Embed Size (px)
Citation preview

Digestive Pathology Lecture 6
Reproduction Prohibited
This file contains original text and images as well as materials adapted from copyrighted sources
For use only as a temporary educational aid
Partially or completely copying or distributing the contents of this file may constitute an infringement of the fair use exception
for teaching faculty of the U.S. Copyright Law
LSUHSC-New Orleans, 2015
Last updated on September 30, 2015
--

The liver III
11. Inborn errors of metabolism
– Wilson disease
– Alpha1-antitrypsin deficiency
– Hemochromatosis12. Intrahepatic biliary tract diseases
13. Circulatory disorders
14. Hepatic disease in pregnancy
15. Nodular hyperplasias
16. Benign neoplasms
17. Malignant neoplasms

Wilson disease (hepatolenticular degeneration)
Autosomal recessive
Progressive copper accumulation
Oxidative injury Commonly manifest in late adolescence Hepatic manifestations
– Chronic hepatitis, cirrhosis– Massive necrosis, fulminant failure

Wilson disease: Chronic hepatitis, copper stains
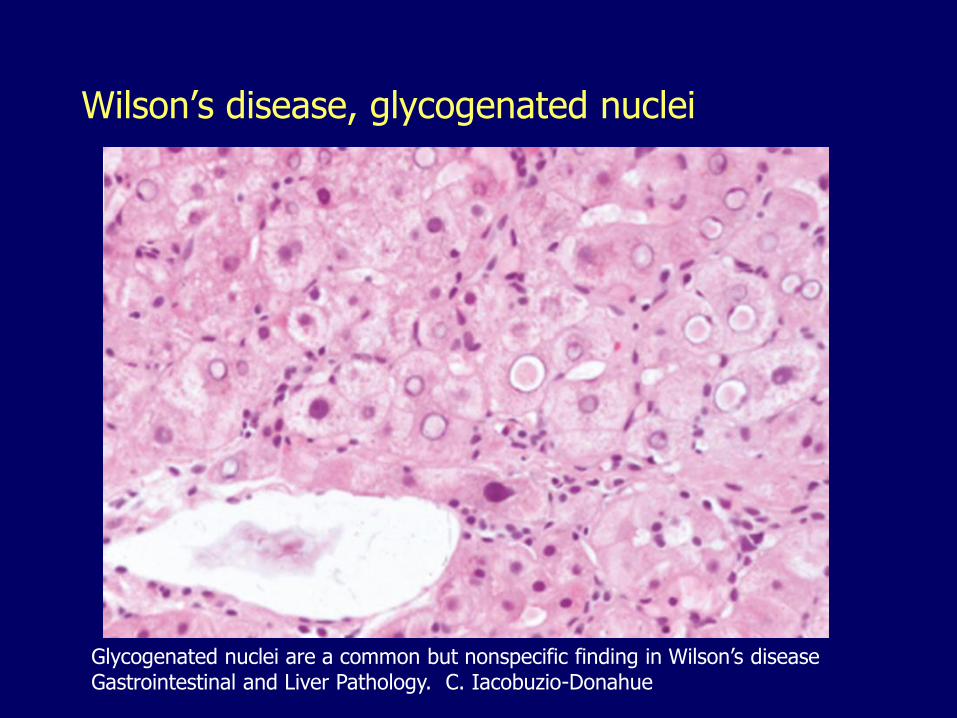
Wilson’s disease, glycogenated nuclei
Glycogenated nuclei are a common but nonspecific finding in Wilson’s diseaseGastrointestinal and Liver Pathology. C. Iacobuzio-Donahue

Wilson disease: Mallory bodies
Numerous Mallory bodies (arrowheads)Scheuer’s Liver Biopsy Interpretation

Wilson, cirrhosis, acute failure
Cirrhotic nodules (N) are surrounded by inflamed fibrous septa Scheuer’s Liver Biopsy Interpretation
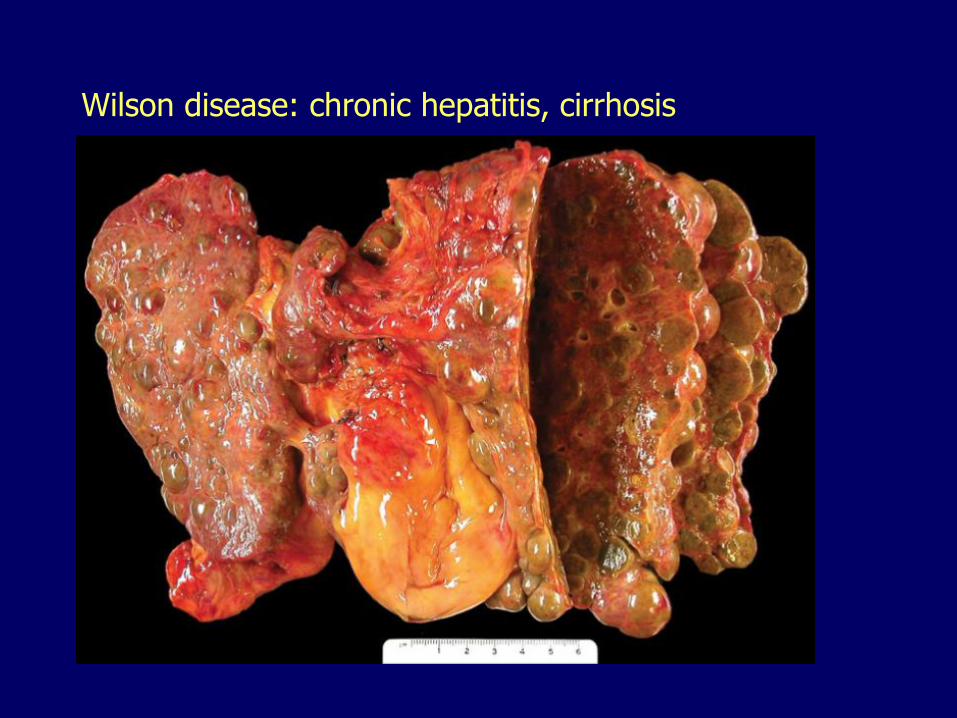
Wilson disease: chronic hepatitis, cirrhosis

