Embed Size (px)
Citation preview

THE JOURNAL OF BIOLOGICAL CHEMISTRY D 1993 by The American Society for Biochemistry and Molecular Biology, Inc. Vol. 268, No. 13, Issue of May 5, p p , 9496-9503,1993
Printed in U.S.A.
Differences in the Properties and Enzymatic Specificities of the Two Active Sites of Angiotensin I-converting Enzyme (Kininase 11) STUDIES WITH BRADYKININ AND OTHER NATURAL PEPTIDES*
(Received for publication, August 3, 1992, and in revised form, December 31, 1992)
Emmanuel Jaspard, Lei Wei, and Franpois Alhenc-Gelas$ From the Institut National de la Sante et de la Recherche Medicale U367 and U36, 17 rue du Fer a Moulin, 75005 Paris, France
Angiotensin I-converting enzyme (ACE, E.C.3.4.15.1) has been recently shown to contain two very similar domains, each of which bears a functional active site hydrolyzing Hip-His-Leu or angiotensin I (AI). The substrate specificity of the two active sites of ACE was compared using wild-type recombinant ACE and mu- tants, where one active site is suppressed by deletion or inactivated by mutations of 2 histidines coordinat- ing an essential zinc atom. Both active sites converted bradykinin (BK) to BK“’ and BK”‘ with similar ki- netics and with KZp at least 30 times lower and kc,,/ K“,“ 10 times higher than for AI. The carboxyl-termi- nal active site, but not the amino-terminal site, was activated by chloride; however, chloride activation was minimal compared with AI. Both domains also hydrolyzed substance P and cleaved a carboxyl-ter- minal protected dipeptide and tripeptide. The car- boxyl-terminal active site was more readily activated by chloride and hydrolyzed substance P faster. Lutein- izing-hormone releasing hormone was hydrolyzed by both active sites, but hydrolysis by the amino-terminal active site was faster. It performed the endoproteolytic amino-terminal cleavage of this peptide at least 30 times faster than the carboxyl-terminal active site. Both active sites cleaved a carboxyl-terminal tripep- tide from luteinizing hormone-releasing hormone. Thus, both active sites of ACE possess dipeptidyl car- boxypeptidase and endopeptidase activities. However, only the carboxyl-terminal active site can undergo a chloride-induced alteration that greatly enhances the hydrolysis of AI or substance P, and the amino-termi- nal active site possesses an unusual amino-terminal endoproteolytic specificity for a natural peptide. This suggests physiologically important differences be- tween the subsites of the two active centers, and dif- ferent substrate specificity, despite the high degree of sequence homology.
Angiotensin I-converting enzyme (dipeptidyl carboxypep- tidase I, kininase 11, EC 3.4.15.1, ACE1) is a widely distributed zinc metallopeptidase that cleaves the carboxyl-terminal di-
* This work was supported by the Institut National de la Sant6 et de la Recherche Medicale and by a grant from the Bristol-Myers- Squibb Institute for Medical Research. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
$ To whom correspondence should be addressed. The abbreviations used are: ACE, angiotensin I-converting en-
zyme; AI, angiotensin I; BK, bradykinin; Hip, hippuryl; HPLC, high performance liquid chromatography; LH-RH, luteinizing hormone releasing hormone; Mes, 2-(N-morpho1ino)ethanesulfonic acid.
peptides of various oligopeptides. It has two well recognized physiological functions: the activation of angiotensin I (AI) to the vasoconstrictor octapeptide angiotensin I1 (I), and the inactivation of the vasodepressor nonapeptide bradykinin (BK) (2). But ACE has an unusual enzymatic specificity in vitro, as it can act either as a dipeptidyl carboxypeptidase or more like an endopeptidase, depending on the substrate. ACE cleaves protected dipeptides or tripeptide from the COOH terminus of some peptides, such as substance P and luteiniz- ing hormone releasing hormone (LH-RH) (3, 4). It can also cleave a tripeptide from BK after the removal of the carboxyl- terminal arginine by carboxypeptidase N. ACE displayed an- other endoproteolytic specificity with LH-RH, as it cleaves an NH2-terminal tripeptide (4). The hydrolysis of AI, sub- stance P, and LH-RH is strongly affected by chloride (1, 4).
The molecular cloning of human endothelial and mouse kidney ACES has revealed the unexpected complexity of the structure of this enzyme. ACE is probably derived from the duplication of an ancestral gene and comprises two very homologous domains (called here the N and C domains) that each contain a putative active site (5, 6). Molecular cloning of the testicular form of ACE, which is encoded by a shorter mRNA presumably corresponding to the ancestral form of the ACE gene but is equally active on AI, has revealed that testicular ACE corresponds to the C domain of endothelial ACE (7-9). This indicates that the C domain is enzymatically active but does not indicate the function(s) of the N domain. The function of the N domain was examined using recombi- nant enzymes expressed in Chinese hamster ovary cells (10, 11). Mutants had one domain suppressed by deletion or one active site inactivated by point mutations on putative critical residues. These experiments first indicated that both the N and C domains are in fact enzymatically active, as each removes the carboxyl-terminal dipeptide of Hip-His-Leu and AI, each with a zinc atom as cofactor. They also showed that the C domain may have higher catalytic constants than the N domain for these two substrates, is more readily activated by chloride, and that the two domains differ in their capacities to bind several competitive ACE inhibitors (11, 12). This strongly suggests structural differences between the two active sites and raises the question of their respective roles in the hydrolysis of BK and the multiple substrates of ACE. We have therefore characterized the substrate specificity of the two active centers of ACE and tested the hypothesis that different substrates may be preferentially cleaved by each active site. The kinetics of BK hydrolysis by ACE were studied in detail, as limited information is available despite the phys- iological importance of ACE in the metabolism of this sub- strate. Substance P and LH-RH were then studied because, unlike the other known ACE substrates, they undergo a primary endoproteolytic cleavage. Substance P, like BK and
9496

Active Sites of Angiotensin I-converting Enzyme 9497
AI, is probably also a true substrate of ACE in vivo (4). The results indicate that the active sites both possess dipeptidyl carboxypeptidase and endopeptidase specificities, hydrolyze BK, AI, and substance P, but differ in the kinetics of hydrol- ysis of these substrates and in the specificity of cleavage for LH-RH.
