Embed Size (px)
Citation preview

Epilepsy Research (2014) 108, 367—378
jo ur nal ho me p ag e: www.elsev ier .com/ locate /ep i lepsyres
Decreased interaction between FoxO3a andAkt correlates with seizure-inducedneuronal death
Yoon Sook Kim, Mee Young Choi, Dong Hoon Lee,Byeong Tak Jeon, Gu Seob Roh, Hyun Joon Kim, Sang Soo Kang,Gyeong Jae Cho, Wan Sung Choi ∗
Department of Anatomy and Neurobiology, School of Medicine, Institute of Health Science, MedicalResearch Center, Gyeongsang National University, 816-15 Jinju-daero, Jinju, Gyeongnam 660-751, SouthKorea
Received 13 April 2013; received in revised form 29 August 2013; accepted 17 January 2014Available online 29 January 2014
KEYWORDSApoptosis;Akt;Bim;FoxO3a;Kainic acid;Status epilepticus
Summary Status epilepticus (SE) leads to neurodegeneration which likely contributes to thedevelopment of chronic temporal lobe epilepsy (TLE). Therefore, neuroprotection following SEis considered as a promising strategy for preventing chronic TLE, but molecular changes thatoccur following SE still remain unclear. The Forkhead homeobox type O (FoxO) family of Fork-head transcription factors mediates cell death in several pathological conditions, but the role ofFoxO in the excitotoxic effects of kainic acid (KA) remains largely unknown. The present studyexamined how FoxO3a and its interaction with other proteins changed in response to excito-toxic stimuli in the mouse hippocampus after SE. Mice were given intraperitoneal injection ofkainate and seizure behavior was monitored for 2 h to ensure SE. Western blot analyses, co-immunoprecipitation experiments, sub-cellular fractionation and double immunofluorescenceanalyses were used to determine changes in levels of FoxO3a, Akt, Bim, cleaved caspase-3and phospho-FoxO3a or phospho-Akt, and their interactions at 6 or 24 h after KA treatment. We
found that SE activated FoxO3a and increased levels of Bim or cleaved caspase-3, and decreasedlevels of phospho-FoxO3a or phospho-Akt in the hippocampus. In addition, we noted extensivehippocampal cell death at 24 h after KA treatment, evidenced by terminal deoxynucleotidyltransferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL), fluoro-jade B oranti-active caspase-3 staining. Furthermore, co-immunoprecipitation experiments revealedthat phospho-Akt interaction with FoxO3a was significantly lowered in the hippocampus at 24 hAbbreviations: Bcl-2, B cell lymphoma; BH3, Bcl-2 homology domain; CA, cornu ammonis; FJB, Fluoro-Jade B; FoxO, Forkhead homeoboxtype O; KA, kainic acid; NeuN, neuron-specific nuclear protein; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick endlabeling.
∗ Corresponding author. Tel.: +82 55 772 8031; fax: +82 55 772 8039.E-mail address: [email protected] (W.S. Choi).
0920-1211/$ — see front matter © 2014 Elsevier B.V. All rights reserved.http://dx.doi.org/10.1016/j.eplepsyres.2014.01.003

368 Y.S. Kim et al.
after KA treatment, paralleling enhanced Bim levels and Bim interaction with Bcl-xL. Moreover,double immunofluorescence analyses showed increased co-localization of FoxO3a or Bim andTUNEL in the hippocampi at 24 h after KA treatment. Identifying molecular mechanism underly-ing SE-induced neuronal death can provide a novel strategy to protect against seizure-inducedneuronal injury. We found that Akt-FoxO3a signaling relates to seizure-induced neuronal death,
rotection following SE.s reserved.
I
SsrnAchirffepaa
iaoise(imao(u2ena
o(fiadAFccrciumsd
M
A
A(A(I(AwMw(DOMUf
A
Mhatsiawegoggggmb48mof animals continued seizing for 2 h after grade IV (theystopped seizing 2—3 h afterwards). In the control group,mice receiving saline injection did not exhibit any of these
providing insight into neurop© 2014 Elsevier B.V. All right
ntroduction
tatus epilepticus (SE) is neurological emergency linked toignificant damage to the hippocampus and other brainegions (Chen and Wasterlain, 2006), and can cause cog-itive deficits and exacerbate epilepsy (Pitkanen, 2002;charya et al., 2008). Despite recent advances in pathologi-al and experimental research of SE, SE remains a feared andazardous condition (Lowenstein, 2005). Our understand-ng of the underlying pathophysiological mechanisms of SEemains limited, and the cellular mechanisms responsibleor seizure-induced neuronal death remain unclear. There-ore, further work and understanding these mechanisms willventually provide new therapeutic strategies to modify orrevent the development of learning and memory deficitsnd behavioral alterations associated with epilepsy (Loschernd Brandt, 2010).
Various signaling pathways are implicated in seizure-nduced neuronal death, including pathways regulatingpoptotic cell death (Engel and Henshall, 2009). Previ-us studies have shown that molecular or pharmacologicalntervention in apoptotic cell death pathways can reduceeizure-induced neuronal loss (Henshall et al., 2002; Royt al., 2002). Forkhead transcription factor of the O classFoxO) proteins is one of the genes involved in apoptoticnjury (Gilley et al., 2003). Three members of the mam-alian Forkhead family have been identified: FoxO1, FoxO3a
nd FoxO4 (Tran et al., 2003). FoxO3a is expressed in vari-us regions of the mouse brain, including the hippocampusHoekman et al., 2006). FoxO transcription factors are reg-lated by the PI3K-AKT pathway (Hannenhalli and Kaestner,009). Phosphorylation of FoxO by Akt leads to nuclearxport (Brunet et al., 2002). In addition, the regulation ofeuronal cell death through the phosphorylation of FoxO3at serine 207 has been reported (Lehtinen et al., 2006).
Previous studies have demonstrated that overexpressionf Akt in the central nervous system prevents apoptosisDatta et al., 1997). Akt may be both necessary and suf-cient for the survival of neurons because expression of
dominant-negative Akt or PI3K resulted in apoptotic celleath (Crowder and Freeman, 1998). Following activation,kt translocates to the nucleus and phosphorylates theoxO transcription factor. FoxO is then exported into theytosol, and this translocation of FoxO inhibits its trans-riptional activities (Brunet et al., 1999). Bim levels areegulated by Forkhead box class O (FoxO) 3a and its trans-riptional control is important (Dijkers et al., 2000), but
ts role in seizure-related neuronal injury remains largelynknown. The identification of novel molecular deter-inants of neuronal death following seizures may haveignificant implications for neuroprotection against seizureamage.
amtia
aterials and methods
ntibodies and chemicals
ntibodies against FoxO3a (sc-11351), phospho-FoxO3aSer253) (sc-101683), phospho-FoxO3a (Thr32) (sc-12357),kt (sc-8312), caspase-3 (sc-7272), Bim (sc-11425), Bcl-xL
sc-7195), lamin A (sc-6214), peroxiredoxin 2 (sc-23967), andgG (sc-2003) were obtained from Santa Cruz BiotechnologySanta Cruz, CA); phospho-Akt (Thr308) (#9275), phospho-kt (Ser473) (#9271), and cleaved caspase-3 (#9661)ere obtained from Cell Signaling Technologies (Beverly,A); neuron specific nuclear antigen (NeuN; MAB377)as obtained from Chemicon (Temecula, CA); Alexa 488
A21206;A11055; A21202), Alexa 594 (A11058; S32356) andAPI (D1306) were obtained from Molecular Probes (Eugene,R); �-tubulin (T5168) was obtained from Sigma (St. Louis,O). KA was obtained from Tocris Cookson Ltd. (Bristol,nited Kingdom); all other chemicals used were obtainedrom Sigma.
