Embed Size (px)
Citation preview

1
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 1
Mon, Wed 11:30 AM - 12:50, on Zoom*Prof: Gill BejeranoCA: Boyoung (Bo) Yoo* Track class on Piazza
CS273A
Gill Lecture 2: RNA Genes, Gene Enrichment
The Human GenomeSource Code
1
• http://cs273a.stanford.edu/– Lecture slides, problem sets, etc.
• Course communications via Piazza– Auditors please sign up too
• HW2 will be posted later today.
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 2
Announcements
2
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 3
Class Topics(0) Genome context:
cells, DNA, central dogma(1) Genome content / genome function:
genes, gene regulation, epigenetics, repeats, SARS-CoV-2(2) Genome sequencing:
technologies, assembly/analysis, technology dependence (3) Genome evolution:
evolution = mutation + selection, main forces of evolution:Neutral evolution, Negative selection, Positive selection
(4) Population genomics:Human migration, paternity testing, forensics, cryptogenomics
(5) Genomics of human disease:personal genomics, GxE disease types, deep dive monogenics
(6) Comparative Genomics :Genomics of amazing animal adaptations, ultraconservation
3

2
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 4
“non coding” RNAs (ncRNA)
4
5
Central Dogma of Biology:
5
6
Active forms of “non coding” RNA
reverse transcription(of eg retrogenes)
long non-codingRNA
microRNA
rRNA, snRNA, snoRNA
6

3
7
What is ncRNA?• Non-coding RNA (ncRNA) is an RNA that functions without being
translated to a protein.• Known roles for ncRNAs:
– RNA catalyzes excision/ligation in introns.– RNA catalyzes the maturation of tRNA.– RNA catalyzes peptide bond formation.– RNA is a required subunit in telomerase.– RNA plays roles in immunity and development (RNAi).– RNA plays a role in dosage compensation.– RNA plays a role in carbon storage.– RNA is a major subunit in the SRP, which is important in protein trafficking.– RNA guides RNA modification.
– RNA can do so many different functions, it is thought in the beginning there was an RNA World, where RNA was both the information carrier and active molecule.
7
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 8
“non coding” RNAs (ncRNA) subtype:
Small structural RNAs (ssRNA)
8
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 9
Remember with protein coding genes?
Gene (DNA) sequence determines protein (AA) sequence,which determines protein (3D) structure,which determines protein’s function.
9

4
10
AAUUGCGGGAAAGGGGUCAACAGCCGUUCAGUACCAAGUCUCAGGGGAAACUUUGAGAUGGCCUUGCAAAGGGUAUGGUAAUAAGCUGACGGACAUGGUCCUAACCACGCAGCCAAGUCCUAAGUCAACAGAUCUUCUGUUGAUAUGGAUGCAGUUCA
It’s the same with small structural RNAs!
P6b
P6a
P6
P4
P5P5a
P5b
P5c
120
140
160
180
200
220
240
260
AAU
UGCGGGAAAG
GGGUCA
ACAGCCGUUCAGU
ACCA
AGUCUCAGGGGAA
ACUUUGAGAU
GGCCUUGCA A A GG
GU A UGGUA
AUA AGCUGACGGACA
UGGUCC
U
A
ACCACGCA
GCCAAGUCC
UAAGUCAACAGAU C U
UCUGUUGAUAU
GGAUGCAGU
UC A
Cate, et al. (Cech & Doudna).(1996) Science 273:1678.
Waring & Davies. (1984) Gene 28: 277.
Computational Challenge: predict 2D & 3D structure from sequence (1D).
Sequence determinesStructure determines Function.
10
For example, tRNA Structure…
11
…Facilitates tRNA Function
Complementation is a “brilliant” way for the Genome to “get things done”.
Replication, Transcription, Translation, …Compliment(Compliment) = Identity
12