MATERIALS AND METHODS
Enzymes and Peptides-Wild-type membrane-bound recombinant and mutated ACEs were produced from Chinese hamster ovary cells transfected with human endothelial ACE cDNA or mutated DNAs (10, 11). The following mutants were studied: the full-length mu- tants in which the 2 zinc-binding histidine residues in either the N or C domain have been replaced by lysine residues (ACEK~OI,~~X, ACEK959,963), and the truncated mutants (N or C fragments) corre- sponding to either the N or C domains (Fig. 1). The enzymes were purified and quantified as described previously (11, 12). [2,3-prolyl- 3,4-3H]BK (specific activity 107 Ci/mmol) was from Du Pont-New England Nuclear; unlabeled BK, substance P, substance P-free acid, and LH-RH were from Bachem (Switzerland). Fragments BK'-7 and BK'-5 were synthesized by Dr. Du (Centre de Pharmacologie, CNRS- INSERM, Montpellier, France). Substance P fragments and LH-RH fragments were either from Bachem or Sigma. Captopril was a gift from the Bristol-Myers-Squibb Institute.
Standard Conditions for Enzyme Assays-All enzymatic reactions were performed at 37 "C, in 50 mM Hepes buffer, pH 7.5, in a final volume of 200 p1 unless otherwise stated. The reactions were stopped by adding 20-40 pl of 12% H3P04 when 0.5-8% substrate had been hydrolyzed.
All HPLC separations of the peptides were done on a reversed- phase column (Harwell, Spherisorb C,, 3-pm particles, 100 X 4 mm) preceded by an RP-8 endcapped guard column (LiChroCART 4-4, Merck, Germany) at a flow rate of 1 ml/min. Hydrolysis products were identified by comparison of retention times with those of au- thentic standards.
Hydrolysis of BK and BIC-7-Experiments with BK were per- formed in the presence of 0.014 p~ tritiated substrate (0.3 pCi). The use of BK tritiated on the Pro residues in position 2 and 3 led to the formation of radiolabeled products of the enzymatic reaction. These products were separated by HPLC under isocratic conditions (aceto- nitri1e:water (13:87), 0.045% trifluoroacetic acid, 0.075% triethyla- mine, pH 3). Unhydrolyzed BK was eluted by increasing the concen- tration of acetonitrile to 57%. The recovery of immunoreactive BK (12-120 ng, 0.05-0.54 p~ in our incubation conditions), as assayed by BK radioimmunoassay (13), was 94-100%. The eluates corre- sponding to the retention times of BK'-7 and BK1-5 were collected in two sets of nine fractions (250 pl). Pico-Fluor 15 (Packard) was added to all fractions, and they were counted in a 1211 Rackbeta liquid scintillation counter (LKB, Wallac, Sweden). The absolute amount
of each product was calculated from the fraction of radioactivity eluting in the corresponding peak (Fig. 2) multiplied by the total quantity of BK added to the incubate. The radioactivity eluted was always over 90% of the radioactivity injected. Initial velocities of ACEs (10-260 PM) for BK hydrolysis were studied over a range of substrate concentrations (0.04-23.29 p~ for the wild-type recombi- nant ACE and A C E K ~ ~ ~ , ~ ~ . 0.04-3.89 p~ for ACE~361.365). The actual
unlabeled BK. As the number of molecules of BK1-7 plus BK'-5 formed substrate concentration was calculated from the sum of tritiated plus
at any moment corresponded to the number of molecules of BK hydrolyzed, the initial rate of BK hydrolysis was calculated from the sum of the two products. The kinetic parameters of BK hydrolysis were determined in 50 mM NaCl, three to four times for each enzyme studied.
The effects of chloride on BK hydrolysis by ACEs were studied by incubating the enzymes (20-50 pM) with 0.13 or 2.34 pM BK (i.e. [SI < FZp or [SI >> K",pp) over a range of sodium chloride concentrations (0-75 mM).
The pH dependence of ACE-catalyzed BK hydrolysis was studied at a single substrate concentration (2.34 VM BK). The buffers used (Mes, Hepes, and sodium borate) were 100 mM, and the chloride concentration was 50 mM. Since ACE activity decreases because of the loss of zinc below pH 7, assays were performed with the appro- priate Zn2+ concentration (14).
Inhibition of BK hydrolysis by captopril was studied by measuring the initial velocities of ACEs (6-24 PM) for 0.13 pM BK over a range of inhibitor concentrations (10"' M-lO-' M ) (12, 15).
The kinetics of BK'-7 hydrolysis were determined by incubating ACE (5 pM wild-type recombinant ACE, 23 pM ACE~361,36, and 9 pM A C E K ~ ~ ~ , ~ ~ ) with a range of substrate concentrations (2-650 p M for the wild-type recombinant ACE and 3-1,620 p~ for ACE~361.365 and ACE~959.963) in a final volume of 300 pl. The amount of BK1-5 formed was determined by UV absorbance at 210 nm.
The inhibition of the wild-type recombinant-catalyzed hydrolysis of BK'-7 by BK was studied over a range of BK'-7 concentrations (12-718 p ~ ) and BK concentrations of 0, 0.1, 0.2, 0.5, 1.0, and 2.0 p ~ . The enzyme concentration was 7 PM. It was assumed that the quantity of BK'-7 formed by the hydrolysis of BK was negligible compared with the quantity of BK'" added.
Hydrolysis of Substance P-For kinetic studies, ACE (0.5 nM wild- type recombinant ACE, 1.3 nM ACEK361,3ffi, 1.0 nM AcE~~~9.96~) was incubated with 150 p~ substance P, in 10 mM NaCl. Samples of the reaction mixture were removed at various times for HPLC. All peptides were separated by a linear gradient of acetonitrile in water (7-38.8% in 32 min). Retention times were 9.8 min for the carboxyl- terminal dipeptide and 12.5 min for the carboxyl-terminal tripeptide.
Chloride activation of substance P hydrolysis was studied with 30 p M substrate and chloride concentrations of 0-100 mM.
The hydrolysis of substance P free acid (15 p ~ ) was studied in the same conditions as substance P but at 50 mM NaCI.
