Embed Size (px)
Citation preview

RESEARCH LETTER
De Novo Duplication of 18p11.21–18q12.1 ina Female With Anorectal MalformationCharlotte Schramm,1,2 Markus Draaken,1,2 Enrika Bartels,1 Thomas M. Boemers,3 Eberhard Schmiedeke,4
Sabine Grasshoff-Derr,5 Stefanie M€arzheuser,6 Stuart Hosie,7 Stefan Holland-Cunz,8
Friederike Baudisch,1,9 Lutz Priebe,1,2 Per Hoffmann,1,2 Alexander M. Zink,1 Hartmut Engels,1
Felix F. Brockschmidt,1,2 Stefan Aretz,1 Markus M. N€othen,1,2 Michael Ludwig,9 and Heiko Reutter1,10*1Institute of Human Genetics, University of Bonn, Bonn, Germany2Department of Genomics, Life and Brain Center, University of Bonn, Bonn, Germany3Department of Pediatric Surgery and Pediatric Urology, Children’s Hospital, Cologne, Germany4Department of Pediatric Surgery and Urology, Center for Child and Adolescent Health, Hospital Bremen-Mitte, Bremen, Germany5Department of Pediatric Surgery, University Hospital W€urzburg, W€urzburg, Germany6Department of Pediatric Surgery, Campus Virchow Clinic, Charit�e University Hospital Berlin, Berlin, Germany7Department of Pediatric Surgery, Klinikum Schwabing, Technische Universit€at M€unchen, M€unchen, Germany8Department of Pediatric Surgery, University of Heidelberg, Heidelberg, Germany9Department of Clinical Chemistry and Clinical Pharmacology, University of Bonn, Bonn, Germany10Department of Neonatology, Children’s Hospital, University of Bonn, Bonn, Germany
Received 15 September 2010; Accepted 1 November 2010
TO THE EDITOR:
Anorectal malformations (ARM) occur in about 1 in 2,500
live births [Cuschieri, 2001]. ARM can be classified into different
groups regarding their diagnostic, therapeutic, and prognostic
features [Levitt and Pe~na, 2007], ranging from ARM without fistula
(e.g., imperforate anus without fistula), with fistula (e.g., recto-
perineal malformation) to complex ARM including cloacal
malformations. Similar to most congenital defects, ARM can occur
as an isolated malformation or associated with other defects or
syndromes. Among the defined syndromic forms are chromosomal
abnormalities (e.g., trisomy 13, 18, and 21; cat eye syndrome),
monogenic syndromes (e.g., Townes–Brocks, Klippel–Feil, or
Pallister–Hall), sequences (Pierre Robin) or associations
(VATER/VACTERL) [Levitt and Pe~na, 2007; Stoll et al., 2007].
Other than the molecular genetic elucidation of a few syndromic
forms, the current knowledge of genetic factors underlying most
ARM is very limited. Since many ARM phenotypes are associated
with reduced reproductive fitness, it is reasonable to assume
that a significant proportion of patients carry new (de novo)
mutations. On this basis we performed a systematic screening
of 13 ARM patients (Table I) and their unaffected parents using
a SNP-array-based genome-wide approach to search for mutations
characterized by a loss or gain of genomic material. The parallel
investigation of the patients’ parents allowed for identification of de
Charlotte Schramm and Markus Draaken contributed equally to this work.
Grant sponsor: German Federal Ministry of Education and Research
(Bundesministerium f€ur Bildung und Forschung, BMBF); Grant
number: 01GM08107; Grant sponsor: University of Bonn; Grant
number: O-149.0093.
*Correspondence to:
Heiko Reutter, Department of Neonatology, Institute of Human Genetics,
University of Bonn, Sigmund-Freud-Str. 25, D-53127 Bonn, Germany.
E-mail: [email protected]
Published online 11 January 2011 in Wiley Online Library
(wileyonlinelibrary.com).
DOI 10.1002/ajmg.a.33820
How to Cite this Article:Schramm C, Draaken M, Bartels E, Boemers
TM, Schmiedeke E, Grasshoff-Derr S,
M€arzheuser S, Hosie S, Holland-Cunz S,
Baudisch F, Priebe L, Hoffmann P, Zink AM,
Engels H, Brockschmidt FF, Aretz S, N€othen
MM, Ludwig M, Reutter H. 2011. De novo
duplication of 18p11.21–18q12.1 in a female
with anorectal malformation.
