Embed Size (px)
Citation preview

Available online at www.sciencedirect.com
ology 95 (2008) 30–41www.elsevier.com/locate/jconhyd
Journal of Contaminant Hydr
Development of a scalable model for predicting arsenic transportcoupled with oxidation and adsorption reactions
Tanja Radu, Anjani Kumar, T. Prabhakar Clement ⁎, Gautham Jeppu, Mark O Barnett
Department of Civil Engineering, 238 Harbert Engineering Center, Auburn University, AL, 36849, United States
Received 5 January 2007; received in revised form 4 July 2007; accepted 6 July 2007Available online 17 July 2007
Abstract
Understanding the fundamentals of arsenic adsorption and oxidation reactions is critical for predicting its transport dynamics ingroundwater systems. We completed batch experiments to study the interactions of arsenic with a common MnO2(s) mineral,pyrolusite. The reaction kinetics and adsorption isotherm developed from the batch experiments were integrated into a scalablereactive transport model to facilitate column-scale transport predictions. We then completed a set of column experiments to test thepredictive capability of the reactive transport model. Our batch results indicated that the commonly used pseudo-first order kineticsfor As(III) oxidation reaction neglects the scaling effects with respect to the MnO2(s) concentration. A second order kineticequation that explicitly includes MnO2(s) concentration dependence is a more appropriate kinetic model to describe arsenicoxidation by MnO2(s) minerals. The arsenic adsorption reaction follows the Langmuir isotherm with the adsorption capacity of0.053μmol of As(V)/g of MnO2(s) at the tested conditions. The knowledge gained from the batch experiments was used to developa conceptual model for describing arsenic reactive transport at a column scale. The proposed conceptual model was integratedwithin a reactive transport code that accurately predicted the breakthrough profiles observed in multiple column experiments. Thekinetic and adsorption process details obtained from the batch experiments were valuable data for scaling to predict the column-scale reactive transport of arsenic in MnO2(s)-containing sand columns.© 2007 Elsevier B.V. All rights reserved.
Keywords: Reactive transport; Ground water modeling; Contaminant transport; Scaling; Numerical modeling
1. Introduction
Management of groundwater systems contaminatedby various natural and anthropogenic arsenic (As)sources has been a major environmental problem thathas received increased attention in recent years. Sincearsenic is a toxic metalloid, high concentrations ofarsenic in groundwater pose considerable risk to human
⁎ Corresponding author. Tel.: +1 334 844 6268; fax: +1 334 8446290.
E-mail address: [email protected] (T.P. Clement).
0169-7722/$ - see front matter © 2007 Elsevier B.V. All rights reserved.doi:10.1016/j.jconhyd.2007.07.004
health (Smith et al., 2000). Currently, there is consid-erable interest in understanding the processes thatcontrol arsenic transport in contaminated groundwatersystems (Zhang and Selim, 2006). Such an understand-ing can be used to design efficient methods to treatcontaminated drinking water sources. Also, a funda-mental understanding of the transport, adsorption andoxidation of arsenic in groundwater systems will helpbetter manage and mitigate the overall risks posed byarsenic contamination.
The predominant forms of arsenic in water and soilare inorganic, which can be present in the form ofarsenate [As(V)] or arsenite [As(III)]. The toxicity of

31T. Radu et al. / Journal of Contaminant Hydrology 95 (2008) 30–41
arsenic depends on its chemical form and As(III) is themore toxic species (Chris Le et al., 2004). The presenceof arsenic in natural waters is controlled by adsorption,ion exchange, dissolution/precipitation, and redoxreactions. In general, As(III) tends to bind more weaklyto soils compared to As(V) and hence is more solubleand difficult to remove from contaminated water(Edwards, 1994). Therefore, many research effortshave focused on oxidizing As(III) to the more easilyremovable As(V) (Katsoyiannis et al., 2004; Wilkie andHering, 1998). The MnO2(s), a well known naturaloxidizing agent, has often been used to oxidize As(III)to As(V) (Chiu and Hering, 2000; Driehaus et al., 1995;Manning et al., 2002).
Mn (hydr)oxides typically appear in natural soils inconcentrations much lower than those of Fe and Al(hydr)oxides (Kent and Fox, 2004); however,MnO2(s) isan excellent oxidant, and it strongly influences arsenicspeciation. MnO2(s) can exist in nature in three poly-morphic forms: birnessite (δ-MnO2(s)), cryptomelane(α-MnO2(s)) and pyrolusite (β-MnO2(s)) (Oscarsonet al., 1983). X-ray diffraction analysis indicates thatboth birnessite and cryptomelane have relatively poorcrystalline structures and relatively high specific surfacearea, while pyrolusite has a highly ordered crystallinestructure (Oscarson et al., 1983). The point of zerocharge (pHpzc) values reported for birnessite, cryptome-lane and pyrolusite, are 2.3, 2.8 and 6.4, respectively(Oscarson et al., 1983). Therefore, pyrolusite would bepositively charged below pH∼ 6.4, while both birnessiteand cryptomelane would both be negatively chargedover the entire pH range of natural waters. Pyrolusite caninteract more strongly with arsenic oxyanions comparedto either birnessite or cryptomelane (Oscarson et al.,1983) hence it is a better adsorbent despite its highlycrystalline structure and low surface area. Pyrolusite iscommonly present in natural soils (Jardine and Taylor,1995; Liakopoulos et al., 2001); therefore, it is anexcellent choice for a fundamental experimental studyinvolving arsenic fate and transport in natural systems.Furthermore, the abundance of pyrolusite in naturalmanganese ores (Chakravarty et al., 2002) indicatethe potential for using pyrolusite to develop low costsolutions for treating arsenic contaminated drinkingwater (Anawar et al., 2003).
Researchers have used pyrolusite in column experi-ments to study its interactions with various environ-mental contaminants including Co(II)-EDTA complexes(Jardine and Taylor, 1995), Cr (Eary and Rai, 1987;Guha et al., 2001), Fe–CN complexes (Rennert et al.,2005), and Pu (Powell et al., 2006). Since the (hydr)oxides of Mn are widely distributed in soils and sedi-
ments as both discrete particles and as coatings on soil/sediment components (Oscarson et al., 1983), arsenicis likely to interact with MnO2(s) in natural environ-ments. Ouvrard et al. (2002a,b) examined the diffusion-controlled adsorption of As(V) on a natural MnO2(s).They reported that the reactive transport process wasaffected by nonlinear adsorption and intra-particle dif-fusion mechanisms.
Most of the MnO2(s)-mediated arsenic oxidationstudies have focused on the oxidative ability of birnessite(Manning et al., 2002; Scott and Morgan, 1995;Tournassat et al., 2002), and only a limited amount ofdata is available for As(III) oxidation by pyrolusiteminerals (Oscarson et al., 1983). Furthermore, nearly allthe published work examined the oxidation of As(III) inbatch experiments (Manning et al., 2002; Scott andMorgan, 1995) and only a few studies have used columnexperiments to examine the interactions between As andMnO2(s) minerals in dynamic systems (Ouvrard et al.,2002a,b). More importantly, to the best of our knowl-edge, no one has addressed the issue of scaling batch-observed arsenic adsorption and oxidation reactionparameters to predict the reactive transport scenarios ina column-scale system.