5
RNP = RNA-protein ComplexesProteins and ncRNA co-operate.For example the ribosome is a complex of proteins+RNAs.
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 13
13
Predicting ssRNA Structure
To a first approximation: Structural RNAs fold into their most energetically stable structure.An RNA structure stores its energy in the formof bonds between complementary nucleotides.
RNA structure prediction challenge: Given a ssRNA sequence, find its most energetically favorable possible structure, or in other words, the structure which maximizes the energy stored in its bond –this is the predicted structure of this ssRNA.
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 14
14
ssRNA structure rules• Canonical basepairs:
– Watson-Crick basepairs:• G ≡ C• A = U
– Wobble basepair:• G − U
• Stacks: continuous nested basepairs. (energetically favorable)
• Non-basepaired loops:– Hairpin loop– Bulge– Internal loop– Multiloop
• Pseudo-knots
15

6
Ab initio RNA structure prediction: Recursive formulation
• Objective: Maximizing # of base pairings (Nussinov et al, 1978)
simple model:w(i, j) = 1 iff GC|AU|GUfancier model:GC > AU > GU
1 2 3 4
16
Dynamic programming§ If you naively try to evaluate S(0,n) using
recursion – it will work, but you will be re-evaluating the same terms over and over.
§ Dynamic programming is a clever way of organizing term calculations, so that each is called & computed exactly once.– Compares a sequence against itself in a dynamic
programming matrix
§ Three steps
17
1. Initialization
Example:
GGGAAAUCC
G G G A A A U C CG 0G 0 0G 0 0A 0 0A 0 0A 0 0U 0 0C 0 0C 0 0
the main diagonal
the diagonal belowL: the length of input sequence
18
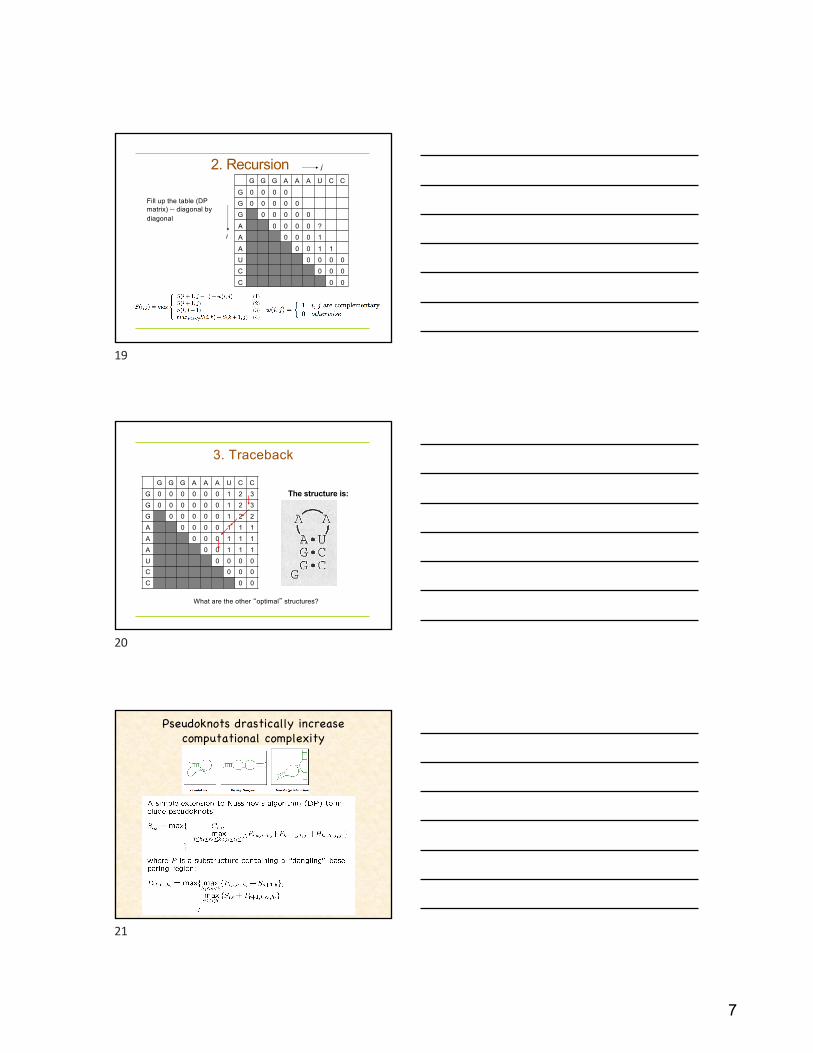
7
2. RecursionG G G A A A U C C
G 0 0 0 0G 0 0 0 0 0G 0 0 0 0 0
A 0 0 0 0 ?A 0 0 0 1
A 0 0 1 1U 0 0 0 0C 0 0 0
C 0 0
Fill up the table (DP matrix) -- diagonal by diagonal
j
i
19
3. Traceback
G G G A A A U C CG 0 0 0 0 0 0 1 2 3G 0 0 0 0 0 0 1 2 3G 0 0 0 0 0 1 2 2A 0 0 0 0 1 1 1A 0 0 0 1 1 1A 0 0 1 1 1U 0 0 0 0C 0 0 0C 0 0
The structure is:
What are the other “optimal” structures?
20
Pseudoknots drastically increase computational complexity
21