Am J Med Genet Part A 155:445–449.
� 2011 Wiley-Liss, Inc. 445

novo mutations. For the present analysis, patients presenting with
distinct clinical syndromes, for example, Currarino syndrome,
Townes–Brocks syndrome, or VACTERL/VATER association, or
with a positive family history were excluded from the analysis.
The study was approved by the local Ethics Committee and
the families provided informed consent. EDTA blood samples
were obtained from each family and isolation of genomic
DNA was carried out using the QIAmp DNA Blood Kit (Qiagen,
Hilden, Germany). Molecular karyotyping was performed using
SNP-based genome-wide arrays (Human660-Quad-v1 BeadChip
and Illumina HumanOmni1-Quad BeadChip [Illumina, San Die-
go, CA] containing 657,366 and 1,140,419 markers, respectively).
All analyses were performed according to the manufacturer’s
protocol. An analysis was considered to have failed if less than
95% of the loci were informative on the BeadChip. To identify
potential copy number variants (CNVs), the SNP fluorescence
intensity data of each individual were analyzed with QuantiSNP
(v2.1, www.well.ox.ac.uk/QuantiSNP/) using an Objective-Bayes
Hidden-Markov model for calling putative CNVs [Colella et al.,
2007]. CNVs with a log Bayes factor below 30 were disregarded, as
recommended. The results were also checked visually using the
GenomeStudio (v2009.2, www.illumina.com/) genotyping module
(see legend for Fig. 1). Filtering of CNVs for de novo events, known
variable regions and gene content was carried out using the
Cartagenia Bench� software (Cartagenia, Leuven, Belgium). Sex
chromosomes were excluded from further analysis.
Molecular karyotyping of the entire sample of 13 cases defined
a duplication of chromosomal region 18p11.21–18q12.1 in a female
patient (case 1; Table I) with anal atresia and rectovesical fistula,
patent ductus arteriosus (PDA) and myopia. No further de novo
chromosomal microaberration containing known genes which
did not correspond to a known variable region listed in the Database
of Genomic Variants (DGV, http://projects.tcag.ca/variation/)
were found in the remaining 12 ARM patients.
The patient (case 1) was born after 41 weeks of gestation as the
first child to a 35-year-old woman (gravida 1, para 1). Pregnancy
was uneventful. The healthy, nonconsanguineous parents were
of Caucasian origin with no family history of malformations. The
patient has a healthy younger sister who was 5 years of age.
At birth, the patient presented with anal atresia and rectovesical
fistula, PDA, slight facial dysmorphism and hypotonia. Because
of the anal atresia, a colostomy was performed. The anal atresia
was surgically corrected at the age of 6 months, and the colostomy
was closed. At the age of 3 years myopia (�7/�4 dpt) was diag-
nosed. Motor development was slightly delayed during infancy, but
treatment was not considered necessary. Cognitive functioning was
evaluated with the Bayley Scales of Infant Development at the age
of 3 years and 3 months [Bayley, 1993] with normal results. In
accordance with this, the girl showed normal development without
any signs of cognitive, language, or social impairments over the
following years. Likewise her gross motor skills were normal with
only slight deficits in her fine motor skills. At the time of assessment
the patient was 7 years of age and successfully attending primary
school, showing no signs of physical illness. Furthermore her slight
facial dysmorphism described in infancy was no longer apparent.
