Embed Size (px)
Citation preview

Universidade Federal de SergipeCentro de Ciências Exatas e Tecnologia
Núcleo de Pós-Graduação em Física
Determinação de estrutura local em cristais dealta simetria dopado com íon Eu3+
por
Heveson Luís Lima de Matos
São Cristóvão, 29 de Janeiro de 2014

Heveson Luís Lima de Matos
Determinação de estrutura local em cristais de altasimetria dopado com íon Eu3+
Dissertação de mestrado apresentada ao Núcleode Pós-Graduação em Física da Universidade Fe-deral de Sergipe, como parte dos requisitos paraobtenção do título de mestre em física
Universidade Federal de Sergipe
Centro de Ciências Exatas e Tecnologia
Núcleo de Pós-Graduação em Física
Orientador: Prof. Dr. Marcos Antonio Couto dos Santos
São Cristóvão
29 de Janeiro de 2014

Heveson Luís Lima de MatosDeterminação de estrutura local em cristais de alta simetria dopado com íon Eu3+/
Heveson Luís Lima de Matos. – São Cristóvão, 29 de Janeiro de 2014-?? p. : il.(alguma color.); 30 cm.
Orientador: Prof. Dr. Marcos Antonio Couto dos Santos
Dissertação de Mestado – Universidade Federal de SergipeCentro de Ciências Exatas e TecnologiaNúcleo de Pós-Graduação em Física, 29 de Janeiro de 2014.
1. Palavra-chave1. 2. Palavra-chave2. I. Orientador. II. Universidade Federal deSergipe. III. Núcleo de Pós-Graduação em Física. IV. Título
CDU 02:141:005.7

Dedico este trabalho as mulheres da minha vida.

Folha de aprovação
Esta dissertação foi julgada adequada como TRABALHO DE MESTRADO no curso demestrado em física, e aprovada em sua forma final pela banca examinadora.
Banca Examinadora:
Prof. Dr. Marcos A. Couto dos Santos
Prof. Dr. Milan Lalic
Prof. Dr. Hélio Anderson Duarte
São Cristóvão, 29 de janeiro de 2014

AgradecimentosAgradeço primeiramente a Deus pela dádiva da vida, do saber e da inteligência. Aos meus pais
Maria e Joildo, não só pela vida, mas pela dedicação e paciência, por terem me formado como
pessoa, por todos os princípios que me ensinaram. Agradeço também à minha querida noiva Max-
sineide pela dedicação, apoio e paciência; por me tolerar durante todos esses anos diante dos meus
esquecimentos constantes. Ao meu orientador professor Marcos Couto pelo incetivo, competên-
cia e paciência no decorrer desses anos de trabalho. Aos meus irmãos Solange, Silmara, Junior,
Heverton e Jefferson por tudo que aprendi com eles e por todo apoio concedido. Aos colegas de
pesquisa Yuri, Adelmo e Ricardo pelas discussões constantes. Aos professores do departamento de
física Mário Everaldo, Francisco, Gêrson, Nelson, Edson e Osmar pelo conhecimento sólido ad-
quirido nas disciplinas. Aos colegas Cledison e Thomas pelas boas discussões sobre física quântica
e estado sólido. Aos amigos do CAFIS pelas diversas descontrações. Aos colegas de sala Socorro,
Maria, Cleverton, Coutinho, Afrânio e Daniela. Aos amigos Mário César, Jonathan e Octávio. Ao
secretário e coordenador do Núcleo de Pós-graduação, Álvaro e Ronaldo, pela atenção e disponibi-
lidade sempre com todos. Ao CNPq, a agência de financiamento deste trabalho. Por fim, agradeço
a todos a quem pude ensinar algo, pois com eles aprendi muito; afinal de contas, de nada vale ter
conhecimento e não disseminá-los.

ResumoUm estudo da estrutura local do sítio luminescente do íon Eu3+ com alta simetria em cristais foi realizado
utilizando a teoria de campo cristalino através do método dos vizinhos equivalentes (MENN). Os cristais
estudados foram: M2O3, SnO2, BaLiF3 e XMgF3 dopados com baixíssimas concentrações do íon Eu3+. A
partir das equações dos parâmetros de campo cristalino, do desdobramento máximo do multipleto 7F1 e do
equilíbrio eletrostático do sítio luminescente foi possível fazer previsões da distância interatômica entre os
íons Eu-O no sítio luminescente do íon Eu3+. Para o cristal M2O3, o recobrimento entre os orbitais 4f e 2p
no intervalo entre 0.07≤ρj≤0.1 e o fator de carga entre 0.55≤gj≤0.9 apresentaram as melhores previsões
para a indicação da estrutura local do sítio luminescente. A neutralidade eletrostática do sítio luminescente
foi satisfeita com o MENN. O Batista-Longo Improved Model (BLIM) foi utilizado como comparativo para
mostrar que a carga do íon Eu3+ pode ser maior que sua própria valência. As previsões dão indicação de
uma simetria pontual S6 ou D3d. Para o óxido SnO2 dois conjuntos de fatores de cargas foram utilizados.
Ambos foram relacionados pela equação do equilíbrio eletrostático do sítio luminescente. Uma constante de
proporcionalidade P=1.0029 relacionou as distâncias interatômicas não equivalentes. O recobrimento entre
os orbitais 4f e 2p no intervalo entre 0.05≤ρj≤0.1 e o fator de carga entre 0.5≤gj≤0.75 apresentaram as
melhores previsões para a indicação da estrutura local do sítio luminescente. As previsões indicam que o íon
Eu3+ substitui o íon Sn4+ em uma simetria pontual C2h ou D2h. Para os cristais BaLiF3 e XMgF3 é suge-
rido que o íon Eu3+ ocupe uma simetria mais baixa D4d com número de coordenação 8. O desdobramento
experimental foi reproduzido com gj = 0.402 e ρj = 0.05. O sinal do B20 foi reproduzido. Este estudo
foi feito baseado no espectro de emissão do íon Eu3+ e de cálculos de estrutura local baseado em dinâmica
molecular. As previsões da distância interatômica Eu-PV e simetria local do sítio luminescente estão em
bom acordo com dados experimentais.
Palavras chaves: cristais, lantanídeos, sítio luminescente, teoria de campo cristalino, espectro de emissão,
simetria, fator de carga.

AbstractA study of the local structure of the Eu3+ ion with high symmetry in crystals was carried out using the crystal
field theory by the method of nearest neighbours (MENN). The crystals studied were: M2O3, SnO2, BaliF3
and XMgF3 doped with very low concentrations of Eu3+ ion. From equations of the crystal field parameters,
the maximum splitting of 7F1 multiplet and the electrostatic equilibrium of luminescent site was possible
to predict the interatomic distance Eu-O ions in the luminescent site. For the M2O3 crystal, the overlap
between the 4f orbitals and 2p in the range 0.07≤ρj≤0.1 and the charge factor between 0.55≤gj≤0.9 gave
better predictions for the indication of the local structure of luminescent site. The electrical neutrality of the
luminescent site was satisfied. The Batista-Longo Improved Model (BLIM) was used to show that the Eu3+
ion charge may be greater than its own valence. The predictions give an indication of a point symmetry S6
or D3d. For the SnO2 oxide two sets of charge factors were used. Both were related by the equation of the
electrostatic equilibrium of luminescent site. A P = 1.0029 proportionality constant related non equivalent
interatomic distances. The overlap between the 4f orbitals and 2p in the range 0.05≤ρj≤0.1 and the charge
factor 0.5≤gj≤0.75 gave better predictions for the indication of the local structure of luminescent site. The
predictions indicate that the Eu3+ ion replaces the Sn4+ ion in a point symmetry C2h or D2h. For the BaliF3
and XMgF3 crystals is suggested that the Eu3+ ion occupies a lowest symmetry D4d with coordination num-
ber eight. The experimental splitting was reproduced with gj = 0.402 and ρj = 0.05. The signal of the B20
was reproduced. This study was done based on the emission spectrum of Eu3+ ion and structure calculations
based on molecular dynamics. The predictions of the Eu-PV interatomic distance and local symmetry of the
luminescent site is in good agreement with experimental data.
Keyword: crystals, lanthanides, luminescent site, crystal field theory, emission spectrum, symmetry, charge
factor.

SumárioSumário . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . i
Lista de ilustrações . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . iv
Lista de tabelas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . v
Lista de abreviaturas e siglas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . vi
Lista de Símbolos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . vii
I Introdução 1
1 Introdução . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
1.1 Considerações Iniciais . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
1.2 História dos Terras Raras . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
1.3 Propriedades Químicas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
II Fundamentação Téorica 9
2 Fundamentação Téorica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
2.1 O Hamiltoniano do Íon Livre . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
2.2 Potencial do Campo Cristalino . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
2.3 Parâmetro de Força e o Máximo Desdobramento do Campo Cristalino . . . . . . . 14
2.4 Posições dos Subníveis de Energia do 7F1 . . . . . . . . . . . . . . . . . . . . . . 16
2.5 Simetria . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
2.5.1 Elementos e Operações de Simetria . . . . . . . . . . . . . . . . . . . . . 17
2.5.2 Grupos de Simetria Pontual . . . . . . . . . . . . . . . . . . . . . . . . . . 18
2.5.3 Eixo de Simetria . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
2.5.4 Simetria e a Quebra de Degenerescência . . . . . . . . . . . . . . . . . . . 20

SUMÁRIO ii
III Modelos e Métodos 23
3 Modelos e Método . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
3.1 Modelo Eletrostático de Cargas Pontuais . . . . . . . . . . . . . . . . . . . . . . . 24
3.2 Modelo de Recobrimento Simples . . . . . . . . . . . . . . . . . . . . . . . . . . 25
3.3 Método dos Vizinhos Equivalentes . . . . . . . . . . . . . . . . . . . . . . . . . . 26
3.4 Metodologia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
IV Desenvolvimento 28
4 Estrutura Local do Íon Eu3+ no Óxido M2O3 . . . . . . . . . . . . . . . . . . . 29
4.1 Considerações Iniciais . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
4.2 Coordenadas Angulares do Sítio Luminescente . . . . . . . . . . . . . . . . . . . 29
4.3 Desdobramento Máximo do Multipleto 7F1 do Íon Eu3+ em Óxidos M2O3 . . . . . 31
4.4 Previsões da Distância Eu-O em Eu3+:M2O3 Através da Carga de Interação . . . . 32
4.5 Simetria do Íon Eu3+ em Óxidos M2O3 . . . . . . . . . . . . . . . . . . . . . . . 36
5 Estrutura Local do Íon Eu3+ no Óxido SnO2 . . . . . . . . . . . . . . . . . . . 39
5.1 Considerações Iniciais . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
5.2 Coordenadas Angulares do Sítio Luminescente . . . . . . . . . . . . . . . . . . . 39
5.3 Desdobramento Máximo do Multipleto 7F1 do Íon Eu3+ no Óxido SnO2 . . . . . 41
5.4 Previsões da Distância Eu-O em Eu3+:SnO2 Através da Carga de Interação . . . . 42
5.5 Simetria do Íon Eu3+ no Óxido SnO2 . . . . . . . . . . . . . . . . . . . . . . . . 47
6 Estrutura Local do Íon Eu3+ nos Cristais BaLiF3 e XMgF3(X=Cs, K, Na, Rb) . 50
6.1 Considerações Iniciais . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
6.2 Estrutura Local dos Cristais BaLiF3 e XMgF3(X=Cs, K, Na, Rb) . . . . . . . . . 50
6.3 Espectro de Emissão do Íon Eu3+ no Cristal BaLiF3 . . . . . . . . . . . . . . . . . 51
6.4 Estrutura Local do Íon Eu3+ em BaLiF3 e XMgF3(X=Cs, K, Na, Rb) . . . . . . . 54

SUMÁRIO iii
V Conclusões e Perspectivas 56
7 Conclusões . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57
8 Perspectivas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58
Referências . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 59
APÊNDICE A Demonstração do ∆E do Multipleto 7F1 . . . . . . . . . . . . . . 62
APÊNDICE B Demonstração do Parâmetro α . . . . . . . . . . . . . . . . . . . . 66
ANEXO A Diagrama de Níveis . . . . . . . . . . . . . . . . . . . . . . . . . . . . 69

iv
Lista de ilustraçõesFigura 1 – Evolução da produção de TR no mundo desde 1970. . . . . . . . . . . . . . . . 5
Figura 2 – Áreas requeridas ao Departamento Nacional de Produção Mineral para TR. . . . 6
Figura 3 – Densidade de probabilidade radial obtida por meio da teoria de Hartree para o
íon Gd+. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
Figura 4 – Molécula de água com ângulo de ligação HOH de 104.5 graus. . . . . . . . . . 20
Figura 5 – Representação esquemática de desdobramentos dos níveis 4f conforme o hamil-
toniano. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
Figura 6 – Distribuição de cargas ao redor do IC. . . . . . . . . . . . . . . . . . . . . . . 24
Figura 7 – Distribuição de cargas ao redor do IC segundo o SOM. . . . . . . . . . . . . . 25
Figura 8 – Ocupação do íon Eu3+ em dois sítios de simetria. . . . . . . . . . . . . . . . . 30
Figura 9 – Eixo de simetria adotado para obtenção das coordenadas angulares. . . . . . . . 31
Figura 10 – Previsões da distância Eu-O em função da carga efetiva no sítio Eu3+:M2O3. . 35
Figura 11 – Elementos de simetria contidos no sítio luminescente Eu3+:M2O3. . . . . . . . 37
Figura 12 – Eixo de simetria adotado para obtenção das (θ, φ) no sítio luminescente. . . . . 40
Figura 13 – Espectro de emissão do íon Eu3+ no sítio Eu3+:SnO2. . . . . . . . . . . . . . 41
Figura 14 – Previsões da distância Eu-O no sítio Eu3+:SnO2 com gEu = 3. . . . . . . . . . 44
Figura 15 – Previsões da distância Eu-O no sítio Eu3+:SnO2 com gEu = 3.3. . . . . . . . . 45
Figura 16 – Previsões da distância Eu-O no sítio Eu3+:SnO2 com gEu = 3.6. . . . . . . . . 45
Figura 17 – Previsões da distância Eu-O no sítio Eu3+:SnO2 com gEu = 3.9. . . . . . . . . 46
Figura 18 – Previsões da distância Eu-O no sítio Eu3+:SnO2 com gEu = 4.2. . . . . . . . . 46
Figura 19 – Elementos de simetria contidos no sítio luminescente Eu3+:SnO2. . . . . . . . 48
Figura 20 – Vizinhança do Bário no cristal BaLiF3. . . . . . . . . . . . . . . . . . . . . . . 51
Figura 21 – Espectro de emissão do íon Eu3+ em BaLiF3. . . . . . . . . . . . . . . . . . . 52
Figura 22 – Vizinhança do íon Eu3+ nos cristais BaLiF3/XMgF3. . . . . . . . . . . . . . . 54
Figura 23 – Representação esquemática das posições dos níveis em repartições simétricas. . 64
Figura 24 – Diagrama de níveis do íon Eu3+. . . . . . . . . . . . . . . . . . . . . . . . . . 69

v
Lista de tabelasTabela 1 – Configuração eletrônica dos elementos lantanídeos bem como dos cátions triva-
lentes. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
Tabela 2 – Raio iônico dos íons Ln3+ com CN 6 e 8. . . . . . . . . . . . . . . . . . . . . . 8
Tabela 3 – Coordenadas angulares dos PV no sítio Eu3+:M2O3. . . . . . . . . . . . . . . 30
Tabela 4 – CFP do sítio 2 do íon Eu3+ em M2O3. . . . . . . . . . . . . . . . . . . . . . . 38
Tabela 5 – Coordenadas angulares dos PV no sítio luminescente Eu3+:SnO2. . . . . . . . 40
Tabela 6 – Posição dos subníveis e desdobramento experimental dos níveis de energia. . . 42
Tabela 7 – CFP do sítio do íon Eu3+ em SnO2. . . . . . . . . . . . . . . . . . . . . . . . 49
Tabela 8 – Coordenadas angulares dos PV em relação aos íons Ba2+ e X+ em BaLiF3 e
XMgF3(X=Cs, K, Na, Rb). . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51
Tabela 9 – CE dos PV em relação ao íon Eu3+ nos compostos BaLiF3 e XMgF3. . . . . . 54
Tabela 10 – CFP do sítio do íon Eu3+ em BaLiF3 e XMgF3. . . . . . . . . . . . . . . . . . 55

vi
Lista de abreviaturas e siglasTR Terras Raras
Ln Lantanídeo
PV Primeiros Vizinhos
TCC Teoria de Campo Cristalino
CC Campo Cristalino
IC Íon Central
Z Número Atômico
CE Coordenadas Esféricas
CN Número de Coordenação ( do inglês Coordination Number)
CFP Parâmetros de Campo Cristalino ( do inglês Crystal Field Parameters)

vii
Lista de símbolosBk
q Parâmetros de Campo Cristalino
Ckq Tensor Esférico de Racah
Ukq Operador tensorial irredutível
Nv Parâmetro de Força do Campo Cristalino
∆E Desdobramento de Energia
ρj Recobrimento entre os orbitais 4f e 2p
gj Fator de carga dos PV
−→Rj Posição dos PV
−→rj Posição dos elétrons 4f
2S+1Lj Representação espectroscópica dos níveis

"Se seus sonhos estão sobre as nuvens, não se preocupe, pois eles estão no lugar certo; agora
construa os alicerces...William Shakespeare"

