Embed Size (px)
Citation preview

Nandkumar Chodankar Ph D TechDIA pACI Chair & DIA Board Member
DIA International Conference
“Quality of Active Pharmaceutical Ingredients”In collaboration with EMEA, EDQM, EU, FDA
& WHO
Disclaimer: The views expressed in this presentation are the personal opinions of the speaker and do not necessarily represent the views of the Organization

Presentation Flow
Objectives of Regulations
History of Indian regulatory requirements
Acts & rules - which regulates Indian Pharmaceutical Industry
Requirements for the Quality of API
Regulatory approval processes
2/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

Objective of Regulation- Quality
The Objective of Drugs & Cosmetics Act is to ensure that public (consumers) are supplied with safe and efficacious drugs and cosmetics.
Responsibility of Manufacturers: Are responsible for quality of drugs manufactured by them.
Responsibility of the Government Regulatory Agencies:Responsible to monitor the quality of drugs by periodic inspections of the manufacturing and sales premises for confirmation to the provisions of Drugs & Cosmetics Act and monitoring the quality of drugs moving in the market by carrying out post market surveillance.
3/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

History: Pre- Independence
1927 Henry Gidney’s resolution to legislate the manufacture and sale of drugs.
1930 Govt. appointed R. N. Chopra Committee to control menaceof spurious, adulterated, misbranded drugs. (Cinchona bark)
1931 Committee submitted Report
1935 After passing Govt. of India Act, Drug became provincial subject.
1939 Introduction of Drug Bill
1940 Drugs Act enacted
1945 In order to provide procedures to the industry rules were made under Drug Act
4/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

1948 Pharmacy Act
1954 Drugs and Magic Remedies (Objectionable advertisement Act)
1955 Amendments to Drugs & Cosmetics Act, extension of definitions
1960 Amendments --- DTAB, Powers to central Govt., etc.
1962 Amendments ---- Cosmetics manufacture regulated, misbranded, import prohibited etc.
1964 Amendments ---- provision for Ayurvedic, Siddha.
1982 Widened scope & definition, empowered
1985 Narcotics Drugs and Psychotropic Act
1986 Powers to consumers to draw samples.
1995 Drugs Price Control Order (DPCO)
History: Post- Independence
5/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

Composition of Drugs and Cosmetics Act
Chapter I Definitions
Chapter II Statutory committees and laboratories
Chapter III Import of drugs and cosmetics
Chapter IV Manufacture of Drugs and Cosmetics
Chapter V Siddha and Unani drugs
Chapter VI Miscellaneous
Books on Standards: Indian Pharmacopoeia (Inclusion of HIV Drugs monographs)
Homeopathic PharmacopoeiaAyurvedic Pharmacopoeia and Indian Pharmacopoeia – Veterinary
6/34Nandkumar Chodankar Ph D Tech6th Sept. 2009
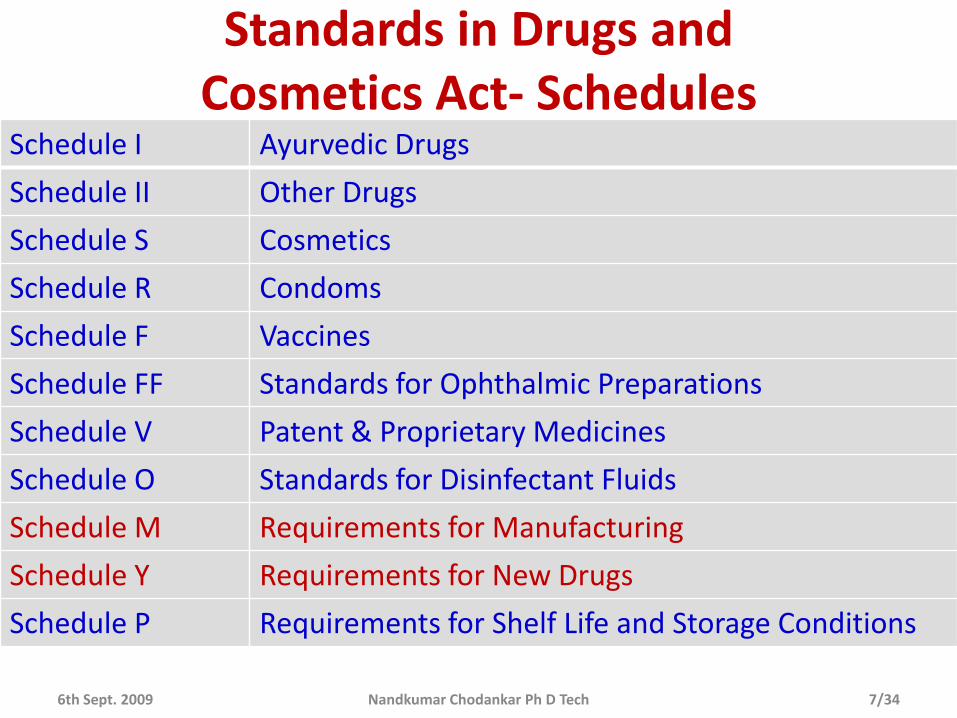
Standards in Drugs and Cosmetics Act- Schedules
Schedule I Ayurvedic Drugs
Schedule II Other Drugs
Schedule S Cosmetics
Schedule R Condoms
Schedule F Vaccines
Schedule FF Standards for Ophthalmic Preparations
Schedule V Patent & Proprietary Medicines
Schedule O Standards for Disinfectant Fluids
Schedule M Requirements for Manufacturing
Schedule Y Requirements for New Drugs
Schedule P Requirements for Shelf Life and Storage Conditions
7/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