nimals and seizure induction
ale ICR mice (age, 7 weeks; weight, 30—35 g) wereoused in a 12/12-h light/dark cycle with food and watervailable ad libitum. Seizures were induced by an intraperi-oneal injection of KA (30 mg/kg body wt) in 0.9% normalaline. Control mice received the same volume of salinentraperitoneally. Seizures were monitored behaviorally fort least 2 h after KA administration. Behavioral seizuresere defined according to a previously published scale (Hut al., 1998) with some modifications: grade 0, no response;rade I, staring, a rigid posture, tail extension, front pawingr hind-limb pawing (scratching) and/or staring; grade II,rade I plus head nodding, rearing, repetitive movement;rade III, grade II plus jumping, wobbling and/or falling;rade IV, continuous seizure activity lasting over 30 min;rade V, death. Within 10 min after KA injection, all ani-als exhibited increasing immobility and rigidity. Seizureehaviors corresponding to grade IV were observed within0 min. Continuous seizures lasted at least 30 min. In total,3% of animals continued seizing for 1—2 h, 17% of ani-als died during KA-induced status epilepticus, and 58%
bnormal behaviors. Mice were graded according to theaximal behavioral response observed within 2 h after KA
reatment, and only mice reaching grade IV were includedn the study. Mice were sacrificed at different time-pointsfter KA administration (each time point n = 5). All animal
Akt-FoxO3a signaling correlates seizure damage 369
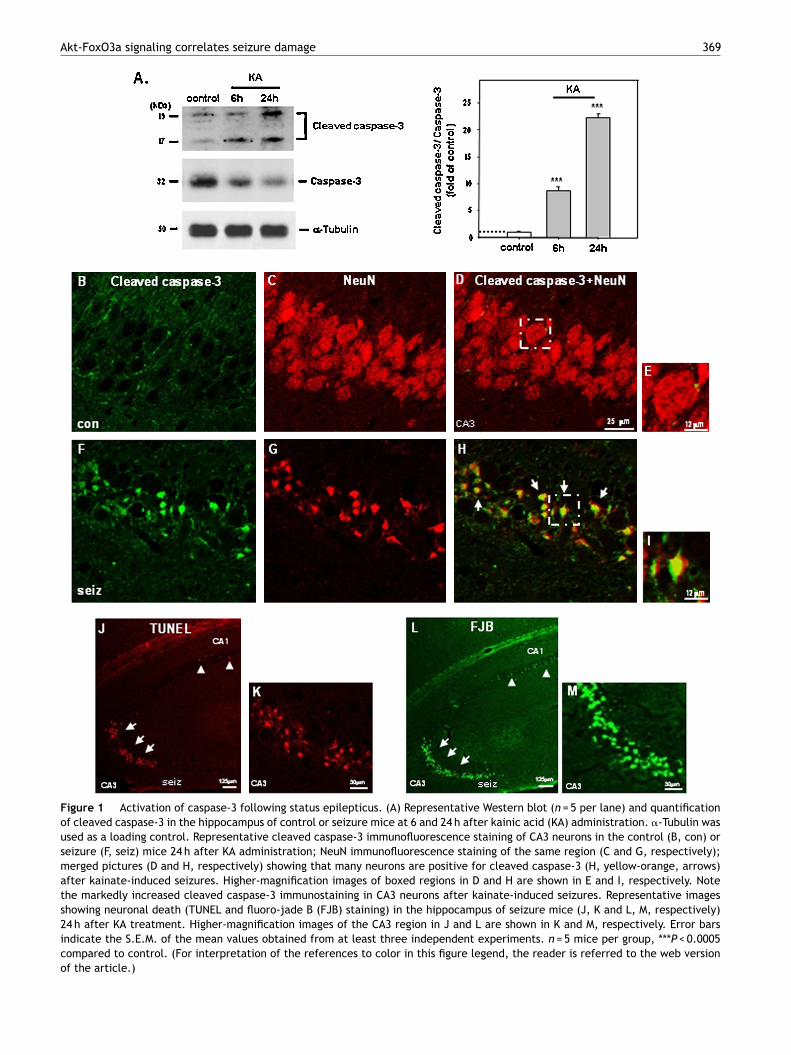
Figure 1 Activation of caspase-3 following status epilepticus. (A) Representative Western blot (n = 5 per lane) and quantificationof cleaved caspase-3 in the hippocampus of control or seizure mice at 6 and 24 h after kainic acid (KA) administration. �-Tubulin wasused as a loading control. Representative cleaved caspase-3 immunofluorescence staining of CA3 neurons in the control (B, con) orseizure (F, seiz) mice 24 h after KA administration; NeuN immunofluorescence staining of the same region (C and G, respectively);merged pictures (D and H, respectively) showing that many neurons are positive for cleaved caspase-3 (H, yellow-orange, arrows)after kainate-induced seizures. Higher-magnification images of boxed regions in D and H are shown in E and I, respectively. Notethe markedly increased cleaved caspase-3 immunostaining in CA3 neurons after kainate-induced seizures. Representative imagesshowing neuronal death (TUNEL and fluoro-jade B (FJB) staining) in the hippocampus of seizure mice (J, K and L, M, respectively)24 h after KA treatment. Higher-magnification images of the CA3 region in J and L are shown in K and M, respectively. Error barsindicate the S.E.M. of the mean values obtained from at least three independent experiments. n = 5 mice per group, ***P < 0.0005compared to control. (For interpretation of the references to color in this figure legend, the reader is referred to the web versionof the article.)