Parte I
Introdução

2
1 Introdução1.1 Considerações Iniciais
O comportamento de íons lantanídeos trivalentes Ln3+ sempre fascinou os físicos da espec-
troscopia experimental ou da espectroscopia teórica. Uma das razões para isso é sua configuração
eletrônica e suas propriedades luminescentes em diferentes materiais. O uso de materiais (vidros,
cristais e complexos) como hospedeiros de espécies opticamente ativas são importantes devido às
aplicações em amplificação e fibras ópticas, óptica não linear e guias de ondas, laser e dispositivos
moleculares [1, 2, 3, 4, 5, 6, 7]. Íons lantanídeos (Ln3+) são candidatos interessantes para aplicação
óptica nas regiões UV próximo – Visível – IV próximo por terem transições radiativas nesta região
tão finas que podem ser interpretadas como monocromáticas [1, 6]. Materiais nanoestruturados
têm recebido extrema atenção, porque é possível produzir sistemas com alta eficiência quântica
do tamanho de nanômetros e, naturalmente, os dispositivos criados com estes sistemas poderão ter
aplicação nas áreas de saúde e em alta tecnologia.
Apesar de hoje em dia existir muito interesse científico em se estudar as propriedades desses
materiais, elas já são conhecidas há muitas décadas. Ainda na primeira década do século XX J.
Becquerel descobriu que em baixa temperatura o espectro de absorção dos íons Ln3+, em um meio
químico, consistia de um grande numero de linhas, assemelhando-se às raias do espectro atômico
[8].
Porém, somente em 1929 com o trabalho de Bethe e o conhecimento da mecânica quântica é
que se pôde compreender melhor o comportamento desses materiais. O modelo proposto por Bethe
para explicar o desdobramento dos níveis de energia dos íons Ln3+ é baseado no fato de que meios
químicos diferentes produzem desdobramentos diferentes. Bethe estimou a ordem de grandeza
da ação desse potencial sobre os elétrons 4f partindo de um modelo puramente eletrostático, no
qual os PV são abordados como cargas pontuais [9]. Entretanto, esta simplificação ocasionou
enormes discrepâncias nas previsões dos cálculos de interação quando comparados com resultados
experimentais.
Após o trabalho proposto por Bethe surgiram alguns trabalhos teóricos com uma série de infor-
mações espectroscópicas sobre os lantanídeos. Van Vleck (1937) discutiu o problema das intensi-
dades espectrais na região do visível contendo íons Ln3+. Para explicar as transições observadas em

Capítulo 1. Introdução 3
materiais contendo íons Ln3+ Van Vleck atribuiu suas origens, em parte, ao mecanismo vibrônico.
Além disso, resultados experimentais indicavam que as transições eletrônicas deveriam realmente
ocorrer entre os níveis de energia 4f . No entanto, os modelos teóricos na época mostravam que
essas transições f-f eram proibidas, segundo a regra de Laporte. Assim, era preciso a elaboração de
um novo modelo para explicá-las de forma satisfatória.
Nas últimas décadas alguns modelos paramétricos surgiram com intuito de descrever melhor
o comportamento de várias grandezas espectroscópicas. Entretanto, a introdução de muitos parâ-
metros não levava a nenhum ganho real[10]. Somente em 1982 Malta [11] propôs o modelo de
recobrimento simples (SOM, do inglês simple overlap model) puramente teórico com o objetivo
de descrever a interação dos íons Ln3+ com os seus PV. Como consequência deste modelo, várias
grandezas puderam ser calculadas satisfatoriamente tais como: parâmetros de campo cristalino,
desdobramento de níveis de energia e parâmetro de força do campo cristalino. Este modelo, ao
contrário dos modelos paramétricos, não necessita de altos custos computacionais para fazer boas
previsões.
Para a aplicação da TCC em materiais contendo íons Ln3+ é necessário que sejam conhecidas
as coordenadas espaciais do sítio luminescente. No entanto, os diagramas de raios-X não mostram
os sítios com baixa concentração e em sistemas cristalinos os lantanídeos normalmente têm bai-
xíssimas concentrações, por razões econômicas, de estabilidade e de eficiência quântica. Alguns
trabalhos fazem uso de simulação computacional, por meio de dinâmica molecular, Hartree Fock
entre outros, para a obtenção da estrutura local destes sítios luminescentes [12]. Entretanto, estes
softwares carregam um elevado custo computacional que, pode ser minimizado por meio de teorias
mais simples.
Assim, neste trabalho o interesse é a determinação da estrutura local do íon Eu3+ em sítios com
alta simetria, para sua série de sistemas (e.g. M2O3 (S6; M=Gd,Y,Lu,In,Sc)[13], BaLiF3(Oh)[14],
XMgF3(X=Na,K,Cs,Rb)(Oh), SnO2 (C2h) [15]), utilizando a TCC através do MENN, pois nestes
casos teremos tantos graus de liberdade quanto equações, que são o desdobramento de níveis com
J≤ 3/2 e o equilíbrio eletrostático do sítio luminescente.

Capítulo 1. Introdução 4
1.2 História dos Terras Raras
Alguns relatos indicam que o início da história dos elementos terras raras ocorreu em 1787,
com a descoberta de um mineral escuro, a iterbita ou gadolinita pelo químico Carl Axel Arrhe-
nius na pequena vila de Ytterby, na Suécia. Entretanto, existem outros relatos sobre a descoberta
desses elementos. Em 1794, Johann Gadolin obteve uma forma impura do óxido de Ítrio deste
mineral. Em seguida, outros elementos foram sendo descobertos no decorrer do tempo[16]. Uma
curiosidade sobre os íons TR é que muitos destes elementos foram descobertos na Suécia, mais es-
pecificamente na vila de Ytterby. Daí a explicação destes elementos serem nomeados como Yttrium
(Y), Ytterbium (Yb), Terbium (Tb) e Erbium (Er) [17].
No entanto, a expressão terras raras é imprópria, porque estes elementos foram inicialmente
conhecidos na natureza na forma de seus óxidos. Além da expressão “terras” ser imprópria para
denominar estes elementos, a expressão “raras” também não é apropriada, pois estes elementos são
abundantes na natureza (com exceção do promécio que não ocorre na natureza) do que muitos ou-
tros elementos. A razão desses elementos até hoje serem chamados de terras raras está relacionada
com questões históricas porque as áreas em que estes elementos foram descobertos não podiam ser
facilmente extraídas devido às condições economicamente desfavoráveis daquela região [16]. Anos
mais tarde, com o desenvolvimento da tecnologia as TR passaram a ganhar novos usos e, hoje em
dia, a gama de aplicações é muito abrangente.
Atualmente estima-se que no ano de 2011, as reservas totais de óxidos de TR (medidas e in-
dicadas) sejam cerca de 158,2 milhões de toneladas e que, até 2016 esse mercado deverá ser de
aproximadamente 258 milhões de toneladas. A figura 1, [18], mostra a evolução da produção de
TR no mundo, entre os anos de 1970 a 2007, sendo que houve um decréscimo da produção dos
EUA, e em outros países, e a China teve um aumento significativo nos últimos anos. Sendo que, a
China possui cerca de 50%, dos quais a maior parte encontra-se no depósito de Bayan Obo (bast-
naesita), na Mongólia, interior da China. A Rússia é apontada como o país que possui a segunda
maior reserva (18%), composta em sua maioria por loparita e apatita. Os EUA ocupam o terceiro
lugar, com 12,4% sendo que as maiores reservas de bastnaesita encontram-se em Mountain Pass
[18].
O Brasil embora não seja um produtor, têm grandes reservas de TR. Estima-se cerca de 31 mil
toneladas, o que representa menos de 1% do total das reservas mundiais [19]. Entretanto, segundo

Capítulo 1. Introdução 5
Figura 1 – Evolução da produção de TR no mundo desde 1970.
dados obtidos pelo serviço geológico dos EUA em 2010, o Brasil possui cerca de 52,6 milhões de
toneladas de TR, que não estão classificados. A figura 2, [18], mostra as áreas requeridas ao De-
partamento Nacional de Produção Mineral para TR como substância principal e como subproduto.
1.3 Propriedades Químicas
Na espectroscopia a expressão terras raras é usada para os elementos Sc (Z=21), Y(Z=39) e
a série que vai do lantânio(Z=57) ao lutécio (Z=71) no qual Z é o número atômico. E o termo
lantanídeo refere-se aos elementos da tabela periódica que vai desde o La[Z=57] ao Lu[Z=71]. Por
encabeçar a série, o lantânio gera o termo lantanídeo, que são os elementos do bloco f, 6o período
da tabela periódica, que possuem os orbitais 4f gradualmente preenchidos e aos quais este trabalho
será doravante dedicado. Apesar de o escândio e o ítrio terem número atômico significativamente
diferente, quando comparado com outros íons TR, sua inclusão na série é explicada com base nas
semelhanças de suas propriedades químicas, pois ambos podem apresenta-se no estado trivalente.
Além disso, o tamanho iônico do cério é de 0,81Å, o que de fato é razoável, uma vez que o último
íon da série dos lantanídeos, Lu3+ têm raio atômico de 0,85Å[20].
Os lantanídeos neutros têm características semelhantes a do gás nobre xenônio [Xe], com dois

Capítulo 1. Introdução 6
Figura 2 – Áreas requeridas ao Departamento Nacional de Produção Mineral para TR.
ou três elétrons no orbital 6s25d1 e uma distribuição parcial no orbital 4f, de acordo com Z. Alguns
destes átomos preferem ter um elétron na camada 5d, ao invés de pôr no orbital 4f. Esta família
de elementos (cuja estado iônico mais estável é 3+), com exceção do lantânio (4f 0) têm a seguinte
configuração eletrônica: [Xe]4fn5s25p65d0−16s2, no qual n é o número de elétrons no orbital
4f(n=0 até 14). Pode−se observar que os elétrons 4f são fortemente blindados pelos orbitais 5s2 e
5p6 mais externos. Isto faz com que a interação do campo cristalino com o meio químico seja fraca
ao ser comparada com as interações descritas pelo hamiltoniano do íon livre. A figura 3 ilustra a
densidade de probabilidade radial eletrônica (P 2(r)) para o íon gadolínio.
Todas os TR podem apresenta-se como cátions trivalentes e, este estado de oxidação é o
mais comum entre os TR devido à estabilidade termodinâmica. Alguns destes elementos podem
apresentar-se nos estados de oxidação 1+, 2+ e 4+. No entanto, estes íons são sempre menos es-
táveis que os cátions trivalentes. Íons divalentes e tetravalentes mais estáveis são formados por
elementos que apresentam a subcamada 4f vazia, semi-cheia ou totalmente preenchida, como por
exemplo, os íons Tb4+ têm configuração f7 e o Yb2+ têm configuração f14.
Do ponto de vista de luminescência, tanto o lantânio quanto o lutécio são opticamente inativos,
já que em um deles não há elétrons na camada 4f e o outro têm a camada 4f completa impossibili-
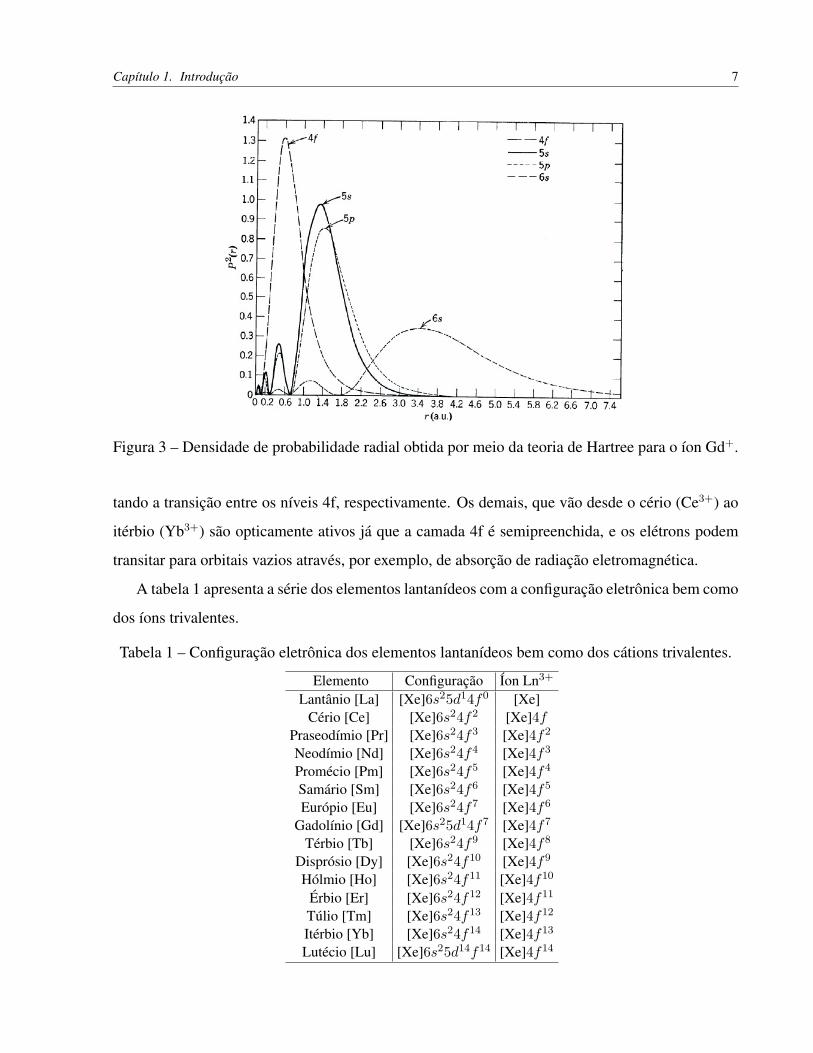
Capítulo 1. Introdução 7
Figura 3 – Densidade de probabilidade radial obtida por meio da teoria de Hartree para o íon Gd+.
tando a transição entre os níveis 4f, respectivamente. Os demais, que vão desde o cério (Ce3+) ao
itérbio (Yb3+) são opticamente ativos já que a camada 4f é semipreenchida, e os elétrons podem
transitar para orbitais vazios através, por exemplo, de absorção de radiação eletromagnética.
A tabela 1 apresenta a série dos elementos lantanídeos com a configuração eletrônica bem como
dos íons trivalentes.
Tabela 1 – Configuração eletrônica dos elementos lantanídeos bem como dos cátions trivalentes.
Elemento Configuração Íon Ln3+
Lantânio [La] [Xe]6s25d14f0 [Xe]Cério [Ce] [Xe]6s24f2 [Xe]4f
Praseodímio [Pr] [Xe]6s24f3 [Xe]4f2
Neodímio [Nd] [Xe]6s24f4 [Xe]4f3
Promécio [Pm] [Xe]6s24f5 [Xe]4f4
Samário [Sm] [Xe]6s24f6 [Xe]4f5
Európio [Eu] [Xe]6s24f7 [Xe]4f6
Gadolínio [Gd] [Xe]6s25d14f7 [Xe]4f7
Térbio [Tb] [Xe]6s24f9 [Xe]4f8
Disprósio [Dy] [Xe]6s24f10 [Xe]4f9
Hólmio [Ho] [Xe]6s24f11 [Xe]4f10
Érbio [Er] [Xe]6s24f12 [Xe]4f11
Túlio [Tm] [Xe]6s24f13 [Xe]4f12
Itérbio [Yb] [Xe]6s24f14 [Xe]4f13
Lutécio [Lu] [Xe]6s25d14f14 [Xe]4f14

Capítulo 1. Introdução 8
Os elementos TR apresentam outra característica bastante interessante chamado de contração
lantanídica, que consiste numa significativa diminuição do tamanho dos átomos e dos íons cau-
sado pelo aumento do número atômico Z. Esse fenômeno pode ser explicado através da blindagem
imperfeita de um elétron com outro no mesmo orbital. Cada átomo possui uma distribuição eletrô-
nica, de modo que, a adição de mais elétrons à subcamada 4f causará um aumento na carga efetiva,
e consequentemente, a redução do volume total da configuração 4fn. A tabela 2, [21], mostra a
diminuição do raio iônico dos elementos lantanídeos conforme o aumento do número atômico para
os íons de número de coordenação (CN) 6 e 8.
Tabela 2 – Raio iônico dos íons Ln3+ com CN 6 e 8.
Número Atômico Símbolo Raio Iônico (CN6) (Å) Raio Iônico (CN8) (Å)57 La 1.032 1.1658 Ce 1.010 1.14359 Pr 0.990 1.12660 Nd 0.983 1.10961 Pm 0.970 1.09362 Sm 0.958 1.07963 Eu 0.947 1.06664 Gd 0.938 1.05365 Tb 0.923 1.04066 Dy 0.912 1.02767 Ho 0.901 1.01568 Er 0.890 1.00469 Tm 0.880 0.99470 Yb 0.868 0.98571 Lu 0.861 0.977

Parte II
Fundamentação Téorica

10
2 Fundamentação Téorica2.1 O Hamiltoniano do Íon Livre
Segundo a mecânica quântica, o hamiltoniano do íon livre, HIL, é composto por uma parte
devido ao campo central, que fornece as energias dos baricentros das configurações eletrônicas,
H0, e por várias outras interações, que são comumente tratadas como efeito perturbativo na apro-
ximação de campo central. Entre estas interações a repulsão coulombiana e a spin-órbita são as
mais relevantes, embora, haja outras interações como, por exemplo, spin-spin e órbita-outra órbita,
que podem ser importantes em determinadas situações mais específicas. Assim, o hamiltoniano do
sistema é escrito como:
HIL = H0 +HRE +HSO (2.1)
O primeiro termo da equação 2.1 descreve o movimento dos elétrons sob a ação do núcleo
de carga Ze. O segundo termo trata da energia de interação entre pares de elétrons e o último
termo o acoplamento spin-órbita. É importante ressaltar que o termo de repulsão coulombiana en-
tre os elétrons faz com que cada elétron não se encontre em um potencial esfero-simétrico. Este
fato ocasiona uma mudança de energia para estados da mesma configuração eletrônica, e conse-
quentemente, a equação de Schrödinger de sistemas monoeletrônicos se diferencia da equação de
Schrödinger para átomos hidrogenoides [22, 23].
Na mecânica quântica a equação de Schrödinger não têm solução exata para sistemas com mais
de um elétron, pois o termo de repulsão coulombiana torna impossível a resolução do problema
analiticamente. Porém, existe atualmente na literatura métodos empíricos, que são capazes de
resolvê-la de forma aproximada. Geralmente os métodos empíricos mais utilizados na obtenção
dos autoestados e autovalores são: Hartree-Fock e DFT [22].
2.2 Potencial do Campo Cristalino
Além do hamiltoniano do íon livre existem outras interações que podem ser consideradas no
hamiltoniano total, H , a depender dos cálculos que estão sendo tratados. Quando o íon Ln é
inserido num meio químico, a próxima interação de maior relevância a ser considerada no H é

Capítulo 2. Fundamentação Téorica 11
a do campo cristalino, Hcc. Esta interação do Hcc é uma pertubação sofrida à nuvem eletrônica
dos elétrons 4f por meio de todos os elétrons do sistema, de maneira que, a simetria local do íon
Ln será imposta pelo meio químico, e sendo assim haverá quebra de degenerescência dos níveis2S+1LJ de acordo com a simetria local. Hcc é considerado como uma pertubação, devido à fraca
interação dos elétrons 4f com o meio químico, tal que, ao ser comparado com os demais termos do
hamiltoniano H0 > HRE > HSO > Hcc [24, 25]. O hamiltoniano do campo cristalino é definido
como:
Hcc =∑i,j
eqj
|ri − Rj|(2.2)
Na equação 2.2 qj é a carga do PV , e é a carga elementar, ri é o vetor posição dos elétrons 4f
e Rj é a distância entre o IC e os PV .
O hamiltoniano total do sistema H pode ser escrito em termos do hamiltoniano do íon livre
mais a contribuição do hamiltoniano do campo cristalino devido à pertubação.
H = HIL +Hcc (2.3)
Para os cálculos de parâmetros de campo cristalino só importa a parcela do hamiltoniano re-
ferente ao campo cristalino, pois esse termo possui uma componente esférica e componentes não
esféricas, sendo a última responsável pelos desdobramentos dos níveis de energia 2S+1LJ do íon
Ln3+. Utilizando o teorema da adição para harmônicos esféricos pode-se expandir o denominador
da equação 2.2 em uma soma infinita em l e m. Então, o denominador da equação 2.2 pode ser
escrito da seguinte forma:
1
|ri − Rj|=
∑l,m
(4π
2l + 1
)rl<rl+1>
Y ∗l,m(Ωj)Yl,m(Ωi) (2.4)
sendo Ωj = (θj, φj) e Ωi = (θi, φi) as respectivas coordenadas espaciais do IC e PV.
Da equação 2.4 fica evidente tanto a componente esférica e as não esféricas, mas também
a separação da parte radial e angular. A parte par (k,q) é responsável pela intensidade do campo
cristalino e a parte ímpar (t,p) pelas transições de dipolo elétrico induzido [26]. Como os elétrons da
configuração eletrônica 4f possuem os mesmos números quânticos [n,l], e as transições eletrônicas
ocorrem dentro do orbital 4f, então as funções de onda |Ψ′Aα⟩ e |ΨAα⟩ possuem mesma paridade.

