Embed Size (px)
Citation preview

JOURNAL OF BACTERIOLOGY, Feb. 1981, p. 814-8230021-9193/81/020814-10$02.00/0
Vol. 145, No. 2
D-Fructose Dehydrogenase of Gluconobacter industrius:Purification, Characterization, and Application to Enzymatic
Microdetermination of D-FructoseMINORU AMEYAMA,* EMIKO SHINAGAWA, KAZUNOBU MATSUSHITA, AND OSAO ADACHI
Laboratory ofApplied Microbiology, Department ofAgricultural Chemistry, Faculty ofAgriculture,Yamaguchi University, Yamaguchi 753, Japan
D-Fructose dehydrogenase was solubilized and purified from the membranefraction of glycerol-grown Gluconobacter industrius IFO 3260 by a procedureinvolving solubilization of the enzyme with Triton X-100 and subsequent frac-tionation on diethylaminoethyl-cellulose and hydroxylapatite columns. The pu-
rified enzyme was tightly bound to a c-type cytochrome and another peptideexisting as a dehydrogenase-cytochrome complex. The purified enzyme was
deemed pure by analytical ultracentrifugation as well as by gel filtration on a
Sephadex G-200 column. The molecular weight of the enzyme complex was
determined to be about 140,000, and sodium dodecyl sulfate-polyacrylamide gelelectrophoresis showed the presence of three components having molecularweights of 67,000 (dehydrogenase), 50,800 (cytochrome c), and 19,700 (unknownfunction). Only D-fructose was readily oxidized by the enzyme in the presence ofdyes such as ferricyanide, 2,6-dichlorophenolindophenol, or phenazine methosul-fate. Nicotinamide adenine dinucleotide, nicotinamide adenine dinucleotide phos-phate, and oxygen did not function as electron acceptors. The optimum pH ofD-fructose oxidation was 4.0. The enzyme was stable at pH 4.5 to 6.0. Stability ofthe purified enzyme was much enhanced by the presence of detergent in theenzyme solution. Removal of detergent from the enzyme solution facilitated theaggregation of the enzyme and caused its inactivation. An apparent Michaelisconstant for D-fructose was observed to be 10-2 M with the purified enzyme. D-Fructose dehydrogenase was shown to be a satisfactory reagent for microdeter-mination of D-fructose.
5-Keto-D-fructose (D-threo-2,5-hexodiulose) isa unique substance which is produced only byacetic acid bacteria. Terada et al. (25-27) incu-bated Gluconobacter cerinus and other Gluco-nobacter spp. strains in a yeast extract rmediumcontaining D-fructose under shaking and ob-served the formation of a strong reducing sub-stance which was not glucosone, a precursor ofkojic acid. The substance was later identified as5-keto-D-fructose. Subsequently, Aida and Ya-mada (5) found an enzyme, 5-keto-D-fructosereductase, which catalyzes the reduction of 5-keto-D-fructose to D-fructose, in D-fructose-grown Gluconobacter albidus. A partially puri-fied preparation of 5-keto-D-fructose reductasewas strictly specific for 5-keto-D-fructose andreduced neither 5-keto-D-gluconate nor 2-keto-D-gluconate. These findings were also confirmedby Avigad et al. (8, 9, 14, 15).Membrane-bound D-fructose 5-dehydrogen-
ase (EC 1.1.99.11) was first detected by Yamadaet al. (30) in G. cerinus. A partially purifiedenzyme preparation oxidized D-fructose, and thereaction product was confirmed to be 5-keto-D-
fructose; other catalytic properties were also re-ported by the same authors (31). They alsoreported another 5-keto-D-fructose-yielding en-zyme, particle-bound L-sorbose dehydrogenasefrom Gluconobacter suboxydans, which did notoxidize D-fructose (23). The participation ofcoenzyme Qlo and cytochrome c in the oxidationof D-fructose by membrane particles was furtherindicated (32). Avigad and Angiuoli briefly re-ported the presence of several kinds of cyto-chrome components in the D-fructose oxidasesystem (G. Avigad and F. Angiuoli, Abstr. Annu.Meet. Am. Soc. Microbiol. 1975, K18, p. 150). Inspite of the efforts mentioned above, detailedmolecular properties of the membrane-boundD-fructose dehydrogenase of acetic acid bacteriahave remained to be elucidated, since the en-zyme has not been purified to homogeneity.
In a series of investigations on carbohydratemetabolism of acetic acid bacteria, we have pu-rified several dehydrogenases to homogeneityfrom membranes and reported on their catalyticproperties (1-4, 18, 19, 24). In this paper, thepurification and the catalytic and molecular
814
on January 21, 2019 by guesthttp://jb.asm
.org/D
ownloaded from

ACETIC ACID BACTERIAL D-FRUCTOSE DEHYDROGENASE 815
properties of D-fructose dehydrogenase fromGluconobacter industrius are reported, alongwith the possible use of the enzyme for thequantitative microdetermination of D-fructose.
MATERIALS AND METHODSMaterials. Yeast extract was a kind gift from the
Oriental Yeast Industrial Co.; DEAE-cellulose was aproduct of the Blood Plasma Corp.; and hydroxylap-atite was prepared by the method of Levin (16). 5-Keto-D-fructose was a product of Kyowa Hakko Ko-gyo Co., and all other chemicals used in this studywere commercial products of analytical grade.
Bacterial strains and culture conditions. Bac-terial strains of the genus Gluconobacter, listed inTable 1, were a generous donation from the Institutefor Fermentation, Osaka (IFO). Stock cultures weremaintained on potato-glycerol slants. This mediumwas prepared as follows. Freshly sliced potato (200 g)was boiled in 1 liter of tap water and autoclaved for 10min at 2 kg/cm2. The autoclaved gruel was centrifugedat 12,000 x g for 20 min, and a light yellow supernatantwas obtained. To the supernatant were added driedyeast extract (10 g), polypeptone (10 g), glycerol (20g), and D-glucose (5 g). In the case of potato-glycerolslants, agar powder was further added to 1.5%.The culture medium for Gluconobacter spp. con-
sisted of 4 g of glycerol, 6 g of sodium glutamate, 500mg each of KH2PO4 and K2HPO4, 200 mg of MgSO4.7H20, and 10 mg each of FeSO4.7H20, NaCl, andMnSO4. 7H20 in 1 liter of tap water. Also, 400 pg eachof thiamin hydrochloride, nicotinic acid, and calciumpantothenate and 100 ,g ofp-aminobenzoic acid wereadded per liter as supplements. Gluconobacter spp.cultures grown on potato-glycerol slants were inocu-lated into 100 ml ofthe medium in 500-ml shake flasks,and cultivation was carried out at 30°C for 24 h withreciprocal shaking. In the case of a large-scale culture,25 liters of the medium was inoculated into a 50-literjar fermentor at 30°C under vigorous agitation (500rpm) and aeration (25 to 30 liters of air per min).Bacterial celLs were harvested at the late exponentialphase.
