Embed Size (px)
Citation preview

1 © 2017 Vertex Pharmaceuticals Incorporated
CYSTIC FIBROSIS (CF):THE CHALLENGE OF EARLY, SYSTEMIC PROGRESSIONA Program for the CF Center Care Team
Vertex Pharmaceuticals Incorporated, 50 Northern Avenue, Boston, MA 02210. Vertex and the Vertex triangle logo are registeredtrademarks for Vertex Pharmaceuticals Incorporated. © 2017 Vertex Pharmaceuticals Incorporated | VXR-US-20-01898 | 10/2017

2 © 2017 Vertex Pharmaceuticals Incorporated
Program ObjectivesImprove your understanding of:
• What causes CF and its cellular effects
• How CF progresses in different organs of the body
• The importance of monitoring disease progression throughout a patient’s life

3 © 2017 Vertex Pharmaceuticals Incorporated
CF and its Progression
A challenge throughout the patient’s care journey

4 © 2017 Vertex Pharmaceuticals Incorporated
In CF, mutations in the CFTR gene affect CFTR protein production• 281 known CFTR mutations can result in CF due to too few and/or defective CFTR proteins1-3
–A person needs 2 CF-causing mutations to have CF2
• With too few and/or defective CFTR proteins, the balance of water and salt at epithelial cell surfaces is disrupted2,4-6
• Mucus becomes thick and sticky in organs throughout the body, and it can clog small passages2,4-6
–This interferes with the proper function of the lungs, pancreas, gastrointestinal system, sinuses, liver, and reproductive system
CFTR, cystic fibrosis transmembrane conductance regulator.
References: 1. Castellani C et al. J Cyst Fibros. 2008;7(3):179-196. 2. Zielenski J. Respiration. 2000;67(2):117-133. 3. The Clinical and Functional TRanslation of CFTR (CFTR2); available at http://www.cftr2.org. List of CFTR2 mutations. https://www.cftr2.org/sites/default/files/CFTR2_17March2017.xlsx. Accessed July 31, 2017. 4. Davis PB. Am J Respir Crit Care Med. 2006;173(5):475-482. 5. Welsh MJ et al. Membrane transport disorders: cystic fibrosis. In: Valle D, Beaudet A, Vogelstein B, et al, eds. The Online Metabolic & Molecular Bases of Inherited Disease. The McGraw-Hill Companies, Inc.; 2004:part 21, chap 201. 6. O’Sullivan BP, Freedman SD. Lancet. 2009;373(9678):1891-1904.
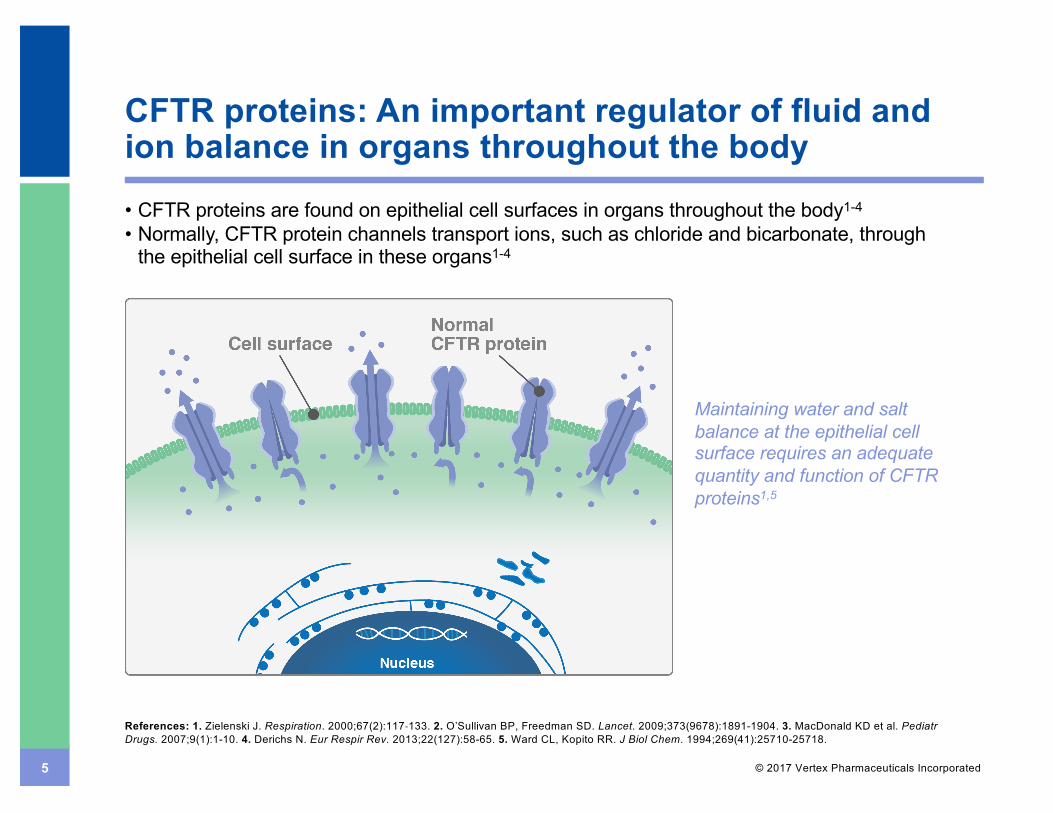
5 © 2017 Vertex Pharmaceuticals Incorporated
CFTR proteins: An important regulator of fluid and ion balance in organs throughout the body• CFTR proteins are found on epithelial cell surfaces in organs throughout the body1-4
• Normally, CFTR protein channels transport ions, such as chloride and bicarbonate, through the epithelial cell surface in these organs1-4
Maintaining water and salt balance at the epithelial cell surface requires an adequate quantity and function of CFTR proteins1,5
References: 1. Zielenski J. Respiration. 2000;67(2):117-133. 2. O’Sullivan BP, Freedman SD. Lancet. 2009;373(9678):1891-1904. 3. MacDonald KD et al. PediatrDrugs. 2007;9(1):1-10. 4. Derichs N. Eur Respir Rev. 2013;22(127):58-65. 5. Ward CL, Kopito RR. J Biol Chem. 1994;269(41):25710-25718.
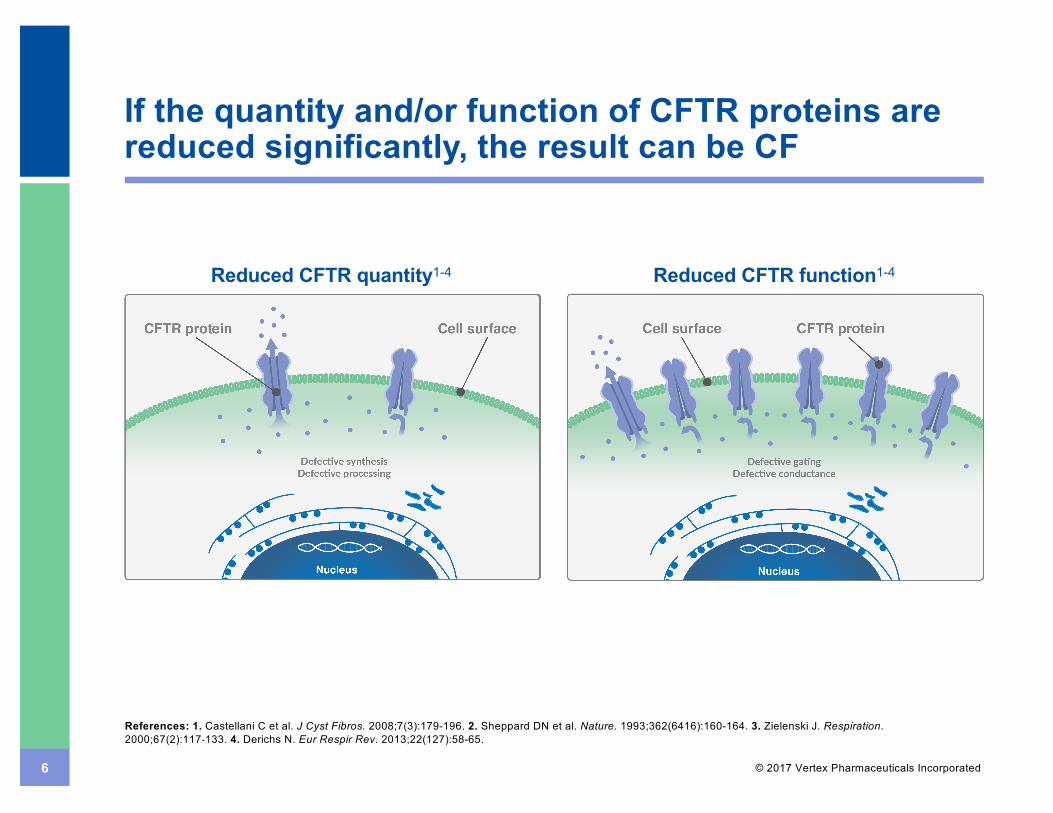
6 © 2017 Vertex Pharmaceuticals Incorporated
If the quantity and/or function of CFTR proteins are reduced significantly, the result can be CF
References: 1. Castellani C et al. J Cyst Fibros. 2008;7(3):179-196. 2. Sheppard DN et al. Nature. 1993;362(6416):160-164. 3. Zielenski J. Respiration. 2000;67(2):117-133. 4. Derichs N. Eur Respir Rev. 2013;22(127):58-65.
Reduced CFTR quantity1-4 Reduced CFTR function1-4

