Embed Size (px)
Citation preview

Eur. J. Biochem. 60, 109- 116 (1975)
Depolymerisation of F-Actin to G-Actin and Its Repolymerisation in the Presence of Analogs of Adenosine Triphosphate Hans G. MANNHERZ, Helga BREHME, and Uli LAMP
Max-Planck-Institut fur Medizinische Forschung, Abteilung Biophysik, Heidelberg
(Received May 16, 1975)
A number of ATP analogs were tested for their ability to depolymerize F-actin and interact with it. It was found that those analogues which are hydrolysable are incorporated into the F-actin polymer whereas the non-hydrolysable ones are displaced from the nucleotide binding site during the poly- merization process.
The thin filament of the striated muscle cell consists of up to 60% of actin, which in the globular form has a molecular weight of 42000, according to the sequence data of Elzinga [l]. G-actin is only stable at low ionic strength and contains either triphosphate or diphosphate nucleotides firmly bound in a 1:l molar ratio. After raising the ionic strength by addi- tion of monovalent or divalent metal ions, G-actin is able to polymerize to a double-stranded helical polymer, F-actin, thereby hydrolysing the bound nucleotide ATP to ADP, which remains bound to the polymer. Polymerization of G-actin to F-actin is also possible when ADP is bound or even if no nucleotide is bound at all [2,3]. The rate of polymerization, however, is increased when ATP is bound, but appreciably decreased when ADP is the nucleotide or no nucleotide is bound at all [4]. During muscle contraction, F-actin interacts with myosin, thereby increasing the steady-state rate of the Mg2+-dependent ATP hydrolysis by myosin up to 200-fold, whereas G-actin is believed not to be able to activate the myo- sin ATPase as F-actin does [ 5 ] . Because of this, one speculates that during polymerization actin either undergoes a conformational change or that more than one actin monomer form the active site along the polymer.
Abbreviations. AMP-P(NH)P, adenosine 5‘-(,L?,y-imino)triphos- phate; AMP-PP(S), adenosine 5’-(3-thio)triphosphate; AMP(CH,)- PP, adenosine 5’-(cc,j-methylene)triphosphate ; AMP-P(CH,)P, ade- iiosine 5‘-(j,y-methylene)triphosphate; AMP-PP(CH,), adenosine 5’-triphosphate y-methyl ester. G-actin, globular actin, filamentous actin.
Enzymes. Myosin ATPase or ATP phosphohydrolase (EC 3.6.1.3) ; pyruvate kinase or ATP : pyruvate phosphotransferase (EC 2.7.1.40); lactate dehydrogenase or L-lactate : NAD oxido- reductase (EC 1.1.1.27).
The polymerization process is thought to be trig- gered by metal ions and to be facilitated by the hydro- lysis of bound ATP to ADP and Pi. Therefore, a comparison of the effects of ATP analogs which are hydrolysed by actin during polymerization to non- hydrolysable ones should give information about the correlation of ATP hydrolysis and rate of polymeriza- tion, assuming that G-actin can bind ATP analogs at all. We have depolymerized F-actin to G-actin in the presence of the following ATP analogs: AMP-
(CH2)PP and AMP-PP(S). Among those analogs AMP-P(CH,)P and AMP-PP(CH3) are certainly not hydrolysable. After addition of 1 mM MgCl, or 0.1 M KC1 the repolymerization of NTP . G-actin was monitored by measuring either the increase in viscosity or the ability to activate the ATPase of myosin sub- fragment 1. The latter parameter appears to be well justified by the now generally accepted view that G-actin itself does not accelerate the ATP-splitting rate of myosin subfragment 1 at low concentrations (0.1 mgiml), so that the increasing ability of actin after raising the ionic strength is directly correlated to the rate of polymerization (Offer et al. [5]) .
A preliminary report of these results has been given earlier [6] and quite recently Cooke and Murdoch [7] published results which are in general agreement to ours.
P(NH)P, AMP-P(CH,)P, AMP-PP(CH,), AMP-
MATERIALS AND METHODS
Actin Preparation
Actin was prepared from acetone powder according to Spudich and Watt [8] and in most cases further

110 Depolymerisation of F-Actin to G-Actin
purified on a Sephadex G-200 column also to eliminate excess salt. Purity was checked by electrophoresis on 10 % polyacrylamide gels in the presence of 0.1 % sodium dodecylsulfate as described by Weber and Osborn [9], and only tropomyosin-free preparations were used.
Myosin Subjiagment 1
Subfragment 1 was obtained from the papain digest of soluble rabbit back muscle myosin according to Lowey et al. [lo] after chromatography on a DEAE- cellulose column.
Protein
Protein concentration was determined by measur- ing the absorbance using A'% = 6.6 at 290nm for actin and A'" = 7.6 at 280 nm for subfragment 1.
Measurement of ATPase of Myosin Subfragment 1
The Mg2+ -dependent subfragment 1 ATPase and its activation by actin was measured by following the oxidation of NADH to NAD+ at 340 nm using a coupled enzyme system as described by Trentham et al. [Ill. In this system the ADP produced is re- phosphorylated by pyruvate kinase forming pyruvate from phosphoenolpyruvate, which is reduced by lactate dehydrogenase with concurrent oxidation of NADH to NAD'. The reaction was carried out in a I-ml quartz cuvette containing 0.1 mg subfragment 1, 1 mM MgATP, 50 mM KC1, 0.1 mg lactate dehydro- genase and pyruvate kinase, 2.5 mM phosphoenol- pyruvate and 0.8 mM NADH to which various amounts of actin could be added to give a final volume of 1 ml. In those experiments where the rate of re- polymerization of the various NTP . G-actins was followed directly the reaction solution contained 2 mM MgC1,. All enzymes and substances necessary for this test were purchased from Boehringer Mann- heim, Germany.