Preparation of cell homogenate. Cells were col-lected by centrifugation at 12,000 x g for 20 min andwashed twice with ice-cold water. The cell paste wassuspended in distilled water and passed twice througha French presure cell press at 1,000 kg/cm2. Intactcells were removed by centrifugation at 5,000 x g for10 min. The resulting supernatant was designated thecell homogenate.Preparation of membrane fraction. The cell
homogenate was centrifuged at 68,000 x g for 90 min.The relting supernatant was collected for the sourceof 5-keto-D-fructose reductase, as was reported previ-ously (M. Ameyama, K. Mieno, E. Shinagawa, K.Matsushita, and 0. Adachi, Abstr. Annu. Meet. Agric.Chem. Soc. Jpn. 1980, p. 507). The sedimented pellet,designated the membrane fraction, could be stored at-20°C for over 6 months without an appreciable lossof enzyme activity.Assay ofD-fructose dehydrogenase. The follow-
ing assay systems were available: D-fructose was oxi-dized by D-fructose dehydrogenase, and the reactionrate was estimated (i) by spectrometry in the presence
TABLE 1. Distribution of D-fructose dehydrogenase,D-glucose dehydrogenase, and alcohol
dehydrogenase in various Gluconobacter spp.a
Strain
G. melanogenus IFO 3293G. cerinuw IFO 3262 ......G. cerinus IFO 3264 ......G. cerinus IFO 3265 ......G. cerinus IFO 3266 ......G. cerinus IFO 3267 ......G. cerinus IFO 3268 ......G. cerinus IFO 3270 ......G. gluconicus IFO 3171G. gluconicus IFO 3285 ...G. gluconicus IFO 3286 ...G. albidus IF0 3251.G. albidus IFO 3253 ......G. suboxydans F03172..G. suboxydans IFO 3289.G. suboxydans IFO 3290..G. suboxydans IFO 3291..G. suboxydans var. a IFO3254 .............
G. suboxydans var. a IFO3256.
G. suboxydans var. a IFO3258.
G. industrius 170 3260 ...G. nonoxygluconicus IFO3275 .........
G. rubigino8us IFO 3244..
Protein(mg)107681768720152152190126929693781128097117
74
97
10445
1120
FDH(U)0000o0160
Thb000000000
0
0
035
00
GDH(U)17988194144471912282307415819278
156112667353
TRc
TR'
TRc27
1826
ADH(U)14514852843575154527171403208582251247312191143373
73
67
1445
2240
'Each strain of Gluconobacter spp. was inoculated intothree flasks containg 100 ml of the medium and incubatedat 30°C for 24 h under reciprocal shaking. Each enzymeactivity was assayed with cell homogenate under the standardasay conditions. Abbreviations: FDH, D-fuctose dehydrogen-as; GDH, D-glucose dehydrogenase; and ADH, alcohol de-hydrogenase.
Total enzyme activity was less than 0.1 U.'Total enzyme activity was les than 5.0 U.
of 2,6-dichlorophenolindophenol and phenazine meth-osulfate, (ii) by colorimetry in the presence of potas-sium ferricyanide, (iii) by polarometry with an oxygenelectrode, or (iv) by manometry in a conventionalWarburg apparatus. In this study, the potassium fer-ricyanide assay was employed mainly because of itssimplicity for routine assay. This method was de-scribed by Wood et al. (29) in the assay of D-gluconatedehydrogenase of Pseudomonas fluorescens with po-tassium ferricyanide as an electron acceptor. Somemodifications were made as described below, and therate of reduction of ferricyanide to ferrocyanide wascorrelated to the rate of D-fructose oxidation. Thereaction mixture contained 10 pimol of potassium fer-ricyanide, 0.5 ml of Mcllvaine buffer (pH 4.5), enzymesolution, and 100 pMol of D-fructose in a total volumeof 1.0 mL The reaction was carried out at 25°C by theaddition of D-fructose and stopped by adding 0.5 ml offerric-Dupanol reagent. Then, 3.5 ml of water wasadded to the reaction mixture. The resulting Prussian-blue color was measured by a photometer at 660 nmafter standing at 25°C for 20 min. One unit of enzymeactivity was defined as the amount of enzyme catalyz-ing the oxidation of 1 pmol of D-fructose per min underthese conditions; 4.0 absorbance units equaled 1 pimolof D-fructose oxidized.
VOL. 145, 1981
on January 21, 2019 by guesthttp://jb.asm
.org/D
ownloaded from

816 AMEYAMA ET AL.
Alcohol dehydrogenase and D-glucose dehydrogen-ase in cell homogenates were assayed under essentiallythe same conditions as D-fructose dehydrogenase, ex-cept that the substrates were changed to ethanol andD-glucose, respectively.
Protein determination. Protein content was de-termined by the method of Lowry et al. (17) withbovine serum albumin as the standard. A modifiedmethod described by Dulley and Grieve (13) was em-ployed for samples which contained Triton X-100.
Analytical ultracentrifugation. Estimation ofsedimentation velocity and examination of purity ofthe purified enzyme were carried out with a Hitachimodel 282 analytical ultracentrifuge at 59,780 rpm byusing schlieren optics (11). Temperature was main-tained at 200C throughout the measurements.
Electrophoresis. Polyacrylamide gel electropho-resis was performed under essentially the conditionsdescribed by Davis (12) with 7.5% polyacrylamide geland Tris-glycine buffer (pH 8.3). Polyacrylamide gelelectrophoresis in the presence of sodium dodecylsulfate (SDS) was performed by the method of Weberand Osborn (28). Protein solution was made 1% inSDS and 1% in 2-mercaptoethanol, heated in a boilingwater bath for 5 min, and subjected to electrophoresis(7 mA per gel for 3 to 5 h). Protein was stained withCoomassie brilliant blue in 7% acetic acid. Enzymeactivity was detected by dipping the gel in a mediumcontaining D-fructose (10 mM), phenazine methosul-fate (0.14 mg/ml), and nitroblue tetrazolium (0.3 mg/ml) in 10-fold-diluted Mcllvaine buffer (pH 4.5). Afterdevelopment of a dark purple band upon incubationat 4°C for 5 to 10 min in the dark, enzyme activity wasterminated by transferring the gel into 7% acetic acid.
Sucrose density gradient centrifugation. Su-crose gradients were prepared with the following com-position: 1 ml each of 21, 17, 13, 9, and 5% (wt/wt)sucrose, each prepared in 20-fold-diluted Mcllvainebuffer (pH 6.0) containing 1 mM 2-mercaptoethanol.One gradient contained Triton X-100 (1%). The gra-dients were left to stand at 40C for about 20 h beforeuse. The sample of D-fructose dehydrogenase plusstandard marker proteins ofyeast alcohol dehydrogen-ase (molecular weight, 151,000) and catalase (molecu-lar weight, 247,500) in a total volume of 0.1 ml waslayered on the top of the gradients and centrifuged at36,000 rpm for 16 h in a Hitachi RPS 40A rotor at 40C.
RESULTSDistribution of D-fructose dehydrogen-
ase in Gluconobacter spp. Enzyme activitywas surveyed with cell homogenates of variousstrains of Gluconobacter spp. (Table 1). G. in-dustrius IFO 3260 was found to be the highestin D-fructose dehydrogenase of the strainstested. Alcohol dehydrogenase and D-glucose de-hydrogenase were also checked with the samecell homogenates by replacing D-fructose withethanol and D-glucose as substrate. Alcohol de-hydrogenase and D-glucose dehydrogenase wereproduced in most strains of Gluconobacter spp.In G. industrius, the level of alcohol dehydro-genase was exceptionally low under the culture
conditions employed in our experiments. Thus,the use of G. industrius for further purificationof D-fructose dehydrogenase was advantageousfrom the standpoint of less contamination withother enzyme proteins. Yamada et al. (30, 31)used the strain of G. cerinus IFO 3267 for thesource of D-fructose dehydrogenase. We alsochecked the same strain and G. cerinu IFO3270; however, these two strains usually con-tained substantial amounts of alcohol dehydro-genase and D-glucose dehydrogenase.
