Embed Size (px)
Citation preview

ValidationValidation -- Process validationProcess validation is the establishmentis the establishmentofof documented evidencedocumented evidence,, which provide a high degree of assurancewhich provide a high degree of assurancethat a specific process (manufacturing of pharmaceutical dosagethat a specific process (manufacturing of pharmaceutical dosageform) will consistently produce a product meeting itsform) will consistently produce a product meeting its predeterminedpredeterminedspecificationsspecifications..
DEFINITION:
VALIDATION
DOCUMENTS
Nitro P
DF Tria
l
www.nitro
pdf.c
om

It reduces risk of regulatory non-compliance.
Reduction of time to the market for the new products.
Eliminates the scrap & reduces the defect cost.
Reduces the chances of product re-call from market.
The final release of the product batch would beexpedited.
It requires less in-process control & end process testing.
Parametric release of batch can be achieved invalidation.
Nitro P
DF Tria
l
www.nitro
pdf.c
om

QA
The emphasis on validation began in the late 1970s, the requirement hasbeen set at 1963 as CGMP regulations for finished pharmaceuticals.
Validation is an integral part of QualityAssurance& its meaning is “Action of providing an evidence”.
Validation is necessarily include process qualification (qualificationof rawmaterials,equipment,system) under the section21 CFR 211.100 which states:
“There shall be written procedures for production and processcontrol designed to assure that the drug products have the identity,strength,quality,and purity”.
Nitro P
DF Tria
l
www.nitro
pdf.c
om

The Kefauver-Harris Amendments to the FD&C Act wereapproved in1962 with Section 501(a)(2)(B) as an amendment.
The result of The Kefauver–Harris drug amendments,Provided an additional powerful regulatory tool to FDA tostop particular manufacturing process when the drug productis deemed to adulteration.
The Drug Product Quality Assurance Program of the 1960s and1970s involved first conducting a massive samplingand testing program of finished batches.
The investigation of clinical failures of several products(including Digoxin, Digitoxin, Prednisolone, and Prednisone)by FDA found significant content uniformity problems that werethe result of poorly controlled manufacturing processes.
Nitro P
DF Tria
l
www.nitro
pdf.c
om

COUNTRY: REGULATORY AUTHORITY:
INDIA
UNITEDKINGDOM
SOUTHAFRICA
CANADA
AUSTRALIA
Indian FDA.
MHRA-Medicine Health& Regulatory Agency.
MCC-Medicinal Control Council.
HPB-Health Protection Branch.
TGA-Therapeutic Goods Administration.
Nitro P
DF Tria
l
www.nitro
pdf.c
om

NETHERLANDS.
UGANDA.
WORLD-WIDE.
BRAZIL.
IDA-International DispensaryAssociation.
NDA-National Drug Authority.
WHO & ICH.
ANVISA-National Agency forVigilance Sanitare.
Nitro P
DF Tria
l
www.nitro
pdf.c
om

The regulatory basis -validation program
of process validation is embodied within the regulations & guidelinesprovided by cGMP & FDA.
The ultimate legal authority is Sec501(a)(2)(B) by the FD&C Act,which states “Drug is deemed to be adulterated due to the methods/facilities used for the manufacturing, processing, packing/holding fails toadminister in conformity – CGMP”
Validation-Process validation is not just an FDA or U.S. requirement.Similar requirements included in the World Health Organization (WHO),the Pharmaceutical Inspection Co-operation Scheme (PIC/S), and theEuropean Union(EU).
Nitro P
DF Tria
l
www.nitro
pdf.c
om

Section211.100(a): Written procedures/deviations.
“There shall be written procedures for production
and process control designed to assure that the drug productshave the identity, strength, quality,and purity.”
Section 211.110: Sampling and testing of in-process materials anddrug products.
"....control procedures shall be established tomonitor the output and Validate the performance of those
manufacturing processes that may be responsible for causingvariability in the characteristics of in-process material and the drugproduct”
REGULATIONS FOR VALIDATION PROCESS
UNDER U.S.FDA & CGMP :
Nitro P
DF Tria
l
www.nitro
pdf.c
om

21CFR211.133: Control of Microbiological Contamination.
" Appropriate written procedures,designed to preventmicrobiological contamination of drug products purporting to besterile, shall be established and followed. Such procedures shall includeValidation of any sterilization process.“
FDA must inspect every drug manufacturing establishment at least onceevery 2 years .
Nitro P
DF Tria
l
www.nitro
pdf.c
om

The first CGMP regulations, based largely on the PharmaceuticalManufacturers Association’s- manufacturing control guidelines .
Validation under document of cGMP covers procedure, processqualification, equipment,& facilities.
211.68: validation of automated process.211.84(d)(2): validation of supplier’s test results for components.
211.84(d)(3): validation of supplier’s test results for containers& closures.
211.110(a): validation of manufacturing process to ensurecontent uniformity& integrity.
211.1113(b): validation of sterilization process.211.165: validation of analytical methods.
REGULATORY REQUIREMENTS FOR VALIDATION. UNDER CGMP:
Nitro P
DF Tria
l
www.nitro
pdf.c
om