Capítulo 2. Fundamentação Téorica 12
Então, os valores de l com paridade ímpar (t,p) se anula em todo espaço, resultando apenas a parte
par (k,q). Em compostos contendo íons Ln3+, dado o fato de que os orbitais 4f são contraídos e
têm uma extensão radial da ordem de 1Å, justifica-se considerar r< = ri. Escrevendo o harmônico
esférico Yk,q(Ωi) em termos do tensor esférico de Racah Ckq (Ωi), têm-se a seguinte equação:
Ckq (Ωi) =
√4π
2k + 1Yk,q(Ωi) (2.5)
Reescrevendo o hamiltoniano Hcc:
Hcc =∑k,q,i
γkq r
ki C
kq (Ωi) (2.6)
donde,
γkq =
∑j
√4π
2k + 1e2gj
Y ∗k,q(Ωj)
Rk+1j
(2.7)
A soma em k vai de k>0 e −k6q6k e gj é a valência dos PV . Note que, ao representar o po-
tencial de interação do campo cristalino em termos dos harmônicos esféricos, consegue-se separar
a contribuição do campo cristalino que pertence ao IC da parte do potencial do campo cristalino que
pertence aos PV. A dependência de γkq , está relacionada apenas com a vizinhança(PV) e rki C
kq (Ωi)
depende somente do IC. Uma vez que, conhecendo a estrutura cristalográfica do sistema, é possível
obter as coordenadas angulares Ωj = (θj, φj) e a distância entre o IC e os PV, a grandeza γkq pode
ser calculada.
A posição dos subníveis de energia devido à pertubação do campo cristalino podem ser calcu-
lados por meio da diagonalização da matriz do campo cristalino. |ΨAα⟩ descreve a base de estados
para calcular os elementos de matriz ⟨Ψ′Aα|Hcc|ΨAα⟩ [26, 27]. E Aα representa todo o conjunto de
bons números quânticos de uma configuração eletrônica 4fn.
Uma vez que a função de onda dos estados 4f depende da parte radial e da parte angular
podemos fazer uma separação de variáveis e escrever a função dos estados 4f da seguinte forma:
|ΨAα⟩ = |ri⟩|Φ⟩ (2.8)
|ri⟩ representa a parte radial de todos elétrons 4f e |Φ⟩ representa a função de onda angular dos
elétrons 4f , que está relacionada com a base de bons números quânticos (l,ml, s,ms).

Capítulo 2. Fundamentação Téorica 13
Utilizando as expressões 2.6 e 2.8 no elemento de matriz ⟨ΨAα|Hcc|ΨAα⟩ temos que:
⟨ΨAα|Hcc|ΨAα⟩ = ⟨ri|⟨Φ|∑k,q,i,j
γkq (j)r
ki C
kq (i)|ri⟩|Φ⟩ =
∑k,q,j
⟨rk⟩γkq (j)⟨Φ|
∑i
Ckq (i)|Φ⟩ (2.9)
Como os elétrons do IC estão todos no mesmo orbital 4f , podem ser considerados como equi-
valentes, e sendo assim o índice i da parte radial é removido na equação 2.9. Logo, o potencial
devido à pertubação do campo cristalino é escrito como:
Vcc =∑k,q,i
BkqC
kq (Ωi) (2.10)
Da equação 2.10 Bkq = ⟨rk⟩γk
q é o parâmetro de campo cristalino que é função apenas nas
coordenadas dos PV e o Ckq é o tensor esférico de Racah que opera nas coordenadas angulares do
elétron.
⟨rk⟩ é o valor esperado da posição dos elétrons 4f , o qual são obtidos através do método de
Dirac-Fock, e estão calculadas no trabalho de Freemann-Desclaux [28]. A parte angular repre-
sentada pelo operador tensorial de Racah pode ser calculada associando-se ao operador tensorial
irredutível unitário Ukq , o qual utiliza a representação dos símbolos 3j. Escrevendo o operador de
Racah em termos do operador unitário temos que:
⟨Φ|∑i
C(k)q (i)|Φ⟩ =
∑k,q
⟨Φ|U (k)q |Φ⟩⟨l∥C(k)∥l⟩ (2.11)
O primeiro termo do lado direito da equação é o operador tensorial irredutível unitário U(k)q , que
está tabelado no trabalho de Carnall e Crosswhite [29] e o segundo termo é o elemento de matriz
reduzido, que pode ser obtido através dos símbolos 3j desenvolvido por Wigner-Eckart, que pode
ser visto em [22]. O último termo pode ser reescrito como:
⟨l∥C(k)∥l⟩ = (−1)l[(2l + 1)(2l + 1)]0.5
l k l
−Mj q Mj
(2.12)
Examinando-se as regras de triangularidade contidas no elemento de matriz reduzido, conclui-
se que, os símbolos 3j será diferente de zero apenas no caso em que, −Mj + q +Mj = 0 e k≤2l,

Capítulo 2. Fundamentação Téorica 14
visto que para os elétrons 4f , l = 3, então 2.12 pode ser reescrita como:
⟨3∥C(k)∥3⟩ = (−1)3[(2·3 + 1)(2·3 + 1)]0.5
3 k 3
0 0 0
= −7
3 k 3
0 0 0
(2.13)
Portanto, conclui-se que, do ponto de vista de quebra de degenerescência apenas os parâmetros
de campo cristalino de posto k=2,4,6 serão considerados. Como discutido anteriormente, o valor
de k=0, corresponde a componente esférica do campo cristalino e, portanto, não contribui para
o desdobramento dos níveis de energia, visto que o parâmetro B00 desloca igualmente os estados
eletrônicos.
2.3 Parâmetro de Força e o Máximo Desdobramento do Campo
Cristalino
Ao estudar o efeito do campo cristalino em uma matriz hospedeira, vários são os parâmetros
considerados neste estudo. O número de parâmetros, usualmente, está restrito a uma dada simetria
local do sítio luminescente, por exemplo, em uma simetria C2h, há 9 parâmetros não nulos a serem
considerados. Baseado nisto, vários autores tentaram simplificar a descrição do campo cristalino,
reduzindo o número de parâmetros. Auzel e Malta [30] foram os primeiros a fazer essa simplifica-
ção, introduzindo o conceito de parâmetro de força do campo cristalino, Nv, que é uma grandeza
que caracteriza a intensidade do campo cristalino em qualquer tipo de simetria. O parâmetro de
força do campo cristalino Nv é definido em termos dos parâmetros de campo cristalino Bkq como:
Nv =
[∑k,q
(Bkq )
1/2
(4π
2k + 1
)]1/2
(2.14)
onde Nv é uma métrica, no qual representa a distância no espaço gerado pelos harmônicos esféricos
Y kq .
Auzel e Malta utilizando algumas aproximações, através do qual, assume-se, que os níveis de
energia estão igualmente espaçados e, desenvolvendo alguns cálculos (ver apêndice A), consegui-
ram relacionar o Nv com o desdobramento máximo do campo cristalino ∆E do multipleto 2S+1LJ ,

Capítulo 2. Fundamentação Téorica 15
dado por,
∆E =
[3g2a
g(ga + 2)(ga + 1)π
]1/2∏k
|⟨J∥∑i
Cki ∥J
′⟩|1/3
Nv (2.15)
Da equação 2.15 ga é a degenerescência efetivamente removida por meio do campo cristalino,
de acordo com a simetria local do sítio luminescente(g = ga para J par e g = 2ga pra J ímpar), e g
é a degenerescência total do multipleto 2S+1LJ .
Para o caso J=1 do multipleto 7F1 do íon Európio, não é difícil mostrar que (ver apêndice A), a
equação 2.15 se reduz a seguinte expressão:
∆E =
[gag
⟨7F1∥C2∥7F1⟩π(2 + α)
]1/2Nv (2.16)
A equação 2.16 descreve o máximo desdobramento do multipleto 7F1 do íon Európio, no qual
α representa o quanto o valor médio difere do nível intermediário, para o caso de J = 1, e seu
valor está compreendido entre 0≤α≤1. α é obtido a partir dos níveis de energia com J = 1. Esta
expressão foi inicialmente proposta por Malta [7] e é dado por:
α =Ea − (E< +∆E)
∆E/2(2.17)
Da equação 2.17, Ea é a energia do nível Stark intermediário no intervalo ∆E e E< é a energia
do nível Stark mais baixo. Entretanto, a forma de expressar a equação pra α, ainda é algo discutido
na literatura. A expressão mais recente é proposta por Couto [31], e é dado por:
α = ±√6Eb − (E< +∆E)
∆E/2(2.18)
Eb é a energia do baricentro dos níveis Stark. A equação 2.18 é obtida a partir de algumas manipu-
lações matemáticas (ver apêndice B).
O elemento de matriz reduzido expresso na equação 2.16 pode ser obtido por meio da relação
com o operador tensorial irredutível unitário, Uk, conforme descrito na seção 2.2. Alguns dos
valores dos elementos de matriz reduzido são dados por:
⟨3∥C2∥3⟩ = −14√105
≈−1.37 (2.19)

Capítulo 2. Fundamentação Téorica 16
⟨3∥C4∥3⟩ =√14√11
≈1.13 (2.20)
⟨3∥C6∥3⟩ = −70√3003
≈−1.28 (2.21)
2.4 Posições dos Subníveis de Energia do 7F1
Para o momento angular orbital total, J = 1, há três valores possíveis de mj , que podem ser
desdobrados por meio do efeito do campo cristalino. A remoção total ou parcial da degenerescência
está diretamente relacionado com a simetria local do sítio luminescente, uma vez que, o número de
níveis observados dará indicação da simetria local do íon Ln. Por exemplo, em sítios de alta simetria
(hexagonal,trigonal, tetragonal) a degenerescência não é totalmente removida, enquanto que, em
sítios de baixa simetria (ortorrômbica, monoclínica ou triclínica) a degenerescência é totalmente
removida. Segundo [24], para simetrias ortorrômbica, monoclínica ou triclínica a posição dos
subníveis de energia do 7F1, depois da quebra de degenerescência é descrito por um parâmetro
de campo cristalino adicional, B22 . Diagonalizando a matriz de energia encontram-se os seguintes
níveis de energia:
E|0⟩ =2√14
15U2B2
0 (2.22)
E|−1,1⟩ =−√14
15U2B2
0 +7√2
5√3U2B2
2 (2.23)
E|1,1⟩ =−√14
15U2B2
0 −7√2
5√3U2B2
2 (2.24)
|±1⟩ é uma abreviação para i√2(|−1⟩±|1⟩) e U2 corresponde ao primeiro termo da equação 2.11.
A partir do conjunto de equações 2.22,2.23 e 2.24 é possível obter a posição dos subníveis de
energia do 7F1 não degenerado, sendo que estas expressões dependem apenas dos parâmetros B20
e B22 . As posições dos subníveis de energia em relação ao baricentro está relacionado com o sinal
do parâmetro B20 . Considere os três subníveis de energia do 7F1 como não degenerado. Se o nível
|0⟩ está em uma energia mais alta do que os níveis |±1⟩, o sinal do parâmetro B20 é positivo, caso
contrário o sinal do parâmetro B20 é negativo.

Capítulo 2. Fundamentação Téorica 17
Para simetrias mais baixas do que a ortorrômbica ou para sítios com simetria distorcida o pa-
râmetro B21 é não nulo, e as equações 2.22,2.23 e 2.24 já não são mais válidas, uma vez que o
parâmetro B21 não é mais desprezível ao resolver o determinante secular da energia. Entretanto, se
a magnitude do B21≪B2
0 , ainda assim, é possível descrever as posições dos subníveis de energia
do 7F1, apenas com dois parâmetros B20 e B2
2 , e as equações 2.22,2.23 e 2.24 podem ser utilizadas
como uma boa aproximação [24].
2.5 Simetria
As manifestações de simetria podem ser observadas em vários ramos da ciência, que vai desde
a química, física até a botânica. A teoria de grupos têm sido uma ferramenta bastante útil na des-
crição da simetria microscópica de moléculas e simetria macroscópica de cristais [32]. Esta é,
frequentemente, utilizada no estudo de estados eletrônicos e na descrição da simetria local de cris-
tais. Nesta seção, serão apresentados alguns conceitos básicos sobre elementos de simetria, grupos
de simetria pontual bem como será discutido a escolha do eixo de simetria e o desdobramento
máximo dos níveis de energia de acordo com a simetria local.
2.5.1 Elementos e Operações de Simetria
Existe uma diferença clara entre as entidades: elementos de simetria e operações de simetria.
Um elemento de simetria é uma entidade geométrica que pode ser um ponto, uma reta ou um
plano com relação a qual se efetua uma ou mais operações de simetria [33, 34, 35]. No estudo
de sistemas infinitos como, por exemplo, moléculas, cristais, sólidos, aglomerados de moléculas
existem apenas quatro tipos de elementos de simetria:
−→ Plano de simetria - são elementos que dividem uma estrutura em metades simétricas. Estes
planos são classificados em:
• Horizontal (σh) - plano de reflexão perpendicular ao eixo de simetria.
• Vertical (σv) - plano de reflexão paralelo ao eixo de simetria.
• Diedral (σd) - plano de reflexão paralelo ao eixo de simetria e é bissetor aos dois eixos binários
perpendiculares ao eixo de simetria.

Capítulo 2. Fundamentação Téorica 18
−→ Centro de simetria - são centros de inversão onde cada átomo da molécula têm um contra-
ponto simétrico em relação a este centro.
−→ Eixo próprio - são elementos através dos quais uma ou mais rotações de 2π/n ao redor do
eixo leva a molécula a uma configuração geométrica equivalente à original. O n indica o grau de
simetria da molécula.
−→ Eixo impróprio - são elementos de simetria através do quais uma rotação própria e em se-
guida uma reflexão num plano perpendicular ao eixo de rotação leva a estrutura a uma configuração
geométrica equivalente à original.
Uma operação de simetria consiste em mover um corpo (moléculas, sólidos, cristais, etc.), de tal
maneira, que sua configuração final após o movimento seja indistinguível da configuração inicial.
As operações de simetria são classificados em:
−→ Identidade (E) - são operações que levam a mesma configuração.
−→ Rotações (Cn) - são operações de 2π/n ao redor de um eixo fixo.
−→ Reflexões (σ) - são operações que transforma um ponto simétrico em relação a um outro
ponto.
−→ Inversões (i) - são operações mediante transformação das coordenadas espaciais de um
objeto que leva (x,y,z) para (-x,-y,-z).
−→ Roto-reflexões (Sn) - são operações que combinam uma rotação própria seguida de uma
reflexão num plano perpendicular ao eixo de rotação.
2.5.2 Grupos de Simetria Pontual
Os grupos de simetria pontual são formados por elementos de simetria, que mediante operações
de simetria deixe o objeto (como por exemplo, moléculas, cristais, sólidos) invariante. Na natureza
existem 32 grupos pontuais cristalográficos, aos quais abrangem todas as possíveis simetria de um
cristal. Estes grupos podem ser divididos em grupos de: alta simetria, baixa simetria e lineares.
Estes grupos pontuais são definidos como [34, 35]:
⋄ Cn - são grupos de simetria consistindo de um único eixo de ordem n (n=1,2,3,4,6).
⋄ Ci - são grupos de simetria compostos de uma inversão e uma identidade.
⋄ Cnv - são grupos constituídos de planos de reflexão σv mais um eixo Cn, (n=2,3,4,6).