Viscosity Measurements
Viscometry was performed by using an Ostwald- type viscometer with a buffer outflow time of 56 s at 25 "C.
Analytical Ult racen tr ifuga t ion
For determination of the sedimentation constant of AMP-P(NH)P . G-actin a Beckman model E ana- lytical ultracentrifuge equipped with a photoelectric scanner was used. Samples of various NTP-G-actin preparations were run in a 4-hole rotor at 42000 rev./ min at 20 "C. Thus the sedimentation velocity of three
different actin preparations could be compared under identical conditions.
ATP Analogs
AMP-P(CH,)P and AMP(CH,)PP were purchased from Miles Biochemicals Lab. U.S.A. AMP-P(NH)P was obtained from Boehringer Mannheim, Germany. AMP-PP(S) was prepared according to Goody and Eckstein [l2] and AMP-PP(CH,) according to Goody (unpublished) ; both substances were generous gifts of Dr R. S. Goody. AMP(CH,)P[32P]P was prepared as described by Glynn and Chapel1 [14] for ATP. [3H]AMP-PP(S) was prepared according to Goody et al. [15] and [,H]AMP-P(NH)P was prepared by making use of the pyrophosphate exchange reaction catalysed by lysine-tRNA aminoacyltransferase (puri- fied by R. M. Dittgen) [16] starting from [3H]ATP and AMP-P(NH)P. Both substances were generous gifts of Dr R. S. Goody. [3H]ATP was obtained from New England Nuclear Comp. U.S.A.
Ail radioactive labelled nucleotides were purified by chromatography on DEAE-cellulose and their purity was checked by thin-layer chromatography on polyethyleneimine-impregnated cellulose plates ob- tained from Machery and Nagel, Germany, after development in 0.75 M KH2P04 pH 3.4.
Radioactivity of the separated nucleotides was determined in a Packard Tri-Carb scintillation counter, after being cut into small strips and eluted with 2 ml 2 N HC1 and addition of 2 ml Instagel scintillation mix from Packard Comp., U.S.A.
RESULTS
Depolymerization and Repolymerization of Actin in the Presence of ATP Analogs
At low ionic strength and in the presence of 0.2 mM of all ATP analogs included in this study F-actin depolymerizes to G-actin as judged by measurement of the relative viscosity and the inability of ATP- analog . G-actin to activate the MgZ+-dependent ATPase of myosin subfragment 1. Furthermore, for some AMP-P(NH)P . G-actin and AMP(CH,)PP . G- actin preparations the sedimentation velocity was compared to that of ATP . G-actin as described in Methods and found to have identical values of 3 S. Complete depolymerization could normally be achiev- ed after incubating F-actin in a solution containing, besides 0.2 mM of the ATP analog in question, 2 mM Tris-HC1 buffer pH 8.2, 0.1 mM dithioerythritol and 0.1 mM CaC1, after 12 to 24 h stirring at 4 "C. A kinetic analysis of the rates of depolymerization for the various ATP analogs was not undertaken; how- ever, at 0.2 mM analog or ATP and an actin concen- tration of 0.1 mM (4 mg/ml) virtually 100 % of the
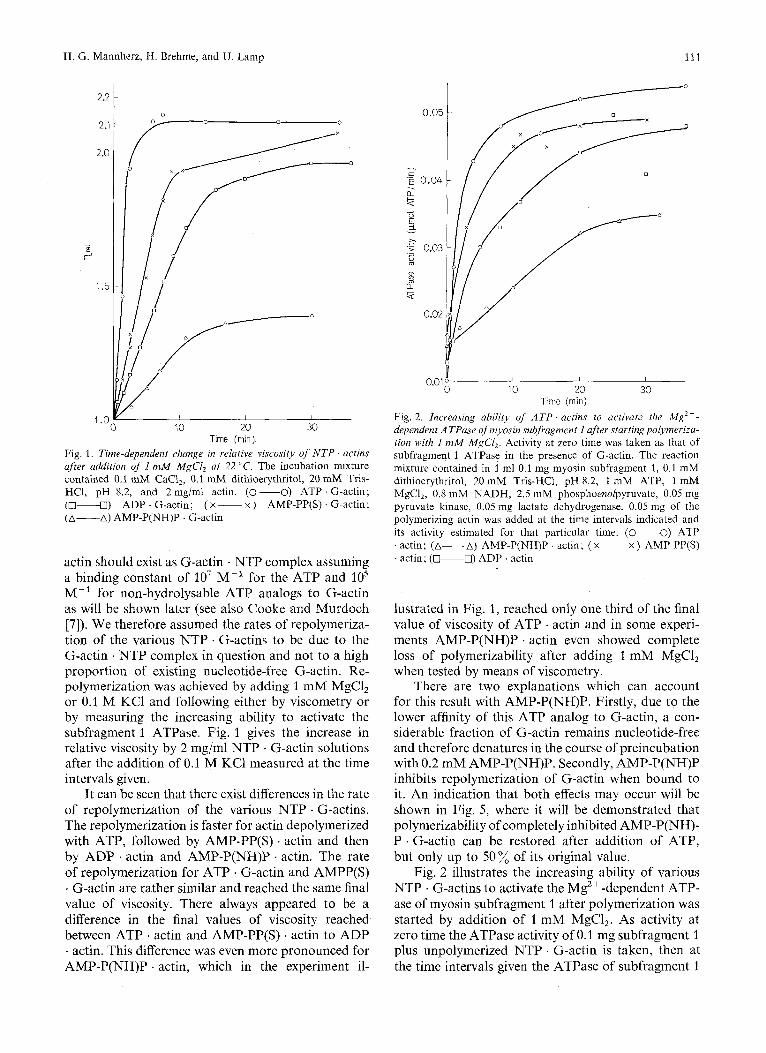
H. G. Mannherz, H. Brehme, and U. Lamp 111
2.2
2.1
2.0
- s 1.5
/ / /
I 1 I
0 10 20 30