7 © 2017 Vertex Pharmaceuticals Incorporated
Too few or defective CFTR proteins means a cascade of disease and lung damage• The cascade of CF in the lungs can result in inflammation, infection, and lung damage, even
before the loss of lung function is detected by spirometry1-4
–Damage can be cumulative over time1,2,5,6
• A similar cascade of events with CFTR dysfunction causes thickening of secretions, blocking of small passages, inflammation, and damage in other organs1,2,7-9
References: 1. O’Sullivan BP, Freedman SD. Lancet. 2009;373(9678):1891-1904. 2. Elborn JS. Lancet. 2016;388(10059):2519-2531. 3. Rao S, Grigg J. Arch Dis Child.2006;91:786–788. 4. Levy H et al. Pediatr Pulmonol. 2007;42(3):256–262. 5. Farrell PM et al. Radiology. 2009;252(2):534-543. 6. Smith L et al. Am J Respir Crit Care Med. 2017. doi:10.1164/rccm.201705-0894LE. [Epub ahead of print.] 7. Cunningham JC, Taussig LM. An introduction to cystic fibrosis for patients and their families. Cystic Fibrosis Foundation. 2013. 8. Orenstein DM et al. Cystic fibrosis: a guide for patient and family. 4th Ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2012. 9. Ooi CY, Durie PR. J Cyst Fibros. 2012;11(5):355-362.
THE CFTR DYSFUNCTION CASCADE IN THE LUNGS2