8
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 22
Objective: Minimize Secondary StructureFree Energy at 37 OC:
C G U U U GG GUUCACAAACG
-2.0
-2.1
-0.9
-0.9
-1.8
-1.6
+5.0
DG°helix = DG°C GG Céëê
ùûú + DG°
G UC Aéëê
ùûú + 2DG°
U UA Aéëê
ùûú + DG°
U GA Céëê
ùûú =
-2.0 kcal/mol - 2.1 kcal/mol + 2x(-0.9) kcal/mol - 1.8 kcal/mol = -7.7 kcal/mol
DG°hairpin loop = DG°initiation (6 nucleotides) + DG°mismatchG GC Aéëê
ùûú
=
5.0 kcal/mol - 1.6 kcal/mol = 3.4 kcal/mol
DG°total = DG°hairpin + DG°helix = 3.4 kcal/mol - 7.7 kcal/mol = -4.3 kcal/mol
Mathews, Disney, Childs, Schroeder, Zuker, & Turner. 2004. PNAS 101: 7287.
Instead of d(i, j), measure and sum energies:
22
Zuker’s algorithm MFOLD: computing loop dependent energies
23
Bafna 1
RNA structure: other representations
• Base-pairing defines a secondary structure. The base-pairing is usually non-crossing.
• In this example the pseudo-knot makes for an exception.
24

9
S à aSuS à cSgS à gSc
S à uSaS à a
S à cS à gS à u
S àSS
1. A CFG
S è aSuè acSguè accSggu
è accuSagguè accuSSaggu
è accugScSagguè accuggSccSagguè accuggaccSaggu
è accuggacccSgagguè accuggacccuSagaggu
è accuggacccuuagaggu2. A derivation of “accuggacccuuagaggu” 3. Corresponding structure
Stochastic context-free grammar
25
ssRNA transcription• ssRNAs like tRNAs are usually encoded by short
“non coding” genes, that transcribe independently.• Found in both the UCSC “known genes” track, and
as a subtrack of the RepeatMasker track
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 26
(In HW2 we’ll see other tracks to find
tRNAs in hg38)
26
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 27
“non coding” RNAs (ncRNA) subtype:
microRNAs (miRNA/miR)
27
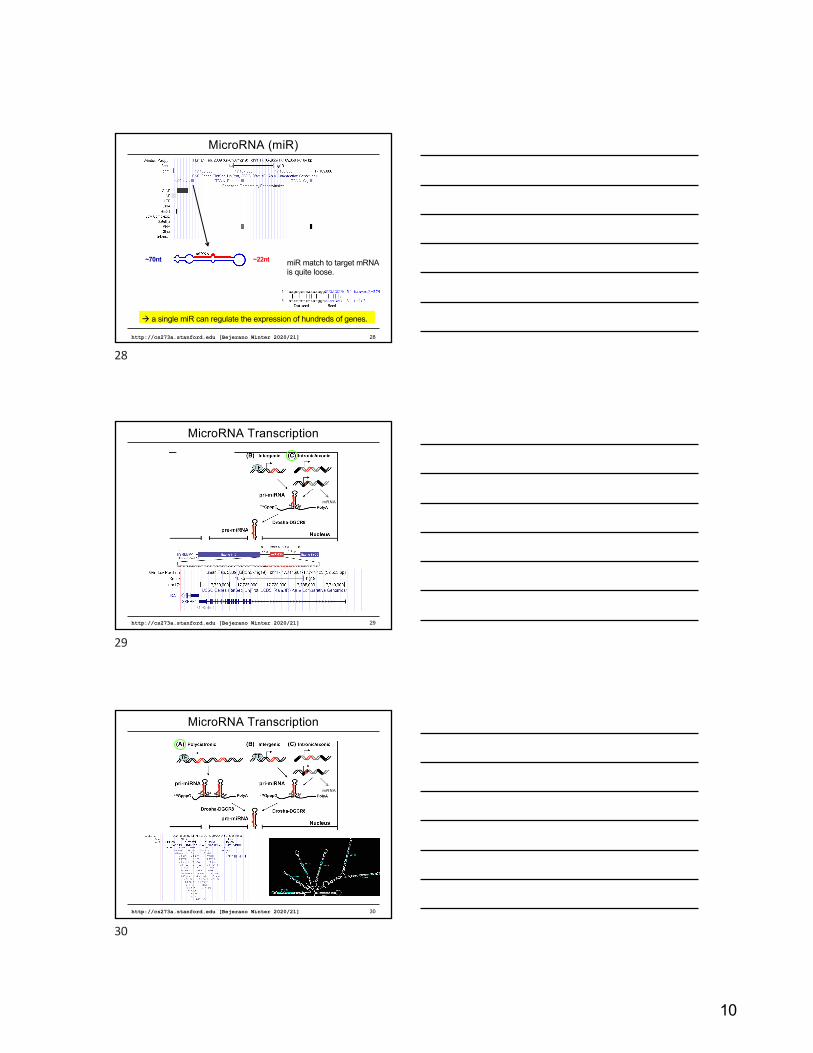
10
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 28
MicroRNA (miR)
mRNA
~70nt ~22nt miR match to target mRNAis quite loose.
à a single miR can regulate the expression of hundreds of genes.
28
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 29
MicroRNA Transcription
mRNA
29
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 30
MicroRNA Transcription
mRNA
30