Hydrolysis of LH-RH and LH-RH4-l0"ACE (6.0 nM wild-type
@ ACE
HEMGH HEMGH
N fragment c fragment
1 BRADYKININ Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe ~ A r g
SUBSTANCE P Arg-Pro-Lys-Pro-Gln-Gln-Phe-Phe-Gly-Leu-Met-NH2
LH-RH Pyro-Glu-His-Trp-Ser -TyrCly-Leu-Arg-Pro-Gly-NH2
1 1 9
1 1 1 l 1
1 1 1 10
FIG. 1. Panel A, ACE structure. The wild-type ACE (ACE) contains the signal peptide (left black box), the two large homologous domains (shaded boxes), and the transmembrane domain (right black box) near the carboxyl terminus. The sequence containing the 2 zinc-binding histidines is shown under each domain. The truncated mutant, N fragment, contains the signal peptide and the N domain. The truncated mutant, C fragment, contains the signal peptide, the C domain, and the carboxyl-terminal region, including the transmembrane and intracellular domains. The 2 zinc-binding histidines have been replaced by lysines in the two full-length mutants. The position of the mutated histidines is indicated in the mutant name (Wei et al. (11)). Panel B, amino acid sequences of the peptides studied. Arrows denote sites of cleavage by ACE.

9498 Active Sites of Angiotensin I-converting Enzyme
- I % E Q U
-
60000 - 160000
40000 - 1 4 5 I 5 5 1 6 5 1 7 5
T l m r 8 m l n
20000 -
0 - 8.0 9.0 10.0 11.0 12.0 13.0 14-0
Time (min) FIG. 2. Reversed-phase HPLC of ACE-catalyzed BK hydrolysis products. Wild-type recombinant ACE (66 PM) was incubated at
37 "C, with 2.33 p~ unlabeled BK and 0.014 p~ [3H] BK, in 50 mM Hepes buffer, pH 7.5, in 50 mM NaC1. 50 p1 of reaction mixture was applied to a Spherisorb Cs column, and the hydrolysis products were separated as described under "Materials and Methods." [3H] measurements of the eluates: 0 min (U), 5 min (+), 30 min (A), 90 min (m). I d B K , fragment of BK ArgI-Phe'; I-7BK, fragment of BK Arg'-Pro7. Inset, [3H] measurements of unhydrolyzed BK.
recombinant ACE, 0.4 nM A C E K ~ ~ ~ , M , 0.1 nM A C E K ~ ~ ~ , ~ , 3.1 nM N fragment, and 0.3 nM C fragment) was incubated with LH-RH: 10- 490 p~ for the wild-type recombinant ACE and the N fragment, 30- 700 pM in the case of ACE~969,m and C fragment, and 60-1000 pM for AcE~381,~~. The carboxyl-terminal tripeptide of LH-RH was first eluted in the absence of acetonitrile with a retention time of 7.8 min. The other fragments were then eluted by increasing the concentration of acetonitrile in water to 8.8% over 22.3 min. Retention times were 22.7 min for the amino-terminal tripeptide and 28.2 min for LH-RHe lo, which were well separated from other LH-RH fragments (not shown). The concentration of acetonitrile was increased to 32% to elute unhydrolyzed LH-RH.
Chloride activation of the hydrolysis of LH-RH was studied with 100 p~ substrate and chloride concentrations of 0-400 mM. The concentrations of wild-type recombinant ACE, A C E K ~ ~ ~ . ~ , and the N and the C fragments were 1.9, 1.2, 1.1, and 1.0 nM, respectively.
The hydrolysis of LH-RH"" by the wild-type recombinant ACE (6.2 nM) and the N fragment (3.5 nM) was studied at LH-RH"" concentrations of 0-1,000 p ~ . The effect of captopril on the hydrolysis was checked by incubating ACES with M captopril for 1 h at 4 "C before adding LH-RH"" (final concentration 490 pM, LH-RH, lo-' M captopril).
RESULTS
Kinetic Parameters of BK and BET-7 Hydrolysis-ACE- catalyzed BK hydrolysis proceeded by the cleavage of the carboxyl-terminal dipeptide of BK (Phes-Arg) producing BK"7, followed by the removal of the new carboxyl-terminal dipeptide Ser6-Pro7 to form BK1-5 (2, 16). BK"5 was not hydrolyzed further even after prolonged incubation. Attempts to quantify the products of BK hydrolysis by measuring initial velocity by UV spectrophotometry failed because this tech- nique was not sensitive enough to follow the hydrolysis at substrate concentrations below 10 p~ under our experimental conditions. The use of tritiated BK allowed determination of initial rates of BK hydrolysis with substrate concentration as low as 0.04 p ~ , one-fifh the Pzp value (see below).
The kinetic parameters of BK and BK1-7 hydrolysis, ob- tained from Lineweaver-Burk plots (Fig. 3), are shown in Table I. The kcat value for BK is 2 orders of magnitude lower than the kcat value for BK"7. Therefore the conversion of BK
0.025 -
- 0.020- E 0 0.015 -
.- c - >
- 1 t
"s -4 -2 0 2 4 6 8 10 12 1k
1 / [BK] (pM") FIG. 3. Lineweaver-Burk plots of BK hydrolysis by the
wild-type recombinant ACE Q, A c E ~ s 6 l . s ~ (a), and ACEKe6g,ma (A). Enzyme assays were carried out with 0.04-23.29 p M BK (wild-type recombinant ACE and ACE~959.w) or 0.04-3.89 pM BK (ACEK361,365) in 50 mM Hepes buffer, pH 7.5, 50 mM NaCl, at 37 "C. Initial rates of BK hydrolysis were calculated from the sum of BK"? and BKId formed.
to BK"7 is the limiting step in ACE-catalyzed BK hydrolysis. The inhibition of BK hydrolysis by BK"7 indicated that
hydrolysis of BK was inhibited only above 1 p~ BK"7. In the case of the inhibition of BK"7 hydrolysis by BK, a Dixon plot of l/Vo uersm [BK] (where Vo was the initial velocity of BK"' hydrolysis) gave a K , value of 3 p ~ . The plots indicated an apparent competitive inhibition of BK"7 hydrolysis by BK (not shown).
