Embed Size (px)
Citation preview

D G Kanhere, Center for Simulation and modelingPune University,
Mastani School- IISER Pune ,July 2014

What is Molecular Dynamics (An Overview)
Ab Initio MD Born Oppenheimer Dynamics Car Parrinello Dynamics
General Comment on “ How to set up dynamics run”
Thermodynamics of clustersMultiple histogram methodsResults of clusters – As Examples

System Many particles interacting with each other
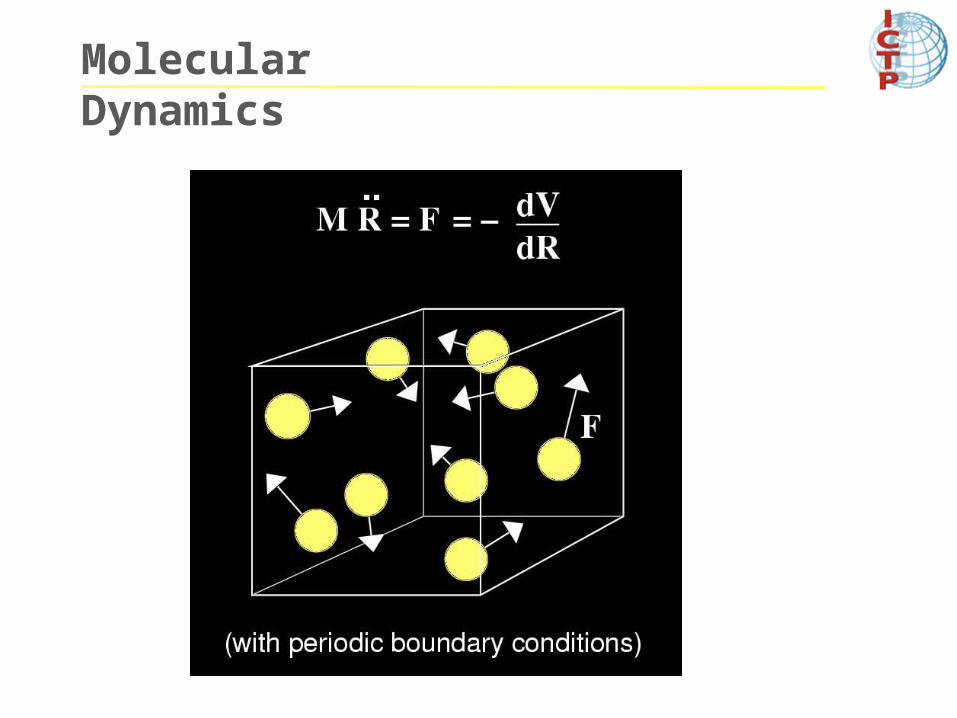
Solid/liquid : Box of Volume V, Temperature T and Pressure P: Periodic BC
Finite size systems clusters :Free BC Given the forces acting on all the ions,
and initial state at time t=0, Compute Trajectories of all the particles as a function of
time t, by using Newton's laws: Essentially exact
We have entire phase space trajectories! Therefore all the information to compute various statistical quantities from microscopic description.

Molecular Dynamics








Consider N particles interacting VIA Lennard Jones Potential (N ~ 10-13)
For given co-ordinates {Ri}write down total energy : V ( R1,R2,…)
Compute Force on atom i.Write a subroutine which takes
coordinates as input and returns total potential energy and forces on each of the atoms.
You are now ready to do simulation experiments after one more step.

)(2
1)()()()1( 32 tOt
M
FttvtRttR
I
IIII
)(2
1)()()()2( 32 tOt
M
FttvtRttR
I
IIII
)()(2)()()2()1( 42 tOtM
FtRttRttR
I
IIII
)(txI
tt
)()()(2)( 42 tOtM
FttRtRttR
I
IIII
II
I Fdt
RdM
2
2
Verlet’s algorithm
How to integrate Newton’s equations

Initialize: select starting atomic positions and velocities as close aspossible to thermal equilibrium
Integrate: compute all forces and determine new positions using Verlet’s algorithm
Equilibrate: let the system loose memory of the initial configurations,i.e. let it reach thermal equilibrium
Average: accumulate averages of observables of interest (A)
PdRdE
PdRdEPRAA
)exp(
)exp(),(
T
dttPtRAT
A0
))(),((1
Average in Molecular Dynamics = Average in Statistical Mechanics
A typical molecular dynamics protocol








