Embed Size (px)
Citation preview

DIFFRAZIONE DEI RAGGI X
La determinazione della struttura di sistemi molecolari, viene
effettuata attraverso l’uso di metodologie notevolmente diversificate, quali l’assorbimento UV, il dicroismo circolare e la spettroscopia vibrazionale IR. Attualmente le metodologie più utilizzate per definire i parametri molecolari e strutturali sono la risonanza magnetica nucleare NMR e la diffrazione dei Raggi x.
La diffrazione dei Raggi X, utilizzata per la determinazione delle tensioni residue nei materiali metallici, è una metodologia che ha scarsa diffusione. Probabilmente ciò è dovuto al fatto che la diffrazione dei Raggi X viene impiegata soprattutto nel settore della ricerca chimica e farmaceutica per determinare la struttura di composti inorganici ed organici, per la comprensione delle funzioni e dei meccanismi molecolari.
La diffrazione dei Raggi X studia e misura gli effetti d’interazione tra un fascio di Raggi X e la materia cristallina, policristallina.
La cristallografia, dall’analisi dei dati di diffrazione dei Raggi X, permette la determinazione della struttura. Il termine struttura comprende gli aspetti costituzionali, cioè il modo in cui gli atomi sono interconnessi tra loro, facendo distinzione tra legami semplici e multipli, configurazionali indicando i diversi possibili arrangiamenti spaziali dei suoi atomi, trascurando quelli che derivano da rotazioni attorno ai legami semplici, e conformazionali, in riferimento ai diversi arrangiamenti spaziali di una molecola conseguenti alla rotazione attorno ad un legame singolo.
La diffrazione dei Raggi X è una tecnica non distruttiva ormai consolidata nell'ambito della chimica dello stato solido. Attraverso la diffrazione si riesce a determinare in modo non ambiguo la struttura completa corrispondente alla conformazione in un minimo energetico.
Generalmente esiste una unica soluzione consistente dal punto di vista chimico-fisico, che sia capace di interpretare il set di riflessi indipendenti osservati per un particolare cristallo singolo in ciascun esperimento di diffrazione.
1

PRINCIPI DEL METODO DIFFRATTOMETRICO 1. La struttura cristallina I solidi cristallini sono caratterizzati da una geometria ripetitiva che si
estende nello spazio tridimensionale. Esiste un motivo nel cristallo che si ripete indefinitamente per traslazione semplice nelle tre dimensioni. Si può individuare nel motivo che si ripete, costituito da ioni e atomi, una porzione di spazio che contiene tale motivo e traslata che genera il cristallo: cella elementare (quella con il volume ‘minimo’ e la forma associata alla massima simmetria). Una cella elementare può essere più o meno simmetrica (possedere degli elementi di simmetria).
In un reticolo tridimensionale
tm = m1a + m2b + m3c
i tre vettori elementari definiscono il parallelepipedo che descrive la cella elementare (unit cell, cella unitaria). Le direzioni ( x, y e z), specificate dai vettori a, b e c chiamati assi cristallografici, gli angoli fra gli assi sono indicati con α, β e γ dove si sceglie sempre ( Figura 1) α opposto ad a, β opposto a b e γ opposto a c. 2

Figura 1. Cella elementare.
Il volume V della cella elementare è dato da V = a . (b x c)
L'orientazione dei tre assi cristallografici è scelta in modo da dare una terna destrorsa. Si indicano con A, B e C le facce della cella elementare opposte ad a, b, c. Se la cella elementare è primitiva (contiene un singolo nodo), i valori mi sono vincolati ad essere interi per qualunque nodo del reticolo. Se è centrata allora i valori di mi saranno in genere numeri razionali. Per la caratterizzazione della cella basterà ricordare che un nodo ai vertici della cella le appartiene per 1/8, un nodo su uno spigolo le appartiene per 1/4, un nodo su una faccia le appartiene per 1/2.
L’insieme degli elementi di simmetria (e delle traslazioni) costituisce il Gruppo Spaziale. L’unità asimmetrica è il più piccolo sottoinsieme della cella elementare necessario per generare l’intera cella elementare (mediante gli opportuni operatori di simmetria).
Dobbiamo considerare il fatto che vi sono restrizioni di simmetria che devono essere applicate agli assi e agli angoli della cella elementare di un reticolo. La presenza di certi assi vincola la geometria del reticolo. Queste restrizioni danno origine ai sette sistemi cristallini. E’ infatti conveniente raggruppare classi di simmetria che hanno delle somiglianze: in tal modo i cristalli corrispondenti potranno essere descritti con uno stesso tipo di cella elementare. Questa a sua volta potrà essere scelta in modo da evidenziare la simmetria presente.
3

I sette sistemi cristallini
Trimetrici
E o i Triclino a ≠ b ≠ c
1 o –1 α ≠ β ≠ γ
C2 o σ Monoclino a ≠ b ≠ c
2 o -2 α = β = 90° ≠ γ
α = γ = 90° ≠ β
C2 o σ Ortorombico a ≠ b ≠ c
2 o -2 α = β = γ = 90°
Dimetrici
C4 o S4 Tetragonale a = b ≠ c
4 o -4 α = β = γ = 90°
C6 o S3 Esagonale a = b ≠ c
6 o -6 α = β = 90° γ = 120°
C3 o S6 Trigonale come l’esagonale
3 o -3 (Romboedrico) a = b = c
α = β = γ ≠ 90°)
Monometrico
4 assi ternari Cubico a = b = c
α = β = γ = 90°
4

Dopo aver associato ad ogni sistema cristallino una cella elementare primitiva compatibile con i gruppi puntuali afferenti al sistema, dobbiamo esaminare la possibile esistenza di altri reticoli non primitivi. Esistono infatti altri tipi di reticolo, basati su celle non primitive, che non possono essere ricondotti ai precedenti.
Centratura dei reticoli. Si procede in modo diretto. Si aggiungono punti a ciascun reticolo dei diversi sistemi cristallini e si valuta se: (a) l’insieme dei nodi è ancora un reticolo (l’intorno di ogni nodo deve essere lo stesso), e (b) si tratta di un nuovo reticolo nello stesso sistema cristallino.
Le possibili centrature di una cella primitiva sono disposte (a partire dall’origine) in:
(I) a corpo centrato (a/2 + b/2 + c/2)
(F) a facce centrate (a/2 + b/2), (a/2 + c/2), e (b/2 + c/2)
(A) A-centrata (b/2 + c/2) (B) B-centrata (a/2 + c/2) (C) C-centrata (a/2 + b/2)
L’analisi sistema per sistema porta ad accertare che esistono solo sette nuovi reticoli non primitivi (centrati).
Questi si sommano ai sette reticoli primitivi (P) a dare i 14 reticoli di Bravais (Figura 2), da Auguste Bravais, che per primo li enumerò nel 1850. La cella romboedrica è primitiva ma per convenzione viene indicata con R invece che con P. La cella convenzionale trigonale ha l’asse ternario parallelo a c, ed è come la cella esagonale. La presenza dell’asse ternario introduce la possibilità per alcune sostanze di presentare una cella P a forma di romboedro.
5

Figura 2. I 14 reticoli di Bravais
Il ferro e le sue leghe presentano una struttura cristallina caratterizzata, a seconda della temperatura, da celle elementari:
cubica a corpo centrato, cubica a facce centrate e, nuovamente, cubica a corpo centrato.
6

Gruppi spaziali I 7 sistemi cristallini, i 14 reticoli di Bravais e le 32 classi cristalline ci
consentono di classificare la geometria reticolare e la simmetria di un cristallo. Tuttavia, per comprendere a pieno una struttura cristallina dobbiamo esaminare la distribuzione spaziale della densità elettronica (la disposizione spaziale degli atomi). E’ necessario descrivere questa distribuzione solo nell’ambito della cella elementare perchè le operazioni traslazionali reticolari generano l’intero cristallo.
Una descrizione della distribuzione elettronica dal punto di vista della simmetria richiede l’uso dei gruppi spaziali. Questi illustrano in modo completo la simmetria di ogni arrangiamento di atomi associati ad un qualunque reticolo.
Formalmente, un gruppo spaziale è il gruppo che contiene tutte le operazioni di simmetria spaziale degli atomi nel cristallo. Per la presenza di operazioni di simmetria traslazionali non varrà più, ovviamente, che tutti gli elementi di simmetria devono passare per un punto.
Gli elementi di simmetria possono generare più oggetti, equivalenti per simmetria, che coesistono all’interno della cella elementare, anche se primitiva. Chiameremo unità asimmetrica la minima porzione di cella elementare che, per applicazione delle operazioni di simmetria, genera l’intero contenuto della cella (non è in genere univocamente definita).
Possiamo dire, a questo punto, che un gruppo spaziale è un gruppo di operazioni che converte una unità asimmetrica in tutte le unità equivalenti dentro e fuori la cella elementare. Il suo simbolo pertanto descriverà sia il tipo di reticolo di Bravais che gli elementi di simmetria presenti.
Queste considerazioni sono quelle normalmente usate dai cristallografi. I fisici dello stato solido preferiscono utilizzare una descrizione alquanto diversa, e, invece di fare riferimento all’unità asimmetrica, considerano l’intero contenuto atomico di una cella, detto base. La base è la collezione di atomi attaccata ad ogni nodo reticolare. Un cristallo completo è generato dal reticolo e dalla base.
7

Classificazione dei gruppi spaziali. Vi sono 230 gruppi spaziali (non magnetici). Vediamo le convenzioni più
rilevanti in accordo con la notazione di Hermann-Mauguin. Al primo posto appare sempre il simbolo del reticolo di Bravais. Dopo di
questo: a) nei gruppi monoclini appare il simbolo dell'asse di simmetria e, se
presente, dopo una barra appare il simbolo del piano o slittopiano normale ad esso;
b) nei gruppi ortorombici i simboli degli elementi di simmetria si riferiscono alle direzioni a, b, c nell'ordine (gli elementi sono paralleli ad essi se assi propri, perpendicolari se piani);
c) nei gruppi afferenti al sistema tetragonale appare per primo il simbolo dell'asse quaternario e, quando presente, dopo una barra appare il simbolo del piano o slittopiano normale ad esso. Subito dopo appare il simbolo dell'elemento di simmetria che si riferisce alla direzione a (e quindi b) e poi quello dell’elemento relativo alle diagonali della maglia normale all’asse quaternario;
d) nei gruppi afferenti al sistema trigonale ed esagonale appare per primo il simbolo dell’asse ternario o senario. Nei gruppi esagonali, quando presente, appare dopo una barra il simbolo del piano normale ad esso. Il primo dei simboli successivi si riferisce all'asse a (e quindi a b e quindi alla diagonale corta della maglia normale all'asse ternario), il secondo alla diagonale lunga della maglia;
e) nei gruppi del sistema cubico i simboli degli elementi di simmetria si riferiscono nell'ordine ad a (e quindi a b e a c), alle diagonali principali della cella (asse ternario), alle diagonali delle facce della cella.
I 230 gruppi spaziali furono determinati alla fine del secolo scorso, attraverso i lavori matematici di Fedorov (1891) e Schoenflies (1891). Tutte le informazioni sui gruppi spaziali sono contenute nelle International Tables for X-Ray Crystallography.
8

I 230 gruppi spaziali includono 11 coppie enantiomorfe: P31 (P32), P3112 (P3212), P3121 (P3221), P41 (P43), P4122 (P4322), P41212 (P43212), P61 (P65), P62
(P64), P6122 (P6522), P6222 (P6422), P4132 (P4332). Se l’isomero (+) di una specie otticamente attiva cristallizza in uno dei due gruppi enantiomorfi, l’isomero (-) cristalizza nell’altro. Le molecole biologiche sono enantiomorfe, quindi cristallizzeranno in gruppi spaziali enantiomorfi, cioè privi di centro o piani di simmetria.
Vediamo ora come si rappresenta graficamente la simmetria completa di un gruppo spaziale secondo le convenzioni internazionali stabilite nelle
International Tables for X-Ray Crystallography, vol. 1 (1983). Il diagramma di un gruppo spaziale è una proiezione lungo c. L'origine
della cella (in alto a sinistra) è posta su un centro di simmetria, se è presente, altrimenti su qualche altro elemento di simmetria o alla loro intersezione, o in altra posizione comunque indicata. Accanto ai simboli grafici degli elementi di simmetria paralleli al piano del diagramma è scritta la quota (come frazione del periodo c).
Come esempi sono riportate nelle Figure le rappresentazioni di due gruppi spaziali: il monoclino P21/c (n. 14) e l’ortorombico Cmm2 (n. 35).
Le posizioni atomiche sono espresse in termini di coordinate frazionarie
(x, y, z), cioè come frazioni della lunghezza degli assi cristallografici a, b e c. Un atomo posto in (x, y, z) dista dall’origine di un vettore r = xa + yb + zc. Un atomo al centro della cella elementare ha coordinate (½, ½, ½), mentre uno posto al centro della faccia C ha coordinate (½, ½, 0).
9

Nelle Tabelle viene riportata la lista delle posizioni equivalenti generali
o speciali (numero e tipo). Queste ultime si applicano ad atomi che occupano posizioni speciali, giacciono cioè su elementi di simmetria (piani, assi propri, centri e loro combinazioni).
Le posizioni equivalenti generali nel gruppo P21/c sono quattro:
1) x, y, z 2) -x, -y, -z 3) -x, ½ + y, ½ - z 4) x, ½ - y, ½ + z
Un atomo in posizione generale (x, y, z) verrà ripetuto nelle altre
posizioni equivalenti.
10