Requirement for the Quality of API
Schedule M discusses about the GMP operations
(especially Chapter IV)
Rule No. 69, Rule No.122 and Schedule Y covers the filing requirements for a manufacturing license of New Drug / approval
8/34Nandkumar Chodankar Ph D Tech6th Sept. 2009
“Assumption can lead to disaster”

Schedule M1.General Requirements
Location & surroundings, Building and premises, Water System, Disposal of waste
2. Warehousing Area
3. Production area
4. Ancillary Areas
5. Quality Control Area
6. Personnel
7. Health, clothing andsanitation of workers
8. Manufacturing Operations and Controls
9. Sanitation in theManufacturing Premises
10. Raw Materials
11. Equipment
12. Documentation and Records
13. Labels and other Printed Materials
14. Quality Assurance
9/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

Schedule M15. Self Inspection and
Quality audit
16. Quality Control System
17. Specifications
18. Master Formula Records
19. Packing Records
20.Batch Packaging Records
21.Batch Processing Records
22.SOPs & Records for Receipt of materials, Sampling, Batch numbering, Testing
23. Reference Samples
24. Reprocessing and Recoveries
25. Distribution records
26. Validation and Process validation
27. Product Recalls
28. Complaints and Adverse Reactions
29. Site Master Files
10/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

Buildings & Facilities
Designed and constructed– To facilitate cleaning,
sanitizing, maintenance (incl. repairing), appropriate manufacturing operations (defined in physical areas or other control systems)
–To prevent mix-ups or contamination(chemical and microbiological)
Utilities: e.g. Water– “Quality of the water used
in the manufacture of APIs should be demonstrated to be suitable, for its intended use”
– Increasing purity of the API along the process increasing quality of the water- Finally Purified water
11/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

Process Equipment
Equipment shall be located, designed, constructed, adapted and maintained to suit the operations to be carried out
The layout and design of the equipment shall aim to minimize the risk of errors and permit effective cleaning and maintenance in order to avoid cross-contamination, build-up of dust or dirt
To avoid accidental contamination, wherever possible, non-toxic / edible grade lubricants shall be used
12/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