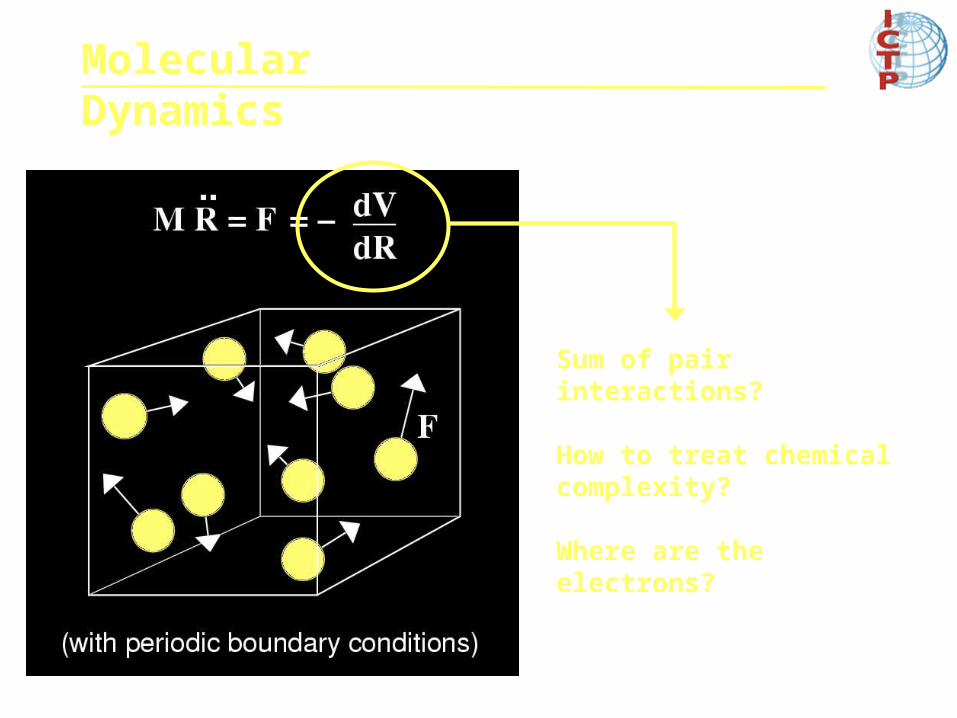
Sum of pair interactions?
How to treat chemical complexity?
Where are the electrons?
Molecular Dynamics

“Empirical” potentials
Starting point: Pair potentials (Lennard-Jones, Born-Mayer, Coulomb, etc)
+ three-body corrections
+ density dependent terms (embedded atom models)
+ atomic distorsion terms (includes polarization)
+ charge transfer terms
...,,426
0
12
0 etceR
Ze
R
R
R
R R
JI RRR
I
J
K
KJIVKJI ˆ3/1cos),,( 0)3(
Parameters are determined from “empirical” data, such as experimental EOS, vibrations, phase diagrams, dynamical properties, etc.

Quantum simulations: The “standard model”
HE
Schroedinger equationfor electrons
“Molecular dynamics” for atoms
Ma = F = -dE/dR
e--e- interactions: Density Functional Theorye--nuclei interactions: Pseudopotentials
“Ab-initio” molecular dynamics = Classical molecular dynamics in the potential energy surface generated by the electrons in their quantum ground state
R. Cohen

Electrons respond much faster thannuclei to external forces, because of their lighter mass, therefore:
Electrons are always in their instantaneous quantum mechanicalground state, for each given {R}.
Consequence: Every MD step requires the calculation of the QM ground state of the electrons
Where are the electrons? The adiabatic approximation
II
II dR
RdEF
dt
RdM
})({2
2
The electronic Hamiltonian He depends parametrically on thenuclear positions {R}
Nuclei
e-
e-
e-
e-
e-e-
eee RHRE

Density functional theory
Electron-electron (many-body)interaction
Density-functional theory [W. Kohn et al. 1964-1965] states that the e-e interaction can be written as a one-electron “effective” term:
However, the exact functional form of Vxc is not (yet) known.Current approximations to the exact Vxc go under different names (LDA, BLYP, BP, GGA, “hybrid”, et al). While GGA and hybrid functionals provide slightly better results than other approximations, the choice of the Vxc is often made in such a way to improve agreement with exps (“ab fine”??).