11
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 31
MicroRNA (miR)
mRNA
~70nt ~22nt miR match to target mRNAis quite loose.
à a single miR can regulate the expression of hundreds of genes.
Computational challenges:Predict miRs.Predict miR targets.
31
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 32
MicroRNA Therapeutics
mRNA
~70nt ~22nt miR match to target mRNAis quite loose.
à a single miR can regulate the expression of hundreds of genes.
Idea: bolster/inhibit miR production tobroadly modulate protein productionHope: “right” the good guys and/or
“wrong” the bad guysChallenge: and not vice versa.
32
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 33
Other Non Coding Transcripts
33

12
lncRNAs (long non coding RNAs)
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 34
Don’t seem to fold into clear structures (or only a sub-region does).Diverse roles only now starting to be understood.Example: bind splice site, cause intron retention.
à Challenges: 1) detect and 2) predict function computationally
34
X chromosome inactivation in mammals
X X X Y
X
Dosage compensation
35
Xist – X inactive-specific transcript
Avner and Heard, Nat. Rev. Genetics 2001 2(1):59-67
36

13
lincRNAs & mRNAs often have structural motifs
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 37
37
SARS-CoV-2 Too…
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 38
Same bases both code for protein and participate in RNA structures
RNA genome!
38
System output measurementsWe can measure non/coding gene expression!(just remember – it is context dependent)1. First generation mRNA (cDNA) and EST sequencing:
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 39
In UCSC Browser:
39

14
2. Gene Expression Microarrays (“chips”)
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 40
40
3. RNA-seq“Next” (2nd) generation sequencing.
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 41
41
Fun computational challenges abound
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 42
42

15
Non/Coding Prediction/Testing
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 43
43
Still Plenty “weird” transcripts out there
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 44
44
Common denominators still worked out..
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 45
Central Dogmahad just mRNAs..
45

16
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 46
The million dollar question
Human Genome
Transcribed* (Tx)
Tx from both strands*
* True size of set unknown
Leaky tx?
Functional?
In HW2 you’ll measure the sizes of some of these entities yourselves!
46
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 47
Gene Finding II: technology dependenceChallenge:“Find the genes, the whole genes, and nothing but the genes”
We started out trying to predict genes directly from the genome.When you measure gene expression, the challenge changes:
Now you want to build gene models from your observations.These are both technology dependent challenges.The hybrid: what we measure is a tiny fraction of the space-time state
space for cells in our body. We want to generalize from measured states and improve our predictions for the full compendium of states.
47
Circle of Life
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 48
To a first approximation this is all your genome cares about:Making the right genes, at the right amount, at the right time, In the right cells.One genome, ten trillion cells, hundred years of life.
also!
also!
48