Double-reciprocal plots for BK hydrolysis by the full-length mutants are shown in the Fig. 3, and the PzP and kcat values derived from these plots are summarized in Table I. The hat values for the two active sites were similar. The substrate activation that occurred with ACEK969,963 (Fig. 3) may be due to the binding of BK molecules to the mutated carboxyl- terminal active site, inducing conformational changes in the amino-terminal active site. As in the case of the wild-type
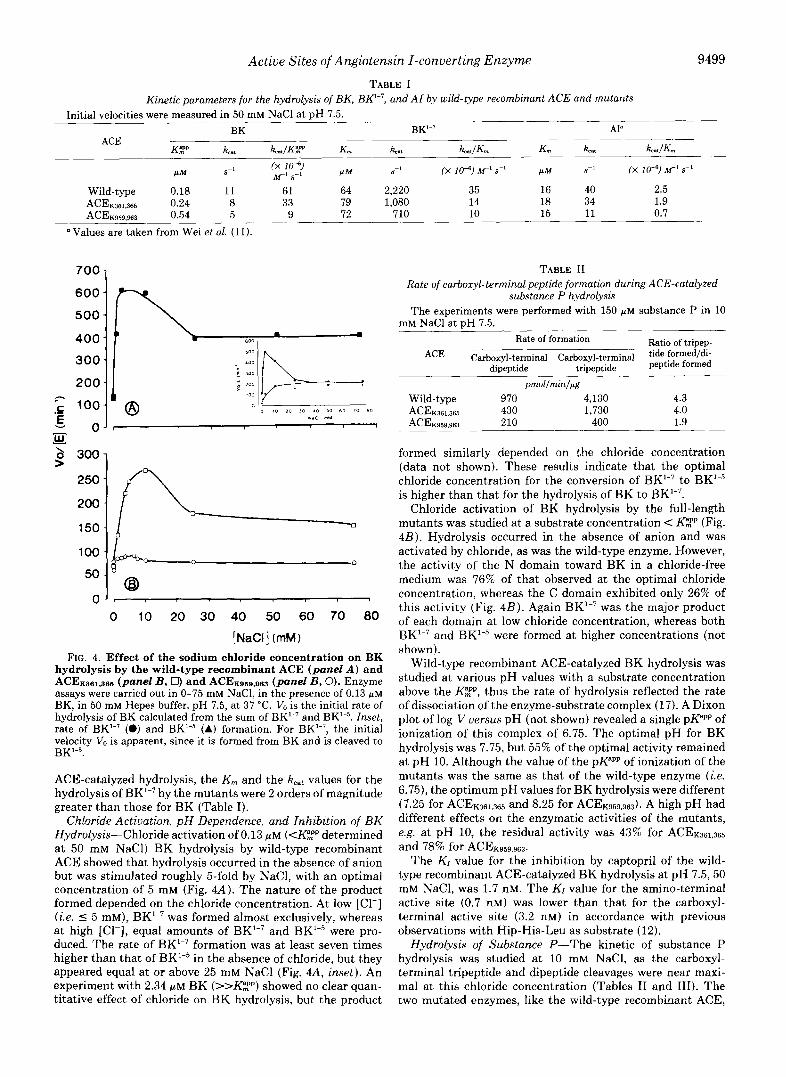
Active Sites of Angiotensin I-converting Enzyme 9499
TABLE I Kinetic parameters for the hydrolysis of BK, BK2-7, and AI by wild-type recombinant ACE and mutants
-
~
~
Initial velocities were measured in 50 mM NaCl at pH 7.5. BK
ACE K$P kat kcat/KF' K ,
S"
Wild-type 0.18 11 61 64 ACE~m.365 0.24 8 33 79 ACEKQSP sm 0.54 5 9 72
Values are taken from Wei e t al. (11).
700
600
500
400 300
200
.E
A!L
> a 300 250
200
150
100
50
0
7 100 v € 0 -
0 10 20 30 40 50 60 70 80 [ NaCl ] (rnM 1
FIG. 4. Effect of the sodium chloride concentration on BK hydrolysis by the wild-type recombinant ACE (panel A ) and ACE~sa1,sea (panel B, 0) and A c E ~ s a s . ~ (panel B, 0). Enzyme assays were carried out in 0-75 mM NaC1, in the presence of 0.13 FM BK, in 50 mM Hepes buffer, pH 7.5, a t 37 "C. VO is the initial rate of hydrolysis of BK calculated from the sum of BK'-7 and BK1-5. Inset, rate of BK'-7 (0) and BK1-5 (A) formation. For BK'", the initial velocity Vo is apparent, since it is formed from BK and is cleaved t o BK'-5.
ACE-catalyzed hydrolysis, the K,,, and the kcst values for the hydrolysis of BK"7 by the mutants were 2 orders of magnitude greater than those for BK (Table I).
Chloride Activation, pH Dependence, and Inhibition of BK Hydrolysis-Chloride activation of 0.13 p~ (<K"mpp determined a t 50 mM NaCl) BK hydrolysis by wild-type recombinant ACE showed that hydrolysis occurred in the absence of anion but was stimulated roughly 5-fold by NaCl, with an optimal concentration of 5 mM (Fig. 44). The nature of the product formed depended on the chloride Concentration. At low [Cl-] (i.e. 5 5 mM), BK'-7 was formed almost exclusively, whereas at high [Cl-1, equal amounts of BK"7 and BK"5 were pro- duced. The rate of BK'-7 formation was at least seven times higher than that of BK"5 in the absence of chloride, but they appeared equal at or above 25 mM NaCl (Fig. 4A, inset). An experiment with 2.34 p~ BK (>>K",Pp) showed no clear quan- titative effect of chloride on BK hydrolysis, but the product
S-l (X 10-6) M' sf' P M S" (X 10-7 MI s-1
2,220 35 16 40 2.5 1,080 14 18 34 1.9
710 10 15 11 0.7
TABLE I1 Rate of carboxyl-terminal peptide formation during ACE-catalyzed
substance P hydrolysis The experiments were performed with 150 FM substance P in 10
mM NaCl at DH 7.5. Rate of formation Ratio of tripep-
ACE Carboxyl-terminal Carboxyl-terminal tide formed/di- dipeptide tripeptide peptide formed
prnol/rnin/@g
Wild-type 970 4,130 4.3 ACE~361.365 430 1,730 4.0 AcE~sr s 96s 210 400 1.9
formed similarly depended on the chloride concentration (data not shown). These results indicate that the optimal chloride concentration for the conversion of BK'-7 to BK"5 is higher than that for the hydrolysis of BK to BK"7.
Chloride activation of BK hydrolysis by the full-length mutants was studied at a substrate concentration c E " (Fig. 4B). Hydrolysis occurred in the absence of anion and was activated by chloride, as was the wild-type enzyme. However, the activity of the N domain toward BK in a chloride-free medium was 76% of that observed at the optimal chloride concentration, whereas the C domain exhibited only 26% of this activity (Fig. 4B). Again BK'-7 was the major product of each domain a t low chloride concentration, whereas both RK'-7 and BK'-5 were formed at higher concentrations (not shown).