The documented act of provingany procedure,process,equipment,material, activity orsystem which actually leads tothe expected results.
WHO (World Health Organization) cGMP GuidelinesstateValidation studies are an essential part of current good manufacturingpractice (CGMP) and should be conducted in accordance with predefinedprotocols.
WHO validation definition:
Strategies of validation under WHO includes:
DQ: Design Qualification IQ: Installation Qualification
OQ : Operational Qualification PQ:Performance Qualification
Nitro P
DF Tria
l
www.nitro
pdf.c
om

DQ:The compliance of the basic design (location plan) with the
user requirements & regulatory requirements should be submitted &documented.
IQ:Documentary evidence to prove that the premises &
equipment have been built & installed in compliance with theirspecifications. IQ include:
1.Preventive maintenance.2 .Equipment info.3. Calibration.4.Verification of the equipment.
OQ:A series of tests to measure the performance capability of
equipment. The OQ for HPLC system is the operation of pump,injector & detector will be tested at this stage.
Nitro P
DF Tria
l
www.nitro
pdf.c
om
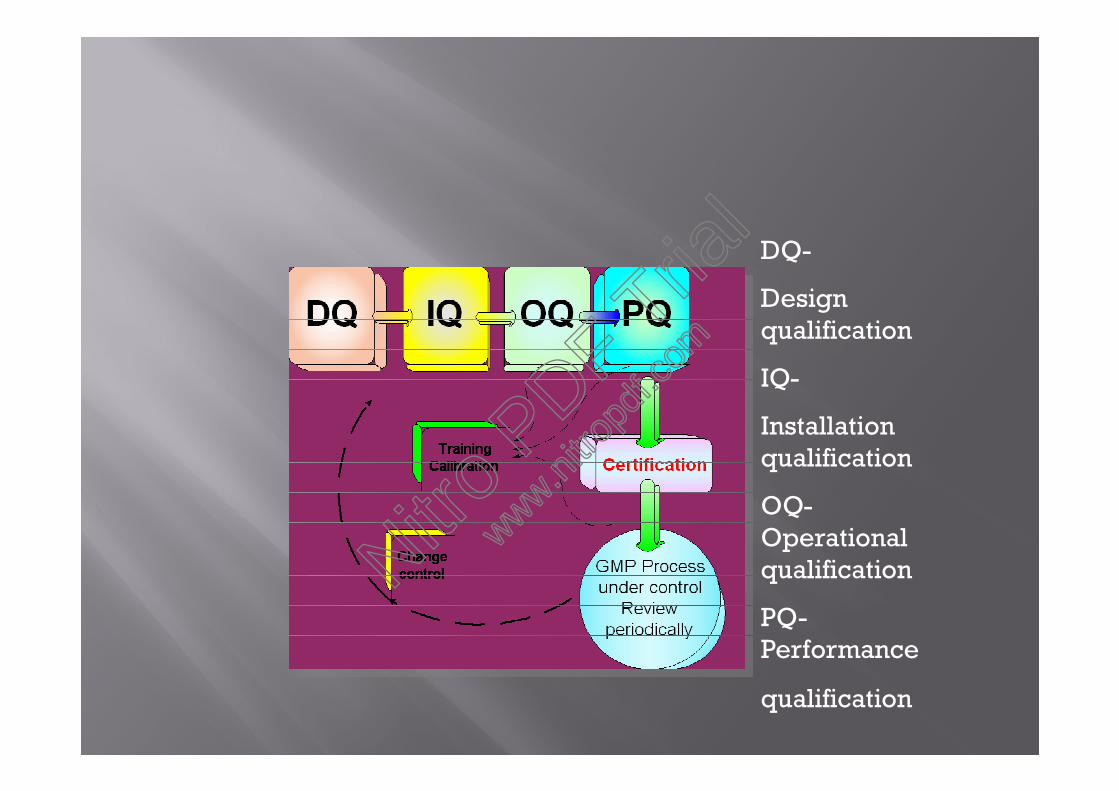
DQ-
Designqualification
IQ-
Installationqualification
OQ-Operationalqualification
PQ-Performance
qualification
Nitro P
DF Tria
l
www.nitro
pdf.c
om

A Dossier is a collection of document submitted to a foreigncountry for marketing of a drug product.
For dossier submission purpose, Process validation protocol shouldinclude such requirements according to dossier guidelines.
Typical OQ test for HPLC system are:
Pump-flow rate accuracyDetector-response,linearity,wavelength accuracyColumn-resolution,HETP.
PQ:
Process to verify that the system is repeatable & capablefor consistently producing a quality product.
Ex: HPLC system-resolution can be tested by injectingrepeatedly a standard mixtures of analytes like phenol ,toulene etc & bymeasuring RT, Peak area etc .
Nitro P
DF Tria
l
www.nitro
pdf.c
om

