Embed Size (px)
Citation preview

D1 Dopamine Receptor Mediates Dopamine-induced Cytotoxicityvia the ERK Signal Cascade*
Received for publication, April 7, 2004, and in revised form, May 21, 2004Published, JBC Papers in Press, July 6, 2004, DOI 10.1074/jbc.M403891200
Jun Chen‡, Milan Rusnak‡, Robert R. Luedtke§, and Anita Sidhu‡¶
From the ‡Department of Pediatrics, Georgetown University, Washington, D. C. 20007 and the §Department ofPharmacology and Neuroscience, University of North Texas Health Science Center, Fort Worth, Texas 76107
Postsynaptic striatal neurodegeneration occursthrough unknown mechanisms, but it is linked to highextracellular levels of synaptic dopamine. Dopamine-mediated cytotoxicity of striatal neurons occursthrough two distinct pathways: autoxidation and the D1dopamine receptor-linked signaling pathway. Here weinvestigated the mitogen-activated protein kinase(MAPK) signaling pathways activated upon the acutestimulation of D1 dopamine receptors. In SK-N-MC neu-roblastoma cells, endogenously expressing D1 dopaminereceptors, dopamine caused activation of phosphoryl-ated (p-)ERK1/2 and of the stress-signaling kinases, p-JNK and p-p38 MAPK, in a time- and dose-dependentmanner. Selective stimulation of D1 receptors with theagonist SKF R-38393 caused p-ERK1/2, but not p-JNK orp-p38 MAPK activation, in a manner sensitive to thereceptor-selective antagonist SCH 23390, protein kinaseA inhibition (KT5720), and MEK1/2 inhibition (U0126 orPD98059). Activation of ERK by D1 dopamine receptorsresulted in oxidative stress and cytotoxicity. In cellstransfected with a catalytically defective mutant ofMEK1, the upstream ERK-specific kinase, both dopa-mine- and SKF R-38393-mediated cytotoxicity was mark-edly attenuated, confirming the participation of theERK signaling pathway. Cell fractionation studiesshowed that only a small amount of p-ERK1/2 was trans-located to the nucleus, with the majority retained in thecytoplasm. From coimmunoprecipitation studies, p-ERKwas found to form stable heterotrimeric complexes withthe D1 dopamine receptor and �-arrestin2. In cellstransfected with the dominant negative mutant of �-ar-restin2, the formation of such complexes was substan-tially inhibited. These data provide novel mechanisticinsights into the role of ERK in the cytotoxicity medi-ated upon activation of the D1 dopamine receptor.
Striatal neurodegeneration is found in several human disor-ders involving postsynaptic dopamine neuronal dysfunction,such as the L-dopa-unresponsive parkinsonism subtype of mul-tiple system atrophy (1), methamphetamine-induced neurotox-icity (2, 3), and secondary dopamine dysfunction in Hunting-ton’s disease (4), in which dopaminergic transmission isinterrupted by progressive loss of the striatal neurons bearingthe postsynaptic D1 and D2 dopamine receptors (5). Although
all of these diseases are characterized by elevated dopaminelevels in the synapse, the underlying pathological mecha-nism(s) promoting striatal degeneration remains enigmatic.Recent studies suggest a role for the participation of bothreactive oxygen species and reactive nitrogen species in thedegeneration of striatal neurons (6–9). Reactive oxygen speciesand reactive nitrogen species may be produced through autox-idation of extracellular dopamine in the synapse and may beespecially important in the striatal degeneration seen in theL-dopa-unresponsive parkinsonism subtype of multiple systematrophy (1, 10).
Dopamine in the synapse causes activation of the postsyn-aptic D1 and D2 subtypes of the dopamine receptor. Accumu-lating evidence in primates indicates that blockage of D1 do-pamine receptors with selective antagonists has strongneuroprotective and anti-parkinsonian effects (11, 12), sup-porting a coparticipatory role of the D1 receptors in the main-tenance and/or pathogenesis of postsynaptic neurodegenera-tion. In both rat striatal primary cultures (13) and in SK-N-MCneuroblastoma cells (14), which endogenously express a func-tional D1 dopamine receptor (15), we have shown recently thatthe chronic treatment of these cells with dopamine promotesincreased expression of the nitric-oxide synthases, neuronalnitric-oxide synthase and inducible nitric-oxide synthase, ac-companied by increased nitric oxide production, oxidativestress, and cytotoxicity. These effects of dopamine were onlypartially mimicked by the oxidant H2O2 and by direct stimu-lation of the D1 dopamine receptor with the selective agonistSKF R-38393. Dopamine effects were only partly blocked by theantioxidant sodium metabisulfite (SMBS)1 and by the D1 re-ceptor antagonist SCH 23390. However, together these com-pounds completely ablated the actions of dopamine (13, 14).These data suggest that dopamine-mediated responses are me-diated by at least two distinct and nonoverlapping pathways:through extracellular autoxidation of the compound andthrough chronic activation of the D1 dopamine receptor.
The current studies were undertaken to examine further theprocytotoxic signaling pathways activated by D1 dopamine re-ceptors. We examined the role of mitogen-activated proteinkinases (MAPKs), the downstream mediators of signal trans-duction from the cell surface receptors to the nucleus. MAPKshave been implicated in a wide variety of cellular processessuch as proliferation, differentiation, and apoptotic cell death.In mammals, three major groups of MAPKs have been identi-fied: extracellular signal-regulated kinases 1 and 2 (ERK1/2)(16), p38 MAPK (17), and c-Jun N-terminal kinase (JNK) (18).* This work was supported by National Institutes of Health Grant
NS-34914. The costs of publication of this article were defrayed in partby the payment of page charges. This article must therefore be herebymarked “advertisement” in accordance with 18 U.S.C. Section 1734solely to indicate this fact.
¶ To whom correspondence should be addressed: Laboratory of Mo-lecular Neurochemistry, Research Bldg., Rm. W222, 3970 Reservoir Rd.NW, Washington, D. C. 20007. Tel.: 202-687-0282; Fax: 202-687-0279;E-mail: [email protected].
1 The abbreviations used are: SMBS, sodium metabisulfite; ERK1/2,extracellular signal-regulated kinases 1 and 2; GPCR, G protein-cou-pled receptor; JNK, c-Jun N-terminal kinase; MAPK, mitogen-activatedprotein kinase; MEK1/2; MAPK/ERK kinase 1 and 2; p-, phosphoryl-ated; p38 MAPK, 38-kDa mitogen-activated protein kinase.
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 279, No. 38, Issue of September 17, pp. 39317–39330, 2004© 2004 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A.
This paper is available on line at http://www.jbc.org 39317
by guest on April 3, 2019
http://ww
w.jbc.org/
Dow
nloaded from
The ERK-linked signaling pathways are stimulated by receptortyrosine kinases (19) and G protein-coupled receptors (GPCRs)(20) and generally lead to a mitogenic and proliferative re-sponse (21). JNK and p38 MAPK are stimulated by cellularstresses, such as free radicals and inflammatory agents, lead-ing to apoptotic cell death (22). In the present studies, theactivation of MAPKs was assessed by examining the expressionlevels and function of the phosphorylated MAPKs: p-JNK, p-p38 MAPK, and p-ERK1/2. Instead of observing an elevation ofthe archetypical proapoptosis MAPKs, p-JNK and p-p38MAPK, we surprisingly found that D1 dopamine receptors se-lectively stimulated the ERK1/2 signaling cascade. Moreover,the activated p-ERK1/2 was primarily retained in the cyto-plasm and formed a heterotrimeric complex with the D1 recep-tor and �-arrestin2, with only low levels seen in the nuclei,
suggesting that the failure of p-ERK to be translocated into thenucleus may be linked to the subsequent cytotoxic response.Our findings underscore a novel and selective mechanism bywhich stimulation of D1 dopamine receptors promotes oxida-tive stress and cytotoxicity while providing a heuristic view inwhich to evaluate alternate roles of p-ERK1/2 in mediatingthese cellular responses.
EXPERIMENTAL PROCEDURES
Materials—Human SK-N-MC neuroblastoma cells were obtainedfrom the American Type Culture Collection (Rockville, MD). Dopamine,hydrogen peroxide (H2O2), SMBS, SKF R-38393, SCH 23390, and for-skolin were purchased from Sigma. KT5720, U0126, SB203580, andPD98059 were from Calbiochem, and SB600125 was from Biomol Re-search Laboratories, Inc.
Cell Culture, Drug Treatment, and Cell Viability—Human neuroblasto-
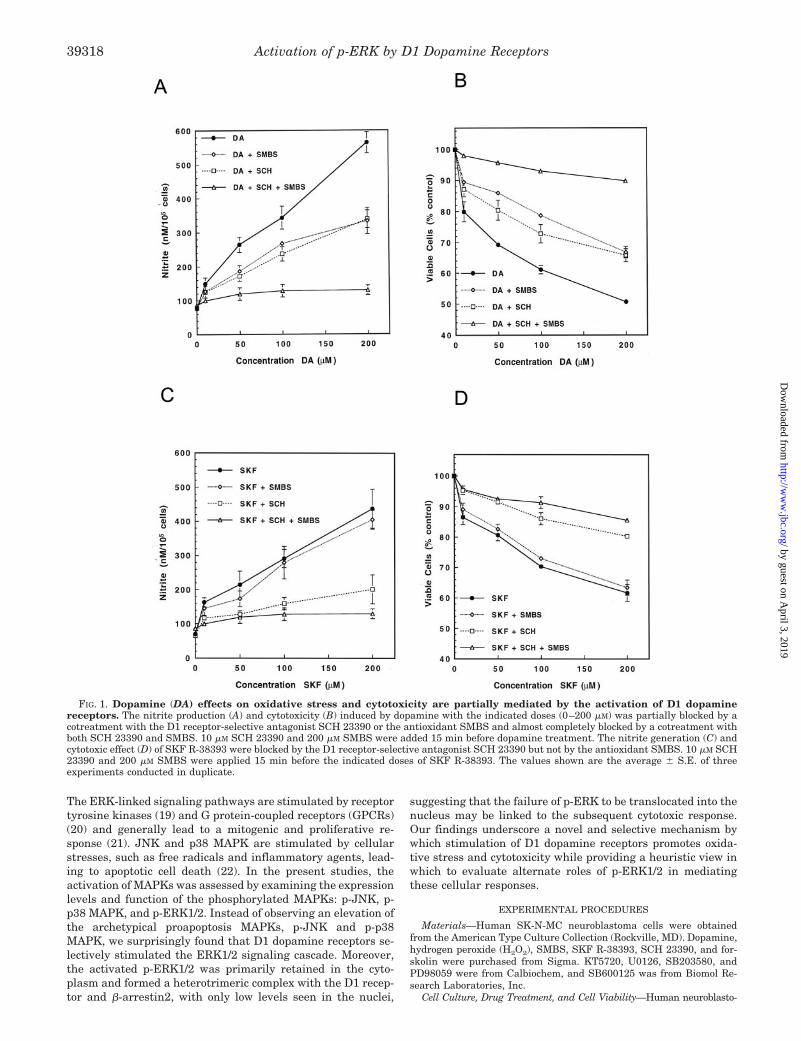
FIG. 1. Dopamine (DA) effects on oxidative stress and cytotoxicity are partially mediated by the activation of D1 dopaminereceptors. The nitrite production (A) and cytotoxicity (B) induced by dopamine with the indicated doses (0–200 �M) was partially blocked by acotreatment with the D1 receptor-selective antagonist SCH 23390 or the antioxidant SMBS and almost completely blocked by a cotreatment withboth SCH 23390 and SMBS. 10 �M SCH 23390 and 200 �M SMBS were added 15 min before dopamine treatment. The nitrite generation (C) andcytotoxic effect (D) of SKF R-38393 were blocked by the D1 receptor-selective antagonist SCH 23390 but not by the antioxidant SMBS. 10 �M SCH23390 and 200 �M SMBS were applied 15 min before the indicated doses of SKF R-38393. The values shown are the average � S.E. of threeexperiments conducted in duplicate.