Capítulo 2. Fundamentação Téorica 19
⋄ Cnh - são grupos que contém um plano de reflexão σh e um eixo Cn, (n=1,2,3,4,6). O grupo
C1h é constituído somente dos elementos [E,σh] e é também conhecido como Cs.
⋄ C∞v - esse é o grupo de moléculas lineares sem centro de inversão.
⋄ Sn - esse grupo contém uma rotação imprópria de ordem n (n=4,6).
⋄ Dn - esse grupo possui n eixos duplos perpendicular ao eixo principal Cn
⋄ Dnd - esse grupo possui o elemento Dn seguido de uma reflexão σd.
⋄ Dnh - esse grupo possui o elemento Dn seguido de uma reflexão σh.
⋄ D∞h - esse é o grupo de moléculas lineares com centro de inversão.
⋄ Td - esse grupo possui simetria tetraedral, no qual contém 24 elementos de simetria.
⋄ Th - esse grupo é formado de uma simetria tetraedral seguido de uma reflexão σh.
⋄ Oh - esse grupo possui simetria octaedral, que contém 48 elementos de simetria.
⋄ Ih - esse grupo possui simetria icosaedral.
⋄ Kh - esse grupo possui simetria esférica.
2.5.3 Eixo de Simetria
Durante a seção 2.5 foi mostrado o porquê de alguns objetos serem mais simétricos do que
outros. Essa relação está diretamente ligado com o número de elementos de simetria, que um
dado objeto possui, por exemplo, a esfera é o objeto mais simétrico do que qualquer outro, pois,
apresenta o maior número de elementos de simetria em relação ao eixo principal.
Para descrever o grupo de simetria pontual de um determinado objeto (como por exemplo, mo-
léculas, cristais, sólidos) é preciso inicialmente adotar um eixo de simetria, que mediante operações
de simetria em torno desse eixo principal, o objeto volte à sua posição original. Entretanto, em de-
terminadas situações o objeto pode apresentar mais de um eixo de simetria. Quando isso acontece,
surge a seguinte pergunta: qual o melhor eixo de simetria a ser adotado? O parâmetro chave que
precisamos conhecer para responder essa pergunta, é o momento de inércia do objeto (como por
exemplo, moléculas, cristais, sólidos). O momento de inércia de um objeto é definido como a massa
mi do i-ésimo átomo multiplicado pelo quadrado da distância ri perpendicular ao eixo de rotação,
que passa pelo centro de massa do objeto [36, 37],
I =∑i
mir2i (2.25)

Capítulo 2. Fundamentação Téorica 20
Em geral, as propriedades de rotação em torno de um eixo principal num objeto (como por
exemplo, moléculas, cristais, sólidos) podem ser expressas em termos do momento de inércia I
sobre os eixos perpendiculares do objeto [36]. Considere, como exemplo, o caso simples de uma
molécula de água H2O, conforme mostra a figura 4. Tomando os eixos de simetria Ia e Ib, pode-
se mostrar de forma trivial, a partir da equação 2.25, que o momento de inércia Ib>Ia. Sendo
que, o eixo de simetria Ib contém o maior número de elementos de simetria, e o átomo H têm um
contraponto simétrico, que representa uma inversão sobre o eixo principal. Então, pode-se concluir,
que o melhor eixo de simetria a ser adotado é o eixo que possui o maior momento de inércia. Esse
eixo, eventualmente, dará indicação da simetria pontual do objeto.
Figura 4 – Molécula de água com ângulo de ligação HOH de 104.5 graus.
2.5.4 Simetria e a Quebra de Degenerescência
O campo cristalino é uma pertubação responsável por distorcer a simetria esférica do íon livre.
Esta distorção provocada na componente esférica do íon livre é bastante útil no estudo das propri-
edades espectroscópicas desses materiais. Algumas delas são, o grande número de transições entre
os estados 4f do espectro (absorção e emissão) e a largura das linhas bastante estreitas, que podem
ser consideradas como monocromáticas.
Quando o íon lantanídeo opticamente ativo é inserido em um meio, os níveis de energia do
multipleto 2S+1LJ são desdobrados de acordo com a simetria local do sítio luminescente e, passa a
ocupar a simetria do sítio. Para sistemas de alta simetria a degenerescência é parcialmente remo-
vida, enquanto que para sistemas de baixa simetria a degenerescência é totalmente removida. A
figura 5 apresenta a ordem de grandeza, de cada um dos termos do hamiltoniano total:

Capítulo 2. Fundamentação Téorica 21
Figura 5 – Representação esquemática de desdobramentos dos níveis 4f conforme o hamiltoniano.
O primeiro desdobramento é devido à repulsão eletrônica, HRE , a segunda é a interação spin-
órbita, HSO, e por último a interação do campo cristalino (efeito Stark), HCC . Note que o efeito
Stark desdobra os níveis de energia da ordem de 102 cm−1. Já a interação spin-órbita e à repulsão
eletrônica apresentam ordens de grandeza maiores do que a do campo cristalino. O campo cristalino
é, então, tratado como efeito perturbativo.
As transições que são permitidas entre os níveis de energia do campo cristalino podem ser ob-
tidas, utilizando as regras de seleção da mecânica quântica. Entretanto, os mecanismos de maior
relevância para que estas transições possam ocorrer são: mecanismo por dipolo elétrico e meca-
nismo por dipolo magnético. De acordo com a regra de Laporte a transição por dipolo elétrico
entre estados (transições 4f-4f) de mesma paridade é proibida, já que o operador de dipolo elétrico
têm paridade ímpar. Porém, os espectros de emissão mostravam que esta transição ocorria, e até
mais intensa que a transição por dipolo magnético. No entanto, as teorias da época não conseguiam
explicar tal efeito. Somente em 1962, Judd e Ofelt, independentemente, mostraram que a regra de
Laporte pode ser violada, quando o sistema atômico se encontra na presença de um campo crista-
lino, cuja simetria não têm centro de inversão. Isto ocorre porque a distorção causada no íon livre,
por meio da parte ímpar do campo cristalino mistura as configurações eletrônicas de paridades
opostas.
Em 1996, K. Binnemans e C. Görller-Walrand publicaram um trabalho relacionando os pa-
râmetros de campo cristalino não nulos no potencial de interação com a simetria local do sítio
luminescente em questão. Neste trabalho, também é mostrado um diagrama para determinação do

Capítulo 2. Fundamentação Téorica 22
grupo pontual em cristais simples, baseado em regras de seleção do íon Eu3+. Em 2010, Peter
Tanner [38] publicou um trabalho de revisão, no qual aprimora o diagrama que relaciona o número
de linhas da transição 5D0−→7Fj com a simetria local do sítio luminescente do íon Eu3+. A partir
deste diagrama é possível fazer previsões da simetria do sítio observando o espectro de emissão
(ver anexo A).

Parte III
Modelos e Métodos

24
3 Modelos e Método3.1 Modelo Eletrostático de Cargas Pontuais
O Modelo Eletrostático de Cargas Pontuais, (PCEM), do inglês Point Charge Electrostatic Mo-
del, baseado no trabalho de Bethe de 1929, foi o primeiro modelo proposto para descrever a inte-
ração entre o íon lantanídeo e sua vizinhança. Este modelo assume que a contribuição de cada PV
é levado em conta separadamente. Ele consiste em considerar, que o campo elétrico sentido pelo
IC é gerado por um conjunto de cargas ou dipolos, no qual essa carga está localizada, exatamente,
na posição do PV, e têm magnitude em módulo igual à valência do PV. Portanto, assume-se que a
ligação é puramente iônica, no qual a contribuição covalente é negligênciada. A figura 6 ilustra um
sítio de coordenação 6 baseado nos pressupostos do PCEM, no qual −→ri é vetor posição do i-ésimo
elétron do IC,−→Rj é vetor posição do j-ésimo elétron do PV, gj é a valência do j-ésimo PV e o
potencial gerado pelos PV sobre o IC é dado pela seguinte expressão [24]:
Hcc =∑i,j
e2gj
|ri − Rj|(3.1)
Figura 6 – Distribuição de cargas ao redor do IC.
O PCEM têm sido utilizado a bastante tempo no estudo das propriedades dos íons Ln, entre-
tanto, apenas sob certos aspectos é bem sucedido. Do ponto de vista qualitativo têm feito boas
previsões, tais como, indicação de simetria local. Entretanto, do ponto de vista quantitativo os
resultados obtidos são bem discrepantes dos valores experimentais e fenomenológicos. Bezerra

Capítulo 3. Modelos e Método 25
et. al. em 2008 [13], mostrou para um sistema de óxidos M2O3(M = Gd,Lu, Y, In, Sc), que as
previsões realizadas com o PCEM é da ordem de três vezes maior que os valores obtidos experi-
mentalmente.
3.2 Modelo de Recobrimento Simples
Devido à algumas discrepâncias existentes do ponto de vista quantitativo no PCEM como mos-
trado na seção 3.1, Malta em 1982 [11], propôs o SOM, do inglês Simple Overlap Model. Este
modelo leva em consideração a covalência, ao introduzir o recobrimento na ligação entre o íon Ln
e sua vizinhança.
Além do caráter covalente da ligação, é proposto que a carga efetiva não deve ser necessaria-
mente igual a valência do PV, e não deverá estar exatamente na posição do PV, mas sim em uma
região intermediária entre o IC e o PV. O SOM têm como pressuposto as seguintes premissas:
i) A energia potencial dos elétrons 4f , devido à presença de um ambiente químico, é produzido
por cargas uniformemente distribuídas em pequenas regiões centradas em torno da meia distância
entre o IC e cada PV.
ii) A carga em cada região é dada por −gjeρj , onde ρj é a integral de recobrimento entre os
orbitais 4f e a camada de valência do PV j, e gj é um fator de carga.
A figura 7 ilustra uma representação esquemática das premissas do SOM:
Figura 7 – Distribuição de cargas ao redor do IC segundo o SOM.
A partir da primeira premissa do SOM, o hamiltoniano do campo cristalino é dado por:
Hcc =∑i,j
gje2ρj
|ri − (Rj/2βj)|(3.2)

Capítulo 3. Modelos e Método 26
no qual −→ri é a posição do i-ésimo elétron do IC,−→Rj é a posição do j-ésimo PV em relação ao IC e
gje2ρj é a carga total em cada região de interação.
O SOM utiliza apenas os PV nos cálculos dos Bkq e ∆E, uma vez que as linhas do espectro de
emissão indicam, exatamente, a simetria local do sítio luminescente. Considerando que a distribui-
ção de carga não esteja exatamente a uma meia distância entre o íon lantanídeo e os PV, nesse caso,
Rj/2βj . βj leva em conta as situações em que o baricentro da região de recobrimento está mais
deslocado para o lado do PV ou para o lado do IC, e é dado por [11]:
βj =1
1±ρj(3.3)
O (+) se aplica quando o raio do IC é maior que o raio do PV, e o sinal (-) se aplica no caso
contrário. Esses são os raios que dão limite a densidade eletrônica. O recobrimento ρj é definido
como [11]:
ρj = ρ0
(Rmin
Rj
)3.5
(3.4)
ρ0=0.05 e Rmin é a menor das distâncias Rj .
Calculando o valor esperado do hamiltoniano da equação 3.2, é possível obter uma relação entre
os parâmetros de campo cristalino do PCEM e do SOM dado por:
Bkq (SOM) = ρ(2β)k+1Bk
q (PCEM) (3.5)
A correção feita em 3.5 evolui exatamente no sentido de corrigir as discrepâncias existentes
entre os valores dos Bkq s obtidos com o PCEM e os obtidos fenomenologicamente. Vale ressaltar,
que a partir de um modelo puramente teórico é possível obter bons resultados, sem precisar utilizar
altos custos computacionais.
3.3 Método dos Vizinhos Equivalentes
Embora as correções aplicadas pelo SOM tenham conseguido apresentar resultados satisfató-
rios para os parâmetros de campo cristalino com k = 2, bem como, reproduzir o desdobramento
experimental do nível de energia com J = 1, em alguns casos o modelo falha. Haja vista, que
o somatório de cargas utilizado na reprodução do desdobramento de energia é bem maior que a

Capítulo 3. Modelos e Método 27
valência do IC. Diante dos aspectos apresentados pelo SOM, Couto em 2008 propôs o MENN do
inglês Method of Nearest Neighbors, que é baseado nas seguintes premissas.
i) Os PV devem ser identificados. A primeira consideração é o equilíbrio eletrostático do sítio
luminescente. As condições de simetria juntamente com o ambiente químico dos PV devem ser
levados em consideração relembrando os conceitos de equivalência a partir da teoria de grupos a
ponto de refinar a identificação;
ii) O desdobramento ∆E experimental deve ser previsto por um conjunto de cargas gj ainda
fenomenológico;
iii) A soma∑
j gj deve ser igual à valência do IC.
Este modelo é baseado no SOM. A partir das condições apresentados por Couto, é possível
descrever satisfatoriamente os parâmetros de campo de cristalino, e desdobramento de energia,
assegurando-se o princípio eletrostático do sítio luminescente, conforme a condição iii).
3.4 Metodologia
Neste trabalho foi desenvolvido um estudo sistemático em sítios com alta simetria contendo íon
Eu3+, tais como: [M2O3(M=Gd,Y,Lu,In,Sc)(S6) [13], BaLiF3(Oh) [14], XMgF3(Oh), SnO2(C2h)
[15]]. O estudo teórico foi feito utilizando a TCC através do MENN, pois neste caso teremos tantos
graus de liberdade quanto equações, que são o desdobramento máximo de níveis com J≤ 3/2 e o
equilíbrio eletrostático do sítio luminescente. As previsões foram comparadas com resultados da
literatura para uma possível indicação da simetria local do íon Eu3+, num dado intervalo da carga
de interação entre o IC e os PV.

Parte IV
Desenvolvimento

29
4 Estrutura Local do Íon Eu3+ no Óxido
M2O34.1 Considerações Iniciais
Neste capítulo serão apresentados e discutidos previsões da estrutura local do sítio luminescente
do íon Eu3+ em uma série de óxidos do tipo M2O3(M=Gd,Y,Lu,In,Sc). Todos esses sítios têm
características semelhantes, tais como número de coordenação, similaridade química dos PV e
tipo de simetria, diferenciando-se apenas as coordenadas esféricas dos PV. Entretanto, devido à
semelhança dos elementos Ln, tais como raio iônico e configuração eletrônica, as coordenadas
esféricas dos PV nos sítios luminescentes dos óxidos M2O3 não se distinguem muito uns dos outros.
Inicialmente serão discutidas as coordenadas angulares dos PV em relação ao IC para toda a
série de óxidos, quando o íon Eu3+ é inserido nesta matriz hospedeira. Na segunda parte serão
expostas as previsões da estrutura média local do sítio luminescente, baseado na equação do desdo-
bramento máximo com J≤3/2, do equilíbrio eletrostático do sítio luminescente, e do recobrimento
entre os orbitais 4f e 2p. Em um último instante serão analisados a simetria local do sítio lumines-
cente do íon Eu3+. Esta análise será realizada por meio da teoria de grupos, do espectro de emissão
e dos cálculos dos CFP.
4.2 Coordenadas Angulares do Sítio Luminescente
Quando é inserido na matriz hospedeira M2O3, o íon Eu3+ passa a ocupar dois sítios de simetria
com número de coordenação 6, e não equivalentes com simetria pontual C2 e S6(D3d). Os dois
sítios luminescentes diferem um do outro em relação ao íon Eu3+ devido a vacância do oxigênio,
conforme mostra a figura 8 [39]. Um desses sítios luminescentes S6(D3d) apresenta simetria com
centro de inversão, ao contrário do outro sítio que apresenta simetria C2. Entretanto, o íon Eu3+
também pode ocupar uma simetria D3d ao invés de uma simetria S6, como será discutido em seções
posteriores.
Neste trabalho as previsões da estrutura média local do sítio luminescente do íon Eu3+ em
óxidos M2O3 com simetria pontual S6(D3d) serão realizadas, através das equações dos subníveis de
energia do 7F1 já conhecido na literatura, e de um conjunto de fatores de cargas já bem testado em

Capítulo 4. Estrutura Local do Íon Eu3+ no Óxido M2O3 30
Figura 8 – Ocupação do íon Eu3+ em dois sítios de simetria.
outros trabalhos. As coordenadas angulares foram obtidas por meio de uma média geral ponderada
das coordenadas angulares dos PV em relação ao íon M nos cristais M2O3. A diferença entre os
raios iônicos dos elementos Ln com número de coordenação 6 é menor do que dez por cento, o
que justifica-se considerar em média, as mesmas coordenadas angulares para toda a série de óxidos
dopados com íon Eu3+. Quando é inserido nesta matriz hospedeira, o íon Eu3+ substitui o íon M .
Apesar de haver distorção local, não há quebra de simetria, a tal ponto de baixar a simetria pontual
do sítio luminescente, uma vez que os raios iônicos e as propriedades químicas destes elementos
são bem semelhantes. A tabela 3 apresenta uma sugestão das coordenadas angulares dos PV para
toda a série.
Tabela 3 – Coordenadas angulares dos PV no sítio Eu3+:M2O3.
θ φ
O1 62.62 0O2 117.22 60O3 62.62 120O4 117.22 180O5 62.62 240O6 117.22 300
Os valores da tabela 3 foram obtidos tomando o átomo O1 como o eixo x, e o eixo z foi tomado
como o eixo de maior momento de inércia em relação ao centro de massa do sítio luminescente.
As coordenadas angulares (θ, φ) com relação ao eixo de simetria adotado é ilustrada na figura 9.
Os ângulos mostrados na tabela 3 juntamente com a distância IC-PV descrevem a força do
campo cristalino em uma simetria S6(D3d). Estes ângulos serão posteriormente utilizados nas
equações dos parâmetros de campo cristalino, para serem feitas as previsões da distância Eu-O no