370 Y.S. Kim et al.
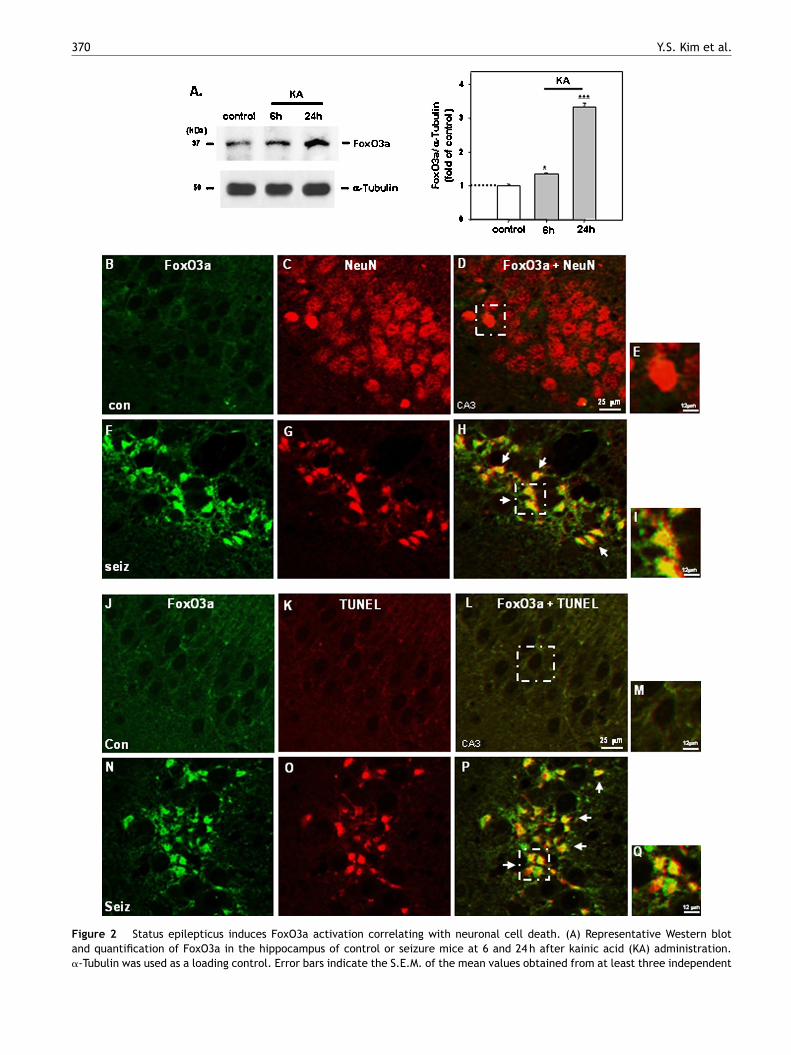
Figure 2 Status epilepticus induces FoxO3a activation correlating with neuronal cell death. (A) Representative Western blotand quantification of FoxO3a in the hippocampus of control or seizure mice at 6 and 24 h after kainic acid (KA) administration.�-Tubulin was used as a loading control. Error bars indicate the S.E.M. of the mean values obtained from at least three independent

Tbwwaompd
HH(wFfbTtbnefpFPs3isctarrdhcAcndkawbi
Akt-FoxO3a signaling correlates seizure damage
procedures were performed in accordance with NIH guide-lines and were approved by the Institutional Animal Careand Use Committee of Gyeongsang University School ofMedicine.
Western blot analysis
Western blotting was performed as previously described(Kim et al., 2012). Briefly, hippocampi were collected 6or 24 h after KA administration and homogenized in lysisbuffer and protease inhibitors. Lysates were cleared bycentrifugation at 14,000 × g for 20 min and protein concen-trations were determined using Bradford assay (Bio-Rad,Hercules, CA). Then, 30-�g samples were separated in 10%sodium dodecyl sulfate-polyacrylamide gel electrophoresis(SDS-PAGE) gels and transferred onto a nitrocellulose mem-brane. The membranes were blocked with 5% skim milk andsequentially incubated with the following primary antibod-ies: anti-cleaved caspase-3 (rabbit, 1:1000), anti-caspase-3(mouse, 1:1000), anti-Bim (rabbit, 1:1000), and anti-Bcl-xL (rabbit, 1:1000). To confirm equality of protein loading,membranes were reprobed with an antibody against �-tubulin. Membranes were then incubated with horseradishperoxidase (HRP)-conjugated secondary antibodies (CaltagLaboratories, Burlington, CA) followed by ECL detection(Pierce, Rockford, IL). The intensity analysis was carried outusing Sigma Gel 1.0 software (Jandel Scientific, Germany).
Subcellular fractionationCollected hippocampal tissue was homogenized in 100 �Llysis buffer [10 mM HEPES-KOH (pH 7.9), 1.5 mM MgCl2,10 mM KCl], placed on ice for 10 min, and centrifuged at12,000 × g for 1 min at 4 ◦C. After collecting the super-natant (cytoplasmic fraction), pellets were resuspended in70 �L cold buffer [20 mM HEPES-KOH (pH 7.9), 1.5 mM MgCl2,420 mM NaCl, 0.2 mM EDTA], incubated on ice for 20 min,and then centrifuged at 12,000 × g for 11 min at 4 ◦C. Thesupernatant (nuclear fraction) was stored at −80 ◦C. Theefficiency of fractionation was analyzed by immunoblottingusing peroxiredoxin 2 or lamin A antibody as a cytoplasmicor nuclear marker, respectively.
Immunoprecipitation
Protein extracts from hippocampi were mixed with proteinA/G agarose beads (Santa Cruz Biotechnology), incubated
for 1 h at 4 ◦C, and then centrifuged at 12,000 × g for 1 min.The supernatant was incubated with 2 �g of the immu-noprecipitation (IP) antibodies overnight at 4 ◦C and thenincubated with protein A/G agarose beads for 2 h at 4 ◦C.esi1
experiments. n = 5 mice per group, *P < 0.05 and ***P < 0.0005 compared
of FoxO3a in the CA3 region of control (B, con) and seizure (F, seiz) mice
the same region (C and G, respectively); merged pictures (D and H, respeorange, arrows) after kainate-induced seizures. Higher magnification imaRepresentative images of immunofluorescence staining of FoxO3a (gree24 h after KA treatment; TUNEL (red) staining of the same region (K andthat many TUNEL-positive cells are FoxO3a positive (P and Q, arrows). Hin M and Q, respectively. (For interpretation of the references to color inarticle.)
371
he negative control was prepared with protein A/G agaroseeads without the antibody. The protein—bead complexas then washed and collected by centrifugation, samplesere boiled in loading buffer to remove the agarose beads,nd the protein (20 �g) was then separated by SDS-PAGEn 10% acrylamide gels. Proteins were then transferred toembranes, probed with antibodies against the interactingrotein of interest, and processed for Western blotting asescribed above.
istological analysis and immunohistochemistryistological analysis was performed as previously described
Kim et al., 2012). Briefly, mice were deeply anesthetizedith intraperitoneal zolazepam (Zoletil; Virbac, Carros,rance) and transcardially perfused with heparinized saline,ollowed by 4% paraformaldehyde in 0.1 M phosphate-uffered saline (PBS, pH 7.4) at 24 h after the KA injection.he brains were removed immediately and postfixed withhe same fixation solution overnight at 4 ◦C. Post-fixedrains were embedded in paraffin and sectioned coro-ally at a thickness of 5 �m. Three different sections forach hippocampus were collected from five different miceor each group for each stain at the same level of hip-ocampus, starting at 2.8 mm posterior to the bregma.ollowing de-paraffinization, rehydration, and washing inBS, sections were blocked with 1% (v/v) normal goaterum and then treated with an anti-cleaved caspase-- (1:200 dilution) or NeuN antibody at 4 ◦C overnightn a humidified chamber. After washing in PBS, theseections were incubated with a secondary antibody (biotin-onjugated anti-rabbit IgG, 1:200) for 90 min at roomemperature. Finally, sections were incubated with anvidin—biotinylated-HRP complex (ABC; Vector Laborato-ies, Burlingame, CA) for 60 min at room temperature,insed in PBS and then developed using 0.027% 3,3-iaminobenzidine tetrahydrochloride (Sigma) with 0.003%ydrogen peroxide. Immunofluorescent staining for cleavedaspase-3 or NeuN was performed with Alexa 488- orlexa-594-labeled secondary antibodies. Terminal deoxynu-leotidyl transferase-mediated deoxyuridine triphosphateick-end labeling (TUNEL) staining was performed toetect DNA fragmentation using a commercially availableit (Roche Molecular Biochemicals, Mannheim, Germany)ccording to the manufacturer’s instructions. Briefly, afterashing in PBS (twice for 5 min each), sections were incu-ated with a blocking solution (3% hydrogen peroxiden methanol) for 10 min at room temperature to quench
ndogenous peroxidase (POD) activity. After quenching,ections were washed in PBS (twice for 5 min each) andncubated in a permeabilization solution (0.1% Triton X-00 in 0.1% sodium citrate) on ice for 2 min. The sectionsto control. Representative images of immunofluorescence staining24 h after KA administration. NeuN immunofluorescence staining ofctively) showing that many neurons are FoxO3a-positive (H, yellow-ges of boxed regions in D and H are shown in E and I, respectively.
