Proteomics Bioinformatics 11: 96–104.https://doi.org/10.1016/j.gpb.2012.11.003
Laurent, J. M., J. H. Young, A. H. Kachroo, and E. M. Marcotte,2016 Efforts to make and apply humanized yeast. Brief. Funct.Genomics 15: 155–163. https://doi.org/10.1093/bfgp/elv041
LaVail, M. M., S. Nishikawa, R. H. Steinberg, M. I. Naash, J. L.Duncan et al., 2018 Phenotypic characterization of P23H andS334ter rhodopsin transgenic rat models of inherited retinal de-generation. Exp. Eye Res. 167: 56–90. https://doi.org/10.1016/j.exer.2017.10.023
Lek, M., K. J. Karczewski, E. V. Minikel, K. E. Samocha, E. Bankset al., 2016 Analysis of protein-coding genetic variation in60,706 humans. Nature 536: 285–291. https://doi.org/10.1038/nature19057
Lin, J. H., H. Li, D. Yasumura, H. R. Cohen, C. Zhang et al.,2007 IRE1 signaling affects cell fate during the unfolded pro-tein response. Science 318: 944–949. https://doi.org/10.1126/science.1146361
Liu, M. Y., J. Liu, D. Mehrotra, Y. Liu, Y. Guo et al., 2013 Thermalstability of rhodopsin and progression of retinitis pigmen-tosa: comparison of S186W and D190N rhodopsin mutants.J. Biol. Chem. 288: 17698–17712. https://doi.org/10.1074/jbc.M112.397257
Liu, R., D. Nahon, B. le Roy, E. B. Lenselink, and A. P. IJzerman,2015 Scanning mutagenesis in a yeast system delineatesthe role of the NPxxY(x)(5,6)F motif and helix 8 of the aden-osine A(2B) receptor in G protein coupling. Biochem. Phar-macol. 95: 290–300. https://doi.org/10.1016/j.bcp.2015.04.005
Liu, R., W. Wong, and A. P. IJzerman, 2016 Human G protein-coupled receptor studies in Saccharomyces cerevisiae. Biochem.Pharmacol. 114: 103–115. https://doi.org/10.1016/j.bcp.2016.02.010
Luo, D. G., W. W. Yue, P. Ala-Laurila, and K. W. Yau, 2011 Activationof visual pigments by light and heat. Science 332: 1307–1312.https://doi.org/10.1126/science.1200172
Macke, J. P., C. M. Davenport, S. G. Jacobson, J. C. Hennessey, F.Gonzalez-Fernandez et al., 1993 Identification of novel rho-dopsin mutations responsible for retinitis pigmentosa: implica-tions for the structure and function of rhodopsin. Am. J. Hum.Genet. 53: 80–89.
Maeda, S., D. Sun, A. Singhal, M. Foggetta, G. Schmid et al.,2014 Crystallization scale preparation of a stable GPCR sig-naling complex between constitutively active rhodopsin andG-protein. PLoS One 9: e98714. https://doi.org/10.1371/journal.pone.0098714
Marsili, S., S. Genini, R. Sudharsan, J. Gingrich, G. D. Aguirreet al., 2015 Exclusion of the unfolded protein response inlight-induced retinal degeneration in the canine T4R RHOmodel of autosomal dominant retinitis pigmentosa. PLoSOne 10: e0115723. https://doi.org/10.1371/journal.pone.0115723
Mendes, H. F., J. van der Spuy, J. P. Chapple, and M. E. Cheetham,2005 Mechanisms of cell death in rhodopsin retinitis pigmen-tosa: implications for therapy. Trends Mol. Med. 11: 177–185.https://doi.org/10.1016/j.molmed.2005.02.007
Min, K. C., T. A. Zvyaga, A. M. Cypess, and T. P. Sakmar,1993 Characterization of mutant rhodopsins responsible forautosomal dominant retini