Wild-type recombinant ACE-catalyzed BK hydrolysis was studied a t various pH values with a substrate concentration above the FZP, thus the rate of hydrolysis reflected the rate of dissociation of the enzyme-substrate complex (17). A Dixon plot of log V versus pH (not shown) revealed a single p P P p of ionization of this complex of 6.75. The optimal pH for BK hydrolysis was 7.75, but 55% of the optimal activity remained at pH 10. Although the value of t h e p P P p of ionization of the mutants was the same as that of the wild-type enzyme (i.e. 6.75), the optimum pH values for BK hydrolysis were different (7.25 for ACEK36L,365 and 8.25 for ACEK959,963). A high pH had different effects on the enzymatic activities of the mutants, e.g. at pH 10, the residual activity was 43% for ACEK361,365 and 78% for A c E ~ ~ ~ 9 . 9 ~ ~ .
The Kr value for the inhibition by captopril of the wild- type recombinant ACE-catalyzed BK hydrolysis at pH 7.5,50 mM NaCl, was 1.7 nM. The KI value for the amino-terminal active site (0.7 nM) was lower than that for the carboxyl- terminal active site (3.2 nM) in accordance with previous observations with Hip-His-Leu as substrate (12).
Hydrolysis of Substance P-The kinetic of substance P hydrolysis was studied a t 10 mM NaCl, as the carboxyl- terminal tripeptide and dipeptide cleavages were near maxi- mal at this chloride concentration (Tables I1 and 111). The two mutated enzymes, like the wild-type recombinant ACE,

9500 Active Sites of Angiotensin I-converting Enzyme
TABLE I11 Chloride dependence of the ACE-catalyzed hydrolysis of substance P
The experiments were performed with 30 PM substance P at pH 7.5. Carboxyl-terminal dipeptide Carboxyl-terminal tripeptide
ACE Optimala Activity at 0 Mb Activity at 10 mM ~,~~ Optimal Activity at 0 M Activity at 10 mM
[Cl-I K - 1 [Cl-l [cl-1 [CI-I K - 1 K'. mM % % mM mM % % mM
30 Wild-type 30 26 78 6 35 NDd
ACE~969.m 15 90 95 6 10 86 97 1.5
79 4 ACE~aelaea 53 73 16 40 26 76 8
Values for optimal chloride concentration and the activity in the absence of chloride and in 10 mM chloride were calculated from u uersus [Cl-] plots.
* Activity as a percentage of the activity measured at optimal [Cl-1, K'. values were calculated from 1 / V - V, uersus l/[Cl-] plots. ND, not detectable (i.e. below 2 pmol/min/pg).
exhibited carboxytripeptidase and carboxydipeptidase activ- ity. The carboxyl-terminal tripeptide was the major product of the primary cleavage in all cases. Substance P was then further cleaved by both domains with the appearance of multiple fragments (not shown). The apparent initial rate values calculated from the kinetic curves are summarized in Table 11. The C domain hydrolyzed substance P faster than the N domain.
The initial velocities of wild-type recombinant ACE and mutant-catalyzed substance P hydrolysis were measured at various chloride concentrations. The Kh values (Table 111) were calculated from double-reciprocal plots of 1/V - Vo uersus l/[Cl-] (not shown), where Vo was the rate of hydrol- ysis in the chloride-free medium and V was the rate of hydrolysis in the presence of various chloride concentrations. The C domain exhibited a greater chloride dependence than the N domain.
In substance P-free acid there was no apparent tripeptide cleavage, and the three enzymes exhibited only the dipepti- dase activity (not shown). The rate of dipeptide formation (in nmol of dipeptide formed/min/pg of ACE), in the same con- ditions of hydrolysis as substance P, was 12.3 for the wild- type recombinant ACE, 1.3 for A C E K ~ ~ ~ , ~ ~ ~ , and 4.0 for A C E K B ~ , ~ ~ ~ .
Hydrolysis of LH-RH-LH-RH was cleaved at its carboxyl- terminal extremity to form the carboxyl-terminal tripeptide Ar$-Prog-Gly"-NH2 and LH-RH"7. As expected (3), this latter fragment was further hydrolyzed to the amino-terminal tripeptide PyroG1u'-His2-Trp3. LH-RH was also cleaved at the amino-terminal extremity, and this initial cleavage led to the formation of the amino-terminal tripeptide and
The results presented here indicate that LH-RH4"' is also hydrolyzed by ACE. A peptide with the same retention time as the carboxyl-terminal tripeptide was released from LH-
captopril. The KBzp, the kcat, and the kCat/KBzp values for the wild-type recombinant ACE were 1410 p ~ , 0.6 s-', and 0.4
pc"' s-', respectively. The corresponding values for the N fragment were 275 FM, 0.9 s-', and 3.3 p"' s-'. The C fragment produced no hydrolysis over this concentration range, and cleavage was detectable only above 1,300 p~ LH- RH4"'. Therefore no kinetic parameters were determined for this fragment.
The initial velocities of LH-RH hydrolysis by the five enzymes were measured at various substrate concentrations. Since both the amino-terminal and the carboxyl-terminal tripeptide were stable products that could be formed from an initial amino- or carboxyl-terminal cleavage of LH-RH, the amounts of each of these tripeptides gave information on the
LH-RH4"0.
RH4-10 , and the reaction was completely inhibited by M
rate of ACE-catalyzed LH-RH hydrolysis. However, it was easier to separate and thus quantify the amino-terminal tri- peptide. The kinetic parameters calculated from Lineweaver- Burk plots (not shown) are summarized in Table IV. Although LH-RH4"O was hydrolyzed by ACE, the specificity constants for this peptide were roughly 10 times lower than those for LH-RH. The substrate concentration also remained in large excess over that of the products under conditions of initial velocity for LH-RH hydrolysis. Under these conditions, the hydrolysis of LH-RH4"O formed from LH-RH was probably minimal. The rate of appearance of this peptide was therefore considered to reflect the extent of the primary cleavage be- tween Trp3 and Ser4. Hence the kinetic parameters for LH- RH hydrolysis were also calculated from the amount of LH- RH4-'' formed.
The results indicate that LH-RH was hydrolyzed by both active sites but faster by the amino-terminal active site. The N fragment had a kcat roughly 10 times higher than the C fragment. The primary amino-terminal cleavage, as reflected by the formation of LH-RH4-", occurred with both active sites but was much faster with the N fragment, which had a kcat for this reaction at least 30 times higher than the C fragment. Comparison of the kinetic parameters for the amino-terminal tripeptide (reflecting the overall rate of hy- drolysis of LH-RH, i.e. both the primary amino- and carboxyl- terminal cleavages) and for LH-RH4-'' (reflecting only the primary amino-terminal cleavage) indicated that the amino- terminal active site performed both of these primary cleav- ages, whereas the carboxyl-terminal active site hydrolyzed LH-RH mainly by a primary carboxyl-terminal cleavage. The kinetic parameters for the overall rate of hydrolysis of LH- RH by each active site were of the same order of magnitude, suggesting that both active sites of the wild-type enzyme hydrolyze LH-RH (Table IV).