n) in the CA3 neurons of control (J, con) or seizure mice (N, seiz) O, respectively); merged pictures (L and P, respectively) showingigher magnification images of boxed regions in L and P are shown
this figure legend, the reader is referred to the web version of the
372 Y.S. Kim et al.
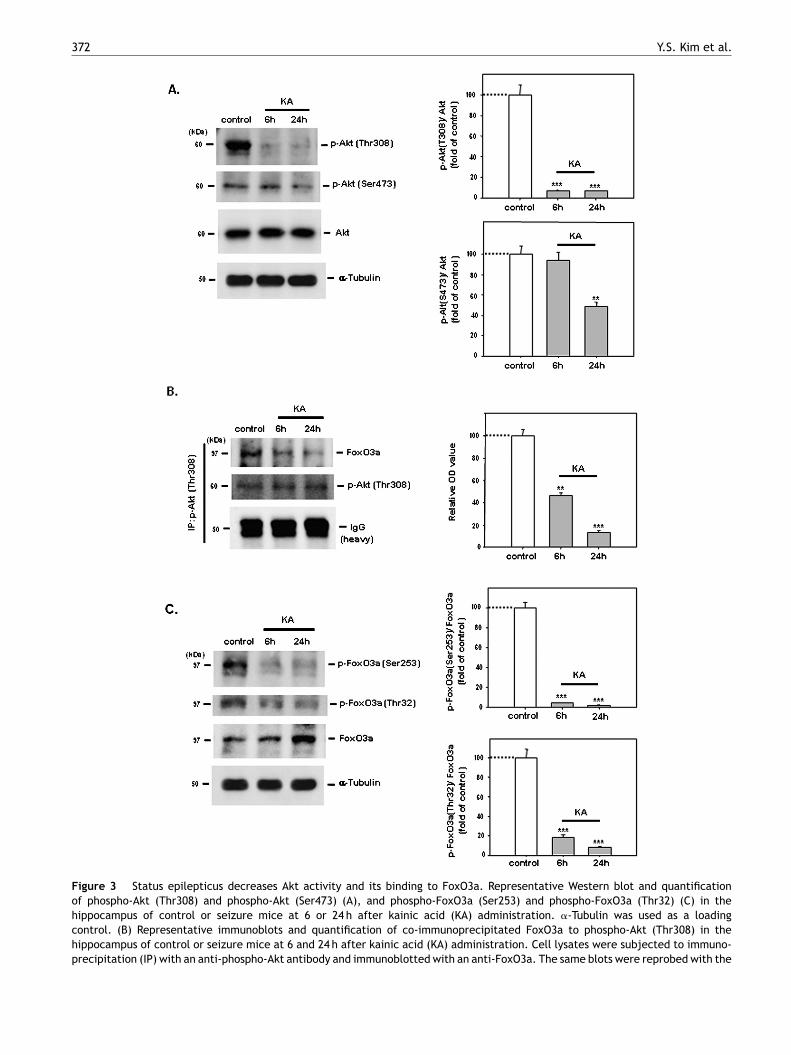
Figure 3 Status epilepticus decreases Akt activity and its binding to FoxO3a. Representative Western blot and quantificationof phospho-Akt (Thr308) and phospho-Akt (Ser473) (A), and phospho-FoxO3a (Ser253) and phospho-FoxO3a (Thr32) (C) in thehippocampus of control or seizure mice at 6 or 24 h after kainic acid (KA) administration. �-Tubulin was used as a loadingcontrol. (B) Representative immunoblots and quantification of co-immunoprecipitated FoxO3a to phospho-Akt (Thr308) in thehippocampus of control or seizure mice at 6 and 24 h after kainic acid (KA) administration. Cell lysates were subjected to immuno-precipitation (IP) with an anti-phospho-Akt antibody and immunoblotted with an anti-FoxO3a. The same blots were reprobed with the
Akt-FoxO3a signaling correlates seizure damage 373
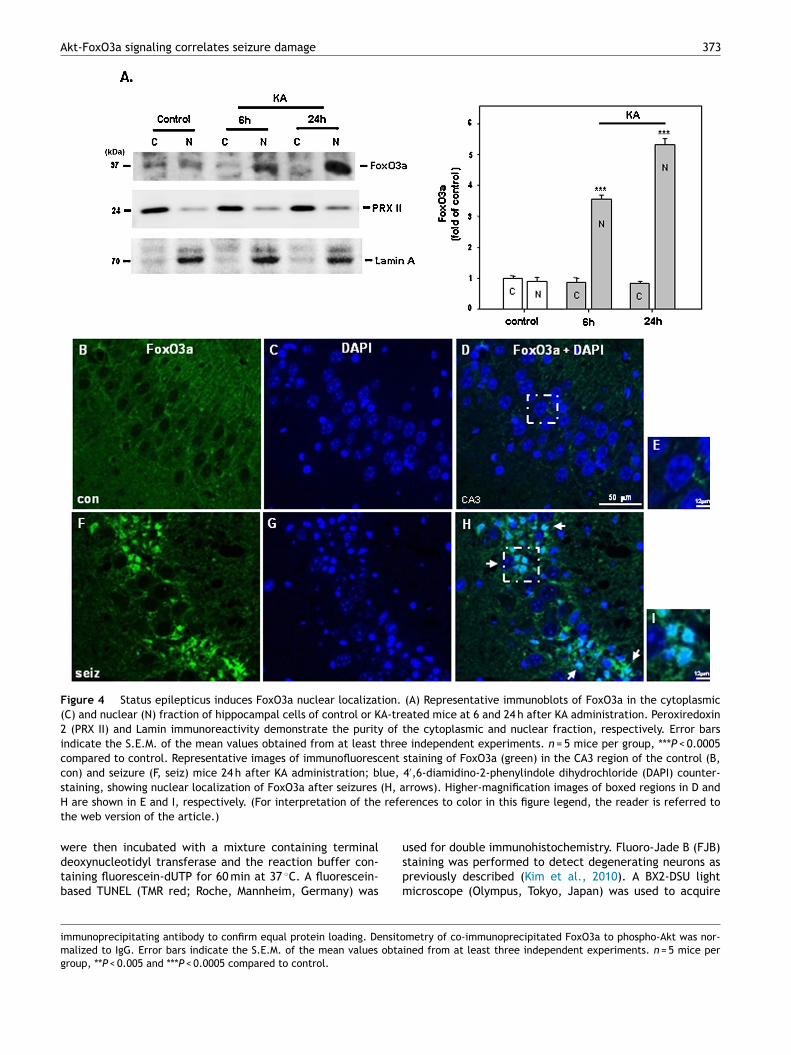
Figure 4 Status epilepticus induces FoxO3a nuclear localization. (A) Representative immunoblots of FoxO3a in the cytoplasmic(C) and nuclear (N) fraction of hippocampal cells of control or KA-treated mice at 6 and 24 h after KA administration. Peroxiredoxin2 (PRX II) and Lamin immunoreactivity demonstrate the purity of the cytoplasmic and nuclear fraction, respectively. Error barsindicate the S.E.M. of the mean values obtained from at least three independent experiments. n = 5 mice per group, ***P < 0.0005compared to control. Representative images of immunofluorescent staining of FoxO3a (green) in the CA3 region of the control (B,con) and seizure (F, seiz) mice 24 h after KA administration; blue, 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) counter-
(H, a refe
staining, showing nuclear localization of FoxO3a after seizures
H are shown in E and I, respectively. (For interpretation of thethe web version of the article.)