The overall aim of this research effort was to studythe interactions of solid MnO2(pyrolusite) and arsenic ata batch scale and use the knowledge to develop ascalable reactive transport model that can utilize batch-scale data to predict column-scale transport scenarios.Therefore, the objective of the first phase of this studywas to complete batch experiments to evaluate thekinetics of As(III) oxidation using pyrolusite, and alsocharacterize pyrolusite's ability to adsorb variousarsenic species. The objective of the second phase wasto develop a column-scale reactive transport modelusing the reaction information obtained from the batch-scale experiments. The final objective was to completea set of column experiments to test the predictivecapability of the reactive transport model.
2. Experimental details
2.1. Materials and methods
The 13μM As stock solutions were prepared usingNaAsO2 (Fisher Scientific) for As(III) solutions, andAs2O5 (Alfa Aesar) for the As(V) solutions. A constantionic strength of 0.01M was achieved by the addition ofNaNO3. The pH value was adjusted to 4.5 using HCl. Inall experiments, ionic strength and pH were keptconstant. This pH was chosen because it allowed us tomaintain a relatively constant pH throughout the course

32 T. Radu et al. / Journal of Contaminant Hydrology 95 (2008) 30–41
of the experiments without the use of pH buffers thatcould potentially influence As and/or MnO2(s) chemis-try. The water used in all experiments was double-deionized water passed through a MILLI-Q system.
The MnO2(s) used in our experiments was a syntheticpyrolusite manufactured by J. T. Baker Inc. The powderX-ray diffraction method confirmed the modification ofthe oxide to be pyrolusite. The column experimentswere completed in a system packed with pure pyrolusiteand various mixtures of quartz sand (Iota 6, Unimin)and pyrolusite. The Mn contents in three samples ofpyrolusite-sand mixtures were determined by the aciddigestion method (EPA, 1996) to ensure uniformmixing. The porosities of pure sand and pyrolusitewere obtained by gravimetric analyses using a glasspycnometer.
Aqueous samples were analyzed for total arsenicconcentration by atomic absorption spectrometry usinga Perkin Elmer HGA-600 Graphite Furnace and 3110Perkin Elmer Atomic Absorption Spectrometer (AAS)with an electrodeless discharge lamp (EDL) as theradiation source at a wavelength of 193.7nm. The sameinstrument was used for analyzing Mn concentrationswith a wavelength of 279.5nm and HCL lamp. All thesamples were filtered prior to analysis with a 0.45μmfilter (Fisher Scientific). Mn samples were diluted, ifrequired, prior to AAS analysis.
Separation of As(III) and As(V) species was obtainedusing an ion exchange column containing Dowex 1X50–100 mesh Cl− form resin (Ficklin, 1983). The As(V)species were adsorbed to the resin while As(III) passedthrough the resin column. The inlet and outlet solutionswere analyzed using AAS to estimate the concentrationvalues of Astotal and As(III), respectively. The concen-tration of As(V) was calculated as the differencebetween these two values. The arsenic species concen-trations in the column inlet solution were verified priorto each experiment and at the end of the experiments.
2.2. Batch experiments
Stock solutions of 13μM of As(III) and As(V) wereused for all batch experiments. The kinetics of As(III)oxidation was examined in four separate experimentswith 13μM As(III) and 0.25, 0.5, 2.0, and 10.0g/l ofMnO2(s), while As(III) and As (V) adsorption kineticexperiments were performed with 100g/l of MnO2(s).Experimental vials were shaken and samples were takenat selected time intervals. The As(V) adsorptionisotherm was evaluated from the data collected frombatch adsorption experiments. One gram of MnO2(s)was allowed to equilibrate with a liter of As(V) solutions
at various concentrations. After 48h of shaking, thecontents of the vials were filtered and analyzed forarsenic concentration.
2.3. Column experiments
Various combinations of sand and pyrolusite were dry-packed in a 7cm long and 1cm diameter glass column.Columns were packed with either pure pyrolusite or witha sand-pyrolusite mixture. Previous experiments in ourlaboratory (results not shown) had indicated that theadsorption of arsenic on the sand (IOTA-6 UNIMINquartz sand) used in our columns was negligible. Thecolumns were oriented vertically and solutions wereintroduced from the bottom up at a constant flow rate atthe ambient temperature ≈ 22–23°C. All the inletsolutions were open to the atmosphere. Solutions werepumped through the column using an Acuflow Series IIhigh performance liquid chromatography pump. Thecolumn was pre-conditioned to remove trapped air bypumping As-free, 0.01M NaNO3 solution with a pH of4.5 for 24h. Later, a solution containing 13μM As(III) orAs(V) was pumped through the column for 35 porevolumes. Effluent was collected using a Spectra/ChromCF-1 Fraction Collector and later analyzed to estimatearsenic concentration levels.
3. Results of batch experiments
3.1. Oxidation of As(III) by MnO2(s)
The observed concentrations of As and Mn species inthe batch kinetic experiments conducted using variousdoses of MnO2(s) are presented in Fig. 1. In all thesebatch experiments, as the As(III) concentration de-creased with time an equivalent amount of As(V)appeared in the solution; the total arsenic concentrationremained constant in all the experiments. This clearlyindicates that the decrease of As(III) in solution iscaused by the direct oxidation process which convertedAs(III) to As(V). Furthermore, since the total amountof arsenic in the solution remained constant over thetime, there was negligible adsorption of either As(III) orAs(V) on the MnO2(s). This data also indicated close toan one-to-one ratio between As(III) depletion and As(V)release. Scott and Morgan (1995) made similarobservations in their experimental study where adsorp-tion of the oxidation product As(V) onto the MnO2
surface (birnessite) was minimal. The observed half lifeof As(III) decay in our experiments ranged from 2daysin the 0.25g/l MnO2(s) experiments to 1h in the 10g/l ofMnO2(s) experiments.