MaterialsMaterial management
13/34Nandkumar Chodankar Ph D Tech6th Sept. 2009
- All incoming materials shall be purchased from
approved sources
- Evaluation of suppliers of critical materials shall
be based on Historical experience,
Questionnaire, Analytical verification of samples,
Audit of the supplier
Receipt Quarantine Sampling Testing (min. Identity)
Reject ApprovedStorageReevaluation
Indian rules are somewhat silent on “acceptance of hazardous materials”
and does not define the starting material
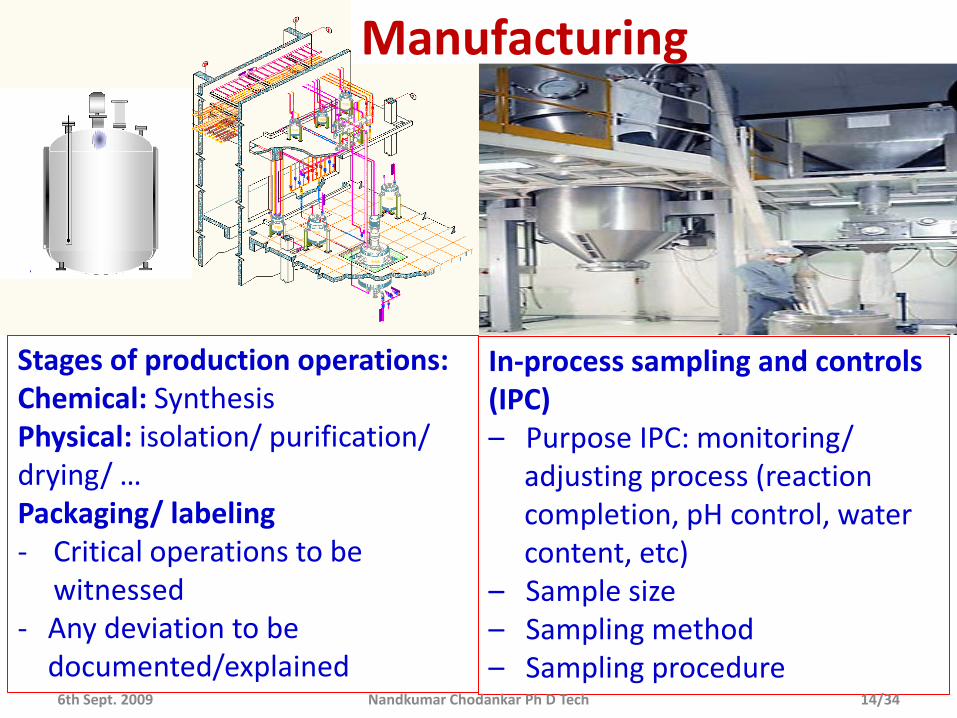
Stages of production operations:Chemical: SynthesisPhysical: isolation/ purification/ drying/ …Packaging/ labeling- Critical operations to be
witnessed- Any deviation to be
documented/explained
In-process sampling and controls (IPC)– Purpose IPC: monitoring/
adjusting process (reaction completion, pH control, water content, etc)
– Sample size– Sampling method– Sampling procedure
14/34Nandkumar Chodankar Ph D Tech6th Sept. 2009
Manufacturing

Blending of intermediates or APIs“Combining materials with the same specifications to produce a homogeneous product” Blend should
– be traceable back to all individual batches– only contain individual batches that conform to
specifications No blending of OOS batches Blending should include testing of
parameters that may be affected: Particle size distribution Bulk density Tapped density
(May be we should include Color)
Manufacturing
15/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

Where reprocessing is necessary, written procedures shall be established and approved by the QA that shall specify the conditions and limitations of repeating chemical reactions. Such reprocessing shall be validated. (Scientific Rational is important)
If the product batch has to be reprocessed, an investigation shall be carried out into the causes necessitating re-processing and appropriate corrective measures shall be taken for prevention of recurrence.
Re-processed batch shall be subjected to stability evaluation. Recovery of the product residue may be carried out, if permitted,
in the master production and control records by incorporating it in subsequent batches of the product.
Reprocessing and Recoveries
16/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

Quality controlOut of specifications (OOS) results to be investigated according to SOP including e.g.,
– checklist of potential defects in laboratory– checklist of potential defects in production– check list of sampling and sampling devices– guidance on re-sampling and re-testing– testing of control (reserve) sample [RS, WRS and storage]
Standards
– Reagents and standard solutions with “use before” date– Primary reference standard: from documented source. If from
official source and if stored as stipulated: no testing necessary
– Secondary reference standards (working standards): compared (by testing) to primary standard prior to first use
17/34Nandkumar Chodankar Ph D Tech6th Sept. 2009
Schedule L1 Effective from Nov. 2010 will answer many of the questions

Quality control
Stability monitoring Purpose: to confirm appropriate
storage conditions and retest or expiry date Samples stored in containers simulating market
containerNormally, first 3 commercial production batches, then
1 batch/year placed on the on-going stability program
18/34Nandkumar Chodankar Ph D Tech6th Sept. 2009
Mention Expiration period instead of Retest period for APIs (?)