were then incubated with a mixture containing terminaldeoxynucleotidyl transferase and the reaction buffer con-taining fluorescein-dUTP for 60 min at 37 ◦C. A fluorescein-based TUNEL (TMR red; Roche, Mannheim, Germany) was
uspm
immunoprecipitating antibody to confirm equal protein loading. Densitomalized to IgG. Error bars indicate the S.E.M. of the mean values obtagroup, **P < 0.005 and ***P < 0.0005 compared to control.
rrows). Higher-magnification images of boxed regions in D andrences to color in this figure legend, the reader is referred to
sed for double immunohistochemistry. Fluoro-Jade B (FJB)taining was performed to detect degenerating neurons asreviously described (Kim et al., 2010). A BX2-DSU lighticroscope (Olympus, Tokyo, Japan) was used to acquire
metry of co-immunoprecipitated FoxO3a to phospho-Akt was nor-ined from at least three independent experiments. n = 5 mice per
374 Y.S. Kim et al.
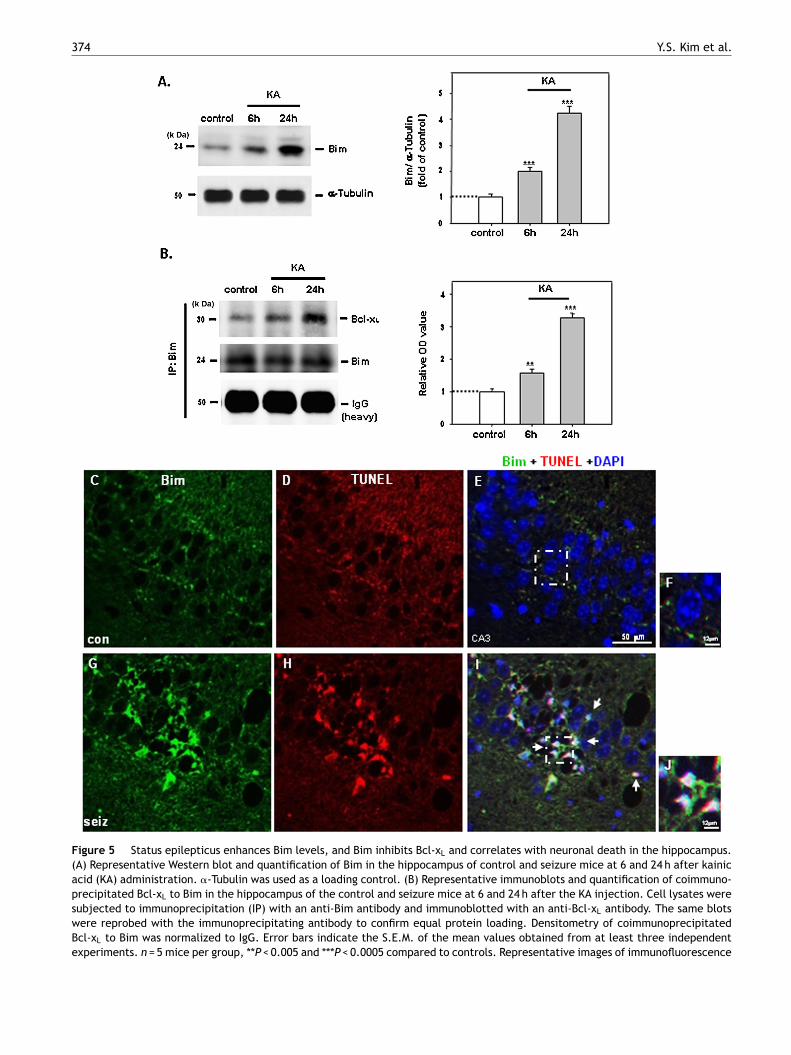
Figure 5 Status epilepticus enhances Bim levels, and Bim inhibits Bcl-xL and correlates with neuronal death in the hippocampus.(A) Representative Western blot and quantification of Bim in the hippocampus of control and seizure mice at 6 and 24 h after kainicacid (KA) administration. �-Tubulin was used as a loading control. (B) Representative immunoblots and quantification of coimmuno-precipitated Bcl-xL to Bim in the hippocampus of the control and seizure mice at 6 and 24 h after the KA injection. Cell lysates weresubjected to immunoprecipitation (IP) with an anti-Bim antibody and immunoblotted with an anti-Bcl-xL antibody. The same blotswere reprobed with the immunoprecipitating antibody to confirm equal protein loading. Densitometry of coimmunoprecipitatedBcl-xL to Bim was normalized to IgG. Error bars indicate the S.E.M. of the mean values obtained from at least three independentexperiments. n = 5 mice per group, **P < 0.005 and ***P < 0.0005 compared to controls. Representative images of immunofluorescence

Sc
WitcsKaiwp(mi((
Si
Vpi2p(solAsaaeslwFapis2(W
Akt-FoxO3a signaling correlates seizure damage
images within the CA3 region or hippocampus at a similarlocation in different animals.
Double immunohistochemistryFor double staining of cleaved caspase-3 plus NeuN (a neu-ronal marker) or FoxO3a plus NeuN, cleaved caspase-3 orFoxO3a and NeuN were labeled with Alexa 488 and Alexa594, respectively. Immunofluorescence staining for cleavedcaspase-3 or FoxO3a was followed by NeuN immunostaining.For double staining of FoxO3a or Bim plus TUNEL, FoxO3aor Bim was labeled with Alexa 488, and immunofluorescentstaining for FoxO3a or Bim was followed by TUNEL staining.A BX2-DSU light microscope (Olympus, Tokyo, Japan) wasused to acquire images, and captured images were mergedto reveal co-distribution sites.
Statistical analysis
Data were presented as the mean ± standard error of themean (S.E.M.) from three separate experiments. A total ofthree experiments were repeated with five different micefor each group per experiment. Data were analyzed usingone-way ANOVA or Student’s t-test. Results were consideredsignificant at P < 0.05.