Capítulo 4. Estrutura Local do Íon Eu3+ no Óxido M2O3 31
Figura 9 – Eixo de simetria adotado para obtenção das coordenadas angulares.
sítio luminescente Eu3+:M2O3 através do MENN.
4.3 Desdobramento Máximo do Multipleto 7F1 do Íon Eu3+
em Óxidos M2O3
A equação do desdobramento máximo do multipleto 7F1 do íon Eu3+ na presença de um campo
cristalino é uma ferramenta bastante útil para indicação da estrutura local do sítio luminescente.
Uma vez que, o desdobramento máximo dos níveis estão relacionados com os aspectos de simetria.
Por exemplo, não há desdobramento de níveis em sistemas de altíssima simetria, Oh .
A equação 2.15 descrita na seção 2.3 descreve o desdobramento máximo de um sistema de
níveis múltiplos de J, e que estão em repartições aproximadamente simétricas. Entretanto, para J
pequeno a equação 2.15 não pode ser utilizada [30]. Para J=1, Mj pode assumir no máximo três
valores possíveis. Com isto Malta et. al.[7] descrevem um procedimento, no qual permite mostrar
a linearidade entre ∆E e Nv para um sistema de óxidos M2O3 contendo íon Eu3+.
Sendo assim, o desvio médio quadrático dos níveis de energia com J pequeno ∆ε2 será escrito

Capítulo 4. Estrutura Local do Íon Eu3+ no Óxido M2O3 32
em termos dos parâmetros de campo cristalino da seguinte forma,
∆ε2 =1
g
∑k,q
|Bkq |2
2k + 1|⟨J∥Ck
i ∥J⟩|2 (4.1)
da equação 4.1, o desvio médio quadrático pode ser escrito em termos do desdobramento máximo
dos múltiplos de J como:
∆ε2 =1
ga
(∆E
2
)2
(1 + α22 + · · ·+ α2
ga−2 + 1) (4.2)
Para J pequeno, ou mais especificamente J=1 para o caso do multipleto 7F1, apenas o termo
com posto k=2 será necessário na equação 4.1. Combinando as equações 4.1 e 4.2 obtêm-se:
∆E =
[gag
0.2875
(2 + α2)
]1/2Nv (4.3)
A degenerescência totalmente removida ga = 2, pois em uma simetria S6(D3d), o campo cris-
talino produzido pelo meio químico não é suficiente para remover totalmente a degenerescência do
multipleto J.
Baseado na equação 4.3 Malta et. al. [7] obtiveram o desdobramento máximo do nível 7F1
do íon Eu3+ em óxidos M2O3. O íon Eu3+ inserido no óxido de escândio foi o composto que
apresentou o máximo desdobramento dentre todos os óxidos.
Para realizar previsões da distância Eu-O no sítio luminescente Eu3+:M2O3 foi utilizado o
máximo desdobramento ∆E do multipleto 7F1 em toda a série de óxidos, obtido no trabalho de
Malta et. al.[7].
4.4 Previsões da Distância Eu-O em Eu3+:M2O3 Através da
Carga de Interação
O sítio luminescente com simetria S6(D3d) tem 6 PV em sua vizinhança. Todos os PV estão
localizados a uma mesma distância interatômica em relação ao IC. Além disso, as coordenadas
angulares estão relativamente bem organizadas, como mostram a figura 9 e a tabela 3.
Segundo as premissas apresentadas pelo MENN justifica-se considerar todos os PV como equi-
valentes, uma vez que estão a uma mesma distância em relação ao IC no sítio luminescente. Sendo

Capítulo 4. Estrutura Local do Íon Eu3+ no Óxido M2O3 33
assim, todos os PV podem ser tratados com o mesmo fator de carga, de forma que teremos um
sistema com apenas um grau de liberdade, o que facilita a descrição do sistema em estudo.
Como as coordenadas angulares e o desdobramento máximo do multipleto 7F1 são conhecidos,
e utilizando a terceira premissa do MENN, que é o equilíbrio eletrostático do sítio luminescente
é possível fazer previsões da distância Eu-O utilizando a equação do desdobramento máximo do
multipleto 7F1 para J=1 com apenas uma variável, que é a carga de interação entre o IC e seus PV.
Reescrevendo a equação 4.3 em termos dos parâmetros de campo cristalino obtêm-se:
∆E =
[gag
0.2875
(2 + α2)
]1/2 [4π
5
∑q
|B2q |2
]1/2
(4.4)
α é uma quantidade que dá a intensidade dos níveis, dentro de um intervalo ∆E em unidades
de ∆E2
como foi mostrado na seção 2.3 por meio da equação 2.18. O valor mais recente de α foi
proposto por Couto em 2008 [31], e seu valor é dado por α =√
23. Entretanto, a equação 2.18 é
válida apenas para sistemas com baixa simetria. Neste trabalho a equação 2.18 foi utilizado para
um sistema de alta simetria como uma aproximação, e apresentou bons resultados.
Os parâmetros de campo cristalino de posto k=2 dependem exclusivamente das componentes
angulares dos PV, que são representadas pelos harmônicos esféricos Y 2q , da carga de interação
gje entre o IC-PV, do valor médio das integrais radias dos elétrons 4f ⟨r2⟩, do recobrimento ρj
entre os orbitais 2p e 4f e do inverso cúbico da distância interatômica Eu-O no sítio luminescente
(1/R3). Destas grandezas, apenas o fator de carga gj e a distância interatômica Eu-O (1/R3) não
são conhecidas. Então a expressão dos CFP pode ser escrita como:
B2q t =
g
R3B2
q t(g = R3 = 1) (4.5)
Para o posto k=2, a grandeza q pode assumir 3 valores possíveis. Sendo assim, a equação 4.5
pode ser reescrita em termos das componentes q como:
B2q t =
g
R3
(B2
0t+ 2B21t+ 2B2
2t)(g = R3 = 1) (4.6)
O fator multiplicativo nos dois últimos termos da equação 4.6 se refere a dupla contribuição
quando q é diferente de zero, uma vez que de acordo com as regras de triangularidade −k≤q≤k.
Para sistemas com alta simetria a degenerescência não é totalmente removida, de forma que
para J=1, um dos estados quânticos é duplamente degenerado, já que o efeito do campo cristalino

Capítulo 4. Estrutura Local do Íon Eu3+ no Óxido M2O3 34
não é suficiente para remover totalmente a degenerescência. Sendo assim, apenas o parâmetro B20t
será diferente de zero na equação 4.6. Isto também pode ser verificado por meio das equações 2.22,
2.23 e 2.24.
Combinando as equações 4.6 e 4.4 obtêm-se a seguinte relação envolvendo R, ∆E e g:
R =
[(4π5
)1/2gB2
0t
∆E/Fa
]1/3
(4.7)
sendo Fa dependente de α e do elemento tensorial reduzido de Racah de posto k=2 dado por:
Fa =
[2
3
0.2875
(2 + α2)
]1/2(4.8)
A partir da equação 4.7 é possível fazer previsões da distância Eu-O no sítio luminescente, por
meio de um conjunto satisfatório de cargas, uma vez que as outras grandezas são conhecidas e,
foram discutidas em seções anteriores. Para fazer essa análise é preciso definir um intervalo de
cargas gj . Como o sítio luminescente possui 6 PV equivalentes em sua vizinhança, o fator de carga
gj não pode ser menor do que 0.5 [31], de acordo com a terceira premissa do MENN, que é a
neutralidade eletrostática do sítio luminescente. Além disso, cálculos de química quântica (SMLC)
através do modelo Sparkle têm estimado que a carga do oxigênio para complexos com íons Ln3+ é
gj < 1 [3, 5, 6]. Baseado nestas informações, as previsões da distância Eu-O no sítio luminescente
de alta simetria serão feitas com um intervalo de cargas entre 0.5≤gj < 1.
A carga efetiva egjρj têm uma dependência direta com as integrais de recobrimento entre os
orbitais 4f e 2p. egjρj é o principal responsável por afetar o comportamento dos elétrons 4f . En-
tretanto, as integrais de recobrimento indicam a contribuição do efeito de covalência em uma dada
ligação química entre íons e, significativamente, a influência do campo cristalino experimentado
pelo IC [40]. A contribuição do efeito de covalência em íons lantanídeos é extremamente pequena.
Malta e Couto têm frequentemente utilizado um valor de ρ0 = 0.05, que está explícito na equação
3.4, como recobrimento mínimo entre os orbitais 4f e 2p. Entretanto, o resultado obtido no tra-
balho de Axe e Burns (1966)[41] para o recobrimento entre os orbitais do Ln3+-F−1 excede esse
valor a depender da separação internuclear entre os íons e do número de elétrons na camada 4f do
íon Ln. Baseado nestes pressupostos foi utilizado seis valores diferentes para o recobrimento entre
os orbitais 4f e 2p num intervalo de 0.05≤ρ≤0.1, com o objetivo de analisar a percentagem de
covalência na ligação entre os íons Eu-O. Sendo 0.1 o máximo recobrimento entre estes orbitais.

Capítulo 4. Estrutura Local do Íon Eu3+ no Óxido M2O3 35
A partir das discussões realizadas nos dois últimos parágrafos e utilizando a equação 4.7 com
∆E = 470cm−1 foi obtido as previsões da distância Eu-O no sítio luminescente Eu3+:M2O3 como
mostra a figura 10:
0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,00,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
0,05 0,06 0,07 0,08 0,09 0,1
Dis
tanc
ia E
u-O
no
sitio
Eu3+
:M2O
3 (Ang
stro
m)
Carga efetiva
Figura 10 – Previsões da distância Eu-O em função da carga efetiva no sítio Eu3+:M2O3.
As curvas em cores diferentes na figura 10 representa cada um dos recobrimentos utilizados nas
previsões da distância interatômica Eu-O no sítio luminescente. Observe que a partir do MENN é
possível fazer boas previsões por meio de um modelo puramente teórico, e relativamente simples
de ser trabalho, sem a necessidade de altos custos computacionais.
Shannon em 1976 publicou um trabalho de revisão mostrando o raio iônico efetivo para um
série de íons em função do número de coordenação. De acordo com o trabalho de Shannon, o raio
iônico do íon Eu3+ com CN=6 é de RIEu = 0.947 e do íon O−2 com CN=3 é de RIO = 1.36 [21].
A distância interatômica entre os íons Eu-O (R) no sítio luminescente pode ser definida como:
R = RIEu +RIO = 2.307 (4.9)

Capítulo 4. Estrutura Local do Íon Eu3+ no Óxido M2O3 36
Observe que os valores obtidos na figura 10 estão de acordo com o valor calculado na equação
4.9, obtidos do trabalho de Shannon. A linha tracejada mostra uma boa indicação da distância Eu-O
no sítio luminescente. O recobrimento entre os orbitais 4f e 2p no intervalo entre 0.07≤ρj≤0.1 e o
fator de carga no intervalo entre 0.55≤gj≤0.9 apresentaram as melhores previsões para a indicação
da estrutura local do sítio luminescente. A região no qual gj apresentou as melhores previsões, a
soma das cargas∑
j gj é maior que a valência do íon Eu3+.
Em ordem de comparar a carga do íon Eu3+, gEu, com o somatório de cargas dos PV∑
j gj
The Batista-Longo Improved Model (BLIM) [42] foi utilizado. O BLIM é baseado no seguinte
pressuposto: a blindagem imperfeita da carga nuclear é devido a extensão radial dos orbitais fe-
chado 5s e 5p dos íons Ln. Assim, dependendo da distância interatômica Ln3+-PV, diferentes PV
experimentará diferentes cargas efetiva nuclear. gEu têm a seguinte dependência radial:
gEu = 3 + 14e−Ar2 (4.10)
A = 0.5/a20 e r é dado em unidades de raio de Bohr, a0. Essa função gaussiana reproduz
satisfatoriamente a gEu em complexos. O BLIM é válido para sistemas complexos, entretanto,
foi utilizado como um comparativo em sistemas de alta simetria para mostrar que a carga do íon
Eu3+ pode ser maior que sua própria valência, a depender da distância interatômica entre os íons
Eu3+-PV. Sendo assim, o princípio de neutralidade eletrostática do sítio luminescente é satisfeita,
de acordo com as premissas do MENN.
4.5 Simetria do Íon Eu3+ em Óxidos M2O3
O íon Eu3+ ocupa dois sítios de simetria no composto M2O3. A literatura sugere que o íon
Eu3+ ocupe um sítio de simetria S6 no sítio 2, entretanto, de acordo com a estrutura cristalográfica
do sítio luminescente, o íon Eu3+ também pode ocupar uma simetria D3d neste sítio.
A partir das previsões obtidas para a estrutura média local do íon Eu3+ no sítio luminescente
Eu3+:M2O3, e utilizando os conceitos de simetria pontual discutido na seção 2.5 é possível fazer
previsões da simetria local ocupada pelo íon Eu3+ no sítio 2. Uma vez que as coordenadas an-
gulares em relação a um dado eixo de simetria e o espectro de emissão também são conhecidos, e

Capítulo 4. Estrutura Local do Íon Eu3+ no Óxido M2O3 37
calculando os CFP é possível fazer previsões. A figura 11 mostra os elementos de simetria contidos
no sítio 2.
Figura 11 – Elementos de simetria contidos no sítio luminescente Eu3+:M2O3.
A figura 11 mostra que uma rotação C3 seguida de uma reflexão horizontal σh em torno do
eixo principal de simetria deixa a estrutura invariante. Esta operação caracteriza uma simetria
S6. Entretanto, a figura também mostra três eixos duplos perpendiculares ao eixo principal, o que
caracteriza um elemento de simetria D3. Uma rotação D3 seguido de uma reflexão σd também
deixa a estrutura invariante. Dessa forma, a partir dos aspectos apresentados por meio da teoria de
grupo, ambas as simetrias são possíveis.
A segunda e última análise a ser feita será por meio dos CFP, uma vez que os CFP estão
diretamente relacionados com os aspectos de simetria. A tabela 4 mostra os CFP calculados a
partir das coordenadas angulares da tabela 3, e utilizando R = 2.315, gj = 0.67 e ρj = 0.1.
A tabela 4 apresenta os CFP em dois sítios de simetria diferentes S6 e D3d. Os valores dos
CFP designados pela simetria S6 foram calculados mediante uma rotação de 5 graus na coordenada
ϕ em torno do eixo principal de simetria, C3. Uma rotação em ϕ fez com que, a contribuição
imaginária dos CFP aparecesse, e sendo esta, a responsável por alterar a simetria da estrutura,
embora não altere a magnitude do desdobramento máximo do multipleto 7F1. Sendo assim, é
sugerido neste trabalho que íon Eu3+ ocupe a simetria S6 ou D3d em óxidos M2O3. De acordo,
com as duas análises abordadas, ambas as possibilidades são permitidas, embora, a simetria D3d
seja mais satisfatória devido os CFP serem reais. Alguns trabalhos atribuem os CFP reais a simetria
S6 [39], entretanto, de acordo com o trabalho de revisão [24], a contribuição dos CFP reais são
atribuídos a simetria D3d, e a contribuição dos CFP imaginários são atribuídos a simetria S6. As
coordenadas angulares utilizadas nos cálculos de CFP compõem o conjunto dos chamados "ângulos

Capítulo 4. Estrutura Local do Íon Eu3+ no Óxido M2O3 38
Tabela 4 – CFP do sítio 2 do íon Eu3+ em M2O3.
S6 D3d
B20 496 496
B21 0 0
B22 0 0
B40 -1321 -1321
B41 0 0
B42 0 0
B43 2764+741i 2862
B44 0 0
B60 2297 2297
B61 0 0
B62 0 0
B63 -952-255i -985
B64 0 0
B65 0 0
B66 1407+812i 1624
bons"[24], que juntamente com a distância interatômica entre os íon Eu-PV descrevem a força do
campo cristalino em uma simetria pontual S6(D3d).

39
5 Estrutura Local do Íon Eu3+ no Óxido
SnO25.1 Considerações Iniciais
Neste capítulo serão apresentados e discutidos previsões da estrutura local do íon Eu3+, quando
inserido na matriz hospedeira do óxido de Estanho, SnO2.
Inicialmente serão discutidos as coordenadas angulares dos PV em relação ao IC (Eu3+), quando
o íon Eu3+ entra no sítio do Estanho. Na segunda parte serão expostas as previsões da estrutura
média local do sítio luminescente, a partir da equação do desdobramento máximo com J≤3/2 e
do equilíbrio eletrostático do sítio luminescente. Na terceira parte serão analisados os intervalos de
recobrimento entre os orbitais 4f e 2p, ρj , e o fator de carga gj que apresenta as melhores previsões
para a distância interatômica entre os íons Eu-O no sítio luminescente Eu3+:SnO2. Em um último
momento serão analisados a simetria local, quando o íon Eu3+ substitui o íon Sn4+ na matriz hos-
pedeira SnO2, por meio da teoria de grupos, dos cálculos de CFP e do espectro de emissão do sítio
luminescente Eu3+:SnO2.
5.2 Coordenadas Angulares do Sítio Luminescente
O íon Sn4+ possui número de coordenação 6 e o O2− possui número de coordenação 3 na matriz
hospedeira SnO2. Trabalhos preveem que íon Sn4+ ocupa uma simetria C2h(D2h) no cristal SnO2
[43]. Gonçalves et. al. observaram que quando incorporado ao óxido SnO2, o íon Eu3+ substitui
o íon Sn4+, para concentrações de dopante menor do que 0.05mol% [15]. Apesar do íon Eu3+ ter
raio iônico cerca de 30% maior do que o raio iônico do íon Sn4+, a sua ocupação na matriz SnO2
substituindo o íon Sn4+, se deve a baixa concentração do íon Eu3+ na matriz hospedeira. Quando
é inserido nesta matriz hospedeira, o íon Eu3+ causa distorções localmente, por substituir um íon
de raio iônico menor, entretanto, não há quebra de simetria local devido à baixa concentração de
dopante. Apesar dos diagramas de raio-X (DRX) não identificar o sítio luminescente do íon Eu3+
em baixíssimas concentrações, o espectro de emissão do íon Eu3+ reportado por Gonçalves et. al.
[15] mostra que o íon Eu3+ ocupa um sítio de simetria com centro de inversão, no qual as linhas
do espectro de emissão estão bem definidas.