The chloride effect on the hydrolysis of LH-RH was studied at a substrate concentration of 100 WM, below the K T p (Table V). The primary amino-terminal cleavage and the overall hydrolysis of LH-RH by the wild-type recombinant ACE were markedly stimulated by NaC1. The hydrolysis of LH-RH by the full-length mutants possessing only one functional active site was also stimulated by chloride. Supraoptimal chloride concentrations inhibited the enzymes except the C fragment.
DISCUSSION
Our previous studies indicate that Hip-His-Leu and AI are cleaved by the two homologous domains of ACE, and therefore each of these domains includes a functional active site. Mu- tations of the two histidines binding a zinc atom, in either the N or C domain, lead to complete inactivation of the corresponding active site, the other domain remaining func-

Active Sites of Angiotensin I-converting Enzyme 9501
TABLE IV Kinetic parameters for the hydrolysis of LH-RH by wild-type recombinant ACE and its mutants
Initial velocities of the formation of the amino-terminal tripeptide and LH-RH4"' were measured in 50 mM NaCl at pH 7.5. Amino-terminal tripeptide LH-RH'""
ACE
Wild-type 265 1.8 ACE~361.366 520 1.1 C fragment 160 0.8 A C E K B ~ ~ , ~ 760 8.4 N fragment 235 8.5
Considered as a stable product in these experimental
7 215 2 210 5 85
11 960 36 190
conditions (see "Materials and Methods").
0.50 2.35 0.01 0.05 0.08 0.95 2.50 2.60 2.50 13.00
TABLE V Chloride dependence of the ACE-catalyzed hydrolysis of LH-RH
The experiments were performed with 100 p~ LH-RH at pH 7.5. Amino-terminal tripeptide LH-RHC'O"
ACE Optimal Activity at Activity at 0 M b Optimal Activity at
optimal optimal Activity at 0 M [Cl-] W I K1-1 K 1 - 1 Icl-1 K - 1
mM pmol /min /g % rnM pmol/min/@ % Wild-type 50 103 6 300 70 ND'
C fragment 400 198 25 30 43 31 K9sg.w 10 371 76 20 114 44 N fragment 25 1,006 3 75 402 35
K361,365 500 66 64 ND ND ND
' Considered as a stable Droduct in these experimental conditions. Activity as a percentage of the activity measured at optimal [Cl-] ND, not detectable (i.e. below 2 pmol/min/pg).
tional (11). However, the two active sites of ACE do not hydrolyze Hip-His-Leu and AI equally and differ in their susceptibility to anion activation. The amino-terminal active site has a lower optimal chloride concentration and is less activated by chloride than is the carboxyl-terminal active site. Consequently, at or above the optimal chloride concentration for these substrates the C domain hydrolyzes Hip-His-Leu roughly 10 times faster and AI 3 times faster than does the N domain (11). Anion activation is an unusual property of ACE (1) and depends on the nature of the anion (chloride is the most efficient), on the pH, and also on the nature and con- centration of the substrate (14, 18). As the hydrolysis of BK by ACE is less chloride-dependent than that of other sub- strates, such as AI (16), we determined whether BK, unlike AI, was preferentially cleaved by the N domain. The results indicate that this is not so. BK is well hydrolyzed by both active sites. The kcat values of the amino- and the carboxyl- terminal active sites are similar at pH 7.5 and 50 mM NaC1, above the optimal chloride concentration. The Pzp is lower for the carboxyl-terminal active site but remains in the same order of magnitude as for the amino-terminal active site (Table I). Moreover, the chloride activation of the carboxyl- terminal active site for BK hydrolysis is much smaller than for AI; AI hydrolysis is activated roughly 100 times, whereas BK is activated less than 5 times in similar conditions (Fig. 4 and Ref. 11). Chloride has little influence on the hydrolysis of BK by the N domain.
These results indicate that the previously observed differ- ences in the chloride activation profile for the hydrolysis of AI or BK by ACE can be ascribed mostly to the differing chloride sensitivities of the C domain for these two substrates. The molecular basis of anion activation remains almost un- known. Chloride is believed to react with a lysine residue located at or near the active site to induce conformational changes that reduce the dissociation rate of the enzyme-
substrate complex (19). This occurs for the carboxyl-terminal active site and improves its efficiency for AI hydrolysis more than for BK, which is a more tightly bound substrate. On the other hand, studies with BK and substrates that have less favorable kinetic parameters, or those with competitive inhib- itors (12), all indicate that the amino-terminal active site probably undergoes less conformational change than does the C domain in the ACE molecule when the chloride concentra- tion is varied. Synthetic tripeptide substrates of ACE can display large differences in chloride sensitivity and have been divided in three classes according to their chloride sensitivity (14). Our studies with BK and AI indicate that this probably reflects different sensitivities of these substrates to the chlo- ride-induced conformational changes of the C domain rather than differential hydrolysis by the amino- or carboxyl-termi- nal active site.
BK is the natural substrate for which the enzyme displays the most favorable kinetic parameters (16, 20). BK has the lowest PZp among natural ACE substrates, and the kcat/PZP at pH 7.5, 50 mM NaCl, is roughly 25 times higher than that for AI. Even at a high chloride concentration, BK is a better substrate than AI for each of the two active sites. The hy- drolysis of BK proceeds in two steps, the intermediate prod- uct, BK"7, being in turn a substrate for the two active sites of ACE (Table I). BK"7 is also a better substrate than AI for both domains of ACE and displays specificity constants sim- ilar to those of BK. The removal of the carboxyl-terminal dipeptide of BK induces a dramatic increase in kcat (Table I). This suggests that the peptide bond Phe5-Ser' is more rapidly hydrolyzed than Pro7-Phe8, and this may be explained by the presence of a Phe residue in position P1 (21) in BK"7, instead of a Pro residue in BK which distorts the polypeptide chain (22). We found that the nature of the final product of BK hydrolysis depends strongly on the chloride concentration. BK"7 accumulates in a low chloride medium (<25 mM),

9502 Active Sites of Angiotensin I-converting Enzyme
whereas both BK"7 and BK"5 are produced a t higher chloride concentration. A similar observation has been made for the hydrolysis of BK by ACE in rat cerebrospinal fluid (23 ) . The conversion of BK"7 to BK"5 is more chloride-dependent than the hydrolysis of BK into BK"7. This is consistent with the loss of a positively charged carboxyl-terminal extremity in the case of BK"7, since such an extremity seems to be a feature of substrates that are well hydrolyzed by both the basal and the chloride-induced conformations of the enzyme (14).