Results
Status epilepticus induced apoptosis ofhippocampal neurons
To test whether status epilepticus caused neuronal deathvia apoptotic cell death pathways, we measured the levelsof active caspase-3 in the susceptible hippocampal CA3subfield after kainate-induced status epilepticus. Activecaspase-3 immunoreactivity was significantly increased inthe hippocampus of SE mice after kainate-induced seizurescompared to control mice (Fig. 1A, P < 0.0005, eachgroup n = 5). Furthermore, much of the cleaved caspase-3immunoreactivity was neuronal, evidenced by immunoflu-orescence double staining for cleaved caspase-3 and NeuN(a neuronal marker) (Fig. 1H and I, arrows). In addition,numerous TUNEL-positive cells were found in the CA3 regionafter KA administration (Fig. 1J and K, arrows), while afew TUNEL-positive cells were observed in the CA1 region(Fig. 1J), and no TUNEL-positive cells at all in the controlmice (data not shown). Brains from control and SE mice
were analyzed with Fluoro-Jade B (FJB) staining. SE miceshowed many FJB-positive cells in the hippocampus after KAadministration (Fig. 1L and M, arrows), and no FJB-positivecells at all in the control mice (data not shown).ps2(
staining of Bim (green) in the CA3 neurons of control (C, con) and seizuthe same region (D or H, respectively); merged pictures (E or I, respectJ, arrows); blue, 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) cand I are shown in F and J, respectively. (For interpretation of the refeweb version of the article.)
375
tatus epilepticus activates FoxO3a, a neuronalell death factor
e examined the association of FoxO3a with seizure-nduced cell death in hippocampal neurons after KAreatment. Western blot analysis showed that FoxO3a wasonstitutively expressed and that the levels of FoxO3a wereignificantly enhanced in the hippocampus at 6 and 24 h afterA treatment compared to control mice (Fig. 2A; P < 0.05nd P < 0.0005, respectively; each group n = 5). Further,mmunohistochemical analysis demonstrated that FoxO3aas greatly increased in the CA3 region of the hippocam-us of SE mice (Fig. 2F—I, arrows) compared to control miceFig. 2B—E), supporting the Western blot data. Moreover,any FoxO3a-positive cells also appeared TUNEL-positive
n SE mice, indicated by co-staining of FoxO3a and TUNELFig. 2N—Q, arrows), while they did not in control miceFig. 2J—M).
tatus epilepticus regulates Akt activity thatnteracts with FoxO3a
arious molecules in serine and threonine kinase Aktathways have been implicated in mechanisms underly-ng epileptic seizures (Zeng et al., 2009; Backman et al.,001). Increased Akt phosphorylation has been reported torevent hippocampal cell death following seizures in micePiermartiri et al., 2009). Therefore, we tested whethereizures have an effect on Akt activity in the hippocampusf mice after KA treatment. We found significantly loweredevels of activated phospho-Akt (Thr308) and phospho-kt (Ser473) in the hippocampus 24 h after KA-inducedeizures compared with control mice (Fig. 3A; P < 0.0005nd P < 0.005, respectively; each group n = 5). Furthermore,ctivated Akt phosphorylates various downstream targets,ventually changing their cellular function. FoxO3a is oneuch protein, and Akt-mediated phosphorylation of FoxO3aeads to its inactivation (Biggs et al., 1999). Therefore,e examined whether activated Akt directly interacts withoxO3a and whether FoxO3a phosphorylation by Akt wasltered in the hippocampus after seizures. We found thathospho-Akt interacted with FoxO3a, evidenced by a co-mmunoprecipitation experiment, and this interaction wasignificantly lowered in the hippocampus of mice at 6 and4 h after KA administration compared with control miceFig. 3B; P < 0.005 and P < 0.0005, respectively; each n = 5).estern blot analysis consistently showed that levels of
hospho-FoxO3a (Ser253) or phospho-FoxO3a (Thr32) wereignificantly lowered in the hippocampus of SE mice 6 and4 h after KA administration compared with control miceFig. 3C, P < 0.0005; each group n = 5).re mice (G, seiz) 24 h after KA injection; TUNEL (red) staining ofively) showing that many Bim-positive cells are TUNEL positive (I,ounterstaining. Higher-magnification images of boxed regions in Erences to color in this figure legend, the reader is referred to the

3
SF
TiatFotbb2wsonttipa
Li
Wrlra2baiopnbopa(BaimSBib6mglaBKcsF
aoac
D
HFaaptooi
Fevisdl2wislaawaspacBsoBfeoimawfd
psbctv
76
eizures enhance the nuclear localization ofoxO3a
he major mechanism of FoxO regulation occurs via changesn its subcellular localization (Calnan and Brunet, 2008),nd therefore we determined whether FoxO3a localiza-ion was changed after prolonged seizures. We measuredoxO3a levels using Western blotting after the separationf hippocampal extracts into cytoplasmic and nuclear frac-ions. The cytoplasmic and nuclear extracts were separatedy sequential centrifugation. Fraction purity was assessedy Western blotting using antibodies against peroxiredoxin
or Lamin A. An immunoreactive band at 24 or 70 kDaas identified in the cytoplasmic or nuclear extract exclu-
ively, the expected molecular weight for peroxiredoxin 2r Lamin A, respectively. We observed a strong increase inuclear FoxO3a after KA treatments compared with con-rol mice (Fig. 4A, P < 0.0005; each n = 5) correspondingo the nuclear labeling found by immunocytochemistry,ndicated by co-staining for FoxO3a plus 4′,6-diamidino-2-henylindole dihydrochloride (DAPI), a nuclear dye (Fig. 4Hnd I, arrows).
evels of Bim and its interaction with Bcl-xL arencreased after status epilepticus
ith evidence suggesting that FoxO3a correlates with neu-onal cell death after SE, we next measured Bim proteinevels. FoxO3a may promote apoptotic injury through theegulation of Bcl-2 interacting mediator of cell death (Bim),
pro-apoptotic BH3-only Bcl-2 family member (Chong et al.,005; Dijkers et al., 2000). Overexpression of FoxO3a haseen reported to enhance Bim expression during cell stressnd lead to programmed cell death that requires Bim activ-ty (Gilley et al., 2003). Western blotting showed that levelsf Bim were significantly increased after seizures com-ared with control mice (Fig. 5A, P < 0.0005; each group
= 5). The molecular mass of Bim identified by Westernlotting was ∼24 kDa (Fig. 5A). There are three isoformsf Bim protein: BimS, BimL, and BimEL. These forms areroduced by alternative splicing (Putcha et al., 2001),nd the extra-long (EL) form is most common in neuronsShibata et al., 2002). Based on its molecular mass, theim protein recognized on our Western blots was identifieds BimEL. Bim has also been shown to bind and neutral-ze the pro-survival protein Bcl-xL enabling Bax-mediateditochondrial outer membrane permeabilization (Youle and
trasser, 2008). Therefore, we examined Bim binding tocl-xL in the hippocampus after prolonged seizures. Co-
mmunoprecipitation experiments revealed that Bim-Bcl-xL
inding was significantly enhanced in the hippocampus at and 24 h after KA administration compared with controlice (Fig. 5B; P < 0.005 and P < 0.0005, respectively; each
roup n = 5). Additionally, to determine whether Bim corre-ates with neuronal death after seizures, co-staining of Bimnd TUNEL was performed. Immunofluorescence staining ofim showed greatly increased Bim in the CA3 region of the
A-treated mice 24 h after the KA administration (Fig. 5G)ompared with control mice (Fig. 5C). This finding is con-istent with the results of Western blot analyses (Fig. 5A).urthermore, many Bim-positive cells in the CA3 region were(ra(
Y.S. Kim et al.
lso TUNEL-positive in the CA3 region of the hippocampusf seizure mice 24 h after KA administration (Fig. 5I and J,rrows). Conversely, there were few Bim- or TUNEL-positiveells in control mice (Fig. 5C—F).
iscussion
ere, we show the activation of the transcription factoroxO3a in the cell death susceptible hippocampal CA3 regionfter kainate-induced SE in mice. This FoxO3s activationnd its nuclear localization may be due to the decreasedhosphorylation of FoxO3a by Akt. As well as the activa-ion and new localization of FoxO3a, enhanced expressionf Bim, its inhibition of Bcl-xL, and caspase-3 cleavage werebserved, suggesting their relation to apoptotic cell deathn the hippocampus following SE.