Because of her congenital anomalies, a karyotype (G-banding)
had been performed when she was a neonate. This revealed a 46,XX
karyotype with the presence of a small duplication on chromosome
TABLE I. Phenotypes of the Investigated ARM Patients
Case Chip array Sex ARM Other major malformations/deformations1a 660 F þ Slight facial dysmorphism in infancy, myopia, PDA2 660 M þ Myelomeningocele, Arnold–Chiari malformation II,
glandular hypospadias, right-sided renal agenesis3 660 M þ Craniosynostosis (coronal and lambdal synostosis),
bilateral pre-auricular tags, hemifacial microsomia4 660 M þ Left-sided renal dysplasia, bilateral VUR IV�,
cryptorchidism, persistent urachus5 660 M þ Prune-belly syndrome, right-sided cystic renal dysplasia,
caudal regression syndrome, costal bone deformity6 660 F þ Hyperopia, oral cutaneous fistula, left-sided pre-auricular fistula,
left-sided UPJ obstruction, left-sidedhydronephrosis, Hypomelanosis Ito
7 Omni F þ Right-sided VUR II–III�, left-sided VUR I�, atrial septal defect type II8 Omni M þ Left-sided renal agenesis, right-sided UPJ obstruction,
right-sided hydronephrosis, glandular hypospadias9 Omni F þ PFO, right-sided UPJ obstruction,
right-sided hydronephrosis, bicornuate uterus10 Omni F þ Vaginal atresia, bicornuate uterus, sacral dysplasia11 Omni F þ gibbus, missing teeth12 Omni M þ Phimosis, cryptorchidism, left-sided VUR II–III�
13 Omni M þ Left-sided renal agenesis, glandular hypospadias
VUR, vesico-ureteral reflux; PDA, patent ductus arteriosus; UPJ obstruction, ureteropelvic junction obstruction; PFO, persistent foramen ovale.aPatient with de novo duplication of chromosomal region 18p11.21–18q12.1.
446 AMERICAN JOURNAL OF MEDICAL GENETICS PART A
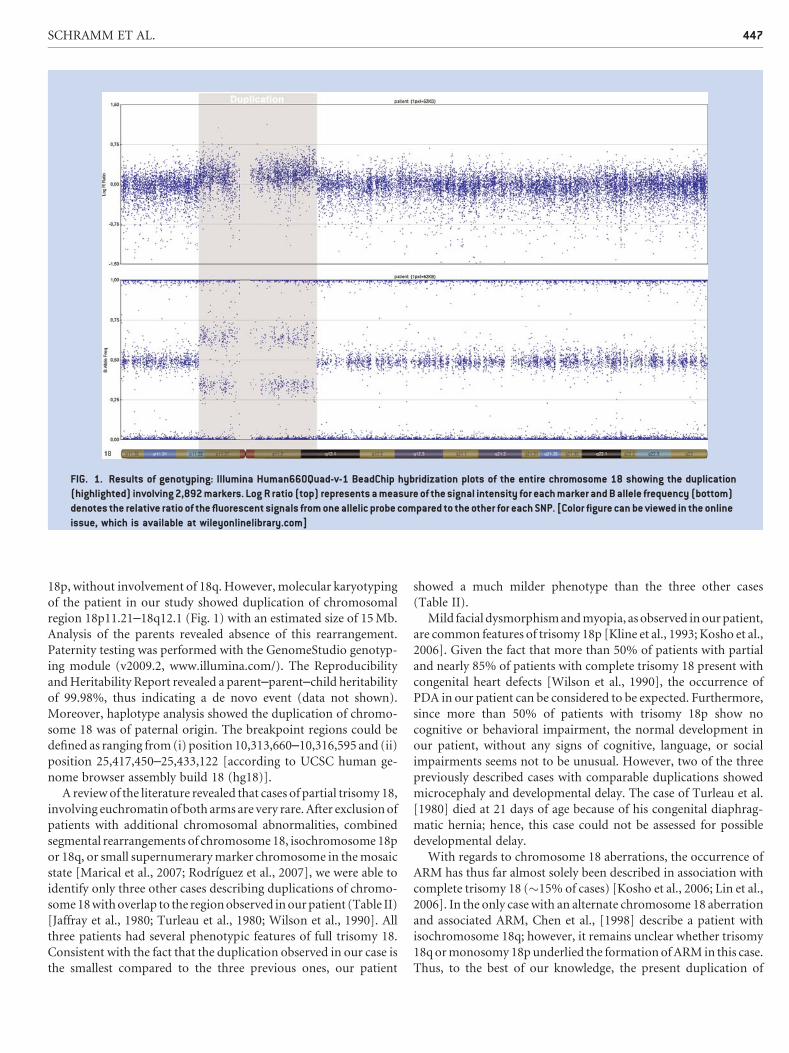
18p, without involvement of 18q. However, molecular karyotyping
of the patient in our study showed duplication of chromosomal
region 18p11.21–18q12.1 (Fig. 1) with an estimated size of 15 Mb.
Analysis of the parents revealed absence of this rearrangement.