La notazione internazionale per piani e assi di simmetria è illustrata nelle Figure seguenti.
11
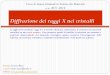
Direzioni cristallografiche. Come abbiamo visto, i cristalli sono anisotropi: sarà quindi necessario
specificare in modo semplice le direzioni nelle quali si manifestano determinate proprietà fisiche.
Esistono in un reticolo infiniti filari paralleli, ciascuno definito da due punti nodali, che sono caratterizzati da uno stesso periodo di ripetizione (Figura 3. ).
Figura 3. Filari paralleli
I filari definiscono una direzione cristallografica. Se un filare passa per
l’origine la sua direzione sarà definita dai valori mi di uno qualunque dei nodi del filare. La direzione viene indicata con [m1 m2 m3]. La stessa direzione è indicata anche da un multiplo del tipo [nm1 nm2 nm3].
Per una cella primitiva la direzione [222] è quella della diagonale di corpo, ma lo è anche [111].
12

Per convenzione si dividono i valori mi per il massimo comune divisore, ottenendo così il più piccolo set. La direzione [936] diventa quindi [312]. Filari che non passano per l’origine hanno sempre un filare parallelo centrale, passante cioè per l’origine.
Se la cella non è primitiva i valori mi sono numeri razionali, esprimibili cioè come rapporti di numeri interi. La direzione corrispondente ad una diagonale di faccia in un reticolo F può essere [½½0].
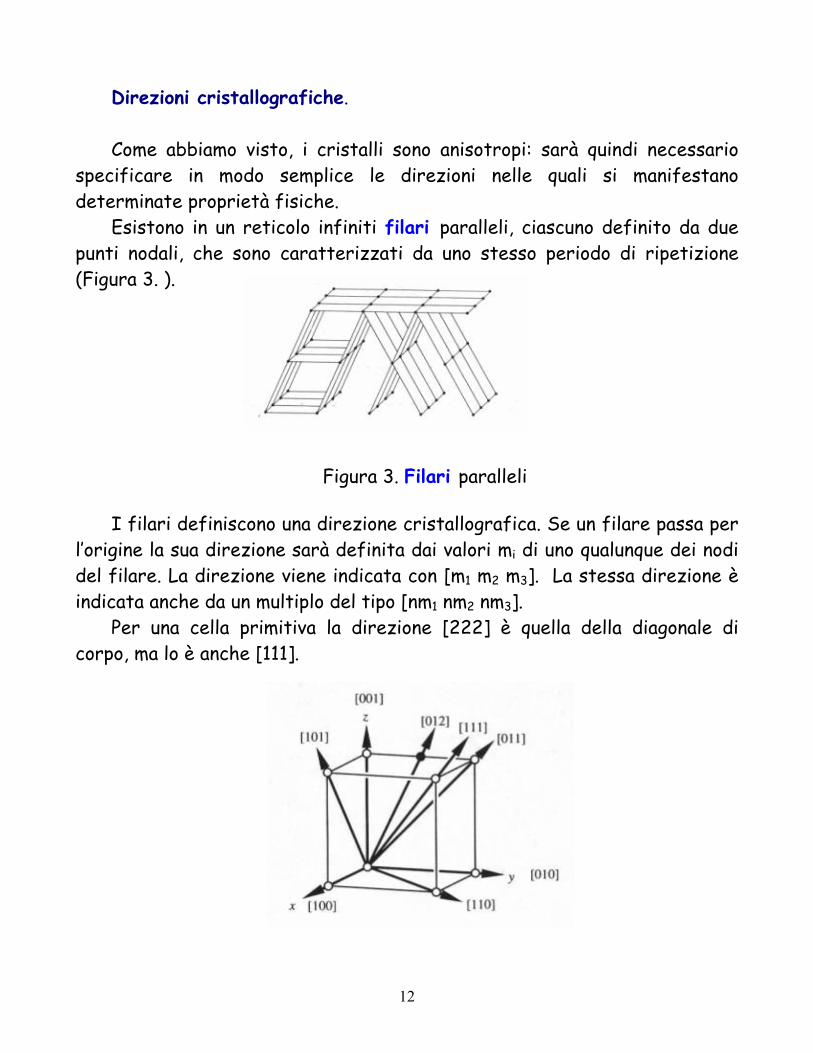
Piani cristallografici.
L’orientazione di un piano cristallografico è definita in termini di indici di Miller (hkl). Tre nodi individuano un piano cristallografico. Se un piano incontra i tre assi cristallografici nei tre nodi (m1, 0, 0), (0, m2, 0) e (0, 0, m3), gli indici (m1, m2, m3) forniscono l’orientazione del piano.
Si preferiscono però gli indici di Miller del piano, che sono numeri interi e primi fra loro, inversamente proporzionali alle intercette del piano con gli assi, cioè
h : k : l = m1-1 : m2
-1: m3-1
In Figura sono mostrati alcuni piani con i loro indici di Miller. Se mi = ∞ il corrispondente indice di Miller è 0. Un simbolo (hkl) viene usato per definire un numero infinito di piani paralleli equidistanti.
13
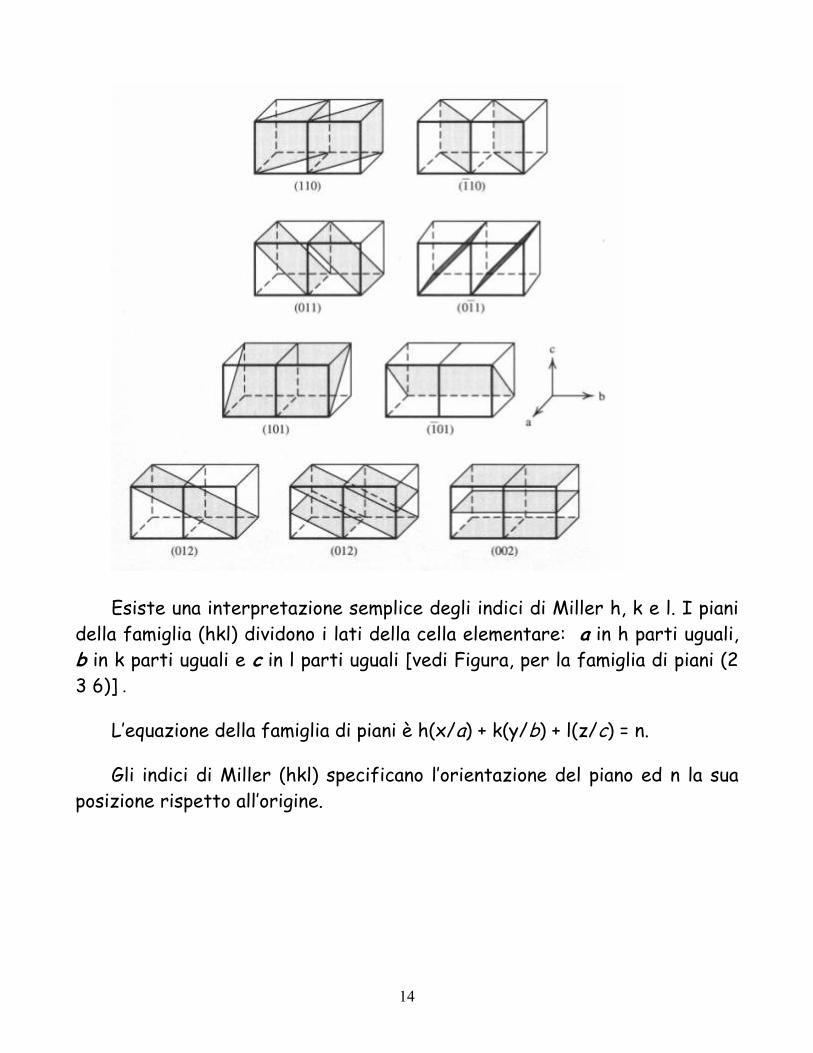
Esiste una interpretazione semplice degli indici di Miller h, k e l. I piani della famiglia (hkl) dividono i lati della cella elementare: a in h parti uguali, b in k parti uguali e c in l parti uguali [vedi Figura, per la famiglia di piani (2 3 6)] .
L’equazione della famiglia di piani è h(x/a) + k(y/b) + l(z/c) = n.
Gli indici di Miller (hkl) specificano l’orientazione del piano ed n la sua posizione rispetto all’origine.
14
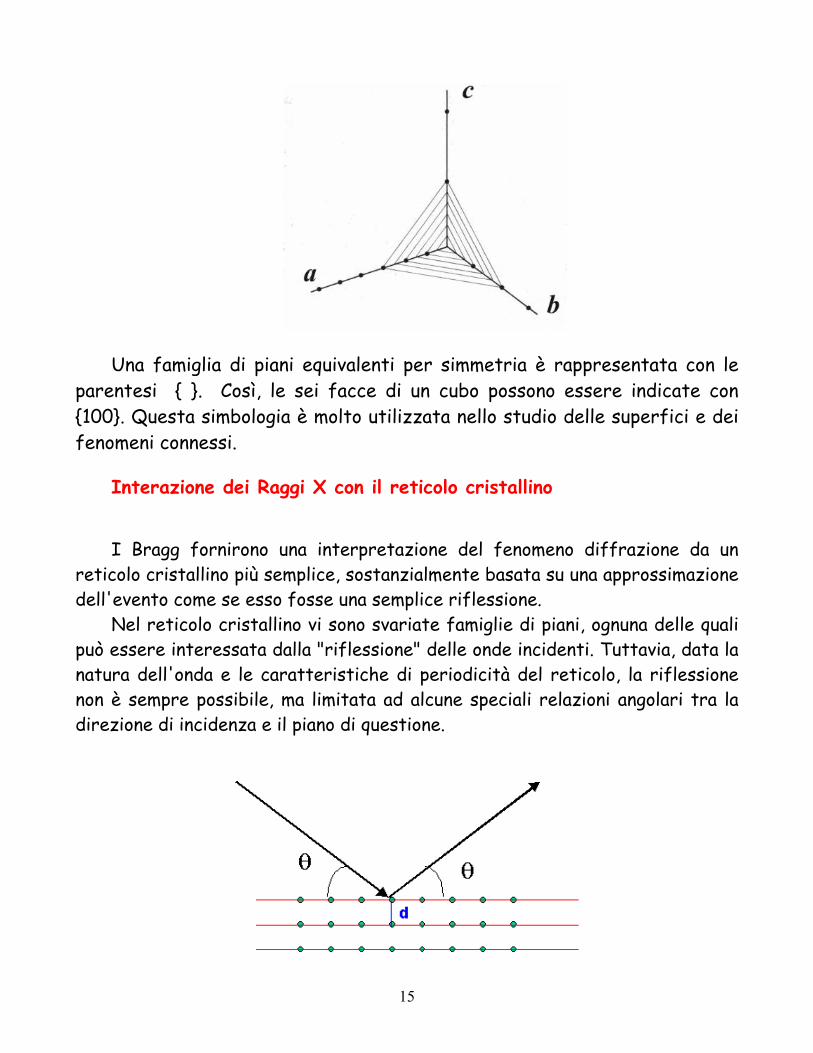
Una famiglia di piani equivalenti per simmetria è rappresentata con le parentesi { }. Così, le sei facce di un cubo possono essere indicate con {100}. Questa simbologia è molto utilizzata nello studio delle superfici e dei fenomeni connessi.
Interazione dei Raggi X con il reticolo cristallino
I Bragg fornirono una interpretazione del fenomeno diffrazione da un
reticolo cristallino più semplice, sostanzialmente basata su una approssimazione dell'evento come se esso fosse una semplice riflessione.
Nel reticolo cristallino vi sono svariate famiglie di piani, ognuna delle quali può essere interessata dalla "riflessione" delle onde incidenti. Tuttavia, data la natura dell'onda e le caratteristiche di periodicità del reticolo, la riflessione non è sempre possibile, ma limitata ad alcune speciali relazioni angolari tra la direzione di incidenza e il piano di questione.
15
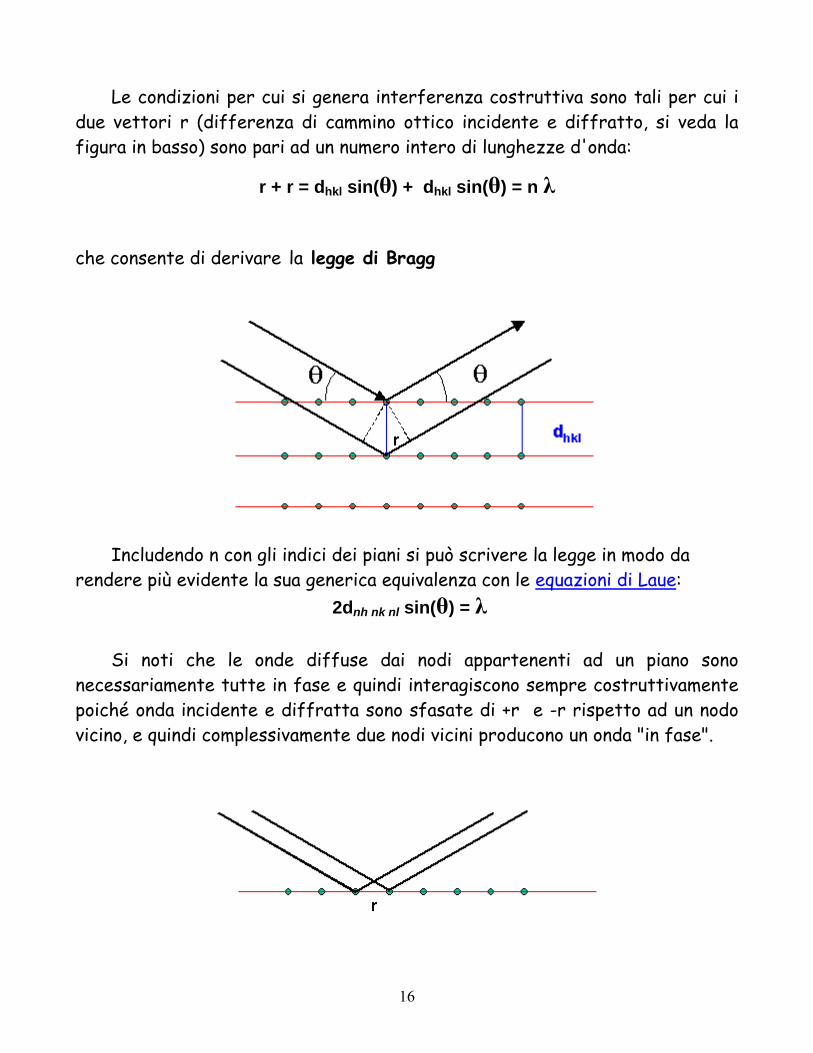
Le condizioni per cui si genera interferenza costruttiva sono tali per cui i due vettori r (differenza di cammino ottico incidente e diffratto, si veda la figura in basso) sono pari ad un numero intero di lunghezze d'onda:
r + r = dhkl sin(θ) + dhkl sin(θ) = n λ
che consente di derivare la legge di Bragg
Includendo n con gli indici dei piani si può scrivere la legge in modo da rendere più evidente la sua generica equivalenza con le equazioni di Laue:
2dnh nk nl sin(θ) = λ
Si noti che le onde diffuse dai nodi appartenenti ad un piano sono necessariamente tutte in fase e quindi interagiscono sempre costruttivamente poiché onda incidente e diffratta sono sfasate di +r e -r rispetto ad un nodo vicino, e quindi complessivamente due nodi vicini producono un onda "in fase".
16
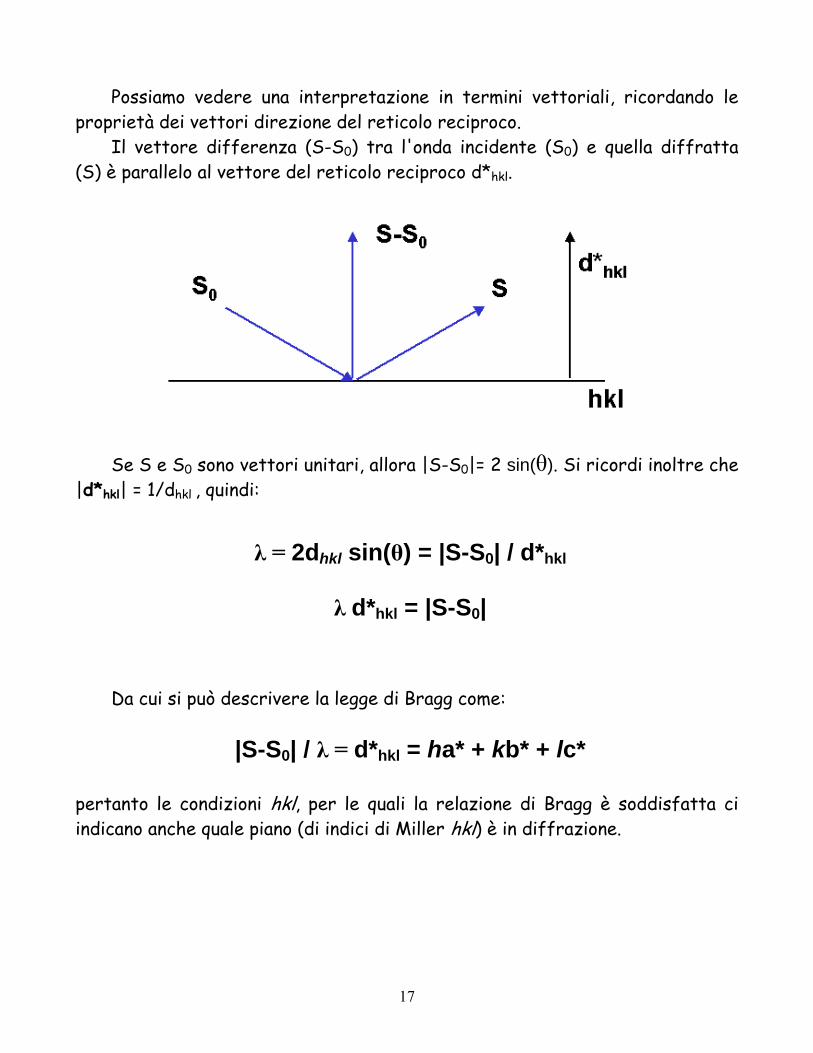
Possiamo vedere una interpretazione in termini vettoriali, ricordando le proprietà dei vettori direzione del reticolo reciproco.
Il vettore differenza (S-S0) tra l'onda incidente (S0) e quella diffratta (S) è parallelo al vettore del reticolo reciproco d*hkl.
Se S e S0 sono vettori unitari, allora |S-S0|= 2 sin(θ). Si ricordi inoltre che |d*hkl| = 1/dhkl
, quindi:
λ = 2dhkl sin(θ) = |S-S0| / d*hkl
λ d*hkl = |S-S0|
Da cui si può descrivere la legge di Bragg come:
|S-S0| / λ = d*hkl = ha* + kb* + lc*
pertanto le condizioni hkl, per le quali la relazione di Bragg è soddisfatta ci indicano anche quale piano (di indici di Miller hkl) è in diffrazione.
17