Of note, the lowered phospho-Akt interaction withoxO3a may cause decreased levels of phospho-FoxO3a andventually the nuclear localization of FoxO3a because acti-ation of Akt causes the phosphorylation of FoxO factors;n the case of FoxO3a, at the threonine32, serine253, anderine315 sites (Brunet et al., 1999). Location is the majoreterminant of how FoxO transcription factors are regu-ated in response to external stimuli (Calnan and Brunet,008), and nuclear localization of FoxO3a may correlateith the timing of dephosphorylation of protein, where bind-
ng between Akt and FoxO3a may decrease. Moreover, toupport our microscopy observations, we examined FoxO3aevels in the nuclear compartment after subcellular fraction-tion. Eventually, FoxO3a is activated by dephosphorylationnd drives Bim upregulation (Dijkers et al., 2000). Indeed,e found that Bim, which promotes caspase-dependentpoptosis mainly via neutralizing antiapoptotic Bcl-xL, wasignificantly upregulated in the hippocampus after SE com-ared with control mice, evidenced by Western blot analysesnd immunohistochemistry. Furthermore, a coimmunopre-ipitation assay showed that the interaction of Bim withcl-xL was significantly enhanced in the hippocampus ofeizure mice compared with control mice. That is, activationf Bim inhibits the protective function of Bcl-xL, enablingax-mediated MOMP (Youle and Strasser, 2008). Indeed, weound that Bcl-xL interaction with Bax was significantly low-red after seizures (Kim et al., 2012). Evidence in supportf this finding showed that Bim has a causal role in seizure-nduced neuronal death in vivo in the intra-amygdala KAodel (Murphy et al., 2010), where hippocampal damage
fter SE was significantly reduced in bim−/− mice comparedith wild-type animals. Thus, Bim may be a possible target
or protection of the brain from seizure-induced neuronalamage.
We found that levels of phospho-FoxO3a (Thr32) orhospho-FoxO3a (Ser253) were significantly lowered aftereizures, which correlates with the lowered interactionetween Akt and FoxO3a in the hippocampus compared withontrol mice. Phosphorylation of FoxO factors by Akt causeshe sequestration of FoxO factors in the cytoplasm, pre-enting FoxO factors from transactivating their target genes
Brunet et al., 1999). Although the major mechanism of FoxOegulation occurs via changes in its subcellular localization,ltering FoxO levels also have great effects in the organismCalnan and Brunet, 2008). Therefore, in contrast to changes
B
B
B
C
C
C
C
C
D
D
E
G
H
H
H
H
K
K
Akt-FoxO3a signaling correlates seizure damage
in subcellular localization, changes in FoxO3a protein levelscould be more permanent and may have a profound impacton FoxO3a’s functions. Ultimately, transcriptional controlof Bim by FoxO3a may be involved in neuronal death afterseizures, where interactions between Bcl-2 family proteinsresult in Bax/Bak oligomerization, conformational change,and insertion into the outer mitochondrial membrane totrigger mitochondrial outer membrane permeabilization(MOMP) (Chipuk et al., 2010). Indeed, we found that Biminhibits the protective function of Bcl-xL in the hippocampusof seizure mice, shown by co-immunoprecipitation, and cor-relates with neuronal cell death, evidenced by co-stainingof Bim and TUNEL. A previous study demonstrated that SEevoked by intra-amygdala KA upregulated Bim protein levelsin the hippocampus of rats (Shinoda et al., 2004). However,other studies showed that bim−/− mice were not protectedagainst neuronal death in an in vivo model of excitotoxi-city (Theofilas et al., 2009). Bim levels did not increasein the neocortex of mice after seizures, despite the pres-ence of cell death, indicating the regional importance ofBim (Murphy et al., 2010). Therefore, Bim may contributeto neuronal death in some but not all seizure models.
Assessment of changes in FoxO3a-Akt or Bim-Bcl-xL inter-action may be required to determine activation of FoxO3aor Bim because FoxO3a or Bim is constitutively expressedin the brain, and altered levels of FoxO3a or Bim (and evensubcellular location) may be insufficient to determine theiractivity. In addition, while Bim is known to regulate apo-ptosis after seizures via its interaction with Bcl-w (Shinodaet al., 2004), we report for the first time that Bim may pro-mote apoptosis via its interaction with Bcl-xL. Although welooked at the association of proteins with seizure-inducedcell death, but did not conduct a cause and effect study, ourdata show that the altered interaction between FoxO3a andAkt is associated with FoxO3a activation and apoptotic celldeath in the hippocampus after SE. While we observed muchof apoptotic cell death in the hippocampus of SE mice, otherpatterns of cell death may occur that may be functionallysignificant.
Conclusions
The activation of transcription factor FoxO3a in the celldeath susceptible hippocampal CA3 subfield along with theinteraction of Bim with Bcl-xL and caspase-3 activation cor-relates with neuronal death after SE. As a possible activationmechanism we suggest the decreased phosphorylation ofFoxO3a by Akt, providing insight into neuroprotection fol-lowing SE.Acknowledgement
This work was supported by National Research Foundationof Korea grants (NRFK grant, R13-2005-012-01001-0).
References
Acharya, M.M., Hattiangady, B., Shetty, A.K., 2008. Progress in neu-roprotective strategies for preventing epilepsy. Prog. Neurobiol.
84, 363—404.Backman, S.A., Stambolic, V., Suzuki, A., Haight, J., Elia, A., Pre-torius, J., Tsao, M.S., Shannon, P., Bolon, B., Ivy, G.O., et al.,2001. Deletion of Pten in mouse brain causes seizures, ataxia
L
377
and defects in soma size resembling Lhermitte-Duclos disease.Nat. Genet. 29, 396—403.
iggs 3rd., W.H., Meisenhelder, J., Hunter, T., Cavenee,W.K., Arden, K.C., 1999. Protein kinase B/Akt-mediatedphosphorylation promotes nuclear exclusion of the winged helixtranscription factor FKHR1. Proc. Natl. Acad. Sci. U. S. A. 96,7421—7426.
runet, A., Bonni, A., Zigmond, M.J., Lin, M.Z., Juo, P., Hu, L.S.,Anderson, M.J., Arden, K.C., Blenis, J., Greenberg, M.E., 1999.Akt promotes cell survival by phosphorylating and inhibiting aforkhead transcription factor. Cell 96, 857—868.
runet, A., Kanai, F., Stehn, J., Xu, J., Sarbassova, D., Fran-gioni, J.V., Dalal, S.N., DeCaprio, J.A., Greenberg, M.E., Yaffe,M.B., 2002. 14-3-3 transits to the nucleus and participates indynamic nucleocytoplasmic transport. J. Cell Biol. 156, 817—828.
