Embed Size (px)
Citation preview

1
¿Cuando sospecho una Hemoglobinuria
Paroxística Nocturna?
Andrés L. Brodsky, MD
Hospital de Clínicas
Universidad de Buenos Aires

2
¿Qué es la H.P.N.?
Enfermedad clonal no maligna de la hemopoyesis
Poco frecuente: 1,3 casos con clon HPN/1.000.000 hab./año
Clon HPN > 10%: 0,56/1.000.000 hab./año
Caracterizada por
1) Anemia hemolítica
2) Hemoglobinuria
3) + grado variable de fallo medular
4) + trombofilia

3
¿Cómo se origina y que genera el clon HPN?
Una mutación en el gen PIG-A en una
stem cell hemopoyética impide la síntesis
del ancla glicosilfosfatidilinositol (GPI)1
1. Brodsky RA. Hematology - Basic Principles and Practices. 4th ed. Philadelphia, PA: Elsevier Churchill
Livingstone; 2005:419-427. 2. Kelly R, et al. Ther Clin Risk Manag. 2009;5:911-921. 3. Rother RP, et al. JAMA.
2005;293(13):1653-1662. 4. Hill A, et al. Blood. 2013;121(25):4985-4996. 5. Bessler M, et al. Hematology Am Soc
Hematol Educ Program. 2008:104-110.
PIG-A PIG-A mutado
Ancla
GPI
Proteína con
anclaje GPI
Amplificación local sin
control del complemento
Células
intactas
Hemólisis3
Depleción de óxido nítrico3
Trombosis4
Daño de órganos blanco 5
Las células HPN carecen de proteínas
con anclaje GPI (GPI-AP), incluyendo
los reguladores del complemento CD55 y
CD591
La ausencia de los reguladores de
membrana genera una amplificación
local del complemento fuera de control2
1.
2.
3.
4. Morbilidades
generadas por
Esta sobreactivación local del
complemento produce múltiples daños
celulares y orgánicos propios de la HPN2
Células
deficientes en
GPI-AP

4
¿Por qué el clon HPN persiste pese a ser tan
sensible al complemento?
Las mutación de PIG-A es necesaria pero no suficiente para causar la HPN
Las stem cells hemopoyéticas deficientes en GPI-AP persisten y se expanden en una
médula ósea patológica
Inoue N, et al. Int J Hematol. 2003;77(2):107-112.
Gs Rs
Monocitos
Plaquetas
Granulocitos
Linfocitos
Paso 1 Paso 2
Stem cells
hemopoyéticas
Stem cells deficientes
en GPI-AP Células
seleccionadas
Mutación somática en PIG-A
Ataque inmunológico
Selección inmune del clon HPN
Deficiente en GPI-AP
Intacta

5
¿Por qué se expande el clon HPN hasta dar
manifestaciones clínicas?
La necesidad de ambas, selección y expansión, explicaría la rareza de la HPN
Adaptado de: Inoue N et al. Int J Hematol 2003;77:107.
Stem Cells
Hematopoyéticas
Normales
Deficiencia del
Ancla GPI-
Célula Deficiente en
GPI- Células Seleccionadas Células Expandidas
Selección
Inmunológica
Expansión símil tumor
benigno
±3er Paso
2a Mutación:
Ventaja
Proliferativa
2do Paso
Ataque
Inmunológico
Daño Selectivo
1er Paso
Mutación
Somática
del PIG-A

6
Guías Argentinas: tamaño del clon HPN y
formas clínicas

8
Consecuencias de la falta de reguladores del
complemento en las células HPN
Eritrocitos H.P.N.: HEMOLISIS INTRAVASCULAR
Anemia
Hemoglobina libre en plasma
Disminución de NO
Sobrecarga renal de hierro y pigmentos
Plaquetas H.P.N.: activación y generación de micropartículas
Gs. Bs. H.P.N.: activación
Generación de micropartículas
Producción de citokinas
Complejo de Ataque de Membrana
(MAC) en un Eritrocito HPN Foto: W. Rosse. Reproducida con Permiso.

