Embed Size (px)
Citation preview

Polyhedron Vol. 11, No. II, pp. 1343-1348, 1992 Printed in Great Britain
0277-5387/92 SS.oO+.OO
0 1992 Pergamon Press Ltd
CRYSTAL STRUCTURE AND EPR SPECTRUM OF BISPYRIDINIUM BIS(CITRATO)CHROMIUM(III)
TETRAHYDRATE
MIGUEL QUIROS,* DAVID M. L. GOODGAME? and DAVID J. WILLIAMS
Chemistry Department, Imperial College of Science, Technology and Medicine, London SW7 2AY, U.K.
(Received 12 November 1991; accepted 23 January 1992)
Abstract-Citric acid reacts with chromium(V1) in aqueous solution leading to the for- mation of chromium(V), easily detectable by EPR. However, the redox reaction goes further with the formation of a chromium(II1) citrate complex. This has been crystallized as the pyridinium salt and analysed by EPR and X-ray diffraction. The structure contains discrete Cr(Cit)22- units, in which the chromium atom is octahedrally coordinated by two tridentate citrate anions. The anions and the solvate water molecules are linked by an extensive set of hydrogen bonds. The X-band and Q-band EPR spectra are well resolved, showing a large and highly anisotropic zero-field splitting.
The carcinogenic and mutagenic properties of chromium(V1) have prompted a continuing interest in the interactions of chromium(V1) with biological molecules and in the chromium(V) species derived therefrom. ‘,2 In terms of chromium(VI)/substrate reactions of direct ecological relevance, relatively long-lived chromium(V) species have been observed right from the initial chromium(VI)/soil humus stage 3*4 through to their presence in plant roots and leaves.’ Additionally, a number of naturally occurring compounds such as glutathione,2*“8 ascorbic acid,’ D-galactouronic acid” and ribo- nucleotides” have been identified as being able to produce chromium(V) species from chromium(V1) precursors. Such chromium(V) species eventually undergo further redox reaction to yield chromium (III) compounds.
In continuation of our own investigations on such systems we report here some EPR studies on the reaction of chromium(V1) with citric acid, HO2 CH2C(OH)(COIH)CH1C02H, and the structure of a chromium(II1) citrate complex which was isolated as the ultimate product from the chromium(VI)/ citric acid redox reaction sequence. Citric acid is
* Present address : Departamento de Quimica Inor- ginica, Facultad de Ciencias, 18071 Granada, Spain.
t Author to whom correspondence should be addressed.
one of the most widely spread plant acids. As well as occurring in e.g. lemon juice, it is found in many other fruits (e.g. in cranberries and redcurrants), in beetroot juice and in wine, and also in animals.
EXPERIMENTAL
Synthesis of bispyridinium bis(citrato)chromium(III) tetrahydrate
Pyridinium dichromate (0.376 g, 1 mmol) and citric acid (0.96 g, 5 mmol) were dissolved in water (50 cm3), and the solution was stored in a closed flask at room temperature for 1 week. After that, 1 cm3 of pyridine was added and the solution allowed to evaporate in the open air. When the volume had been reduced to a few cm3, the product separated as yellowish-grey crystalline plates, which were filtered off and washed with water and acetone. Found : C, 40.5; H, 4.5; N, 3.9. Calc. for CrC22H3,N20,8: C, 39.8; H, 4.7; N, 4.2%. Selected IR bands (cm-‘): 3416, v(O-H) water ; 1737, v(c---O) of protonated carboxylate ; 1622, v(C=O) of anionic carboxylate ; 1539, 1488 and 1418, pyridinium cation.