Fig. 1. Time-dependent change in relative viscosity of NTP . actins after addition of I mM MgCl, at 22 "C. The incubation mixture contained 0.1 mM CaCI,, 0.1 mM dithioerythritol, 20 mM Tris- HC1, pH 8.2, and 2 mg/ml actin. (0-0) ATP G-actin; (0-0) ADP . G-actin; ( x ~ x ) AMP-PP(S) . G-actin; (A-A) AMP-P(NH)P . G-actin
I .u
Time (min)
actin should exist as G-actin . NTP complex assuming a binding constant of lo7 M-' for the ATP and lo5 M-' for non-hydrolysable ATP analogs to G-actin as will be shown later (see also Cooke and Murdoch [7]). We therefore assumed the rates of repolymeriza- tion of the various NTP . G-actins to be due to the G-actin . NTP complex in question and not to a high proportion of existing nucleotide-free G-actin. Re- polymerization was achieved by adding 1 mM MgCl, or 0.1 M KC1 and following either by viscometry or by measuring the increasing ability to activate the subfragment 1 ATPase. Fig. 1 gives the increase in relative viscosity by 2 mg/ml NTP . G-actin solutions after the addition of 0.1 M KC1 measured at the time intervals given.
It can be seen that there exist differences in the rate of repolymerization of the various NTP . G-actins. The repolymerization is faster for actin depolymerized with ATP, followed by AMP-PP(S) . actin and then by ADP . actin and AMP-P(NH)P . actin. The rate of repolymerization for ATP . G-actin and AMPP(S) . G-actin are rather similar and reached the same final value of viscosity. There always appeared to be a difference in the final values of viscosity reached between ATP . actin and AMP-PP(S) . actin to ADP . actin. This difference was even more pronounced for AMP-P(NH)P . actin, which in the experiment il-
0.05
- c 'E 0.04 a k . - e
0.01 1 I
0 10 20 30
Fig. 2. Increasing ability of ATP . actins to activure the Mg2+- dependent ATPase of myosin subfragment I after startingpolyrneriza- tion with I mM MgCl,. Activity at zero time was taken as that of subfragment 1 ATPase in the presence of G-actin. The reaction mixture contained in 1 ml 0.1 mg myosin subfragment 1, 0.1 mM dithioerythritol, 20 mM Tris-HC1, pH 8.2, 1 mM ATP, 1 mM MgCl,, 0.8 mM NADH, 2.5 mM phosphoenolpyruvate, 0.05 mg pyruvate kinase, 0.05 mg lactate dehydrogenase. 0.05 mg of the polymerizing actin was added at the time intervals indicated and its activity estimated for that particular time. (-0) ATP . actin; (A-A) AMP-P(NH)P . actin; ( x ~ x ) AMP-PP(S) . actin; (-0) ADP . actin
Time (min)
lustrated in Fig. 1, reached only one third of the final value of viscosity of ATP . actin and in some experi- ments AMP-P(NH)P . actin even showed complete loss of polymerizability after adding 1 mM MgCl, when tested by means of viscometry.
There are two explanations which can account for this result with AMP-P(NH)P. Firstly, due to the lower affinity of this ATP analog to G-actin, a con- siderable fraction of G-actin remains nucleotide-free and therefore denatures in the course of preincubation with 0.2 mM AMP-P(NH)P. Secondly, AMP-P(NH)P inhibits repolymerization of G-actin when bound to it. An indication that both effects may occur will be shown in Fig. 5, where it will be demonstrated that polymerizability of completely inhibited AMP-P(NH)- P . G-actin can be restored after addition of ATP, but only up to 50% of its original value.