9
Factores que afectan la hemólisis en HPN
La severidad de la hemólisis mediada por complemento1 en la HPN depende de
1. Hill A, et al. Br J Haematol. 2007;137(3):181-192. 2. Brodsky RA. Hematology - Basic Principles and Practices. 4th ed.
Philadelphia, PA: Elsevier Churchill Livingstone; 2005:419-427. 3. Parker CJ. Hematology Am Soc Hematol Educ Program.
2011;2011:21-29. 4. Rosse WF. Blood. 1971;37(5):556-562. 5. Rachidi S, et al. Eur. J. Intern. Med. 2010;21(4):260-267. 6.
Pu JJ, et al. Clin. Transl Sci. 2011;4(3):219-224.
La expansión del clon deficiente en GPI difiere entre los pacientes
Expresión de CD59 Ninguna
(Células tipo III)
Vida media aproximada 4,5
Reducida
(Células tipo II)
Normal
(Células tipo I)
8-10 días 30-40 días 90-120 días
Proporción de
hematíes deficientes
en el ancla GPI3
Grado de deficiencia
del ancla GPI3
Nivel de
activación del
complemento 6
Activación continua espontánea “tick-over” (vía alterna)
Eventos que amplifican el complemento: infecciones,
transfusiones de sangre, cirugías, ingesta alcohólica, ejercicios
extenuantes, embarazo, traumatismos, quemaduras
3.
2.
1. 10% 50% Tamaño clonal 90%

13
La activación no controlada del complemento causa
vasoconstricción y trombosis por múltiples vías
Adapted from: Gladwin MT et al. Free Rad Biol & Med 2004;36:707–717; Rother RP et al. JAMA 2005;293:1653–1662.
COAGULO
Activación
Plaquetaria
Activación
Leucocitaria
Activación
Crónica
no controlada del
complemento
C5a C5b-9
C5b-9 [↓↓NO]
Inflamación
Agregación
Plaquetaria
Hemólisis
Crónica
Inflamación
Injuria celular
endotelial
Activación
plaquetaria
Regulación alterada del
músculo liso
Vasoconstricción local
Efecto pro-inflamatorio
en células endoteliales
C5b-9
C5a
Citokinas

14
Características clínicas de la trombosis en HPN
FRECUENTE: 20-40% de los pacientes sufre trombosis1
GRAVE: Responsable del 40%-67% de los decesos con causa
conocida en HPN1
El primer evento trombótico puede ser fatal1
– Si sobrevive, el riesgo de muerte aumenta 5 a 10 veces
EN SITIOS INUSUALES: Localizaciones frecuentes
incluyen las venas intra-abdominales y cerebrales2
– Las venas hepáticas (sme. de Budd-Chiari) son afectadas en
entre el 7,25% y el 25% de los pacientes
– La TVP de miembros inferiores afecta ~33% de los pacientes
– La incidencia de trombosis arterial (cerebral y coronaria) está
aumentada
REFRACTARIA: La anticoagulación no suele prevenirlas1,2,3
Angiorresonancia
magnética de paciente con
HPN y trombosis de senos
longitudinal superior y
transverso izquierdo que
recibe circulación
colateral3
1. Hill A, et al. Blood. 2013;121(25):4985-4996.
2. Hillmen P, et al. Blood. 2007;110(12):4123-4128.
3. Brodsky A, Mazzocchi O, Sanchez F, et al. Exp Hematol Oncol. 2012;1:26.