CRESCITA DI CRISTALLI L’ottenimento dei cristalli è il punto cruciale per intraprendere uno
studio diffrattometrico e talvolta è il limite di questa tecnica. Anche se si utilizza una piccola quantità di materiale, essa deve avere in indice di purezza superiore al 99%.
La cristallizzazione di molecole da una soluzione è un fenomeno di equilibrio reversibile e le caratteristiche termodinamiche e cinetiche di questo processo dipendono dalla natura di soluto e solvente. Quando un sistema formato da un soluto disciolto in un solvente è sovrasaturo, esso tende ad uno stato di equilibrio in cui il soluto è ripartito fra la fase in soluzione e quella solida. In sistemi molto semplici come sali inorganici o piccole molecole organiche in soluzione, i fattori che intervengono durante il processo di cristallizzazione sono ben conosciuti e relativamente facili da prendere in esame. Per quanto riguarda le macromolecole biologiche questi meccanismi sono molto più complicati e più difficilmente descrivibili.
La strategia che si usa per la cristallizzazione di una molecola è quella di portare il sistema molto lentamente verso un minimo di solubilità e in questo modo ottenere cristalli adatti all’analisi cristallografica, cioè che siano non solo cristalli singoli, ma di dimensioni almeno dell’ordine di 0.3-0.4 mm per ogni lato. Avere una soluzione sovrasatura della molecola che interessa è fondamentale per poter ottenere dei cristalli. Ci sono vari metodi per ottenere la sovrasaturazione ma sono due quelli più utilizzati: l’uso della tecnica di microdialisi e di quella della diffusione di vapore, che hanno anche il vantaggio di permettere l’utilizzo di microquantità di soluzione, cosa molto importante quando si lavora con molecole biologiche. La diffusione di vapore sfrutta l’equilibrio di vapore che si viene a creare fra due soluzioni della stessa sostanza a concentrazioni diverse.
Ci sono due diversi modi di procedere: utilizzando il metodo della “sitting drop” (goccia appoggiata) o quello della “hanging drop” (goccia pendente).
18

Il metodo della sitting drop (Fig.2) consiste nel porre una goccia di 10-40 µl di una soluzione contenente la macromolecola da fare cristallizzare e il precipitante all’interno di un microbridge tramite una micropipetta. Tali gocce vengono poi sigillate all’interno di contenitori trasparenti che contengono già una certa quantità, variabile fra 0,5 e 3 ml, di una soluzione di precipitante (reservoir) ad una concentrazione maggiore da quella delle gocce. Attraverso la fase vapore la concentrazione del precipitante nelle gocce si equilibra con quella del reservoir e se viene raggiunta la condizione di sovrasaturazione si possono ottenere i cristalli.
Quando viene utilizzata la precipitazione con PEG o con sali inorganici, le gocce di soluzione devono contenere una concentrazione di precipitante minore (solitamente la metà) rispetto a quella della soluzione che viene aggiunta. Se invece si utilizzano solventi volatili come ad esempio l’Etanolo o l’Acetone, non c’è bisogno di aggiungerne all’interno della goccia. Nel primo caso la precipitazione avviene con lo spostamento dell’acqua dalla goccia al reservoir, nel secondo caso invece si ha uno spostamento di acqua dalla goccia al recipiente e di agente precipitante in senso opposto.
Questo metodo ha il vantaggio di richiedere solo piccole quantità di campione ed è inoltre l’ideale per provare diverse condizioni di cristallizzazione su più campioni contemporaneamente. Per quest’ultimo scopo viene soprattutto utilizzata la tecnica dell’hanging drop (Fig.3).
Una microgoccia di soluzione della macromolecola (massimo 5 µl) viene posta su un vetrino coprioggetti da microscopio che viene successivamente sospeso su un pozzetto contenente 1 ml della soluzione precipitante. E’ importante che il vetrino venga sigillato al pozzetto con del silicone, in modo da impedire l’evaporazione del solvente dalle gocce o l’entrata di umidità dall’esterno. Questo tipo di esperimenti è facilitato dall’uso di piattini particolari; sono piastre per colture di cellule (Linbro plates), formati da 24 pozzetti cilindrici (1.7 cm di diametro e 1.6 cm di altezza) che possono con facilità essere chiusi con i vetrini e siliconati.
Sono costruiti in plexiglas e questo permette il controllo dell’esperimento con il microscopio.
19
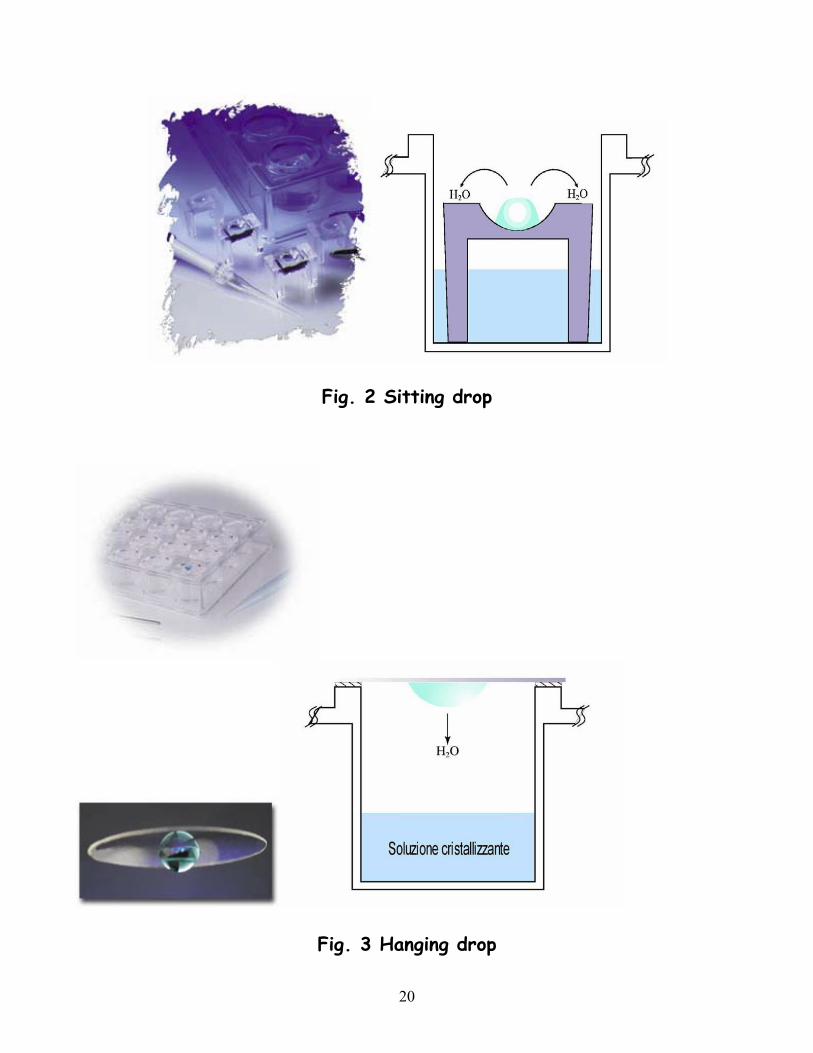
Fig. 2 Sitting drop
Fig. 3 Hanging drop
20