X-ray study
The crystal data are : C22H31CrN20,8, M, = 663.5, triclinic, a = 7.584(l), b = 10.070(2), c =
1343

1344 M. QUIROS etal.
10.389(2) A, CI = 88.01(2)“, /!? = 69.81(2)“, y = 72.60(l)“, V = 708.5(2) A3, space group Pi, 2 = 1 (the structure is disposed about a centre of sym- metry), D, = 1.56 g cmm3, Cu-K, radiation (2 = 1.54178 A), p(Cu-K,) = 41 cm-‘, F(OOO) = 345. Data were measured on a Nicolet R3m diffrac- tometer (graphite monochromator) using o-scans.
A yellow-grey crystal of dimensions 0.04 x 0.06 x0.10 mm was used. 1913 independent reflections (28 < 116”) were measured, of which 1798 had IF,] > 3o(]F,]) and were considered observed. The data were corrected for Lorentz and polarization factors ; a numerical absorption cor- rection (face-indexed crystal) was applied ; maxi- mum and minimum transmission factors, 0.874 and 0.608.
The structure was solved by direct methods. The non-hydrogen atoms were refined anisotropically. The positions of all the hydrogen atoms, including those of partial occupancy on both the hydroxyl groups and the water molecules, were determined from a AF map. The OH hydrogen atoms were refined isotropically subject to an O-H distance constraint (0.90 A). The remaining hydrogen atom positions were idealized, C-H 0.96 A, and allowed to ride on their parent atoms. Refinement was by full-matrix least-squares to R = 0.035, R, = 0.038 [w- ’ = o’(F) + 0.0004F2]. The maximum and mini- mum residual electron densities in the final AF map were 0.24 and - 0.25 ek 3, respectively. The mean
and maximum shift/error in the final refinement were 0.004 and 0.021, respectively. Computations were carried out on an IBM PS/2 Model 70 386PC, using the SHELXTL PC program system. I2 Selec- ted bond lengths and angles are listed in Table 1.
EPR studies
EPR spectra were measured at room temperature on a Varian E-9 X-band (ca 9.5 GHz) and a Bruker 200D-SRC Q-band (ca 34 GHz) spectrometer. An Heraeus flat cell was used for solution work.
For monocrystal work, a crystalline plate of bis- pyridinium bis(citrato)chromate(III) was attached to the bottom of a Q-band tube and this was intro- duced in the cavity in such a way that the crystal was placed at the height where one of the nodes of the cavity microwave mode takes place. Spectra were taken in several positions rotating the rod with the tube and the crystal around a vertical axis.
RESULTS AND DISCUSSION
The room temperature EPR spectrum of an aque- ous solution of potassium chromate (O.lM) and citric acid (0.2M) comprised a very sharp signal at g = 1.977, with four 53Cr hyperfine satellites (coup- ling constant 18.7 G) (Fig. 1). Such spectra are clearly assignable to chromium(V). The intensity of the signal grew during the first two days after
Table 1. Bond lengths (A) and angles (“) in the Cr(Cit)22- anion
Cr-0( 11) Cr-O(61) C(l)--o(l2) C(2)-C(3) C(3)--c(4) C(4FC(5) C(5)--0(52) ‘7+-0(62)
1.987(2) 1.967(2) 1.248(3) 1.537(4) 1.522(3) 1.499(4) 1.309(3) 1.228(4)
Cr-O( 3) C(l)_-o(ll) C(l)-C(2) C(3)_-0(3) C(3)_C(6) C(5)_-0(51) C(6)_-0(61)
1.965(l) 1.268(3) 1.514(4) 1.442(3) 1.543(4) 1.188(3) 1.289(3)
0( 1 l)-Cr-O(3) O(3)-Cr-0(61) O( 1 l)-C(l)-C(2) Cr-O(lI)-C(1) C(2t-c(3)_-0(3) O(3)-C(3)-C(4) O(3)-C(3)-C(6) Cr-0(3)-C(3) C(4)--c(5)_-0(5 1) 0(51)-C(S)-0(52) C(3)-C(6V(62) Cr-0(61)--C(6)
88.6(l) 81.5(l)
121.1(2) 130.8(2) 109.4(2) 111.1(2) 107.3(2) 107.8(l) 126.6(2) 122.3(3) 119.6(2) 113.3(2)
0( 1 l)-Cr-0(61) 0(11)-C(l)-O(12) 0(12)--W&--C(2) C(l)-C(2)-C(3) C(2)_C(3)-C(4) C(2)_C(3)--c(6) C(4)_C(3)-C(6) C(3)-C(4)-C(5) C(4)-C(5)-0(52) C(3)--C(6)_-0(61) 0(61)-C(6)-0(62)
88.7(l) 121.6(2) 117.3(2) 115.7(2) 109.1(2) 108.8(2) 111.1(3) 114.5(2) 111.2(2) 115.4(3) 125.0(Z)

Bispyridinium bis(citrato)chromium(III) tetrahydrate 1345
r-lmT--l
Fig. 1. Room temperature X-band EPR spectrum of an aqueous solution of potassium chromate (O.lM) and citric acid (0.2M). The s3Cr hypefine peaks were recorded at an increased ( x 12.5) gain
setting.