8 © 2017 Vertex Pharmaceuticals Incorporated
Affects many of the patient’s organs, throughout their lifetime
CF and its Progression
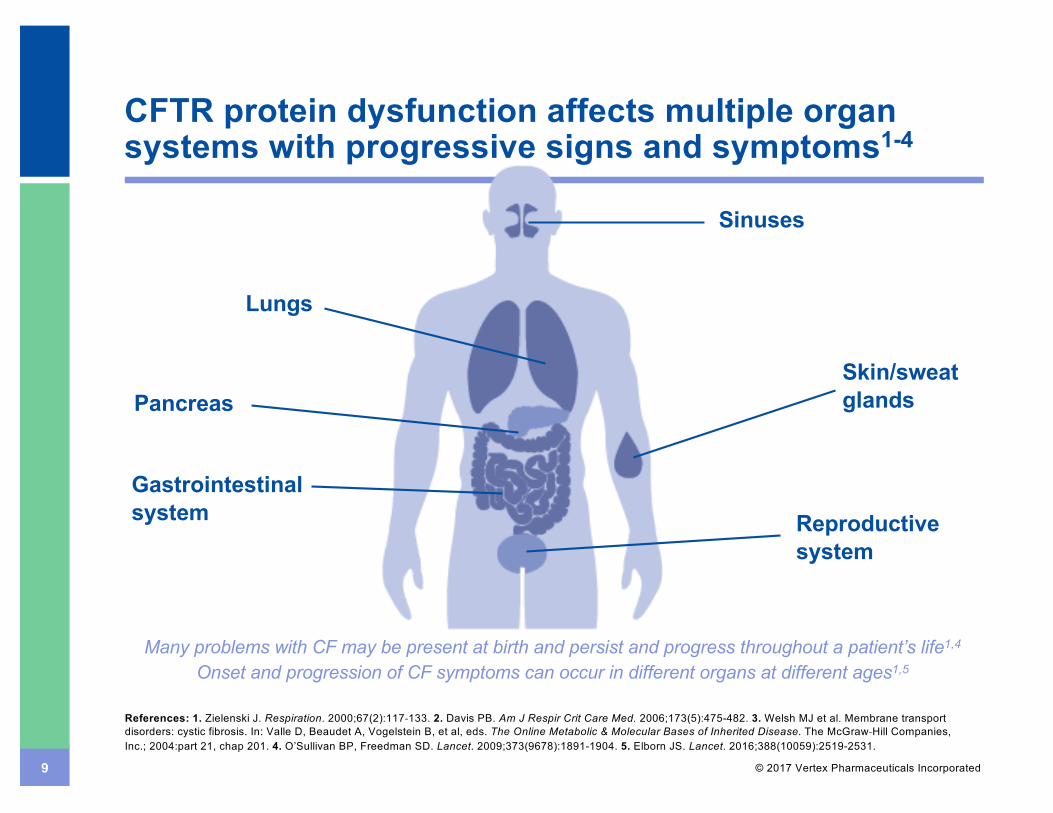
9 © 2017 Vertex Pharmaceuticals Incorporated
CFTR protein dysfunction affects multiple organ systems with progressive signs and symptoms1-4
References: 1. Zielenski J. Respiration. 2000;67(2):117-133. 2. Davis PB. Am J Respir Crit Care Med. 2006;173(5):475-482. 3. Welsh MJ et al. Membrane transport disorders: cystic fibrosis. In: Valle D, Beaudet A, Vogelstein B, et al, eds. The Online Metabolic & Molecular Bases of Inherited Disease. The McGraw-Hill Companies, Inc.; 2004:part 21, chap 201. 4. O’Sullivan BP, Freedman SD. Lancet. 2009;373(9678):1891-1904. 5. Elborn JS. Lancet. 2016;388(10059):2519-2531.
Sinuses
Gastrointestinal system Reproductive
system
Skin/sweat glandsPancreas
Lungs
Many problems with CF may be present at birth and persist and progress throughout a patient’s life1,4
Onset and progression of CF symptoms can occur in different organs at different ages1,5