Capítulo 5. Estrutura Local do Íon Eu3+ no Óxido SnO2 40
Sendo assim, é sugerido que no sítio luminescente do íon Eu3+, os PV tenham as mesmas
coordenadas angulares dos PV na matriz SnO2 em relação ao íon Sn4+, uma vez que o íon Eu3+
substitui o íon Sn4+ nesta matriz hospedeira. A tabela 5 apresenta uma sugestão das coordenadas
angulares dos PV na vizinhança do íon Eu3+ obtido do trabalho de [44]:
Tabela 5 – Coordenadas angulares dos PV no sítio luminescente Eu3+:SnO2.
θ φ
O1 0 0O2 180 0O3 90 0O4 90 78.15O5 90 180O6 90 258.15
Os valores da tabela 5 foram obtidos tomando o átomo O3 como o eixo x, e o eixo z foi tomado
como o eixo de simetria, que possui o maior momento de inércia em relação ao centro de massa do
sítio luminescente, sendo este o átomo O1. As coordenadas angulares (θ, φ) em relação ao eixo de
simetria adotado é ilustrado na figura 12 [44]:
Figura 12 – Eixo de simetria adotado para obtenção das (θ, φ) no sítio luminescente.
As coordenadas angulares apresentadas na tabela 5 juntamente com a distância interatômica
entre os íons Eu-O descrevem a força do campo cristalino em uma simetria com centro de inversão
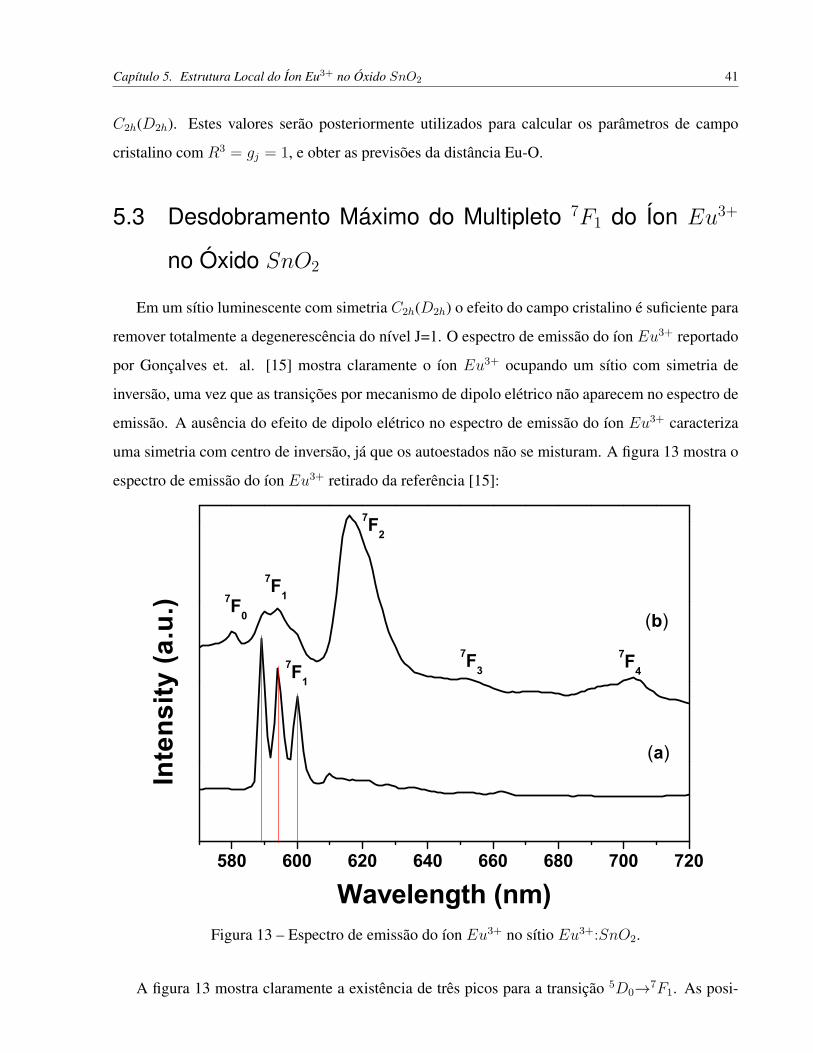
Capítulo 5. Estrutura Local do Íon Eu3+ no Óxido SnO2 41
C2h(D2h). Estes valores serão posteriormente utilizados para calcular os parâmetros de campo
cristalino com R3 = gj = 1, e obter as previsões da distância Eu-O.
5.3 Desdobramento Máximo do Multipleto 7F1 do Íon Eu3+
no Óxido SnO2
Em um sítio luminescente com simetria C2h(D2h) o efeito do campo cristalino é suficiente para
remover totalmente a degenerescência do nível J=1. O espectro de emissão do íon Eu3+ reportado
por Gonçalves et. al. [15] mostra claramente o íon Eu3+ ocupando um sítio com simetria de
inversão, uma vez que as transições por mecanismo de dipolo elétrico não aparecem no espectro de
emissão. A ausência do efeito de dipolo elétrico no espectro de emissão do íon Eu3+ caracteriza
uma simetria com centro de inversão, já que os autoestados não se misturam. A figura 13 mostra o
espectro de emissão do íon Eu3+ retirado da referência [15]:
580 600 620 640 660 680 700 720
7F47F3
7F2
7F0
7F1
7F1
(b)
(a)
Inte
nsity
(a.u
.)
Wavelength (nm)Figura 13 – Espectro de emissão do íon Eu3+ no sítio Eu3+:SnO2.
A figura 13 mostra claramente a existência de três picos para a transição 5D0→7F1. As posi-

Capítulo 5. Estrutura Local do Íon Eu3+ no Óxido SnO2 42
ções dos subníveis de energia do multípleto 7F1 mostrado na tabela 6 foram obtidos a partir dos
picos adjacentes das transições 5D0→7F0,1 da figura 13, e em seguida utilizados para calcular o
desdobramento experimental:
Tabela 6 – Posição dos subníveis e desdobramento experimental dos níveis de energia.7F0 0cm−1
268.23cm−1
7F1 410.99cm−1
584.73cm−1
∆Eexp 316.5cm−1
Posteriormente, o valor do desdobramento experimental dado na tabela 6 será utilizado na equa-
ção 4.4 para realizar previsões da distância Eu-O no sítio luminescente Eu3+:SnO2. Esses resulta-
dos comprovarão que o íon Eu3+ realmente substitui o íon Sn4+ no sítio luminescente.
5.4 Previsões da Distância Eu-O em Eu3+:SnO2 Através da
Carga de Interação
Este sítio luminescente têm 6 PV em sua vizinhança. Entretanto, os PV equivalentes localizados
sobre o eixo de simetria (eixo z) têm distâncias interatômicas diferentes dos PV localizados no
plano xy, conforme mostra a figura 12. As coordenadas angulares dos PV em relação ao IC é
relativamente bem organizada, como mostrados na figura 12 e tabela 5.
De acordo com as premissas do MENN teremos dois conjuntos diferentes de PV. Sendo destes,
quatro PV equivalentes no plano xy e 2 PV equivalentes com direção coincidindo ao eixo z. Sendo
assim, os dois PV paralelos ao eixo z podem ser tratados com um mesmo fator de carga, g1, e os
quatro PV perpendiculares ao eixo z serão tratados com o outro fator de carga, g2. Desta forma,
a equação do desdobramento máximo com J = 1 dependerá de duas variáveis, que são g1 e g2.
Entretanto, o problema de um grau de liberdade descrito na seção 4.4 de forma exata, se estende a
um problema com dois graus de liberdade, o que dificulta um pouco mais a descrição do sistema
em estudo. Uma forma conveniente de abordar tal problema é utilizar o equilíbrio eletrostático do
sítio luminescente, uma vez que tal propriedade foi satisfeita no problema com um único grau de
liberdade.

Capítulo 5. Estrutura Local do Íon Eu3+ no Óxido SnO2 43
Escrevendo os CFP em termos de R1, R2, g1 e g2. Sendo R1 e g1 as respectivas distância inte-
ratômica Eu-O e o fator de carga dos dois PV paralelos ao eixo z, e R2 e g2 a distância interatômica
Eu-O e o fator de carga dos quatro PV perpendicular ao eixo z. Então, os CFP com posto k=2
podem ser escritos da seguinte forma:
B2q t =
(g1R3
1
+g2R3
2
)B2
q t(g1 = g2 = R31 = R3
2 = 1) (5.1)
t=0,1,...,5 representa o número de PV na vizinhança do IC.
A distância R1 pode ser relacionada com R2 a menos de uma constante P a ser definida. Ba-
seado na distância interatômica Sn-O no cristal SnO2 é sugerido neste trabalho, que P = 1, 0029
para R1 = PR2. Sendo assim, a equação 5.1 pode ser reescrita como:
B2q t =
(g1
(PR2)3+
g2R3
2
)B2
q t(g1 = g2 = R32 = 1) (5.2)
Observe da equação 5.2, que g1 está associado aos PV paralelos ao eixo z, e consequentemente
aos termos de B2q t com t=0,1. Da mesma forma, g2 está associado aos termos de B2
q t com t=2,3,4,5.
Fazendo uma substituição de variáveis de forma a separar as contribuições de g1 e g2, e sabendo
que em uma simetria C2h(D2h) apenas os CFP B20 e B2
2 de posto k=2 são diferentes de zero. Então,
a equação 5.2 pode ser reescrita da seguinte forma:
B2q t =
1
R32
(A1
g1P 3
+ A2g2 + 2C1g1P 3
+ 2g2C2
)(g1 = g2 = R3
2 = 1) (5.3)
A1 e A2 são as contribuições do B20t, e C1 e C2 são as contribuições do B2
2t.
Relacionando as equações 4.4 e 5.3 obtêm-se uma expressão relacionando R2 com g1, g2 e ∆E
dada por:
R2 =
[(4π5
)1/2 [| g1P 3 (A1 + 2C1) + g2(A2 + 2C2)|
]∆E/Fa
]1/3
(5.4)
sendo ga = 3 na expressão 4.8, uma vez que a degenerescência é totalmente removida.
Da equação 5.4 apenas R, g1 e g2 são grandezas variáveis, todas as outras grandezas são co-
nhecidas, conforme discutido em seções anteriores. Entretanto, há um pequeno problema a ser

Capítulo 5. Estrutura Local do Íon Eu3+ no Óxido SnO2 44
resolvido, pois temos uma equação com duas variáveis. Uma forma inteligente de abordar este
problema é utilizar o equilíbrio eletrostático do sítio luminescente, por meio da seguinte expressão:
g1 =gEu
2− 2g2 (5.5)
sendo gEu a carga do íon Eu3+.
Conhecendo a carga do íon Eu3+ é possível reduzir o problema de dois graus de liberdade a
um problema de um único grau de liberdade. Entretanto, como visto na seção 4.4 a carga do íon
Eu3+ têm uma dependência radial, que está relacionado com a extensão radial dos orbitais 5s e
5p. Baseado nos resultados obtidos na seção 4.4 para o∑
j(gj), foram utilizados cinco valores
diferentes para a carga do íon Eu3+ num intervalo 3≤gEu≤4.2, e recobrimento entre os orbitais
2p e 4f num intervalo 0.05≤ρj≤0.1. As figuras 14, 15, 16,17 e 18 apresentam as previsões da
distância Eu-O no sítio luminescente Eu3+:SnO2:
0,35 0,40 0,45 0,50 0,55 0,60 0,65 0,70 0,75 0,80
1,5
2,0
2,5
3,0
3,5
4,0
Soma das cargas igual a 3
0,05 0,06 0,07 0,08 0,09 0,1
Dis
tânc
ia E
u-O
em
Eu3+
:SnO
2(Ang
stro
m)
Fator de Carga
Figura 14 – Previsões da distância Eu-O no sítio Eu3+:SnO2 com gEu = 3.

Capítulo 5. Estrutura Local do Íon Eu3+ no Óxido SnO2 45
0,45 0,50 0,55 0,60 0,65 0,70 0,75
1,0
1,5
2,0
2,5
3,0
3,5
4,0
Soma das cargas igual a 3,3
0,05 0,06 0,07 0,08 0,09 0,1
Dis
tânc
ia E
u-O
em
Eu3+
:SnO
2(Ang
stro
m)
Fator de Carga
Figura 15 – Previsões da distância Eu-O no sítio Eu3+:SnO2 com gEu = 3.3.
0,50 0,55 0,60 0,65 0,70 0,75 0,800,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
Soma das cargas igual a 3,6
0,05 0,06 0,07 0,08 0,09 0,1
Dis
tânc
ia E
u-O
em
Eu3+
:SnO
2(Ang
stro
m)
Fator de Carga
Figura 16 – Previsões da distância Eu-O no sítio Eu3+:SnO2 com gEu = 3.6.

Capítulo 5. Estrutura Local do Íon Eu3+ no Óxido SnO2 46
0,55 0,60 0,65 0,70 0,75 0,80 0,85
0,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
Soma das cargas igual a 3,9
0,05 0,06 0,07 0,08 0,09 0,1
Dis
tânc
ia E
u-O
em
Eu3+
:SnO
2(Ang
stro
m)
Fator de Carga
Figura 17 – Previsões da distância Eu-O no sítio Eu3+:SnO2 com gEu = 3.9.
0,60 0,65 0,70 0,75 0,80 0,85 0,900,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
Soma das cargas igual a 4,2
0,05 0,06 0,07 0,08 0,09 0,1
Dis
tânc
ia E
u-O
em
Eu3+
:SnO
2(Ang
stro
m)
Fator de Carga
Figura 18 – Previsões da distância Eu-O no sítio Eu3+:SnO2 com gEu = 4.2.

Capítulo 5. Estrutura Local do Íon Eu3+ no Óxido SnO2 47
A linha tracejada nas figuras 14, 15, 16, 17, 18 mostram boas previsões para a distância Eu-O
no sítio luminescente Eu3+:SnO2, por meio de um conjunto satisfatório de fatores de carga num
intervalo entre 0.5≤gj≤0.75. O conjunto de cargas obtido nas previsões para a distância Eu-O é
uma das duas possíveis soluções existentes, conforme mostra a equação 5.5. Note que este conjunto
de fatores de carga é bem semelhante ao conjunto de gj obtido nas previsões do sítio luminescente
Eu3+:M2O3. Esta semelhança, certamente deve está relacionando com o CN e a similaridade
química da vizinhança do IC, bem como, em ambas as matrizes hospedeiras o íon Eu3+ ocupa
uma simetria com centro de inversão. A linha tracejada nas figuras 14, 15, 16, 17, 18 dão indicação
de que o íon Eu3+ substitui o íon Sn4+ sem haver quebra de simetria.
Todos os valores utilizados para o recobrimento entre os orbitais 2p e 4f num intervalo entre
0.05≤ρj≤0.1 apresentaram boas previsões para estrutura local do sítio luminescente Eu3+:SnO2.
5.5 Simetria do Íon Eu3+ no Óxido SnO2
A simetria ocupada pelo íon Eu3+ quando inserido na matriz hospedeira SnO2 em pequenas
concentrações é algo discutido na literatura. Rogéria et. al. sugerem que o íon Eu3+ ocupe uma
simetria com centro de inversão C2h [15]. Brik et. al. sugerem que o íon Eu3+ pode ocupar dois
tipos de simetria com centro de inversão C2h e D2h [43].
A partir das previsões obtidas para a estrutura local do íon Eu3+ no sítio luminescente Eu3+:SnO2,
e utilizando os conceitos de simetria pontual discutidos na seção 2.5 é possível prevê a simetria ocu-
pada pelo íon Eu3+. Uma vez que, as coordenadas angulares dos PV em relação ao um dado eixo
de simetria também são conhecidas, e calculando os CFP pode-se identificar a simetria ocupada
pelo íon Eu3+. A figura 19 mostra os elementos de simetria contidos no sítio luminescente do íon
Eu3+:SnO2:
A figura 19 mostra que uma rotação C2 seguida de uma reflexão horizontal σh em torno do eixo
de simetria deixa a estrutura invariante. Essa operação caracteriza uma simetria C2h. Entretanto,
a figura também mostra 2 eixos C ′2 e C2” perpendiculares a um eixo C2, o que caracteriza um
elemento de simetria D2. Uma rotação D2 seguido de uma reflexão horizontal σh também deixa
a estrutura invariante, o que caracteriza uma simetria D2h. Dessa forma, a partir dos aspectos
apresentados por meio da teoria de grupos pontuais ambas as simetrias podem ser possíveis.
Agora será analisada a simetria ocupada pelo íon Eu3+ na matriz hospedeira SnO2 por meio

Capítulo 5. Estrutura Local do Íon Eu3+ no Óxido SnO2 48
Figura 19 – Elementos de simetria contidos no sítio luminescente Eu3+:SnO2.
do espectro de emissão, uma vez que as linhas do espectro dão indicação da simetria local do sítio
luminescente. De acordo com a figura 13 a transição 5D0→7F1 apresenta três picos, e todas as
outras transições não apresentam nenhum pico. De acordo com o diagrama de Tanner (ver anexo
A) [38] três simetrias são possíveis: C2h, D2h e Ci.
A terceira e última análise será feita por meio dos CFP. A tabela 7 mostra os CFP calculados
a partir das coordenadas angulares da tabela 5 que compõem o conjunto dos chamados "ângulos
bons"[24], e utilizando R1 = 2.308, R2 = 2, 301 com g1 = 0.68, g2 = 0.56 e ρj = 0.09:
A tabela 7 apresenta os CFP do íon Eu3+ em dois sítios de simetria. Os valores dos CFP desig-
nados pela simetria D2h foram calculados mediante uma rotação de 51 graus na coordenada ϕ em
torno do eixo de simetria C2, ou seja, a rotação em ϕ removeu a contribuição imaginária dos CFP,
e sendo assim, altera a simetria do sítio luminescente, embora não altere as outras propriedades
físicas, tal como o desdobramento máximo. Sendo assim é sugerido neste trabalho que o íon Eu3+
ocupe a simetria C2h ou D2h na matriz hospedeira SnO2. De acordo com as três análises abordadas
nos parágrafos anteriores, ambas as possibilidades são permitidas, embora a simetria D2h seja mais
satisfatória devido os CFP serem todos reais. Certamente, deve haver uma maior preferência de
ocupação do íon Eu3+ na matriz hospedeira SnO2, em uma das duas simetrias. Entretanto, não foi

Capítulo 5. Estrutura Local do Íon Eu3+ no Óxido SnO2 49
Tabela 7 – CFP do sítio do íon Eu3+ em SnO2.
C2h D2h
B20 496 496
B21 0 0
B22 137+653i -667
B40 2879 2879
B41 0 0
B42 -50-237i 242
B43 0 0
B44 926-407i -1012
B60 928 928
B61 0 0
B62 46+220i -225
B63 0 0
B64 -1006+442i 1099
B65 0 0
B66 549+769i 945
possível identificar em qual simetria o íon Eu3+ têm maior preferência de ocupação, por meio do
MENN. Alguns trabalhos [45] atribuem, que o íon Eu3+ em baixíssimas concentrações têm uma
maior preferência de ocupação em sítios de baixa simetria devido à força do campo cristalino ser
maior. Entretanto, tanto a simetria C2h quanto a D2h têm a mesma magnitude.