These observations may have physiological relevance. ACE hydrolyzes BK well if this peptide is released into the circu- lation, even in the presence of AI (24). The present kinetic studies indicate that this is by a preferential hydrolysis of BK over AI by both active sites rather than by a differential hydrolysis of these two peptides by each active site. The physiological significance of the differential chloride sensitiv- ity of the ACE-catalyzed hydrolysis of BK, BK"7, and AI hydrolysis is less evident at the present time. The effect of chloride is more apparent when the substrate concentration is low, which is probably the case in uiuo, as the main effect of chloride is to lower the Pzp of the reaction (19). The chloride concentration in the extracellular fluid is above the optimum for AI, BK, and BK"7, ensuring optimal hydrolysis of all of these substrates. ACE however has been detected intracellularly (4, 25, 26). The chloride concentration in the intracellular vesicles is not known exactly, but it is less than 10 mM in the cytoplasm (27). It has been reported that endothelial cells maintained in serum-free medium, or per- fused arteries, release BK after treatment with ACE inhibi- tors, suggesting that BK can be produced intracellularly (28, 29). If BK is indeed produced in cells, it can well be hydrolyzed by ACE in spite of a low chloride concentration, and the main product would be BK"7. However, no physiological effect has been yet reported for this peptide.
Substance P was studied because it is probably a true substrate for ACE in the brain and in the upper respiratory tract, where accumulation of this peptide during ACE inhi- bition is believed to be responsible for ACE inhibitor-induced cough (4, 30). Furthermore, substance P, an undecapeptide with a protected carboxyl-terminal extremity, undergoes a novel endoproteolytic cleavage by ACE, which releases either a dipeptide or a tripeptide from its carboxyl-terminal extrem- ity (3). The results indicate that the two active sites can catalyze these endoproteolytic cleavages of substance P, al- though the carboxyl-terminal active site performs them faster. The hydrolysis of the Phe8-Glyg bond by the wild-type enzyme appears to be more chloride-dependent than that of the Gly9- Leu" bond, as reported earlier (4), and this was again ex- plained by different chloride sensitivity of the carboxyl-ter- minal active site for these two cleavages (Table 111). The observation that substance P-free acid was hydrolyzed by both active sites, with a primary cleavage occurring only between Gly9 and Leu", is in agreement with previous obser- vations made on native ACE and suggests that the model proposed for the interaction of substance P with ACE is valid for both active sites (4). This model underlines the critical role of an active site arginine, essential for the interaction with either the carbonyl of the penultimate peptide bond (in the case of substance P) or a free carboxyl-terminal amino acid (in the case of substance P-free acid).
LH-RH is a decapeptide with blocked amino- and carboxyl- terminal extremities. Skidgel and Erdos (3) demonstrated that LH-RH is cleaved by ACE. The primary cleavages occur between Trp3 and Ser4 (releasing the amino-terminal tripep- tide and LH-RH4"') and between Leu7 and Ar$ (releasing the carboxyl-terminal tripeptide). Although ACE hydrolyzes
LH-RH in vitro and is present in the pituitary, the role of the enzyme in the metabolism of LH-RH in uiuo is debated (3, 4, 31). However, as LH-RH undergoes unusual amino-terminal and carboxyl-terminal endoproteolytic cleavages by ACE, we have determined whether these unusual specificities of the enzyme are due to one or both active sites. Ehlers and Riordan (32) reported recently that the kidney ACE performs the primary amino-terminal endoproteolytic cleavage of LH-RH much faster than testicular ACE, which contains only the carboxyl-terminal active site. They suggested that the amino- terminal active site is responsible for the amino-terminal cleavage of LH-RH by kidney ACE. LH-RH4"' was detected after incubation of LH-RH with both the amino- and the carboxyl-terminal active sites. Therefore, both active sites cleave LH-RH at the Trp3-Ser4 bond. However, the kinetic studies indicate that the amino-terminal active site cleaves much faster than the carboxyl-terminal active site (Table IV). Hence the amino-terminal active site is mainly responsible for the primary amino-terminal endoproteolytic cleavage of LH-RH by ACE, whereas the primary carboxyl-terminal cleavage is performed by both active sites. The observation that a primary endoproteolytic cleavage between Trp3 and Ser4 of LH-RH is specific to the N domain indicates that this domain possesses specific subsites for interaction with this peptide.
The N and C fragments of ACE have lower FZp values (and identical Kcat values) for LH-RH than the corresponding full- length mutants (Table IV). This phenomenon was not ob- served with other substrates. It may indicate some interaction between the two domains of ACE which can slightly reduce the accessibility of LH-RH to each active site. The lack of interdomain interactions in the truncated mutants may in- duce some conformational changes, compared with the full- length molecule (33), altering the binding of LH-RH. This may also explain the differences in the chloride activation of full-length and truncated enzymes observed with this sub- strate. An interaction between the two domains, involving the two zinc atoms, and altered by chloride, may also perhaps explain the loss of endoproteolytic activity of the wild- type enzyme in a chloride-free medium, consistent with previous reports (3,4), whereas each active site still possesses a residual activity (Tables I11 and V).
In conclusion, BK and substance P are hydrolyzed, like AI, by the two domains of ACE, each of which possesses a functional active site with multiple specificity. This indicates that it would probably not be possible to dissociate the inhi- bition of AI conversion from the potentiation of BK or sub- stance P in uiuo by designing inhibitors specific to each active site. The two active sites of ACE have dipeptidyl carboxypep- tidase activity and also endoproteolytic activity. They hydro- lyze BK almost equally but differ in their capacity to cleave other less tightly bound substrates because the carboxyl- terminal active site is able to undergo an anion-induced conformational change that greatly enhances the binding and hydrolysis of these substrates. However, although the car- boxyl-terminal active site can hydrolyze AI and substance P faster, the amino-terminal active site hydrolyzes LH-RH faster, and this is due at least in part to the specificity of this active site for a primary amino-terminal tripeptide endopro- teolytic cleavage. Compared with the carboxyl-terminal active site (which is present in both the somatic and the germinal forms of the enzyme), the amino-terminal active site (which is specific to the somatic ACE) has lost most of the capacity of anion activation and possesses a novel endoproteolytic specificity for at least one natural peptide. This suggests that the N domain may have a different substrate specificity in

Active Sites of Angiotensin I-converting Enzyme 9503 uiuo than the c domain, although a truly specific substrate 9. Kumar, R. S., Kusari, J., ROY, S. N., Soffer, R. L., and Sen, G. C. (1989) J.