10 © 2017 Vertex Pharmaceuticals Incorporated
Potentially irreversible lung damage can begin early in life
References: 1. Elborn JS. Lancet. 2016;388(10059):2519-2531. 2. Sly PD et al. N Engl J Med. 2013;368(21):1963-1970. 3. Byrnes CA et al. Thorax. 2013;68(7):643-651. 4. Kraemer R et al. Am J Respir Crit Care Med. 2005;171(4):371-378. 5. Rowan SA et al. Am J Respir Crit Care Med. 2014;189(5):586-592.
• Mucus plugging and thickening of airways (bronchiectasis) often occurs in infants and young children (age 0-5 years)1,2
–Bronchiectasis advances as children age and is typically established before age 20 years1
• Pulmonary exacerbations become more common (age 6-10 years)2,3
• Small airway damage has been seen in infants and children, often despite normal-appearing lung function2,4,5
• The predominant infection in small children is Staphylococcus aureus, but increasingly becomes Pseudomonas aeruginosa beginning in adolescence1
• In adult patients, established bronchiectasis with hemoptysis and pneumothorax may occur1
• Eventually, patients develop progressive respiratory failure and may need lung transplant1
Childhood Mucus plugging, infections, beginning lung damage1
Adolescence Progressive lung disease1
Adulthood Eventual lung failure1
Lifelong Pulmonary exacerbations2,3
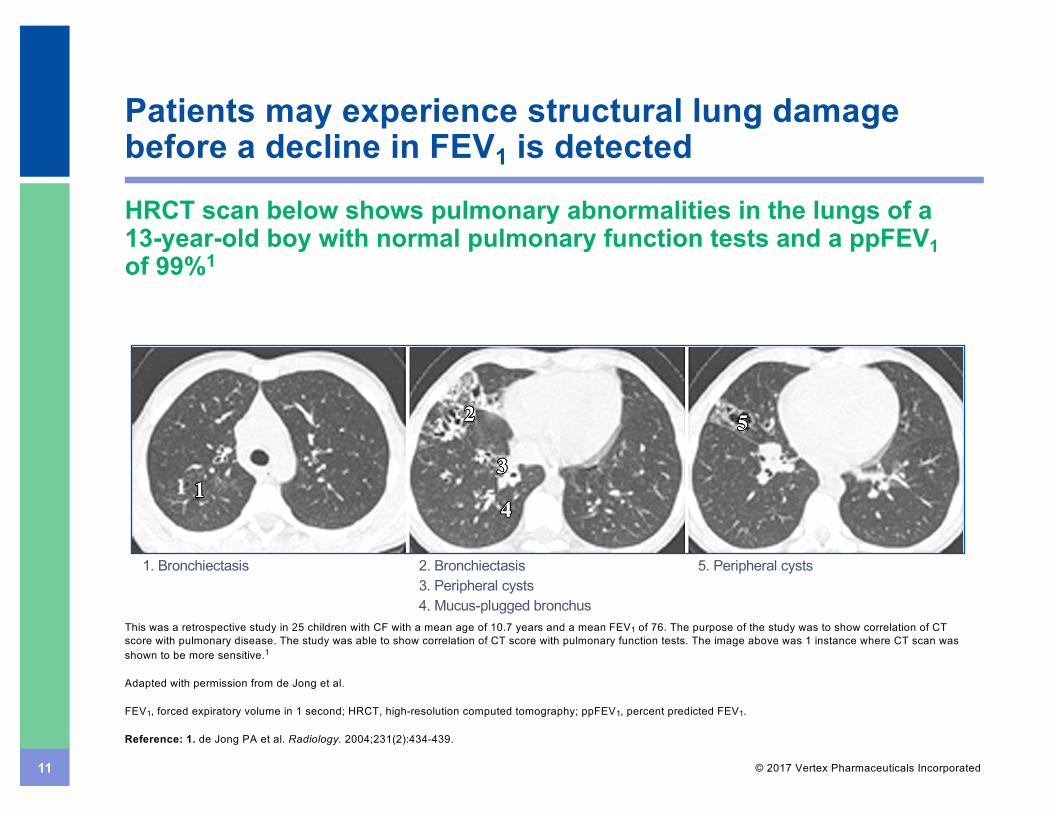
11 © 2017 Vertex Pharmaceuticals Incorporated
Patients may experience structural lung damage before a decline in FEV1 is detected
HRCT scan below shows pulmonary abnormalities in the lungs of a 13-year-old boy with normal pulmonary function tests and a ppFEV1of 99%1
This was a retrospective study in 25 children with CF with a mean age of 10.7 years and a mean FEV1 of 76. The purpose of the study was to show correlation of CT score with pulmonary disease. The study was able to show correlation of CT score with pulmonary function tests. The image above was 1 instance where CT scan was shown to be more sensitive.1
Adapted with permission from de Jong et al.
FEV1, forced expiratory volume in 1 second; HRCT, high-resolution computed tomography; ppFEV1, percent predicted FEV1.
Reference: 1. de Jong PA et al. Radiology. 2004;231(2):434-439.
1. Bronchiectasis 5. Peripheral cysts2. Bronchiectasis3. Peripheral cysts4. Mucus-plugged bronchus

12 © 2017 Vertex Pharmaceuticals Incorporated
LCI is sensitive at detecting early CF airway disease
A significant linear correlation was observed between CT score and LCI score1*
Reprinted from Respir Med, 104(12), Ellemunter H, Fuchs SI, Unsinn KM, et al, Sensitivity of lung clearance index and chest computed tomography in early CF lung disease, 1834-1842, Copyright 2010, with permission from Elsevier.
*138 patients from the Medical University of Innsbruck were screened to identify patients with normal FEV1, defined as the annual average >80% predicted normal value at a minimum of 4 lung function measurements per year. Forty-one patients were asked to participate. Thirty-four patients age 6-26 years (mean age 14 years) with early lung disease shown on CT scan were assessed; 59% were F508del homozygous and 35% were F508del heterozygous.1
LCI, lung clearance index.
References: 1. Ellemunter H et al. Respir Med. 2010;104(12):1834-1842. 2. Davies JC et al. Thorax. 2008;63(2):96-97. 3. Kent L et al. J Cyst Fibros. 2014;13(2):123-138.
• LCI shows a strong correlation with CT scan for verification of early disease1
• In the same study, FEV1 did not detect the presence of abnormalities observed on CT scan1
–LCI is more sensitive to damage to the small peripheral airways than FEV1
2
• LCI is still considered an exploratory endpoint in clinical trials3
LCI z-score vs CT score1

