Embed Size (px)
Citation preview

International Journal of Mass Spectrometry and Ion Processes. 68 (1986) 35-48
Elsevier Science Publishers B.V., Amsterdam - Printed in The Netherlands
35
DECOMPOSITION THRESHOLDS AND ASSOCIATED TRANSLATIONAL ENERGY RELEASES FOR EIGHT C,H,O +’ ISOMERS
JOHN C. TRAEGER
Department of Chemistry, La Trobe University, Bundoora, Vie. 3083 (Australia)
DAVID J. McADOO
Marine Biomedical Institute, Galveston, TX 77550 2 772 (U.S.A.)
(First received 6 May 1985; in final form 24 June 1985)
ABSTRACT
Appearance energies of C,H,O+ and C,H,O+ produced from a variety of C,HsO+’ ions were measured utilizing a photoionization mass spectrometer. The results demonstrate that this technique is extremely useful for obtaining information about very complex potential surfaces. Except for ionized cyclobutanol, all C4H,0+’ isomers examined with the oxygen on the terminal carbon decompose to C2H30+ and C3H50t with critical energies of 794 f 1 kJ
mol-‘, consistent with earlier conclusions that these ions decompose through common pathways. This energy is about 20 kJ mol-’ greater than AHr(CH,CO+ + ‘C,H,), $0 kJ mall’ greater than AHr(CH&H&O+ + ‘CH,) and equal to AH,(CH,=CHCH=OH+
CH,). Except for ionized 2-butanone, the C,H,O +’ ions having the oxygen on the second carbon and CH,=CHCHOHCH2CD3+‘ also have activation energies for decomposition that are substantially above the heats of formation of the resulting fragments. The ratio of translational energy released to energy present in excess of A H,(products) is 2.5 times higher for CH,CO+ formation than for formation of CH&H&O+ from C,HsO+’ ions. Possible relationships between this observation and previous conclusions that C,HsO+’ ions decom- pose without prior randomization of internal energy are discussed.
INTRODUCTION
Previous studies of the reactions of a large number of C4H80+’ isomers have revealed a very rich and complex chemistry [l]. These ions isomerize extensively on the way to losing a+ methyl group to produce a mixture of CH,CH,CO+ and CH,=CHCH=OH [l(b),l(c),2] and an ethyl group to produce CH,CO+. CH,CO+ and CH,CH,CO+ ions appear to be produced following isom+erization to the 2-butanone ion [l(b)-l(d)], and CH,=CHCH=OH is probably formed by simple cleavage of
0168-1176/86/$03.50 0 1986 Elsevier Science Publishers B.V.

36
CH&H$HCH=bH [l(d),2]. Isomers of C,H*O+’ and related ions re- arrange by 5- and 6-membered ring hydrogen transfers [1,3,4] and 3-mem- bered ring transfers of a variety of moieties [l(d),4(d),4(e),5-71. Parallels between the 5- and 6-membered ring hydrogen transfers in radical positive ions and isomerizations of free radicals have been noted by others [g]. Three-membered ring isomerizations in radical cations, parallels of which are usually highly unfavorable in free radical chemistry [9], have been termed “carbocation-like” reactions [4(d)]. While it is clear that 3- and 5-membered ring isomerizations are important in radical positive ions, little experimental information about their critical energies exists. Irregt$arities in translational energy + releases associated with CH,CH ,C(=OH)CH 2 and CH,- CHC(=OH)CH, decompositions have been interpreted as evidence that those ions decompose without prior randomization of energy [lO,ll]. There- fore, we undertook a photoionization study of C,HsO+’ ions to better define the energetic requirements for their isomerizations and to explore the re- lationship between excess energy in the ions and the translational energy released in their decompositions.
EXPERIMENTAL
Photoionization efficiency curves were obtained with a microcomputer- controlled photoionization mass spectrometer, which has been described in detail elsewhere [12]. The photon beam used in the present experiments was produced from the hydrogen pseudo-continuum with a wavelength resolu- tion of 0.125 nm corresponding to an energy resolution of 0.01 eV at 10 eV photon energy. Calibration of the absolute energy scale with known atomic emission lines indicated it to be accurate to less than 0.003 eV. Experiments were performed at ambient temperature (297 K) with sample pressures of 10e3 Pa in the region of the ion source. Appearance energies, AE, were derived by linear extrapolation to the abscissa of the first rising portion of the curves, ignoring hot bands. Uncertainties in the AE measurements are, at most, kO.02 eV, except for measurements on 3-heptanone and 3-methyl-2- hexanone, which have uncertainties of f0.05 eV stemming from the low intensities of the latter signals. Heats of formation of decomposition prod- ucts were derived from the appearance energies utilizing the stationary electron convention and the relationship [13]
AHlgH(A++ B) = AEm + AH,t,,(AB) + AK,,,
where AH,,,, = A H&(A’) + A H&(B) - 745 R and is obtained by statistical mechanical calculations described elsewhere [13(a),(b)]. Estimated heats of formation of neutral species are thought to be within 5 kJ mall’ of the correct values.

31
Translational energies released were determined by scanning the electric sector following the third field-free region of a mass spectrometer of the geometry electric-sector/magnetic-sector/electric-sector [14,36].
Except for n-butanal, which may have contained small amounts of other substances, all compounds were commercial or synthesized samples [l(b),(c)] shown to be of high purity be gas chromatography.