sample preparation and was then stable for at least a week.
The formation of chromium(V) complexes with citric acid in aqueous solution has been previously observed by means of visible spectroscopy, I3 but the present EPR results suggested that they are rather more stable than was previously inferred. Accordingly, attempts were made to precipitate a solid chromium(V) complex by addition of various cations.
Use of the pyridinium cation afforded some yellowish-grey plate-like crystals, the analysis of which suggested a formulation of bispyridinium bis(citrato)chromium tetrahydrate (1). If all the car- boxylate groups are deprotonated, the chromium atom in 1 would be in the +4 oxidation state. However, this is inconsistent with the X- (v = 9.5 GHz) and Q-band (v = 33.9 GHz) EPR spectra of a polycrystalline sample of the complex (Fig. 2), as chromium(W) compounds would be expected to be EPR-silent. Moreover, the spectra are too complex for chromium(V) (S = l/2), arising from the additional deprotonation of one of the citrate hydroxyl groups. The EPR results pointed rather to the presence of chromium(II1) with the observed complex band system arising from the effects of zero-field splitting. The spectra are unusually well resolved for polycrystalline samples of chromium- (III). Such well resolved spectra are usually only obtained if chromium(II1) is doped in an EPR-silent trivalent metal host lattice.14 The EPR spectra of
powdered chromium(II1) compounds frequently show merely a broad band at geff = 2 with not many distinct features.
A powdered solid with similar X- and Q-band EPR spectra was obtained when potassium dichro- mate and potassium hydroxide were used instead of pyridinium dichromate and pyridine.
As 1 was obtained in crystalline form, it was possible to determine its structure by X-ray methods and thereby to correlate the + 3 oxidation state of the chromium with the extent and nature of the deprotonation of the coordinated citrate groups.