Fig. 2 illustrates the increasing ability of various NTP . G-actins to activate the Mg2 + -dependent ATP- ase of myosin subfragment 1 after polymerization was started by addition of 1 mM MgCl,. As activity at zero time the ATPase activity of 0.1 mg subfragment 1 plus unpolymerized NTP . G-actin is taken, then at the time intervals given the ATPase of subfragment 1
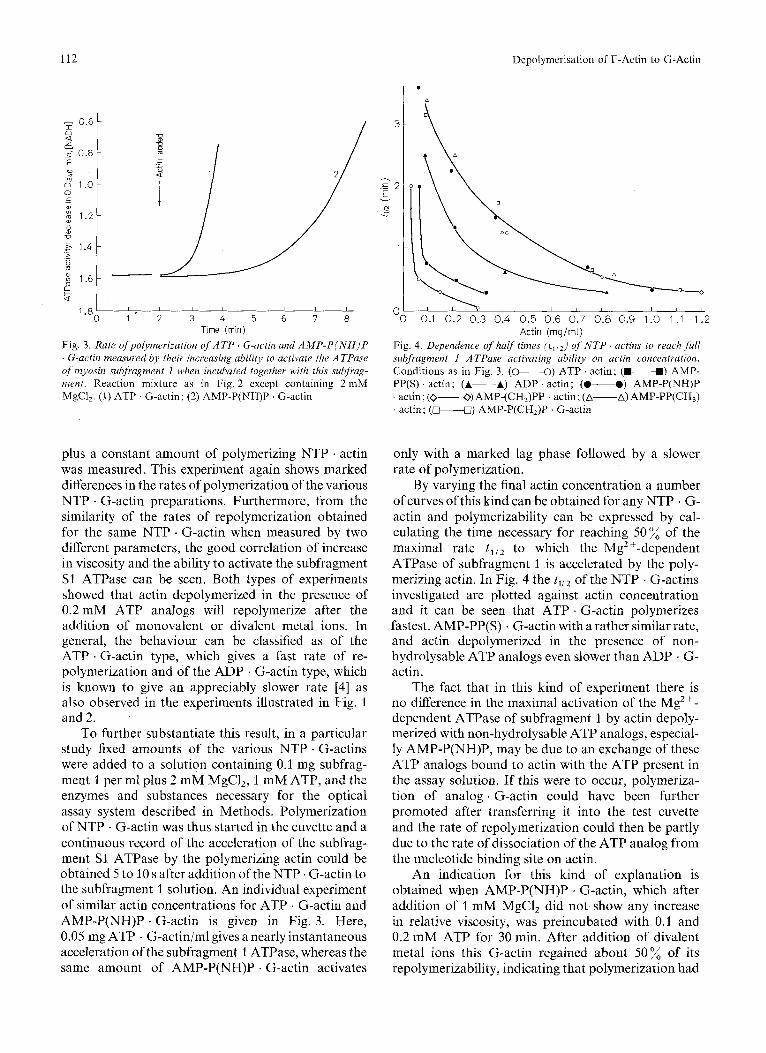
112
n L I I
Depolymerisation of F-Actin to G-Actin
a
3 -
1 -
I I I I 1 I I I
1 " 2 3 4 5 6 7 8 Time (min) Actin (rng/rnl)
Fig. 3 . Rate ofpolymerization of A T P G-actin and A M P - P ( N H ) P . G-actin measured by their increasing ability to activate the ATPase of myosin subfragment 1 when incubated together with this subfrag- ment. Reaction mixture as in Fig. 2 except containing 2 mM MgCl,. (1) ATP G-actin; (2) AMP-P(NH)P . G-actin
Fig. 4. Dependence of halftimes (illZ) of N T P . actins to reach full subfragment I ATPase activating ability on actin concentration. Conditions as in Fig. 3. (0-0) ATP . actin; (W-W) AMP- PP(S) . actin; (A-A) ADP . actin; (.---.) AMP-P(NH)P ' actin ; (0-0) AMP-(CHJPP . actin; (A-A) AMP-PP(CH,) . actin; (*U) AMP-P(CH,)P . G-actin
plus a constant amount of polymerizing NTP . actin was measured. This experiment again shows marked differences in the rates of polymerization of the various NTP . G-actin preparations. Furthermore, from the similarity of the rates of repolymerization obtained for the same NTP . G-actin when measured by two different parameters, the good correlation of increase in viscosity and the ability to activate the subfragment S1 ATPase can be seen. Both types of experiments showed that actin depolymerized in the presence of 0.2 mM ATP analogs will repolymerize after the addition of monovalent or divalent metal ions. In general, the behaviour can be classified as of the ATP . G-actin type, which gives a fast rate of re- polymerization and of the ADP . G-actin type, which is known to give an appreciably slower rate [4] as also observed in the experiments illustrated in Fig. 1 and 2.
To further substantiate this result, in a particular study fixed amounts of the various NTP . G-actins were added to a solution containing 0.1 mg subfrag- ment 1 per ml plus 2 mM MgCI,, 1 mM ATP, and the enzymes and substances necessary for the optical assay system described in Methods. Polymerization of NTP . G-actin was thus started in the cuvette and a continuous record of the acceleration of the subfrag- ment S1 ATPase by the polymerizing actin could be obtained 5 to 10 s after addition of the NTP . G-actin to the subfragment 1 solution. An individual experiment of similar actin concentrations for ATP . G-actin and AMP-P(NH)P . G-actin is given in Fig. 3. Here, 0.05 mg ATP . G-actin/ml gives a nearly instantaneous acceleration of the subfragment 1 ATPase, whereas the same amount of AMP-P(NH)P . G-actin activates
only with a marked lag phase followed by a slower rate of polymerization.
By varying the final actin concentration a number of curves of this kind can be obtained for any NTP . G- actin and polymerizability can be expressed by cal- culating the time necessary for reaching 50% of the maximal rate t I i z to which the Mg2+-dependent ATPase of subfragment 1 is accelerated by the poly- merizing actin. In Fig. 4 the t y , of the NTP . G-actins investigated are plotted against actin concentration and it can be seen that ATP . G-actin polymerizes fastest, AMP-PP(S) . G-actin with a rather similar rate, and actin depolymerized in the presence of non- hydrolysable ATP analogs even slower than ADP . G- actin.