I
e
II
II dR
RHd
dR
RdEF
dt
RdM
00
2
2 })({})({
00
0000
000
000
000
000
00
})({
})({})({
})({})({})({
})({})({})({})({
I
e
II
e
III
e
Iee
II
e
I
e
dR
RdH
dR
dRE
dR
RdH
dR
dRE
dR
dRE
dR
RdH
dR
dRHRH
dR
d
dR
RdH
dR
RHd
“Ab-initio” forces: the Hellmann-Feynman theorem
Forces can be calculated without
recalculating the ground state wave
function for small atomic
displacements

The Born Oppenheimer Dynamics
How to keep electron in the ground state (1)
II
II dR
RdEF
dt
RdM
})({2
2
00 })({})({ RHRE e
jeiji
jie HccH ,
*
ii
ic
If we expand wavefunctions in a basis set
finding the ground state is equivalent tominimizing a quadratic form in the {c}’s(variational principle). So, standard minimizationschemes can be used, e.g. steepest descent:
ee HH

Carr- Parrinello dynamics

How to keep electron in the ground state (2)
II
II dR
RdEF
dt
RdM
})({2
2
00 })({})({ RHRE e
})({RH e
{R}
})({RH e
III dR
RdERM
})({
The Car-Parrinello algorithm
down to the minimumfor each {R}
???

(i) integrate the equations of motion on the (long) time scale set by the nuclear motion but nevertheless
(ii) take intrinsically advantage of the smooth time evolution of the dynamically evolving electronic subsystem as much as possible.
The second point allows to circumvent explicit diagonalization or minimization to solve the electronic structure problem for the next molecular dynamics step as it is done in Born-Oppenheimer Molecular dynamics. Car-Parrinello molecular dynamics is an efficient method to satisfy requirement (ii) in a numerically stable fashion and makes an acceptable compromise concerning the length of the time step (i).
Car-Parrinello Molecular Dynamics

In CPMD a two-component quantum / classical problem is mapped onto a two-component purely classical problem with two separate energy scales at the expense of loosing the explicit time dependence
of the quantum subsystem dynamics.
Now, in classical mechanics the force on the nuclei is obtained from the derivative of a Lagrangian with respect to the nuclear positions. This suggests that a functional derivative with respect to the orbitals, which are interpreted as classical fields, might yield the force on the orbitals, given a suitable Lagrangian. In addition, possible constraints within the set of orbitals have to be imposed, such as e.g.orthonormality (or generalized orthonormality conditions that include an overlap matrix).

Car-Parrinello Lagrangian
1 1| { } ( | )
2 2CP R i i i DFT i ij i i ijR i ij
L M R E
Car and Parrinello (1985) have postulated the following Lagrangian
where i are fictitious masses and i are classical fields. Classical action needs to be minimized which results in equation of motions
( )
( ) ( )
( ) ( )
( , ) ( , )
CP
CP CP
CP CP
i i
S L t dt
d dL t dL t
dt dR dRd dL t dL t
dt d r t d r t
Kinetic Energy Potential Energy Constraints

2[{ },{ }]( , )
| ( ) |
[{ },{ }]( , ) ( , )
( , )
( , ) ( , )
DFT i RR
DFT ii i ij j
ji
DFT i ij jj
E R Z eM R r t dr
R r R t
E Rr t r t
r t
H r t r t
Car-Parrinello Equations of Motions
Car and Parrinello (1985) have derived equations of motions
which are obviously transformed back to Born-Oppenheimer molecular dynamics if fictitious masses for the electrons i
At the eqilibrium, there are no forces on electrons, thereforeGround state of density functional theory is reached with theeigenvalues being the Kohn Sham eigenstates.