Activation of p-ERK by D1 Dopamine Receptors39318
by guest on April 3, 2019
http://ww
w.jbc.org/
Dow
nloaded from
ma-derived SK-N-MC cells were grown in 12-well culture plates (seedingdensity, 1.0 � 105/well) in RPMI 1640 medium without phenol red, supple-mented with 10% (v/v) Nu-serum (Collaborative Biomedical Products, Bed-ford, MA), antibiotics, and 2 mM L-glutamine in a humidified atmosphere of95% air, 5% CO2 at 37 °C until 90% confluent. Cells were then serumstarved overnight with serum-free RPMI medium and incubated with var-ious drugs for 16 h or the indicated times. Control cells were treated with anequal concentration of solvent (0.2% H2O). After incubation, 0.3 ml of me-dium was removed to measure nitrite concentration. Cell viability in 12-wellplates was evaluated by counting viable cells in a Neubauer cell using thetrypan blue exclusion test, as described by the manufacturer’s (Sigma)protocol, whereby viable cells exposed to trypan blue for no more than 15min exclude the dye. Values from each treatment were expressed as apercentage of cell survival relative to control.
Plasmids and Transfection—Wild type MEK1 and catalytically de-fective MEK1 mutant (MEK1-K97M) cDNA were kind gifts from M.Cobb, University of Texas Southwestern Medical Center, Dallas, TX.The MEK1-K97M cDNA construct was subcloned into vector pRSET(Invitrogen), and the wild type MEK1 cDNA construct was subclonedinto vector pCMV5 (Invitrogen). �-Arrestin1 dominant negative mutant(�-arrestin1 V53D) and �-arrestin2 dominant negative mutant (�-ar-restin2 V54D) cDNA constructs (gifts from M. G. Caron, Duke Univer-sity, Durham, NC) were subcloned into the mammalian expressionvector pcDNA1AMP (Invitrogen). SK-N-MC neuroblastoma cells weretransiently transfected (1 �g of DNA/105 cells) by LipofectAMINE 2000(Invitrogen). Briefly, cells were seeded in 12 wells (105 cells seeded/well)and grown to 80% confluence on the day of transfection. DNA-Lipo-fectAMINE 2000 was prepared as follows. Diluted DNA in Opti-MEM(without serum) was brought to a concentration of 1.6 �g/100 �l. Thendiluted LipofectAMINE 2000 reagent in Opti-MEM (without serum)was brought to a concentration of 4.0 �l/100 �l, mixed gently, andincubated for 5 min at room temperature. After a 5-min incubation, thediluted DNA was combined with the diluted LipofectAMINE 2000,mixed gently, and incubated for 20 min at room temperature to allow
DNA-LipofectAMINE 2000 complexes to form. 200 �l of the DNA-LipofectAMINE 2000 complex was added to each well containing cellsand Dulbecco’s modified Eagle’s medium (without serum and antibiot-ics), mixed gently, and incubated at 37 °C and 5% CO2 for 5 h. After the5-h incubation, the medium was replaced with Dulbecco’s modifiedEagle’s medium containing 20% fetal bovine serum and used 48 h aftertransfection. Mock transfected cells were transfected with a pcDNA3plasmid (Invitrogen) that lacked a DNA insert.
Measurement of Nitrite Concentration—The nitrite concentrationwas measured as described previously (13, 14). Briefly, 0.3 ml of Griessreagent (1 part of 0.1% naphthylethylenediamine dihydrochloride inH2O and 1 part of 1% sulfanilamide in 5% H3PO4) and 0.3 ml of culturemedium from treated cells (see above) were mixed. After a 30-minincubation at 45 °C, the absorbance was determined at 550 nm. Thenitrite concentration was measured from a standard curve usingNaNO2 at a range of 0–10 �M. Results were expressed as nM/1.0 � 105
cells or -fold increase of the nitrite level from drug treatment over adistilled water-treated control.
Protein Preparation and Immunoblot Analysis—SK-N-MC cells werelysed in buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM
EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM �-glycerolphos-phate, 1 mM Na3VO4) containing 5 �g/ml leupeptin, 5 �g/ml pepstatin,and 1 mM phenylmethylsulfonyl fluoride. Protein concentrations weredetermined by the Bradford method (23). Immunoblot analysis wasperformed essentially as described previously (24). Briefly, protein (50�g/lane) was separated eletrophoretically using 10% SDS-PAGE andtransferred overnight to polyvinylidene difluoride membranes (MicronSeparations Inc., Westboro, MA). The membranes were blocked with 5%nonfat dried milk in Tris-buffered saline with Tween 20 (TBST; 20 mM
Tris-HCl, pH 7.5, 150 mM NaCl, and 0.05% Tween 20) followed byincubation in primary antibodies. The blots were then washed andincubated with secondary antibody. Enhanced chemiluminescence wascarried out using Renaissance Chemiluminescence Reagent Plus(PerkinElmer Life Sciences). The primary antibodies used were as
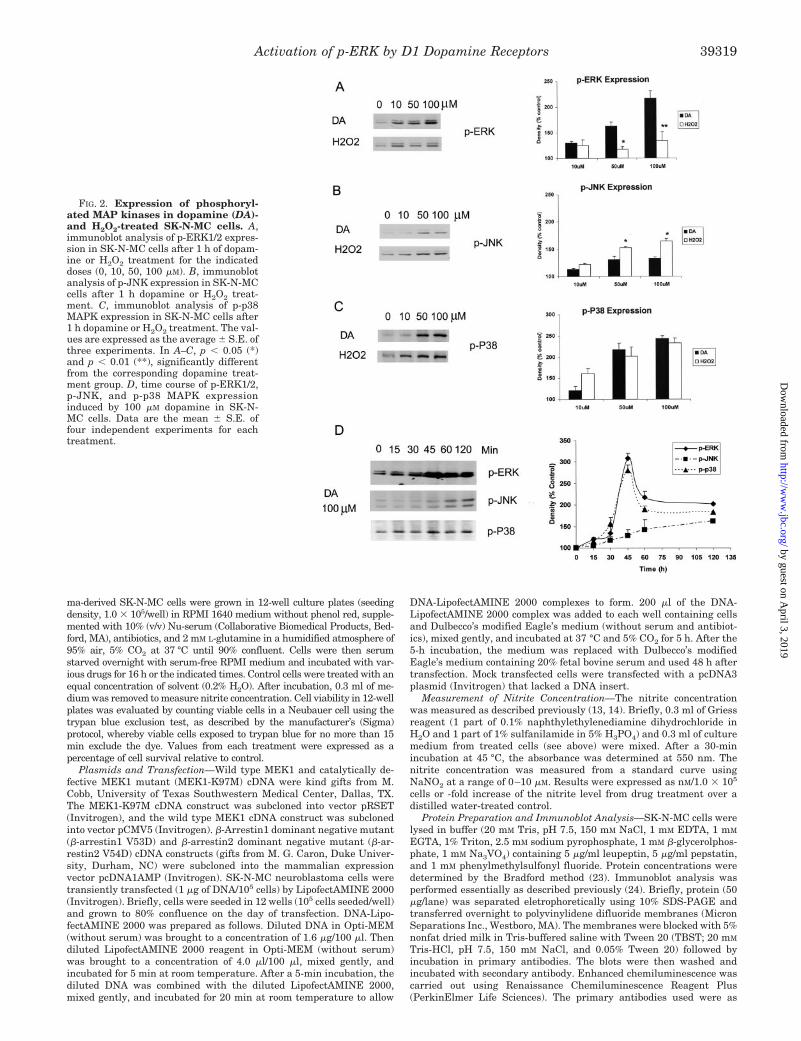
FIG. 2. Expression of phosphoryl-ated MAP kinases in dopamine (DA)-and H2O2-treated SK-N-MC cells. A,immunoblot analysis of p-ERK1/2 expres-sion in SK-N-MC cells after 1 h of dopam-ine or H2O2 treatment for the indicateddoses (0, 10, 50, 100 �M). B, immunoblotanalysis of p-JNK expression in SK-N-MCcells after 1 h dopamine or H2O2 treat-ment. C, immunoblot analysis of p-p38MAPK expression in SK-N-MC cells after1 h dopamine or H2O2 treatment. The val-ues are expressed as the average � S.E. ofthree experiments. In A–C, p � 0.05 (*)and p � 0.01 (**), significantly differentfrom the corresponding dopamine treat-ment group. D, time course of p-ERK1/2,p-JNK, and p-p38 MAPK expressioninduced by 100 �M dopamine in SK-N-MC cells. Data are the mean � S.E. offour independent experiments for eachtreatment.
Activation of p-ERK by D1 Dopamine Receptors 39319
by guest on April 3, 2019
http://ww
w.jbc.org/
Dow
nloaded from
follows: p-ERK1/2, p-JNK and p-p38 MAPK mouse monoclonal antibody(1:1,000, Santa Cruz Biotechnology, Santa Cruz, CA), p-MEK1/2 rabbitpolyclonal antibody (1:1,000, Cell Signaling Technology, Beverly, MA).The optical density of the immunoblots was quantified by NIH Image.
Coimmunoprecipitation Studies—Untransfected or transfected SK-N-MC cells (1–2 � 107 cells) were solubilized with 1% sodium cholate(Sigma) as described previously (25). To the soluble extracts (200–400�l/assay tube, 1–1.5 mg/ml of protein), either of the following antiserawas added (1:100): p-ERK1/2 rabbit monoclonal (Cell Signaling Tech-nology, Inc., Beverly, MA) or pan-�-arrestin rabbit polyclonal (AffinityBioReagents, Golgen, CO) or mouse anti-D1 monoclonal (26). Afterovernight incubation, immune complexes were precipitated with pro-tein A-Sepharose beads (CL-4B, Amersham Biosciences). Pellets werewashed five times and subjected to SDS-PAGE and Western blotting.Blots were probed with antibodies (1:1,000) for either p-ERK1/2 mousemonoclonal (Santa Cruz Biotechnology) or �-arrestin2 mouse mono-clonal (Santa Cruz Biotechnology) or D1 dopamine receptors mousemonoclonal (26). Proteins were visualized using peroxidase-conjugatedsecondary antibodies (1:8,000; Santa Cruz Biotechnology) and en-hanced chemiluminescence (Amersham Biosciences). p-ERK1/2 was de-tected as two bands, about 42 and 44 kDa; �-arrestin2 had a band ofabout 47 kDa, and D1 is about 50 kDa, consistent with the knownmolecular sizes reported in the literature.
Subcellular Fractionation—For the separation of nuclear and cyto-solic pools of endogenous p-ERK1/2, serum-starved cells were stimu-lated with dopamine or SKF R-38393. Monolayers were washed twicewith ice-cold phosphate-buffered saline and solubilized with 2 ml ofice-cold lysis buffer. Cells were incubated on ice for 10 min to allowlysis. The cellular extract was centrifuged at 500 � g for 5 min to pelletnuclei. The supernatants contained the cytosolic fraction. Pellets con-taining cell nuclei were washed with lysis buffer and pelleted again at500 � g for 10 min. Both cytosolic and nuclear fractions were solubilizedin 2 � Laemmli loading buffer, and p-ERK1/2 levels were determinedby immunoblotting. The purity of nuclear and cytosolic fractions wasverified by immunoblotting with antibodies to Elk-1.