Qualification: DQ, IQ, OQ, PQ “Before starting process validation activities, appropriate qualification of facilities (rooms), equipment, systems (e.g., steam, gases, HVAC,...) should be completed”.
Validation of processesProcesses and procedures shall be established on the basis of validation study and undergo periodic revalidation to ensure that they remain capable of achieving the intended results
Validation of analytical methods– Degree of validation to be in relation to purpose of analysis
and stage of production process (see also ICH Q2A, Q2B)– Analytical methods from recognized pharmacopoeia: only
suitability for use
Validation
19/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

Change control
Any system needs to constantly be adapted/ optimized (deviations, OOS,..)Formal (written) proposal for changeApproval of change by Quality UnitImplementation of changeEvaluation of impact of the implemented change
20/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

Documentation All quality relevant activities should be
described (written procedures, instructions) and documented (records)
“Life cycle” of documents according to written procedure
– Issuance, review, approval, distribution, retention, withdrawal, revision history of documents
Records on major equipment use and cleaning including date, signature and– Use: product, batch-no.– Cleaning (sanitization, sterilization): SOP– Maintenance: preventative, during production, repair
Water System Qualification and trending21/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

Records on materials: traceability, i.e., origin and use Master production/ control instructions
– To ensure uniformity from batch to batch– Issued and checked independently
Batch production/ control records– Should include all significant steps
Batch production/ control records review– To be performed accordingly to written procedure– Review of critical steps such as OOS reports and
deviation investigations and their impact on product quality, check for missing records, incomplete or illegible entries, compliance with specifications, … before batch release
22/34Nandkumar Chodankar Ph D Tech6th Sept. 2009
Documentation

A typical Drug Development Cycle
23/34Nandkumar Chodankar Ph D Tech6th Sept. 2009
Discovery
Development
API
Drug Product
Packaged product for market

Regulatory approval processes
Regulatory Authorities in India
Central Drug Regulatory Authorities
- CDSCO – Central Drugs Standard Control Organization
DCGI, DELHI – Drugs Controller General of India- CBN – Central Bureau of Narcotics NCI, GWALIOR –
Narcotics Commissioner of India
State Regulatory Authorities
- Each state is having state licensing authoritye.g., DCA, Andra Pradesh - Director of Drugs Control
AdministrationDC, Tamil Nadu – Director of Drugs Control FDA, PONDY – Licensing Authority,
Food and Drugs Administration
24/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

Drug Approval category in India:
New drug – First time in India
Approved new drug- Approved by DCG(I), but not more than 4 years old
Approved drug - Approved by DCG(I), but more than 4 years old
Approved drugs details are Available at < www.cdsco.nic.in >
25/34Nandkumar Chodankar Ph D Tech6th Sept. 2009
Regulatory approval processes
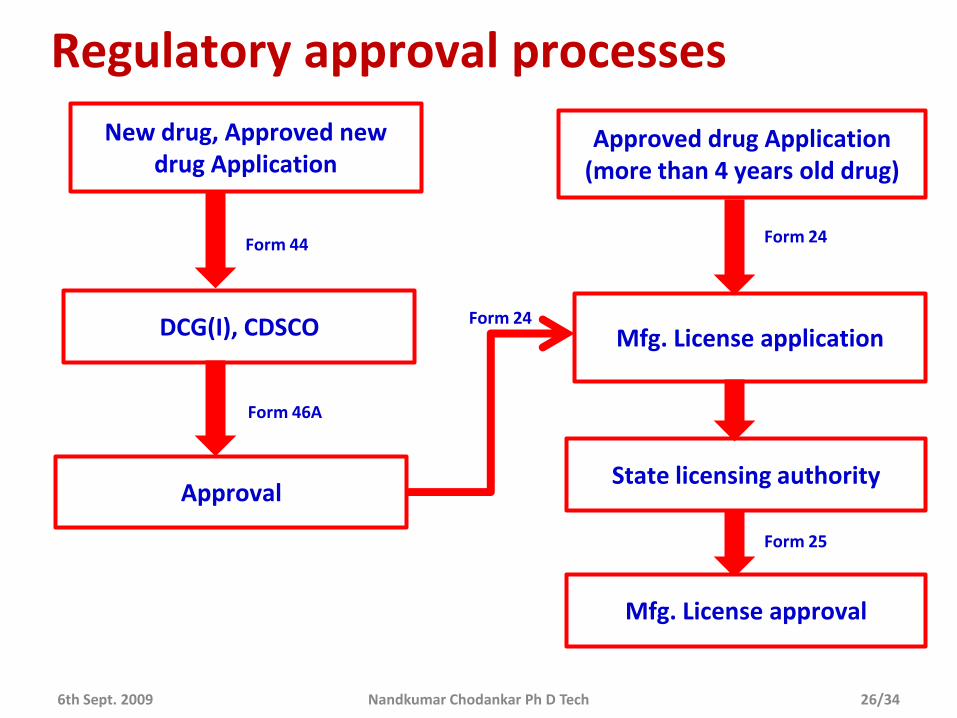
New drug, Approved new drug Application
DCG(I), CDSCO
Form 44
Form 46A
Approval
Mfg. License application
Approved drug Application (more than 4 years old drug)
Form 24
State licensing authority
Mfg. License approval
Form 24
Form 25
26/34Nandkumar Chodankar Ph D Tech6th Sept. 2009
Regulatory approval processes