Why does the Car-Parrinello Method works?
Conserved Energy in CPMD is not a physical energy but supplemented with a small fictitious kinetic term
1{ }
2
1 1| [{ }{ }]
2 2
1|
2
phys R DFT iR
cons R i i i DFT i phys fictR i
fict i i ii
E M R E
E M R E R E T
T
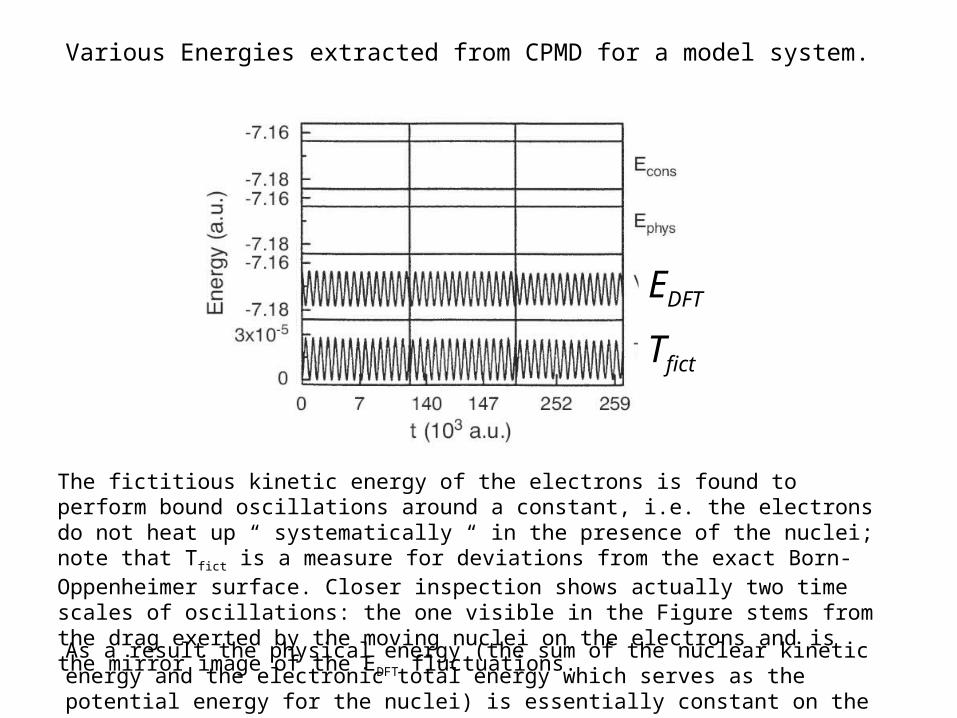
Various Energies extracted from CPMD for a model system.
Tfict
EDFT
The fictitious kinetic energy of the electrons is found to perform bound oscillations around a constant, i.e. the electrons do not heat up “ systematically “ in the presence of the nuclei; note that Tfict is a measure for deviations from the exact Born-Oppenheimer surface. Closer inspection shows actually two time scales of oscillations: the one visible in the Figure stems from the drag exerted by the moving nuclei on the electrons and is the mirror image of the EDFT fluctuations. As a result the physical energy (the sum of the nuclear kinetic energy and the electronic total energy which serves as the potential energy for the nuclei) is essentially constant on the relevant energy and time scales.

Given the adiabatic separation and the stability of the propagation, the centralquestion remains if the forces acting on the nuclei are actually the “correct" onesin Car-Parrinello molecular dynamics. As a reference serve the forces obtainedfrom full self-consistent minimizations of the electronic energy at each time step, i.e. Born-Oppenheimer molecular dynamics with extremely well converged wavefunctions.

How to control adiabaticity?
Since the electronic degrees of freedom are described by much heavier masses than the electronic masses, time step to perform CPMD simulations needs not to be too small as compared to Ehrenfest molecular dynamics.
For a system with a gap in the spectrum, the lowest possible frequency of “fictitious” electronic oscillations
2min
1/ 2
min
~
~
gap
gap
E
E
To guarantee adiabatic separation this frequency should bemuch larger than the typical phonon energy and/or the gapin the spectrum which would make sure that the electronsfollow the nuclei adiabatically. Hence fictitious mass .

At the same time, small fictitious mass would implysmaller and smaller time step because maximum fictitious electronic frequency is proportional to the plane-wave cutoff energy
2
1/ 2
max
1/ 2
max
~
~
~
max cut
cut
cut
E
E
tE
As a result a compromise fictitious mass needs to be foundin CPMD simulations.
For metals gap is zero and zero frequency “fictitious” electronicmodes occur in the spectrum overlapping with the phonon spectrum. Thus, a well-controlled Born-Oppenheimer approach can only be recommended

| |H
n nn
c
2
| |
| | 1
n n m mnm
nnm
c H c
c
Consider variational principle which can be used to find an upper bound for the lowest eigenstates of the hamiltonian
using basis set expansion
CP Method as dynamical solution of DFT equationsCP Method invented a new way to solve Kohn-Shamequations alternative to diagonalization.
CPMD offers a way to determine the coefficients withoutreduction to the eigenvalue problem.