Inoltre i cristalli ottenuti, spesso non sono utilizzabili sia per le
dimensioni (la dimensione ottimale è tra 0.01 e qualche mm) che per la qualità (difetti di geminazione, disordine termico o statistico).
La crescita del cristallo avviene spesso da una soluzione, con molti possibili metodi, quali:
diffusione di un secondo solvente, diffusione di vapore, lenta evaporazione del solvente, applicazione di un qualche gradiente termico, tutti atti a diminuire la solubilità della specie chimica desiderata, che quindi precipita in forma solida e, se le condizioni sono opportune, cristallina.
Vi è anche possibilità di ottenere il cristallo direttamente dal fuso, ma riuscire a crescere monocristalli adeguati ad esperimenti di diffrazione può essere difficile.
Coppie solvente/precipitante comunemente usate sono: acqua/acetone, cloroformio o cloruro di metilene/etere di petrolio o esano.
La qualità del cristallo deve essere controllata prima di iniziare l'esperimento di diffrazione. Spesso si impiega l'analisi con microscopio, e a volte con microscopio a luce polarizzata, che può evidenziare la presenza di difetti estesi, geminazioni ecc.
21

I microscopi a luce polarizzata sono usati anche per evidenziare proprietà anisotropiche dei solidi cristallini
La maggior parte dei solidi hanno proprietà che dipendono dall’orientazione della luce incidente rispetto agli assi cristallografici.
La tecnica del microscopio a luce polarizzata esplora l’interferenza dei raggi divisi dal campione birifrangente quando vengono riunificati dall’analizzatore.
Microscopio a luce polarizzata
22

PREPARAZIONE DEL CRISTALLO
Per un normale esperimento di diffrazione da raggi X con strumentazione da laboratorio universitario, il cristallo deve avere una forma più isotropa possibile (cioè più vicino possibile ad una sfera, per minimizzare gli effetti dell'assorbimento) ed avere dimensione lineare massima di ca. 0.5 mm (poiché il fascio collimato ha spesso questa sezione). Dal momento che la brillanza delle sorgenti usualmente impiegate (tubi a raggi X) non sono elevatissime, dimensioni lineari inferiori a 0.1 mm sono consigliabili sono in presenza di ottima qualità (cioè ottima diffrazione) della specie cristallina prescelta.
23
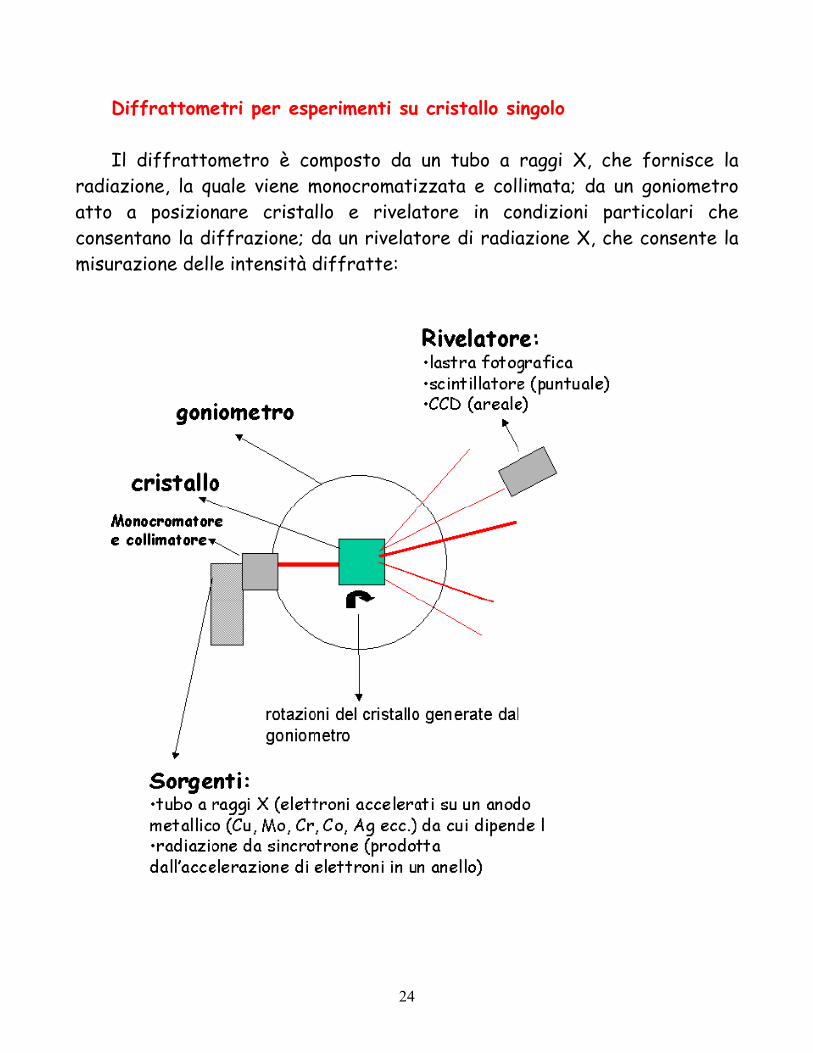
Diffrattometri per esperimenti su cristallo singolo Il diffrattometro è composto da un tubo a raggi X, che fornisce la
radiazione, la quale viene monocromatizzata e collimata; da un goniometro atto a posizionare cristallo e rivelatore in condizioni particolari che consentano la diffrazione; da un rivelatore di radiazione X, che consente la misurazione delle intensità diffratte:
24

DIFFRATTOMETRO MACH3
25

Il diffrattometro utilizzato monta un goniometro a quattro cerchi con geometria euleriana (Figura 2.3). I gradi di libertà di rotazione del cristallo sono tre: ω intorno all’asse verticale, χ intorno ad un’asse orizzontale perpendicolare al cerchio grande, ϕ intorno all’asse della testina goniometrica. Il contatore ruota di 2θ intorno all’asse verticale.
Figura 2.3 -Goniometro a quattro cerchi
Il cristallo viene posto al centro del goniometro in modo da essere
colpito dalla radiazione incidente. Esso viene posizionato su una testina goniometrica, che consente di muovere il cristallo in precise posizioni senza farlo uscire dal fascio incidente.
Testina goniometrica
26

Il rivelatore è una parte essenziale in quanto l'esperimento di diffrazione necessita di accurata misura dell'intensità diffratta dal cristallo.
Storicamente si sono impiegate lastre fotografiche per la misura delle intensità diffratte. Questa tecnica possiede elevate accuratezza risolutiva, ma scarsa accuratezza nella misura dell'intensità
In tempi più moderni (a partire dagli anni 70) si sono impiegati
diffrattometri automatici dotati di rivelatori a scintillazione, ossia strumenti in grado di rivelare un'intensità prodotta da radiazione X, misurando il potere ionizzante. Questi strumenti forniscono accurata misura delle intensità ed anche delle posizioni, ma hanno il difetto di poter misurare una sola intensità diffratta alla volta (in pratica la misura è di tipo seriale, mentre quella di una lastra è di tipo simultaneo o parallelo). Per questo motivo questi strumenti sono particolarmente lenti.
Detector a scintillazione
27

Infine, da alcuni anni sono impiegati strumenti che rappresentano l'evoluzione "fotografica" delle lastre, ossia camere a CCD. Questi strumenti garantiscono la stessa "simultaneità" di una lastra, con una migliore misura delle intensità diffratte. Peccano in potere risolutivo, a causa delle dimensioni (e dei costi di produzione!) dei chip.
La radiazione X viene assorbita prima da uno schermo di fosfori, che
"traduce" il segnale X in segnale di luce visibile e poi immagazziata sul rivelatore a CCD, da cui è possibile leggere l'immagine (fotografia) digitalizzata della diffrazione.
La sorgente dei raggi x è costituita da un tubo a raggi X (tubi di Coolidge). Un fascio di elettroni (ottenuti da un filamento, con correnti dell'ordine di alcune decine di milliAmpere) viene accelerato ad elevato voltaggio (ordine di decine di kilovolts) contro un anodo metallico. L'energia viene principalmente dissipata come calore e in parte minore utilizzata per l'emissione di radiazioni X. Per esperimenti di cristallo singolo la radiazione impiegata è quella prodotta dalle linee caratteristiche di un dato anodo metallico. In particolare è impiegata la radiazione Kα ossia la radiazione emessa dal rilassamento verso lo strato K di un elettrone dello strato L. Una sorgente alternativa e di maggiore efficienza è l'anodo rotante, ossia un sistema in cui l’anodo non sia fisso, bensì in costante rotazione.
28
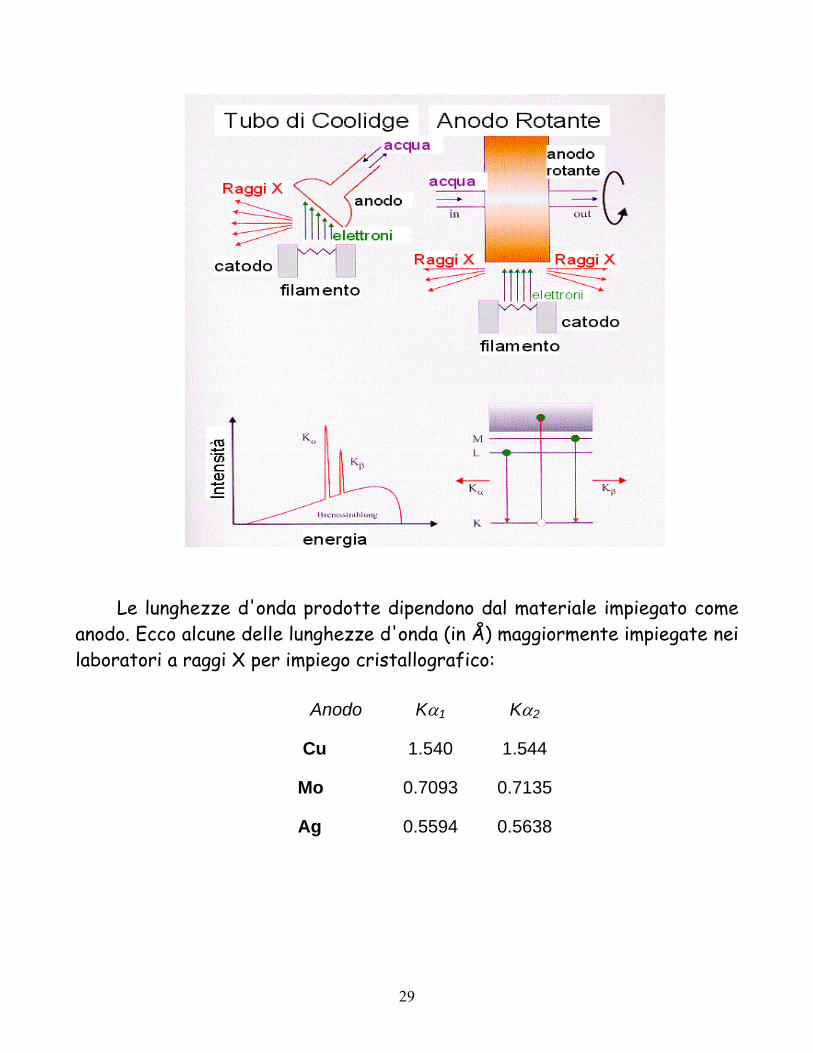
Le lunghezze d'onda prodotte dipendono dal materiale impiegato come
anodo. Ecco alcune delle lunghezze d'onda (in Å) maggiormente impiegate nei laboratori a raggi X per impiego cristallografico:
Anodo Kα1 Kα2
Cu 1.540 1.544
Mo 0.7093 0.7135
Ag 0.5594 0.5638
29
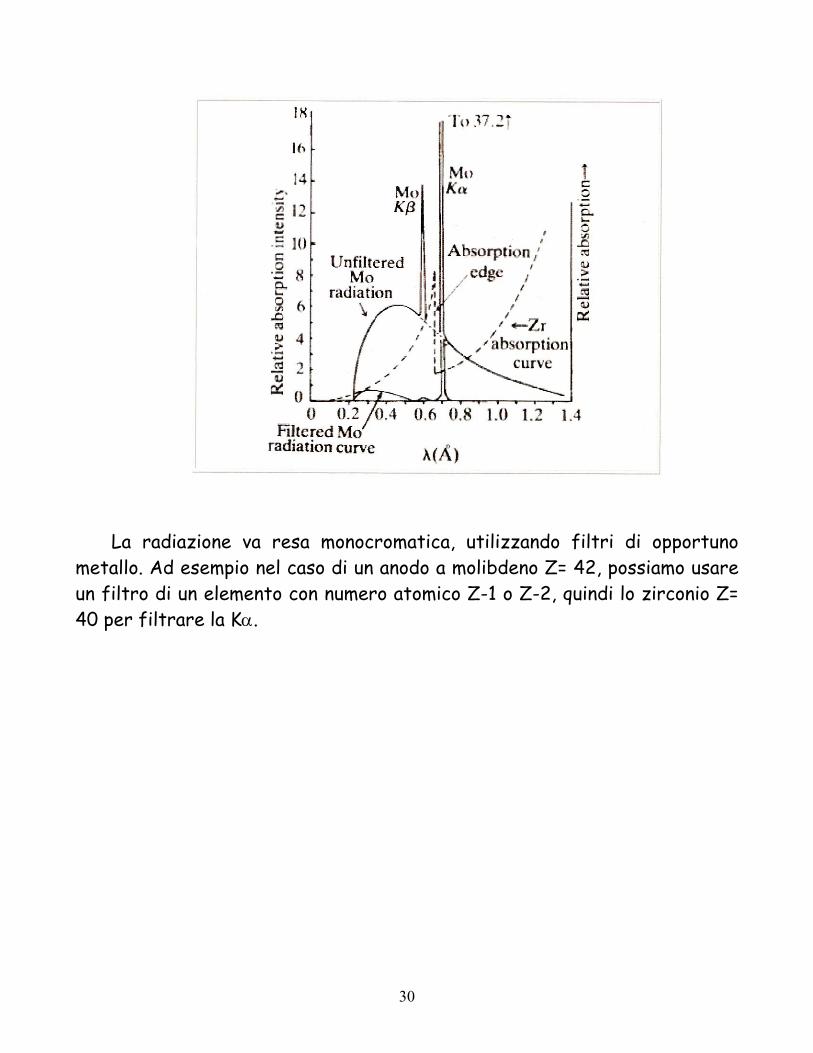
La radiazione va resa monocromatica, utilizzando filtri di opportuno
metallo. Ad esempio nel caso di un anodo a molibdeno Z= 42, possiamo usare un filtro di un elemento con numero atomico Z-1 o Z-2, quindi lo zirconio Z= 40 per filtrare la Kα.
30