Analysis of Data—All statistical analyses values were accomplishedusing Instat Statistical Software (GraphPad, Sorrento Valley, CA).Statistical comparisons were performed with one-factor analysis ofvariance followed by Fisher’s least squares difference test. Values of p �0.05 were considered statistically significant.
RESULTS
Activation of D1 Dopamine Receptors Enhances OxidativeStress and Cytotoxicity—We have shown previously that chronic(16 h) stimulation of D1 dopamine receptors with dopamine or D1
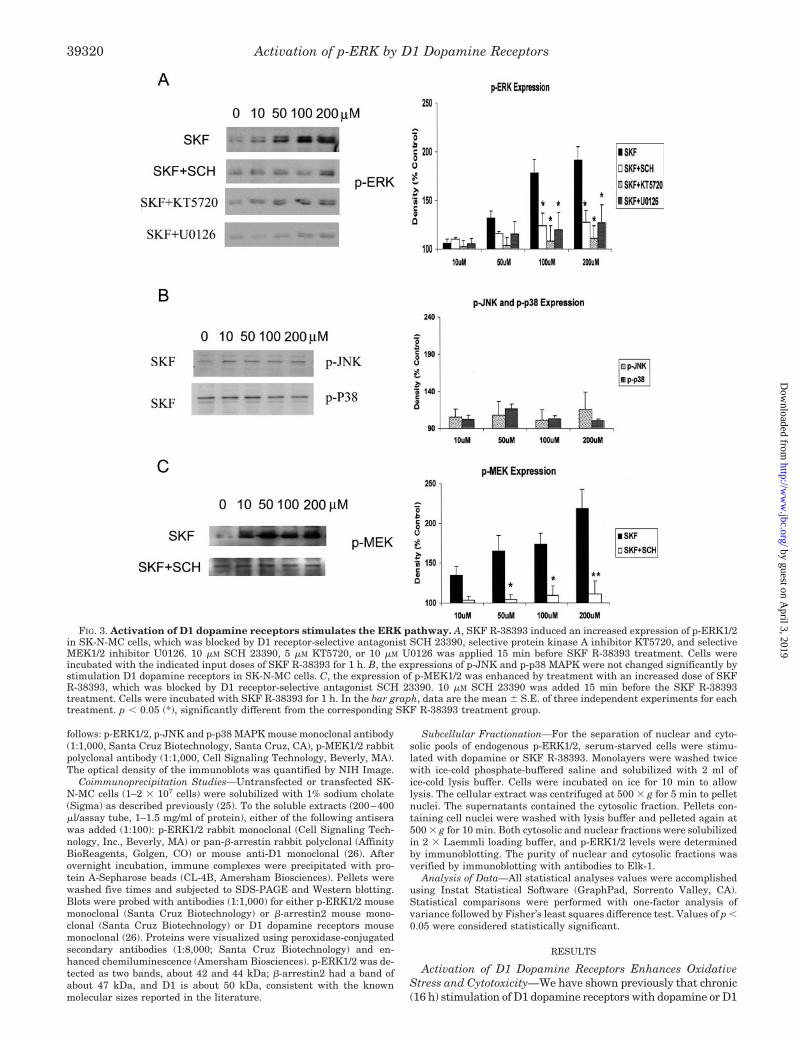
FIG. 3. Activation of D1 dopamine receptors stimulates the ERK pathway. A, SKF R-38393 induced an increased expression of p-ERK1/2in SK-N-MC cells, which was blocked by D1 receptor-selective antagonist SCH 23390, selective protein kinase A inhibitor KT5720, and selectiveMEK1/2 inhibitor U0126. 10 �M SCH 23390, 5 �M KT5720, or 10 �M U0126 was applied 15 min before SKF R-38393 treatment. Cells wereincubated with the indicated input doses of SKF R-38393 for 1 h. B, the expressions of p-JNK and p-p38 MAPK were not changed significantly bystimulation D1 dopamine receptors in SK-N-MC cells. C, the expression of p-MEK1/2 was enhanced by treatment with an increased dose of SKFR-38393, which was blocked by D1 receptor-selective antagonist SCH 23390. 10 �M SCH 23390 was added 15 min before the SKF R-38393treatment. Cells were incubated with SKF R-38393 for 1 h. In the bar graph, data are the mean � S.E. of three independent experiments for eachtreatment. p � 0.05 (*), significantly different from the corresponding SKF R-38393 treatment group.
Activation of p-ERK by D1 Dopamine Receptors39320
by guest on April 3, 2019
http://ww
w.jbc.org/
Dow
nloaded from
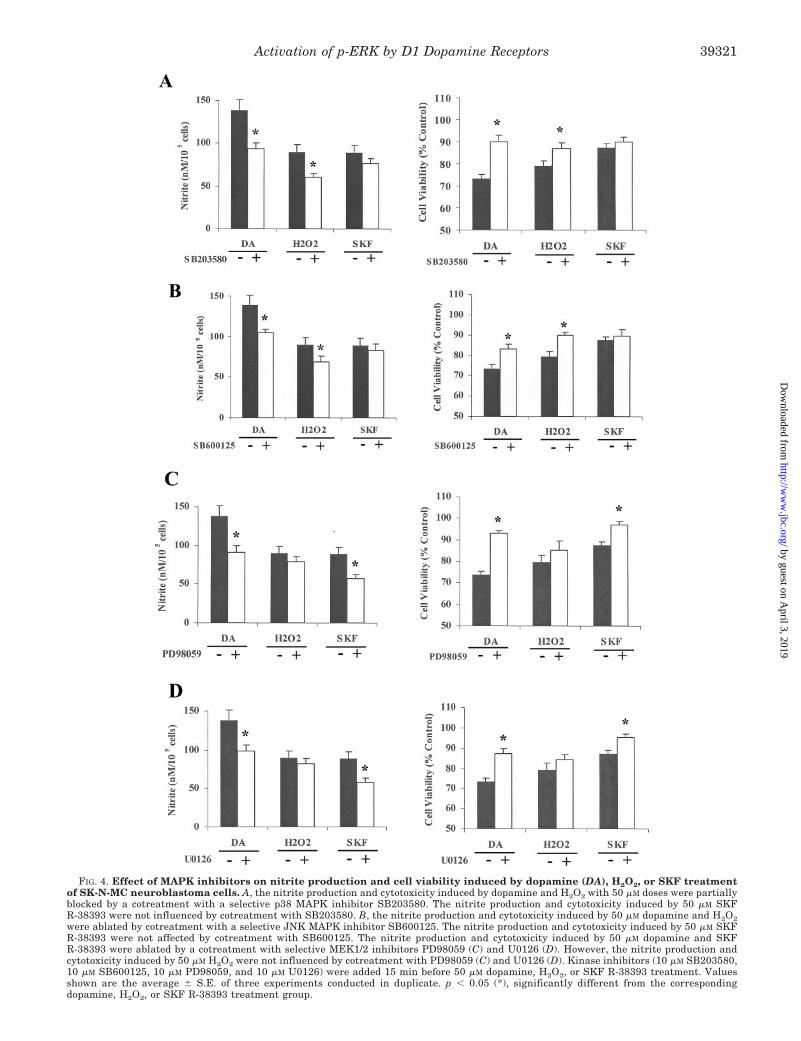
FIG. 4. Effect of MAPK inhibitors on nitrite production and cell viability induced by dopamine (DA), H2O2, or SKF treatmentof SK-N-MC neuroblastoma cells. A, the nitrite production and cytotoxicity induced by dopamine and H2O2 with 50 �M doses were partiallyblocked by a cotreatment with a selective p38 MAPK inhibitor SB203580. The nitrite production and cytotoxicity induced by 50 �M SKFR-38393 were not influenced by cotreatment with SB203580. B, the nitrite production and cytotoxicity induced by 50 �M dopamine and H2O2were ablated by cotreatment with a selective JNK MAPK inhibitor SB600125. The nitrite production and cytotoxicity induced by 50 �M SKFR-38393 were not affected by cotreatment with SB600125. The nitrite production and cytotoxicity induced by 50 �M dopamine and SKFR-38393 were ablated by a cotreatment with selective MEK1/2 inhibitors PD98059 (C) and U0126 (D). However, the nitrite production andcytotoxicity induced by 50 �M H2O2 were not influenced by cotreatment with PD98059 (C) and U0126 (D). Kinase inhibitors (10 �M SB203580,10 �M SB600125, 10 �M PD98059, and 10 �M U0126) were added 15 min before 50 �M dopamine, H2O2, or SKF R-38393 treatment. Valuesshown are the average � S.E. of three experiments conducted in duplicate. p � 0.05 (*), significantly different from the correspondingdopamine, H2O2, or SKF R-38393 treatment group.
Activation of p-ERK by D1 Dopamine Receptors 39321
by guest on April 3, 2019
http://ww
w.jbc.org/
Dow
nloaded from
receptor selective agonist, SKF R-38393, causes profound cyto-toxicity, as indexed by increased oxidative stress and acceleratedcell death (14). SK-N-MC neuroblastoma cells, a postsynapticstriatal cell model endogenously expressing the D1 receptors (15,27), were treated with increasing concentrations of dopamine inthe presence of SCH 23390, a D1-selective antagonist, or SMBS,a potent antioxidant. In the presence of either of these two com-pounds, the cytotoxic effects of dopamine were reduced sharply
by �50%, as indexed by a decrease in both nitrite production(Fig. 1A) and cell death (Fig. 1B). When SCH 23390 and SMBSwere used together, the cytotoxic effects of dopamine were com-pletely prevented, indicating that both autoxidation of dopamineand direct activation of the D1 receptors by dopamine contributeto mediation of dopamine cytotoxicity.