Clinical trial
Chemical and pharmaceutical information.
Animal Toxicology
Animal pharmacology
Regulatory status in other countries
Marketing information
Post-marketing surveillance study
27/34Nandkumar Chodankar Ph D Tech6th Sept. 2009
Data requirements for filing –
Schedule Y
Recently IPR status is also needs to be submitted along with the
Pharmaceutical category and route of administration.

Clinical Trials Clinical trials required to be carried out in the country
before a new drug is approved for marketing depend on the status of the drug in other countries.
If the drug is already approved/marketed and internationally available, Phase III trials are required
For new drug substances discovered in India, clinicaltrials are required to be carried out in India right fromphase I
28/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

Chemical and Pharmaceutical Information
Brief description of the drug and the therapeutic class
Physiochemical data
- Chemical name, Structure,
Empirical formula
Molecular weight
- Physical properties
Description, solubility, rotation, partition coefficient,
dissociation constant, crystalline / amorphous, water of
hydration, flow properties (even micro metrics)
29/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

Chemical and Pharmaceutical Information
Analytical Data
Elemental analysis
Mass spectrum
NMR spectra
IR spectra
UV spectra
Polymorphic identification
Particle size distribution
Crystalline / amorphous,
Water of hydration,
Flow properties (even micro metrics)
5.97
5
13.5
35
25.5
17
28.9
55
36.6
49
40.9
47
AU
0.000
0.005
0.010
0.015
0.020
0.025
0.030
0.035
0.040
Minutes
0.00 5.00 10.00 15.00 20.00 25.00 30.00 35.00 40.00 45.00 50.00
30/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

Chemical and Pharmaceutical Information
Complete monograph specification- Identification - Identity/quantification of impurities - Enantiomeric purity- Assay
Validations- Assay method - Impurity estimation method - Residual Solvents/ OVI estimation method
31/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