DETERMINAZIONE E RAFFINAMENTO DELLE STRUTTURE, CRISTALLO SINGOLO
Il risultato della raccolta dei dati di diffrazione su cristallo singolo è un
set di intensità relative, I, per ogni riflessione h,k,l: I h,k,l. Dalla misura delle intensità dei raggi diffratti, non può essere ottenuto direttamente il calcolo della densità elettronica e quindi la determinazione della struttura. Le intensità Ih,k,l vanno corrette in genere per alcuni fattori geometrici che riguardano il modo in cui sono stati ottenuti (Fattore di Lorentz, Fattore di polarizzazione) e per l’assorbimento se la forma e la grandezza del cristallo non sono “appropriate”.
Il Fattore di Lorentz tiene conto della non perfetta monocromaticità della radiazione incidente, della leggera divergenza tra fascio incidente e diffratto e della conseguente diversa orientazione dei microcristalli nei solidi policristallini e nelle polveri. Sperimentalmente si osserva che in assenza di queste piccole imperfezioni, è estremamente bassa la probabilità di ottenere le figure di diffrazione.
Il Fattore di polarizzazione tiene conto dell’interazione fra il fascio di fotoni X (in genere non polarizzato, il campo e.m. oscilla in tutte le direzioni) e gli elettroni del campione.
La densità elettronica è data dalla trasformata di Fourier dei fattori di struttura (F), che sono quantità caratterizzate da un modulo corrispondente alla radice quadrata delle intensità dei raggi diffratti e da una fase (ϕ) di cui i dati sperimentali non forniscono alcuna informazione. Il valore del fattore di struttura dipenderà dagli indici hkl, dalla natura e dalla posizione degli atomi nella cella elementare.
Se definiamo il fattore di struttura Fh con indice h=(hkl)
∑ ===
N
jjj iFfF
1)e x p ()2e x p ( hhh h r ϕπ
31

La densità elettronica ρ(r) per ogni generico punto r della cella unitaria è definita da:
)2exp()exp()( 1 hrrh
hh iiFV πϕρ −∑= −
Per determinare una struttura (ottenere le posizioni atomiche) bisogna
risolvere il problema della fase (ϕ). I metodi più importanti di procedere sono: “trial and error”, Patterson e
metodi diretti. I “trial and error” sono metodi di tentativo che assegnano valori di
tentativo alle incognite (costituite dalle coordinate e fattori termici degli atomi), valutano di volta in volta i corrispondenti fattori di struttura “calcolati” e li confrontano con ivalori sperimentali fino all’ottenimento di un sostanziale accordo tra tutti i fattori calcolati ed i corrispondenti sperimentali.
I metodi di Patterson sono particolarmente utili quando nella molecola sono contenuti atomi pesanti. Nella funzione di Patterson sono considerati i quadrati dei fattori di struttura, in questo modo la parte immaginaria relativa alla fase scompare, inoltre fa individuare, non gli atomi ma i vettori interatomici. Nel caso di atomi pesanti i massimi di densità elettronica ed i relativi vettori atomo pesante-atomo pesante sono facilmente individuabili e quindi risulta più facile risalire alle loro posizioni equivalenti.
I metodi diretti sulla base della conoscenza della simmetria del contenuto unitario forniscono direttamente la struttura usando relazioni probabilistiche esistenti tra i fattori di struttura.
Dall’analisi strutturale si ottengono le tre coordinate frazionali di ogni atomo riferite agli assi cristallografici della cella unitaria ed un fattore termico (se isotropo, o una serie di sei numeri corrispondenti all’ellisoide se anisotropo). I valori di queste coordinate si ottengono dal raffinamento con
32

una procedura dei minimi quadrati tra fattori di struttura calcolati, basati sul confronto tra i fattori di struttura calcolati(Fhkl)calc e quelli osservati (Fhkl)osserv.
I parametri strutturali vengono modificati fino a quando non si ottiene la minima differenza tra osservazione e calcolo. Il modo di visualizzare tale differenza è il calcolo del fattore R detto indice di disaccordo. L’indice di disaccordo può essere calcolato sui moduli dei fattori di struttura, R = ∑hkl⏐Fo - Fc ⏐2/ ∑hkl Fo oppure sui moduli quadrati, R = ∑hkl⏐Fo
2 - Fc 2⏐2/ ∑hkl Fo
2. A ciascun atomo si assegna una terna di coordinate frazionarie (x, y, z; eventualmente alcune di esse possono essere vincolate dalla simmetria di sito), un parametro termico isotropo, oppure una serie di parametri che descrivono l'anisotropia del moto atomico. Inoltre si deve associare un parametro di scala che consente di equiparare le intensità misurate con quelle calcolate.
33

CALCOLO DELLA DENSITÀ La densità è data da d=m/V M è la massa e V è il volume La massa m è data da m = Z·M/N M = peso molecolare, N = numero di Avogadro, Z= numero di molecole
per cella Quindi d=m/V diventa d= Z ·M/N·V Il volume sarà V= a·b·c·(sen α·sen β·sen γ) Il volume è espresso in Å, bisogna trasformarlo in cm e quindi
moltiplicarlo per 10 -8. 1 Å = 10 -10 m d = 1.66 ·Z·M / V d ≈ 1.2 per sostanze organiche
34

Procedure per un esperimento da cristallo singolo
• Selezione del cristallo
dimensioni opportune
qualità (regolarità delle facce, colore,
presenza di geminazioni ecc.)
• Collocazione del cristallo in diffrazione
disposizione su testina goniometrica
centratura
• Determinazione della cella unitaria
analisi della posizione dei picchi
determinazione del reticolo reciproco
• Misura delle intensità diffratte • Analisi delle intensità
correzione dei dati
estrazione dei fattori di struttura
• Risoluzione della struttura
metodi diretti (statistici)
metodi "indiretti" (riconoscimento di "distanze")
• Affinamento del modello strutturale
35
Rappresentazione di una Struttura Cristallina
36

DIFFRAZIONE DI RAGGI X SU CAMPIONI POLICRISTALLINI
Nel caso in cui il campione si presenta sottoforma di polvere cristallina, formato da un’infinità di microcristalli (con tutte le possibili orientazioni), il metodo di diffrazione utilizza un diffrattometro per polveri.
Nella diffrazione da polveri i raggi diffratti diventano dei coni con centro nell’origine ed asse nella direzione del fascio incidente (coni di Debye-Scherrer); ciò è dovuto alla particolare simmetria della distribuzione delle orientazioni dei microcristalli intorno alla direzione del fascio incidente. Il diffrattometro utilizzato è progettato secondo la geometria di Bragg-Brentano (Figura 2.4) e lavora in riflessione consentendo così una elevata intensità del fascio diffratto. Il fascio incidente viene emesso da un tubo radiogeno e collimato da una fenditura per fargli assumere un percorso quanto più possibile rettilineo. La polvere cristallina viene pressata nell’incavo di una piastrina piatta che montata sul goniometro viene investita dai raggi-X.
Figura 2.4- Diffrattometro con geometria Bragg-Brentano.
37