The European Union requirements for validation is anextract from ICH Q8, Q9 and Q10 documented guidelines and helps to studycontinuous process verification.
Documented evidence that the process,operated within established parameters,can perform effectively and reproducibly,To produce a medicinal product meetingits predetermined specifications andquality attributes.
EU Validation Definition:
Traditional process verification
Contineous process validation.(CPV)
Critical process parameter.(CPP)
Critical quality attributes.(CQA)
Strategies of validation
under EU includes:
Nitro P
DF Tria
l
www.nitro
pdf.c
om
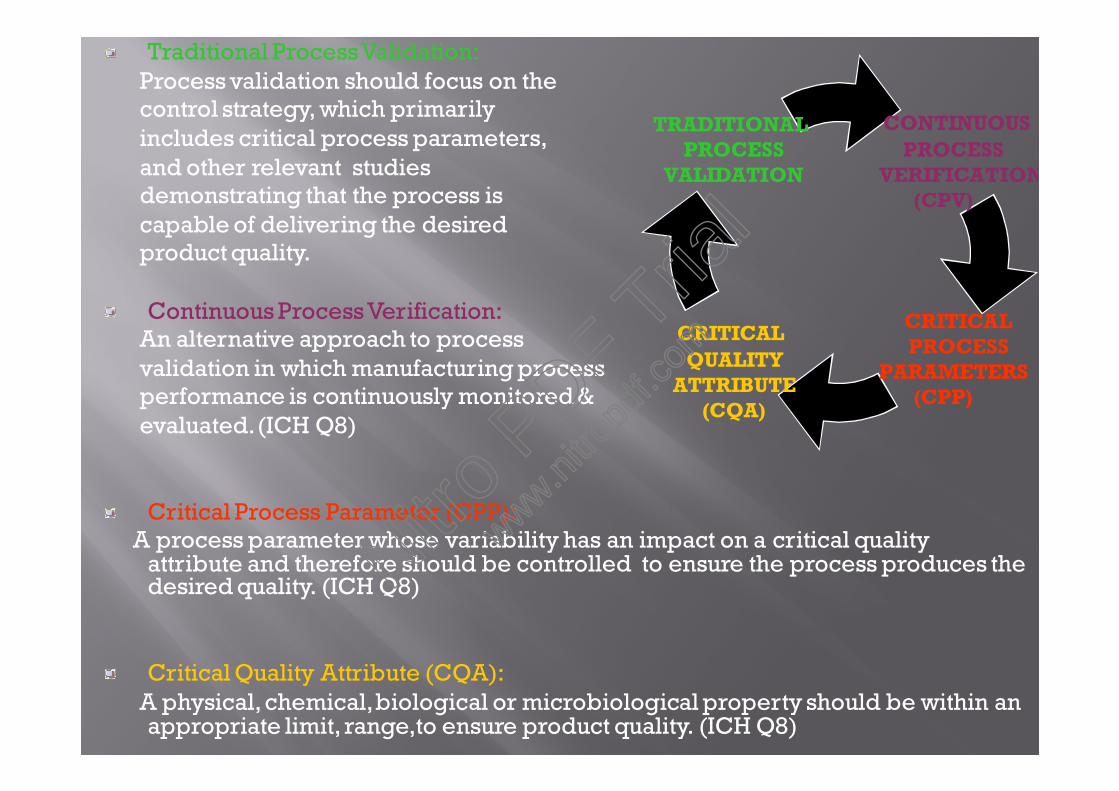
Traditional ProcessValidation:Process validation should focus on thecontrol strategy, which primarilyincludes critical process parameters,and other relevant studiesdemonstrating that the process iscapable of delivering the desiredproduct quality.
Continuous ProcessVerification:An alternative approach to processvalidation in which manufacturing processperformance is continuously monitored &evaluated.(ICH Q8)
Critical Process Parameter (CPP):A process parameter whose variability has an impact on a critical quality
attribute and therefore should be controlled to ensure the process produces thedesired quality. (ICH Q8)
Critical Quality Attribute (CQA):A physical, chemical,biological or microbiological property should be within anappropriate limit, range,to ensure product quality. (ICH Q8)
CONTINUOUSPROCESS
VERIFICATION(CPV)
CRITICALQUALITY
ATTRIBUTE(CQA)
TRADITIONALPROCESS
VALIDATION
CRITICALPROCESS
PARAMETERS(CPP)
Nitro P
DF Tria
l
www.nitro
pdf.c
om

According the EU Guidelines to Good Manufacturing Practicefor Medicinal Products in Annex 15 the principles of qualification &validation of the PIC/S is given under document PIC/S PI 006-3:
This document applies primarily to inspectorates of the PIC/S memberfor whom it is intended as instruction for preparing an inspection, and asan advanced training aid for qualification/validation.
GMP for medicinal products(Recommendations on Validation Master PlanInstallation and Operational QualificationNon-Sterile Process Validation CleaningValidation) can assist with theinterpretation and the implementation.
Doc states:
Nitro P
DF Tria
l
www.nitro
pdf.c
om