The fact that in this kind of experiment there is no difference in the maximal activation of the Mg2+- dependent ATPase of subfragment 1 by actin depoly- merized with non-hydrolysable ATP analogs, especial- ly AMP-P(NH)P, may be due to an exchange of these ATP analogs bound to actin with the ATP present in the assay solution. If this were to occur, polymeriza- tion of analog . G-actin could have been further promoted after transferring it into the test cuvette and the rate of repolymerization could then be partly due to the rate of dissociation of the ATP analog from the nucleotide binding site on actin.
An indication for this kind of explanation is obtained when AMP-P(NH)P . G-actin, which after addition of 1 mM MgC1, did not show any increase in relative viscosity, was preincubated with 0.1 and 0.2 mM ATP for 30 min. After addition of divalent metal ions this G-actin regained about 50% of its repolymerizability, indicating that polymerization had
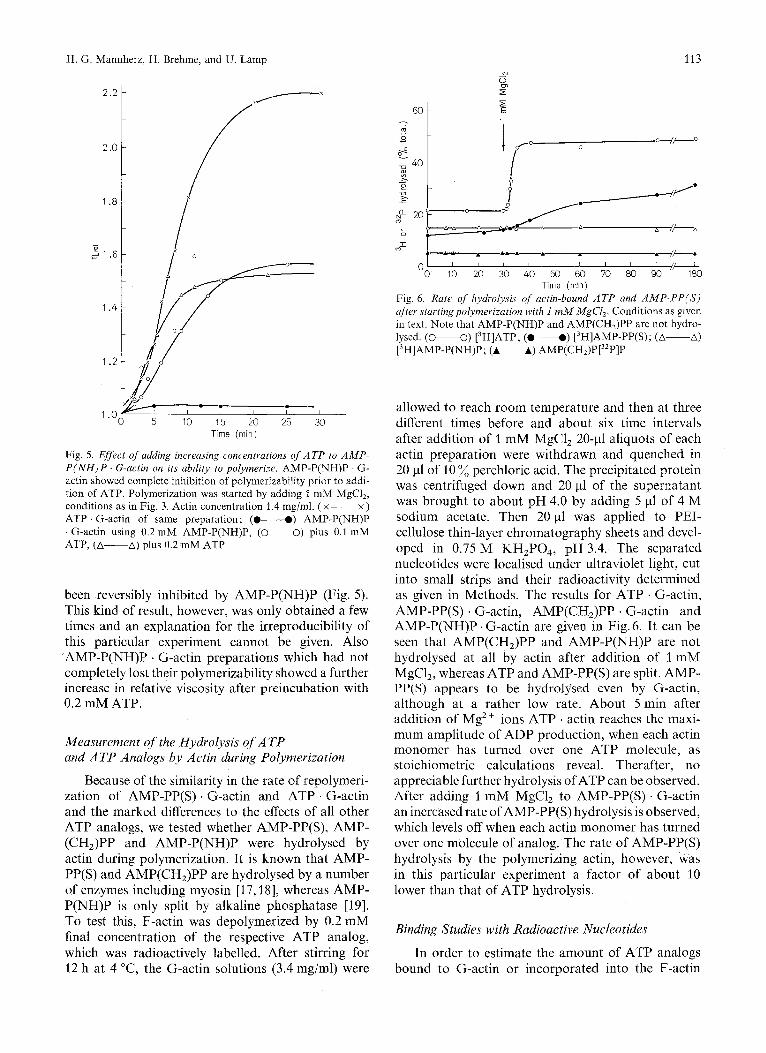
H. G. Mannherz, H. Brehme, and U. Lamp 113
-----.- 01 1'0 do ?A 4'0 ;o $0 ;o 8io do I180
Time (rnin) Fig. 6 . Rate of hydrolysis of actin-bound ATP and AMP-PP(S) after startingpolymerization with I mM MgC1,. Conditions as given in text. Note that AMP-P(NH)P and AMP(CH,)PP are not hydro- lysed. (0-0) [3H]ATP; (0-0) [3H]AMP-PP(S); (A-A) [3H]AMP-P(NH)P; (A-A) AMP(CHJP[32P]P
.-- I .o I I I
0 5 10 15 20 25 30 Time (rnin)
Fig. 5. Effect of adding increasing concentrations of ATP to AMP- P ( N H ) P . G-actin on its ability to polymerize. AMP-P(NH)P . G- actin showed complete inhibition of polymerizability prior to addi- tion of ATP. Polymerization was started by adding 1 mM MgCI,, conditions as in Fig. 3. Actin concentration 1.4 mg/ml. ( x ~ x ) ATP . G-actin of same preparation ; (0-0) AMP-P(NH)P . G-actin using 0.2 mM AMP-P(NH)P, (0-0) plus 0.1 mM ATP, (A-A) plus 0.2 mM ATP
been reversibly inhibited by AMP-P(NH)P (Fig. 5). This kind of result, however, was only obtained a few times and an explanation for the irreproducibility of this particular experiment cannot be given. Also AMP-P(NH)P . G-actin preparations which had not completely lost their polymerizability showed a further increase in relative viscosity after preincubation with 0.2 mM ATP.