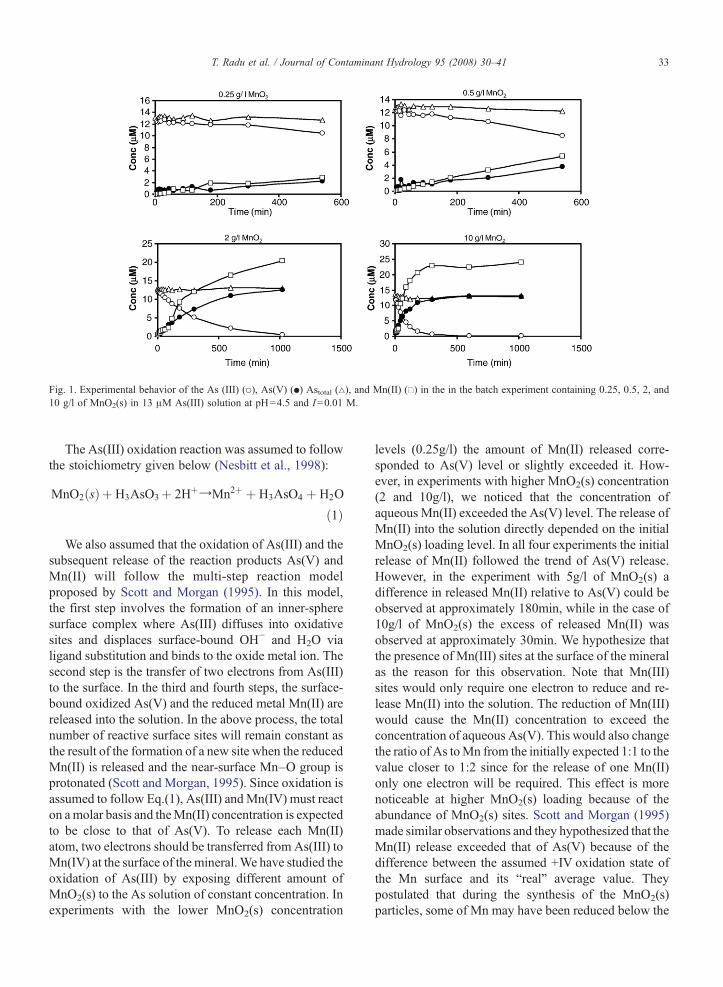
Fig. 1. Experimental behavior of the As (III) (○), As(V) (●) Astotal (△), and Mn(II) (□) in the in the batch experiment containing 0.25, 0.5, 2, and10 g/l of MnO2(s) in 13 μM As(III) solution at pH=4.5 and I=0.01 M.
33T. Radu et al. / Journal of Contaminant Hydrology 95 (2008) 30–41
The As(III) oxidation reaction was assumed to followthe stoichiometry given below (Nesbitt et al., 1998):
MnO2 sð Þþ H3AsO3þ 2HþYMn2þ þ H3AsO4 þ H2O
ð1Þ
We also assumed that the oxidation of As(III) and thesubsequent release of the reaction products As(V) andMn(II) will follow the multi-step reaction modelproposed by Scott and Morgan (1995). In this model,the first step involves the formation of an inner-spheresurface complex where As(III) diffuses into oxidativesites and displaces surface-bound OH− and H2O vialigand substitution and binds to the oxide metal ion. Thesecond step is the transfer of two electrons from As(III)to the surface. In the third and fourth steps, the surface-bound oxidized As(V) and the reduced metal Mn(II) arereleased into the solution. In the above process, the totalnumber of reactive surface sites will remain constant asthe result of the formation of a new site when the reducedMn(II) is released and the near-surface Mn–O group isprotonated (Scott and Morgan, 1995). Since oxidation isassumed to follow Eq.(1), As(III) andMn(IV) must reacton amolar basis and theMn(II) concentration is expectedto be close to that of As(V). To release each Mn(II)atom, two electrons should be transferred from As(III) toMn(IV) at the surface of the mineral.We have studied theoxidation of As(III) by exposing different amount ofMnO2(s) to the As solution of constant concentration. Inexperiments with the lower MnO2(s) concentration
levels (0.25g/l) the amount of Mn(II) released corre-sponded to As(V) level or slightly exceeded it. How-ever, in experiments with higher MnO2(s) concentration(2 and 10g/l), we noticed that the concentration ofaqueous Mn(II) exceeded the As(V) level. The release ofMn(II) into the solution directly depended on the initialMnO2(s) loading level. In all four experiments the initialrelease of Mn(II) followed the trend of As(V) release.However, in the experiment with 5g/l of MnO2(s) adifference in released Mn(II) relative to As(V) could beobserved at approximately 180min, while in the case of10g/l of MnO2(s) the excess of released Mn(II) wasobserved at approximately 30min. We hypothesize thatthe presence of Mn(III) sites at the surface of the mineralas the reason for this observation. Note that Mn(III)sites would only require one electron to reduce and re-lease Mn(II) into the solution. The reduction of Mn(III)would cause the Mn(II) concentration to exceed theconcentration of aqueous As(V). This would also changethe ratio of As toMn from the initially expected 1:1 to thevalue closer to 1:2 since for the release of one Mn(II)only one electron will be required. This effect is morenoticeable at higher MnO2(s) loading because of theabundance of MnO2(s) sites. Scott and Morgan (1995)made similar observations and they hypothesized that theMn(II) release exceeded that of As(V) because of thedifference between the assumed +IV oxidation state ofthe Mn surface and its “real” average value. Theypostulated that during the synthesis of the MnO2(s)particles, some of Mn may have been reduced below the
34 T. Radu et al. / Journal of Contaminant Hydrology 95 (2008) 30–41
+IVoxidation state to the +III or +II oxidation states. Thecomputed apparent oxidation state for their systems were+3 .6 and + 3.42. In the published literature, Kanungoand Mahapatra (1989) reported an average oxidationstate of 3.62 for a similar system.
As(III) oxidation by MnO2(s) is commonly repre-sented by a (pseudo-) first-order kinetic expression ofthe form (Oscarson et al., 1983; Scott and Morgan,1995; Thanabalasingam and Pickering, 1986):
d As IIIð Þ½ �dt
¼ �kobs As IIIð Þ½ � ð2Þ
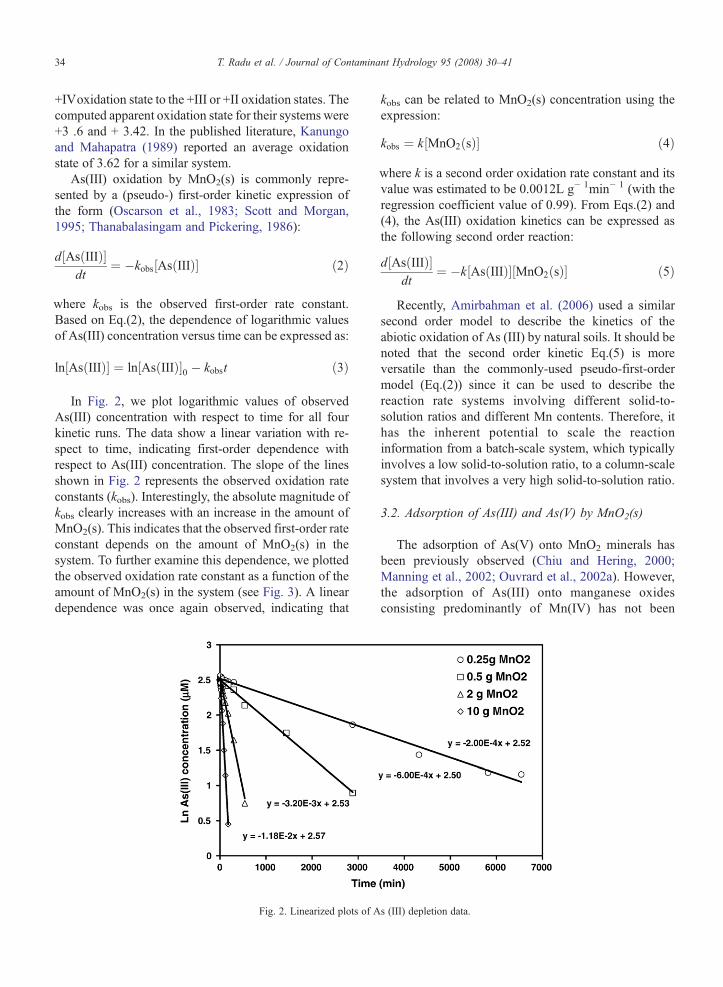
where kobs is the observed first-order rate constant.Based on Eq.(2), the dependence of logarithmic valuesof As(III) concentration versus time can be expressed as:
ln As IIIð Þ½ � ¼ ln As IIIð Þ½ �0 � kobst ð3Þ
In Fig. 2, we plot logarithmic values of observedAs(III) concentration with respect to time for all fourkinetic runs. The data show a linear variation with re-spect to time, indicating first-order dependence withrespect to As(III) concentration. The slope of the linesshown in Fig. 2 represents the observed oxidation rateconstants (kobs). Interestingly, the absolute magnitude ofkobs clearly increases with an increase in the amount ofMnO2(s). This indicates that the observed first-order rateconstant depends on the amount of MnO2(s) in thesystem. To further examine this dependence, we plottedthe observed oxidation rate constant as a function of theamount of MnO2(s) in the system (see Fig. 3). A lineardependence was once again observed, indicating that
Fig. 2. Linearized plots of A
kobs can be related to MnO2(s) concentration using theexpression:
kobs ¼ k MnO2 sð Þ½ � ð4Þ
where k is a second order oxidation rate constant and itsvalue was estimated to be 0.0012L g− 1min− 1 (with theregression coefficient value of 0.99). From Eqs.(2) and(4), the As(III) oxidation kinetics can be expressed asthe following second order reaction:
d As IIIð Þ½ �dt
¼ �k As IIIð Þ½ � MnO2 sð Þ½ � ð5Þ
Recently, Amirbahman et al. (2006) used a similarsecond order model to describe the kinetics of theabiotic oxidation of As (III) by natural soils. It should benoted that the second order kinetic Eq.(5) is moreversatile than the commonly-used pseudo-first-ordermodel (Eq.(2)) since it can be used to describe thereaction rate systems involving different solid-to-solution ratios and different Mn contents. Therefore, ithas the inherent potential to scale the reactioninformation from a batch-scale system, which typicallyinvolves a low solid-to-solution ratio, to a column-scalesystem that involves a very high solid-to-solution ratio.