Experience
Planning
Resources
Understanding & communication
Training
SOP,s instruments & methodologies.
Validation Master Plan.
Data analysis.
Validation report.
Nitro P
DF Tria
l
www.nitro
pdf.c
om
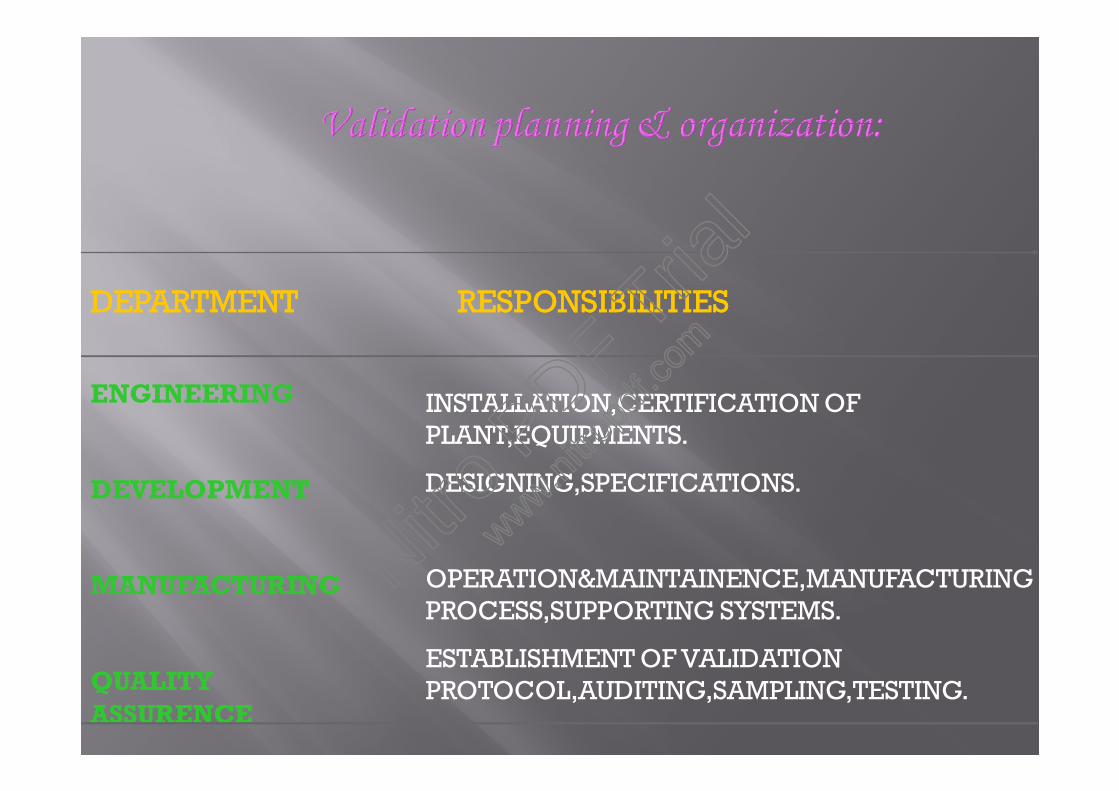
DEPARTMENTDEPARTMENT RESPONSIBILITIESRESPONSIBILITIES
ENGINEERING
DEVELOPMENT
MANUFACTURING
QUALITYASSURENCE
INSTALLATION,CERTIFICATION OFPLANT,EQUIPMENTS.
DESIGNING,SPECIFICATIONS.
OPERATION&MAINTAINENCE,MANUFACTURINGPROCESS,SUPPORTING SYSTEMS.
ESTABLISHMENT OF VALIDATIONPROTOCOL,AUDITING,SAMPLING,TESTING.
Nitro P
DF Tria
l
www.nitro
pdf.c
om
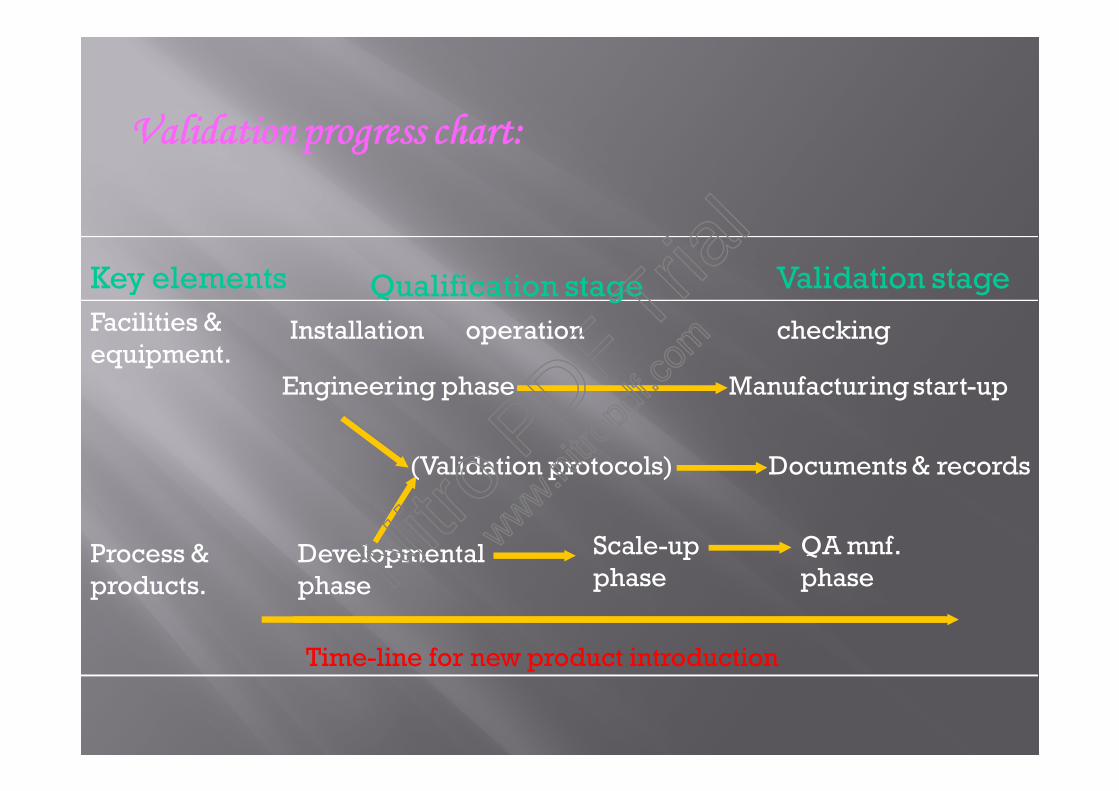
Key elements Qualification stage Validation stageFacilities &equipment.
Process &products.
Installation
Engineering phase Manufacturing start-up
(Validation protocols) Documents & records
Developmentalphase
Scale-upphase
QA mnf.phase
Time-line for new product introduction
operation checking
Validation progress chart:
Nitro P
DF Tria
l
www.nitro
pdf.c
om