for each domain has not been identified. These results, how- 10. Wei, L., Alhenc-Gelas, F., Soubrier, F., Michaud, A,, Corvol, P., and Clauser,
every emphasize that there are different in the two 11. Wei, L., Alhenc-Gelas, F., Corvol, P., and Clauser, E. (1991) J. Biol. Chem. active centers, despite the high degree of sequence homology. 266,9002-9008 One of these subsites may include a previously identified 12. Wei, L., Clauser, E., Alhenc-Gelas, F., and Corvol, P. (1992) J. Biol. Chem.
critical tyrosine residue of the C domain which site-directed 13. Alhenc-Gelas, F., Marchetti, J., Allegrini, J., Corvol, P., and Menard, J. mutagenesis suggests participates in catalysis (34). Identifi- 14. Shapiro, R., Holmquist, B., and Riordan, J. F. (1983) Biocbmistry 2 2 , cation of these subsites will probably have to await structural 3850-3857 studies of crystallized enzymes, but the availability of ACE 15. Ondetti, M. A., Rubin, B., and Cushman, D. W. (1977) Science 196,441-
mutants each containing one intact domain can help to elu- 16. Dorer, F. E., Kahn, J. R., Lentz, K. E., Levine, M., and Skeggs, L. T. (1974) d a t e the &~ctural and functional differences between the 17. Dixon, M., and Webb, E. C. (1979) Enzymes, 3rdEd., Academic Press, New
Biol. Chem. 264,16754-16758
E. (1991) J. Biol. Chem. 266,5540-5546
267,13398-13405
(1981) Biochim. B~ophys. Acta 677,477-488
444
Circ. Res. 3 4 , 824-827
two active sites.
Acknowledgments-We are grateful to Prof. Pierre Corvol, Dr. Jeannine Yon, and Dr. Marie-Therbse Chauvet for discussion and critical reading of the manuscript and to Dr. Eric Clauser for help. We thank Francoise Savoie for technical assistance and Annie De- pardieu for help in preparing the figures.
1.
2.
3.
4.
5.
6.
7.
8.
REFERENCES
Skeggs, L. T., Kahn, J. R., and Shumway, N. P. (1956) J. Exp. Med. 1 0 3 ,
Yang, H. Y. T., Erdos, E. G., and Levin, Y. (1970) Biochim. Biophys. Acta
Skidgel, R. A., and Erdos, E. G. (1985) Proc. Natl. Acad. Sci. U. S. A. 8 2 ,
Skidgel, R. A., Defendini, R., and Erdos, E. G. (1987) in Neuropeptides and
Horwood, Chicfester, U. K. Their Neurope tidases (Turner, A. J., ed) pp. 165-182, V. C. H. Ellis-
Soubrier, F., Alhenc-Gelas, F., Hubert, C., Allegrini, J., John, M., Tregear, G., and Corvol, P. (1988) Proc. Natl. Acad. Sei. U. S. A. 86, 9386-9390
Bernstein, K. E., Martin, B. M., Edwards, A. S., and Bernstein, E. A. (1989) J. Biol. Chem. 264,11945-11951
Lattion, A. L., Soubrier, F., Allegrini J., Hubert, C., Corvol, P., and Alhenc- Gelas, F. (1989) FEES Lett. 262,’99-104
Ehlera, M. R. W., Fox, E. A., Strydom, D. J., and Riordan, J. F. (1989) Proc. Natl. Acad. Sci. U. S. A. 86, 7741-7745
295-299
214,374-376
1025-1029
18. 19.
21. 20.
22.
23. 24.
25.
26.
27.
28.
29.
30.
31.
32. 33. 34.
Biinning, P., and Riordan, J. F. (1983) Biochemistry 2 2 , 110-116 Shapiro, R., and Riordan, J. F. (1983) Biochemistry 22,5315-5321 Stewart, T. A., Weare, J . A., and Erdos, E. G. (1981) Pe tides 2,145-152 Schechter, I., and Berger, A. (1967) Biochem. Biophys. ges. Commun. 27.
York
1.57-1 fi3 L e h n i n p A: L. (1972) Biochemisty,The Molecular Basis of Cell Structure
Yoshida, T. and Nosaka S. (1990) J. Neurochem. 66, 1861-1869 Isbida, H., kcicli, A. G.,’and Carretero, 0. A. (1989) J. Phrmacol. Ezp.
Ther. 261.817-820 Bruneval, P, Hinglais, N., Alhenc-Gelas, F., Tricottet, V., Corvol, P.,
Mbnard, J., Camilleri, J. P., and Bariety, J. (1986) Histochemistry 86, 73-80
Takada, Y., Hiwada, K., Uno, M., and Kokubu, T. (1982) Jpn. Circ. J. 4 6 ,
Alberts, B. Bray D., Lewis, J Raff M., Roberts, K., and Watson J. D. 503-505
Wiemer, G., Sholkens, B. A,, Becker, R. H. A., and Bus& R. (1991) (1983) holecdlar Biology ofthe dell, Garland Publishing Ne; York
M o x u l i , J . V., and VanHoutte, P. M. (1991) J. Cardiouasc. Phrmacol. H rtension 18,558-563
Subissi, A., Guelfi, M., and Criscuoli, M. (1990) Br. J. Phrmacol. 100 , 18,926-928
Kenny, A. J., Ste henson, S. L., and Turner, A. J. (1987) in Mammalian 502-506
Ectoenzymes (zenny, A. J., and Turner, A. J., e&) pp. 169-210, Elsevier Science Publishing Co. Inc., New York
”. ”_
and unctton, 6th Ed., Worth Pu Ilshers, Inc., New York
Ehlers, M. R. W., and Riordan, J. F. (1991) Biochemistry 30,7118-7126 Dautry-Varsat, A., and Garel. J.-R. (1981) Biochemistry 2 0 , 1396-1401 Chen, Y. N., Ehlers, M. R. W., and Riordan, J. F. (1992) Biochem. Biophys.
Res Commun. 184.306-309





