RESULTS AND DISCUSSION
Decomposition thresholds for ions with the oxygen on the terminal carbon
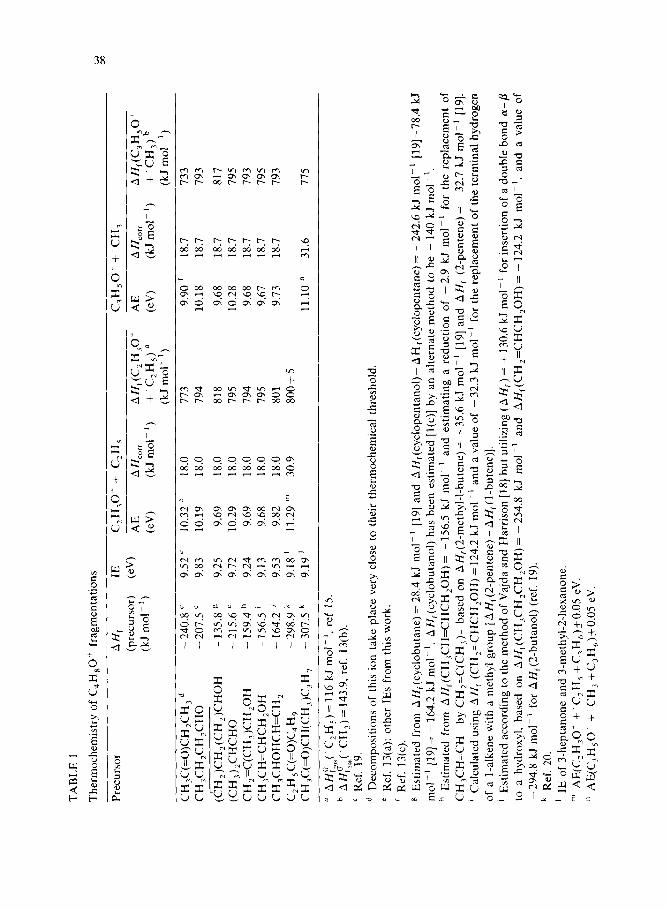
Photoionization appearance energies and the thresholds for fragmentation derived therefrom are given in Table 1. Heats of formation of three C,H80+’ isomers for which precise values were not previously available can be derived
1 from the data in Table 1. These are AN,[(CH2)CH,(CH,)CHC)fi)] = 75’7 kJ
mol-‘, AH,[CH,:C(CH7)CHZdI-I] = 732 kJ mol-’ and
AH,(CH,CH=CHCH,OH) = 724 kJ mol-‘. Previous electron impact mea- surements on the first and last of these ions [l(c)] gave values of 757 and 736
kJ mol-‘, respectively. The agreement for Au,[(CH,)CH,(CH,)CHbII] is
fortuitous, as AH,[(CH,)CH,(CH2)CHbII] was estimated previously [l(c)] to be -140 kJ mol-‘, which is 4 k,J .mol-’ below the value used in the present work. The CH,CH=CHCH,OH discrepancy of 12 kJ mol-’ is the worst of the six comparisons possible between values in ref. l(c) and the present work, validating much of the C,H*O+’ potential diagram in ref. l(c).
Except for ionized cyclobutanol, whose apparent decomposition thresholds were about 24 kJ mol-’ above those of the other ions, both decompositions of all of the Cl ions (ions with the oxygen on the end carbon) had the same threshold, 794 + 1 kJ mol-‘. This strongly supports the previous conclusion [l(b),(c)] that these ions interconvert and decompose by common pathways. Relevant pathways to CH,CO+ and CH,CH,CO+ based on previous work [l] are given in Scheme 1. It is likely that the common high energy point on the potential surface is at the transition state for converting Cl to C2 ions (ions with the oxygen on the second carbon).
The thresholds for CH,CO++ ‘C,H, formation were about 20 kJ mol-’ higher than expected from the known heats of formation of CH3COf (657 kJ mol-‘) [13(a)] + ‘C,H, (116 kJ mol-‘) [15]. However, CH&O+ is the only energetically accessible C,H,O+ isomer in these decompositions (all others are at least 181 kJ mol-’ above CH,CO+ [16]). We believe CH,CO+ can only be formed via the 2-butanone ion, as that is the only C,H80+‘ isomer from which it can be formed by simple fragmentation [17]. The derived C3H50++ ‘CH, heats for formation are about 60 kJ mol-’ higher than expected for CH,CH,CO+ (591 kJ mol-‘) [13(c)]+ ‘CH, (144 kJ
TA
BL
E
1
The
rmoc
hem
istr
y of
C
4H80
+
frag
men
tatio
ns
Prec
urso
r A
H,
IE
C,H
,O+
+
C
,H,
C,H
,O+
+
‘CH
,
(pre
curs
or)
(ev)
A
E
AH
+
A
Hf(
C,H
,O
AE
A
%,,
AH
,(C
,H,O
+
(kJ
mol
-‘)
(eV
) (k
J g:
lF’)
+
.C
,H,)
a
(eV
) (k
J m
ol-‘
) +
.CH
,)
b (k
J m
ol-‘
) (k
J m
ol-‘
)
CH
,C(=
O)C
H,C
H,’
-
240.
8 ’
9.52
’
10.3
2 ’
18.0
77
3 9.
90
i 18
.7
733
CH
,CH
,CH
,CH
O
- 20
7.5
’ 9.
83
10.1
9 18
.0
794
10.1
8 18
.7
793
(CH
,)C
H,(
CH
,)C
HO
H
- 13
5.8
g 9.
25
9.69
18
.0
818
9.68
18
.7
817
(CH
,)
,CH
CH
O
-215
.6’
9.72
10
.29
18.0
79
5 10
.28
18.7
79
5
CH
,=C
(CH
,)C
H,O
H
- 15
9.4
h 9.
24
9.69
18
.0
794
9.68
18
.7
793
CH
,CH
=C
HC
H,O
H
- 15
6.5
’ 9.