3.2. Adsorption of As(III) and As(V) by MnO2(s)
The adsorption of As(V) onto MnO2 minerals hasbeen previously observed (Chiu and Hering, 2000;Manning et al., 2002; Ouvrard et al., 2002a). However,the adsorption of As(III) onto manganese oxidesconsisting predominantly of Mn(IV) has not been
s (III) depletion data.
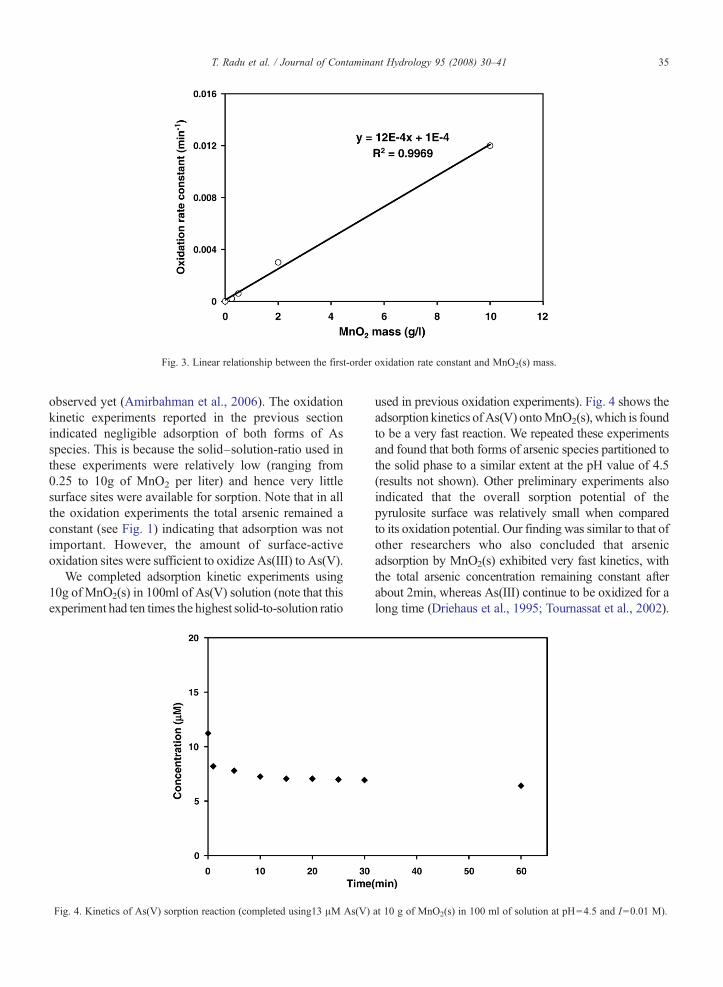
Fig. 3. Linear relationship between the first-order oxidation rate constant and MnO2(s) mass.
35T. Radu et al. / Journal of Contaminant Hydrology 95 (2008) 30–41
observed yet (Amirbahman et al., 2006). The oxidationkinetic experiments reported in the previous sectionindicated negligible adsorption of both forms of Asspecies. This is because the solid–solution-ratio used inthese experiments were relatively low (ranging from0.25 to 10g of MnO2 per liter) and hence very littlesurface sites were available for sorption. Note that in allthe oxidation experiments the total arsenic remained aconstant (see Fig. 1) indicating that adsorption was notimportant. However, the amount of surface-activeoxidation sites were sufficient to oxidize As(III) to As(V).
We completed adsorption kinetic experiments using10g of MnO2(s) in 100ml of As(V) solution (note that thisexperiment had ten times the highest solid-to-solution ratio
Fig. 4. Kinetics of As(V) sorption reaction (completed using13 μM As(V)
used in previous oxidation experiments). Fig. 4 shows theadsorption kinetics ofAs(V) ontoMnO2(s), which is foundto be a very fast reaction. We repeated these experimentsand found that both forms of arsenic species partitioned tothe solid phase to a similar extent at the pH value of 4.5(results not shown). Other preliminary experiments alsoindicated that the overall sorption potential of thepyrulosite surface was relatively small when comparedto its oxidation potential. Our finding was similar to that ofother researchers who also concluded that arsenicadsorption by MnO2(s) exhibited very fast kinetics, withthe total arsenic concentration remaining constant afterabout 2min, whereas As(III) continue to be oxidized for along time (Driehaus et al., 1995; Tournassat et al., 2002).
at 10 g of MnO2(s) in 100 ml of solution at pH=4.5 and I=0.01 M).