Le diffrazioni emergono tutte simultaneamente, e un contatore montato su un cerchio 2θ, analogo a quello del diffrattometro per cristallo singolo, va a raccoglierle una per una, ruotando verso valori di 2θ crescenti o decrescenti.
I dati che si raccolgono sono dunque le intensità e gli angoli di diffrazione. Nello spettro di polveri le intensità sono riportate in funzione di 2θ. In generale il diffrattometro è controllato da un computer che gestisce la parte goniometrica e provvede all'acquisizione dei dati fornitigli dal rilevatore, dotato inoltre di un braccio rotante per acquisire informazioni a diverse angolazioni. Lo spettro di diffrazione raccolto mediante un diffrattometro automatico (Figura 2.5) è lo “squeezing” del reticolo reciproco tridimensionale nello spazio monodimensionale 2 θ. Ogni picco dello spettro è la traccia di un nodo del reticolo reciproco.
Figura 2.5 – Interpretazione dello spettro raccolto mediante un diffrattometro automatico come “squeezing” del reticolo reciproco tridimensionale nello spazio
monodimensionale 2 θ.
38
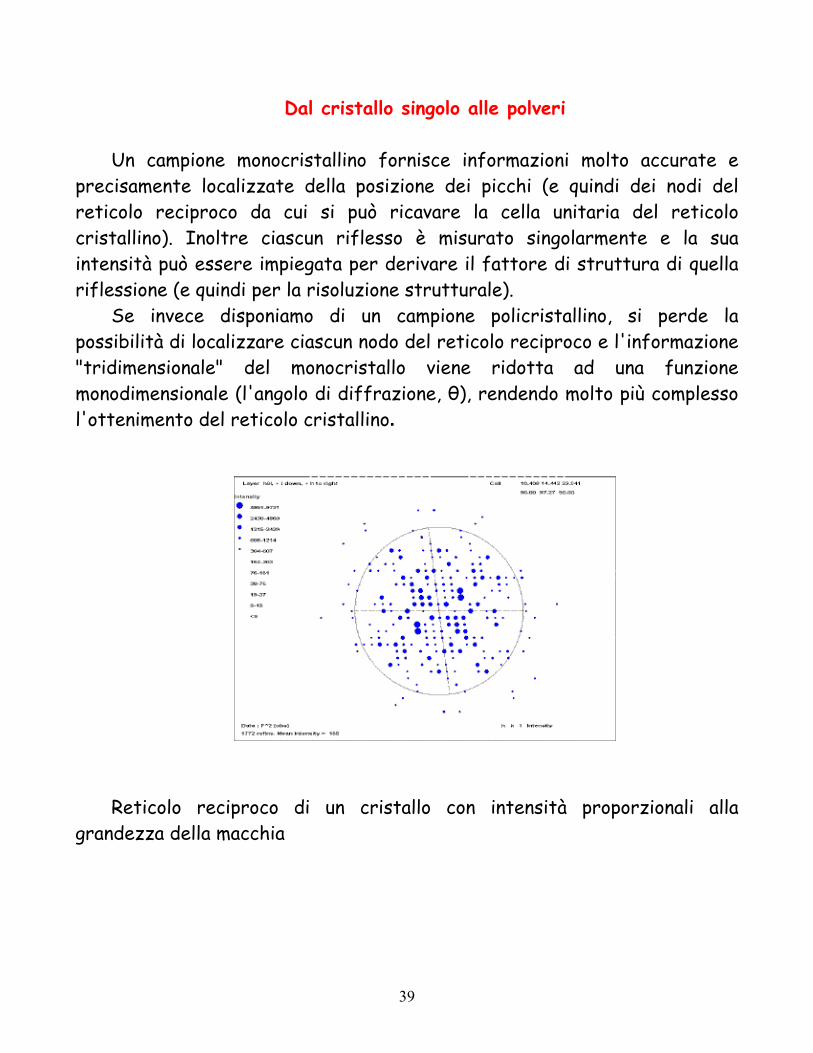
Dal cristallo singolo alle polveri Un campione monocristallino fornisce informazioni molto accurate e
precisamente localizzate della posizione dei picchi (e quindi dei nodi del reticolo reciproco da cui si può ricavare la cella unitaria del reticolo cristallino). Inoltre ciascun riflesso è misurato singolarmente e la sua intensità può essere impiegata per derivare il fattore di struttura di quella riflessione (e quindi per la risoluzione strutturale).
Se invece disponiamo di un campione policristallino, si perde la possibilità di localizzare ciascun nodo del reticolo reciproco e l'informazione "tridimensionale" del monocristallo viene ridotta ad una funzione monodimensionale (l'angolo di diffrazione, θ), rendendo molto più complesso l'ottenimento del reticolo cristallino.
Reticolo reciproco di un cristallo con intensità proporzionali alla
grandezza della macchia
39
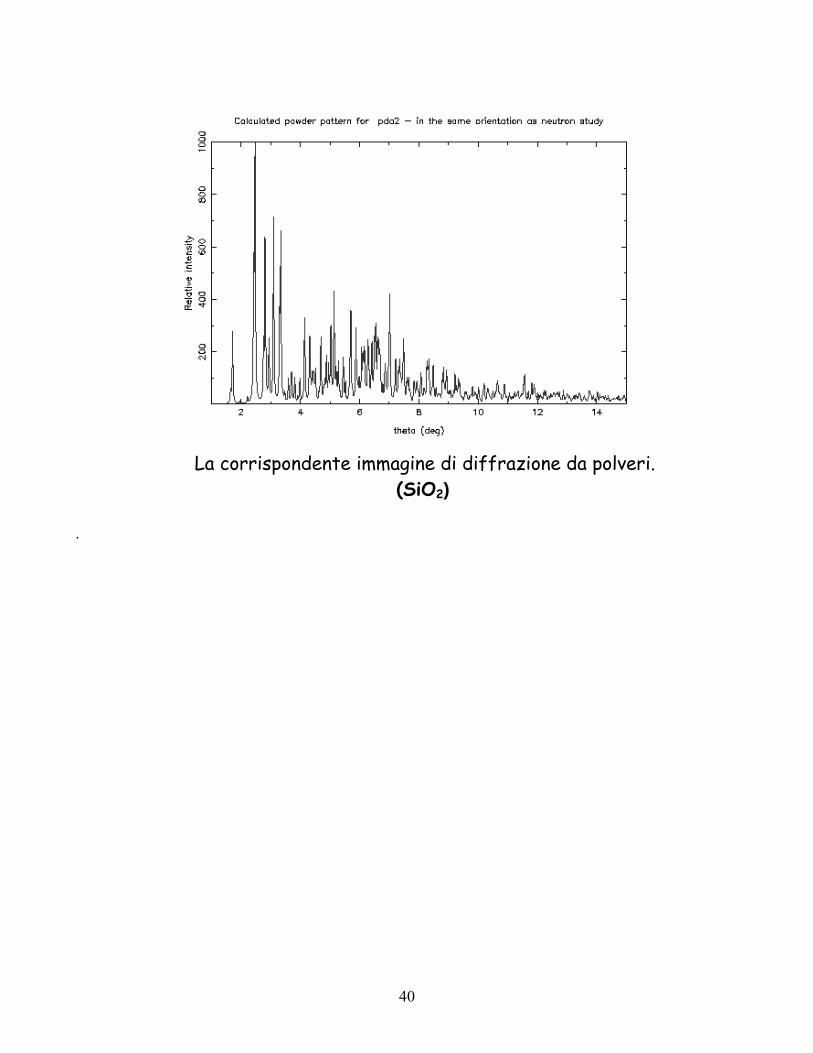
La corrispondente immagine di diffrazione da polveri.
(SiO2)
.
40
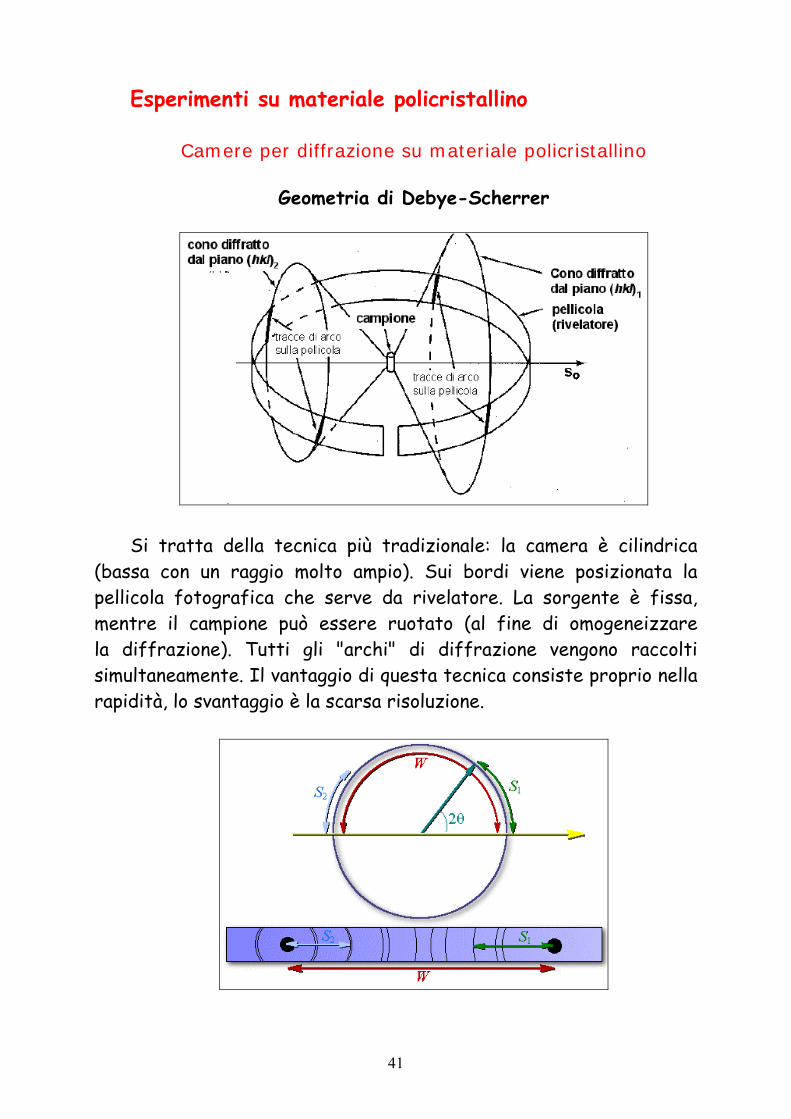
Esperimenti su materiale policristallino
Camere per diffrazione su materiale policristallino
Geometria di Debye-Scherrer
Si tratta della tecnica più tradizionale: la camera è cilindrica
(bassa con un raggio molto ampio). Sui bordi viene posizionata la pellicola fotografica che serve da rivelatore. La sorgente è fissa, mentre il campione può essere ruotato (al fine di omogeneizzare la diffrazione). Tutti gli "archi" di diffrazione vengono raccolti simultaneamente. Il vantaggio di questa tecnica consiste proprio nella rapidità, lo svantaggio è la scarsa risoluzione.
41

La tipica immagine prodotta sulla pellicola avrà la seguente forma:
diffrazione a polvere registrata su pellicola
Un'immagine di una camera di Debye
42

Geometria di Bragg-Brentano Con questa geometria, il campione è sempre in una precisa
posizione "focalizzata", che viene preservata cambiando simultaneamente l'angolo incidente e quello di rivelazione (θ-θ, con sorgente mobile e campione fisso), oppure variando opportunamente l'orientazione del campione e l'angolo di rivelazione (ω-2θ). Il rivelatore è un contatore fotonico.
Diffrattometro con geometria Bragg-Brentano
43

Caratterizzazione del Campione
Procedura sperimentale selezione del campione (microcristallinità) macinatura per migliorare l’omogeneità riducendo le dimensioni delle particelle (ma non troppo per evitare l’allargamento dei picchi) deposizione del campione su supporto centratura del supporto nel goniometro scansione (selezionando il tipo di scansione, la velocità ecc.)
44

45

supporti per depositare il campione microcristallino Come il cristallo singolo, presenta al proprio interno una disposizione periodica
degli atomi, così per un campione policristallino, ad ogni cristallita è associato il
corrisponde reticolo reciproco caratteristico della specie in esame ed orientato in
modo casuale.
46

Il pattern di diffrazione complessivo, dovuto alla diffrazione contemporanea dei
cristalliti, è la 'composizione' della figura di diffrazione di ognuno di essi. Uno spettro
di diffrazione di raggi-X da polveri consente l’acquisizione di almeno tre informazioni:
a) la posizione angolare dei picchi, dipendente dai parametri di cella e
dall’allineamento strumentale;
b) l’intensità dei picchi, influenzata dal contenuto dell’unità asimmetrica e dalla
distribuzione statistica del particolato;
c) il profilo dei picchi, connesso alla geometria ed all’allineamento strumentali
nonché alle dimensioni ed allo strain dei cristalliti.
Utilizzo di banche dati
• PDF: archivio che supporta l’analisi dei dati da diffrazione di
polveri e consente l’analisi qualitativa delle fasi • ICSD: archivio elettronico delle strutture inorganiche; contiene
informazioni raccolte in esperimenti a polveri o cristallo singolo di materiali inorganici (sono esclusi i metalli e leghe)
• CSDS: archivio elettronico dei composti molecolari (organici o
metallorganici). Prevalentemente i dati sono raccolti da esperimenti su cristallo singolo
• PDB: archivio elettronico delle strutture di proteine
47

Analisi qualitativa L’analisi qualitativa si riferisce alla identificazione di fasi presenti
nei materiali a composizione mista oppure al riconoscimento di fasi a componente singolo.
La struttura cristallina di molte fasi solide è nota, perché identificata con metodi diffrattometrici a partire dalla introduzione di queste tecniche, cioè a partire dalla prima metà del XX secolo.
La principale "risorsa" di informazioni per l’identificazione di fasi ignote è il Powder Diffraction File, ossia un archivio elettronico (o cartaceo) dove sono contenute informazioni cristallografiche per più di 300000 fasi inorganiche ed organiche.
La diffrazione è una informazione primaria, che combinata con l’analisi elementare identifica senza ambiguità una certa fase cristallina.
Le impronte di diffrazione sono generalmente note da esperimenti condotti con radiazione X su materiale policristallino (registrate su pellicola oppure da rivelatore fotonico).
Tuttavia è possibile anche calcolare lo "spettro" di diffrazione a partire dalla struttura determinata su cristallo singolo.
In realtà con il termine fase ignota si intende una fase riconosciuta in passato (e quindi già identificata) ma non facilmente identificabile nel campione in esame.
Ovviamente le potenzialità di questo tipo di metodo crescono con il tempo, ossia più fasi nuove vengono identificate attraverso accurate metodologie cristallografiche maggiori sono le possibilità di riconoscere tali fasi in campioni in esame
co-presenza di più fasi
Se in un campione policristallino esistono più fasi, la diffrazione da polveri conterrà picchi corrispondenti a distanze interplanari di tutte le fasi, complicando il riconoscimento delle stesse.
48

Applicazioni
• mineralogia:
determinazioni strutturali
riconoscimento di fasi
determinazioni di composizione chimica
• chimica inorganica e dei materiali
determinazioni strutturali
studi di transizioni di fase (in particolare quelle accompagnate da mutamenti di proprietà, quali conduzione, magnetismo ecc.)
• chimica organica e organometallica:
determinazione di strutture molecolari
studio della dinamica in stato solido
analisi del legame chimico
studi di transizioni di fase e studi di polimorfismo
• bio-cristallografia:
determinazione strutturale di proteine (migliaia di atomi)
49

Applicazioni in ambito biologico - cristallografia di proteine
Le proteine possono essere pensate come polimeri dei 20 amminoacidi naturali, contenenti eventualmente altri gruppi (ad esempio, carboidrati oppure cationi complessati).
Le proteine possono essere di tipo globulare oppure fibroso. Le prime sono adatte all'ottenimento di forme mono-cristalline,
con cui è possibile determinare la struttura mediante diffrazione di raggi X. A differenza delle proteine, gli acidi nucleici sono meno ottenibili in forma cristallina. Data l'enorme dimensione di queste macromolecole, le conformazioni che possono essere assunte sono innumerevoli e questo può complicare lo studio strutturale. Inoltre si deve tenere in considerazione che dalla diffrazione su cristallo si ottiene necessariamente un'immagine statica, nonostante la molecola sia in realtà dominata da effetti dinamici considerevoli.
Dal punto di vista strutturale, possiamo individuare una struttura primaria, formata dai legami covalenti della sequenza di amminoacidi e dove risultano parametri rilevanti le torsioni attorno a due legami. Nonostante non sia possibile prevedere esattamente a partire da una data sequenza di amminoacidi quale sia la struttura tridimensionale della proteina, alcuni importanti fattori incidono sulla struttura:
50

• i gruppi in grado di formare legami ad idrogeno si posizionano sulla "superficie" della proteina in modo da massimizzare l'interazione con il solvente, oppure all'interno in modo da interagire al meglio con gli eventuali donatori
• in una proteina solubile in acqua, i residui polari si posizioneranno preferibilmente alla "superficie"
Oltre alla struttura primaria è sempre riconoscibile una struttura secondaria, dominata dalla presenza di legami ad idrogeno (di tipo N-H...O). Questo impone un limitato numero di conformazioni della proteina:
• elicoidale (alpha-helix) • lineare (beta strands), con accoppiamento parallelo o
antiparallelo con altre catene (beta-sheet) E' anche possibile riconoscere ulteriori livello di arrangiamento
(strutture terziarie, quaternarie, ecc.).
Un aspetto molto importante nell'ambito sperimentale è la crescita di cristalli di proteine adatti agli esperimenti di diffrazione da raggi X. Spesso si usano agenti precipitanti (sali inorganici o organici, solventi organici e glicoli polietilenici) per favorire la nucleazione di cristalli a partire da soluzioni sovra-sature di proteine.
51