13
9.68
18
.0
795
9.67
18
.7
795
CH
,CH
OH
CH
=C
H,
- 16
4.2
’ 9.
53
9.82
18
.0
801
9.73
18
.7
793
C,H
,C(=
O)C
,H,
-298
.9
k 9.
18 ’
11
.29
m
30.9
80
0&5
CH
,C(=
O)C
H(C
H,)
C,H
, -3
07.5
k
9.19
’
11.1
0 ”
31.6
77
5
0 A
HF,
4X(‘
CZ
H5)
=11
6 kJ
mol
-‘,
ref
15.
’ A
HE
q,(
CH
,)
= 1
43.9
, re
f. 13
(b).
’
Ref
. 19
. ’
Dec
ompo
sitio
ns
of
this
io
n ta
ke
plac
e ve
ry
clos
e to
th
eir
ther
moc
hem
ical
th
resh
old.
’
Ref
. 13
(a):
ot
her
IEs
from
th
is
wor
k.
’ R
ef.
13(c
).
8 E
stim
ated
fr
om
AH
,(cy
clob
utan
e)
= 2
8.4
kJ m
ol-’
[1
9] a
nd
AH
,(cy
clop
enta
nol)
- A
H,(
cycl
open
tane
) =
-2
42.6
kJ
m
ol-’
[1
9]-7
8.4
kJ
mol
-’
[19]
=
- 16
4.2
kJ m
ol.
‘.
A H
,(cy
clob
utan
ol)
has
been
es
timat
ed
[l(c
)]
by
an
alte
rnat
e m
etho
d to
be
-
140
kJ m
ol-‘
. h
Est
imat
ed
from
A
H,(
CH
,CH
=C
HC
H,O
H)
= -
15
6.5
kJ
mol
-’
and
estim
atin
g a
redu
ctio
n of
-2
.9
kJ
mol
F’
for
the
repl
acem
ent
of
CH
,CH
=C
H-
by
CH
,=C
(CH
,)-
base
d on
A
H,(
2-m
ethy
l-l-
bute
ne)
= -
35
.6
kJ
mol
-’
[19]
and
A
H,
(2-p
ente
ne)
= -
32
.7
kJ
mol
-’
[19]
. ’
Cal
cula
ted
usin
g A
H,
(CH
,=C
HC
H,O
H)
= 1
24.2
kJ
mol
-’
and
a va
lue
of
- 32
.3 k
J m
ol-’
fo
r th
e re
plac
emen
t of
th
e te
rmin
al
hydr
ogen
of
a
1 -al
kene
w
ith
a m
ethy
l gr
oup
[AH
, (2
-pen
tene
) -
AH
, (I
-bu
tene
)].
’ E
stim
ated
ac
cord
ing
to t
he
met
hod
of V
ajda
an
d H
arri
son
[18]
but
ut
ilizi
ng
(AH
,)
= +
130
.6 k
J m
ol-’
fo
r in
sert
ion
of a
dou
ble
bond
C
Y-/
3 to
a
hydr
oxyl
, ba
sed
on
AH
,(C
H,C
H,C
H,O
H)=
-2
54.8
kJ
m
ol-’
an
d A
H,(
CH
,=C
HC
H,O
H)=
-1
24.2
kJ
m
ol-‘
, an
d a
valu
e of
-
294.
8 kJ
mol
-’
for
AH
,(2-
buta
nol)
(r
ef.
19).
k
Ref
. 20
. ’
IE
of 3
-hep
tano
ne
and
3-m
ethy
l-2-
hexa
none
. m
AE
(C,H
,O+
+
‘C
,H,
+C
,H,)
+_0
.05
eV.
” A
E(C
,H,O
+
+‘C
H,
+C
,H,)
+0.
05
eV.

39
EH2CH2C”~&:: - CH3CHJ CH3CH=CHCHfi)H
+OH .e CH3CH*CH ”
1 +OH
II CH2=C”CH
1 +
t 0+.
E CH3CH2 CH3
/ I+ CHpl* . + +flCCH3 CH3CH*C0 + CH
’ 3
Scheme 1.
mall’) [13(b)], but match that of CH,=CHCH=bH -t ‘CH, (649 +_ 13 kJ mol-’ [18] + 144 kJ mol-‘). The latter species would be expected to dominate rapid, ion-source decompositions at energies above the threshold for its formation due to the len$hy pathway to CH,CH,CO+. However, based+on results for CH2=CHCHOHCH,CD, discussed below, CH,=CHCH=OH may not be formed from C4H80+’ ions until somewhat above the calculated thermochemical threshold. It seems likely that CH,CH,CO+ is generated from Cl ions at the thresholds observed in the present experiments, as its formation would accompany CH,CO+ production via the 2-butanone ion (Scheme 1).
Barring an unusually large error in the estimated Au,(cyclobutanol), the measurements imply an activation energy for getting between ionized cyclo- butanol and the butanal ion. Support for the accuracy of AHr(cyclobutano1) is provided by a previous estimate [l(c)] which is only 4 kJ mol-’ below the present one. The apparent activation energy is surprising, as ring opening might be expecte+d to take place w$hout activation energy, and CH,CH,CH,CH=OH + CH,CH,CH$IIO appears +to be a lower energy process than CH,CH,CH,CHO’+ CH,CHCH,CH=OH [l(c)].