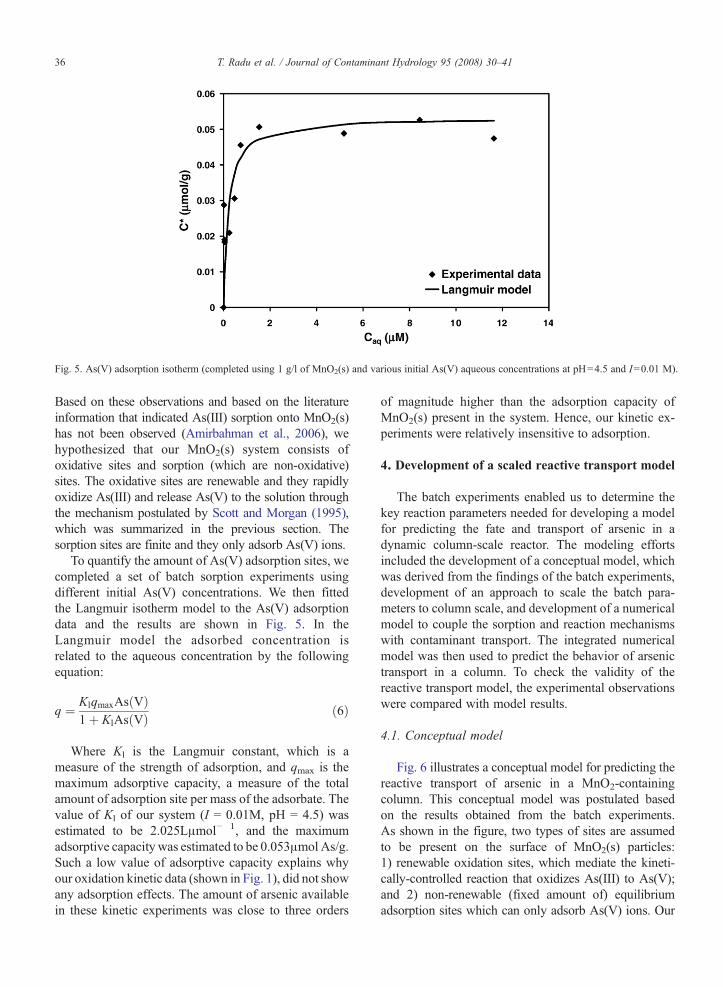
Fig. 5. As(V) adsorption isotherm (completed using 1 g/l of MnO2(s) and various initial As(V) aqueous concentrations at pH=4.5 and I=0.01 M).
36 T. Radu et al. / Journal of Contaminant Hydrology 95 (2008) 30–41
Based on these observations and based on the literatureinformation that indicated As(III) sorption onto MnO2(s)has not been observed (Amirbahman et al., 2006), wehypothesized that our MnO2(s) system consists ofoxidative sites and sorption (which are non-oxidative)sites. The oxidative sites are renewable and they rapidlyoxidize As(III) and release As(V) to the solution throughthe mechanism postulated by Scott and Morgan (1995),which was summarized in the previous section. Thesorption sites are finite and they only adsorb As(V) ions.
To quantify the amount of As(V) adsorption sites, wecompleted a set of batch sorption experiments usingdifferent initial As(V) concentrations. We then fittedthe Langmuir isotherm model to the As(V) adsorptiondata and the results are shown in Fig. 5. In theLangmuir model the adsorbed concentration isrelated to the aqueous concentration by the followingequation:
q ¼ KlqmaxAs Vð Þ1þ KlAs Vð Þ ð6Þ
Where Kl is the Langmuir constant, which is ameasure of the strength of adsorption, and qmax is themaximum adsorptive capacity, a measure of the totalamount of adsorption site per mass of the adsorbate. Thevalue of Kl of our system (I = 0.01M, pH = 4.5) wasestimated to be 2.025Lμmol− 1, and the maximumadsorptive capacity was estimated to be 0.053μmol As/g.Such a low value of adsorptive capacity explains whyour oxidation kinetic data (shown in Fig. 1), did not showany adsorption effects. The amount of arsenic availablein these kinetic experiments was close to three orders
of magnitude higher than the adsorption capacity ofMnO2(s) present in the system. Hence, our kinetic ex-periments were relatively insensitive to adsorption.
4. Development of a scaled reactive transport model
The batch experiments enabled us to determine thekey reaction parameters needed for developing a modelfor predicting the fate and transport of arsenic in adynamic column-scale reactor. The modeling effortsincluded the development of a conceptual model, whichwas derived from the findings of the batch experiments,development of an approach to scale the batch para-meters to column scale, and development of a numericalmodel to couple the sorption and reaction mechanismswith contaminant transport. The integrated numericalmodel was then used to predict the behavior of arsenictransport in a column. To check the validity of thereactive transport model, the experimental observationswere compared with model results.
4.1. Conceptual model
Fig. 6 illustrates a conceptual model for predicting thereactive transport of arsenic in a MnO2-containingcolumn. This conceptual model was postulated basedon the results obtained from the batch experiments.As shown in the figure, two types of sites are assumedto be present on the surface of MnO2(s) particles:1) renewable oxidation sites, which mediate the kineti-cally-controlled reaction that oxidizes As(III) to As(V);and 2) non-renewable (fixed amount of) equilibriumadsorption sites which can only adsorb As(V) ions. Our

Fig. 6. Conceptual model of the reactive transport system.
37T. Radu et al. / Journal of Contaminant Hydrology 95 (2008) 30–41
adsorption experiments showed that the maximumadsorption capacity (at I = 0.01M, pH = 4.5) of thesystem (total amount of sites) is 0.053μmol of As/g ofMnO2(s).
Conceptually, the fate and transport of As(III) within asaturated column can be described as follows: 1) As(III)enters the column and is partially or fully oxidized to As(V); 2) As(V) is then adsorbed onto the MnO2(s) grains;this step continues until the adsorption capacity is fullyexhausted; and 3) the portion of As(III) not oxidized aswell as the portion of As(V) not adsorbed are transportedfurther into the column.
4.2. Reactive transport model
The As(III) transport described in the aboveconceptual model was simulated using the followingreactive-transport equation:
A As IIIð Þ½ �At
¼ DA2 As IIIð Þ½ �Ax2
� vA As IIIð Þ½ �
Axþ Rcolumn As IIIð Þ½ � ð7Þ
where the aqueous-phase concentration of As(III) isgiven in units of [ML− 3], D is the hydrodynamicdispersion coefficient [L2T− 1], v is the pore watervelocity [LT− 1], and Rcolumn[As(III)] represents theremoval rate of As(III) oxidation [ML− 3T− 1] in thecolumn which can be evaluated using the oxidationkinetic Eq.(5). Using (5), Eq.(7) can be written as:
A As IIIð Þ½ �At
¼ DA2 As IIIð Þ½ �Ax2
� vA As IIIð Þ½ �
Ax� k As IIIð Þ½ � MnO2 sð Þ½ � ð8Þ
The reactive transport equation for As(V) species canbe written as:
A As Vð Þ½ �At
þ f qce
A qð ÞAt
¼ DA2 As Vð Þ½ �Ax2
� vA As Vð Þ½ �
Axþ Rcolumn As Vð Þ½ � ð9Þ
where aqueous-phase concentration of As(V) is given inunits of [ML− 3], q is the adsorbed phase concentration ofAs(V) related to the aqueous phase concentration by Eq.(6), ε is the porosity of the column, Rcolumn[As(V)] is theproduction rate of As(V) with magnitude identical toRcolumn[As(III)], ρc is the bulk density of sand–MnO2(s)column, and f is the fraction (by weight) MnO2(s)present in the column. It should be noted that the factor“f ρc” is an estimate of the concentration of MnO2(s)present in the system (i.e., the amount of MnO2(s)present per unit bulk volume). Eqs. (5) and (6) can beused to modify Eq.(9) as:
A As Vð Þ½ �At
1þ f qce
Klqmax
1þ Kl As Vð Þ½ �ð Þ2 !( )
¼ DA2 As Vð Þ½ �Ax2
� vA As Vð Þ½ �
Axþ k As IIIð Þ½ � MnO2 sð Þ½ � ð10Þ
Note that expressing the oxidation reaction rate as asecond order expression allows the kinetics to automat-ically scale with respect to the MnO2(s) concentration toreflect the local reaction conditions. On the other hand ifone employed only the more commonly used pseudo-first order kinetics (Oscarson et al., 1983; Scott and

Table 1Summary of transport parameters
Parameter Value
Inlet As concentration (μM) 13MnO2 column porosity 0.54Sand porosity 0.425Porosity of 50%MnO2 column 0.472Column height (cm) 7Column diameter (cm) 1Flow rate (ml/min) 0.3Longitudinal dispersivity (cm) 1.0Kl (Lμmol−1) 2.025k (Lg−1min−1) 0.0012qmax (μmol As/g of MnO2(s)) 0.053
38 T. Radu et al. / Journal of Contaminant Hydrology 95 (2008) 30–41
Morgan, 1995; Thanabalasingam and Pickering, 1986)it would be impossible to correctly scale the reactionrate.