The direct participation of D1 dopamine receptors in induc-ing cytotoxicity was tested by stimulating the receptor with the
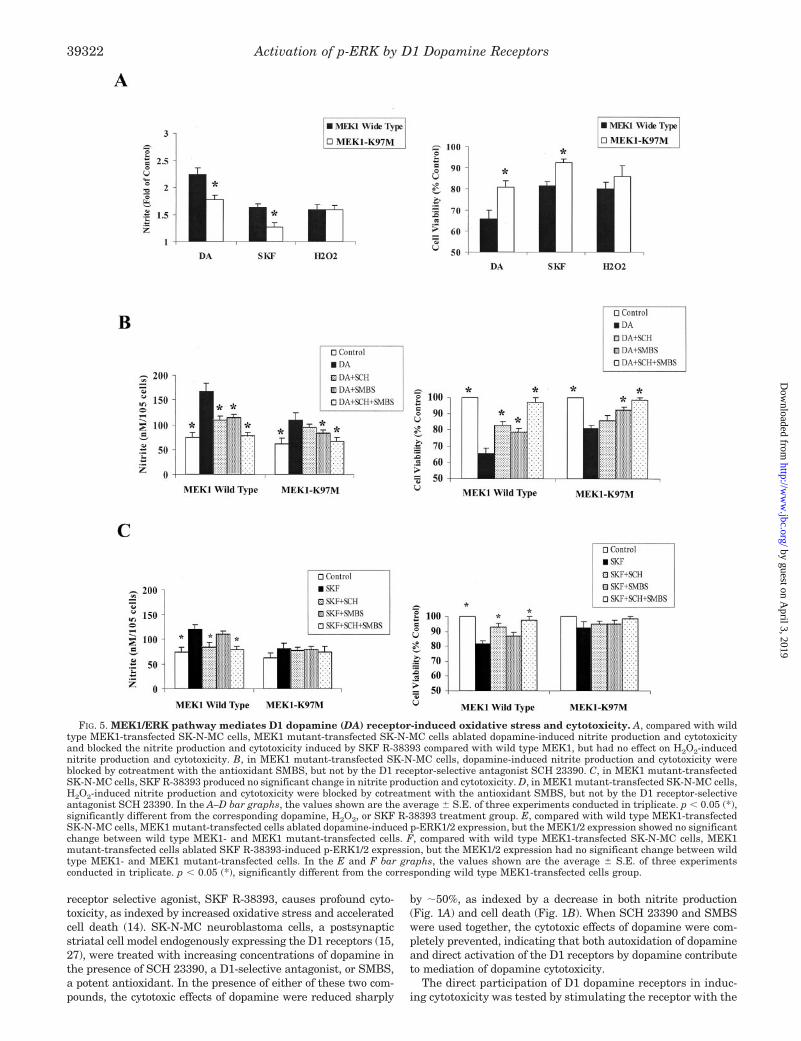
FIG. 5. MEK1/ERK pathway mediates D1 dopamine (DA) receptor-induced oxidative stress and cytotoxicity. A, compared with wildtype MEK1-transfected SK-N-MC cells, MEK1 mutant-transfected SK-N-MC cells ablated dopamine-induced nitrite production and cytotoxicityand blocked the nitrite production and cytotoxicity induced by SKF R-38393 compared with wild type MEK1, but had no effect on H2O2-inducednitrite production and cytotoxicity. B, in MEK1 mutant-transfected SK-N-MC cells, dopamine-induced nitrite production and cytotoxicity wereblocked by cotreatment with the antioxidant SMBS, but not by the D1 receptor-selective antagonist SCH 23390. C, in MEK1 mutant-transfectedSK-N-MC cells, SKF R-38393 produced no significant change in nitrite production and cytotoxicity. D, in MEK1 mutant-transfected SK-N-MC cells,H2O2-induced nitrite production and cytotoxicity were blocked by cotreatment with the antioxidant SMBS, but not by the D1 receptor-selectiveantagonist SCH 23390. In the A–D bar graphs, the values shown are the average � S.E. of three experiments conducted in triplicate. p � 0.05 (*),significantly different from the corresponding dopamine, H2O2, or SKF R-38393 treatment group. E, compared with wild type MEK1-transfectedSK-N-MC cells, MEK1 mutant-transfected cells ablated dopamine-induced p-ERK1/2 expression, but the MEK1/2 expression showed no significantchange between wild type MEK1- and MEK1 mutant-transfected cells. F, compared with wild type MEK1-transfected SK-N-MC cells, MEK1mutant-transfected cells ablated SKF R-38393-induced p-ERK1/2 expression, but the MEK1/2 expression had no significant change between wildtype MEK1- and MEK1 mutant-transfected cells. In the E and F bar graphs, the values shown are the average � S.E. of three experimentsconducted in triplicate. p � 0.05 (*), significantly different from the corresponding wild type MEK1-transfected cells group.
Activation of p-ERK by D1 Dopamine Receptors39322
by guest on April 3, 2019
http://ww
w.jbc.org/
Dow
nloaded from
D1-selective agonist SKF R-38393 in the absence or presence ofSMBS or SCH 23390. Treatment of SK-N-MC cells with SKFR-38393 caused an increase in both nitrite levels (Fig. 1C) andcell death (Fig. 1D). SMBS did not modulate the SKF R-38393-mediated effects, consistent with lack of oxidation of this stablecompound. By contrast, SCH 23390 attenuated the effects ofSKF R-38393 with regard to both nitrite production and celldeath.
Dopamine Enhances Phosphorylation of the ERK1/2 MAPKinases—To gain insight into the mechanistic pathways bywhich dopamine mediates its toxic effects, we analyzed theearly signaling cascades that are activated by examining thephosphorylation of the three major groups of MAP kinases:ERK1/2, JNK, and p38 MAPK, in SK-N-MC cells treated withdopamine or H2O2. Dopamine caused a strong enhancement ofp-ERK1/2 in SK-N-MC cells in a dose-dependent manner, andan increase of 116 � 14.9% was observed at the highest con-centration of dopamine (100 �M) used in these studies (Fig. 2A).By contrast, H2O2 enhancement of p-ERK1/2 was weak, andeven at 100 �M, a modest increase of only 33% was seen. On theother hand, dopamine caused only a weak enhancement (33%)of p-JNK, whereas H2O2 increased p-JNK levels by 65% (Fig.
2B). Dopamine and H2O2 activated p-p38 MAPK to similarlevels, and increases of 143 and 133%, respectively, were ob-served at concentrations of 100 �M (Fig. 2C).
The time course of activation of p-ERK1/2, p-JNK, and p-p38MAPK by 100 �M dopamine was investigated. Maximal activa-tion of p-ERK1/2 and p-p38 MAPK was achieved within 45 minof exposure to dopamine (Fig. 2D), with a slight decrease after60 and 120 min of exposure. The weak activation of p-JNK bydopamine was seen only after 60 min and was sustained up to120 min of exposure to dopamine. These data indicate thatdopamine strongly enhances the phosphorylation of theERK1/2 and p38 MAPKs, with weak activation of JNK.
D1 Dopamine Receptor-mediated Cytotoxicity Proceedsthrough the p-ERK1/2 Signaling Pathway—Because the abilityof dopamine to enhance phosphorylation of ERK1/2 was muchhigher than that of H2O2, we speculated that the increase inp-ERK1/2 levels was not the result of the oxidant properties ofthese compounds, but was a direct consequence of D1 dopaminereceptor stimulation by dopamine. To test for this, we exam-ined p-ERK1/2, p-JNK, and p-p38 MAPK levels after stimulat-ing SK-N-MC cells with the D1 receptor-selective agonist, SKFR-38393. Treatment of cells with increasing doses of SKF
FIG. 5—continued
Activation of p-ERK by D1 Dopamine Receptors 39323
by guest on April 3, 2019
http://ww
w.jbc.org/
Dow
nloaded from

R-38393 selectively enhanced p-ERK1/2 levels (Fig. 3A), withno corresponding increase in either p-JNK or p-p38 MAPKlevels (Fig. 3B). Moreover, the increase in p-ERK1/2 wasblocked by the D1-selective antagonist SCH 23390, proteinkinase A inhibitor KT5720, and by U0126, a selective inhibitorof MEK1/2 (Fig. 3A).
One feature of the ERK signaling cascade is that ERK1/2phosphorylation is exclusively regulated by MEK1/2, the up-stream dual specificity kinase. This dual phosphorylation isnecessary and sufficient for ERK activation. We therefore alsomeasured p-MEK1/2 levels by stimulating SK-N-MC cells withSKF R-38393 and found a dose-dependent increase inp-MEK1/2 which was blocked completely by SCH 23390(Fig. 3C).
To assess further the participation p-ERK1/2 in mediatingdopamine and D1 receptor cytotoxicity, SK-N-MC cells weretreated for 16 h with dopamine or SKF R-38393 (50 �M each) inthe presence or absence of selective inhibitors of the variousMAPKs, and nitrite levels and cell viability were measured.Cells were also treated with 50 �M H2O2 to measure separatelythe role of MAPKs in the oxidative pathway. 10 �M SB203580,a selective inhibitors of p38 MAPK, reduced nitrite productionby both dopamine (47%) and H2O2 (62%), with a significant(p � 0.05) increase in cell viability (Fig. 4A). When cells weretreated with SKF R-38393, SB203580 caused a modest de-crease (21%) in nitrite production, without any significantchanges in cell viability (Fig. 4A). By contrast, inhibition ofp-JNK activity with its selective inhibitor SB600125 had vir-tually no significant effect on SKF R-38393-mediated nitriteproduction or cell death, while significantly reducing nitritelevels produced by dopamine and H2O2 (29 and 44%, respec-tively) (Fig. 4B).
U0126 and PD98059, another selective inhibitor of MEK1/2(10 �M each), blocked (41 and 48%, respectively) dopamine-mediated nitrite production, causing a significant (p � 0.05)increase in cell viability (Fig. 4, C and D). Moreover, bothU0126 and PD98059 almost completely blocked an SKFR-38393-mediated increase in nitrite production (by 86 and84%, respectively), while significantly (p � 0.05) restoring cellviability to near control levels (Fig. 4, C and D). By contrast,H2O2-induced nitrite production was much less affected by thepresence of the MEK1/2 inhibitors, with inhibition of only15–22% with U0126 and PD98059, respectively, and withoutsignificant changes in cell viability (Fig. 4, C and D). Together,these data suggest that the cytotoxicity observed is the result ofthe direct stimulation of D1 receptors and is dependent onactivation of p-ERK1/2, but not p-JNK, with only a modestinvolvement of p-p38 MAPK in these events.
D1 Dopamine Receptor-mediated Cytotoxicity InvolvesMEK1—To confirm the involvement of MEK1 in D1 dopaminereceptor-mediated oxidative stress and cytotoxicity, we trans-fected SK-N-MC cells with wild type MEK1 and its catalyti-cally defective MEK1 mutant, MEK1-K97M. Compared withwild type MEK1-transfected SK-N-MC cells, MEK1-K97M-transfected cells had decreased nitrite production and celldeath upon treatment with both dopamine and SKF R-38393(Fig. 5A) (38 and 56%, respectively; p � 0.05), without affectingH2O2-induced nitrite production and cytotoxicity (Fig. 5A). Toascertain whether MEK1 was involved in the direct receptorstimulation pathway of dopamine action or through productionof free radicals, MEK1-K97M-transfected cells were treatedwith dopamine (Fig. 5B), SKF R-38393 (Fig. 5C), and H2O2
(Fig. 5D) in the absence or presence of SMBS and SCH 23390.In wild type MEK1-transfected cells, dopamine-induced nitriteproduction and cytotoxicity were significantly (p � 0.05) andequally ablated by both SMBS and SCH 23390 (Fig. 5B). How-
ever, in MEK1-K97M-transfected cells, dopamine-induced ni-trite production and cytotoxicity were only blocked upon co-treatment with the antioxidant SMBS, but not by the D1receptor-selective antagonist SCH 23390 (Fig. 5B). Meanwhile,in wild type MEK1-transfected cells, 50 �M SKF R-38393caused a significant (p � 0.05) increase in nitrite productionand cytotoxicity, which were blocked by the D1 receptor-selec-tive antagonist SCH 23390, but not by SMBS (Fig. 5C). Incontrast to MEK1-transfected cells, SKF R-38393 had no sig-nificant effect on either nitrite production or cytotoxicity inMEK1-K97M-transfected SK-N-MC cells (Fig. 5C). On theother hand, in both wild type MEK1- and MEK1-K97M-trans-fected SK-N-MC cells, H2O2-induced nitrite production andcytotoxicity were only blocked by cotreatment with the antiox-idant SMBS (Fig. 5D), but not by the D1 receptor-selectiveantagonist SCH 23390 (Fig. 5D). These results are consistentwith our finding that MEK1/2 is selectively activated by D1dopamine receptors but not through the oxidative stress path-way of dopamine autoxidation.