Ionized 3-buten-2-o/ The results in Table 1 are inconsistent with the proposed intermediacy

40
[l(b),(c)] (Scheme 1) of CH,CHOHCH=CHl. in the conversion of Cl to C2 ions in that the apparent activation energy for the formation of CH,CO+ from CH,CHOHCH=CHi’ is 6-8 kJ mol-’ greater than that for CH,CO+ formation from the Cl ions. This discrepancy could result from an error of several kJ mall’ in the estimation of AH,(CH&HOHCH=CH,) in con- junction with smaller uncertainties in the other values employed in these AH, calculations. However, a second problem is that the threshold for CH,CO+ formation is 8 kJ mol-’ above that for C3HSOf formation from CH,CHOHCH=CH:.. This difference is too large to be due to experimental error, and contrasts with results for the Cl ions. Thus the AE measurements indicate that the Cl ions might not isomerize to C2 ions via CH,CHOHCH=CH,+‘, but do not suggest an alternate pathway. If both decompositions of CH,CHOHCH=CHl’ take place above the thermochem- ical threshold for the higher energy process, then identical appearance energies would be expected. Electron impact measurements [l(c)] indicate that AE(CH&O+)+ may also be about 8 kJ +mol-’ above AE(CIIHgO+) for both CH,CH,C(=OH)CH, and CH,CHC(=OH)CH,, suggesting that such a discrepancy may be a property of C2 ions. It seems unlikely that the difference is due to a competitive shift [21], as such shifts are not observed for either Cl or 2-butanone ions [13(c)].
Ionized I -penten-3-ol-5,5,.5-d,
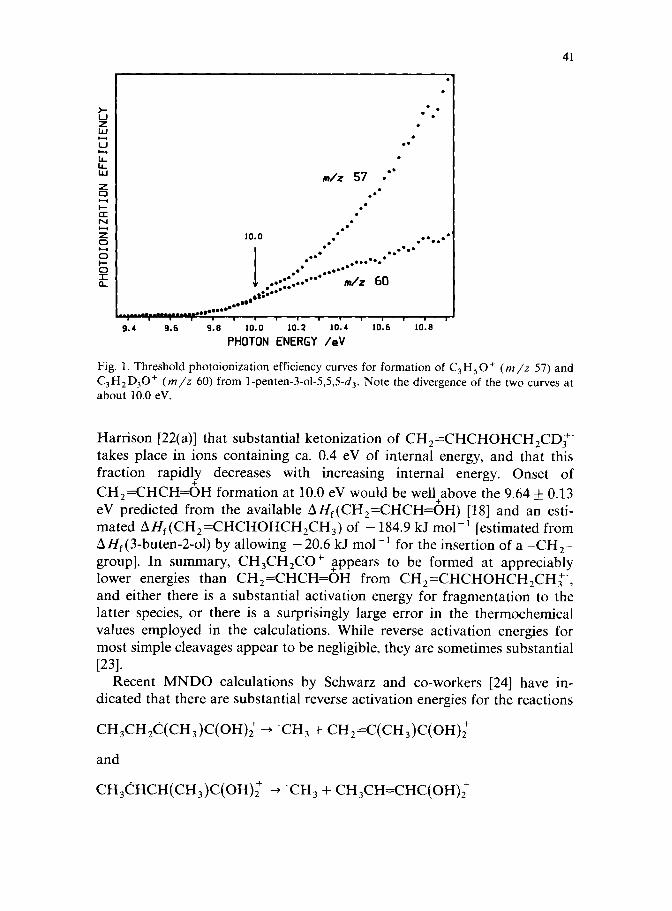
To characterize further the isomerizations of ionized unsaturated alcohols to ketone ions, AE(C3HSO+) and AE(C,H,D,O’) were determined for l-penten-3-01-5,5,5-d,. These AE curves were superimposable from threshold (9.77 eV) to about 10.0 eV, with the curve for C3HSOf rising more steeply at higher energies (Fig. 1). The threshold is above the ionization potential of l-penten-3-01-5,5,5-d,, 9.34 + 0.03 eV (this work). Using AH,(CH,=CH- CHOHCH,CH,) = - 184.9 kJ mol-’ and AH,,, = 20.8 kJ mol-’ [13(c)], leads to AH,(CH,CH,CO+) = 662 kJ mol-‘, which is 71 kJ mol-’ above the thermochemicaj ., value [13(c)], and even 13 kJ mol- ’ above AH,(CH,=CHCH=OH) [18]. This gap is slightly greater than the difference between the observed and thermochemically predicted threshold for CH&H,CO++ ‘CH, formation from CH,CHOHCH=CHl’ (60 kJ mol-‘). Thus, CH,=CHCHOHCH,CHl’ appears to isomerize to the 3-pentanone ion at energies substantially above those required to decompose the latter ion. Previous workers have shown that CH,CH,CO+ is the dominant C3H50+ fragment of ionized 1-penten-3-01 at very low energies [4(c),22]. Thus, we believe that over the range of superposition of the AE curves, CH,CH,CO+ a,nd CD,CH,CO+ are the exclusive products, with the onset of CH,=CHCH=OH formation taking place at the divergence of the curves. This interpretation is consistent with the conclusion of Zwinselman and
41
.
l .
- .
.
.:
.
m/z 57 l ** ..*
. :
.** IO. 0 : . **..a
I
.* ..*
_*.-.* .
l .* l ..-• l . ...**.
..*::... l ..* m/z 60 .*.**
l ..*” ,rw
*.,...a f
9.6 9.8 10.0 10.2 10.4 10.6 10.8
PHOTON ENERGY /eV
Fig. 1. Threshold photoionization efficiency curves for formation of C,H,O+ (m/z 57) and C3H2D,0+ (m/z 60) from l-penten-3-01-5,5,5-d,. Note the divergence of the two curves at about 10.0 eV.