13 © 2017 Vertex Pharmaceuticals Incorporated
Sinusitis can be a lifelong problem
• The sinuses are considered to be a major reservoir for bacteria that later infect the lungs1
–Chronic rhinosinusitis and increased incidence of nasal polyps occur in almost all patients with CF due to increased mucus viscosity–Sinusitis often precedes the development of
progressive lung disease in CF
The sinuses may be a reservoir for lung pathogens
Reference: 1. Illing EA, Woodworth BA. Curr Opin Pulm Med. 2014;20(6):623-631.
Lifelong Chronic rhinosinusitis, nasal polyps1

14 © 2017 Vertex Pharmaceuticals Incorporated
Pancreatic problems are very common and progressive• As many as 90% of people with CF have impaired pancreatic
function (pancreatic insufficiency) at birth or shortly afterward1,2
–The rest are considered pancreatic sufficient3
• In pancreatic insufficiency, mucus obstructs pancreatic ducts, impedes function, and leads to progressive damage2
–Such obstruction can begin even before birth1
–These patients with pancreatic insufficiency have a hard time digesting and absorbing food, resulting in poor growth and health2
–Over time, the pancreas undergoes autolysis (destruction) with replacement of the body of the pancreas with fat2
• Up to 40% of adults with CF will develop CF-related diabetes mellitus1
– Insulin insufficiency and carbohydrate intolerancefollow autolysis, marking the development of CF-relateddiabetes mellitus2
• Among the rest (patients with CF who are pancreatic sufficient), 15% develop pancreatitis by age 20 years3
–By age 35 years, as many as 30% have pancreatitis
References: 1. Elborn JS. Lancet. 2016;388(10059):2519-2531. 2. O’Sullivan BP, Freedman SD. Lancet. 2009;373(9678):1891-1904. 3. Ooi CY, Durie PR. J Cyst Fibros. 2012;11(5):355-362.
Childhood Pancreatic insufficiency, poor nutrition and growth1-3
Adolescence Progressive pancreatic damage1,3
Adulthood CF-related diabetes, pancreatitis1,3