16
La activación descontrolada del complemento
genera daño renal
La hemoglobina libre surgida de la hemólisis intravascular causa daño tubular renal1,2
La sobrecarga de los túbulos renales con hemosiderina genera stress oxidativo
Las trombosis de los pequeños vasos renales contribuyen por isquemia al daño renal
1. Rother RP, et al. JAMA. 2005;293(13):1653-1662.
2. Clark DA, et al. Blood. 1981;57(1):83-89.
Depósitos de hemosiderina
en la corteza renal por RNM
Microfotografía de la biopsia renal de un
paciente con HPN, indicativa de daño vascular2
Fibrosis
intersticial a la
izquierda
Tejido normal
a la derecha

17
Hillmen P, et al. Am J Hematol. 2010;85(8):553-559.
Enfermedad renal crónica en HPN
64% de los pacientes con HPN
presentan estadíos 1 a 5 de ERC
– 21% con estadío 3 o 4 o con fallo
renal (estadío 5)
– Los pacientes que requieren
mínimas transfusiones tienen una
distribución de disfunción renal
similar
El fallo renal es causa del 8%-
18% de los decesos relacionados a
la HPN
0
10
20
30
40
50
60
70
ERC global Estadíos 1-2
de ERC
Estadíos 3-5
de ERC
Sin ERC
Pro
porc
ión
de P
acie
nte
s, %
64%
Distribución de la disfunción renal en los pacientes
con HPN del programa de ensayos clínicos
43%
21%
36%
18
1. Rother R. et al. JAMA. 2005; 293: 1653-1662.
2. Hill A, et al. Br J Haematol. 2012;158(3):409-414.
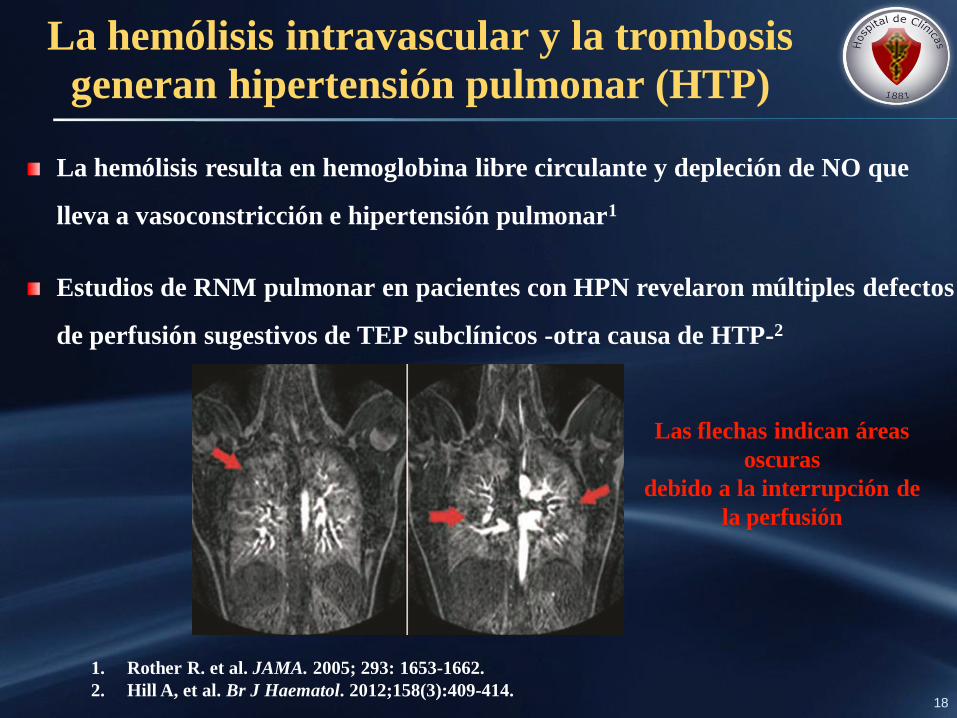
La hemólisis resulta en hemoglobina libre circulante y depleción de NO que
lleva a vasoconstricción e hipertensión pulmonar1
Estudios de RNM pulmonar en pacientes con HPN revelaron múltiples defectos
de perfusión sugestivos de TEP subclínicos -otra causa de HTP-2
La hemólisis intravascular y la trombosis
generan hipertensión pulmonar (HTP)
Las flechas indican áreas
oscuras
debido a la interrupción de
la perfusión