Typical standard Operating Procedure (SOP) format.
Operation Dept :Name: Date:
Compliance:
ABC PHARMACEUTICAL Ltd.
Plant operations.
Page 1-5PREPARED BYPREVIOUS DATEISSUE DATES.O.P No. 258-04,Revision 4DEPT . PLANT.
STANDARD OPERATING PROCEDURE
TITLE:AIR HANDLING SYSTEM 234A-ABC-04CONDITIONS & PROCEDURE COVERINGTHE OPERATION & MAINTAINANCE
Name: Date:
1.PURPOSE:To define the Std. Operating conditions & SOP,sfor air handling system 234A-ABC-04.
2.GENERAL INFORMATION:2.1 Supply Air Fan
Fan Manufacturer: Narayan.Model: 325-567-2222.Capacity: 30,000CMF.RPM: 1500.POWER: 440V,3Phase,60cycles.
Nitro P
DF Tria
l
www.nitro
pdf.c
om

The complete overview of validation operation,organization structure,content &planning in the form of a document isthe VMP.
VMP should contain the fallowing data:
Validation policy of company, location& schedule.List of product,processes&system to be validated.Installation &qualification for new equipment.Key acceptance criteria.Documentation format used for protocols & report.Time planning & scheduling of project.
A VMP Helps: Members of validation team to know their task &responsibility.
Nitro P
DF Tria
l
www.nitro
pdf.c
om
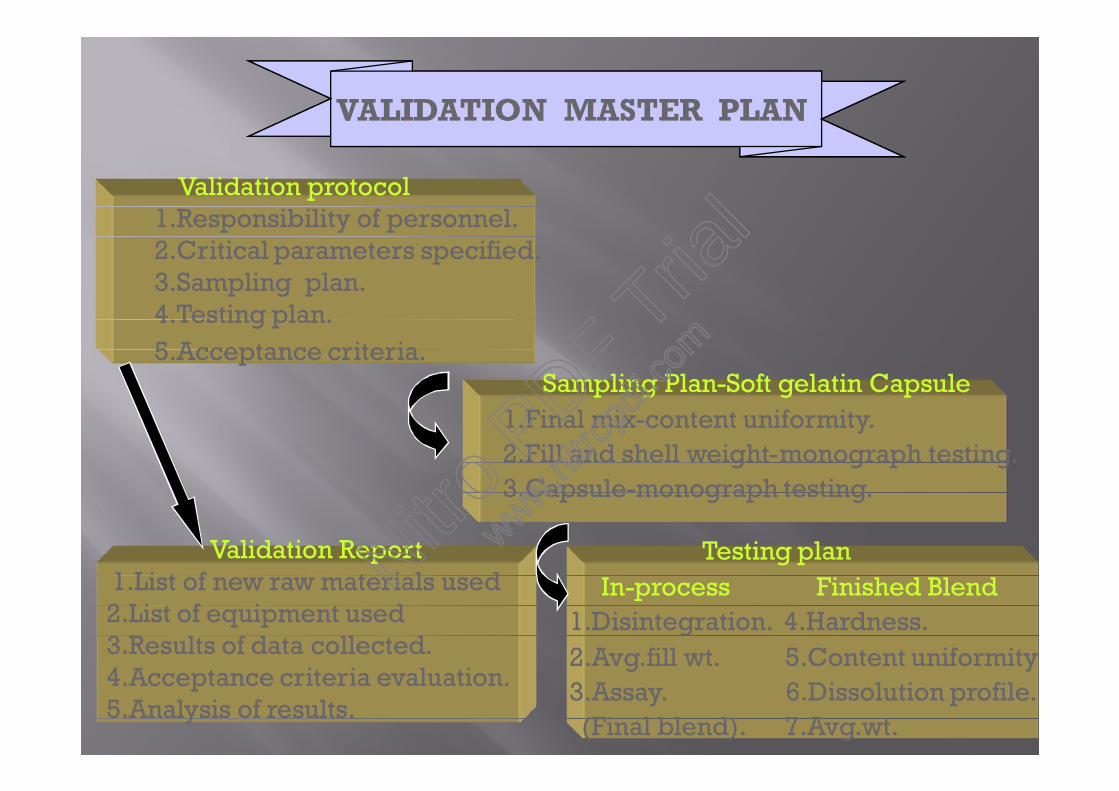
Validation Report1.List of new raw materials used2.List of equipment used3.Results of data collected.4.Acceptance criteria evaluation.5.Analysis of results.
VALIDATION MASTER PLAN
Validation protocol1.Responsibility of personnel.2.Critical parameters specified.3.Sampling plan.4.Testing plan.5.Acceptance criteria.
Sampling Plan-Soft gelatin Capsule1.Final mix-content uniformity.2.Fill and shell weight-monograph testing.3.Capsule-monograph testing.
Testing planIn-process Finished Blend
1.Disintegration. 4.Hardness.2.Avg.fill wt. 5.Content uniformity3.Assay. 6.Dissolution profile.(Final blend). 7.Avg.wt.
Nitro P
DF Tria
l
www.nitro
pdf.c
om