Vista la labilità di queste molecole, le condizioni in cui si conduce una cristallizzazione non devono essere particolarmente estreme. Inoltre, al fine di comprendere l'utilità biologica della molecola in studio, si deve cercare di creare un ambiente non troppo dissimile da quello fisiologico in cui la proteina è attiva.
Infine, la protezione criogenica dei cristalli formati è essenziale per condurre esperimenti sugli stessi (solitamente anche gli esperimenti di diffrazione avvengono a basse temperature).
Le metodologie sperimentali spesso si basano sull'impiego di una radiazione con elevata brillanza, quale quella ottenuta da anelli di sincrotrone oppure, per quanto riguarda strumenti di laboratorio, che siano dotati almeno di anodo rotante. Inoltre, si impiegano tecniche di raffreddamento per preservare i campioni durante la raccolta dati.
Per quanto riguarda i metodi di soluzione delle strutture, il generale problema della fase rimane il principale ostacolo, acuito dalla bassa risoluzione dei set di dati a disposizione (in genere è difficile poter estendere la raccolta dei dati poiché la qualità dei cristalli e l'ampiezza del moto termico sono tali da impedire risoluzioni inferiori a 1 Å). Inoltre, viste le dimensioni delle molecole e la loro natura è molto difficile poter impiegare semplicemente i metodi di soluzione introdotti fino ad ora.
Per proteine completamente nuove, il metodo classicamente più utilizzato e tuttora molto valido è la sostituzione isomorfa: la proteina di cui si vuole determinare la struttura viene modificata andando ad inserire ad esempio nei canali che ospitano solvente, molecole con atomi più pesanti, in modo da poter semplificare il problema della fase. Supponendo che le modificazioni indotte da questa operazione siano trascurabili è possibile correlare la struttura del derivato isomorfo con quella della proteina iniziale (andando ad indagare le differenze di funzioni di Patterson, con le quali si possono inizialmente localizzare gli atomi pesanti). Questa procedura può coinvolgere un solo prodotto di sostituzione (SIR) oppure più prodotti di sostituzione (MIR).
Un metodo di risoluzione alternativo è quello che si basa sull'accentuazione della dispersione anomala di una classe di diffusori. Questo principio è anche alla base del metodo multiple 52

anomalous dispersion (MAD) con cui si impiegano differenti lunghezze d'onda. Ricordando che la componente diffusiva anomala dipende appunto dalla lunghezza d'onda della radiazione incidente si può capire che l'impiego di diverse radiazioni (scelte opportunamente in prossimità di massime e minime variazioni delle componenti immaginaria e reale del fattore diffusivo anomalo) consenta di ricostruire la diffusione anomala ed infine la soluzione del problema della fase, in modo analogo alla sostituzione isomorfa, pur in assenza di elementi pesanti.
Applicazione della diffrazione da polveri nella scienza forense
David Rendle (Forensic Science London Laboratory, UK) in
Industrial application of X-ray diffraction Principali tecniche impiegate da molti anni: • Diffrazione da raggi X • Fluorescenza da raggi X • Radiografie a raggi X Principali ambiti di applicazione: • analisi di medicinali, farmaci, droghe ecc. • frammenti vetrosi • fibre, polveri ecc. • capelli, peli • tessuti • metalli • materiali per costruzioni
Tecniche analitiche complementari
• NMR • FT-IR • microscopia elettronica (SEM) • gas cromatografia e gas cromatografia di massa • cromatografia liquida (HPLC) • UV • microscopia (anche con luce polarizzata)
53

Modalità di utilizzo delle tecniche diffrattometriche
Si utilizzano le più comuni camere per diffrazione da polveri con
procedure convenzionali che privilegiano l’esigenza di rapidità (tipico spettro in ca. 1h), conservazione del campione (tipicamente un’analisi diffrattometrica è di tipo conservativo) e di accuratezza (in genere l’errore nel riconoscimento di fase è basso se si dispone di sufficiente campione e di una buona cristallinità dello stesso).
Un altro vantaggio consiste nella elevata versatilità della tecnica che consente analisi di materiali organici, biologici, inorganici, metalli ecc, disponibili anche in piccola quantità (almeno 20-30 mg.).
Difficoltà nelle analisi possono venire dalle scarse quantità di campione (comunque non riproducibili) e dalla elevata contaminazione.
Esempi Caso 1: Se in una indagine vengono trovati materiali presumibilmente
proibiti (come droghe) queste vengono inviate al FSS per l’analisi. L’analisi preliminare si effettua con FTIR, GC (qualitativa) ; HPLC o
NMR per un’analisi quantitativa; per riconoscimento fase solida si impiega anche la diffrazione da polvere.
Ad esempio si vuole confermare che le polveri cristalline siano fasi solide note delle sostanze sospette.
In un caso, venne riconosciuta eroina (cloroidrata), e NaCl, come sospettato da indagini spettroscopiche. Tuttavia il glucosio (pure trovato per spettroscopia) non venne identificato dall’analisi diffrattometrica. Al contrario si evidenziava una fase (apparentemente) sconosciuta. In seguito si identificò questa fase come una fase mista (glucosio+NaCl), in realtà nota fin dal 1947).
54
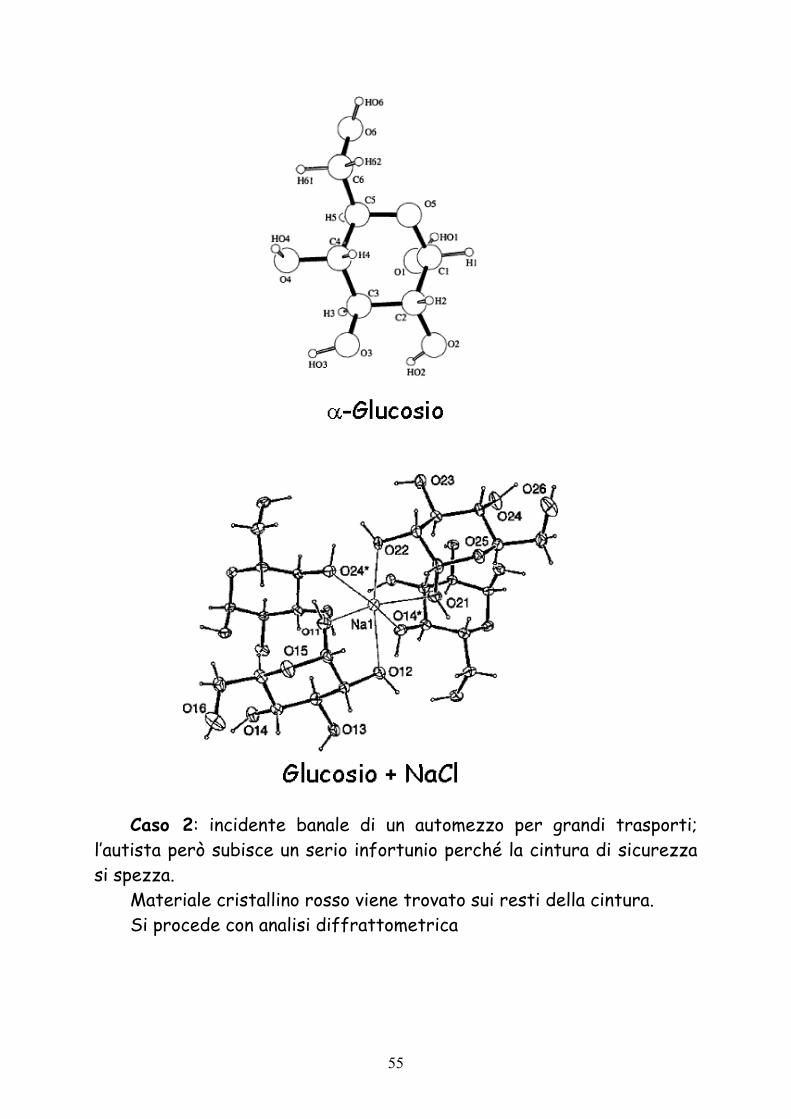
Caso 2: incidente banale di un automezzo per grandi trasporti; l’autista però subisce un serio infortunio perché la cintura di sicurezza si spezza.
Materiale cristallino rosso viene trovato sui resti della cintura. Si procede con analisi diffrattometrica
55
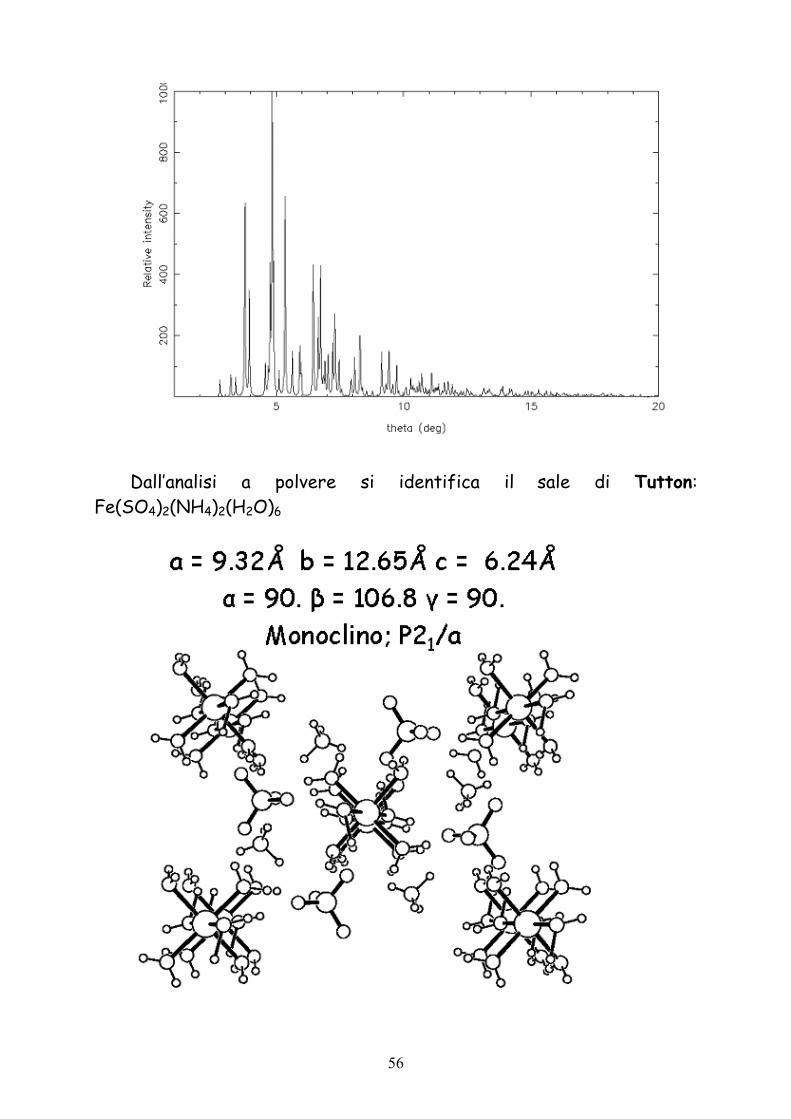
Dall’analisi a polvere si identifica il sale di Tutton: Fe(SO4)2(NH4)2(H2O)6
56

Che cosa è successo? L’autotrasportatore era solito maneggiare, batterie per automobili
(quindi contenenti H2SO4); le cinture di sicurezza sono costituite da poliammidi.
La casa produttrice del automezzo non è responsabile dell’incidente
applicazioni in ambito artistico e archeologico
Sebbene arte e scienza si siano sviluppate in parallelo, esistono spesso alcuni "punti di contatto", sia quando la scienza dei materiali consente nuove forme di espressione artistica, sia quando le tecniche analitiche consentono di analisi di approfondire la conoscenza artistica (storia dell'arte), sia quando la scienza consente di restaurare o conservare più a lungo materiali impiegati nella produzione artistica.
Esistono due metodiche analitiche, una basata sulla visione globale dell'oggetto e l'altra su una visione puntuale. In entrambi i casi si possono utilizzare radiazioni che vanno dai raggi gamma fino alla radiazione infrarossa (si veda lo spettro delle radiazioni).
57

Con le radiazioni X, si impiegano sia metodi di fluorescenza sia di diffrazione. Sebbene le tecniche siano genericamente indicate come non distruttive (ossia non distruggono il campione) in ambito artistico archeologico è chiaro che la preparazione del campione di fatto sia di tipo distruttivo. Metodi di applicazione in situ non sono facilmente applicabili a causa della enorme quantità di radiazione incidente che verrebbe dispersa in un ipotetico sistema aperto. Esiste anche un problema di stabilità dei materiali coloranti sotto l'azione dei raggi X (in particolare negli strati più superficiali, generalmente molto più sensibili alla luce, e che sono soggetti ad ingiallimento). A volte si impiega la microdiffrazione, ossia si dirige sul campione un fascio di raggi X estremamente sottile, evitando così di dover procedere a minuziose preparazioni del campione. Il vantaggio dei metodi microdiffrattometrici consiste nella possibilità di studiare tutti gli strati di un dipinto senza incorrere nei problemi di contaminazione da parte di altri strati.
58