50
6 Estrutura Local do Íon Eu3+ nos Cris-
tais BaLiF3 e XMgF3(X=Cs, K, Na, Rb)6.1 Considerações Iniciais
Neste capítulo serão apresentados e discutidos previsões da estrutura local do íon Eu3+ quando
inserido nas matrizes hospedeiras BaLiF3 e XMgF3(X=Cs, K, Na, Rb). Estes sítios têm proprie-
dades bem semelhantes, tais como CN, distância interatômica e simetria local.
Inicialmente serão discutidas a simetria ocupada pelos íons (Ba, X) e o CN na matriz hospe-
deira, por meio da estrutura atômica destes cristais. Na segunda parte serão analisados a simetria
e o CN, quando o íon Eu3+ é inserido nas matrizes hospedeiras BaLiF3 e XMgF3(X=Cs, K, Na,
Rb) em pequenas concentrações. Esta análise será realizada por meio do espectro de emissão do
íon Eu3+ em BaLiF3, e de cálculos de estrutura baseado em dinâmica molecular. Em um último
momento serão calculados os CFP e o desdobramento máximo do multipleto 7F1 do íon Eu3+,
a partir de um conjunto de fatores de carga satisfatório, de forma a reproduzir o desdobramento
experimental do multipleto 7F1.
6.2 Estrutura Local dos Cristais BaLiF3 e XMgF3(X=Cs, K,
Na, Rb)
Os íons Ba2+ e X+ ocupam um sítio de altíssima simetria Oh com CN 12. Todas as ligações
Ba-F3 e X-F3 têm a mesma distância interatômica nesse cristal. Sendo que a distância interatômica
é em média de 2.862Åpara todo o conjunto de cristais, e as coordenadas angulares dos PV em
relação aos íons Ba2+ e X+ são bem organizadas em toda à vizinhança. A figura 20 mostra uma
representação esquemática da estrutura local desses íons:
A figura 20 mostra a vizinhança do íon Bário no cristal BaLiF3. Todos os íons X+ têm estrutura
local semelhante a figura 20, embora a distância interatômica seja diferente variando em torno de
2.862Å. A distribuição angular dos PV em relação ao íon X+ é a mesma para todos os cristais. A
tabela 8 mostra as coordenadas angulares dos PV em relação ao centro de massa da estrutura local,
que contém o eixo de maior momento de inércia.

Capítulo 6. Estrutura Local do Íon Eu3+ nos Cristais BaLiF3 e XMgF3(X=Cs, K, Na, Rb) 51
Figura 20 – Vizinhança do Bário no cristal BaLiF3.
Tabela 8 – Coordenadas angulares dos PV em relação aos íons Ba2+ e X+ em BaLiF3 eXMgF3(X=Cs, K, Na, Rb).
θ φ
F1 45 0F2 45 90F3 45 180F4 45 270F5 90 45F6 90 135F7 90 225F8 90 315F9 135 0
F10 135 90F11 135 180F12 135 270
6.3 Espectro de Emissão do Íon Eu3+ no Cristal BaLiF3
O raio iônico do íon Ba2+ é de 1.35 Å, enquanto que o raio iônico do íon Eu3+ é de 0.947Å,
ambos com CN 6. Os íons X1+ têm raios iônicos semelhantes ao do íon Ba2+. Sendo que, o raio
iônico do íon Ba2+ é cerca de 30% maior que do íon Eu3+.
Quando é inserido nas matrizes hospedeiras BaLiF3 e XMgF3, o íon Eu3+ passar a ocupar uma
simetria local própria nestes cristais. Em ambos os cristais os íons Ba2+ e X1+ ocupam um sítio de
altíssima simetria com CN 12, conforme mostra a figura 20. Entretanto, não há relatos mostrando
que o íon Eu3+ ocupa sítios de altíssima simetria. Tan e Shi analisaram o comportamento do

Capítulo 6. Estrutura Local do Íon Eu3+ nos Cristais BaLiF3 e XMgF3(X=Cs, K, Na, Rb) 52
íon Eu3+ no cristal BaLiF3, quando dopado em baixíssimas concentrações. A figura 21 mostra o
espectro de emissão do íon Eu3+ em BaLiF3 reportado por Tan e Shi em 1999 [46]:
Figura 21 – Espectro de emissão do íon Eu3+ em BaLiF3.
O espectro de emissão do íon Eu3+ dá indicação da simetria local do sítio luminescente em
uma matriz hospedeira. O espectro de emissão do Eu3+:BaLiF3 apresenta duas linhas referente a
transição 5D0→7F1 e 2, e 3 linhas pouco intensas referente a transição 5D0→7F2, sendo à última
devido ao mecanismo de dipolo elétrico induzido. A primeira conclusão a ser obtida do espectro
de emissão figura (21) é que o íon Eu3+ não substitui exatamente o íon Ba2+ quando dopado
em baixíssimas concentrações na matriz hospedeira BaLiF3, uma vez que a transição 5D0→7F1
apresenta duas linhas bem definidas. Sendo assim, a presença do íon Eu3+ em BaLiF3 distorce a
simetria Oh a tal ponto de haver quebra de simetria, e o íon Eu3+ passa a ocupar uma simetria mais
baixa. Entretanto, esta simetria não deve apresentar centro de inversão, uma vez que a contribuição
do mecanismo de dipolo elétrico aparece no espectro de emissão (figura 21). Segundo o diagrama
de níveis reportado por Tanner (ver anexo A) [38] várias são as simetrias possíveis, no qual o íon
Eu3+ pode ocupar nesta matriz hospedeira.
Couto et. al. em 2003 [14] calcularam a distorção causada ao BaLiF3 por meio da incorporação
do íon Eu3+ utilizando uma técnica de simulação de defeitos. Neste trabalho é mostrado que para
haver compensação de cargas é necessário dois íons Eu3+ para cada vacância de íon Ba2+. A tabela
3 da referência [14] mostra as coordenadas esféricas dos PV em relação ao IC (Eu3+) considerando
a não distorção do sítio do Ba2+, onde o íon Eu3+ é assumido ocupar o sítio do Ba2+ sem nenhuma
distorção. Os resultados mostram que devido a uma compensação de carga, 4 íons F1− distanciam-

Capítulo 6. Estrutura Local do Íon Eu3+ nos Cristais BaLiF3 e XMgF3(X=Cs, K, Na, Rb) 53
se da vizinhança com a presença do íon Eu3+. Com isso, o íon Eu3+ passar a ocupar um sítio de
simetria com CN 8 em BaLiF3. Neste trabalho foram feitas previsões da estrutura local do íon Eu3+
em BaLiF3 por meio do SOM. Segundo estas previsões o íon Eu3+ ocupa um sítio com simetria de
inversão, C2h.
Entretanto, de acordo com o diagrama de níveis reportado por Tanner [38], e do espectro de
emissão do íon Eu3+ em BaLiF3 reportado por Tan e Shi, a simetria local do sítio luminescente
do Eu3+ não pode ser uma C2h, uma vez que em uma simetria C2h o efeito do campo cristalino é
suficiente para remover totalmente a degenerescência dos níveis de energia do multipleto 7F1. E o
espectro de emissão (figura 21) apresenta apenas duas linhas referente a transição 5D0→7F1.
Baseado em tais discussões, neste trabalho será realizado a sugestão da estrutura local do íon
Eu3+ em BaLiF3 utilizando o MENN. Como a estrutura cristalina do XMgF3 é semelhante a do
BaLiF3, diferenciando-se apenas pela distância interatômica entre os íons X-PV, porém, os raios
iônicos são bem semelhantes uns dos outros. Sendo assim, as previsões realizadas para BaLiF3
serão em média as mesmas para XMgF3.
Em sistemas de altíssima simetria o efeito do campo cristalino não desdobra o multipleto 7F1,
e apenas uma linha é observada no espectro de emissão referente a transição 5D0→7F1. Quando
o íon Eu3+ é inserido nessa matriz hospedeira BaLiF3 há uma quebra de simetria devido a uma
compensação de cargas. A transição 5D0→7F0 não apresenta nenhum pico, e a transição 5D0→7F2
apresenta 2 ou 3 picos com intensidade muito fraca, que também pode ser devido às vibrações. De
acordo com o diagrama de níveis reportado por Tanner [38] várias são as simetrias possíveis. Como
a transição 5D0→7F2 é muito fraca, a simetria mais provável é a D4d, uma vez que nesta simetria
os CFP responsáveis pela forte intensidade da transição 5D0→7F2 são zero. Então, a simetria Oh é
reduzida a uma simetria mais baixa D4d, quando o íon Eu3+ é inserido na matriz cristalina BaLiF3.
Os mesmos argumentos discutidos anteriormente é válido para a estrutura do XMgF3, uma vez
que a estrutura local do íon X+ tem a mesma simetria e CN, bem como a distância interatômica
entre os íons Ba-F e X-F são em média da mesma ordem, o que justifica-se considerar que, o íon
Eu3+ ocupa uma simetria D4d na matriz cristalina XMgF3 em baixíssimas concentrações.

Capítulo 6. Estrutura Local do Íon Eu3+ nos Cristais BaLiF3 e XMgF3(X=Cs, K, Na, Rb) 54
6.4 Estrutura Local do Íon Eu3+ em BaLiF3 e XMgF3(X=Cs,
K, Na, Rb)
O íon Eu3+ ocupa a simetria D4d quando inserido nas matrizes hospedeiras BaLiF3 e XMgF3
[24]. A figura 22 mostra uma representação esquemática da vizinhança do íon Eu3+ em uma
simetria D4d com CN 8:
Figura 22 – Vizinhança do íon Eu3+ nos cristais BaLiF3/XMgF3.
Todas as distâncias interatômicas entre os íons Eu-F são iguais. As coordenadas angulares dos
PV em relação ao IC compõem um conjunto dos chamados "ângulos bons"que descreve a força do
campo cristalino em uma simetria pontual D4d. As coordenadas esféricas dos PV em relação ao IC
foram obtidas tomando o eixo z como o eixo de maior momento de inércia em relação ao centro
de massa do sítio luminescente, conforme indica a figura 22. A tabela 9 apresenta as coordenadas
esféricas em uma simetria D4d:
Tabela 9 – CE dos PV em relação ao íon Eu3+ nos compostos BaLiF3 e XMgF3.
R θ φ
F1 2.396 59.19 0F2 2.396 59.19 90F3 2.396 59.19 180F4 2.396 59.19 270F5 2.396 120.81 315.03F6 2.396 120.81 45.03F7 2.396 120.81 135.03F8 2.396 120.81 225.03

Capítulo 6. Estrutura Local do Íon Eu3+ nos Cristais BaLiF3 e XMgF3(X=Cs, K, Na, Rb) 55
A distância interatômica sugerida na tabela 9 é exatamente a soma dos raios iônicos do íon
Eu3+ com CN 8 e do íon F1− com CN 3. Os valores sugeridos na tabela 9 foram utilizados para
calcular os CFP e o desdobramento máximo do multipleto 7F1, conforme mostra a tabela 10:
Tabela 10 – CFP do sítio do íon Eu3+ em BaLiF3 e XMgF3.
D4d (cm−1)B2
0 -346B2
1 0B2
2 0B4
0 -475B4
1 0B4
2 -0B4
3 0B4
4 0B6
0 470B6
1 0B6
2 0B6
3 0B6
4 0B6
5 0B6
6 0∆E 83
∆Eexp 83
Os CFP apresentados na tabela 10 foram calculados utilizando o MENN. Estes valores mostram
o íon Eu3+ ocupando uma simetria D4d nos cristais BaLiF3 e XMgF3. O MENN consegue reprodu-
zir exatamente o sinal negativo do parâmetro B20 , uma vez que o nível duplamente degenerado está
à direita do espectro de emissão (figura 21), ou seja, acima do nível baricentro. O desdobramento
máximo do multipleto 7F1 foi reproduzido utilizando um fator de carga gj = 0.402 e recobrimento
entre os orbitais ρ0 = 0.05. Em ordem de comparação o espectro de emissão do íon Eu3+ em
BaLiF3 (figura 21)foi utilizado para estimar o desdobramento máximo do multipleto 7F1 reportado
por [46].

Parte V
Conclusões e Perspectivas

57
7 ConclusõesNeste trabalho foi realizado um estudo da estrutura local do sítio luminescente do íon Eu3+ com
alta simetria em cristais, utilizando a TCC através do MENN. Os cristais estudados foram: M2O3,
SnO2, BaLiF3 e XMgF3 contendo baixas concentrações do íon Eu3+. As previsões da distância in-
teratômica Eu-O na matriz hospedeira M2O3 foi realizada por meio da equação do desdobramento
máximo do multipleto 7F1 com apenas um grau de liberdade, que é o fator de carga. Além disso, o
recobrimento entre os orbitais 4f e 2p foi analisado em um intervalo 0.05≤ρj≤0.1. O recobrimento
entre os orbitais 4f e 2p no intervalo entre 0.07≤ρj≤0.1 e o fator de carga entre 0.55≤gj≤0.9
apresentaram as melhores previsões para a indicação da estrutura local do sítio luminescente. A
região no qual gj apresentou as melhores previsões, a soma das cargas é maior que valência do íon
Eu3+. Em ordem de comparação o BLIM foi utilizado com boas previsões. A simetria do sítio
luminescente também foi analisado por meio da teoria de grupos e dos CFP. As previsões dão indi-
cação de que o íon Eu3+ ocupa uma simetria pontual S6 ou D3d, sendo que ambas têm as mesmas
possibilidades de ocorrer.
As previsões da distância interatômica Eu-O também foi realizada para o óxido de estanho,
SnO2. Neste caso, dois conjuntos de PV não equivalentes foram identificados. R1 e R2 foram rela-
cionados por meio de uma constante de proporcionalidade P=1.0029. E os fatores de carga g1 e g2
foram relacionados por meio da equação do equilíbrio eletrostático do sítio luminescente. A carga
do íon Eu3+ foi utilizada no intervalo entre 3≤gEu≤4.2. O recobrimento entre os orbitais 4f e 2p
no intervalo entre 0.05≤ρj≤0.1 e o fator de carga entre 0.5≤gj≤0.75 apresentaram as melhores
previsões para a indicação da estrutura local. A simetria do sítio luminescente foi analisada por
meio da teoria de grupos, do espectro de emissão do íon Eu3+ e dos CFP. Os resultados dão indi-
cação de que o íon Eu3+ pode ocupar tanto a simetria pontual C2h quanto a D2h. Isto mostra que o
íon Eu3+ substitui o íon Sn4+ na matriz hospedeira SnO2.
Por último foi feito uma sugestão da estrutura local do íon Eu3+ nos cristais BaLiF3 e XMgF3.
A análise foi realizada com base no espectro de emissão do íon Eu3+ no cristal BaLiF3 e de cálculos
de estrutura por dinâmica molecular. É sugerido que o íon Eu3+ ocupe uma simetria mais baixa
D4d nas matrizes BaLiF3 e XMgF3. Os CFP foram calculados, e o MENN conseguiu reproduzir o
sinal do CFP B20 . O desdobramento máximo do multipleto 7F1 foi reproduzido com um fator de
carga gj=0.402 e recobrimento entre os orbitais de 0.05.

58
8 PerspectivasDesenvolver um estudo mais amplo através da teoria das intensidades e cálculos de eficiência
quântica, para analisar a simetria mais provável do sítio luminescente nos cristais M2O3 e SnO2.
Ampliar o estudo para determinação da estrutura local em sistemas com sítios luminescentes
de baixa simetria;

59
Referências1 KAMINSKII, A. A. Today and tomorrow of laser-crystal physics. Phys. Stat. Sol. A, v. 148, p.9–79, 1995.
2 YANG, J. et al. Optical transitions and upconversion luminescence of Er3+ sobre Yb3+ codopedhalide modified tellurite glasses. J. Appl. Phys., v. 93, p. 977, 2003.
3 BELTRÃO, M. et al. Spectroscopic properties of the Eu-FOD3-Phen-NO incorporatedcarboxylate glass. J. Lumin., v. 116, p. 132–138, 2006.
4 MALTA, O. L. et al. Intensity parameters of 4f-4f transitions in the Eu(dipivaloylmethanate)3,10-phenanthroline complex. J. Lumin., v. 69, p. 77–84, 1996.
5 FREIRE, R. et al. Sparkle model and photophysical studies of europium BiqO2-cryptate. Chem.Phys. Lett., v. 442, p. 488–491, 2007.
6 PORCHER, P.; COUTO DOS SANTOS, M. A.; MALTA, O. L. Relationship betweenphenomenological crystal field parameters and the crystal structure: The simple overlap model.Phys. Chem. Chem. Phys, v. 1, p. 397–405, 1999.
7 MALTA, O. et al. The crystal field strength parameter and the maximum splitting of the 7F1
manifold of the Eu3+ ion in oxides. J. Alloys. Comp., v. 228, p. 41–44, 1995.
8 AUZEL, F. Les íons terres rares et lês télécommunications optiques. Lécho dês Recherches,v. 1, p. 143, 1991.
9 BETHE, H. A. Splitting of terms in crystals. Ann Physik, v. 3, p. 133–206, 1929.
10 CROSSWHITE, H. M. et al. The spectrum of Nd3+: LaCl3. J. Chem. Phys., v. 64, p.1981–1985, 1976.
11 MALTA, O. A simple overlap model in lanthanide crystal-field theory. Chemical PhysicsLetters, v. 87, p. 27–29, 1982.
12 MONTEIL, A. et al. Molecular dynamics simulation of silver nanoparticles in a europiumdoped sodosilicate glass. Chem. Phys. Lett., v. 493, p. 118–120, 2010.
13 BEZERRA, M. et al. Theoretical investigation of the 7F1 level splitting in a series of Eu3+
doped oxides matrixes. Optical Materials, v. 30, p. 1013–1016, 2008.
14 COUTO DOS SANTOS, M. A. et al. Predicting the spectroscopic behaviour of Eu3+ inBaLiF3 via defect modelling and crystal field parameter calculations. Chem. Phys. Lett., v. 369, p.90–94, 2003.
15 GONÇALVES, R. R. et al. Rare earth doped SnO2 nanoscaled powders and coatings:Enhanced photoluminescence in water and waveguiding properties. J. Nanoscience Nanotech.,v. 10, p. 1–7, 2010.
16 MARTINS, T. S.; ISOLANI, P. C. Terras raras: Aplicações industriais e biológicas. Quim.Nova, v. 28, p. 111–117, 2005.