19 Weitz I et al. J Intern Med 2012.
n=29
La mayoría de los pacientes refirió los síntomas de moderados a muy severos
Síntomas frecuentes asociados a
la hemólisis intravascular
59% de los pacientes estaban libres de transfusiones durante al menos 12 meses, o nunca habían sido transfundidos
76% se vieron obligados a modificar sus actividades diarias para manejar su HPN
17% estaban desempleados debido a la HPN

20
Citopenias/evolución clonal:
Evidencia de la mielopatía subyacente en HPN
Citopenias
Hb < 10 g/dL
Plaquetas < 80.000/L
Neutrófilos < 1.000/L
Prevalencia de ≥ 2 citopenias al diagnóstico (AA-HPN): 52 %
Pancitopenia: 39,1 %
Evolución a bi o tricitopenia: 19,2 % a 10 años
Evolución clonal a SMD: 5,2 % a 10 años
Evolución clonal a LMA: 2,4 % a 10 años
Peffault de Latour R. Blood 2008 p. 3102.

22
HPN puede ser difícil de diagnosticar
Las demoras en el diagnóstico oscilan de 1 a más de 10 años1
Signos o Síntomas Tasa de Incidencia (%)
Fatiga, deterioro de la calidad de vida 96%2
Anemia 88%4
Disnea 66%2
Deterioro de la Función Renal 64%3
Dolor Abdominal 57%2
Disfunción Eréctil 47%2
Disfagia 41%2
Trombosis 40%1
Hemoglobinuria (al debut) 26%5
1. Hillmen P et al. N Engl J Med. 1995;333:1253-8. 2. Meyers G et al. Blood. 2007;110(11):Abstract 3683. 3. Hillmen P
et al. Blood (ASH Annual Meeting Abstracts). 2007;110:abstract 3678. 4. Nishimura J et al. Medicine. 2004;83(3):193-
207. 5. Parker C et a. Blood. 2005;106(12):3699-3709.

23
¿A quién buscar clon HPN?: Guías ICCS
Borowitz MJ et al; Clinical Cytometry Society. Cytom B Clin Cytom 2010;78B:211–230
Indicaciones clínicas para el estudio de HPN
1a) Hemólisis intravascular evidenciada por hemoglobinuria o hemoglobina libre en plasma elevada
1b) Hemólisis
Inexplicable y acompañada de:
- Deficiencia de hierro, o
- Dolor abdominal o espasmo esofágico, o
- Trombosis, o
- Granulocitopenia y/o trombocitopenia
Adquirida, Coombs-negativa, no esquistocítica y no infecciosa
2) Trombosis con características inusuales
Sitios inusuales:
- Venas hepáticas (síndrome de Budd-Chiari)
- Otras venas intra-abdominales (portal, esplénica, mesentérica)
- Senos cerebrales
- Venas dérmicas
Con signos de anemia hemolítica acompañante
Con citopenia inexplicable
3) Citopenias por fallo de la médula ósea
Anemia aplásica o hipoplásica sospechada o comprobada
Citopenia refractaria con displasia unilineal
Otras citopenias de etiología desconocida después de un estudio adecuado

24
¿Cómo se diagnostica la H.P.N.?
H.P.N. clínica
1) Histórico: activación de la vía alterna del complemento→lisis de los hematíes
Por ↓ pH: test de Ham
Por ↑ osmolaridad: test de sucrosa
(+) si >20% de los glóbulos rojos son del clon H.P.N.
2) Actual: por citometría de flujo (CMF)
Disminución o ausencia de ≥ 2 antígenos con anclaje GPI en ≥ 2 líneas celulares
hemáticas
(+) si > 1% de los glóbulos son del clon H.P.N.
H.P.N. subclínica
CMF de alta sensibilidad: (+) con clones ≥ 1/10.000 células