4.3. Numerical solution strategy
Both As(III) and As(V) transport equations weresolved by using the fully-implicit finite-differenceapproach (Clement et al., 1997; Clement et al., 1996).The equation for As(V) transport involves nonlin-ear terms in As(V). The Picard iterative scheme wasused to handle this nonlinear term (Clement et al.,1994).
4.4. Reactive transport simulations and the design ofcolumn experiments
Column-scale numerical simulations were completedto simulate multiple hypothetical experimental scenar-ios. From these numerical experiments, the followingreactive transport scenarios were selected for furthertesting in the laboratory.
a) Reactive transport in a column packed with anextremely high level of MnO2(s): this column wasassumed to contain 100% of pure MnO2 solids. Thisextremely high MnO2(s) content system was selectedto study the reactive transport under the maximumextent of oxidation and adsorption. Two types of ex-periments were conducted with the input solution con-taining either As(III) (designated as Exp A) or As(V)(designated as Exp B).b) Reactive transport in a column packed with amedium level of MnO2(s): this column was con-structed to contain 50% (by wt.) of MnO2(s) and 50%sand (IOTA 6, UNIMIN quartz sand). This medium-level MnO2(s)-content system was selected to studytransport in a moderately reactive system. As(III)-containing solution was flushed through these col-umns to determine how the change in sand-MnO2(s)ratio will affect the oxidation and adsorption capacityof the column. This experiment is designated asExp C.c) Reactive transport in a column packed with anextremely low level of MnO2(s): since in all of theabove experiments the numerical model predictedcomplete oxidation of As(III) to As(V), we designed alowMnO2(s)-content experiment to study As(III) tran-sport in a low-level of reactivity system. The MnO2(s)content was designed sufficiently low to guaranteeonly partial oxidization of the influent As(III) solution.The results of this study is expected to test the ability of
our transport model to predict As(III) transport underpartial oxidation conditions. This experiment isdesignated as Exp D.
Three experimental columns of length 7cm and dia-meter 1cm, containing the selected levels of MnO2(s)-sand mixture, were used to test the three reactive tran-sport scenarios discussed above. The extremely highMnO2(s) system was packed with 12.9g of pyrolusiteminerals, the medium level MnO2(s) system was packedwith a 50% pyrolusite-sand mixture containing 5.3g ofMnO2(s) and 5.3g of sand. The extremely low MnO2(s)system was packed with 0.37g of MnO2(s) and 8.2g ofsand. A constant flow rate of 0.3ml/min was used in allthe column experiments. The As(III) or As(V) concen-tration in the influent solutions was 13μM. Other detailsof the column experiments are summarized in Table 1.The estimated values of MnO2(s) concentration (thevalues of “f ρc”) in the high, medium and low MnO2(s)-content experimental systems are: 2.35, 0.96 and0.067g/cm3, respectively (note: these concentrationvalues are expressed in terms of the bulk volume).
4.5. Results and discussion of column data
As(III) and As(V) breakthrough data from columnsthat were packed with 100% pyrolusite (Exp A andExp B) are compared with the model prediction in Fig. 7.Based on the batch data, one can predict complete oxi-dation of As(III) in the 100% MnO2(s) column. Thiswould imply that the breakthrough results from theAs(III)experiment (Exp A) would closely follow the results fromthe As(V) experiment (Exp B). Fig. 7 depicts Exp A andExp B results along with the theoretical model results.Due to complete oxidation, the breakthrough data fromExp A closely matches the data from Exp B. Com-plete oxidation of As(III) was further verified using the
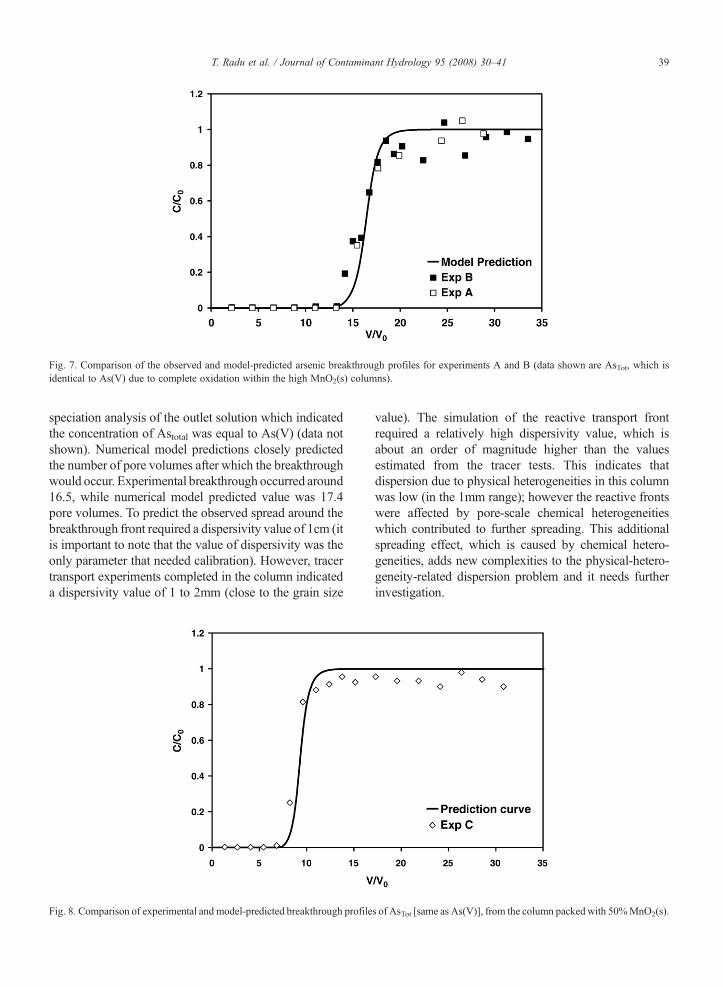
Fig. 7. Comparison of the observed and model-predicted arsenic breakthrough profiles for experiments A and B (data shown are AsTot, which isidentical to As(V) due to complete oxidation within the high MnO2(s) columns).