Measurement of the Hydrolysis of A T P and A T P Analogs by Actin during Polymerization
Because of the similarity in the rate of repolymeri- zation of AMP-PP(S) . G-actin and ATP . G-actin and the marked differences to the effects of all other ATP analogs, we tested whether AMP-PP(S), AMP- (CHJPP and AMP-P(NH)P were hydrolysed by actin during polymerization. It is known that AMP- PP(S) and AMP(CH,)PP are hydrolysed by a number of enzymes including myosin [17,18], whereas AMP- P(NH)P is only split by alkaline phosphatase [19]. To test this, F-actin was depolymerized by 0.2 mM final concentration of the respective ATP analog, which was radioactively labelled. After stirring for 12 h at 4 "C, the G-actin solutions (3.4 mg/ml) were
allowed to reach room temperature and then at three different times before and about six time intervals after addition of 1 mM MgC1, 20-4 aliquots of each actin preparation were withdrawn and quenched in 20 pl of 10 % perchloric acid. The precipitated protein was centrifuged down and 20 pl of the supernatant was brought to about pH 4.0 by adding 5 pl of 4 M sodium acetate. Then 20 pl was applied to PEI- cellulose thin-layer chromatography sheets and devel- oped in 0.75 M KH,P04, pH 3.4. The separated nucleotides were localised under ultraviolet light, cut into small strips and their radioactivity determined as given in Methods. The results for ATP . G-actin, AMP-PP(S) . G-actin, AMP(CH,)PP . G-actin and AMP-P(NH)P . G-actin are given in Fig. 6. It can be seen that AMP(CH,)PP and AMP-P(NH)P are not hydrolysed at all by actin after addition of 1 mM MgCl,, whereas ATP and AMP-PP(S) are split. AMP- PP(S) appears to be hydrolysed even by G-actin, although at a rather low rate. About 5 min after addition of Mg2+ ions ATP ' actin reaches the maxi- mum amplitude of ADP production, when each actin monomer has turned over one ATP molecule, as stoichiometric calculations reveal. Therafter, no appreciable further hydrolysis of ATP can be observed. After adding 1 mM MgCl, to AMP-PP(S) . G-actin an increased rate of AMP-PP(S) hydrolysis is observed, which levels off when each actin monomer has turned over one molecule of analog. The rate of AMP-PP(S) hydrolysis by the polymerizing actin, however, was in this particular experiment a factor of about 10 lower than that of ATP hydrolysis.
Binding Studies with Radioactive Nucleotides
In order to estimate the amount of ATP analogs bound to G-actin or incorporated into the F-actin

114 Depolymerisation of F-Actin to C-Actin
polymer, F-actin was depolymerized in the presence of 0.2 mM rH]AMP-P(NH)P, [3H]AMP-PP(S) and [3H]ATP to give a final actin concentration of 4 mg/ ml. These actin solutions were divided into equal parts and one half was eluted on a Sephadex G-200 column equilibrated with a solution containing 0.1 mM CaCl,, 0.1 mM dithioerythritol, and 2 mM Tris-HC1 buffer, pH 8.2. To the remaining half 1 mM MgCl, was added, left for at least 2 h at room temperature and chromatographed on a Sepharose 4B column equilibrated with the same nucleotide-free solution supplemented with 1 mM MgCl,. In three experi- ments using different actin preparations it was found that after passage through a Sephadex G-200 column, G-actin was only up to 8 % saturated with AMP- P(NH)P, whereas under identical conditions it retained about 70% of the ATP bound initially. After poly- merization of AMP-P(NH)P . G-actin the amount of nucleotide bound decreased by a further factor of 10 after passage through Sepharose 4B, whereas the values obtained for actin polymerized in the presence of ATP tended to increase slightly (from 70 to SO%), but in no case was a decrease in nucleotide (ADP) content found. AMP-PP(S) . actin was tested in the same way and in all respects gave similar results to those obtained for ATP . actin. As equilibration of the columns with 0.2mM AMP-P(NH)P did not significantly alter the results obtained for AMP- P(NH)P. G-actin and its polymerized form, we assume that these results reflect the lower binding constants of AMP-P(NH)P to G-actin and F-actin. From the obtained data of the column experiments a tentative binding constant of ATP to G-actin can be estimated, resulting in about 1.5 . lo7 M-'. It is obvious, however, that this figure can be regarded only as an approxima- tion and further studies involving different approaches are needed to substantiate this figure. This calculation is in quite good agreement with recent determinations of the ATP-binding constant to actin [20,21]. From the same kind of experiment the binding constant of AMP-P(NH)P to G-actin can be provisionally cal- culated to be 6.5 . 104 M-'. A more accurate deter- mination of the binding constants of the ATP analogs used in this study is currently being undertaken (No- wak et d.). From this binding constant for AMP- P(NH)P, it can be calculated that under the conditions used up to 90 % of the G-actin contains bound AMP- P(NH)P. The binding of AMP-P(NH)P is further reduced when G-actin polymerized to F-actin, whereas it is known that there is an increase in binding constant of ADP to actin during the G - F transition [22].