15 © 2017 Vertex Pharmaceuticals Incorporated
Difficulties in the gastrointestinal tract can affect nutrition
DIOS, distal intestinal obstruction syndrome.
Reference: 1. O’Sullivan BP, Freedman SD. Lancet. 2009;373(9678):1891-1904.
• Gastrointestinal consequences in adults with CF include1:–Thickened intestinal secretions–Decreased motility of food through the gut–Nutrient malabsorption
• DIOS or chronic constipation in older patients can occur1
• Nutrient malabsorption especially of fat-soluble vitamins can lead to1:–Acrodermatitis–Anemia –Neuropathy–Night blindness–Osteoporosis–Bleeding disorders
• There is also a risk of focal biliary cirrhosis caused by obstruction of intrahepatic bile ducts1
Childhood Poor gut motility and absorption of nutrients1
Later in life DIOS, focal biliary cirrhosis1
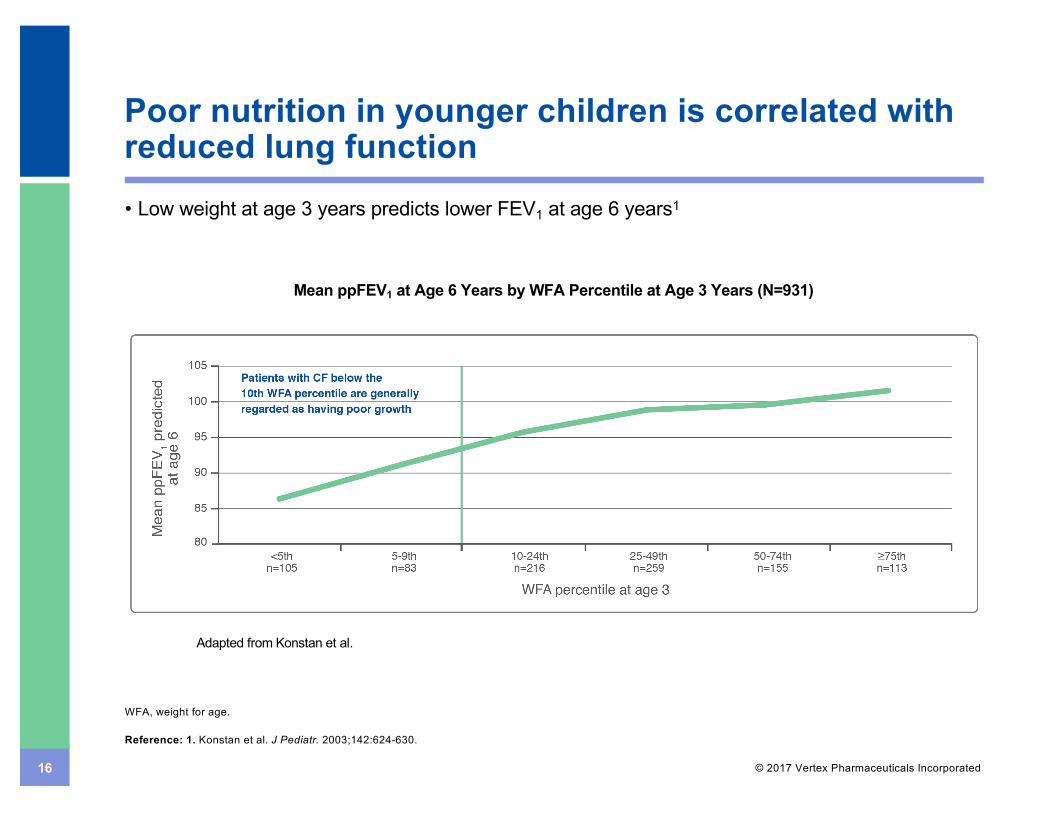
16 © 2017 Vertex Pharmaceuticals Incorporated
Poor nutrition in younger children is correlated with reduced lung function
WFA, weight for age.
Reference: 1. Konstan et al. J Pediatr. 2003;142:624-630.
Adapted from Konstan et al.
Mean ppFEV1 at Age 6 Years by WFA Percentile at Age 3 Years (N=931)
• Low weight at age 3 years predicts lower FEV1 at age 6 years1
17 © 2017 Vertex Pharmaceuticals Incorporated
In the liver, progressive biliary plugging and eventual liver damage can occur
References: 1. O’Sullivan BP, Freedman SD. Lancet. 2009;373(9678):1891-1904. 2. Cañas T et al. Biomed Res Int. 2015;2015:517369. doi:10.1155/2015/517369. [Epub ahead of print.] 3. Elborn JS. Lancet. 2016;388(10059):2519-2531.
• Dysfunctional or absent CFTR proteins appear to alter bile viscosity and cause mucosal obstruction of bile ducts1,2
–This can result in progressive biliary plugging and chronic cholestasis, biliary obstruction, inflammation, and structural damage2
• As early as age 10 years, patients may have abnormal liver function test results3
–5% of patients may develop biliary cirrhosis by age 15 years1,3
• As patients become adults, 5% to 10% additionally develop portal hypertension3
–Liver transplant may be required in some patients (typically age >35 years)
Childhood Abnormal liver function tests3
Adulthood Portal hypertension3
18 © 2017 Vertex Pharmaceuticals Incorporated
CF can cause fertility problems in both sexes
References: 1. Alves MG et al. Curr Drug Targets. 2015;16(9):993-1006. 2. Davis PB. Am J Respir Crit Care Med. 2006;173(5):475-482. 3. Elborn JS. Lancet. 2016;388(10059):2519-2531.
• 97% of males with CF have congenital bilateral absence of the vas deferens (CBAVD) and azoospermia1
–CBAVD can occur in men who do not have clinical CF but have a CFTR mutation
• Women can experience fertility impairment related to thick cervical mucus that fails to undergo the usual mid-cycle thinning2
Lifelong (males)
Infertility1,3
Adulthood (females)
Difficulty conceiving2
19 © 2017 Vertex Pharmaceuticals Incorporated
CF may impact important regulators of bone metabolism
References: 1. Marquette M, Haworth CS et al. Paediatr Respir Rev. 2016;20(suppl 2-5). doi:10.1016/j.prrv.2016.06.003. 2. Jacquot J et al. Osteoporos Int.2016;27(4):1401-1412. 3. Elborn JS. Lancet. 2016;388(10059):2519-2531. 4. O’Sullivan BP, Freedman DS. Lancet. 2009;373(9678):1891-1904.
• CFTR is expressed in bone, and CFTR dysfunction affects bone metabolism1,2
• Emergence of bone disease increases as patients get older3
• Between the ages of 20 and 35 years, patients with CF may demonstrate arthropathy and CF-related bone disease (osteoporosis)3
• Patients with CF are at increased risk of low bone density related trauma fractures2,4
• Nutritional status and chronic inflammation due to other CF-related effects may compound the problem2,4
Adulthood Arthropathy, osteoporosis, fractures2-4