Solubilization and purification of D-fruc-tose dehydrogenase. All operations were car-ried out at 0 to 50C, unless otherwise stated.Mcllvaine buffer of various pH was used. Cellsof G. industrius (50 g, wet wt) were harvestedfrom 25 liters of a 20-h culture, and the mem-brane fraction was prepared. This fraction wassuspended in 20-fold-diluted Mcllvaine buffer(pH 6.0), and the protein concentration was ad-justed to 30 mg/ml. To the suspension, 10%Triton X-100 and 2-mercaptoethanol wereadded to final concentrations of 1% and 1 mM,respectively. The suspension was gently stirredfor 3 h, solubilizing the enzyme from the mem-brane fraction. Cell debris was removed by cen-trifugation at 68,000 x g for 60 min, and a clearrose-red supernatant was obtained. The super-natant solution (340 ml) was applied to a column(2.5 by 20 cm) of DEAE-cellulose which hadbeen equilibrated with 20-fold-diluted Mcllvainebuffer (pH 6.0) containing 0.1% Triton X-100and 1 mM 2-mercaptoethanol. After the columnwas further washed with 300 ml of the samebuffer to wash out nonadsorbable protein, theenzyme was eluted with a descending pH gra-dient composed of 500 ml of 20-fold-dilutedMcllvaine buffer (pH 6.0) and 500 ml of 20-fold-diluted Mcllvaine buffer (pH 4.5). Both buffersolutions were also supplemented with 0.1% Tri-ton X-100 and 1 mM 2-mercaptoethanol. Each10-nil fraction was collected, and the D-fructosedehydrogenase was eluted at about pH 5.2 as arose-red material. Fractions (no. 60 to 90) con-taining enzyme activity were combined (300 ml)and concentrated by membrane filtration (Toyoultra filter UP-50) or dehydration with polyeth-ylene glycol 6,000. The concentrated enzymesolution was dialyzed against 20-fold-dilutedMcllvaine buffer (pH 6.0) containing 0.1% TritonX-100 and 1 mM 2-mercaptoethanol. The di-alyzed enzyme solution was then applied to ahydroxylapatite column (2 by 5 cm) which hadbeen equilibrated with the same buffer used fordialysis. After the column was washed with 100ml of the same buffer, elution of the enzyme wasaccomplished by a gradient of Mcllvaine buffer(pH 6.0). One container was filled with 200 ml of20-fold-diluted buffer, and another was filled
J. BACTERIOL.
on January 21, 2019 by guesthttp://jb.asm
.org/D
ownloaded from
ACETIC ACID BACTERIAL D-FRUCTOSE DEHYDROGENASE 817
with 200 ml of buffer with no dilution. Triton X-100 (0.1%) and 2-mercaptoethanol (1 mM) werealso added to both containers. The enzyme ac-tivity was eluted at 200 to 250 ml. The activefractions in about 75 ml were placed in dialysistubing and concentrated to about 5 ml by de-hydration with polyethylene glycol 6,000. Afterremoval of excess polyethylene glycol in theenzyme solution by extensive dialysis against 20-fold-diluted Mcllvaine buffer (pH 6.0) contain-ing 0.1% Triton X-100 and 1 mM 2-mercaptoeth-anol, insoluble materials were removed by cen-trifugation at 12,000 x g for 20 min. The resultsof a typical purification are given in Table 2.The purified D-fructose dehydrogenase, pre-pared as above, usually had a specific activity ofabout 180 U/mg of protein per min under thestandard assay conditions.The enzyme lost no significant activity when
stored at 0 to 4°C in Mcllvaine buffer (pH 4.0 to5.0) containing 0.1% Triton X-100 and 1 mM 2-mercaptoethanol for at least 2 weeks. The en-zyme activity was completely preserved whenthe enzyme was frozen at -20°C or below. Thepresence of Triton X-100 in the enzyme solutionwas essential for the preservation of activity ofthe purified enzyme preparation, and it becamelabile and less active when the detergent wasnot present. Similar results are seen with othermembrane-bound enzymes such as alcohol de-hydrogenase (2, 3), aldehyde dehydrogenase (4),and D-gluconate dehydrogenase (19, 24) as re-ported previously.Properties of the purified D-fructose de-
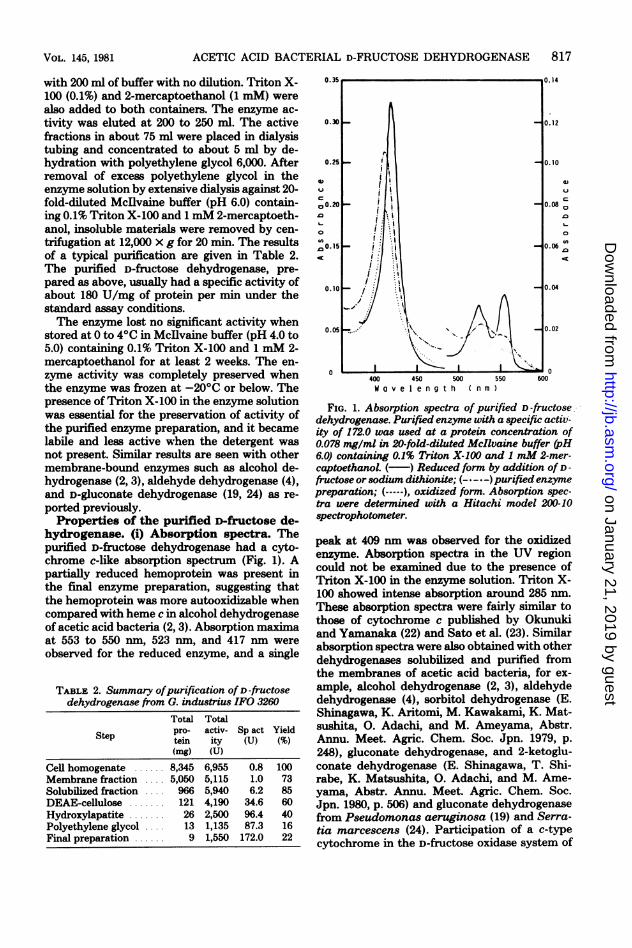
hydrogenase. (i) Absorption spectra. Thepurified D-fructose dehydrogenase had a cyto-chrome c-like absorption spectrum (Fig. 1). Apartially reduced hemoprotein was present inthe final enzyme preparation, suggesting thatthe hemoprotein was more autooxidizable whencompared with heme c in alcohol dehydrogenaseof acetic acid bacteria (2, 3). Absorption maximaat 553 to 550 nm, 523 nm, and 417 nm wereobserved for the reduced enzyme, and a single
TABLE 2. Summary ofpurification of D -fructosedehydrogenase from G. industrius IFO 3260
Total TotalStep pro- activ- Sp act YieldSteP tein ity (U) (%)
(mg) (U)
Cell homogenate ...... 8,345 6,955 0.8 100Membrane fraction .... 5,050 5,115 1.0 73Solubilized fraction .... 966 5,940 6.2 85DEAE-cellulose ....... 121 4,190 34.6 60Hydroxylapatite ....... 26 2,500 96.4 40Polyethylene glycol .... 13 1,135 87.3 16Final preparation ...... 9 1,550 172.0 22
0.35 0.14
0.30 0.12
0.25 0.10
v0.20 0.08
,015 f0.06
0.10 A 0.04
0.05~ ~ ~ I' 0.02
0 0400 450 500 550W a v e I e n g t h ( n m )
600
FIG. 1. Absorption spectra of purified D -fructosedehydrogenase. Purified enzyme with a specific activ-ity of 172.0 was used at a protein concentration of0.078 mg/ml in 20-fold-diluted McIlvaine buffer (pH6.0) containing 0.1% Triton X-100 and 1 mM 2-mer-captoethanol. ( ) Reduced form by addition of D -fructose or sodium dithionite; ( -) purified enzymepreparation; (-----), oxidized form. Absorption spec-tra were determined with a Hitachi model 200-10spectrophotometer.