Born-Oppenheimer –versus- Car-Parrinello AIMD
Born-Oppenheimer Car-Parrinello
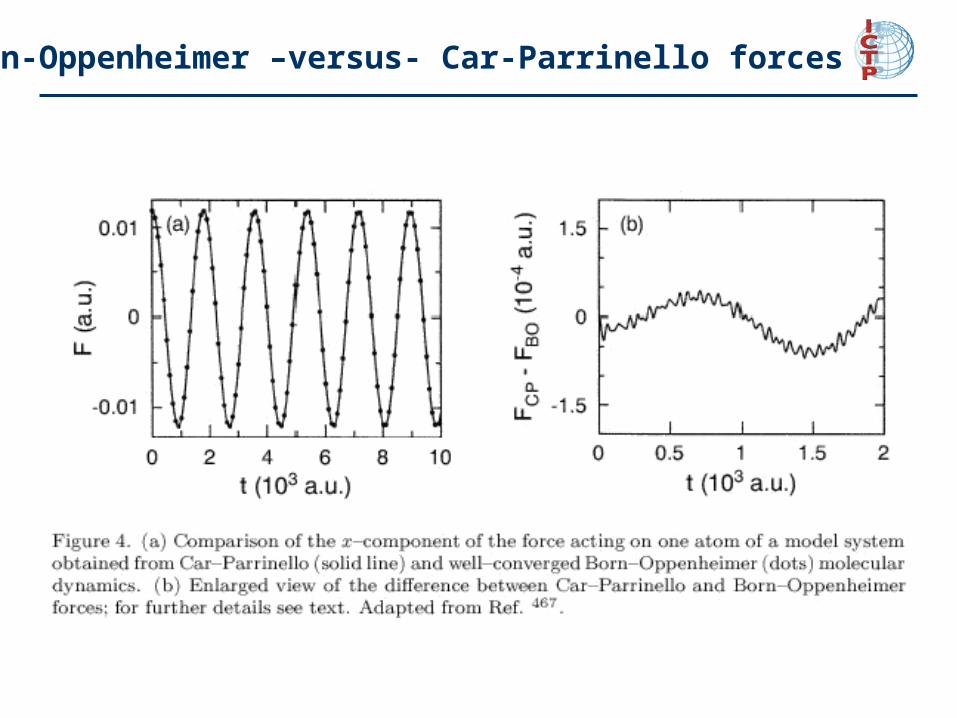
Born-Oppenheimer –versus- Car-Parrinello forces

from M Sprik
Born-Oppenheimer –versus- Car-Parrinello: summary

• R. Car and M. Parrinello, Phys. Rev. Lett. 55, 2471 (1985)
• M. Payne, M. Teter, D. Allan, T. Arias, J. Joannopoulos, Rev. Mod. Phys. 64, 1045 (1992).
• D. Marx, J. Hutter, "Ab Initio Molecular Dynamics: Theory and Implementation", in "Modern Methods and Algorithms of Quantum Chemistry" (p. 301-449), Editor: J. Grotendorst, (NIC, FZ Jülich 2000)
• http://www.theochem.ruhr-uni-bochum.de/research/marx/cprev.en.html
• R. Rousseau and S. Scandolo, “Car-Parrinello Molecular Dynamics”, in “Encyclopedia of Condensed Matter Physics”, edited by G. Bassani, G. Liedl, and P. Wyder, Elsevier, Amsterdam (2005)
Ab-initio Molecular Dynamics: bibliography

Finite Temperature Properties of finite size systems
•D G KanhereD G Kanhere•Pune University Pune University

Lindemann Criteria

Mean Square Displacements

Extracting ionic density of States
•Constant Temperature
•Constant Volume
•Phase space trajectories
•The Multiple Histogram Method
•To
•Extract Density of States









Sodium Clusters
● Simplest of atomic clusters● Jellium model works ( Depends…)● Nice delocalized charge density● Magic Clusters at N=8,20,40,58,92,138,…● Icosahedra for N=13,55,147,…
Chacko et al., Phys. Rev. B 71,155407 (2005)
Lee et al., J. Chem. Phys. 123,164310 (2005)Lee et al., Phys. Rev. B (2007)Shahab et al., Phys Rev B ( 2007)Gazi et al J Chem Phys ( 2008)

The heat Capacities
N=40 : peaked
N=50 : flat
N=55 : very sharp
N=58 : peaked but broad BUT Highest melting Point ( Tm = 375 K )