17
Genes• Gene production is conceptually simple o Contiguous stretches of DNA transcribe (1 to 1) into RNAo Some (coding or non-coding) RNAs are further spliced o Some (m)RNAs are then translated into protein (43 to 20+1)o Other (nc)RNA stretches just go off to do their thing as RNA
• The devil is in the details, but by and large – this is it.
(non/coding) Gene finding - classical computational challenge:1. Obtain experimental data (from limited cell types/conditions)2. Find features in the data (eg, genetic code, splice sites)3. Generalize from features (eg, predict genes yet unseen)4. Predict contexts and functions (eg, via family members)
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 49
49
Cell content is constantly refreshing
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 50
The cell is constantly making new proteins and ncRNAs.
These perform their function for a while,
And are then degraded.
Newly made coding and non coding gene products take their place.
The picture within a cell is constantly “refreshing”.
To change its behavior a cell can change the repertoire of genes and ncRNAs it makes.
50
Cell differentiation
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 51
To change its behavior a cell can change the repertoire of genes and ncRNAs it makes.
That is exactly what happens when cells differentiate during development from stem cells to their different final fates.
51

18
Genes usually work in groupsBiochemical pathways, signaling pathways, etc.We’d like to catalog the functions of every gene.
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 52
52
53
Keyword lists are not enough
Sheer number of terms too much to remember and sort• Need standardized, stable, carefully defined terms• Need to describe different levels of detail• So…defined terms need to be related in a hierarchy
With structured vocabularies/hierarchies• Parent/child relationships exist between terms• Increased depth -> Increased resolution• Can annotate data at appropriate level• May query at appropriate level
Effectively a directed acyclic graph (DAG)• Node levels can be somewhat deceptive, depending on
heterogeneity of curation efforts in different portions of the DAG
organ system
embryo
cardiovascular
heart
… …
… …
… …… …
Anatomy Hierarchy
Organ systemCardiovascular system
Heart
Anatomy keywords
53
TJL-2004 54
Annotate genes to most specific terms
54