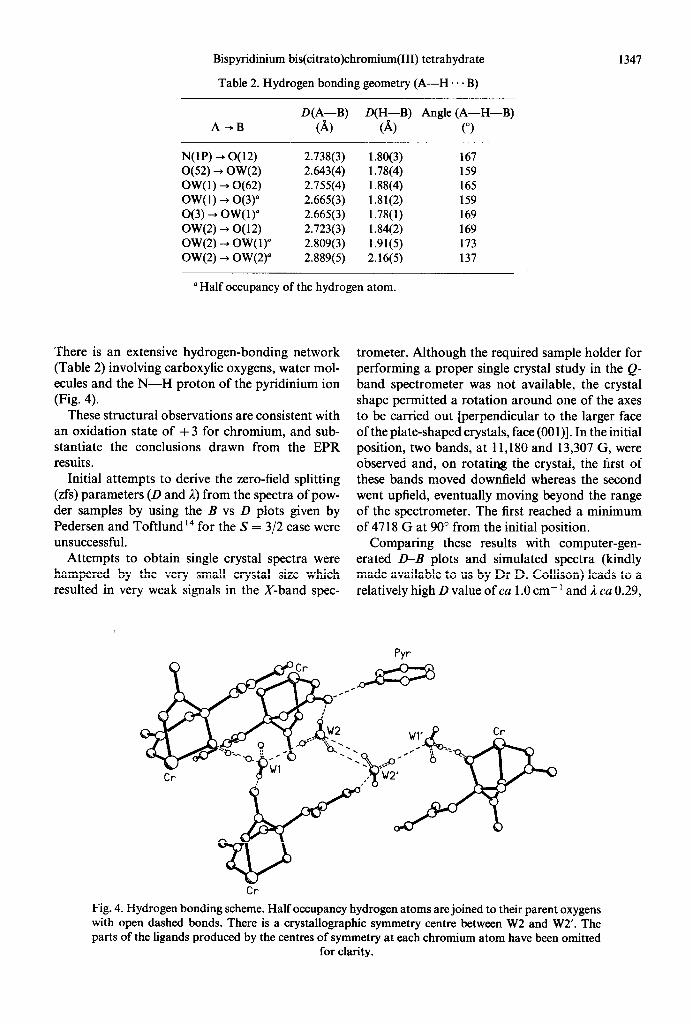
The structure comprises a Cr(Cit),*- anion, two pyridinium cations and four water molecules in the unit cell. The chromium atom lies on a crystallo- graphic symmetry centre and has slightly distorted octahedral geometry (Fig. 3), the angles at chro- mium lying in the range 81.5(1)-88.7(l)“. Each cit- rate ion is tridentate and coordinates to the chro- mium atom via the oxygen atoms 0( 11) and 0(61) of two deprotonated carboxylate groups and by the hydroxyl oxygen atom O(3) ; this coordination mode produces a five- and a six-membered chelate ring (Fig. 3). The Cr-0 bonds within the five- membered ring have the same length [ 1.965( 1) A to O(3) and 1.967(2) A to 0(61)] but that to O(11) is significantly longer [ 1.987(2) A]. This contrasts with the related zinc citrate complex, ’ 5 which displays a greater asymmetry in the Zn-0 bonds (2.052- 2.164 A), but in that case the longest Zn-0 bond