peak at 409 nm was observed for the oxidizedenzyme. Absorption spectra in the UV regioncould not be examined due to the presence ofTriton X-100 in the enzyme solution. Triton X-100 showed intense absorption around 285 nm.These absorption spectra were fairly similar tothose of cytochrome c published by Okunukiand Yamanaka (22) and Sato et al. (23). Similarabsorption spectra were also obtained with otherdehydrogenases solubilized and purified fromthe membranes of acetic acid bacteria, for ex-ample, alcohol dehydrogenase (2, 3), aldehydedehydrogenase (4), sorbitol dehydrogenase (E.Shinagawa, K. Aritomi, M. Kawakami, K. Mat-sushita, 0. Adachi, and M. Ameyama, Abstr.Annu. Meet. Agric. Chem. Soc. Jpn. 1979, p.248), gluconate dehydrogenase, and 2-ketoglu-conate dehydrogenase (E. Shinagawa, T. Shi-rabe, K. Matsushita, 0. Adachi, and M. Ame-yama, Abstr. Annu. Meet. Agric. Chem. Soc.Jpn. 1980, p. 506) and gluconate dehydrogenasefrom Pseudomonas aeruginosa (19) and Serra-tia marcescens (24). Participation of a c-typecytochrome in the D-fructose oxidase system of
VOL. 145, 1981
a4.
c
c.C
uc.C
-4
(D
u
a
on January 21, 2019 by guesthttp://jb.asm
.org/D
ownloaded from
818 AMEYAMA ET AL.
acetic acid bacteria has been indicated by Ya-mada et al. (31, 32) with a partially purifiedenzyme or membranes of G. cerinus. The exist-ence of a c-type cytochrome in the D-fructosedehydrogenase was thus supported by the puri-fied enzyme preparation from G. industrus.
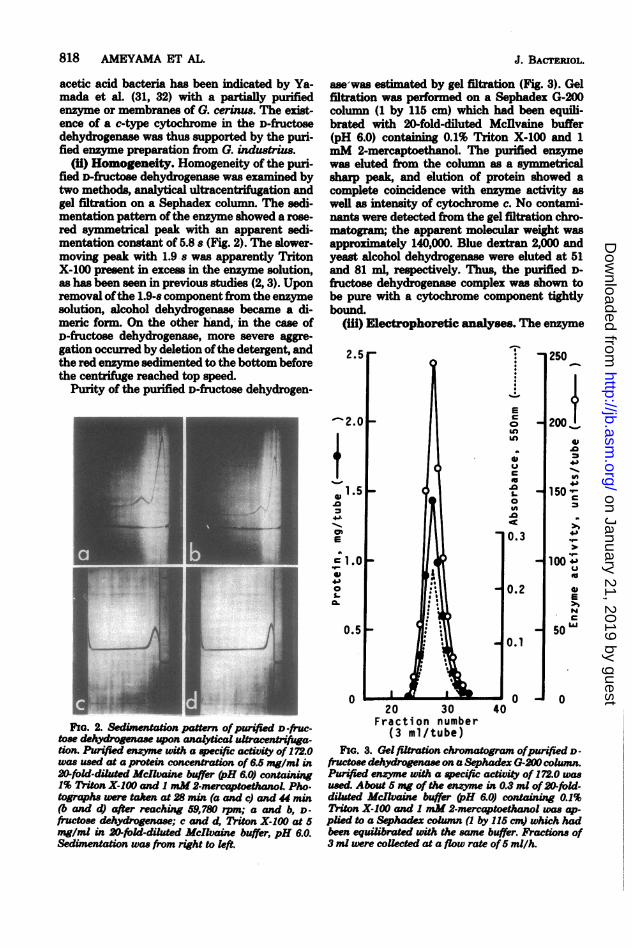
(ii) Homogeneity. Homogeneity of the puri-fied D-fructose dehydrogenase was examined bytwo methods, analytical ultracentrifugation andgel filtration on a Sephadex column. The sedi-mentation pattern of the enzyme showed a rose-red symmetrical peak with an apparent sedi-mentation constant of 5.8 s (Fig. 2). The slower-moving peak with 1.9 s was apparently TritonX-100 present in excess in the enzyme solution,as has been seen in previous studies (2, 3). Uponremoval ofthe 1.9-s component from the enzymesolution, alcohol dehydrogenase became a di-meric form. On the other hand, in the case ofD-fructose dehydrogenase, more severe aggre-gation occurred by deletion ofthe detergent, andthe red enzyme sedimented to the bottom beforethe centrifuge reached top speed.
Purity of the purified D-fructose dehydrogen-
FIG. 2. Sedi_entation pattern ofpUrified D -fruc-tose dehydrogeae upon analytical ultracenifga-tion. Purifed enzyme wih a specific activity of 172.0was used at a protein concentration of 6.5 mg/ml in20-fold-ditd Mcllaine buffer (pH 6.0) containing1% Triton X-100 and I mM2-mercaptoetwnoL Pho.tographs were taken at 28 min (a and c) and 44 min(b and d) after reaching 59,780 rpm; a and b, D-fructose dehydrogenase; c and d, Triton X-100 at 5mg/ml in 20-fold-diluted McIlvaine buffer, pH 6.0.Sedimentation was from right to left.
ase was estimated by gel filtration (Fig. 3). Gelfiltration was performed on a Sephadex G-200column (1 by 115 cm) which had been equili-brated with 20-fold-diluted Mcdlvaine buffer(pH 6.0) containing 0.1% Triton X-100 and 1mM 2-mercaptoethanol. The purified enzymewas eluted from the column as a symmetricalsharp peak, and elution of protein showed acomplete coincidence with enzyme activity aswell as intensity of cytochrome c. No contami-nants were detected from the gel filtration chro-matogram; the apparent molecular weight wasapproximately 140,000. Blue dextran 2,000 andyeast alcohol dehydrogenase were eluted at 51and 81 ml, respectively. Thus, the purified D-fructose dehydrogenase complex was shown tobe pure with a cytochrome component tightlybound.
(iin) Electrophoretic anlalyses. The enzyme
I
250
200 +
0
150 c~4.,
100;E='
50
150L&J
020 30 40
Fraction number(3 ml/tube)
FIG. 3. Gel filtaion chromatogram ofpurified D -fructose dehydrgenase on a Sephadex G-20 column.Prified enzyme wih a specifi activity of 172.0 wasused. About 5 mg of the enzyme in 0.3 ml of20-fold-diluted dMclvaine buffer (H 6.0) containing 0.1%Triton X-100 and 1 mM 2-mercaptoethanol was ap-plied to a Sp oadxcomn (1 by 115 cm) which hadbeen equilbrated with the same buffer. Practions of3 ml were coUected at a flow rate of5 mil/h.