Gli scopi delle tecniche basate su diffrazione di raggi X sono molteplici: analisi qualitativa con riconoscimento di fasi, analisi quantitativa, mappatura della composizione chimica o della distribuzione di fasi. In genere la diffrazione è accoppiata con fluorescenza, microscopia elettronica, spettroscopia UV.
La diffrazione a polveri è condotta utilizzando camere di Gandolfi (oppure anche Bragg-Brentano).
Le analisi su materiali artistici e oggetti di interesse storico si concentra soprattutto su:
1) analisi dei pigmenti al fine di ricostruire le tecniche e le conoscenze in ambito dei coloranti per oggetti artistici.
I pigmenti base noti per le colorazioni sono in realtà abbastanza limitati (circa 100), ma spesso si presentano come composizioni in genere fino a tre componenti. A volte anche piccole tracce di un dato pigmento base rendono tipico il colorante. Ad esempio il lapis lazuli o blu ultramarino. La fase naturale contiene piccole tracce di calcite o di pirite, che però non riescono ad essere introdotte nelle fasi sintetiche. Altre volte i pigmenti differiscono solo per la fase cristallina, non per composizione chimica come avviene per calcite ed aragonite (CaCO3) oppure rutilo e anatasio (TiO2)
esempio: identificazione dei pigmenti in arazzi cinesi nel palazzo di Schönbrunn a Vienna.
Dopo aver grattato pochi frammenti dall'arazzo, si sono identificai i pigmenti Piombo bianco (bianco), azzurrite (blu) e malachite (verde) per via diffrattometrica e confermata la composizione con fluorescenza.
59
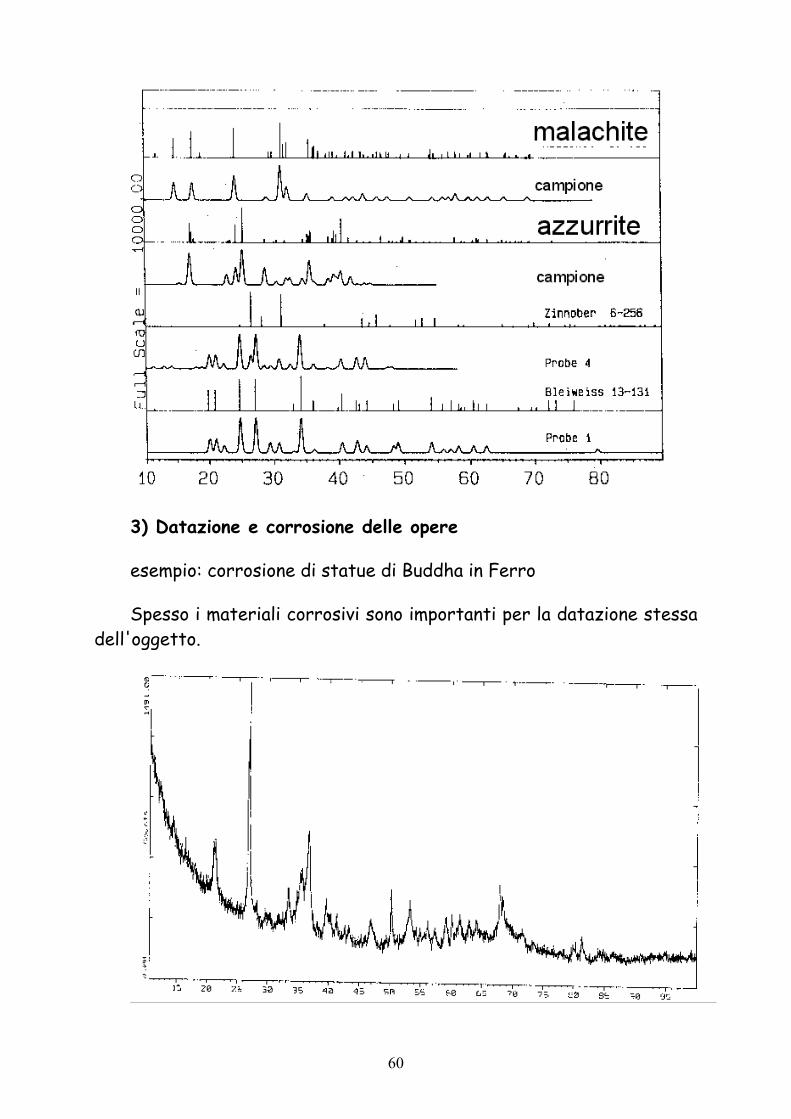
3) Datazione e corrosione delle opere
esempio: corrosione di statue di Buddha in Ferro
Spesso i materiali corrosivi sono importanti per la datazione stessa dell'oggetto.
60

Prodotti della corrosione: α-FeOOH, β-FeOOH, γ-FeOOH, Fe2O3, Fe3O4.
La datazione fa risalire la statua al XV secolo, durante la dinastia Ming.
Analisi delle silici Un esempio delle applicazioni di analisi quantitativa delle fasi è
nell'ambito delle analisi delle silici. Poiché Silicio e Ossigeno sono due degli elementi più abbondanti
sulla crosta terrestre non sorprende che il loro composto più comune (SiO2) sia così comune.
Esistono diverse forme cristalline di SiO2, di cui una sola (α-quarzo) è stabile a temperatura e pressione ambiente:
Quarzo
Cristobalite
Tridimite
61
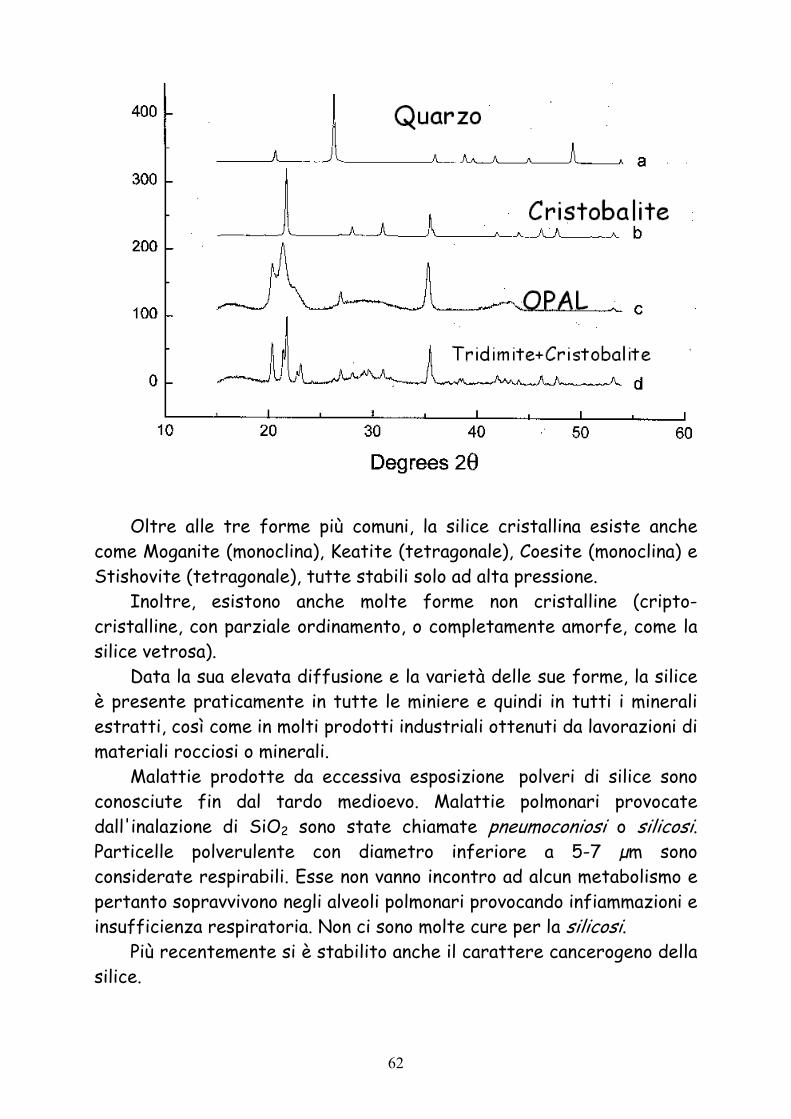
Oltre alle tre forme più comuni, la silice cristallina esiste anche
come Moganite (monoclina), Keatite (tetragonale), Coesite (monoclina) e Stishovite (tetragonale), tutte stabili solo ad alta pressione.
Inoltre, esistono anche molte forme non cristalline (cripto-cristalline, con parziale ordinamento, o completamente amorfe, come la silice vetrosa).
Data la sua elevata diffusione e la varietà delle sue forme, la silice è presente praticamente in tutte le miniere e quindi in tutti i minerali estratti, così come in molti prodotti industriali ottenuti da lavorazioni di materiali rocciosi o minerali.
Malattie prodotte da eccessiva esposizione polveri di silice sono conosciute fin dal tardo medioevo. Malattie polmonari provocate dall'inalazione di SiO2 sono state chiamate pneumoconiosi o silicosi. Particelle polverulente con diametro inferiore a 5-7 µm sono considerate respirabili. Esse non vanno incontro ad alcun metabolismo e pertanto sopravvivono negli alveoli polmonari provocando infiammazioni e insufficienza respiratoria. Non ci sono molte cure per la silicosi.
Più recentemente si è stabilito anche il carattere cancerogeno della silice.
62

Per questi motivi la prevenzione dall'esposizione a polveri di silice è essenziale e le misure di sicurezza adottate dalle industrie devono basarsi anche sull'analisi delle fasi (riconoscimento e quantificazione).
I metodi di analisi quantitativa si basano sia su diffrazione da polveri sia su spettroscopia IR, tuttavia questi metodi non garantiscono accuratezza per presenza di SiO2 al di sotto di 0.1% in peso. Comunque, i metodi diffrattometrici sono quelli che garantiscono la migliore accuratezza nel più ampio spettro di matrici possibili.
Preparazione del campione Bisogna subito distinguere tra analisi di materiale solido e analisi di
materiale disperso in aria. Il materiale "volatile" si può analizzare solo dopo averlo "intrappolato" in filtri appositi con setacci molto fini che separano il gas dal materiale polverulento. I filtri sono normalmente indossati dai lavoratori in apposite apparecchiature. Spesso il quarzo o la cristobailite viene cercato mediante diffrazione direttamente sul filtro oppure dopo trasferimento del materiale su altro substrato.
Analisi qualitativa delle fasi Il riconoscimento delle diverse forme cristalline di silice è
relativamente semplice vista la differente occorrenza dei picchi di diffrazione. L'identificazione delle forme amorfe e cripto-cristalline è invece più difficile. Se una di queste specie è presente, complessivamente l'analisi è resa più difficile.
Un problema aggiuntivo è dato dal grado di cristallinità del campione, che spesso dipende dalla temperatura a cui si è formata la specie (spesso più elevata è la temperatura, maggiore è la cristallinità). Se a cristallinità è bassa, diviene più difficile distinguere fasi cristalline da quelle non cristalline. Ad esempio può essere difficile distinguere campioni a bassa cristallinità di α-cristobalite e campioni di opal (silice idrata, generalmente amorfa).
La differenza è importante visto che l'opal non è considerato cancerogeno, a differenza delle fasi di SiO2. Un modo per distinguere è riscaldare il campione, poiché la silice idrata mostrerà differenze ( a causa dell'acqua presente) mentre la cristobalite non mostra cambiamenti significativi.
Molte forme amorfe si convertono in cristobalite se scaldate oltre 1400°C.
63

Analisi quantitativa delle fasi Il problema è quello di determinare la presenza percentuale di silice
in una certa matrice solida. La silice non è mai la fase più abbondante e spesso è anzi presente in tracce. Una difficoltà sperimentale consiste nel fatto che il campione è depositato su filtri molto sottili. La calibrazioni richiede la preparazione di una serie di standard per confronto. Nel caso di campioni sottili la componente di assorbimento può essere trascurata, semplificando le equazioni.
Viene spesso impiegata la calibrazione esterna con una fase di composizione simile a quella ignota in esame, dal momento che la composizione è spesso quasi costante. Questo metodo è economico perché usa poco tempo-macchina e minimizza i problemi dovuti alla non conoscenza del coefficiente i massa. Inoltre gli standard possono essere preparati una volta sola, ma è necessaria un’analisi chimica dei campioni studiati In altri casi è impiegata la calibrazione interna, molto accurata, ma che richiede molta cura nella preparazione del campione e a volte anche l’uso di un secondo standard.
Difficoltà di integrazione dei picchi Un problema con l’analisi delle silici nasce dalla possibile
sovrapposizione dei picchi di differenti fasi cristalline di SiO2 e molte fasi di minerali presenti in prodotti naturali
Un metodo per eliminare questo problema è l’uso di modellazione del profilo di diffrazione.
Fonti di errore Gli errori nell'analisi quantitativa possono dipendere • da fattori strumentali: errori nel conteggio da parte dei
rivelatori, saturazione, instabilità strumentale, disallineamento; • dal campione: statistica delle particelle, estinzione,
microassorbimento, orientazione preferenziale, errori di misura del peso, errori di campionamento;
• da procedure di trattamento dei dati: interferenza tra picchi, misura dell'area di un picco, misura del fondo, correzione per assorbimento di massa;
Alcuni errori sono determinabili tramite misure multiple anche sullo stesso campione (come fluttuazioni strumentali a breve termine) oppure su campioni diversi (statistica delle particelle orientazione preferenziale ecc.).
64
Altri risultano difficili da correggere con misure ripetute (ad esempio fluttuazioni a lungo termine, presenza di strati amorfi, uso di standard non appropriati, errori di campionamento ecc.).
Analisi diffrattometriche I metodi convenzionali si basano su analisi delle intensità (o delle
intensità integrate) e rapporti tra esse. Tuttavia, le tecniche tradizionali hanno alcuni svantaggi nell'analisi della composizione dei cementi, in particolare, a causa delle sovrapposizioni tra i picchi di diverse fasi che impediscono una accurata determinazione delle abbondanze (poiché i picchi più intensi spesso sono coinvolti nelle sovrapposizioni e le stime quantitative si devono basare sui picchi meno intensi).
Problemi aggiuntivi sono: • la maggior parte delle fasi mostra un ammontare variabile di
sostituzioni atomiche (soluzioni solide) che possono avere una sostanziale influenza nei confronti della misura delle intensità e nel posizionamento dei picchi
• grande varianza del grado di cristallinità delle fasi in esame ( e quindi incertezza sull'ammontare della sovrapposizione tra picchi)
• fasi come C3S mostrano notevole orientazione preferenziale
65