19
1. Annotate at appropriate level, query at appropriate level2. Queries for higher level terms include annotations to lower
level terms 55
General Implementations for Vocabularies
organ system
embryo
cardiovascular
heart
… …
… …
… …… …
Hierarchy DAG
chaperone regulator
molecular function
chaperone activator
… enzyme regulator
enzyme activator… …
Query for this term
Returns things annotated to descendents
55
Genes usually work in groupsWhen a cell changes its behavior to take on a new activity, it stops producing some groups of genes and starts producing other groups of genes, to stop and start the relevant biological processes, respectively.
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 56
56
Let’s compare which genes are active
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 57
57
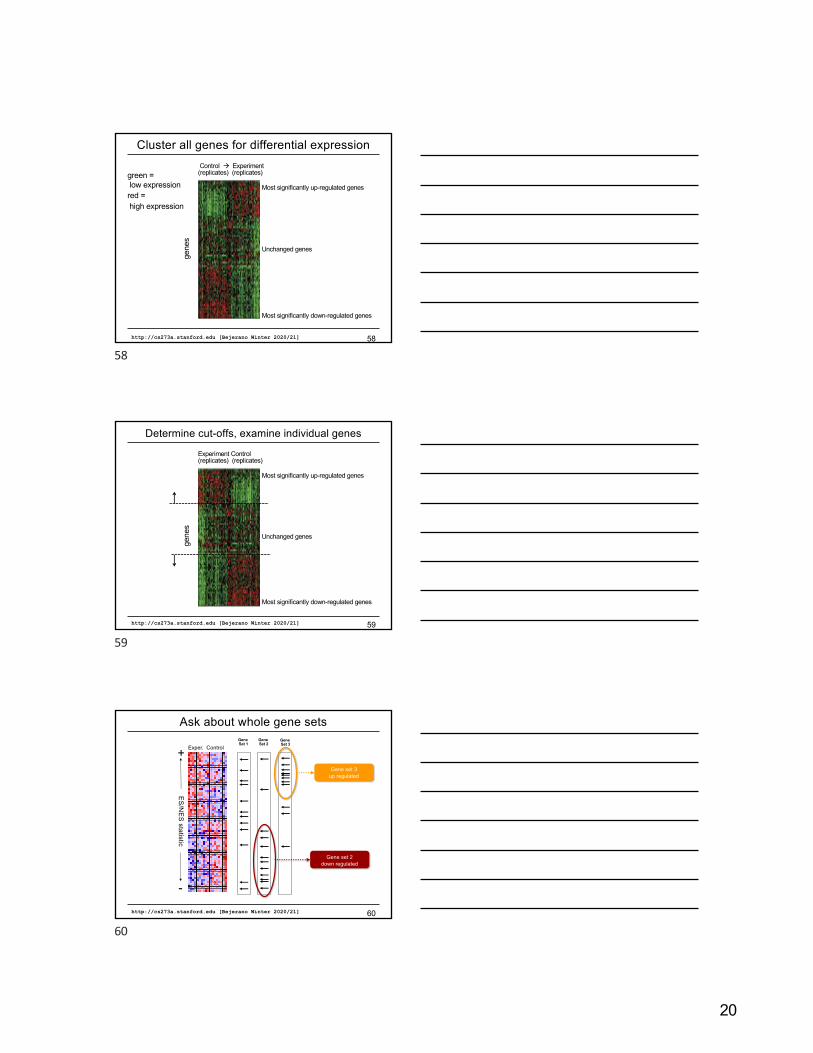
20
Cluster all genes for differential expression
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 58
Most significantly up-regulated genes
Unchanged genes
Most significantly down-regulated genes
Control à Experiment(replicates) (replicates)
gene
sgreen =low expressionred =high expression
58
Determine cut-offs, examine individual genes
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 59
Most significantly up-regulated genes
Unchanged genes
Most significantly down-regulated genes
Experiment Control(replicates) (replicates)
gene
s
59E
S/N
ES
statistic
+
-
Exper. ControlGene Set 1
Gene Set 2
Gene Set 3
Gene set 3up regulated
Gene set 2down regulated
Ask about whole gene sets
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 60
60

21
ES
/NE
S statistic
+
-
Exper. ControlGene Set 3
Gene set 3up regulated
Simplest way to ask: Hypergeometric
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 61
Genes measuredN = 20,000Total genes in set 3K = 11 I’ve picked the topn = 100 diff. expressed genes.Of them k = 8belong to gene set 3.
Under a null of randomlydistributed genes, howsurprising is it?
P-value = Prhyper (k ≥ 8 | N, K, n)
A low p-value, as here, suggests gene set 3 is highly enriched among the diff. expressed genes. Now see what (pathway/process) gene set 3 represents, and build a novel testable model around your observations.
(Test assumes all genes are independent. One can devise more complicated tests)
61
GSEA (Gene Set Enrichment Analysis)
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 62
Dataset distributionN
umber of genes
G ene Expression Level
Gene set 3 distribution
Gene set 1 distribution
62
Multiple Testing Correction
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 63
Note that statistically you cannot just run individual tests on 1,000 different gene sets. You have to apply further statistical corrections, to account for the fact that even in 1,000 random experiments a handful may come out good by chance.(eg experiment = Throw a coin 10 times. Ask if it is biased.If you repeat it 1,000 times, you will eventually get an all heads series from a fair coin. Mustn’t deduce that the coin is biased)
run tool
63

22
RNA-seq“Next” (2nd) generation sequencing.
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 64
64
What will you test?
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 65
Also note that this is a very general approach to test gene lists.Instead of a microarray experiment you can do RNA-seq.Advantage: RNA-seq measures all genes(up to your ability to correctly reconstruct them). Microarrays only measure the probes you can fit on them. (Some genes, or indeed entire pathways, may be missing from some microarray designs).
run tool
65
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 66
Single gene in situ hybridization
Sall1
Recall a human is 1013 cells living for 109 seconds.Which cells will you assay? When?
66

23
Spatial-temporal maps generation
http://cs273a.stanford.edu [Bejerano Winter 2020/21] 67
AI:Robotics,Vision
67





