J. BACTERIOL.
on January 21, 2019 by guesthttp://jb.asm
.org/D
ownloaded from
ACETIC ACID BACTERIAL D-FRUCTOSE DEHYDROGENASE
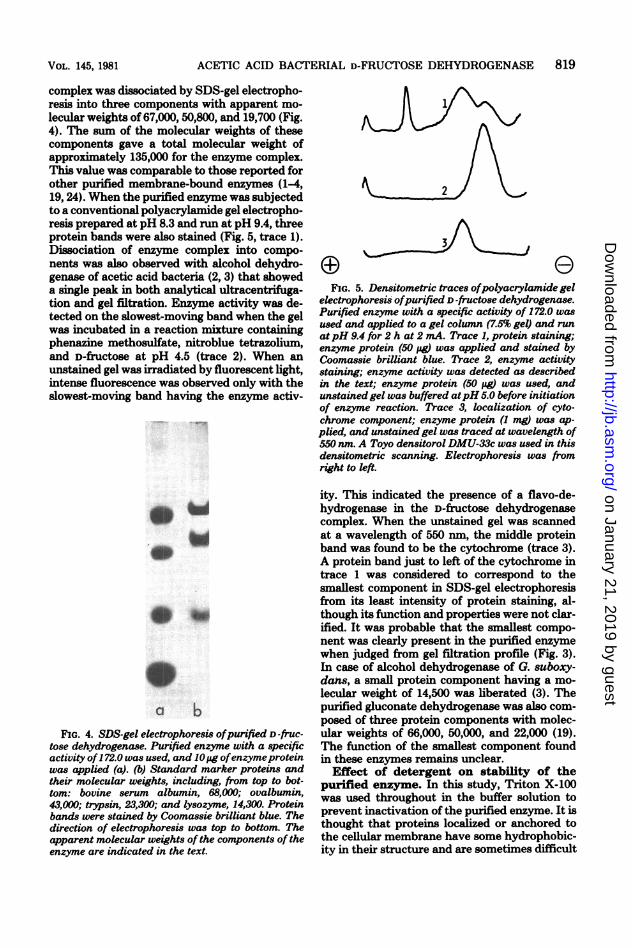
complex was dissociated by SDS-gel electropho-resis into three components with apparent mo-lecular weights of 67,000, 50,800, and 19,700 (Fig.4). The sum of the molecular weights of thesecomponents gave a total molecular weight ofapproximately 135,000 for the enzyme complex.This value was comparable to those reported forother purified membrane-bound enzymes (1-4,19, 24). When the purified enzyme was subjectedto a conventional polyacrylamide gel electropho-resis prepared at pH 8.3 and run at pH 9.4, threeprotein bands were also stained (Fig. 5, trace 1).Dissociation of enzyme complex into compo-nents was also observed with alcohol dehydro-genase of acetic acid bacteria (2, 3) that showeda single peak in both analytical ultracentrifuga-tion and gel filtration. Enzyme activity was de-tected on the slowest-moving band when the gelwas incubated in a reaction mixture containingphenazine methosulfate, nitroblue tetrazolium,and D-fructose at pH 4.5 (trace 2). When anunstained gel was irradiated by fluorescent light,intense fluorescence was observed only with theslowest-moving band having the enzyme activ-
Ut
a bFIG. 4. SDS-gel electrophoresis ofpurified D -fruc-
tose dehydrogenase. Purified enzyme with a specificactivity of172.0 was used, and 10pjg ofenzymeproteinwas applied (a). (b) Standard marker proteins andtheir molecular weights, including, from top to bot-tom: bovine serum albumin, 68,0X0; ovalbumin,43,000; trypsin, 23,300; and lysozyme, 14,300. Proteinbands were stained by Coomassie brilliant blue. Thedirection of electrophoresis was top to bottom. Theapparent molecular weights of the components of theenzyme are indicated in the text.
3
FIG. 5. Densitometric traces ofpolyacrylamide gelelectrophoresis ofpurified D-fructose dehydrogenase.Purified enzyme with a specific activity of 172.0 wasused and applied to a gel column (7.5% gel) and runatpH 9.4 for 2 h at 2 mA. Trace 1, protein staining;enzyme protein (50 pg) was applied and stained byCoomassie brilliant blue. Trace 2, enzyme activitystaining; enzyme activity was detected as describedin the text; enzyme protein (50 pg) was used, andunstained gel was buffered atpH 5.0 before initiationof enzyme reaction. Trace 3, localization of cyto-chrome component; enzyme protein (1 mg) was ap-plied, and unstained gel was traced at wavelength of550 nm. A Toyo densitorol DMU-33c was used in thisdensitometric scanning. Electrophoresis was fromright to left.
ity. This indicated the presence of a flavo-de-hydrogenase in the D-fructose dehydrogenasecomplex. When the unstained gel was scannedat a wavelength of 550 nm, the middle proteinband was found to be the cytochrome (trace 3).A protein band just to left of the cytochrome intrace 1 was considered to correspond to thesmallest component in SDS-gel electrophoresisfrom its least intensity of protein staining, al-though its function and properties were not clar-ified. It was probable that the smallest compo-nent was clearly present in the purified enzymewhen judged from gel filtration profile (Fig. 3).In case of alcohol dehydrogenase of G. suboxy-dans, a small protein component having a mo-lecular weight of 14,500 was liberated (3). Thepurified gluconate dehydrogenase was also com-posed of three protein components with molec-ular weights of 66,000, 50,000, and 22,000 (19).The function of the smallest component foundin these enzymes remains unclear.Effect of detergent on stability of the
purified enzyme. In this study, Triton X-100was used throughout in the buffer solution toprevent inactivation of the purified enzyme. It isthought that proteins localized or anchored tothe cellular membrane have some hydrophobic-ity in their structure and are sometimes difficult
819VOL. 145, 1981
on January 21, 2019 by guesthttp://jb.asm
.org/D
ownloaded from
820 AMEYAMA ET AL.
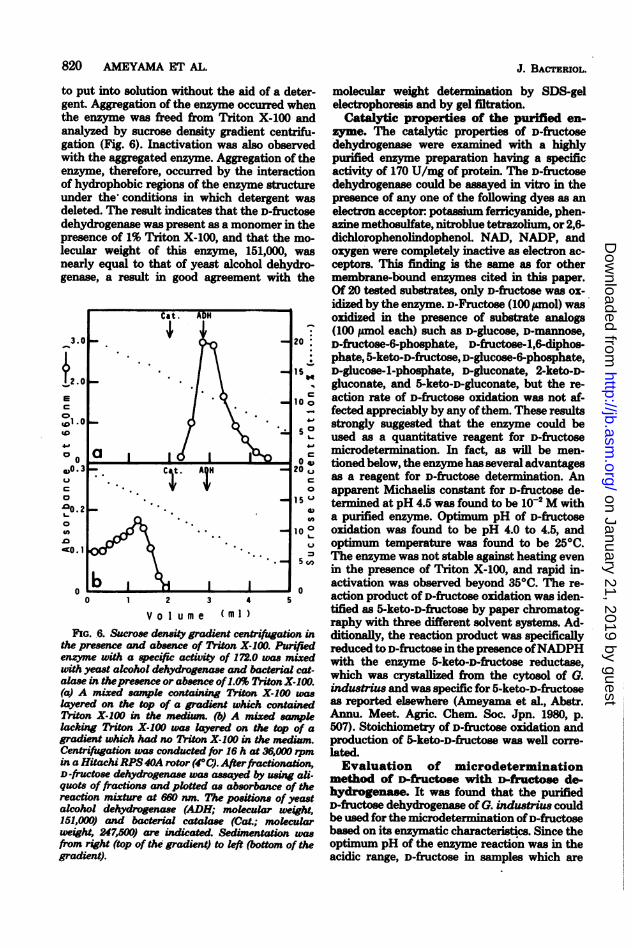
to put into solution without the aid of a deter-gent. Aggregation of the enzyme occurred whenthe enzyme was freed from Triton X-100 andanalyzed by sucrose density gradient centrifu-gation (Fig. 6). Inactivation was also observedwith the aggregated enzyme. Aggregation of theenzyme, therefore, occurred by the interactionof hydrophobic regions of the enzyme stuctureunder the conditions in which detergent wasdeleted. The result indicates that the D-fructosedehydrogenase was present as a monomer in thepresence of 1% Triton X-100, and that the mo-lecular weight of this enzyme, 151,000, wasnearly equal to that of yeast alcohol dehydro-genase, a result in good agreement with the
2 3 4
Volume (ml)FIG. 6. Sucrose density gradient centriftgation in
the presence and absence of Triton X-100. Purifiedenzyme with a specific activity of 172.0 was mixedwith yeast alcohol dehydrogenase and bacterial cat-alase in thepresence or absence of1.0% Triton X-100.(a) A mixed sampl containng Triton X-100 waslayered on the top of a gradient which containedTriton X-100 in the medium. (b) A mixed sampklacking Triton X-100 was layered on the top of agradient which had no Triton X-100 in the medium.Centrifugation was conducted for 16 h at 36,000 rpmin a Hitachi RPS40A rotor (4°C). Afterfractionation,D-fructose dehydrogenase was assayed by usIg al-
quots of fractions and plotted as absorbance of thereaction mixture at 660 nm. The positions of yeastalcohol dehydrogenase (ADH; mokcular ueight,151,000) and bacterial catals (Cat.; mokcularweight, 247,500) are indicated. Seduientation wasfrom right (top of the gradient) to kft (bottom of thegradient).