25
Caso clínico
ANTECEDENTES
Amigdalectomía s/ complicaciones (2003): 17 años
Cirugía de folículo ovárico hemorrágico (26/2/06): se detecta trombocitopenia
Diagnóstico de PTI. Inicia 60 mg/d de meprednisona + ácido fólico

26
Corticoterapia y evolución
7/3/06 9/5/06 6/9/06 13/12/06 30/5/07 21/9/07 3/12/07
Hb 10 13,9 13,9 12,9 13 11,9 11,8
VCM 93 100,5 97 98 99 102 104
Gs Bs 7.380 8.660 6.400 6.300 5.700 4.600 3.380
Ns/µL 4.180 3.516 2.432 2.205 1.824 2.024 1.420
Plaq/µL 45.700 82.500 137.000 118.000 61.000 58.000 42.000
MP mg/d 50 30 30/0 14/0 8/0 3 0

27
Caso clínico
EVOLUCION
Aumento tardío y parcial del rto. plaquetario
Corticoides en lento descenso
Trombocitopenia progresiva
Aparición de macrocitosis
Disminución del recuento leucocitario

28
Estudio y seguimiento del fallo medular
6/2/08 3/4/08 27/1/09 29/10/10
Hb 10,2 9,6 10,5 8,6
VCM 107 107 106 107
Gs Bs 3.500 5.100 7.400 2.300
Ns/µL 735 1.836 2.664 920
Plaq/µL 17.000 18.000 30.000 40.000
LDH (≤480) 284
Medicac. G-CSF
Dexa 10 mg/d
CSA
MP
EPO
EPO
G-CSF
1) ¿Efectuaría una CMF de sangre periférica para
detectar HPN?
2) ¿Qué datos de la paciente apoyan el estudio?
Tricitopenia no explicada (x fallo medular en
plaquetas)

29
Estudio y seguimiento del fallo medular
ESTUDIOS
Biopsia de M. O. (24/1/08): celularidad del 20 a 35%. Diagnóstico: hipoplasia
Citometría de Flujo de M. O. (23/7/08): 0,62% de células CD34 (+). Sin
alteraciones fenotípicas
FISH para +8, -5 y -7 (23/7/08): negativos
Biopsia de M. O. (5/5/10): celularidad del 40%. Disminución de la serie
mieloide y de megacariocitos, sin fibrosis
Citometría de Flujo: 0,38% de células CD34 (+). Sin alteraciones fenotípicas
Citogenético: cariotipo femenino normal 46XX (15)

30
Estudio y seguimiento del fallo medular
6/1/11 1/11/11 11/1/12
Hb 7,8 8,9 7,8
VCM 104 100
Gs Bs 3.400 4.300 4.500
Ns/µL 1.292 2.064 2.520
Plaq/µL 37.000 69.000 80.000
LDH (≤480) 2.681
Medicación
Micofenolato
EPO
G-CSF
Micofenolato Fe + B12 +
Folato
Mejora la
neutropenia
Mejora la
plaquetopenia
Se acentúa la
anemia
Se eleva la LDH

31
EVOLUCION
24/1/12: Astenia + adinamia + epigastralgia
Hb 5,8 g/dL + orinas oscuras
Transfusión de GR
Citometría de Flujo de S. P. (24/1/12)
De hipoplasia medular a HPN
Tipo III Tipo II Tipo I
Granulocitos (CD24; 16; 66b) 93% 7%
Monocitos (CD14) 93% 7%
Glóbulos Rojos (CD59) 74% 8% 18%
¡¡BUSCAR CLONES HPN EN APLASIAS MEDULARES!!

35
¡¡MUCHAS GRACIAS!!
POR SU ATENCION:





