Paternity testing was performed with the GenomeStudio genotyp-
ing module (v2009.2, www.illumina.com/). The Reproducibility
and Heritability Report revealed a parent–parent–child heritability
of 99.98%, thus indicating a de novo event (data not shown).
Moreover, haplotype analysis showed the duplication of chromo-
some 18 was of paternal origin. The breakpoint regions could be
defined as ranging from (i) position 10,313,660–10,316,595 and (ii)
position 25,417,450–25,433,122 [according to UCSC human ge-
nome browser assembly build 18 (hg18)].
A review of the literature revealed that cases of partial trisomy 18,
involving euchromatin of both arms are very rare. After exclusion of
patients with additional chromosomal abnormalities, combined
segmental rearrangements of chromosome 18, isochromosome 18p
or 18q, or small supernumerary marker chromosome in the mosaic
state [Marical et al., 2007; Rodr�ıguez et al., 2007], we were able to
identify only three other cases describing duplications of chromo-
some 18 with overlap to the region observed in our patient (Table II)
[Jaffray et al., 1980; Turleau et al., 1980; Wilson et al., 1990]. All
three patients had several phenotypic features of full trisomy 18.
Consistent with the fact that the duplication observed in our case is
the smallest compared to the three previous ones, our patient
showed a much milder phenotype than the three other cases
(Table II).
Mild facial dysmorphism and myopia, as observed in our patient,
are common features of trisomy 18p [Kline et al., 1993; Kosho et al.,
2006]. Given the fact that more than 50% of patients with partial
and nearly 85% of patients with complete trisomy 18 present with
congenital heart defects [Wilson et al., 1990], the occurrence of
PDA in our patient can be considered to be expected. Furthermore,
since more than 50% of patients with trisomy 18p show no
cognitive or behavioral impairment, the normal development in
our patient, without any signs of cognitive, language, or social
impairments seems not to be unusual. However, two of the three
previously described cases with comparable duplications showed
microcephaly and developmental delay. The case of Turleau et al.
[1980] died at 21 days of age because of his congenital diaphrag-
matic hernia; hence, this case could not be assessed for possible
developmental delay.
With regards to chromosome 18 aberrations, the occurrence of
ARM has thus far almost solely been described in association with
complete trisomy 18 (�15% of cases) [Kosho et al., 2006; Lin et al.,
2006]. In the only case with an alternate chromosome 18 aberration
and associated ARM, Chen et al., [1998] describe a patient with
isochromosome 18q; however, it remains unclear whether trisomy
18q or monosomy 18p underlied the formation of ARM in this case.
Thus, to the best of our knowledge, the present duplication of
FIG. 1. Results of genotyping: Illumina Human660Quad-v-1 BeadChip hybridization plots of the entire chromosome 18 showing the duplication
(highlighted) involving 2,892 markers. Log R ratio (top) represents a measure of the signal intensity for each marker and B allele frequency (bottom)
denotes the relative ratio of the fluorescent signals from one allelic probe compared to the other for each SNP. [Color figure can be viewed in the online