Harrison [22(a)] that substantial ketonization of CH,=CHCHOHCH,CDc. takes place in ions containing ca. 0.4 eV of internal energy, and that this fraction rapidly decreases with increasing internal energy. Onset of
CH,=CHCH=GH formation at 10.0 eV would be well above the 9.64 f 0.13 eV predicted from the available AH,(CH,=CHCH=6H) [18] and an esti- mated AH,(CH,=CHCHOHCH,CH,) of - 184.9 kJ mol-’ [estimated from AH,(3-buten-2-01) by allowing -20.6 kJ mol-’ for the insertion of a -CH,- group]. In summary, CH$H&O+ tappears to be formed at appreciably lower energies than CH,=CHCH=OH from CH,=CHCHOHCH,CH,f’, and either there is a substantial activation energy for fragmentation to the latter species, or there is a surprisingly large error in the thermochemical values employed in the calculations. While reverse activation energies for most simple cleavages appear to be negligible, they are sometimes substantial
]231. Recent MNDO calculations by Schwarz and co-workers [24] have in-
dicated that there are substantial reverse activation energies for the reactions
CH,CH,C(CH,)C(OH): + ‘CH, + CH,=C(CH,)C(OH);
and
CH,CHCH(CH,)C(OH); - ‘CH, + CH,CH=CHC(OH);

42
with the reverse activation energy for the latter being higher. The similarity of the products of the reaction
CH,=CHCH(&)CH&H, + CH,=CHCH=&H + ‘CH,CH,
to those of the above reactions makes it likely that simple cleavage of ionized l-penten-3-01 would also have an appreciable reverse activation energy.
CHICH2C(=6H)(?H2 and CH$?HC(=hH)CH, decompositions The appearance energy for CH,CH; + CH,CH=CH, + CH,CO+ from
3-heptanone +was measured in order to get a critical energy for CH,CH,C(=OH)CH, decomposition. The simple cleavage of ionized 3- heptanone to CH3CI11CO+ prevented meaningful measurements on C3H50+ from CH,CI$,C(=OH)CH2. The derived transition state energy for CH,CH,C(=OH)CH2 -+ CH,CO++ ‘C,H,, 800 + 5 kJ mol-‘, is between previously derived electron impact values, 791 kJ mol-’ [l(c)] and 816 ) 4 kJ mol-’ [lo]. It coincides with the value for the corresponding decomposi- tion of CH,=CHCHOHCH:’ and slightly exceeds the energies required for CH,CO+ formation from the Cl ions. The appearance energy for C,H,O’ + ‘CH, + CH,CH=CH, from 3-methyl-2-hexanone places the high point on the path from CH,CHC(=OH)CH, to CH,CH,C(=0)CH3 at 775 + 5 kJ mol-‘, about 42 kJ mol-’ abov$AH,(CH,CH,CO++ ‘C$13). The transition state energy for CH3CH,C(=OH)CH, --j CH,CH,C(=OH)CH, probably determines the associated AE, a,s the remaining steps are common with the decompositions of CH,CHC(=OH)CH, (Scheme 2), which appears to de-
Scheme 2.

43
compose at significantly lower energies. The +apparently higher critical energy for CH,CH,C(=OH)CH, + CH,CHJC(=OH)CH, confirms predictions [l(a)] that this causes CH,CH,C(=OH)CH3 to isomerize exclu$vely to CH,CH,C(=O).CH, rather than to the more stabie CH,CH,C(=OH)cH, [l(a)]. The CH&H,C(:OH)CH, and CH,CHC(=OH)CH, ions which iso- merize to CH3CH,C(=O)‘CH, clearly contain more energy than they need to decompose, confirming the conclusion [l(a)] that isomerizations over barriers determine the decomposition rates of these ions.
Relationship between translational energy releases and excess energy in the decomposing ions
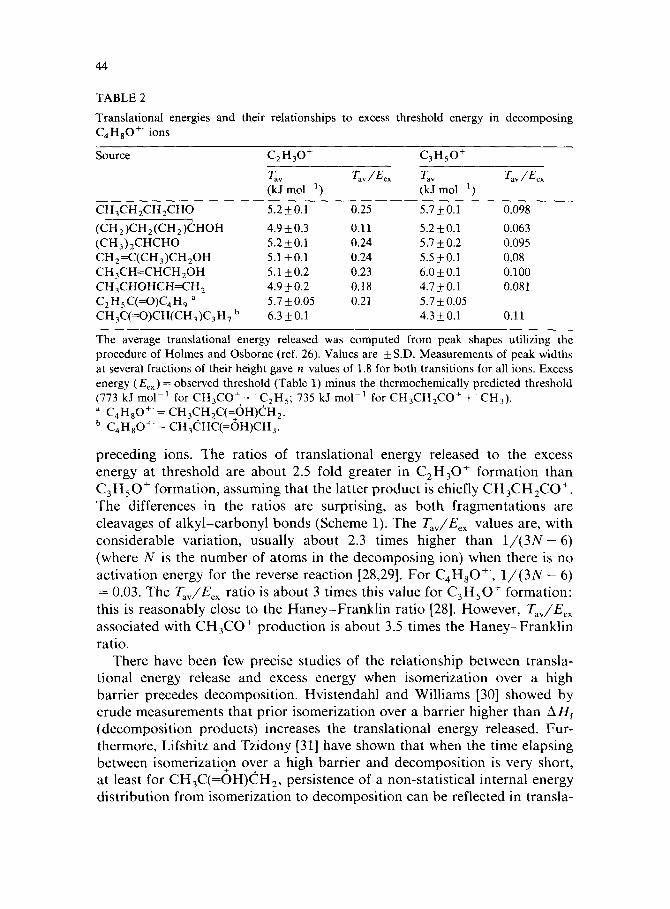
A bimodal distribution in t+he kinetic energy released in the formation of CH,CO+ from CH,CH,C(=OH)CH, with one component going to much higher energies than predicted statistically has been taken as evidence [lo] for the violation of the energy randomization postulate of the quasi-equi- librium theory [25]. In another study, a greater energy release in the formation of C,H,O+ and a $sser energy release associated with CH,CO+ formation from CH,CH,C(=OH)CH, relative to CH,CHC(=OH)CH, was also interpreted as evidence for non-randomization of energy [ll]. The relationships between average translational energies released (T,,) in the decomposition products and the excess energies (E,,) present above the heats of formation of the decomposition products are therefore of consider- able interest.