The validation report should be approved prior to productdistribution and kept permanently on file in quality assurance.
The validation report should have a conclusion that explains themanufacturing specialist’s (preparer’s) statement and opinion.
The validation report should contain the approved validationprotocol, tabulated or graphical results, process monitoring (forms),and all analytical results of the validation batches.
Nitro P
DF Tria
l
www.nitro
pdf.c
om

Nitro P
DF Tria
l
www.nitro
pdf.c
om

Process validation is defined as the collectionand evaluation of data, from the process design stagethrough commercial production, which establishes scientificevidence that a process is capable of consistently deliveringquality product.
A series of activities taking place over the lifecycle ofthe product and process.
stage1: Process design.stage2: Process qualification.stage3: Continued process verification.
Nitro P
DF Tria
l
www.nitro
pdf.c
om
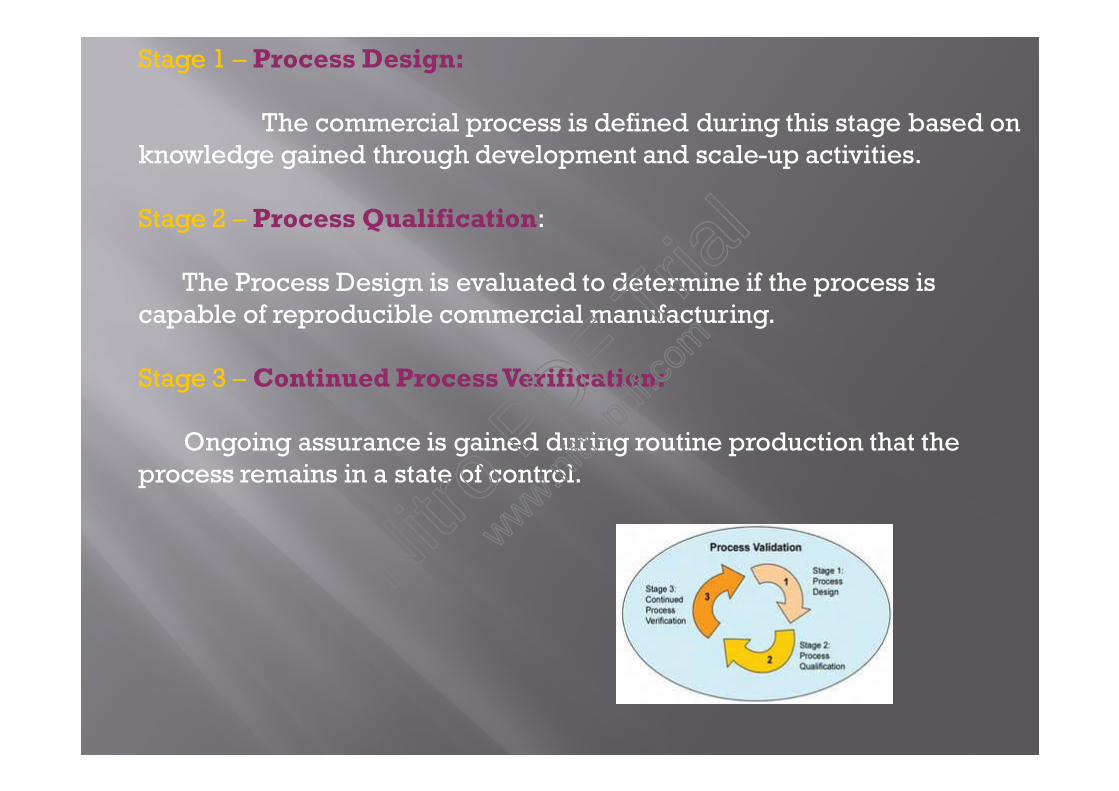
Stage 1 – Process Design:
The commercial process is defined during this stage based onknowledge gained through development and scale-up activities.
Stage 2 – Process Qualification:
The Process Design is evaluated to determine if the process iscapable of reproducible commercial manufacturing.
Stage 3 – Continued Process Verification:
Ongoing assurance is gained during routine production that theprocess remains in a state of control.
Nitro P
DF Tria
l
www.nitro
pdf.c
om