J. BACTERIOL.
molecular weight determination by SDS-gelelectrophoresis and by gel filtration.Catalytic properties of the purified en-
zyme. The catalytic properties of D-fruCtosedehydrogenase were examined with a highlypurified enzyme preparation having a specificactivity of 170 U/mg of protein. The D-fructosedehydrogenase could be asayed in vitro in thepresence of any one of the following dyes as anelectron acceptor: potasium ferricyanide, phen-azine methosulfate, nitroblue tetrazolium, or 2,6-dichlorophenolindophenol. NAD, NADP, andoxygen were completely inactive as electron ac-ceptors. This finding is the same as for othermembrane-bound enzymes cited in this paper.Of 20 tested substrates, only D-fuctose was ox-idized by the enzyme. D-Fructose (100 uimol) wasoxidized in the presence of substrate analogs(100 ,umol each) such as D-glucose, D-mannose,D-fructose-6-phosphate, D-fructose-1,6-diphos-phate, 5-keto-D-fructose, D-glucose-6phosphate,D-glucose-l-phosphate, D-gluconate, 2-keto-D-gluconate, and 5-keto-D-gluconate, but the re-action rate of D-fructose oxidation was not af-fected appreciably by any ofthem. These resultsstrongly suggested that the enzyme could beused as a quantitative reagent for D-fructosemicrodetermination. In fact, as will be men-tioned below, the enzyme has several advantagesas a reagent for D-fructose determination. Anapparent Michaelis constant for D-fructose de-termined at pH 4.5 was found to be 10-2 M witha purified enzyme. Optimum pH of D-fructoseoxidation was found to be pH 4.0 to 4.5, andoptimum temperature was found to be 25°C.The enzyme was not stable against heating evenin the presence of Triton X-100, and rapid in-activation was observed beyond 350C. The re-action product of D-fructose oxidation was iden-tified as 5-keto-D-fructose by paper chromatog-raphy with three different solvent systems. Ad-ditionally, the reaction product was specificallyreduced to D-fructose in the presence ofNADPHwith the enzyme 5-keto-D-fructose reductase,which was crystafized friom the cytosol of G.industris and was specific for 5-keto-D-fructoseas reported elsewhere (Ameyama et al., Abstr.Annu. Meet. Agric. Chem. Soc. Jpn. 1980, p.507). Stoichiometry of D-fructose oxidation andproduction of 5-keto-n-fructose was well corre-lated.Evaluation of microdetermination
method of Dfructose with D-fructose de-hydrogenase. It was found that the purifiedD-fuctose dehydgenase of G. industrius couldbe usd for the microdetermination ofD-fructosebased on its enzymatic characteristics. Since theoptimum pH of the enzyme reaction was in theacidic range, D-fructose in samples which are
on January 21, 2019 by guesthttp://jb.asm
.org/D
ownloaded from
ACETIC ACID BACTERIAL D-FRUCTOSE DEHYDROGENASE 821
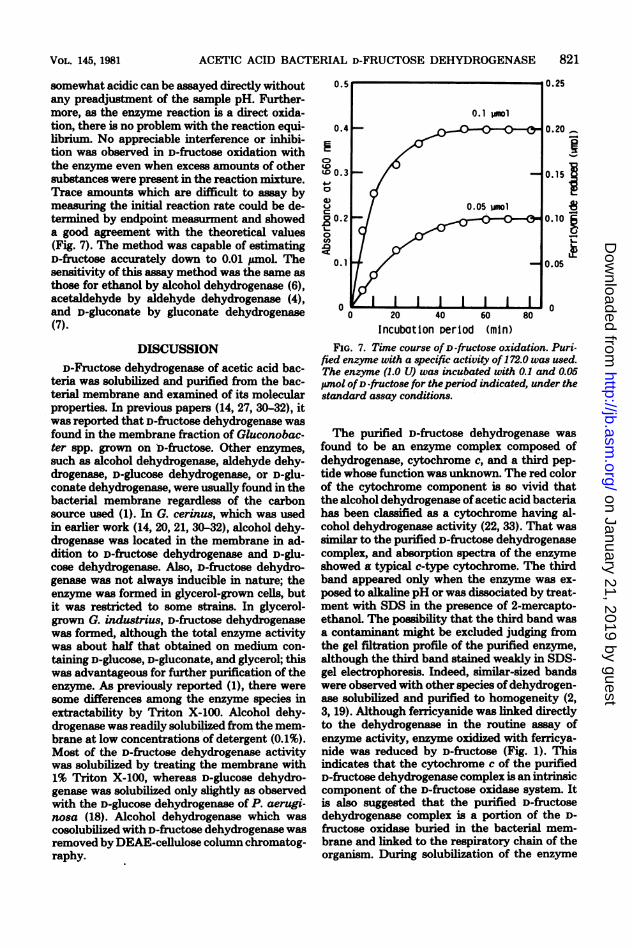
somewhat acidic can be assayed directly withoutany preadjustment of the sample pH. Further-more, as the enzyme reaction is a direct oxida-tion, there is no problem with the reaction equi-librium. No appreciable interference or inhibi-tion was observed in D-fructose oxidation withthe enzyme even when excess amounts of othersubstances were present in the reaction mixture.Trace amounts which are difficult to assay bymeasuring the initial reaction rate could be de-termined by endpoint measurment and showeda good agreement with the theoretical values(Fig. 7). The method was capable of estimatingD-fructose accurately down to 0.01 umol. Thesensitivity of this assay method was the same asthose for ethanol by alcohol dehydrogenase (6),acetaldehyde by aldehyde dehydrogenase (4),and D-gluconate by gluconate dehydrogenase(7).
DISCUSSIOND-Fructose dehydrogenase of acetic acid bac-
teria was solubilized and purified from the bac-terial membrane and examined of its molecularproperties. In previous papers (14, 27, 30-32), itwas reported that D-fructose dehydrogenase wasfound in the membrane fraction of Gluconobac-ter spp. grown on D-fructose. Other enzymes,such as alcohol dehydrogenase, aldehyde dehy-drogenase, D-glucose dehydrogenase, or D-glu-conate dehydrogenase, were usually found in thebacterial membrane regardless of the carbonsource used (1). In G. cerinus, which was usedin earlier work (14, 20, 21, 30-32), alcohol dehy-drogenase was located in the membrane in ad-dition to D-fructose dehydrogenase and D-glu-cose dehydrogenase. Also, D-fructose dehydro-genase was not always inducible in nature; theenzyme was formed in glycerol-grown cells, butit was restricted to some strains. In glycerol-grown G. industrius, D-fructose dehydrogenasewas formed, although the total enzyme activitywas about half that obtained on medium con-taining D-glucose, D-gluconate, and glycerol; thiswas advantageous for further purification of theenzyme. As previously reported (1), there weresome differences among the enzyme species inextractability by Triton X-100. Alcohol dehy-drogenase was readily solubilized from the mem-brane at low concentrations of detergent (0.1%).Most of the D-fructose dehydrogenase activitywas solubilized by treating the membrane with1% Trton X-100, whereas D-glucose dehydro-genase was solubilized only slightly as observedwith the D-glucose dehydrogenase of P. aerugi-nosa (18). Alcohol dehydrogenase which wascosolubilized with D-fructose dehydrogenase wasremoved by DEAE-cellulose column chromatog-raphy.