Sodium Clusters
● Simplest of atomic clusters● Jellium model works ( Depends…)● Nice delocalized charge density● Magic Clusters at N=8,20,40,58,92,138,…● Icosahedra for N=13,55,147,…
Chacko et al., Phys. Rev. B 71,155407 (2005)
Lee et al., J. Chem. Phys. 123,164310 (2005)Lee et al., Phys. Rev. B (2007)Shahab et al., Phys Rev B ( 2007)Gazi et al J Chem Phys ( 2008)

Application II
ThermodynamicsSize sensitive specific heats
Magic meltersHigher than Bulk Melting
temperatures

The Case of Gallium and Aluminum clusters
● Gallium and aluminum clusters show extreme size sensitivity
● Gallium clusters melt at Higher than their Bulk Tm
● Magic Melters ----- Non - Melter

Gallium Clusters: Heat Capacity
Experimental Data from Indiana group
*
N=30 Flat
N=31 Peaked
*
Tm Shifts by 200K @ N=45
And by 350K across the series

Calculations
• Density functional with LDA/GGA• Born Oppenheimer MD• Delta T 100 au• Total simulation time per temp 100 ps or
more• Soft pseudo potentials, plane waves• Extensive search for equilibrium
geometries. Simulated annealing, Basin Hopping …

The Heat Capacity : Ga30- Ga31
Joshi et al. Phys. Rev. Lett. 96,135703 (2006)

The MSD : Ga30 and Ga31

Finite Size Effect : Amorphous Clusters
• In amorphous cluster each atom may have different environment.
• Atoms may be bonded with the rest of the cluster with different strength.
• When heated, they will begin diffusive motion at different temperatures
• This may result in continuous phase change.
• No sharp peak in specific heat, very broad transition.
• Cluster with large island having local order will show a peak in the heat capacity ..Most of the atoms “melt” together.

Thermodynamics: Ga27Si3Ghazi and Kanhere J Phys Chem (2012)

Ga13 – Ga12C and Al13 - Al12C
Ga13 is Decahedra
Ga12C is a Perfect Icosahedra

Calculated Specific Heat Ga
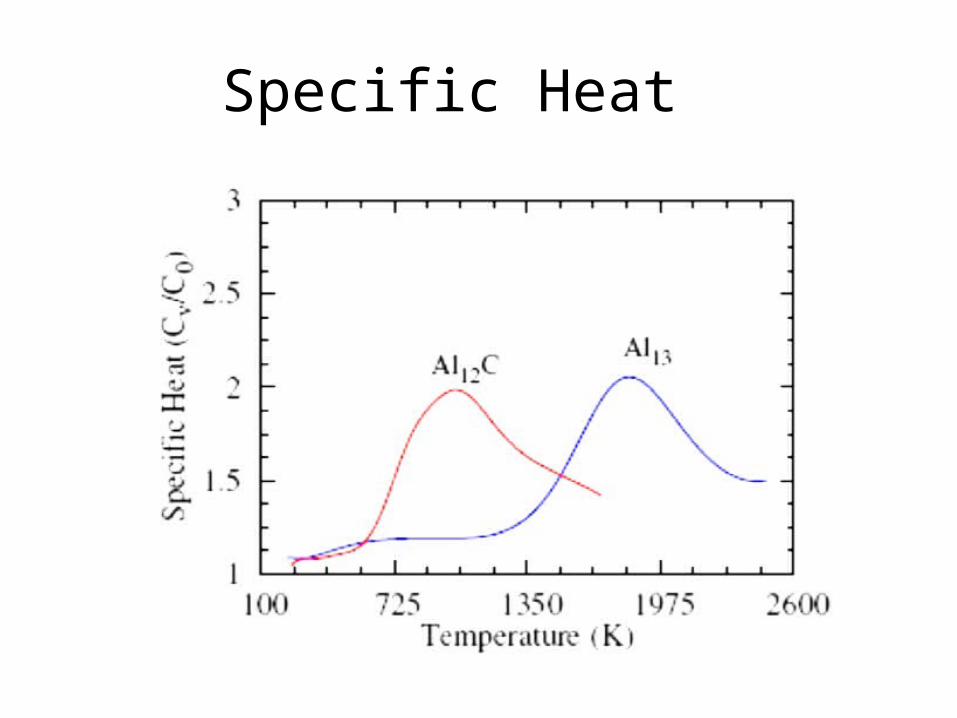
Specific Heat