It can not at the present time be decided whether this change in binding constant of AMP-P(NH)P to actin during polymerization, is a necessary prerequi- site of the polymerization process, i.e. whether AMP- P(NH)P has to be displaced from the binding site for polymerization to occur. The exact binding constant
of AMP-P(NH)P to F-actin however, should give information on whether the F-actin polymer obtained in the presence of AMP-P(NH)P contains bound
Therefore the following experiment was done to estimate the binding constant of AMP-P(NH)P to F-actin. Nucleotide-free F-actin was prepared by polymerization of AMP-P(NH)P . G-actin by adding 1 mM MgCl, and subsequently the free AMP-P(NH)P was removed by repeated treatments with Dowex 1 according to the method given by Asakura [20]. The F-actin was then centrifuged for 3 h at 100000 x g and the pellets were taken up in a nucleotide-free solution to give an F-actin concentration of 3 mg/ml. The amount of residual nucleotide was determined after perchloric acid precipitation of a 0.2-ml aliquot and found to be less than 1 pM by measuring the absorbance at 260 nm. Then 3-ml parts of this F-actin solution were incubated with increasing concentra- tions of AMP-P(NH)P ranging from 50 pM to 10 mM supplemented with [3H]AMP-P(NH)P and 32P04 as volume marker to give 12000 counts 32P min-l m1-l. The phosphate concentration employed as volume marker was about 10 nM and preliminary experi- ments had shown no binding to F-actin at that concen- tration. After 6 h of incubation at room temperature the F-actin samples were centrifuged at 100 000 x g for 3 h. The supernatant was carefully removed and the pellets were taken up in 2 rnl of 10% perchloric acid and both were counted for [3H]AMP-P(NH)P and 32P0,. There was no measurable increase in [3H]AMP-P(NH)P counts in the pellets up to 0.1 mM AMP-P(NH)P; at 1 mM the AMP-P(NH)P incorpor- ated into the polymer was about 1 % of the amount of actin monomers and at 10 mM about 18 %. Un- fortunately, in the experiment described higher con- centrations of AMP-P(NH)P could not be used because of dilution problems of the [3H]AMP-P(NH)P with unlabelled nucleotide. Therefore the binding constant of this analog to F-actin could not be deter- mined accurately, but using the amount of AMP- P(NH)P bound at a concentration of 10 mM, a binding constant of 10 M-' can be calculated. As polymeriza- tion was normally performed at 0.2 mM AMP- P(NH)P it appears quite unlikely that any of this analog could have been incorporated into the F-actin polymer. Cooke and Murdoch looked for F-actin- bound AMP-P(NH)P at 2.5 mM and also at this concentration the amount incorporated into the polymer cannot be higher than 2.5% of the actin Concentration. One therefore has to conclude that during the G - F transition of actin the binding constant of AMP-P(NH)P to it decreased appreciably, and the F-actin obtained at the concentrations of this analog employed in this study and in the work described by Cooke and Murdoch [7] is essentially nucleotide-free.
AMP-P(NH)P.

H. G. Mannherz, H. Brehme, and U. Lamp 115
DISCUSSION
The present study indicates the F-actin depoly- merizes in the presence of a number of ATP analogs carrying the modification at the polyphosphate chain. Binding of at least two analogs, namely AMP-PP(S) and AMP-P(NH)P, to G-actin could be proved. Furthermore, the idea that hydrolysis of ATP facilita- tes repolymerization of G-actin is supported by the observed correlation of fast repolymerization and concomitant hydrolysis of ATP or AMP-PP(S) and slow repolymerization to lack of hydrolysis of the remaining ATP analogs included in this study. It could be shown that AMP-P(NH)P is displaced from its binding site on G-actin during polymerization, and it appears probable that this occurs with all non- hydrolysable ATP analogs during the G - F transition of actin. The product of ATP or AMP-PP(S) hydro- lysis, ADP, is incorporated into the actin polymer. In this respect our results are in direct contradiction to those reported by Cooke and Murdoch [7], who reported an incorporation of AMP-P(NH)P into the F-actin polymer at a 1 : 1 molar ratio and an essentially similar rate of repolymerization of ATP . G-actin and AMP-P(NH)P . G-actin. They followed poly- merization by measuring the increase in absorbance at 232 nm, which was reported by Higashi and Oosawa [2] to occur during polymerization. However, from the original paper by Higashi and Oosawa [2] one can see that the parallelism between change in absorbance and polymerization is only complete for ATP . G- actin, whereas for ADP . G-actin a larger change in absorbance is associated with polymerization. Cooke and Murdoch [7] reported a difference in the observed rates of polymerization of AMP-P(NH)P . G-actin and ADP . G-actin using the optical method, whereas in our experiments the rates of polymerization of AMP-P(NH)P . G-actin and ADP . G-actin are simi- lar, taking the increasing ability of actin to activate the Mg2+-dependent ATPase of myosin subfragment 1 and its change of viscosity as parameters, which we regard as more direct ones. The second fundamental difference to Cooke and Murdoch’s report [7] con- cerns the incorporation of AMP-P(NH)P into the F-actin polymer and appears easier to resolve. They polymerized AMP-P(NH)P . G-actin in the presence of a 60-fold excess of AMP-P(NH)P over actin, but apparently did not correct for free AMP-P(NH)P in solution by using a volume marker. Therefore, we assume that in their experiment the centrifuged pellet still contained enough free AMP-P(NH)P to produce the result they described.