20 © 2017 Vertex Pharmaceuticals Incorporated
Monitoring plays a critical role in disease management
All Patients with CF Experience Progression
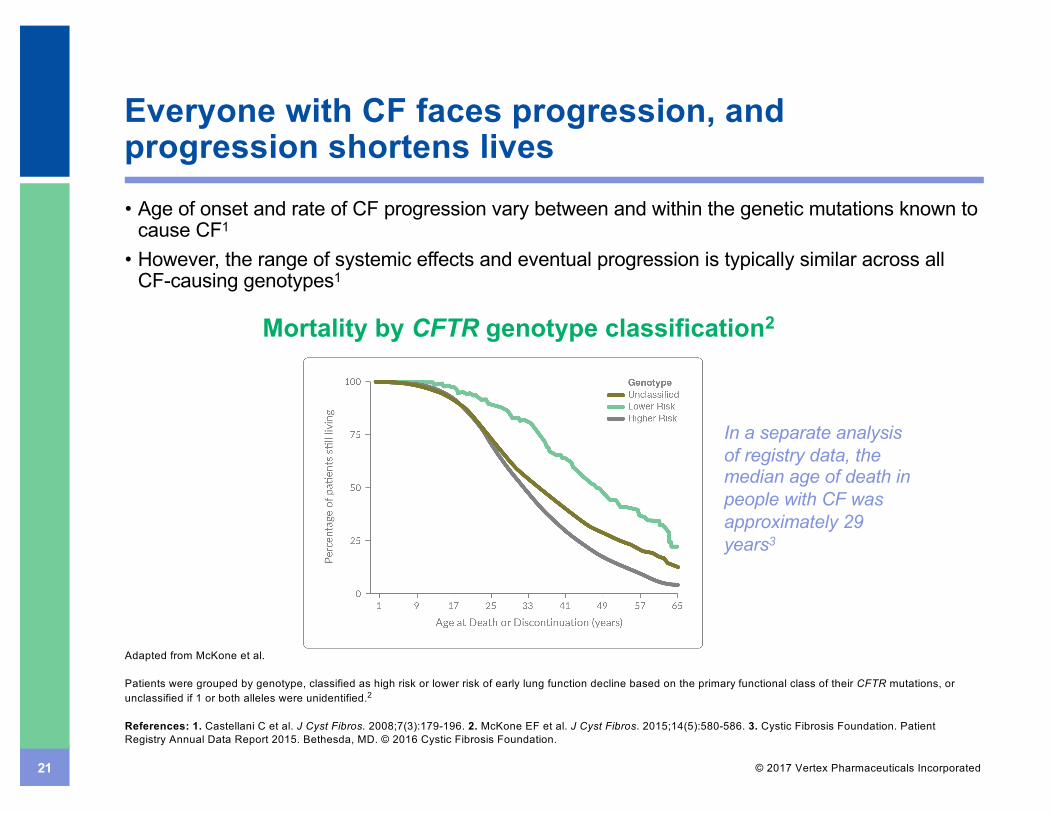
21 © 2017 Vertex Pharmaceuticals Incorporated
Everyone with CF faces progression, and progression shortens lives• Age of onset and rate of CF progression vary between and within the genetic mutations known to
cause CF1
• However, the range of systemic effects and eventual progression is typically similar across all CF-causing genotypes1
Adapted from McKone et al.
Patients were grouped by genotype, classified as high risk or lower risk of early lung function decline based on the primary functional class of their CFTR mutations, or unclassified if 1 or both alleles were unidentified.2
References: 1. Castellani C et al. J Cyst Fibros. 2008;7(3):179-196. 2. McKone EF et al. J Cyst Fibros. 2015;14(5):580-586. 3. Cystic Fibrosis Foundation. Patient Registry Annual Data Report 2015. Bethesda, MD. © 2016 Cystic Fibrosis Foundation.
Mortality by CFTR genotype classification2
In a separate analysis of registry data, the median age of death in people with CF was approximately 29 years3

22 © 2017 Vertex Pharmaceuticals Incorporated
Lung assessments remain fundamental to helping monitor disease progression
• HRCT–Radiation burden with frequent imaging may be a concern, especially in young children1
• Magnetic resonance imaging–Similar accuracy to CT without radiation risk2
–Relatively new technology; unproven in clinical practice2
• Spirometry (eg, FEV1, FVC)3-5
–Standard assessment of lung function, specifically large airways–Young children (<6 years) may find it difficult to perform–May be normal despite underlying small airway structural damage
• LCI3,6-8
–May be easier for young children (<6 years), requiring only passive breathing–Can detect underlying small airway lung damage even with normal FEV1–LCI is still considered an exploratory endpoint in clinical trials
Considerations when choosing lung assessments
FVC, forced vital capacity.
References: 1. Davies JC et al. Thorax. 2008;63(2):96-97. 2. Roach DJ et al. Ann Am Thorac Soc. 2016;13(11):1923-1931. 3. Lahiri T et al. Pediatrics. 2016 Apr;137(4). pii: e20151784. doi: 10.1542/peds.2015-1784. Epub 2016 Mar 23. 4. Jat KR. Prim Care Respir J. 2013;22(2):221-229. 5. Mall MA et al. Pediatr Pulmonol. 2016;51(S44):S49-S60. 6. Benseler A. Respirology. 2015;20(3):459-466. doi: 10.1111/resp.12470. Epub 2015 Jan 21. 7. Aurora P et al. Thorax. 2004;59:1068-1073. 8. Kent L et al. J Cyst Fibros. 2014;13(2):123-138.