Translational energies released and the associated T,,/E,, ratios are given in Table 2. The actual fractions of excess energy released are somewhat lower than the ratios in Table 2, as portions of each metastable C,H,O+’ popula- tion contained energies in excess of their decomposition thresholds. [Kinetic energies of up, to 50 kJ mol-’ are released in the loss of an ethyl group from CH,CH,C(=OH)CH, [lo], whereas the excess energy at threshold is only about 27 kJ mol-’ (Table l)]. An 8% CH+2=CHCH=ODf contribution to the C3H4DOf intensity from CH&H,C(=OD)CHCH, [22(b)] had no detecta- ble influence on the associated transla$onal energy release [27]. Fractions of C,H,O+ formed from CH,=CHCH=OH in the first field-free region of the instrument used have been estimated to be 10% for CH,CH@,CHO+‘, 17% for (CH,)CH,(CH,)CHOH+‘, 18% for CH,CH=CHCH,OH, 7% for CH,CHOHCH=CHi’, 5% for CH,CH,C(=OH)CH, and 1% for CH,CHC(=OH)CH, [l(c)]. Based on observations on C,HiOO+’ ions [22(b)], these fractions may decrease between the first and third field-free regions of the mass spectrometer. Energy releases were measured in the latter region. Thus, we feel justified in treating the energy release as though CH,CH,CO+ were the only product formed, at least from the first and the last three
44
TABLE 2
Translational energies and their relationships to excess threshold energy in decomposing C4Hs0+’ ions
Source C2H,0t C,H,O+
CH,CH,CH,CHO I
(CH,)CH,(CH,)CHOH (CH,),CHCHO CH,=C(CHa)CH,OH CH,CH=CHCH,OH CH,CHOHCH=CH, C,H,C(=O)C,H, a CH,C(=O)CH(CH,)C,H, b
Tav /J&x mol-‘)
5.2kO.l 0.25
4.9f0.3 0.11 5.2kO.l 0.24 5.1 _t 0.1 0.24 5.1 f0.2 0.23 4.9kO.2 0.18 5.7 * 0.05 0.21 6.3kO.l
T (lZ mol-‘)
L /&
5.7kO.l 0.098
5.2 +0.1 0.063 5.7 + 0.2 0.095 5.5 * 0.1 0.08 6.0 +O.l 0.100 4.7kO.1 0.081 5.7kO.05 4.3 * 0.1 0.11
The average translational energy released was computed from peak shapes utilizing the procedure of Holmes and Osborne (ref. 26). Values are f S.D. Measurements of peak widths at several fractions of their height gave n values of 1.8 for both transitions for all ions. Excess energy (E,,) = observed threshold (Table 1) minus the thermochemically predicted threshold (773 kJ mol-’ for CH,CO+++ ‘C2Hs; 735 kJ mol-’ for CH,CH,CO+ + CH,). a C4Ha0+‘= CH,CH,C(;OH)CH,. b C,H,O+‘= CH,CHC(=OH)CH,.
preceding ions. The ratios of translational energy released to the excess energy at threshold are about 2.5 fold greater in C2H,0+ formation than C,H,O+ formation, assuming that the latter product is chiefly CH,CH,CO+. The differences in the ratios are surprising, as both fragmentations are cleavages of alkyl-carbonyl bonds (Scheme 1). The 7’.‘.,/E,, values are, with considerable variation, usually about 2.3 times higher than 1/(3N - 6) (where N is the number of atoms in the decomposing ion) when there is no activation energy for the reverse reaction [28,29]. For C,H,O+‘, 1/(3N - 6) = 0.03. The TJE,, ratio is about 3 times this value for C3HSO+ formation: this is reasonably close to the Haney-Franklin ratio [28]. However, T,,/E,,
associated with CH,CO+ production is about 3.5 times the Haney-Franklin ratio.
There have been few precise studies of the relationship between transla- tional energy release and excess energy when isomerization over a high barrier precedes decomposition. Hvistendahl and Williams [30] showed by crude measurements that prior isomerization over a barrier higher than AH, (decomposition products) increases the translational energy released. Fur- thermore, Lifshitz and Tzidony [31] have shown that when the time elapsing between isomerizatio+n over a high barrier and decomposition is very short, at least for CH,C(=OH)CH,, persistence of a non-statistical internal energy distribution from isomerization to decomposition can be reflected in transla-

45
tional energy release. This explanation has been extrapolated to C,H,O+’ ions to explain why more energy is released during the release of ,an ethyl group and less during the loss 9f a methyl group from CH,CHC(=OH)CH, in comparison to CH,CH,C(=OH)CH, [ll). This observation is difficult to rationalize by conventional thinking because of the identity of the corre- sponding fragmentation steps for the two ions (Scheme 2). However, it could be somehow related to the differing T,,/E,, ratios associated with CH,CO+ and CH,CH,CO+ formation. Unfortunately, in the present studies, interfer- ing processes prevented two of the four relevant AE measurements being made. The+ relatively low AE for C,H,O+ formation from CH,CHC(=OH)CH, provides an explanation for the low T,, associated with that process. The AE for CH,CO+ formation from CH,CHC(==OH)CH,, the process on which the non-statistical interpretation most heavily depends [l(c)], can be estimated by assuming AE(CH,CO+) from 3-methyl- 2-hexanone is 8 kJ mol-’ above the associated AE(C,H,O+) (justified above). This gives a TJE,, ratio of 0.63, well above any ratio in Table 2. This AE would have to exceed AE(C3HSO+) by 26 kJ mol-’ to bring Ta,/Eex down to the ratios for the other CH,CO+ formations in Table 2.