The F-actin polymer obtained after polymeriza- tion in the presence of AMP-P(NH)P is nucleotide-free, but in many respects does not differ from nucleotide- containing (ADP) F-actin. Preliminary electron micro- scopical investigations revealed no difference in their
structural appearance and their ability to form para- crystals in the presence of 20 mM MgCl, of F-actin formed after polymerization from ATP . G-actin or AMP-P(NH)P . G-actin. This study, however, has shown that the hydrolysis of bound ATP by actin during polymerization guarantees a fast rate of this process and furthermore the speed of ATP turnover by actin might provide a mechanism which allows only the formation of polymers of definite length.
The extremely low binding constant of AMP- P(NH)P to F-actin raises the suspicion that F-actin polymers cannot be formed as long as they contain a bound triphosphate nucleotide. Therefore if some way could be found to bind a non-hydrolysable ATP analog to G-actin, which is not displaced after the addition of 1 mM MgCl,, inhibition of the G-F transition of actin should be achieved.
In fact, we observed in some of our experiments that actin depolymerized with AMP-P(NH)P exhibited complete inhibition of repolymerization after raising the ionic strength as judged from viscometry or the lack of activation of the ATPase of subfragment S1. At present we cannot decide whether in those cases the lack of repolymerization was due to denaturation of the AMP-P(NH)P . G-actin or a real effect of this ATP analog. In several cases, however, the activity of ‘inhibited’ AMP-P(NH)P ‘ G-actin could be restored partially be exchanging the ATP analog against ATP as described earlier and shown in Fig. 5 . An investiga- tion into this aspect of the interaction of ATP analogs with actin is presently being undertaken.
We thank Dr S. Morris for performing the elctron microscopy for us and Dr A. G. Szent-Gyorgyi for advice with the actin prep- arations. For reading the manuscript and helpful discussions we thank Dr R. S. Goody.
REFERENCES
2. 3.
4.
5.
6.
7.
8. 9.
10.
11.
12.
Elzinga, M. & Collins, J. H. (1972) Cold Spring Harbor Symp.
Higashi, S . & Oosawa, F. (1965) 1. Mol. Bid. 12, 843-865. Kasai, M., Nakano, E. & Oosawa, F. (1965) Biochim. Biophys.
Hayashi, T. & Rosenbluth, R. (1960) Biol. Bull. (Woods Hole)
Offer, G., Baker, H. & Baker, L. (1972) J . Mol. Biol. 66, 435-
Brehme, H., Goody, R. S. & Mannherz, H. G. (1973) Hoppe
Cooke, R. & Murdoch, L. (1973) Biochemistry, 12, 3927-
Spudich, J. A. &Watt, S. (1971) J . Bid. Chem. 246,4866-4871. Weber. K. & Osborn, M. (1969) J . Biol. Chem. 244,4406-4412. Lowey, S., Slayter, H. S., Weeds, A. G. & Baker, H. (1969)
Trentham, D. R., Bardsley, R. G., Eccleston, J. F. & Weeds,
Goody, R. S. & Eckstein, F. (1971) J . Am. Chem. Soc. 93,
Quant. Bid. 37, 1-7.
Arta, 94,494- 503.
119, 290 - 294.
444.
Seyler’s Z. Physiol. Chem. 354, 234.
3932.
1. Mol. Bid. 42, 1 - 29.
A. G. (1972) Biochem. J . 126, 635-644.
6252 - 6257.

116 H. G. Mannherz, H. Brehme, and U. Lamp Depolymerisation of F-Actin to G-Actin
13. Reference deleted. 14. Glynn, I. M. & Chapell, J. B. (1964) Biochem. J . 90, 147- 150. 15. Goody, R. S., Eckstein, F. & Schirmer, R. H. (1972) Biochim.
Biophys. Acta, 276, 157- 161. 16. Leberman, R., v. Schutz, H., Dittgen, R. & Ulniar, G. (1973)
Anal. Biochem. 51, 111-115. 17. Bagshaw, C. R., Eccleston, J. F., Trentham, D. R., Yates,
D. W. & Goody, R. S. (1972) Cold Spring Harbor Symp. Quant. Biol. 37, 127- 135.
18. Mannherz, H. G., Barrington Leigh, J., Holmes, K. C. & Rosenbaum, G. (1973) Nat. New Biol. 241, 226-229.
19. Yount, R. G., Babcock, D., Ballantyne, W. & Ojala, D. (1971) Biochemistry, 10, 2484- 2489.
20. Bender, N. & Rack, M. (1974) Abstr. Commun. 9th Meet. Fed. Eur. Biochem. Soc. s 1 c 1.
21. Thames, K. E., Cheung, H. C. & Harvey, S. C. (1974) Biochem. Biophys. Res. Commun. 60, 1252- 1257.
22. Grubhofer, V. N. & Weber, H. H. (1961) 2. Naturfousch. 16b, 435-444.
H. G. Mannherz, H. Brehme, and U. Lamp, Abteilung Biophysik, Max-Planck-Institut fur Medizinische Forschung, D-6900 Heidelberg, JahnstraDe 29, Federal Republic of Germany


![Review Actin-targeting natural products: structures ... · actin-binding proteins actively break or ‘sever’ actin filaments [e.g. actin-depolymerizing factor (ADF) and cofilin]](https://img.pdfslide.net/doc/110x75/5f0f85bd7e708231d44494d0/review-actin-targeting-natural-products-structures-actin-binding-proteins-actively.jpg)


