23 © 2017 Vertex Pharmaceuticals Incorporated
A wide range of tests can help assess CF progression across organ systems
Organ Commonly used measures Considerations
Pancreas
Body mass index Standard assessment for the nutritional impact of CF, along with weight and height measurements1
Immunoreactive trypsinogen Increasingly used in neonatal screening for CF2,3
Fecal elastase-1 Easy to use for all ages4
Liver Transaminase levels
• Transaminases vary over time in patients with CF-related liver disease5
• Consistently high transaminase levels occur only with advanced liver disease6,7
Gastrointestinal Signs and symptoms (dysmotility, constipation, etc)
Monitoring may help avoid effects that may compound malnourishment due to pancreatic insufficiency8
SinusNasal endoscopy Useful for detecting chronic rhinosinusitis and polyps, which
occur in almost all patients and may precede development of CF lung disease9,10CT radiographic imaging
Bone Dual-energy x-ray absorptiometry Radiation burden may be a concern11
References: 1. Lahiri T et al. Pediatrics. 2016 Apr;137(4). pii: e20151784. doi: 10.1542/peds.2015-1784. Epub 2016 Mar 23. 2. Tardelli ACS et al. J Pediatr GastroenterolNutr. 2013;56(2):178–181. 3. Daftary A et al. J Cystic Fibros. 2006;5(2):71-76. 4. Paracchini V et al. JIMD Rep. 2012;4:17-23. doi: 10.1007/8904_2011_55. Epub 2011 Nov 4. 5. Cañas T et al. Biomed Res Int. 2015;2015:517369. doi:10.1155/2015/517369. Epub 2015 Nov 2. 6. Woodruff SA et al. J Cyst Fibros. 7. Stonebraker JR et al. Clin Gastroenterol Hepatol. 2016;14(8):1207-1215. 2017;16(1):129-145. 8. Sathe MN, Freeman AJ. Pediatr Clin North Am. 2016;63(4):679-698. 9. Illing EA et al. CurrOpin Pulm Med. 2014;20(6):623-631. 10. Savastano V et al. Eur Rev Med Pharmacol Sci. 2014;18(14):1985-1989. 11. Stalvey MS, Clines GA. Curr Opin EndocrinolDiabetes Obes. 2013;20(6):547-552.
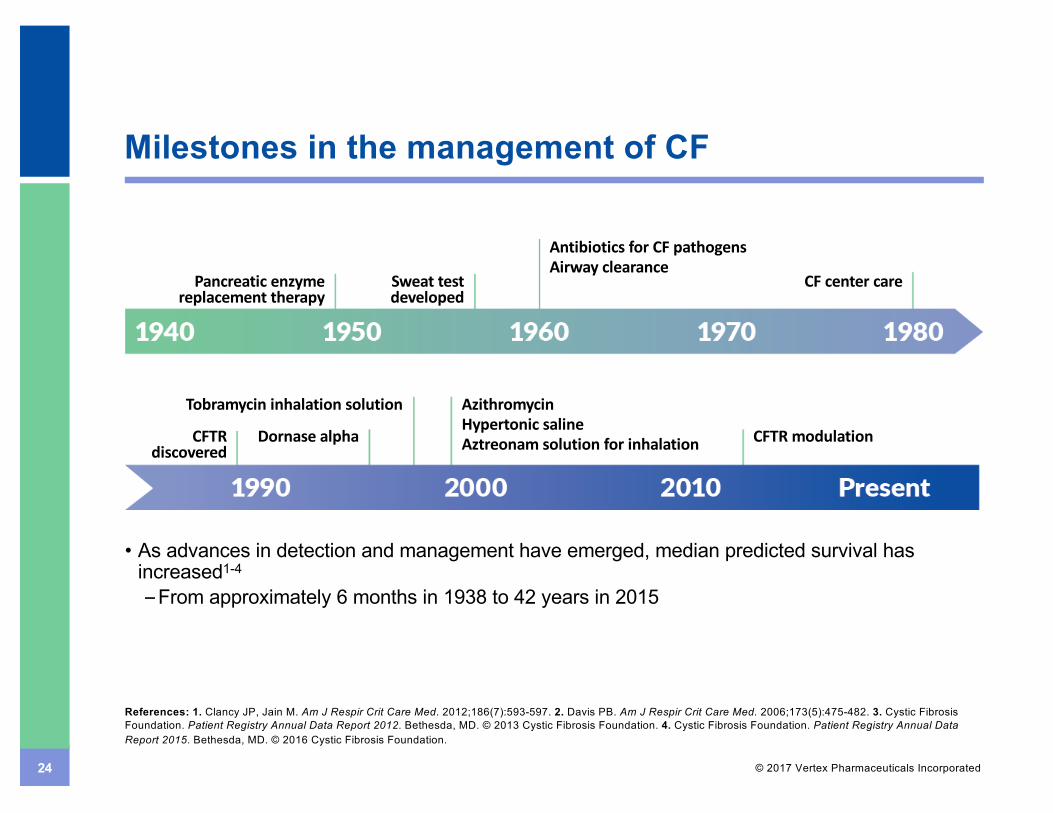
24 © 2017 Vertex Pharmaceuticals Incorporated
Milestones in the management of CF
• As advances in detection and management have emerged, median predicted survival has increased1-4
–From approximately 6 months in 1938 to 42 years in 2015
References: 1. Clancy JP, Jain M. Am J Respir Crit Care Med. 2012;186(7):593-597. 2. Davis PB. Am J Respir Crit Care Med. 2006;173(5):475-482. 3. Cystic Fibrosis Foundation. Patient Registry Annual Data Report 2012. Bethesda, MD. © 2013 Cystic Fibrosis Foundation. 4. Cystic Fibrosis Foundation. Patient Registry Annual Data Report 2015. Bethesda, MD. © 2016 Cystic Fibrosis Foundation.
Pancreaticenzymereplacementtherapy
Sweattestdeveloped
AntibioticsforCFpathogensAirwayclearance
CFcentercare
CFTRdiscovered
Dornase alpha
Tobramycininhalationsolution AzithromycinHypertonicsalineAztreonam solutionforinhalation CFTRmodulation

25 © 2017 Vertex Pharmaceuticals Incorporated
Summary

26 © 2017 Vertex Pharmaceuticals Incorporated
CF progression: Understanding the cause and its impact on your patients• CF is a multi-organ, progressive, genetic disease that affects the whole body, and many problems
with CF may be present at birth1,2
–Onset and progression of CF symptoms can occur in different organs at different ages
• Multisystemic damage can occur before symptoms emerge3
• Our ability to detect and monitor CF progression in different organs has increased over time with improved understanding of the disease4
• All patients with CF experience progression; however, CF management has improved over time in concert with scientific and medical discoveries1,5
References: 1. Elborn JS. Lancet. 2016;388(10059):2519-2531. 2. Zielenski J. Respiration. 2000;67(2):117-133. 3. Schram CA. Can Fam Physician. 2012;58:1342-1345. 4. Clancy JP, Jain M. Am J Respir Crit Care Med. 2012;186(7):593-597. 5. McKone EF et al. J Cyst Fibros. 2014;14(5):580-586.





