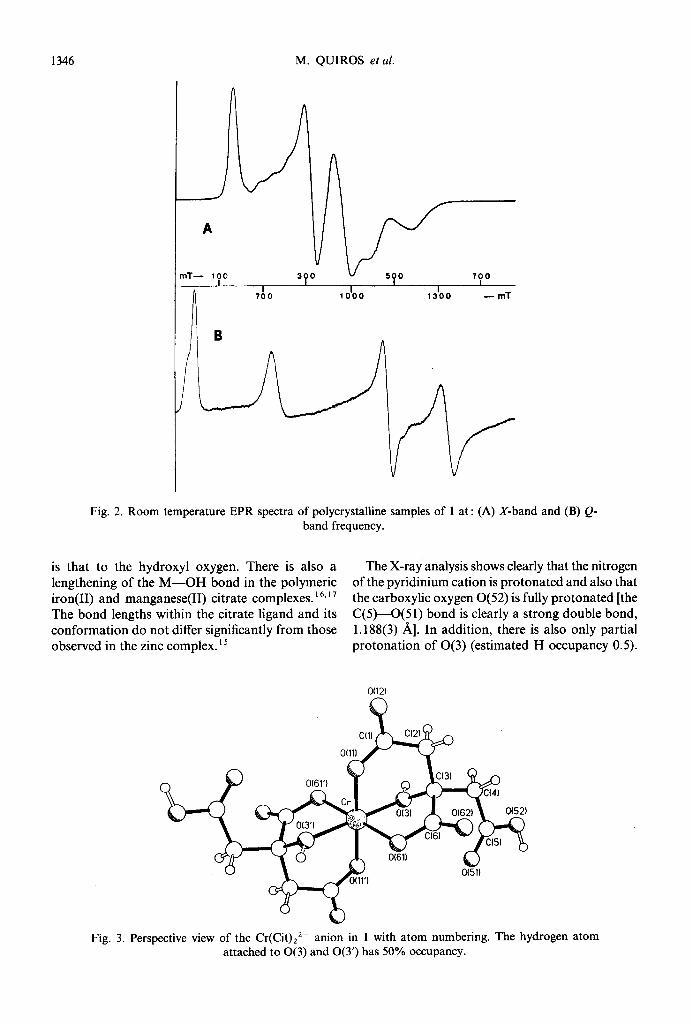
1346 M. QUIROS et al.
” ?T- 100 3 0 u 50 700
n
I I I 700 1000 1300 -mT
Fig. 2. Room temperature EPR spectra of polycrystalline samples of 1 at : (A) X-band and (B) Q- band frequency.
is that to the hydroxyl oxygen. There is also a The X-ray analysis shows clearly that the nitrogen lengthening of the M-OH bond in the polymeric of the pyridinium cation is protonated and also that iron(I1) and manganese(I1) citrate complexes. I6317 the carboxylic oxygen 0(52) is fully protonated [the The bond lengths within the citrate ligand and its C(5)-0(51) bond is clearly a strong double bond, conformation do not differ significantly from those 1.188(3) A]. In addition, there is also only partial observed in the zinc complex.‘5 protonation of O(3) (estimated H occupancy 0.5).
O(12)
Fig. 3. Perspective view of the Cr(Cit)22- anion in 1 with atom numbering. The hydrogen atom attached to O(3) and 0(3’) has 50% occupancy.
Bispyridinium bis(citrato)chromium(III) tetrahydrate 1347
Table 2. Hydrogen bonding geometry (A-H. . . B)
A-+B
N(lP) -, 0(12) O(52) + OW(2) OW( 1) + 0(62) OW(1) --t O(3) O(3) --) OW(1) OW(2) -+ O( 12) OW(2) -+ OW( 1) OW(2) + OW(2)
D(A-B) @H-B) Angle (A-H-B)
(A) (A) (“)
2.738(3) 1.80(3) 167 2.643(4) 1.78(4) 159 2.755(4) 1.88(4) 165 2.665(3) 1.81(2) 159 2.665(3) 1.78(l) 169 2.723(3) 1.84(2) 169 2.809(3) 1.91(5) 173 2.889(5) 2.16(5) 137
” Half occupancy of the hydrogen atom.
There is an extensive hydrogen-bonding network (Table 2) involving carboxylic oxygens, water mol- ecules and the N-H proton of the pyridinium ion (Fig. 4).
These structural observations are consistent with an oxidation state of +3 for chromium, and sub- stantiate the conclusions drawn from the EPR results.
Initial attempts to derive the zero-field splitting (zfs) parameters (D and 1) from the spectra of pow- der samples by using the B vs D plots given by Pedersen and Toftlund14 for the 5’ = 312 case were unsuccessful.
Attempts to obtain single crystal spectra were hampered by the very small crystal size which resulted in very weak signals in the X-band spec-
trometer. Although the required sample holder for performing a proper single crystal study in the Q- band spectrometer was not available, the crystal shape permitted a rotation around one of the axes to be carried out [perpendicular to the larger face of the plate-shaped crystals, face (OOl)]. In the initial position, two bands, at 11,180 and 13,307 G, were observed and, on rotating the crystal, the first of these bands moved downfield whereas the second went upfield, eventually moving beyond the range of the spectrometer. The first reached a minimum of 4718 G at 90” from the initial position.
Comparing these results with computer-gen- erated D-B plots and simulated spectra (kindly made available to us by Dr D. Collison) leads to a relatively high D value of ca 1 .O cm- ’ and 2 ca 0.29,
Fig. 4. Hydrogen bonding scheme. Half occupancy hydrogen atoms are joined to their parent oxygens with open dashed bonds. There is a crystallographic symmetry centre between W2 and W2’. The parts of the ligands produced by the centres of symmetry at each chromium atom have been omitted
for clarity.