In most ionic decompositions, T,, increases smoothly with increasing ion internal energy [32]. However, T,,/E,, may increase markedly with decreas- ing internal energy near threshold 1331, and actual increases in translational energy with decreasing internal energy have been reported [34]. This may be due to a rapid decrease in the ability of internal modes in the fragments to absorb energy with decreasing E,, [33], a suggestion which could explain the difference in TJE,, between the two metastable decompositions of the C,H,O+’ iyns. The energy released in CH,CO+ formation from CH,CHC(=OD)CH, at higher energies generated by collision (T,,* = 60
meV) is 50% greater than that released in the accompanying C,H,DO+ formation (T-,,* = 41 meV), an apparent enhancement of the unimolecular pattern, or in+the collision-induced losses of both methyl and C,H,D from CH,CH,C(=OD)CH, ( Tl,* = 42 and 40 meV, respectively) [ll]. The average energy released is also much higher for the formation of CH,CO+ (T,,* = 37 meV) than for CH$HICOf (T,,,* = 14 meV) in collision-induced dissocia- tions of ionized 2-butanone [ll]. These higher energy patterns appear to indicate that the energies released in the decompositions of 2-butanone ions depend on features of the generation of those ions other than the amount of energy they contain, casting doubt on the possibility that the differing energy release patterns are simply due to an irregular variation of Tav with internal energy. As the amounts of energy deposited in the ions by collision is uncertain, determination of kinetic energy release distributions associated with the decompositions of 2-butanone ions of defined internal energies are needed to explore this.

46
It is tempting to conclude that T’.,/E,, is greater for CH,CO+ formation than for CH,CH,CO+ formation because energy is concentrated in the C,H, moiety following isomerization to CHJ(=O+‘)CH&H, and remains there until decomposition. However, the unequal energy releases in the collision-induced decompositions of ionized 2-butanone which does not isomerize before decomposing suggests that this may not be so. An unex- pectedly large fraction of the excess energy required to convert CH&(OH)l to CH,CO,H” also appears to be released upon subsequent decomposition [35], suggesting that the C4HsO+’ results may not be very unusual. Thus, the relationships between excess energy present and translational energy released for decompositions following high energy isomerizations seems a ripe area for future study. As the present work suggests, this will require utilization of systems whose mechanisms and energetic requirements for decomposition are both well characterized.
ACKNOWLEDGEMENTS
We thank C. Hudson for helpful discussions and assistance in obtaining some of the compounds used in this work, Prof. H. Svec for helpful comments, D. Pavlu and P. Waldrop for typing, Prof. M. Gross and the Midwest Center for Mass Spectrometry (NSF Grant CHE 78-18572) for use of the MS 50 TA mass spectrometer, and the Australian Research Grants Scheme and the Robert A. Welch Foundation (Grant H-609) for financial support.
REFERENCES
1 (a) D.J. McAdoo, F.W. McLafferty and T.E. Parks. J. Am. Chem. Sot., 94 (1972) 1601. (b) D.J. McAdoo, C.E. Hudson and D.N. Witiak, Org. Mass Spectrom., 14 (1979) 350. (c) D.J. McAdoo and C.E. Hudson, Org. Mass Spectrom.. 18 (1983) 466. (d) C.E. Hudson and D.J. McAdoo. Org. Mass Spectrom., 20 (1985) 402.
2 G. Bouchoux. Y. Hoppilliard. R. Flammang, A. Mayuestiau and P. Meyrant, Org. Mass
Spectrom., 18 (1983) 340. 3 F.W. McLafferty, Interpretation of Mass Spectra, University Science Books, Mill Valley,
CA, 3rd edn., 1980. pp. 60, 155. 4 (a) D.J. McAdoo and D.N. Witiak. Org. Mass Spectrom.. 13 (1978) 499. (b) D.J. McAdoo,
D.N. Witiak, F.W. McLafferty and J.D. Dill. J. Am. Chem. Sot., 100 (1978) 6639. (c) D.J. McAdoo, W. Farr and C.E. Hudson, J. Am. Chem. Sot., 102 (1980) 5165. (d) D.J. McAdoo. C.E. Hudson, F.W. McLafferty and T.E. Parks, Org. Mass Spectrom., 19 (1984) 353. (e) C.E. Hudson and D.J. McAdoo, Org. Mass Spectrom., 19 (1984) 1.
5 (a) P.H. Hemberger, J.C. Kleingeld, K. Levsen, N. Mainzer, A. Mandelbaum, N.M.M. Nibbering, H. Schwarz, R. Weber, A. Weisz and C. Wesdemiotis, J. Am. Chem. Sot.. 102 (1980) 3736. (b) E. Goksu, T. Weiske, H. Halim and H. Schwarz, J. Am. Chem. Sot.. 106 (1984) 1167.