*D 0.3 0.15
0.05 jInol0.2 0.100
0.1 0.05
0V1 1 00 20 40 60 80
Incubation Period (min)FIG. 7. Time course of D-fructose oxidation. Puri-
fied enzyme with a specific activity of 172.0 was used.The enzyme (1.0 U) was incubated with 0.1 and 0.05Wmol ofD-fructose for theperiod indicated, under thestandard assay conditions.
The purified D-fructose dehydrogenase wasfound to be an enzyme complex composed ofdehydrogenase, cytochrome c, and a third pep-tide whose function was unknown. The red colorof the cytochrome component is so vivid thatthe alcohol dehydrogenase ofacetic acid bacteriahas been classified as a cytochrome having al-cohol dehydrogenase activity (22, 33). That wassimilar to the purified D-fructose dehydrogenasecomplex, and absorption spectra of the enzymeshowed a typical c-type cytochrome. The thirdband appeared only when the enzyme was ex-posed to alkaline pH or was dissociated by treat-ment with SDS in the presence of 2-mercapto-ethanol. The possibility that the third band wasa contaminant might be excluded judging fromthe gel filtration profile of the purified enzyme,although the third band stained weakly in SDS-gel electrophoresis. Indeed, similar-sized bandswere observed with other species ofdehydrogen-ase solubilized and purified to homogeneity (2,3, 19). Although ferricyanide was linked directlyto the dehydrogenase in the routine assay ofenzyme activity, enzyme oxidized with ferricya-nide was reduced by D-fructose (Fig. 1). Thisindicates that the cytochrome c of the purifiedD-fructose dehydrogenase complex is an intrinsiccomponent of the D-fructose oxidase system. Itis also suggested that the purified D-fructosedehydrogenase complex is a portion of the D-fructose oxidase buried in the bacterial mem-brane and linked to the respiratory chain of theorganism. During solubilization of the enzyme
VOL. 145, 1981
on January 21, 2019 by guesthttp://jb.asm
.org/D
ownloaded from

822 AMEYAMA ET AL.
from the membrane fraction, Triton X-100caused release of the D-fructose dehydrogenasecomplex from the membrane. The enzyme wassolubilized in the presence of Triton X-100, likethe alcohol dehydrogenases of G. suboxydans(3) and Acetobacter aceti (2), the aldehyde de-hydrogenase of G. suboxydans (4), and gluco-nate dehydrogenases of P. aeruginosa (19) andS. marcescens (24). In the presence of Triton X-100, the purified enzyme exhibited a molecularweight of approximately 140,000. The purifiedenzyme was apparently monomeric in solutioncontaining at least 0.1% Triton X-100 and tendedto aggregate when Triton X-100 was removedfrom the enzyme solution, causing decrease ofthe enzyme activity. These results suggest thatD-fructose dehydrogenase has a hydrophobicand probably membrane-lipid-interacting struc-ture and is thus an intrinsic membrane protein,although it remains unclear which polypeptideof the three components causes the hydropho-bicity.The specific activity of the purified enzyme
was approximately 170 to 180 ,tmol of D-fructoseoxidized per mg of protein per min. Yamada etal. (31) reported on the partial purification ofthe enzyme from G. cerinus and got a finalenzyme preparation having a specific activity of300 after DEAE-cellulose chromatography.They assayed the enzyme activity with 2,6-di-chlorophenolindophenol and defined one en-zyme unit as the amount of enzyme catalyzing a0.01 decrease in absorbance at 590 nm at pH 6.0.When converted to the enzyme units used in thispaper, the activity is about 30,umol of D-fructoseoxidized per mg of protein per min, assuming anextinction coefficient of 2,6-dichlorophenolindo-phenol of 10 mM-'.cm-' at 590 nm (pH 6.0).With their enzyme preparation, several sub-strates were oxidized, and the reaction rate ofD-glucose oxidation was two times higher thanthat of D-fructose. It is quite probable that theenzyme preparation of Yamada et al. was stillcontaminated with some enzymes predomi-nantly found in the bacterial membrane of theorganism. The results so far obtained indicatethat the membrane-bound dehydrogenases showstrict substrate specificity, that is, D-gluconateoxidation is only catalyzed by D-gluconate de-hydrogenase, glycerol oxidation is only catalyzedby glycerol dehydrogenase, alcohol oxidation isonly catalyzed by alcohol dehydrogenase, alde-hyde oxidation is only catalyzed by aldehydedehydrogenase, and D-glucose oxidation is onlycatalyzed by D-glucose dehydrogenase (1-4, 18,19, 24). The purified D-fructose dehydrogenasecatalyzed the oxidation of D-fructose exclusively.
In this study, it was indicated that D-fructosedehydrogenase could be useful as an analytical
reagent for D-fructose determination. An enzy-matic method of D-fructose determination hasbeen reported by Bernt and Bergmeyer (10)using a coupled reaction composed of hexoki-nase (EC 2.7.1.1), phosphoglucose isomerase(D-glucose-6-phosphate ketol isomerase, EC5.3.1.9) and D-glucose-6-phosphate dehydrogen-ase (EC 1.1.1.49). This assay method has beenevaluated as the most sensitive enzymaticmethod so far reported, but it depends largelyon the purity of the reagents used and is ratherexpensive. Difficulty in preparing the requiredenzymes without any contaminants could makethis assay system complicated and potentiallytroublesome. Several advantages were found inthe use of the D-fructose dehydrogenase for thepurpose of enzymatic D-fructose determination.D-Fructose was oxidized at an acidic pH, and noappreciable interference was observed when thesystem was tested in the presence of varioussusbtances at high concentrations. As D-fructoseoxidation was catalyzed only by the D-fructosedehydrogenase, the bacterial membrane of G.industrius could be used for the purpose, ifsamples for D-fructose determination wereknown to contain only D-fructose. We have al-ready reported the uses of membrane-bounddehydrogenases to assay ethanol (6), acetalde-hyde (4), and D-gluconate (7). In the same way,the D-fructose dehydrogenase could be used forthe enzymatic microdetermination of D-fructose.Regarding the reaction product of D-fructose
dehydrogenase, Mowshowitz et al. (20, 21) haveproposed that 5-keto-D-fructose has a role as theoxidant ofNADPH during growth on D-fructose.Recently, we have succeeded in the crystalliza-tion of 5-keto-D-fructose reductase (Ameyamaet al., Abstr. Annu. Meet. Agric. Chem. Soc. Jpn.1980, p. 507). The role of 5-keto-D-fructose re-ductase and the fate of 5-keto-D-fructose will bediscussed elsewhere.
ACKNOWLEDGMENTS
The authors wish to thank Keiko Mieno for her skillfultechnical assistance during the course of this work. We alsothank S. Oka, Department of Technology, Hiroshima Univer-sity, for his kind advice in analytical ultracentrifugation.
This work has been supported in part by Scientific Re-search Grant 486028 from the Ministry of Education, Scienceand Culture of Japan (1979-1980).