We examined the p-ERK1/2 expression levels after transfect-ing cells with wild type MEK1 and the MEK1 mutant. In wildtype MEK1-transfected SK-N-MC cells, dopamine and SKFR-38393 caused a strong enhancement of p-ERK1/2 in a dose-dependent manner. Maximal increases of 172.7 � 17.6% and94.7 � 13.5% were observed at the highest concentration ofdopamine (100 �M) and SKF R-38393 (100 �M), respectively,used in these studies (Fig. 5, E and F). However, in MEK1-K97M-transfected cells, dopamine- and SKF R-38393-inducedp-ERK1/2 expressions were significantly (p � 0.05) reduced byabout 45 and 73% at the highest concentration of dopamine(100 �M) and SKF R-38393 (100 �M), respectively (Fig. 5, E andF). In addition, total MEK1/2 expression was detected with nosubstantial difference in wild type MEK1- and MEK1-K97M-transfected cells, indicating that the reduction in p-ERK1/2levels was not caused by changes in MEK1/2 protein levels (Fig.5, E and F). These data clearly indicate the participation ofMEK1/2 and thereby the p-ERK1/2 pathway upon activation ofthe D1 dopamine receptor.
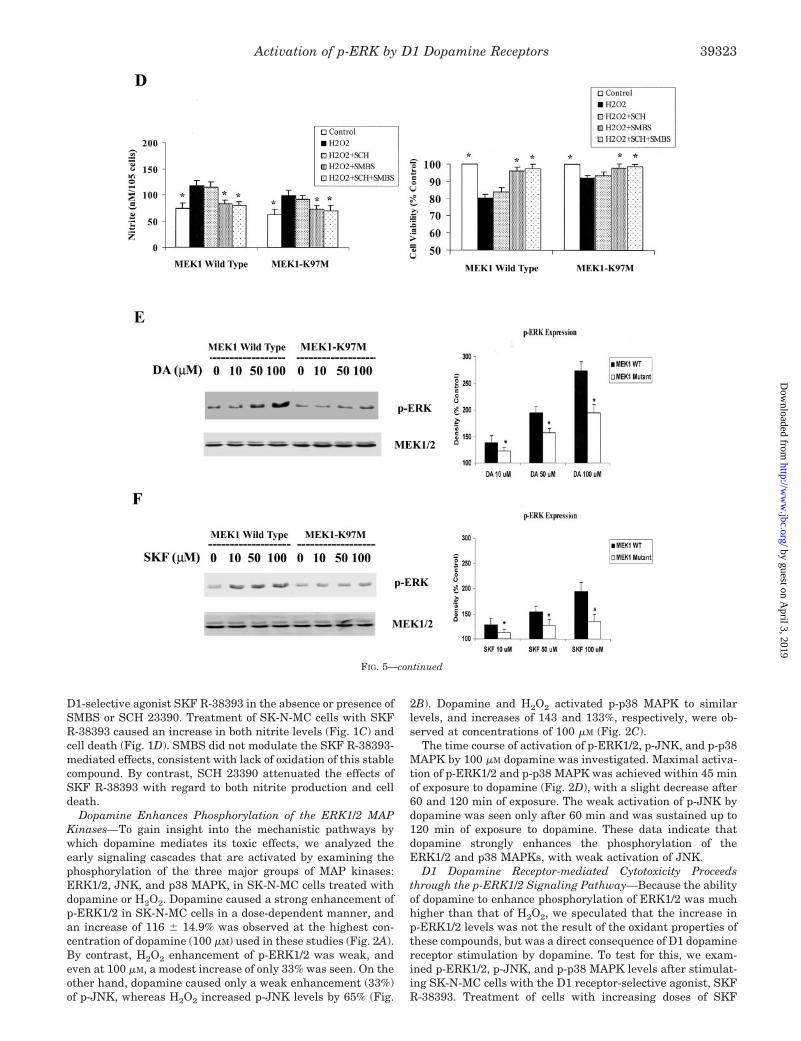
p-ERK Is Retained in the Cytoplasm in a HeterotrimericComplex—p-ERK elicits a wide range of biological functionsthrough phosphorylation of nuclear and cytoplasmic substrates(28), and the translocation of ERK from the cytoplasm to thenucleus is mandatory for efficient regulation of cell survivaland proliferation (29, 30). Because our results showed paradox-ically that p-ERK promoted a cytotoxic response, we conductedstudies to determine whether p-ERK was translocated in anappropriate manner into the nucleus. Accordingly, SK-N-MCcells were treated with either dopamine or SKF R-38393 in adose- and time-dependent manner, the cells fractionated intocytoplasm and nucleus fractions, and levels of p-ERK1/2 inthese fractions were assessed by Western blots. Dopamine andSKF R-38393 (100 �M each) caused a time-dependent increasein p-ERK1/2 levels in both the cytoplasm and the nuclei frac-tions, reaching maximal levels in 60 min (Fig. 6A). However,the relative increase in p-ERK1/2 in the nuclei was signifi-cantly (p � 0.05) below that seen in the cytoplasm at all times.Only 35% of p-ERK1/2 was translocated to the nuclei, whereas65% of p-ERK1/2 was retained in the cytoplasm after a 60-mintreatment with dopamine or SKF R-38393 (Fig. 6A). Similarly,both dopamine and SKF R-38393 caused an increase inp-ERK1/2 in a dose-dependent manner, but there were clearlymuch higher levels (�65%) of p-ERK1/2 in the cytoplasm com-pared with the nuclei (Fig. 6B). As a positive control, SK-N-MCcells were treated with forskolin, a cAMP-elevating agent,which caused a dose-dependent increase in p-ERK1/2 levels. Incontrast to our findings with dopamine and SKF R-38393, in
Activation of p-ERK by D1 Dopamine Receptors39324
by guest on April 3, 2019
http://ww
w.jbc.org/
Dow
nloaded from
forskolin-treated cells 72% of p-ERK1/2 was translocated to thenuclei, and only 28% of p-ERK1/2 was retained in the cyto-plasm after a 30-min 10 �M forskolin treatment (Fig. 6C).
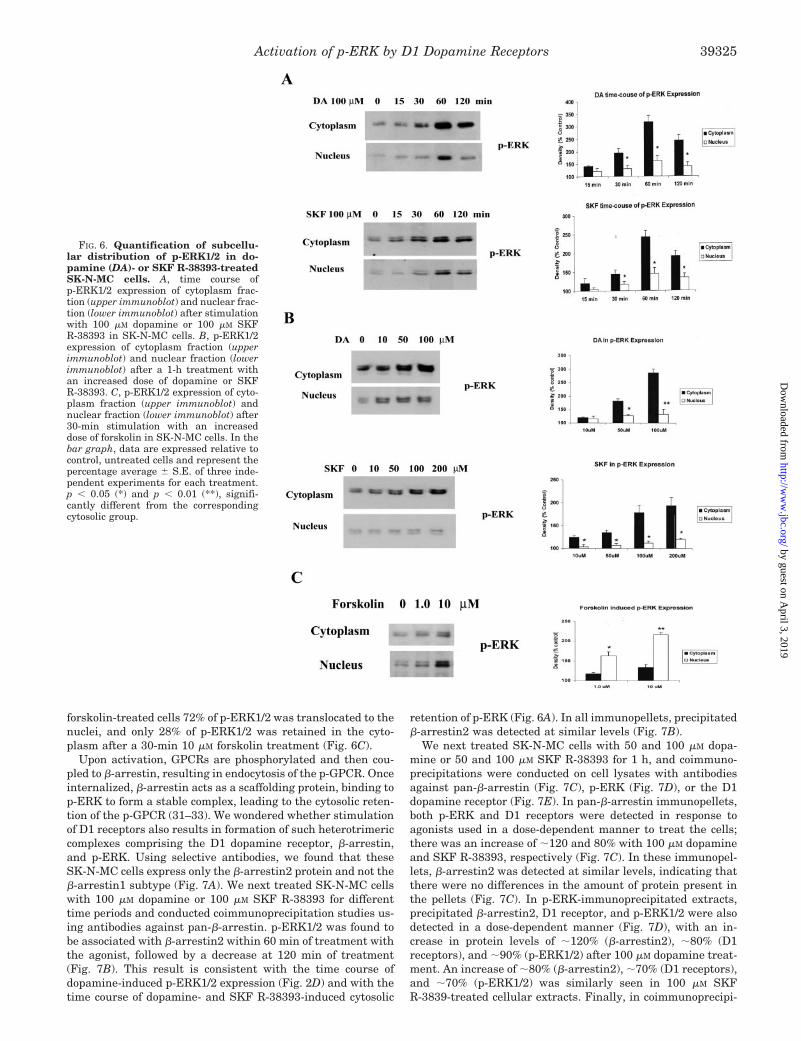
Upon activation, GPCRs are phosphorylated and then cou-pled to �-arrestin, resulting in endocytosis of the p-GPCR. Onceinternalized, �-arrestin acts as a scaffolding protein, binding top-ERK to form a stable complex, leading to the cytosolic reten-tion of the p-GPCR (31–33). We wondered whether stimulationof D1 receptors also results in formation of such heterotrimericcomplexes comprising the D1 dopamine receptor, �-arrestin,and p-ERK. Using selective antibodies, we found that theseSK-N-MC cells express only the �-arrestin2 protein and not the�-arrestin1 subtype (Fig. 7A). We next treated SK-N-MC cellswith 100 �M dopamine or 100 �M SKF R-38393 for differenttime periods and conducted coimmunoprecipitation studies us-ing antibodies against pan-�-arrestin. p-ERK1/2 was found tobe associated with �-arrestin2 within 60 min of treatment withthe agonist, followed by a decrease at 120 min of treatment(Fig. 7B). This result is consistent with the time course ofdopamine-induced p-ERK1/2 expression (Fig. 2D) and with thetime course of dopamine- and SKF R-38393-induced cytosolic
retention of p-ERK (Fig. 6A). In all immunopellets, precipitated�-arrestin2 was detected at similar levels (Fig. 7B).
We next treated SK-N-MC cells with 50 and 100 �M dopa-mine or 50 and 100 �M SKF R-38393 for 1 h, and coimmuno-precipitations were conducted on cell lysates with antibodiesagainst pan-�-arrestin (Fig. 7C), p-ERK (Fig. 7D), or the D1dopamine receptor (Fig. 7E). In pan-�-arrestin immunopellets,both p-ERK and D1 receptors were detected in response toagonists used in a dose-dependent manner to treat the cells;there was an increase of �120 and 80% with 100 �M dopamineand SKF R-38393, respectively (Fig. 7C). In these immunopel-lets, �-arrestin2 was detected at similar levels, indicating thatthere were no differences in the amount of protein present inthe pellets (Fig. 7C). In p-ERK-immunoprecipitated extracts,precipitated �-arrestin2, D1 receptor, and p-ERK1/2 were alsodetected in a dose-dependent manner (Fig. 7D), with an in-crease in protein levels of �120% (�-arrestin2), �80% (D1receptors), and �90% (p-ERK1/2) after 100 �M dopamine treat-ment. An increase of �80% (�-arrestin2), �70% (D1 receptors),and �70% (p-ERK1/2) was similarly seen in 100 �M SKFR-3839-treated cellular extracts. Finally, in coimmunoprecipi-
FIG. 6. Quantification of subcellu-lar distribution of p-ERK1/2 in do-pamine (DA)- or SKF R-38393-treatedSK-N-MC cells. A, time course ofp-ERK1/2 expression of cytoplasm frac-tion (upper immunoblot) and nuclear frac-tion (lower immunoblot) after stimulationwith 100 �M dopamine or 100 �M SKFR-38393 in SK-N-MC cells. B, p-ERK1/2expression of cytoplasm fraction (upperimmunoblot) and nuclear fraction (lowerimmunoblot) after a 1-h treatment withan increased dose of dopamine or SKFR-38393. C, p-ERK1/2 expression of cyto-plasm fraction (upper immunoblot) andnuclear fraction (lower immunoblot) after30-min stimulation with an increaseddose of forskolin in SK-N-MC cells. In thebar graph, data are expressed relative tocontrol, untreated cells and represent thepercentage average � S.E. of three inde-pendent experiments for each treatment.p � 0.05 (*) and p � 0.01 (**), signifi-cantly different from the correspondingcytosolic group.