issue, which is available at wileyonlinelibrary.com]
SCHRAMM ET AL. 447
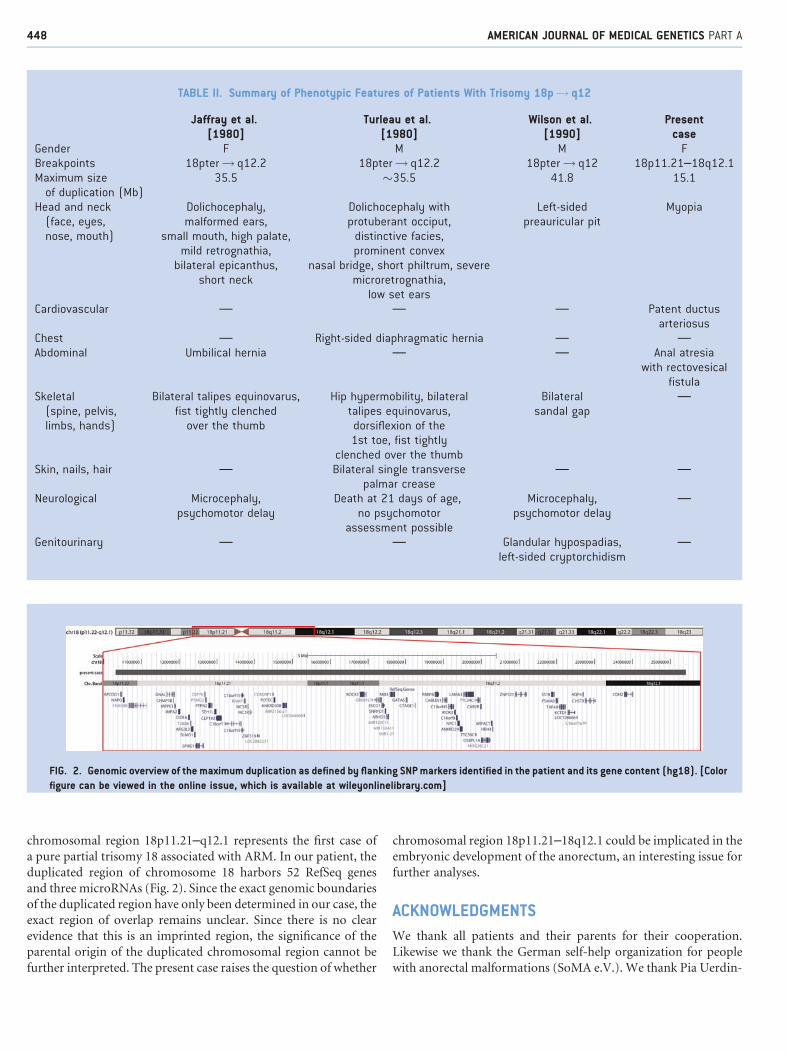
chromosomal region 18p11.21–q12.1 represents the first case of
a pure partial trisomy 18 associated with ARM. In our patient, the
duplicated region of chromosome 18 harbors 52 RefSeq genes
and three microRNAs (Fig. 2). Since the exact genomic boundaries
of the duplicated region have only been determined in our case, the
exact region of overlap remains unclear. Since there is no clear
evidence that this is an imprinted region, the significance of the
parental origin of the duplicated chromosomal region cannot be
further interpreted. The present case raises the question of whether
chromosomal region 18p11.21–18q12.1 could be implicated in the
embryonic development of the anorectum, an interesting issue for
further analyses.
ACKNOWLEDGMENTS
We thank all patients and their parents for their cooperation.
Likewise we thank the German self-help organization for people
with anorectal malformations (SoMA e.V.). We thank Pia Uerdin-
FIG. 2. Genomic overview of the maximum duplication as defined by flanking SNP markers identified in the patient and its gene content (hg18). [Color
figure can be viewed in the online issue, which is available at wileyonlinelibrary.com]
TABLE II. Summary of Phenotypic Features of Patients With Trisomy 18p! q12
Jaffray et al.[1980]
Turleau et al.[1980]
Wilson et al.[1990]
Presentcase
Gender F M M FBreakpoints 18pter! q12.2 18pter! q12.2 18pter! q12 18p11.21–18q12.1Maximum size
of duplication (Mb)35.5 �35.5 41.8 15.1
Head and neck(face, eyes,nose, mouth)
Dolichocephaly,malformed ears,
small mouth, high palate,mild retrognathia,
bilateral epicanthus,short neck
Dolichocephaly withprotuberant occiput,
distinctive facies,prominent convex
nasal bridge, short philtrum, severemicroretrognathia,
low set ears
Left-sidedpreauricular pit
Myopia
Cardiovascular — — — Patent ductusarteriosus
Chest — Right-sided diaphragmatic hernia — —Abdominal Umbilical hernia — — Anal atresia
with rectovesicalfistula
Skeletal(spine, pelvis,limbs, hands)
Bilateral talipes equinovarus,fist tightly clenched
over the thumb
Hip hypermobility, bilateraltalipes equinovarus,
dorsiflexion of the1st toe, fist tightly
clenched over the thumb
Bilateralsandal gap
—
Skin, nails, hair — Bilateral single transversepalmar crease
— —
Neurological Microcephaly,psychomotor delay
Death at 21 days of age,no psychomotor
assessment possible
Microcephaly,psychomotor delay
—
Genitourinary — — Glandular hypospadias,left-sided cryptorchidism
—
448 AMERICAN JOURNAL OF MEDICAL GENETICS PART A

gen for her excellent technical assistance and Dr. Christine Schmael
for her expert advice on the manuscript. C.S., M.D., E.B., T.M.B.,
E.S., S.G.-D., S.H., S.H.-C., F.B., M.M.N., M.L., and H.R. are
members of the ‘‘Network for the Systematic Investigation of
the Molecular Causes, Clinical Implications, and Psychosocial
Outcome of Congenital Uro-Rectal Malformations (CURE-Net)’’
which is supported by a research grant (01GM08107) from
the German Federal Ministry of Education and Research
(Bundesministerium f€ur Bildung und Forschung, BMBF). C.S. is
supported by the BONFOR program of the University of Bonn,
grant number O-149.0093.