alnan, D.R., Brunet, A., 2008. The FoxO code. Oncogene 27,2276—2288.
hen, J.W., Wasterlain, C.G., 2006. Status epilepticus: pathophysi-ology and management in adults. Lancet Neurol. 5, 246—256.
hipuk, J.E., Moldoveanu, T., Llambi, F., Parsons, M.J., Green, D.R.,2010. The BCL-2 family reunion. Mol. Cell 37, 299—310.
hong, Z.Z., Li, F., Maiese, K., 2005. Oxidative stress in the brain:novel cellular targets that govern survival during neurodegener-ative disease. Prog. Neurobiol. 75, 207—246.
rowder, R.J., Freeman, R.S., 1998. Phosphatidylinositol 3-kinaseand Akt protein kinase are necessary and sufficient for the sur-vival of nerve growth factor-dependent sympathetic neurons. J.Neurosci. 18, 2933—2943.
atta, S.R., Dudek, H., Tao, X., Masters, S., Fu, H., Gotoh, Y.,Greenberg, M.E., 1997. Akt phosphorylation of BAD couplessurvival signals to the cell-intrinsic death machinery. Cell 91,231—241.
ijkers, P.F., Medema, R.H., Lammers, J.W., Koenderman, L., Cof-fer, P.J., 2000. Expression of the pro-apoptotic Bcl-2 familymember Bim is regulated by the forkhead transcription factorFKHR-L1. Curr. Biol. 10, 1201—1204.
ngel, T., Henshall, D.C., 2009. Apoptosis, Bcl-2 family proteins andcaspases: the ABCs of seizure-damage and epileptogenesis? Int.J. Physiol. Pathophysiol. Pharmacol. 1, 97—115.
illey, J., Coffer, P.J., Ham, J., 2003. FOXO transcription factorsdirectly activate bim gene expression and promote apoptosis insympathetic neurons. J. Cell Biol. 162, 613—622.
annenhalli, S., Kaestner, K.H., 2009. The evolution of Fox genesand their role in development and disease. Nat. Rev. Genet. 10,233—240.
enshall, D.C., Araki, T., Schindler, C.K., Lan, J.Q., Tiekoter,K.L., Taki, W., Simon, R.P., 2002. Activation of Bcl-2-associateddeath protein and counter-response of Akt within cell popula-tions during seizure-induced neuronal death. J. Neurosci. 22,8458—8465.
oekman, M.F., Jacobs, F.M., Smidt, M.P., Burbach, J.P., 2006. Spa-tial and temporal expression of FoxO transcription factors inthe developing and adult murine brain. Gene Expr. Patterns 6,134—140.
u, R.Q., Koh, S., Torgerson, T., Cole, A.J., 1998. Neuronal stressand injury in C57/BL mice after systemic kainic acid administra-tion. Brain Res. 810, 229—240.
im, Y.S., Choi, M.Y., Kim, Y.H., Jeon, B.T., Lee, D.H., Roh, G.S.,Kang, S.S., Kim, H.J., Cho, G.J., Choi, W.S., 2010. Protein kinaseCdelta is associated with 14-3-3 phosphorylation in seizure-induced neuronal death. Epilepsy Res. 92, 30—40.
im, Y.S., Choi, M.Y., Ryu, J.H., Lee, D.H., Jeon, B.T., Roh, G.S.,Kang, S.S., Kim, H.J., Cho, G.J., Choi, W.S., 2012. Clusterin
interaction with Bcl-xL is associated with seizure-induced neu-ronal death. Epilepsy Res. 99, 240—251.ehtinen, M.K., Yuan, Z., Boag, P.R., Yang, Y., Villen, J., Becker,E.B., DiBacco, S., de la Iglesia, N., Gygi, S., Blackwell, T.K.,

3
L
L
M
P
P
P
R
S
S
T
T
Y
78
et al., 2006. A conserved MST-FOXO signaling pathway medi-ates oxidative-stress responses and extends life span. Cell 125,987—1001.
oscher, W., Brandt, C., 2010. Prevention or modification of epilep-togenesis after brain insults: experimental approaches andtranslational research. Pharmacol. Rev. 62, 668—700.
owenstein, D.H., 2005. Treatment options for status epilepticus.Curr. Opin. Pharmacol. 5, 334—339.
urphy, B.M., Engel, T., Paucard, A., Hatazaki, S., Mouri, G.,Tanaka, K., Tuffy, L.P., Jimenez-Mateos, E.M., Woods, I., Dun-leavy, M., et al., 2010. Contrasting patterns of Bim inductionand neuroprotection in Bim-deficient mice between hippocam-pus and neocortex after status epilepticus. Cell Death Differ. 17,459—468.
iermartiri, T.C., Vandresen-Filho, S., de Araujo Herculano, B., Mar-tins, W.C., Dal’agnolo, D., Stroeh, E., Carqueja, C.L., Boeck,C.R., Tasca, C.I., 2009. Atorvastatin prevents hippocampal celldeath due to quinolinic acid-induced seizures in mice by increas-ing Akt phosphorylation and glutamate uptake. Neurotox. Res.16, 106—115.
itkanen, A., 2002. Drug-mediated neuroprotection and antiepilep-togenesis: animal data. Neurology 59, S27—S33.
utcha, G.V., Moulder, K.L., Golden, J.P., Bouillet, P., Adams, J.A.,Strasser, A., Johnson, E.M., 2001. Induction of BIM, a proapo-ptotic BH3-only BCL-2 family member, is critical for neuronalapoptosis. Neuron 29, 615—628.
Z
Y.S. Kim et al.
oy, M., Hom, J.J., Sapolsky, R.M., 2002. HSV-mediated delivery ofvirally derived anti-apoptotic genes protects the rat hippocam-pus from damage following excitotoxicity, but not metabolicdisruption. Gene Ther. 9, 214—219.
hibata, M., Hattori, H., Sasaki, T., Gotoh, J., Hamada, J., Fuku-uchi, Y., 2002. Temporal profiles of the subcellular localization ofBim, a BH3-only protein, during middle cerebral artery occlusionin mice. J. Cereb. Blood Flow Metab. 22, 810—820.
hinoda, S., Schindler, C.K., Meller, R., So, N.K., Araki, T.,Yamamoto, A., Lan, J.Q., Taki, W., Simon, R.P., Henshall, D.C.,2004. Bim regulation may determine hippocampal vulnerabilityafter injurious seizures and in temporal lobe epilepsy. J. Clin.Invest. 113, 1059—1068.
heofilas, P., Bedner, P., Huttmann, K., Theis, M., Steinhauser, C.,Frank, S., 2009. The proapoptotic BCL-2 homology domain 3-only protein Bim is not critical for acute excitotoxic cell death.J. Neuropathol. Exp. Neurol. 68, 102—110.
ran, H., Brunet, A., Griffith, E.C., Greenberg, M.E., 2003. Themany forks in FOXO’s road. Sci. STKE 2003, RE5.
oule, R.J., Strasser, A., 2008. The BCL-2 protein family: opposingactivities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9,47—59.
eng, L.H., Rensing, N.R., Wong, M., 2009. The mammaliantarget of rapamycin signaling pathway mediates epileptogen-esis in a model of temporal lobe epilepsy. J. Neurosci. 29,6964—6972.





