Activation of p-ERK by D1 Dopamine Receptors 39325
by guest on April 3, 2019
http://ww
w.jbc.org/
Dow
nloaded from
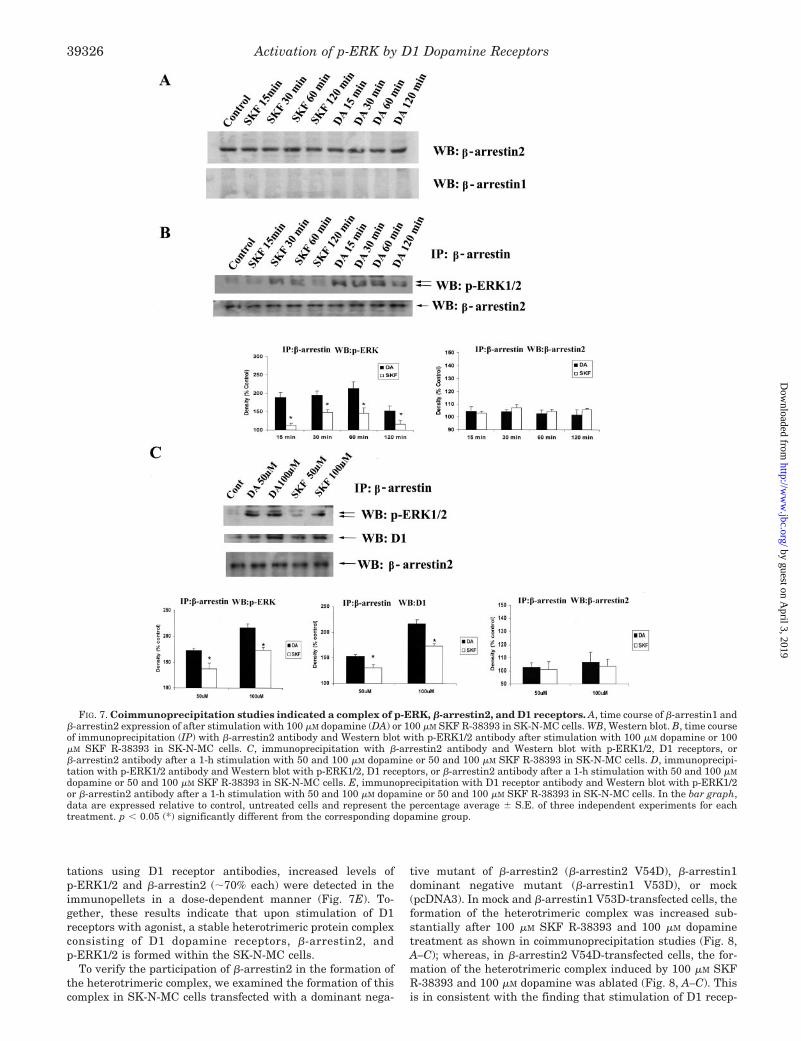
tations using D1 receptor antibodies, increased levels ofp-ERK1/2 and �-arrestin2 (�70% each) were detected in theimmunopellets in a dose-dependent manner (Fig. 7E). To-gether, these results indicate that upon stimulation of D1receptors with agonist, a stable heterotrimeric protein complexconsisting of D1 dopamine receptors, �-arrestin2, andp-ERK1/2 is formed within the SK-N-MC cells.
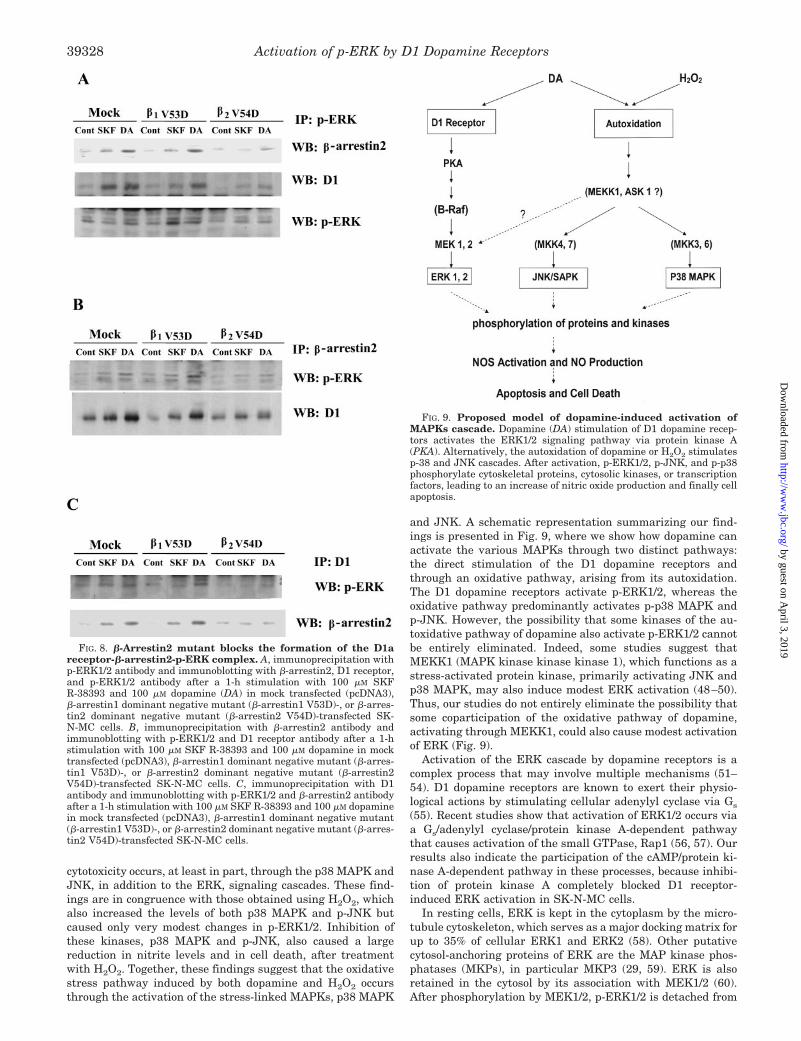
To verify the participation of �-arrestin2 in the formation ofthe heterotrimeric complex, we examined the formation of thiscomplex in SK-N-MC cells transfected with a dominant nega-
tive mutant of �-arrestin2 (�-arrestin2 V54D), �-arrestin1dominant negative mutant (�-arrestin1 V53D), or mock(pcDNA3). In mock and �-arrestin1 V53D-transfected cells, theformation of the heterotrimeric complex was increased sub-stantially after 100 �M SKF R-38393 and 100 �M dopaminetreatment as shown in coimmunoprecipitation studies (Fig. 8,A–C); whereas, in �-arrestin2 V54D-transfected cells, the for-mation of the heterotrimeric complex induced by 100 �M SKFR-38393 and 100 �M dopamine was ablated (Fig. 8, A–C). Thisis in consistent with the finding that stimulation of D1 recep-
FIG. 7. Coimmunoprecipitation studies indicated a complex of p-ERK, �-arrestin2, and D1 receptors. A, time course of �-arrestin1 and�-arrestin2 expression of after stimulation with 100 �M dopamine (DA) or 100 �M SKF R-38393 in SK-N-MC cells. WB, Western blot. B, time courseof immunoprecipitation (IP) with �-arrestin2 antibody and Western blot with p-ERK1/2 antibody after stimulation with 100 �M dopamine or 100�M SKF R-38393 in SK-N-MC cells. C, immunoprecipitation with �-arrestin2 antibody and Western blot with p-ERK1/2, D1 receptors, or�-arrestin2 antibody after a 1-h stimulation with 50 and 100 �M dopamine or 50 and 100 �M SKF R-38393 in SK-N-MC cells. D, immunoprecipi-tation with p-ERK1/2 antibody and Western blot with p-ERK1/2, D1 receptors, or �-arrestin2 antibody after a 1-h stimulation with 50 and 100 �M
dopamine or 50 and 100 �M SKF R-38393 in SK-N-MC cells. E, immunoprecipitation with D1 receptor antibody and Western blot with p-ERK1/2or �-arrestin2 antibody after a 1-h stimulation with 50 and 100 �M dopamine or 50 and 100 �M SKF R-38393 in SK-N-MC cells. In the bar graph,data are expressed relative to control, untreated cells and represent the percentage average � S.E. of three independent experiments for eachtreatment. p � 0.05 (*) significantly different from the corresponding dopamine group.
Activation of p-ERK by D1 Dopamine Receptors39326
by guest on April 3, 2019
http://ww
w.jbc.org/
Dow
nloaded from
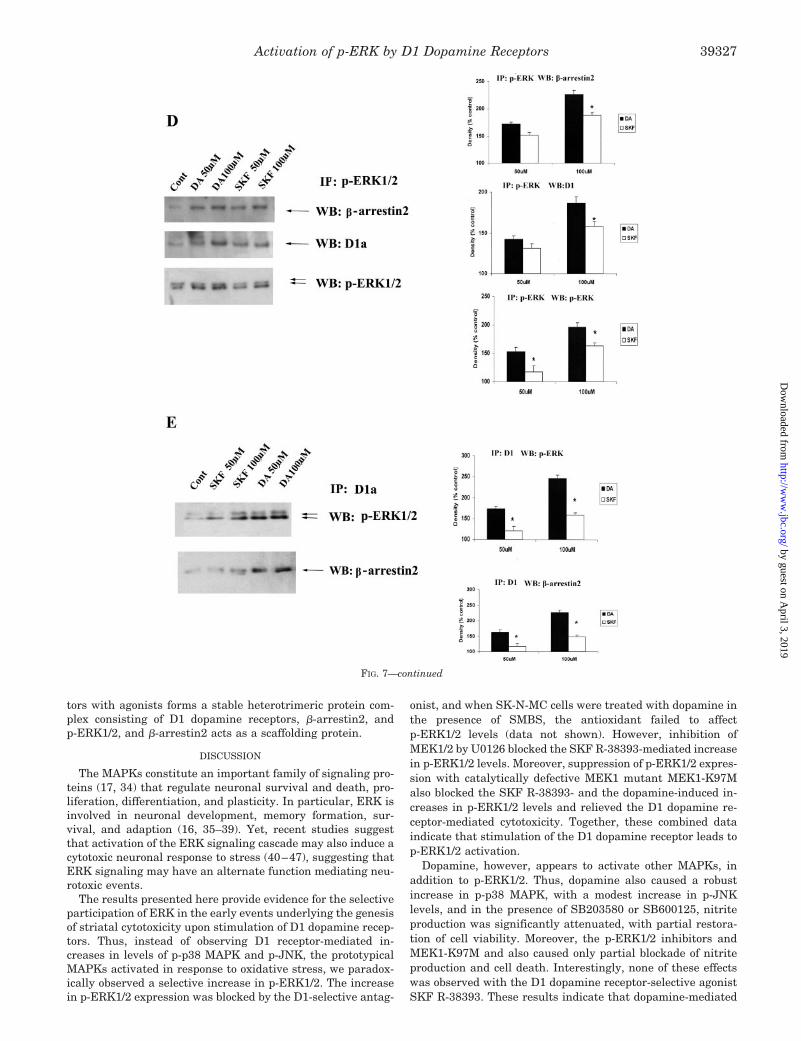
tors with agonists forms a stable heterotrimeric protein com-plex consisting of D1 dopamine receptors, �-arrestin2, andp-ERK1/2, and �-arrestin2 acts as a scaffolding protein.