REFERENCES
Bayley N. 1993. Barley scales of infant development. 2nd edition.Psychological Corporation: San Antonio, TX.
Chen CP, Chern SR, Lee CC, Town DD. 1998. Isochromosome 18q in afetus with congenital magacystis, intra-uterine growth retardation andcloacal dysgenesis sequence. Prenat Diagn 18:1068–1074.
Colella S, Yau C, Taylor JM. 2007. QuantiSNP: An Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation usingSNP genotyping data. Nucleic Acids Res 35:2013–2025.
Cuschieri A. 2001. EUROCAT Working Group. Descriptive epidemiologyof isolated anal anomalies: A survey of 4.6 million births in Europe. Am JMed Genet 103:207–215.
Jaffray JY, Geneix A, Goumy P, Perissel B, Menut G, Malet P. 1980. Trisomiepartielle 18(pter! q122) d’origine maternelle. Ann G�en�et 23:224–227.
Kline AD, White ME, Wapner R, Rojas K, Biesecker LG, Kamholz J, ZackaiEH, Muenke M, Scott CI Jr, Overhauser J. 1993. Molecular analysis of the
18q� syndrome and correlation with phenotype. Am J Hum Genet52:895–906.
Kosho T, Nakamura T, Kawame H, Baba A, Tamura M, Fukushima Y. 2006.Neonatal management of trisomy 18: Clinical details of 24patients receiving intensive treatment. Am J Med Genet Part A140A:937–944.
Levitt MA, Pe~na A. 2007. Anorectal malformations. Orphanet J Rare Dis2:33.
Lin HY, Lin SP, Chen YJ, Hung HY, Kao HA, Hsu CH, Chen MR, Chang JH,Ho CS, Huang FY, Shyur SD, Lin DS, Lee HC. 2006. Clinical character-istics and survival of trisomy 18 in a medical center in Taipei, 1988–2004.Am J Med Genet Part A 140A:945–951.
Marical H, Le Bris MJ, Douet-Guilbert N, Parent P, Descourt JP, Morel F,De Brakeleer M. 2007. 18p trisomy: A case of direct 18p duplicationcharacterized by molecular cytogenetic analysis. Am J Med Genet Part A143A:2192–2195.
Rodr�ıguez L, Liehr T, Mrasek K, Mansilla E, Mart�ınez-Fern�andez ML,Garcia A, Mart�ınez-Fr�ıas ML. 2007. Small supernumerary chromosomemarker generating complete and pure trisomy 18p, characterized bymolecular cytogenetic techniques and review. Am J Med Genet Part A143A:2727–2732.
Stoll C, Alembik Y, Dott B, Roth MP. 2007. Associated malformations inpatients with anorectal anomalies. Eur J Med Genet 50:281–290.
Turleau C, Chavin-Colin F, Narbouton R, Asensi D, de Grouchy J. 1980.Trisomy 18q-. Trisomy mapping of chromosome 18 revisited. Clin Genet18:20–26.
Wilson GN, Heller KB, Elterman RD, Schneider NR. 1990. Partial trisomy18 with minimal anomalies: Lack of correspondence between phenotypicmanifestations and triplicated loci along chromosome 18. Am J MedGenet 36:506–510.
SCHRAMM ET AL. 449




![Epididymitis Due to Bilateral Ectopic Vas Deferens …orchitis episodes. Discussion Epididymitis is associated with anorectal malformation in 1.2-6.1% [2]. This uncommon urologic condition](https://img.pdfslide.net/doc/110x75/5f37581ebe40af7a227a9d32/epididymitis-due-to-bilateral-ectopic-vas-deferens-orchitis-episodes-discussion.jpg)
