1348 M. QUIROS et al.
i.e. close to the limiting value of 0.33 and well out of the range reported by Pedersen and Toftlund.14 According to this, the x axis of the D tensor was parallel to the magnetic field in the initial position of the crystal and the two bands observed cor- respond well with the features at both sides of geff = 2 in the Q-band powder spectrum, which are also related to the bands at 3 100 and 3800 G in the X-band powder spectrum. A 1 value of 0.29 can be estimated from the position of these bands.
Spanish Ministry of Education and Science for the Flem- ing Fellowship to MQ and SERC for the EPR spec- trometers.
These approximate zfs parameters explain all the main features of both X- and Q- band powder spec- tra, except for the 7282 G in the Q-band spectrum, which could be an off-axis or a forbidden transition. An accurate value of D is, in any case, quite difficult to determine, since when 1 is large, the position of the bands is rather insensitive to changes in large D
values. It is interesting that, although the asym- metry of the metal environment shown by the X- ray study is not unduly severe, it is sufficient to produce quite large zfs parameters.
In summary, the combination of X-ray and EPR studies has provided a good characterization of the final product from the redox reaction between chrom- ium(W) and citric acid. Whereas a simplistic assess- ment of the observed stoichiometry of the solid obtained from the original chromium(V) species in solution pointed to the presence of chromium (IV), the EPR results indicated a chromium(II1) oxidation state. The X-ray determination was sufficiently clear-cut to permit a detailed analysis of the degrees of protonation of the oxygen donor atoms, thus permitting a rationalization of the stoi- chiometry with the spectroscopic results.
Acknowledgements-We thank Dr David Collison of the University of Manchester for his computer-generated simulated EPR spectra, to the British Council and the
1.
2.
3.
4.
5.
9.
10.
11.
12.
13.
14.
15.
16.
17.
REFERENCES
S. Kawanishi, S. Inoue and S. Sano, J. Biol. Chem. 1986, 261, 5952 and refs therein. S. L. Brauer and K. E. Wetterhahn, J. Am. Chem. Sot. 1991,113,3001 and refs therein. D. M. L. Goodgame, P. B. Hayman and D. E. Hath- way, Inorg. Chim. Acta 1984,91, 113. S. L. Boyko and D. M. L. Goodgame, Znorg. Chim. Acta 1986, 123, 189. G. Micera and A. Dessi, J. Znorg. Biochem. 1988,34, 157. D. M. L. Goodgame and A. M. Joy, J. Znorg. Biochem. 1986, 26,219. S. Kitagawa, H. Seki, F. Kametani and H. Sakurai, Znorg. Chim. Actu 1988, 152,251. P. O’Brien, J. Pratt, F. J. Swanson, P. Thornton and G. Wang, Znorg. Chim. Acta 1990, 169, 265 and refs therein. D. M. L. Goodgame and A. M. Joy, Znorg. Chim. Acta 1987, 135, 115. M. Branca, G. Micera and A. Dessi, Znorg. Chim. Acta 1988, 153, 61. M. Quiros and D. M. L. Goodgame, Polyhedron 1992, 11, 1. G. M. Sheldrick, SHELXTL PC, Version 4.2, Siemens Analytical Instruments Inc. (1990). M. Krumpolc and J. Rocek, J. Am. Chem. Sot. 1976, 98, 872. E. Pedersen and H. Toftlund, Znorg. Chem. 1974, 13, 1603. R. Swanson, W. H. Ilsley and A. G. Stanislowski, J. Znorg. Biochem. 1983, 18, 187. J. Strouse, S. W. Layten and C. E. Strouse, J. Am. Chem. Sot. 1977,99,562. H. L. Carrel and J. P. Glusker, Acta Cryst. 1973, B29, 638.





