DISCUSSION
The MAPKs constitute an important family of signaling pro-teins (17, 34) that regulate neuronal survival and death, pro-liferation, differentiation, and plasticity. In particular, ERK isinvolved in neuronal development, memory formation, sur-vival, and adaption (16, 35–39). Yet, recent studies suggestthat activation of the ERK signaling cascade may also induce acytotoxic neuronal response to stress (40–47), suggesting thatERK signaling may have an alternate function mediating neu-rotoxic events.
The results presented here provide evidence for the selectiveparticipation of ERK in the early events underlying the genesisof striatal cytotoxicity upon stimulation of D1 dopamine recep-tors. Thus, instead of observing D1 receptor-mediated in-creases in levels of p-p38 MAPK and p-JNK, the prototypicalMAPKs activated in response to oxidative stress, we paradox-ically observed a selective increase in p-ERK1/2. The increasein p-ERK1/2 expression was blocked by the D1-selective antag-
onist, and when SK-N-MC cells were treated with dopamine inthe presence of SMBS, the antioxidant failed to affectp-ERK1/2 levels (data not shown). However, inhibition ofMEK1/2 by U0126 blocked the SKF R-38393-mediated increasein p-ERK1/2 levels. Moreover, suppression of p-ERK1/2 expres-sion with catalytically defective MEK1 mutant MEK1-K97Malso blocked the SKF R-38393- and the dopamine-induced in-creases in p-ERK1/2 levels and relieved the D1 dopamine re-ceptor-mediated cytotoxicity. Together, these combined dataindicate that stimulation of the D1 dopamine receptor leads top-ERK1/2 activation.
Dopamine, however, appears to activate other MAPKs, inaddition to p-ERK1/2. Thus, dopamine also caused a robustincrease in p-p38 MAPK, with a modest increase in p-JNKlevels, and in the presence of SB203580 or SB600125, nitriteproduction was significantly attenuated, with partial restora-tion of cell viability. Moreover, the p-ERK1/2 inhibitors andMEK1-K97M and also caused only partial blockade of nitriteproduction and cell death. Interestingly, none of these effectswas observed with the D1 dopamine receptor-selective agonistSKF R-38393. These results indicate that dopamine-mediated
FIG. 7—continued
Activation of p-ERK by D1 Dopamine Receptors 39327
by guest on April 3, 2019
http://ww
w.jbc.org/
Dow
nloaded from
cytotoxicity occurs, at least in part, through the p38 MAPK andJNK, in addition to the ERK, signaling cascades. These find-ings are in congruence with those obtained using H2O2, whichalso increased the levels of both p38 MAPK and p-JNK butcaused only very modest changes in p-ERK1/2. Inhibition ofthese kinases, p38 MAPK and p-JNK, also caused a largereduction in nitrite levels and in cell death, after treatmentwith H2O2. Together, these findings suggest that the oxidativestress pathway induced by both dopamine and H2O2 occursthrough the activation of the stress-linked MAPKs, p38 MAPK
and JNK. A schematic representation summarizing our find-ings is presented in Fig. 9, where we show how dopamine canactivate the various MAPKs through two distinct pathways:the direct stimulation of the D1 dopamine receptors andthrough an oxidative pathway, arising from its autoxidation.The D1 dopamine receptors activate p-ERK1/2, whereas theoxidative pathway predominantly activates p-p38 MAPK andp-JNK. However, the possibility that some kinases of the au-toxidative pathway of dopamine also activate p-ERK1/2 cannotbe entirely eliminated. Indeed, some studies suggest thatMEKK1 (MAPK kinase kinase kinase 1), which functions as astress-activated protein kinase, primarily activating JNK andp38 MAPK, may also induce modest ERK activation (48–50).Thus, our studies do not entirely eliminate the possibility thatsome coparticipation of the oxidative pathway of dopamine,activating through MEKK1, could also cause modest activationof ERK (Fig. 9).
Activation of the ERK cascade by dopamine receptors is acomplex process that may involve multiple mechanisms (51–54). D1 dopamine receptors are known to exert their physio-logical actions by stimulating cellular adenylyl cyclase via Gs
(55). Recent studies show that activation of ERK1/2 occurs viaa Gs/adenylyl cyclase/protein kinase A-dependent pathwaythat causes activation of the small GTPase, Rap1 (56, 57). Ourresults also indicate the participation of the cAMP/protein ki-nase A-dependent pathway in these processes, because inhibi-tion of protein kinase A completely blocked D1 receptor-induced ERK activation in SK-N-MC cells.
In resting cells, ERK is kept in the cytoplasm by the micro-tubule cytoskeleton, which serves as a major docking matrix forup to 35% of cellular ERK1 and ERK2 (58). Other putativecytosol-anchoring proteins of ERK are the MAP kinase phos-phatases (MKPs), in particular MKP3 (29, 59). ERK is alsoretained in the cytosol by its association with MEK1/2 (60).After phosphorylation by MEK1/2, p-ERK1/2 is detached from
FIG. 8. �-Arrestin2 mutant blocks the formation of the D1areceptor-�-arrestin2-p-ERK complex. A, immunoprecipitation withp-ERK1/2 antibody and immunoblotting with �-arrestin2, D1 receptor,and p-ERK1/2 antibody after a 1-h stimulation with 100 �M SKFR-38393 and 100 �M dopamine (DA) in mock transfected (pcDNA3),�-arrestin1 dominant negative mutant (�-arrestin1 V53D)-, or �-arres-tin2 dominant negative mutant (�-arrestin2 V54D)-transfected SK-N-MC cells. B, immunoprecipitation with �-arrestin2 antibody andimmunoblotting with p-ERK1/2 and D1 receptor antibody after a 1-hstimulation with 100 �M SKF R-38393 and 100 �M dopamine in mocktransfected (pcDNA3), �-arrestin1 dominant negative mutant (�-arres-tin1 V53D)-, or �-arrestin2 dominant negative mutant (�-arrestin2V54D)-transfected SK-N-MC cells. C, immunoprecipitation with D1antibody and immunoblotting with p-ERK1/2 and �-arrestin2 antibodyafter a 1-h stimulation with 100 �M SKF R-38393 and 100 �M dopaminein mock transfected (pcDNA3), �-arrestin1 dominant negative mutant(�-arrestin1 V53D)-, or �-arrestin2 dominant negative mutant (�-arres-tin2 V54D)-transfected SK-N-MC cells.
FIG. 9. Proposed model of dopamine-induced activation ofMAPKs cascade. Dopamine (DA) stimulation of D1 dopamine recep-tors activates the ERK1/2 signaling pathway via protein kinase A(PKA). Alternatively, the autoxidation of dopamine or H2O2 stimulatesp-38 and JNK cascades. After activation, p-ERK1/2, p-JNK, and p-p38phosphorylate cytoskeletal proteins, cytosolic kinases, or transcriptionfactors, leading to an increase of nitric oxide production and finally cellapoptosis.
Activation of p-ERK by D1 Dopamine Receptors39328
by guest on April 3, 2019
http://ww
w.jbc.org/
Dow
nloaded from

this cytosolic anchor and rapidly translocated into the nucleus(61), in a process that requires dimerization and phosphateincorporation into the regulatory Thr and Tyr residues of atleast one of the ERKs in the dimer (62). Several sites on ERKhave been identified to be important in both its nuclear trans-location (residues 321–327) and its cytosolic retention (residues312–320), with the three acidic residues Asp-316, Asp-319, andGlu-320 being especially important (63). Once in the nucleus,p-ERK1/2 can phosphorylate substrates, such as Elk-1 (64),thereby transmitting signals originally received by cell surfacereceptors to the nucleus.
In our studies, we found that the majority of p-ERK1/2 wasretained in the cytoplasm, with only a modest amount trans-located into the nucleus. The failure of p-ERK1/2 to translocateinto the nucleus, coupled with its retention in the cytoplasm,provides some evidence for a possible mechanism by whichERK activation may trigger a cytotoxic, rather than a mito-genic, response upon activation of the D1 dopamine receptor. Inour coimmunoprecipitation studies, we have shown thatp-ERK1/2 forms a stable heterotrimeric complex with the D1dopamine receptor and �-arrestin2. The arrestin proteins en-able the internalization and trafficking of certain phosphoryl-ated GPCRs, including the D1 dopamine receptor (65), awayfrom the plasma membrane, terminating receptor-dependentsignals by precluding receptor-G protein coupling (66). Block-ade of the expression of �-arrestin2 with its dominant negativemutant almost completely prevented the formation of the het-erotrimeric complex, while simultaneously blocking the phos-phorylation of pERK1/2.
A similar observation has also been made for a limited num-ber of GPCRs. Thus, activation of the protease-activated recep-tor type 2 (67), vasopressin V2 (68), and the angiotensin AT1areceptor (31) leads to the cytosolic retention of p-ERK, with lowtranscription activity and lack of a mitogenic response. In all ofthese studies, the cytosolic pool of p-ERK was shown to formstable complexes with the activated receptors and �-arrestin(69). In other studies with vasopressin V2 receptors, it wasshown that the stability of the receptor-�-arrestin complex, andnot the specificity of the G protein coupling, was paramount indetermining the subcellular distribution of ERK and its reten-tion in the cytosol (31, 70). Interestingly, in human embryonickidney-293 cells, the rat variant of D1 dopamine receptors wasfound to form only transient complexes with �-arrestin,whereas both the AT1a and the vasopressin V2 receptors wereshown to form stable complexes (32). Yet, our data clearly showthat at least in SK-N-MC cells, the D1 dopamine receptor isable to form stable complexes with both �-arrestin2 andp-ERK1/2.
If abnormally retained in the cytoplasm, �-arrestin-boundp-ERK1/2 may phosphorylate multiple plasma membrane, cy-toplasmic, and cytoskeletal substrates (34), resulting in al-tered, and perhaps inappropriate, series of cellular events. Thesignificance of cytosolic retention of p-ERK1/2 is underscoredby recent findings in postmortem tissue of neurodegenerativediseases, where neuronal inclusion bodies were found to con-tain substantially high levels of aggregated p-ERK1/2. Thus,from immunohistochemical analyses, granular precipitates ofp-ERK1/2 were seen in the cytoplasm of neurons exhibitingearly tau deposition in Alzheimer’s disease, in neurons withPick bodies in Pick’s disease, and in neurons in progressivesupranuclear palsy and corticobasal degeneration (71). More-over, p-ERK1/2, but not p-p38 MAPK or p-JNK, was found inLewy bodies in Parkinson’s disease and in dementia with Lewybodies (72), which were shown to be located primarily in thecytoplasm and not in the nucleus (73). Therefore, elucidation ofthe precise mechanisms that cause cytosolic retention of
p-ERK1/2 is likely to provide important insights into the etiol-ogy of multiple neurodegenerative diseases.
Acknowledgment—We thank Dr. Christophe Wersinger for assist-ance in preparation of the striatal neurons.