Nitro P
DF Tria
l
www.nitro
pdf.c
om
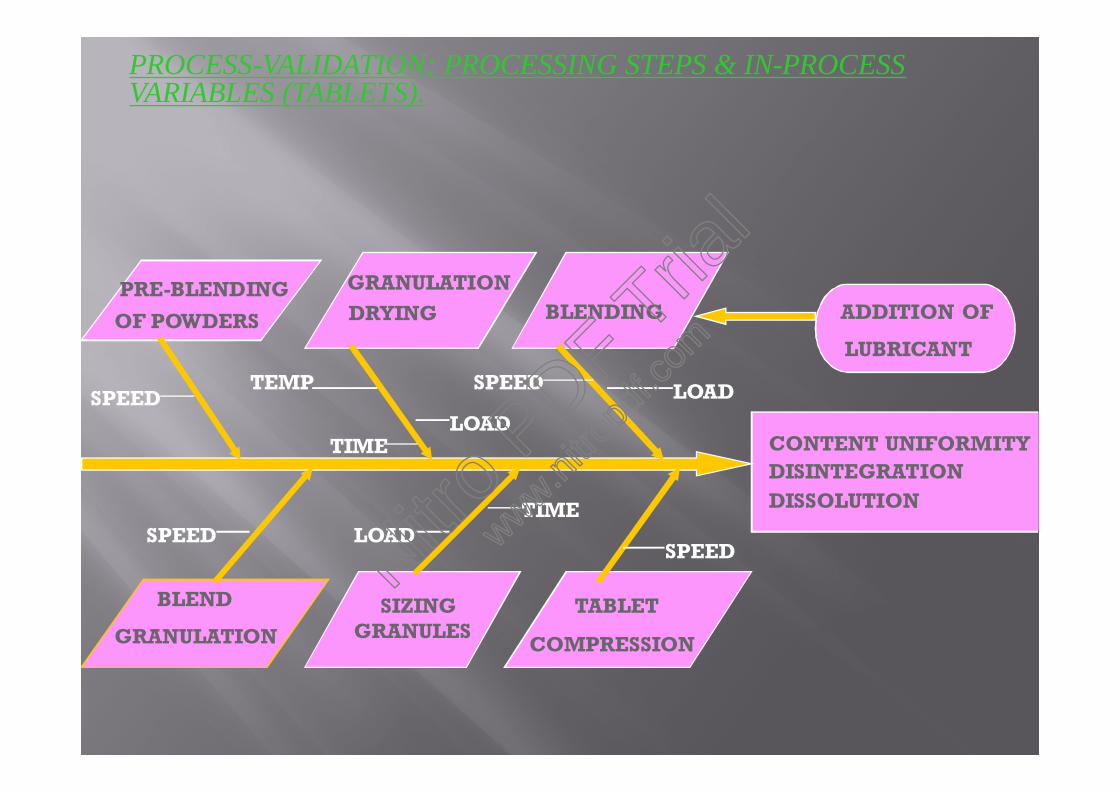
PROCESS-VALIDATION: PROCESSING STEPS & IN-PROCESSVARIABLES (TABLETS).
CONTENT UNIFORMITYDISINTEGRATIONDISSOLUTION
GRANULATIONDRYING
PRE-BLENDINGOF POWDERS
BLEND
GRANULATIONSIZING
GRANULESTABLET
COMPRESSION
BLENDING ADDITION OF
LUBRICANT
SPEEDTEMP SPEED
SPEEDSPEED
LOAD
LOAD
LOAD
TIME
TIME
Nitro P
DF Tria
l
www.nitro
pdf.c
om