Chemical and Pharmaceutical Information
Stability studies
- Study on final release specification
3 batches:
Long term 30°C ± 2°C/ 65%RH ± 5%RH – 12 months
Accelerated 40°C ± 2°C/ 75%RH ± 5%RH – 6 months
Photo stability study on min of 1 batch is required
32/34Nandkumar Chodankar Ph D Tech6th Sept. 2009
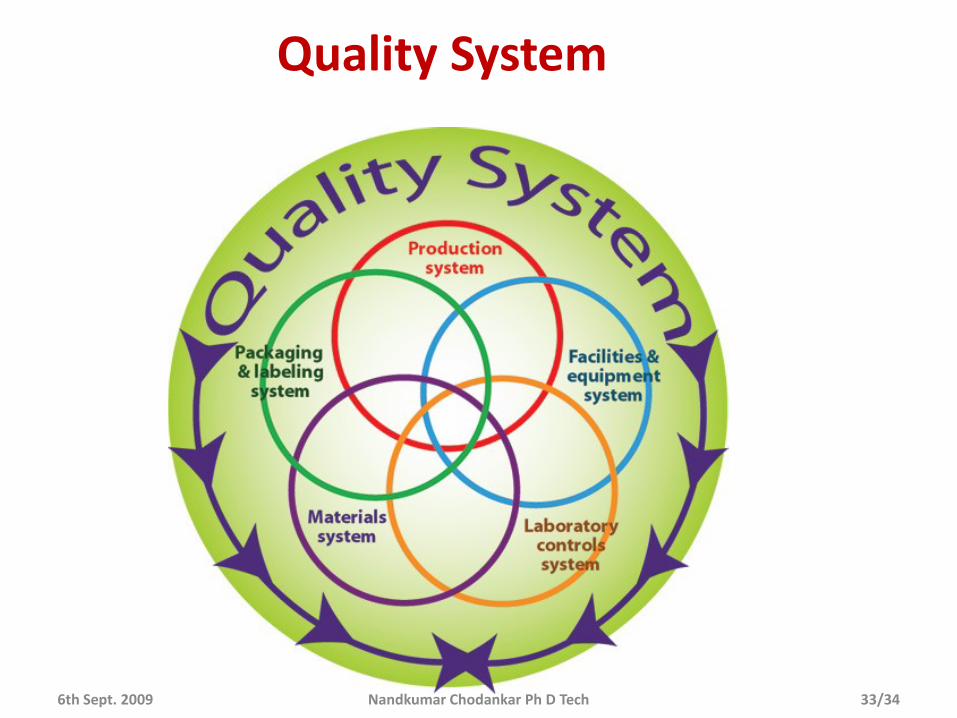
Quality System
33/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

Thank you!!!
Nandkumar Chodankar Ph D [email protected]
34/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

Back up Slides

GROWTH OF PHARMACEUTICAL INDUSTRY
1950-1960: Imports, Simple Formulations
(tinctures and mixtures)
1970-1980: APIs by chemical synthesis and
fermentation, different dosage forms
1980-1990: Growth and sophistication
1990-2000: Research, Biotechnology and Exports
36/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

REASONS FOR INITIATIVES
A) Globalization
B) New Technologies
C) New Therapies
37/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

REASONS FOR INITIATIVES CONTD..
A) Globalization
Opportunities in:
–Contract Manufacturing
–Contract Clinical Research
–Contract R & D.
38/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

REASONS FOR INITIATIVES CONTD..
–Contract Documentation & Testing.
–Neutraceuticals
–Herbal Medicines
39/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

REASONS FOR INITIATIVES CONTD..
B) New Technologies
–Vaccines
–Recombinant DNA
–Tissue Culture
–Cell Biology
–Fermentation
–Chemical Synthesis
40/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

REASONS FOR INITIATIVES CONTD..
C)New Therapies
–Stem Cells
–Genomics
41/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

INITIATIVES TAKEN BY THE GOVERNMENT
Upgradation of G.M.P. requirements (Schedule M)
Issuance of guidelines for clinical trials and bio-equivalance studies (GCPs)
42/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

INITIATIVES TAKEN BY THE GOVERNMENT CONTD..
Good Laboratory Practices for the Government as well as Private Testing Laboratories.
Concepts of DMF for drugs imported into the country.
G.M.P. for Ayurvedic Drugs.
43/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

IMPORTANT COMMITTEES CONSTITUTED BY THE GOVERNMENT
• HATHI COMMITTEE
• SPECIAL TASK FORCE AND
• MASHELKAR COMMITTEE
44/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

INITIATIVES IN THE PIPELINE
Guidelines for manufacture, sale and distribution of medical devices.
Guidelines for good distribution and storage practices for the hospitals.
Guidelines for manufacture of biotech products.
45/34Nandkumar Chodankar Ph D Tech6th Sept. 2009

INITIATIVES IN THE PIPELINE CONTD..
Guidelines for some of the important G.M.P. components i.e. stability testing, validation requirements etc.
Guidelines for Neutraceuticals.
46/34Nandkumar Chodankar Ph D Tech6th Sept. 2009





