LITERATURE CITED1. Adachi, 0. 1979. Studies on enzymes in carbohydrate
metabolism of acetic acid bacteria. J. Agric. Chem. Soc.Jpn. 53:R77-R86.
2. Adachi, O., E. Miyagawa, E. Shinagawa, K. Mat-sushita, and M. Ameyama. 1978. Purification andproperties of particulate alcohol dehydrogenase fromAcetobacter aceti. Agric. Biol. Chem. 42:2331-2340.
3. Adachi, O., K. Tayama, E. Shinagawa, K. Mat-sushita, and M. Ameyama. 1978. Purification andcharacterization of particulate alcohol dehydrogenase
J. BACTERIOL.
on January 21, 2019 by guesthttp://jb.asm
.org/D
ownloaded from

ACETIC ACID BACTERIAL D-FRUCTOSE DEHYDROGENASE 823
from Giuconobacter suboxydans. Agric. Biol. Chem.42:2045-2056.
4. Adachi, O., K. Tayama, E. Shinagawa, K. Mat-sushita, and M. Ameyama. 1980. Purification andcharacterization of membrane-bound aldehyde dehy-drogenase from Gluconobacter suboxydans. Agric. Biol.Chem. 44:503-515.
5. Aida, K., and Y. Yamada. 1964. A new enzyme, 5-ketofructose reductase. Agric. Biol. Chem. 28:74-75.
6. Ameyama, ML, K. Tayama, E. Miyagawa, E. Shina-gawa, K. Matsushita, and 0. Adachi. 1978. A new
enzymatic microdetermination procedure for ethanolwith particulate alcohol dehydrogenase from acetic acidbacteria. Agric. Biol. Chem. 42:2063-2069.
7. Ameyama, AL, K. Tayama, E. Shinagawa, K. Mat-sushita, and 0. Adachi. 1978. New enzymatic deter-mination of D-gluconate with particulate D-gluconatedehydrogenase. Agric. Biol. Chem. 42:2347-2354.
8. Avigad, G., and S. Englard. 1965. 5-Keto-D-fructose. I.Chemical characterization and analytical determinationof the dicarbonyl-hesose produced by Gluconobactercerinus. J. Biol. Chem. 240:2290-2296.
9. Avigad, G., S. Englard, and S. Pfko. 1966. 5-Keto-D-fructose. IV. A specific reduced nicotine adenine dinu-cleotide phosphate-linked reductase from Gluconobac-ter cerinus. J. Biol. Chem. 241:373-378.
10. Bernt, E., and H. U. Bergmeyer. 1974. D-Fructose.Methods Enzymatic Anal. 3:1304-1307.
11. Chervenka, C. H. 1970. A manual of methods for theanalytical ultracentrifuge, p. 23. Beckman InstrumentCo., Fullerton, California.
12. Davis, B. J. 1964. Disc electrophoresis. H. Methods ofapplication to human serum proteins. Ann. N. Y. Acad.Sci. 121:404-427.
13. Dulley, J. R., and P. A. Grieve. 1975. A simple techniquefor eliminating interference by detergents in the Lowrymethod of protein determination. Anal. Biochem. 64:136-141.
14. Englard, S., and G. Avigad. 1965. 5-Keto-D-fructose. II.Patterns of formation and of associated dehydrogenaseactivities in Gluconobacter cerinus. J. Biol. Chem. 240:2297-2301.
15. Englard, S., G. Avigad, and L. Prosky. 1965. 5-Keto-D-fructose. m. Proofofstructure based on stereospecificpatters of enzymatic reduction. J. Biol. Chem. 240:2302-2307.
16. Levin, 0. 1962. Column chromatography of proteins: cal-cium phosphate. Methods Enzymol. 5:27-32.
17. Lowry, O. H, N. J. Rosebrough, A. L. Farr, and R. J.Randall. 1951. Protein measurement with the Folinphenol reagent. J. Biol. Chem. 193:265-275.
18. Matsushita, K., Y. Ohno, E. Shinagawa, 0. Adachi,and M. Ameyama. 1980. Membrane-bound D-glucosedehydrogenase from Pseudomonas sp. Solubilization,purification and characterization. Agric. Biol. Chem.44:1505-1512.
19. Matsushita, K., E. Shinagawa, 0. Adachi, and M.
Ameyama. 1979. Membrane-bound D-gluconate dehy-drogenase from Pseudomonas aeruginosa. Purificationand structure of cytochrome binding form. J. Biochem.85:1173-1181.
20. Mowshowitz, S., G. Avigad, and S. Englard. 1974. 5-Keto-D-fructose: formation and utilization in the courseof D-fructose assimlation by Gluconobacter cerinus. J.Bacteriol. 118:1051-1058.
21. Mowshowitz, S., S. Englard, and G. Avigad. 1974.Metabolic consequence of a block in the synthesis of 5-keto-D-fructose in a mutant of Gluconobacter cerinus.J. Bacteriol. 119:363-370.
22. Okunuki, K., and T. Yamanaka. 1970. Cytochromes, p.3. Asakura-shoten, Tokyo.
23. Sato, K., Y. Yamada, A. Aida, and T. Uemura. 1969.Enzymatic oxidation of sugar and sugar alcohol. VIII.Particle-bound L-sorbose dehydrogenase from Glu-conobacter suboxydans. J. Biochem. 66:521-527.
24. Shinagawa, E., K. Matsushita, 0. Adachi, and M.Ameyama. 1978. Membrane-bound D-gluconate dehy-drogenase of Serratia marcescens. Agric. Biol. Chem.42:2355-2361.
25. Terada, O., S. Suzuki, and S. Kinoshita. 1961. For-mation of 5-dehydrofructose by members of Acetobac-ter species. Part HI. Characterization of the unknownsubstance. J. Agric. Chem. Soc. Jpn. 35:178-182.
26. Terada, O., K. Tomizawa, and S. Kinoshita. 1961.Formation of 5-dehydrofructose by members of Aceto-bacter species. Part I. Formation of a new reducingsubstance from fructose and its discrimination fromglucosone. J. Agric. Chem. Soc. Jpn. 35:127-130.
27. Terada, 0., K. Tomizawa, S. Suzuki, and S. Kino-shita. 1961. Formation of 5-dehydrofructose by mem-bers of Acetobacter species. Part II. Studies on thefermentation, purification and properties of crystal. J.Agric. Chem. Soc. Jpn. 35:131-134.
28. Weber, K., and M. Osborn. 1969. The reliability ofmolecular weight determinations by dodecylsulfate-polyacrylamide gel electrophoresis: J. Biol. Chem. 244:4406-4412.
29. Wood, W. A., R. A. Fetting, and B. C. Hertlein. 1962.Gluconic dehydrogenase from Pseudomonas fluores-cens. Methods Enzymol. 5:287-291.
30. Yamada, Y., K. Aida, and T. Uemura. 1966. A newenzyme, D-fructose dehydrogenase. Agric. Biol. Chem.30:95-96.
31. Yamada, Y., K. Aida, and T. Uemura. 1967. Enzymaticstudies on the oxidation of sugar and sugar alcohol. L.Purification and properties of particle-bound fructosedehydrogenase. J. Biochem. 61:636-646.
32. Yamada, Y., K. Aida, and T. Uemura. 1968. CoenzymeQlo in the respiratory chain linked to fructose dehydro-genase of Gluconobacter cerinus. Agric. Biol. Chem.32:532-534.
33. Yamanaka, T., and K. Okunuki. 1975. Cytochromes ofmicroorganisms, p. 10 and 104. Kohdansha-scientific,Tokyo.
VOL. 145, 1981
on January 21, 2019 by guesthttp://jb.asm
.org/D
ownloaded from





