47
6 G. Bouchoux and Y. Hoppilliard, Int. J. Mass Spectrom. Ion Processes, 55 (1983/1984) 47.
7 (a) J.J. Zwinselman, N.M.M. Nibbering, C.E. Hudson and D.J. McAdoo, Int. J. Mass Spectrom. Ion Phys., 47 (1983) 129. (b) D.J. McAdoo, C.E. Hudson, J.J. Zwinselman and N.M.M. Nibbering, J. Chem. Sot. Perkin Trans. 2, in press.
8 M.M. Green, Tetrahedron, 36 (1980) 2687. 9 C. Walling, in P. de Mayo (Ed.) Molecular Rearrangements, Interscience, New York,
1963, pp. 407.
10 C. Lifshitz, P. Berger and E. Tzidony, Chem. Phys. Lett., 95 (1983) 109. 11 D.J. McAdoo and C.E. Hudson, J. Phys. Chem., 87 (1983) 2451. 12 (a) J.C. Traeger and R.G. McLaughlin, Int. J. Mass Spectrom. Ion Phys., 27 (1978) 319.
(b) J.C. Traeger, Int. J. Mass Spectrom. Ion Processes, 58 (1984) 259. 13 (a) J.C. Traeger, R.G. McLaughlin and A.J.C. Nicholson, J. Am. Chem. Sot., 104 (1982)
5318. (b) J.C. Traeger and R.G. McLaughlin, J. Am. Chem. Sot., 103 (1981) 3647. (c) J.C. Traeger, Org. Mass Spectrom., 20 (1985) 223.
14 M.L. Gross, E.K. Chess, P.A. Lyon, F.W. Crow, S. Evans and H. Tudge, Int. J. Mass Spectrom. Ion Phys., 42 (1982) 243.
15 A.L. Castelhano and D. Griller, J. Am. Chem. Sot., 104 (1982) 3655. 16 R.H. Nobes, W.J. Bouma and L. Radom, J. Am. Chem. Sot., 105 (1983) 309. 17 D.J. McAdoo and C.E. Hudson. Int. J. Mass Spectrom. Ion Processes, 62 (1984) 269. 18 J.H. Vajda and A.G. Harrison, Int. J. Mass Spectrom. Ion Phys., 30 (1979) 293. 19 J.B. Pedley and J. Rylance, Sussex-NPL Computer-Analysed Thermochemical Data:
Organic and Organometallic Compounds, University of Sussex, 1977. 20 J.L. Holmes, M. Fingas and F.P. Lossing, Can. J. Chem., 59 (1981) 80. 21 C. Lifshitz and F.A. Long, J. Chem. Phys., 41 (1964) 2468. 22 (a) J.J. Zwinselman and A.G. Harrison, Org. Mass Spectrom., 19 (1984) 573. (b) D.J.
McAdoo and C.E. Hudson, Org. Mass Spectrom., 18 (1983) 159. 23 P.C. Burgers, J.L. Holmes and J.K. Terlouw, 32nd Annu. Conf. Mass Spectrom. Allied
Top., San Antonio, TX, 1984. 24 T. Weiske, H. Halim and H. Schwarz, Chem. Ber., 118 (1985) 495. 25 H.M. Rosenstock, M.B. Wallenstein, A.L. Wahrhaftig and H. Eyring, Proc. Natl. Acad.
Sci., 38 (1952) 667. 26 J.L. Holmes and A.D. Osborne, Org. Mass Spectrom., 16 (1981) 236. 27 There are no detectable differences in the translational energy releases associated with the
formations of C3H50+ and C3H4DOt from CH2DCH,C(=OH)t?HCH,. CH,CH,C (=dD)cHCH, and CH,CH,C(=i)H)cHCHzD [unpublished obserzations made in as- sociation with+the results in ref. 22(b)]. No detectable CH,=CHCH=OH was formed from CH,CH,C(=OD)cHCH, in that study, whereas 8% of the C3H4DO+ produced in the third field-free region was estimated to be CH,=CHCH=OD [22(b)].
28 M.A. Haney and J.L. Franklin, J. Chem. Phys., 48 (1968) 4093. 29 J.L. Holmes and J.K. Terlouw, Org. Mass Spectrom., 15 (1980) 383. 30 G. Hvistendahl and D.H. Williams, J. Am. Chem. Sot., 97 (1975) 3097. 31 (a) C. Lifshitz and E. Tzidony. Int. J. Mass Spectrom. Ion Phys., 39 (1981) 181. (b) C.
Lifshitz, J. Phys. Chem.. 87 (1983) 2304. 32 (a) D.M. Mintz and T. Baer, J. Chem. Phys., 65 (1976) 2407. (b) D.M. Mintz and T. Baer.
Int. J. Mass Spectrom. Ion Phys.. 25 (1977) 39. (c) I. Powis and C.J. Danby, Int. J. Mass Spectrom. Ion Phys., 32 (1979) 27.
33 P.C. Burgers and J.L. Holmes, Int. J. Mass Spectrom. Ion Processes, 58 (1984) 15.

48
34 (a) I.W. Griffiths, ES. Mukhtar, F.M. Harris and J.H. Beynon, Int. J. Mass Spectrom. Ion Phys.., 38 (1981) 333. (b) P.W. Harland and J.L. Franklin, J. Chem. Phys., 61 (1974) 1621.
35 D.J. McAdoo, C.E. Hudson and L.L. Griffin, J. Phys. Chem., 88 (1984) 1481. 36 Kratos Analytical Instruments, 170 Williams Drive, Ramsey, NJ 07446, U.S.A., Model
MSSOTA.














![Isomers [compatibility mode]](https://img.pdfslide.net/doc/110x75/5590bc1e1a28abbf308b46da/isomers-compatibility-mode-5593e8f124020.jpg)