39T. Radu et al. / Journal of Contaminant Hydrology 95 (2008) 30–41
speciation analysis of the outlet solution which indicatedthe concentration of Astotal was equal to As(V) (data notshown). Numerical model predictions closely predictedthe number of pore volumes after which the breakthroughwould occur. Experimental breakthrough occurred around16.5, while numerical model predicted value was 17.4pore volumes. To predict the observed spread around thebreakthrough front required a dispersivity value of 1cm (itis important to note that the value of dispersivity was theonly parameter that needed calibration). However, tracertransport experiments completed in the column indicateda dispersivity value of 1 to 2mm (close to the grain size
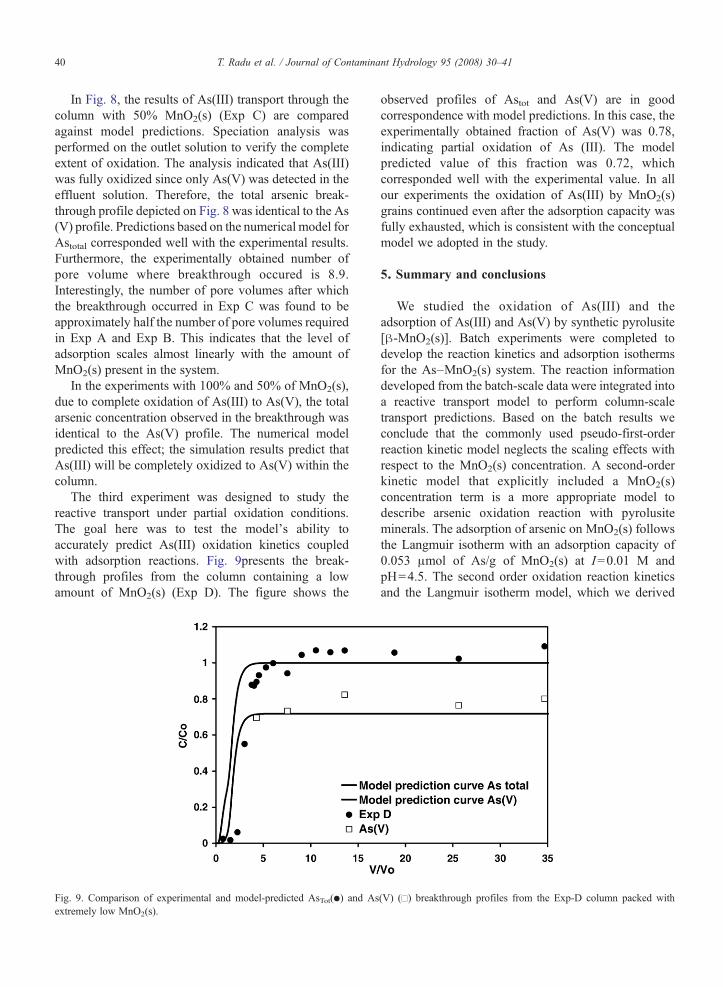
Fig. 8. Comparison of experimental and model-predicted breakthrough profile
value). The simulation of the reactive transport frontrequired a relatively high dispersivity value, which isabout an order of magnitude higher than the valuesestimated from the tracer tests. This indicates thatdispersion due to physical heterogeneities in this columnwas low (in the 1mm range); however the reactive frontswere affected by pore-scale chemical heterogeneitieswhich contributed to further spreading. This additionalspreading effect, which is caused by chemical hetero-geneities, adds new complexities to the physical-hetero-geneity-related dispersion problem and it needs furtherinvestigation.
s of AsTot [same as As(V)], from the column packed with 50%MnO2(s).
40 T. Radu et al. / Journal of Contaminant Hydrology 95 (2008) 30–41
In Fig. 8, the results of As(III) transport through thecolumn with 50% MnO2(s) (Exp C) are comparedagainst model predictions. Speciation analysis wasperformed on the outlet solution to verify the completeextent of oxidation. The analysis indicated that As(III)was fully oxidized since only As(V) was detected in theeffluent solution. Therefore, the total arsenic break-through profile depicted on Fig. 8 was identical to the As(V) profile. Predictions based on the numerical model forAstotal corresponded well with the experimental results.Furthermore, the experimentally obtained number ofpore volume where breakthrough occured is 8.9.Interestingly, the number of pore volumes after whichthe breakthrough occurred in Exp C was found to beapproximately half the number of pore volumes requiredin Exp A and Exp B. This indicates that the level ofadsorption scales almost linearly with the amount ofMnO2(s) present in the system.
In the experiments with 100% and 50% of MnO2(s),due to complete oxidation of As(III) to As(V), the totalarsenic concentration observed in the breakthrough wasidentical to the As(V) profile. The numerical modelpredicted this effect; the simulation results predict thatAs(III) will be completely oxidized to As(V) within thecolumn.
The third experiment was designed to study thereactive transport under partial oxidation conditions.The goal here was to test the model's ability toaccurately predict As(III) oxidation kinetics coupledwith adsorption reactions. Fig. 9presents the break-through profiles from the column containing a lowamount of MnO2(s) (Exp D). The figure shows the
Fig. 9. Comparison of experimental and model-predicted AsTot(●) and Asextremely low MnO2(s).
observed profiles of Astot and As(V) are in goodcorrespondence with model predictions. In this case, theexperimentally obtained fraction of As(V) was 0.78,indicating partial oxidation of As (III). The modelpredicted value of this fraction was 0.72, whichcorresponded well with the experimental value. In allour experiments the oxidation of As(III) by MnO2(s)grains continued even after the adsorption capacity wasfully exhausted, which is consistent with the conceptualmodel we adopted in the study.
5. Summary and conclusions
We studied the oxidation of As(III) and theadsorption of As(III) and As(V) by synthetic pyrolusite[β-MnO2(s)]. Batch experiments were completed todevelop the reaction kinetics and adsorption isothermsfor the As–MnO2(s) system. The reaction informationdeveloped from the batch-scale data were integrated intoa reactive transport model to perform column-scaletransport predictions. Based on the batch results weconclude that the commonly used pseudo-first-orderreaction kinetic model neglects the scaling effects withrespect to the MnO2(s) concentration. A second-orderkinetic model that explicitly included a MnO2(s)concentration term is a more appropriate model todescribe arsenic oxidation reaction with pyrolusiteminerals. The adsorption of arsenic on MnO2(s) followsthe Langmuir isotherm with an adsorption capacity of0.053 μmol of As/g of MnO2(s) at I=0.01 M andpH=4.5. The second order oxidation reaction kineticsand the Langmuir isotherm model, which we derived
(V) (□) breakthrough profiles from the Exp-D column packed with

41T. Radu et al. / Journal of Contaminant Hydrology 95 (2008) 30–41
from the batch-scale data, were coupled to a transportmodel to predict column-scale reactive transport. Theresults from the scaled reactive transport modelaccurately predicted the breakthrough profiles observedin multiple column experiments. Therefore, we con-clude that the oxidation and adsorption reactioninformation obtained from batch-scale experiments arevaluable data and, if appropriately scaled, they candescribe column-scale reactive transport.