Validation
Prospective
Concurrent
RetrospectiveProspective validation:
The Validation that has been made before an entry of a newproduct or employing a new formula/process & also is been carried out ifthere is a change in the manufacturing process is called–PV.
Concurrent validation:
validation that has to be carried out for the existing productsthat is taken from manufacturing line in a batch wise manner .extensivetesting during validation may verify quality attributes.
Nitro P
DF Tria
l
www.nitro
pdf.c
om
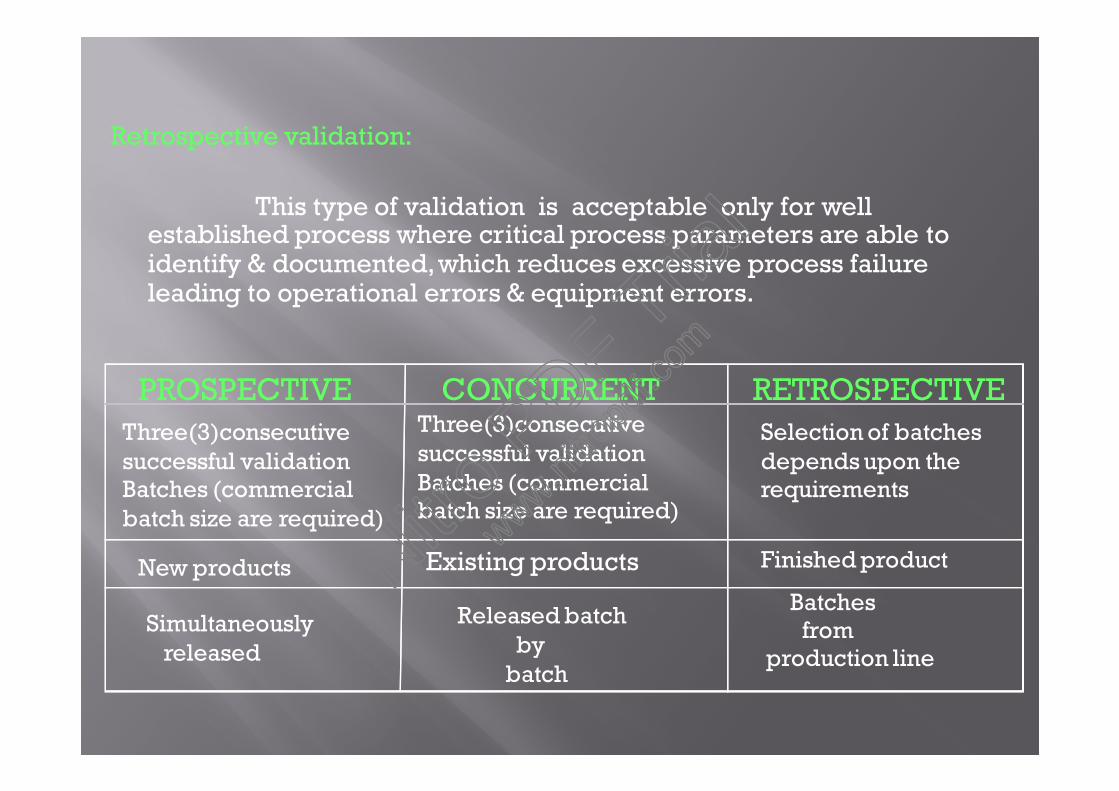
Retrospective validation:
This type of validation is acceptable only for wellestablished process where critical process parameters are able toidentify & documented,which reduces excessive process failureleading to operational errors & equipment errors.
PROSPECTIVE CONCURRENT RETROSPECTIVEThree(3)consecutivesuccessful validationBatches (commercialbatch size are required)
Three(3)consecutivesuccessful validationBatches (commercialbatch size are required)
New products Existing products
Simultaneouslyreleased
Released batchby
batch
Selection of batchesdepends upon therequirements
Finished product
Batchesfrom
production line
Nitro P
DF Tria
l
www.nitro
pdf.c
om

Regulatory authorities working on strategies to reduce the cost of processvalidation and incorporate validation consideration during product designand development.
New technologies under development for 100% analysis of drug productsand other innovations in pharmaceutical industry may also have a significanteffect on Validation & basic regulatory authority's acceptance.
The future of process validation is also of great interest, especially with theworldwide expansion of pharmaceutical manufacturing & for harmonizing ininternational standards and requirements.
Nitro P
DF Tria
l
www.nitro
pdf.c
om

Pharmaceutical Process Validation by R. BerryPharmaceutical Process Validation by R. Berry& Robert A. Nash.& Robert A. Nash.
www.fda.gov/cder/guidance/pv.htm (US FDA).www.fda.gov/cder/guidance/pv.htm (US FDA).
Remington’s pharmaceutical science.Remington’s pharmaceutical science.
PharmaceuticalPharmaceutical preformulationspreformulations byby j.jj.j. Wells.. Wells.
www.emea.eu.int/ (EU Audit Agency)www.emea.eu.int/ (EU Audit Agency).
Nitro P
DF Tria
l
www.nitro
pdf.c
om

THANK YOU!THANK YOU!
Nitro P
DF Tria
l
www.nitro
pdf.c
om



















![Parenteral Process Validation[1]](https://img.pdfslide.net/doc/110x75/543fd712b1af9f620a8b4b80/parenteral-process-validation1.jpg)