Referências 60
17 GREEDAN, J. E.; ZHENG, Z. Rare earth elements and materials. Encyc. Of Phys. Scie. AndTech, v. 3, p. 1–22, 2001.
18 LIMA, P. C. R. Terras-raras: Elementos estratégicos para o Brasil. [S.l.]: Consultorialegislativa, 2012.
19 LOUREIRO, F. E. V. L. Terras-raras no Brasil: depósitos, recursos identificados, reservas.[S.l.]: MCT,CNPq, CETEM, 1994.
20 BRANDÃO, A. Química e tecnologia de terras raras. [S.l.]: Cetem/CNPq, 1994.
21 SHANNON, R. D. Revised effective ionic radii and systematic studies of interatomicdistances in halides and chalcogenides. Acta Crystallographica Section A, v. 32, n. 5, p. 751–767,1976.
22 WYBOURNE, B. G. Spectroscopic properties of rare earths. [S.l.]: Interscience, 1965.
23 LIU, G.; JACQUIER, B. Spectroscopic properties of rare earths in optical materials. [S.l.]:Springer, 2005.
24 GÖRLLER-WALRAND, C.; BINNEMANS, K. Handbook on the Physics and Chemistry ofrare earths. [S.l.]: Elsevier Science B, 1996.
25 OLIVEIRA, Y. A. R. Estudo sistemático de parâmetros de campo cristalino em complexos deions Eu3+. Dissertação (Mestrado) — Universidade federal de Sergipe, 2012.
26 SOUZA, A. Cristais fluoretos contendo íons lantanídeos: estudo dos parâmetros de campocristalino e dos níveis de energia. Dissertação (Mestrado) — Universidade Federal de Sergipe,2008.
27 JESUS, R. Aplicação de modelos de campo cristalino a materiais microporosos dopado comeurópio. Dissertação (Mestrado) — Universidade Federal de Sergipe, 2011.
28 FREEMAN, A. J.; DESCLAUX, J. P. Dirac-fock studies of some electronic properties ofrare-earth ions. Journal of Magnetism and Magnetic Materials, v. 8, p. 119–129, 1978.
29 CARNALL, W.; CROSSWHITE, H.; CROSSWHITE, H. Energy level structures andtransition probabilities of the trivalent lanthanides in LaF3. The johns hopkins university, v. 1,p. 34, 1970.
30 AUZEL, F.; MALTA, O. A scalar crystal field stregth parameter for rare-earth ions: meaningand usefulness. Journal Physique, v. 44, p. 201–206, 1983.
31 COUTO DOS SANTOS, M. A. Electrostatic equilibrium and charge factors in lanthanidecomplexes. Chemical Physics Letters, v. 455, p. 339–342, 2008.
32 LADD, M. F. C. Symmetry in molecules and crystals. [S.l.]: Ellis Horwood, 1989.
33 MCWEENY, R. Symmetry: An introduction to group theory and its applications. [S.l.]: Doverpublications, 2002.

Referências 61
34 FAZZIO, A.; WATARI, K. Introdução a teoria de grupos: com aplicações em moléculas esólidos. [S.l.]: Ufsm, 1998.
35 INUI, T.; TANABE, Y.; ONODERA, Y. Group theory and its applications in physics. [S.l.]:Springer, 1996.
36 ATKINS, P. W. Physical Chemistry. [S.l.]: Oxford University Press, 1992.
37 HEINE, V. Group Theory in Quantum Mechanics: an introduction to its present usage. [S.l.]:Pergamon Press, 1960.
38 TANNER, P. A. Lanthanide luminescence in solids. In: HäNNINEN, P.; HäRMä, H.(Ed.). Lanthanide Luminescence. [S.l.]: Springer Berlin Heidelberg, 2011, (Springer Series onFluorescence, v. 7). p. 183–233.
39 ANTIC-FIDANCEV, E.; HÖLSÄ, J.; LASTUSAARI, M. Crystal field energy levels of Eu3+
and Yb3+ in the C2 and S6 sites of the cubic C-type R2O3. Jornal of Physics: Condensed Matter,v. 15, p. 863–876, 2003.
40 Brik, M. G.; Avram, N. M. Numerical calculations of the overlap integrals between the wavefunctions of 3d ions and ligands. In: Proceeding of the physics conference TIM-08. [S.l.: s.n.],2009.
41 AXE, J. D.; BURNS, G. Infuence of covalency upon rare-earth ligand field splittings. PhysicalReview, v. 152, p. 331–340, 1966.
42 BATISTA, H. J.; LONGO, R. L. Improved point-charge model within the indo/s-ci methodfor describing the ligand excited states of lanthanide coordination compounds. InternationalJournal of Quantum Chemistry, v. 90, p. 924–932, 2002.
43 BRIK, M. et al. Spectroscopic and crystal-field analysis of energy levels of Eu3+ in SnO2 incomparison with ZrO2 and TiO2. Journal of Alloys and Compounds, v. 509, n. 8, p. 3441–3451,2011.
44 BAUR, W. H.; KHAN, A. A. Rutile-type compounds. iv. SiO2, GeO2 and a comparison withother rutile-type structures. Acta Crystallographica Section B, v. 27, n. 11, p. 2133–2139, 1971.
45 GRILL, A.; SCHIKBER, M. Magnetic susceptibilities of cubic mixed europium oxides.Physical review b, v. 1, p. 2241–2242, 1970.
46 TAN, Y.; SHI, C. Ce3+ → Eu2+ energy transfer in BaLiF3 phosphor. Journal of Physics andChemistry of Solids, v. 60, p. 1805–1810, 1999.

62
APÊNDICE A – Demonstração do ∆E do
Multipleto 7F1Ao calcularmos os elementos de uma matriz nxn, apenas os elementos da diagonal serão dife-
rentes de zero, uma vez que, as funções de onda são ortogonais entre si. O desvio médio quadrático
de um número de níveis E1, E2,..., En g vezes degenerado é dado pela seguinte expressão:
(∆ε)2 =1
gtr (P0υP0υ) (A.1)
onde P0 é a projeção em todo subespaço do estado do íon livre e υ é o potencial perturbativo.
O traço é definido como:
tr (P0υP0υ) =∑M
⟨JM |∑M ′
|JM ′⟩⟨JM ′|υ∑M ′′
|JM ′′⟩⟨JM ′′|υ|JM⟩ (A.2)
Atribuindo a relação de recorrência∑M ′
|JM ′⟩⟨JM ′| = I na expressão A.2, de forma a ser
escrita como:
tr (P0υP0υ) =∑M,M ′
⟨JM |υ|JM ′⟩⟨JM ′|υ|JM⟩ (A.3)
Substituindo a equação 2.10 devido à perturbação do campo cristalino em A.3:
tr (P0υP0υ) =∑
M,M ′,k,k′,q,q′
BkqB
k′
q′ ⟨JM |C(k)q (i)|JM ′⟩⟨JM ′|C(k′)
q′ (i)|JM⟩ (A.4)
Utilizando o teorema de Wigner-Eckart para os estados JM em A.4, no qual é dado pela seguinte
expressão:
⟨JM |T (k)q |J ′M ′⟩ = (−1)J−M
J k J ′
−M q M ′
⟨J∥T (k)∥J ′⟩ (A.5)
onde ⟨J∥T (k)∥J ′⟩ é o elemento de matriz reduzido. Utilizando A.5 em A.4 obtêm:

APÊNDICE A. Demonstração do ∆E do Multipleto 7F1 63
tr (P0υP0υ) =∑
M,M ′,k,k′,q,q′
(−1)J−M+J−M ′Bk
qBk′
q′
J k J
−M q M ′
J k′ J
−M ′ q′ M
. ⟨J∥
∑i
C(k)(i)∥J⟩⟨J∥∑i
C(k′)(i)∥J⟩ (A.6)
Utilizando as condições de triangularidade dos símbolos 3-J, no qual M = q + M ′ e k≤2J .
Para que essa condição de triangularidade seja satisfeita k = k′ e q = q′. Outra relação importante
dos símbolos 3-J é a condição de ortonormalidade dada por:
δ(J, J, k)
2k + 1=
∑M,M ′
(−1)J−M+J−M ′
J k J
−M q M ′
J k J
−M ′ q M
(A.7)
Então a expressão A.6 pode ser reescrita como:
tr (P0υP0υ) =∑k,q
|Bkq |2
2k + 1δ(J, J, k)|⟨J∥
∑i
C(k)(i)∥J⟩|2 (A.8)
O desvio médio ∆ε também pode ser escrito em termos do desdobramento máximo ∆E como:
(∆ε)2 =1
ga
n∑i
(Ei)2 (A.9)
onde Ei é o i-ésimo nível Stark com respecto a posição do íon livre, e ga é degenerescência efe-
tivamente removida por meio do campo cristalino: ga = g se J for inteiro e ga = g/2 se J for
semi-inteiro. Assumindo uma repartição simétrica entre os níveis de energia, A.9 pode ser escrita
como:
∆ε2 =1
ga
(∆E
2
)2
(1 + α22 + · · ·+ α2
ga−2 + 1) (A.10)
onde αi é uma quantidade 0 < αi < 1, que dar a intensidade dos níveis em um intervalo ∆E em
unidades de ∆E2
. Considerando a repartição simétrica entre os níveis, podemos observar da figura
23 que cada intervalo é equivalente a ∆E/ga. Sendo assim, a equação A.10 pode ser escrita como:
(∆ε)2 =1
ga
∑n
(∆E
ga
)2
n2 =2
ga
(∆E
2
)2ga/2∑
n=1
n2
(2
ga
) (A.11)

APÊNDICE A. Demonstração do ∆E do Multipleto 7F1 64
Figura 23 – Representação esquemática das posições dos níveis em repartições simétricas.
Como n é um valor inteiro, então A.11 pode ser rescrita como:
(∆ε)2 =
(2
ga
)2(∆E
2
)21
6
(ga2
+ 1)(ga + 1) =
1
12g2a(∆E)2 (ga + 2) (ga + 1) (A.12)
Combinando A.1 e A.12 obtêm-se:
(∆E)2 =12g2a
g(ga + 1)(ga + 1)tr(P0υP0υ) (A.13)
Combinado A.8 e A.12 obtêm:
(∆E)2 =12g2a
g(ga + 1)(ga + 1)
∑k,q
|Bkq |2
2k + 1δ(J, J, k)|⟨J∥
∑i
C(k)(i)∥J⟩|2 (A.14)
Note que o termo à direita de A.14 pode ser visto como uma função quadrática de uma superfície
quadrática no sub-espaço k. Então, pode ser escrito em termos da métrica de uma esfera de raio R.
∑k,q
|Bkq |2
2k + 1|⟨J∥
∑i
C(k)(i)∥J⟩|2∼=∑k,q
|Bkq |2
2k + 1
[∏k′
|⟨J∥∑i
C(k′)(i)∥J⟩|2]1/3
(A.15)
Definindo o parâmetro de força do campo cristalino Nv como:
Nv =
[∑k,q
4π
2k + 1|Bk
q |2]1/2
(A.16)
Combinando A.15 e A.16 obtêm-se:
∆E =
[3g2a
g(ga + 1)(ga + 1)π
]1/2 [∏k
|⟨J∥∑i
C(k)(i)∥J⟩|
]1/3
Nv (A.17)

APÊNDICE A. Demonstração do ∆E do Multipleto 7F1 65
A partir da desigualdade triangular, k≤2J , temos que para J≤3/2 apenas o parâmetro k=2
contribui na equação do desdobramento máximo. Portanto, a expressão A.16 pode ser escrita
como:
Nv2 =
[∑q
4π
5|B2
q |2]1/2
(A.18)
E a equação A.10 se reduz a seguinte expressão:
∆ε2 =1
ga
(∆E
2
)2
(2 + α2) =1
gtr(P0υP0υ) (A.19)
Combinando A.8, A.18 e A.19 obtêm-se a seguinte expressão para o desdobramento máximo
com J = 1:
∆E =
[gag
|⟨J∥C(2)(i)∥J⟩|2
π(2 + α2)
]1/2Nv2 (A.20)
onde o elemento de matriz reduzido está tabelado em [29, 22].
Para o multipleto 7F1 do íon Eu3+ apenas o elemento de matriz com k = 2 contribui para o
desdobramento máximo, no qual é dado por:
∆E =
[gag
0.2875
π(2 + α2)
]1/2Nv2 (A.21)

66
APÊNDICE B – Demonstração do Parâ-
metro αO parâmetro αj (0<αj<1) dá a intensidade dos níveis num intervalo ∆E em unidades de ∆E
2,
com repartições igualmente simétricas. Para um sistema de três níveis não degenerado, o parâmetro
αi é reduzido a um único parâmetro, α, como pode ser visto em A.21, e o desvio médio quadrático
em relação ao baricentro pode ser escrito como:
∆ε2 =
[(Eb − E>)
2 + (Eb − Ea)2 + (Eb − E<)
2
ga
](B.1)
onde Eb = (E> +Ea +E<)/3. Sendo, Eb, Ea, E<, E> a energia do nível baricentro, a energia do
nível intermediário, a energia do nível menos energético abaixo do baricentro, a energia do nível
mais energético acima do baricentro do multípleto 7F1, e ∆E = E> − E< é o desdobramento
máximo. Sendo assim, podemos obter as seguintes relações:
E> = ∆E + E< (B.2)
Ea = 3Eb − (E> + E<) (B.3)
Escrevendo B.3 em função do desdobramento:
Ea = 3Eb − (E< +∆E + E<) = 3Eb − (∆E + 2E<) (B.4)
Fazendo uma substituição de variáveis, de tal forma, a simplificar a equação B.1:
I = (Eb − E>)2 = E2
b − 2EbE> + E2> (B.5)
Utilizando B.2 em B.5:
I = E2b − 2Eb(∆E + E<) + (∆E + E<)
2
= E2b − 2Eb∆E − 2EbE< + (∆E)2 + 2∆EE< + (E<)
2 (B.6)

APÊNDICE B. Demonstração do Parâmetro α 67
II = (Eb − Ea)2 = E2
b − 2EbEa + E2a (B.7)
Utilizando B.4 em B.7 obtêm-se:
II = 4E2b − 4Eb∆E − 8EbE< + (∆E)2 + 4E<∆E + 4E2
< (B.8)
III = (Eb − E<)2 = E2
b − 2EbE< + E2< (B.9)
Somando as equações B.6, B.8 e B.9 obtêm-se:
I + II + III = 6E2b + 2(∆E)2 + 6E2
< + 6E<∆E − 12EbE< − 6Eb∆E (B.10)
Rearrumando B.10 em termos de ∆E2
:
I + II + III = 6E2b − 6Eb
∆E
2− 6EbE< + 6E<
∆E
2+ 6E2
<
− 6EbE< + 6E<∆E
2− 6Eb
∆E
2+ 8
(∆E)2
4(B.11)
I + II + III = 6Eb
[Eb −
(∆E
2+ E<
)]− 6E<
[Eb −
(∆E
2+ E<
)]− 6
∆E
2
[Eb −
(∆E
2+ E<
)]+ 2
(∆E
2
)2
(B.12)
∆ε2 =1
ga
[6
[Eb −
(∆E
2+ E<
)]2+ 2
(∆E
2
)2]
(B.13)
Combinando A.20 e B.13 obtêm a seguinte expressão:
1
ga
(∆E
2
)2 (2 + α2
)=
1
ga
[6
[Eb −
(∆E
2+ E<
)]2+ 2
(∆E
2
)2]
(B.14)
(2 + α2) = 6
[Eb −
(∆E2
+ E<
)∆E/2
]2
+ 2 (B.15)
α = ±√6
[Eb −
(∆E2
+ E<
)∆E/2
](B.16)

APÊNDICE B. Demonstração do Parâmetro α 68
Observe que, o lado direito da equação B.16 pode ser escrito como:
[Eb −
(∆E2
+ E<
)∆E/2
]=
[(Ea+E<+E>
3
)−(∆E2
+ E<
)∆E/2
]
=Ea
3+ 1
3(E< − E>) +
∆E2
∆E/2
=13(Ea −∆E + 2∆E)
∆E/2(B.17)
Considerando que o nível intermediário esteja exatamente na posição do baricentro:
[Eb −
(∆E2
+ E<
)∆E/2
]=
∆E/6
∆E/2=
1
3(B.18)
Combinando as equações B.18 e B.16:
α = ±√
6
9= ±
√2
3(B.19)

69
ANEXO A – Diagrama de Níveis
O número de linhas do espectro de emissão indica a simetria local do íon Eu3+ em uma matriz
hospedeira (cristal, vidro e complexo ). Uma vez que, o espectro de emissão é conhecido é possível
prevê a simetria do sítio luminescente. A figura 24 ilustra um diagrama de níveis para o íon Eu3+
reportado por Tanner em 2010 [38].
Figura 24 – Diagrama de níveis do íon Eu3+.





