Acknowledgements
This research was supported by the Office of Science(BER), U.S. Department of Energy, Grant no. DE-FG02–06ER64213 and also by the Strategic Environ-mental Research Program (SERDP) under the directionof Dr Andrea Leeson.
References
Amirbahman, A., Kent, D.B., Curtis, G.P., Davis, J.A., 2006. Kineticsof sorption and abiotic oxidation of arsenic(III) by aquifermaterials. Geochimica et Cosmochimica Acta 70 (3), 533–547.
Anawar, H.M., et al., 2003. Geochemical occurrence of arsenic ingroundwater of Bangladesh: sources and mobilization processes.Journal of Geochemical Exploration 77 (2–3), 109–131.
Chakravarty, S., Dureja, V., Bhattacharyya, G., Maity, S., Bhattachar-jee, S., 2002. Removal of arsenic from groundwater using low costferruginous manganese ore. Water Research 36 (3), 625–632.
Chiu, V.Q., Hering, J.G., 2000. Arsenic adsorption and oxidation atmanganite surfaces. 1. Method for simultaneous determination ofadsorbed and dissolved arsenic species. Environmental Science &Technology 34 (10), 2029–2034.
Chris Le, X., Lu, X., Li, X.-F., 2004. Arsenic speciation. AnalyticalChemistry 27–33.
Clement, T.P., Peyton, B.M., Skeen, R.S., Jennings, D.A., Petersen, J.N.,1997. Microbial growth and transport in porous media underdenitrification conditions: experiments and simulations. Journal ofContaminant Hydrology 24 (3–4), 269–285.
Clement, T.P., Wise, W.R., Molz, F.J., 1994. A physically-based, 2-dimensional, finite-difference algorithm for modeling variablysaturated flow. Journal of Hydrology 161 (1–4), 71–90.
Clement, T.P., Wise, W.R., Molz, F.J., Wen, M.H., 1996. A comparisonof modeling approaches for steady-state unconfined flow. Journalof Hydrology 181 (1–4), 189–209.
Driehaus, W., Seith, R., Jekel, M., 1995. Oxidation of arsenate(III)with manganese oxides in water-treatment. Water Research 29 (1),297–305.
Eary, L.E., Rai, D., 1987. Kinetics of chromium(III) oxidation tochromium(VI) by reaction with manganese dioxide. Environmen-tal Science and Technology 21 (12), 1178–1184.
Edwards, M., 1994. Chemistry of arsenic removal during coagulationand Fe–Mn oxidation. Journal American Water Works Association86 (9), 64–78.
EPA, 1996. Method 3050b: Acid digestion of sediments, sludges, andsoils.
Ficklin, W.H., 1983. Separation of arsenic(III) and arsenic(V) inground waters by ion-exchange. Talanta 30 (5), 371–373.
Guha, H., Saiers, J.E., Brooks, S., Jardine, P., Jayachandran, K., 2001.Chromium transport, oxidation, and adsorption in manganese-coatedsand. Journal of Contaminant Hydrology 49 (3–4), 311–334.
Jardine, P.M., Taylor, D.L., 1995. Kinetics and mechanisms of Co(II)EDTA oxidation by pyrolusite. Geochimica et Cosmochimica Acta59 (20), 4193–4203.
Kanungo, S.B., Mahapatra, D.M., 1989. Interfacial properties of somehydrous manganese dioxides in 1–1 electrolyte solution. Journal ofColloid and Interface Science 131 (1), 103–111.
Katsoyiannis, I.A., Zouboulis, A.I., Jekel, M., 2004. Kinetics of bacterialAs(III) oxidation and subsequent As(V) removal by sorption ontobiogenic manganese oxides during groundwater treatment. Industrial& Engineering Chemistry Research 43 (2), 486–493.
Kent, D.B., Fox, P.M., 2004. The influence of groundwater chemistryon arsenic concentrations and speciation in a quartz sand andgravel aquifier. Geochemical Transactions 5 (1), 1–12.
Liakopoulos, A., Glasby, G.P., Papavassiliou, C.T., Boulegue, J., 2001.Nature and origin of the Vani manganese deposit, Milos, Greece:an overview. Ore Geology Reviews 18 (3–4), 181–209.
Manning, B.A., Fendorf, S.E., Bostick, B., Suarez, D.L., 2002. Arsenic(III) oxidation and arsenic(V) adsorption reactions on syntheticbirnessite. Environmental Science & Technology 36 (5), 976–981.
Nesbitt, H.W., Canning, G.W., Bancroft, G.M., 1998. XPS study ofreductive dissolution of 7 angstrom-birnessite by H3AsO3, withconstraints on reaction mechanism. Geochimica Et CosmochimicaActa 62 (12), 2097–2110.
Oscarson, D.W., Huang, P.M., Liaw, W.K., Hammer, U.T., 1983.Kinetics of oxidation of arsenite by various manganese dioxides.Soil Science Society of America Journal 47 (4), 644–648.
Ouvrard, S., Simonnot, M.O., de Donato, P., Sardin, M., 2002a.Diffusion-controlled adsorption of arsenate on a natural manga-nese oxide. Industrial & Engineering Chemistry Research 41 (24),6194–6199.
Ouvrard, S., Simonnot, M.O., Sardin, M., 2002b. Reactive behaviourof natural manganese oxides toward the adsorption of phosphateand arsenate. Industrial & Engineering Chemistry Research 41(11), 2785–2791.
Powell, B.A., et al., 2006. Plutonium oxidation and subsequentreduction by Mn(IV) minerals in Yucca Mountain tuff. Environ-mental Science & Technology 40 (11), 3508–3514.
Rennert, T., Pohlmeier, A., Mansfeldt, T., 2005. Oxidation offerrocyanide by birnessite. Environmental Science and Technology39 (3), 821–825.
Scott, M.J., Morgan, J.J., 1995. Reactions at oxide surfaces.1.Oxidation of As(III) by synthetic birnessite. EnvironmentalScience & Technology 29 (8), 1898–1905.
Smith, A.H., Lingas, E.O., Rahman, M., 2000. Contamination ofdrinking-water by arsenic in Bangladesh: a public health emergency.Bulletin of the World Health Organization 78 (9), 1093–1103.
Thanabalasingam, P., Pickering, W.F., 1986. Effect of pH oninteraction between arsenic(III) or arsenic(V) and manganese(IV)oxide. Water, Air, and Soil Pollution 29 (2), 205–216.
Tournassat, C., Charlet, L., Bosbach, D., Manceau, A., 2002. Arsenic(III) oxidation by birnessite and precipitation of manganese(II)arsenate. Environmental Science & Technology 36 (3), 493–500.
Wilkie, J.A., Hering, J.G., 1998. Rapid oxidation of geothermalarsenic(III) in streamwaters of the eastern Sierra Nevada.Environmental Science and Technology 32 (5), 657–662.
Zhang, H., Selim, H.M., 2006. Modeling the transport and retention ofArsenic (V) in soils. Soil Science Society of America Journal 70,1677–1687.





