REFERENCES
1. Ghorayeb, I., Fernagut, P. O., Aubert, I., Bezard, E., Poewe, W., Wenning,G. K., and Tison, F. (2000) Movement Disorders 15, 531–536
2. Stephans, S., and Yamamoto, B. (1996) Neuroscience 72, 593–6003. LaVoie, M. J., and Hastings, T. G. (1999) J Neurosci. 19, 1484–14914. Drago, J., Padungchaichot, P., Wong, J. Y., Lawrence, A. J., McManus, J. F.,
Sumarsono, S. H., Natoli, A. L., Lakso, M., Wreford, N., Westphal, H., Kola,I., and Finkelstein, D. I. (1998) J. Neurosci. 18, 9845–9857
5. Hantraye, P. (1998) Nucl. Med. Biol. 25, 721–7286. Imam, S. Z., el-Yazal, J., Newport, G. D., Itzhak, Y., Cadet, J. L., Slikker, W.,
Jr., and Ali, S. F. (2001) Ann. N. Y. Acad. Sci. 939, 366–3807. Kitazawa, M., Wagner, J. R., Kirby, M. L., Anantharam, V., and Kanthasamy,
A. G. (2002) J. Pharmacol. Exp. Ther. 302, 26–358. Junn, E., and Mouradian, M. M. (2001) J. Neurochem. 78, 374–3839. Spencer, J. P., Whiteman, M., Jenner, P., and Halliwell, B. (2002) J. Neuro-
chem. 81, 122–12910. Stefanova, N., Puschban, Z., Fernagut, P. O., Brouillet, E., Tison, F., Reindl,
M., Jellinger, K. A., Poewe, W., and Wenning, G. K. (2003) Acta Neuro-pathol. 106, 157–166
11. Cools, A. R., Lubbers, L., van Oosten, R. V., and Andringa, G. (2002) Neurop-harmacology 42, 237–245
12. Andringa, G., Stoof, J. C., and Cools, A. R. (1999) Psychopharmacology 146,328–334
13. Wersinger, C., Chen, J., and Sidhu, A. (2004) Mol. Cell. Neurosci. 25, 124–13714. Chen, J., Wersinger, C., and Sidhu, A. (2003) J. Biol. Chem. 278, 28089–2810015. Sidhu, A., and Fishman, P. H. (1990) Biochem. Biophys. Res. Commun. 166,
574–57916. Sweatt, J. D. (2001) J. Neurochem. 76, 1–1017. Johnson, G. L., and Lapadat, R. (2002) Science 298, 1911–191218. Davis, R. J. (2000) Cell 103, 239–25219. Cobb, M. H., and Goldsmith, E. J. (1995) J. Biol. Chem. 270, 14843–1484620. Post, G. R., and Brown, J. H. (1996) FASEB J. 10, 741–74921. Seger, R., and Krebs, E. G. (1995) FASEB J. 9, 726–73522. Paul, A., Wilson, S., Belham, C. M., Robinson, C. J., Scott, P. H., Gould, G. W.,
and Plevin, R. (1997) Cell. Signalling 9, 403–41023. Bradford, M. M. (1976) Anal. Biochem. 72, 248–25424. Kimura, K., White, B. H., and Sidhu, A. (1995) J. Biol. Chem. 270,
14672–1467825. Sidhu, A., Kimura, K., Uh, M., White, B. H., and Patel, S. (1998) J. Neurochem.
70, 2459–246726. Luedtke, R. R., Griffin, S. A., Conroy, S. S., Jin, X., Pinto, A., and Sesack, S. R.
(1999) J. Neuroimmunol. 101, 170–18727. Sidhu, A., Olde, B., Humblot, N., Kimura, K., and Gardner, N. (1999) Neuro-
science 91, 537–54728. Formstecher, E., Ramos, J. W., Fauquet, M., Calderwood, D. A., Hsieh, J. C.,
Canton, B., Nguyen, X. T., Barnier, J. V., Camonis, J., Ginsberg, M. H., andChneiweiss, H. (2001) Dev. Cell 1, 239–250
29. Brunet, A., Roux, D., Lenormand, P., Dowd, S., Keyse, S., and Pouyssegur, J.(1999) EMBO J. 18, 664–674
30. Hochholdinger, F., Baier, G., Nogalo, A., Bauer, B., Grunicke, H. H., andUberall, F. (1999) Mol. Cell. Biol. 19, 8052–8065
31. Tohgo, A., Choy, E. W., Gesty-Palmer, D., Pierce, K. L., Laporte, S., Oakley,R. H., Caron, M. G., Lefkowitz, R. J., and Luttrell, L. M. (2003) J. Biol.Chem. 278, 6258–6267
32. Oakley, R. H., Laporte, S. A., Holt, J. A., Caron, M. G., and Barak, L. S. (2000)J. Biol. Chem. 275, 17201–17210
33. Tohgo, A., Pierce, K. L., Choy, E. W., Lefkowitz, R. J., and Luttrell, L. M. (2002)J. Biol. Chem. 277, 9429–9436
34. Pearson, G., Robinson, F., Beers Gibson, T., Xu, B. E., Karandikar, M.,Berman, K., and Cobb, M. H. (2001) Endocr. Rev. 22, 153–183
35. Segal, R. A., and Greenberg, M. E. (1996) Annu. Rev. Neurosci. 19, 463–48936. Xia, Z., Dickens, M., Raingeaud, J., Davis, R. J., and Greenberg, M. E. (1995)
Science 270, 1326–133137. Atkins, C. M., Selcher, J. C., Petraitis, J. J., Trzaskos, J. M., and Sweatt, J. D.
(1998) Nat. Neurosci. 1, 602–60938. English, J. D., and Sweatt, J. D. (1996) J. Biol. Chem. 271, 24329–2433239. Davis, R. J. (1993) J. Biol. Chem. 268, 14553–1455640. Alessandrini, A., Namura, S., Moskowitz, M. A., and Bonventre, J. V. (1999)
Proc. Natl. Acad. Sci. U. S. A. 96, 12866–1286941. Namura, S., Iihara, K., Takami, S., Nagata, I., Kikuchi, H., Matsushita, K.,
Moskowitz, M. A., Bonventre, J. V., and Alessandrini, A. (2001) Proc. Natl.Acad. Sci. U. S. A. 98, 11569–11574
42. Stanciu, M., Wang, Y., Kentor, R., Burke, N., Watkins, S., Kress, G., Reynolds,I., Klann, E., Angiolieri, M. R., Johnson, J. W., and DeFranco, D. B. (2000)J. Biol. Chem. 275, 12200–12206
43. Kulich, S. M., and Chu, C. T. (2001) J. Neurochem. 77, 1058–106644. Kulich, S. M., and Chu, C. T. (2003) J. Biosci. 28, 83–8945. Oh-hashi, K., Maruyama, W., Yi, H., Takahashi, T., Naoi, M., and Isobe, K.
(1999) Biochem. Biophys. Res. Commun. 263, 504–50946. Perry, G., Roder, H., Nunomura, A., Takeda, A., Friedlich, A. L., Zhu, X.,
Raina, A. K., Holbrook, N., Siedlak, S. L., Harris, P. L., and Smith, M. A.(1999) Neuroreport 10, 2411–2415
47. Stanciu, M., and DeFranco, D. B. (2002) J. Biol. Chem. 277, 4010–401748. Minden, A., Lin, A., McMahon, M., Lange-Carter, C., Derijard, B., Davis, R. J.,
Johnson, G. L., and Karin, M. (1994) Science 266, 1719–172349. Yan, M., Dai, T., Deak, J. C., Kyriakis, J. M., Zon, L. I., Woodgett, J. R., and
Activation of p-ERK by D1 Dopamine Receptors 39329
by guest on April 3, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Templeton, D. J. (1994) Nature 372, 798–80050. Karandikar, M., Xu, S., and Cobb, M. H. (2000) J. Biol. Chem. 275,
40120–4012751. Brami-Cherrier, K., Valjent, E., Garcia, M., Pages, C., Hipskind, R. A., and
Caboche, J. (2002) J. Neurosci. 22, 8911–892152. Liu, J. C., Baker, R. E., Sun, C., Sundmark, V. C., and Elsholtz, H. P. (2002)
J. Biol. Chem. 277, 35819–3582553. Gerfen, C. R., Miyachi, S., Paletzki, R., and Brown, P. (2002) J. Neurosci. 22,
5042–505454. Zhen, X., Zhang, J., Johnson, G. P., and Friedman, E. (2001) Mol. Pharmacol.
60, 857–86455. Sidhu, A., and Niznik, H. B. (2000) Int. J. Dev. Neurosci. 18, 669–67756. Vossler, M. R., Yao, H., York, R. D., Pan, M. G., Rim, C. S., and Stork, P. J.
(1997) Cell 89, 73–8257. Schmitt, J. M., and Stork, P. J. (2002) Mol. Cell 9, 85–9458. Reszka, A. A., Seger, R., Diltz, C. D., Krebs, E. G., and Fischer, E. H. (1995)
Proc. Natl. Acad. Sci. U. S. A. 92, 8881–888559. Camps, M., Nichols, A., Gillieron, C., Antonsson, B., Muda, M., Chabert, C.,
Boschert, U., and Arkinstall, S. (1998) Science 280, 1262–126560. Fukuda, M., Gotoh, Y., and Nishida, E. (1997) EMBO J. 16, 1901–190861. Chen, R. H., Sarnecki, C., and Blenis, J. (1992) Mol. Cell. Biol. 12, 915–92762. Khokhlatchev, A. V., Canagarajah, B., Wilsbacher, J., Robinson, M., Atkinson,
M., Goldsmith, E., and Cobb, M. H. (1998) Cell 93, 605–61563. Rubinfeld, H., Hanoch, T., and Seger, R. (1999) J. Biol. Chem. 274,
30349–3035264. Gille, H., Kortenjann, M., Thomae, O., Moomaw, C., Slaughter, C., Cobb,
M. H., and Shaw, P. E. (1995) EMBO J. 14, 951–96265. Kim, O. J., Gardner, B. R., Williams, D. B., Marinec, P. S., Cabrera, D. M.,
Peters, J. D., Mak, C. C., Kim, K. M., and Sibley, D. R. (2004) J. Biol. Chem.279, 7999–8010
66. Hall, R. A., and Lefkowitz, R. J. (2002) Circ. Res. 91, 672–68067. DeFea, K. A., Zalevsky, J., Thoma, M. S., Dery, O., Mullins, R. D., and
Bunnett, N. W. (2000) J. Cell Biol. 148, 1267–128168. Shenoy, S. K., and Lefkowitz, R. J. (2003) J. Biol. Chem. 278, 14498–1450669. Miller, W. E., and Lefkowitz, R. J. (2001) Curr. Opin. Cell Biol. 13, 139–14570. Oakley, R. H., Laporte, S. A., Holt, J. A., Barak, L. S., and Caron, M. G. (1999)
J. Biol. Chem. 274, 32248–3225771. Ferrer, I., Blanco, R., Carmona, M., Puig, B., Barrachina, M., Gomez, C., and
Ambrosio, S. (2001) J. Neural Transm. 108, 1383–139672. Ferrer, I., Blanco, R., Carmona, M., Ribera, R., Goutan, E., Puig, B., Rey, M. J.,
Cardozo, A., Vinals, F., and Ribalta, T. (2001) Brain Pathol. 11, 144–15873. Zhu, J. H., Kulich, S. M., Oury, T. D., and Chu, C. T. (2002) Am. J. Pathol. 161,
2087–2098
Activation of p-ERK by D1 Dopamine Receptors39330
by guest on April 3, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Jun Chen, Milan Rusnak, Robert R. Luedtke and Anita SidhuSignal Cascade
D1 Dopamine Receptor Mediates Dopamine-induced Cytotoxicity via the ERK
doi: 10.1074/jbc.M403891200 originally published online July 6, 20042004, 279:39317-39330.J. Biol. Chem.
10.1074/jbc.M403891200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/279/38/39317.full.html#ref-list-1
This article cites 73 references, 35 of which can be accessed free at
by guest on April 3, 2019
http://ww
w.jbc.org/
Dow
nloaded from





