Wilson disease (hepatolenticular degeneration)
Neurologic manifestations– Injury of basal ganglia– Incoordination, dystonia
• muscle spasms/cramps, involuntary twisting, jerking, speech and gait disturbances
– Psychiatric disorders• Depression, bipolar disorder, psychosis
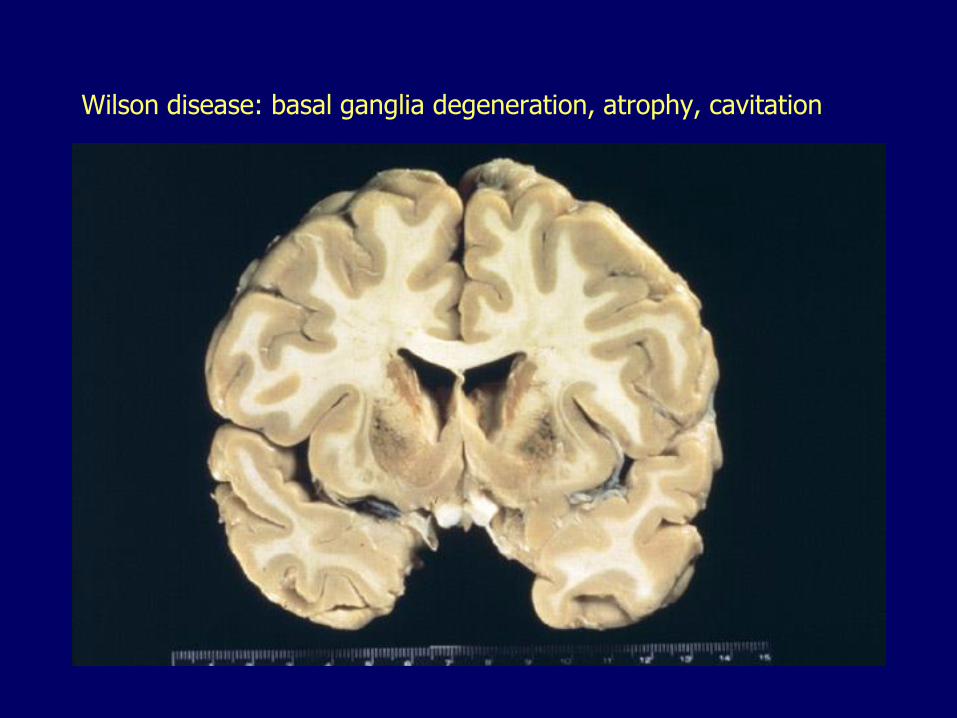
Wilson disease: basal ganglia degeneration, atrophy, cavitation

Wilson disease (hepatolenticular degeneration)
Hematologic manifestations– Hemolytic anemia
Ophthalmologic manifestations– Kayser-Fleischer rings (corneal limbus)
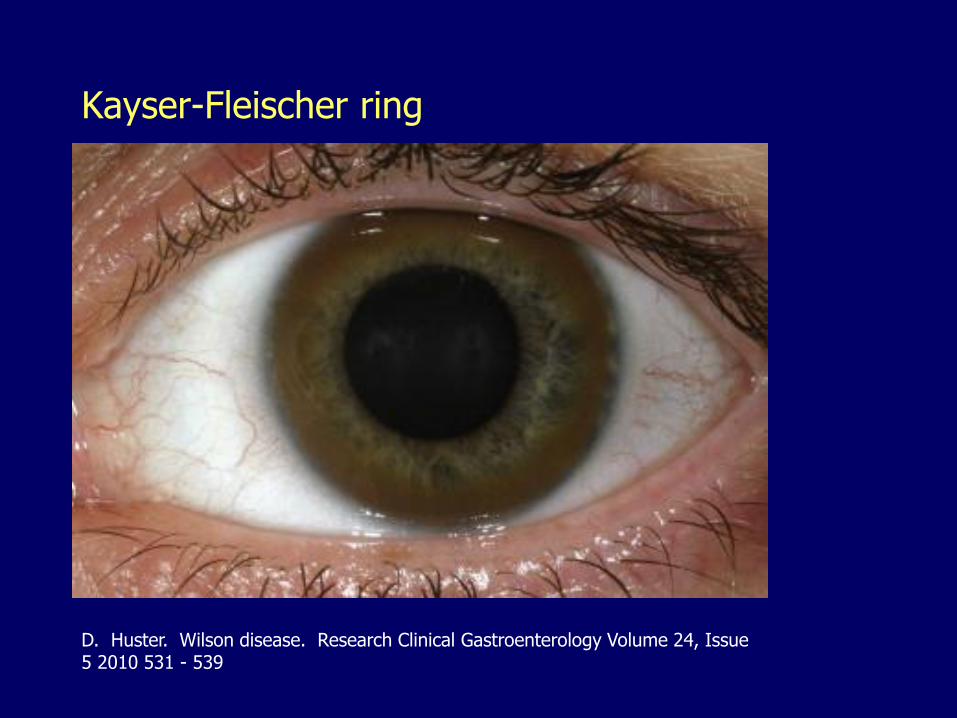
Kayser-Fleischer ring
D. Huster. Wilson disease. Research Clinical Gastroenterology Volume 24, Issue 5 2010 531 - 539
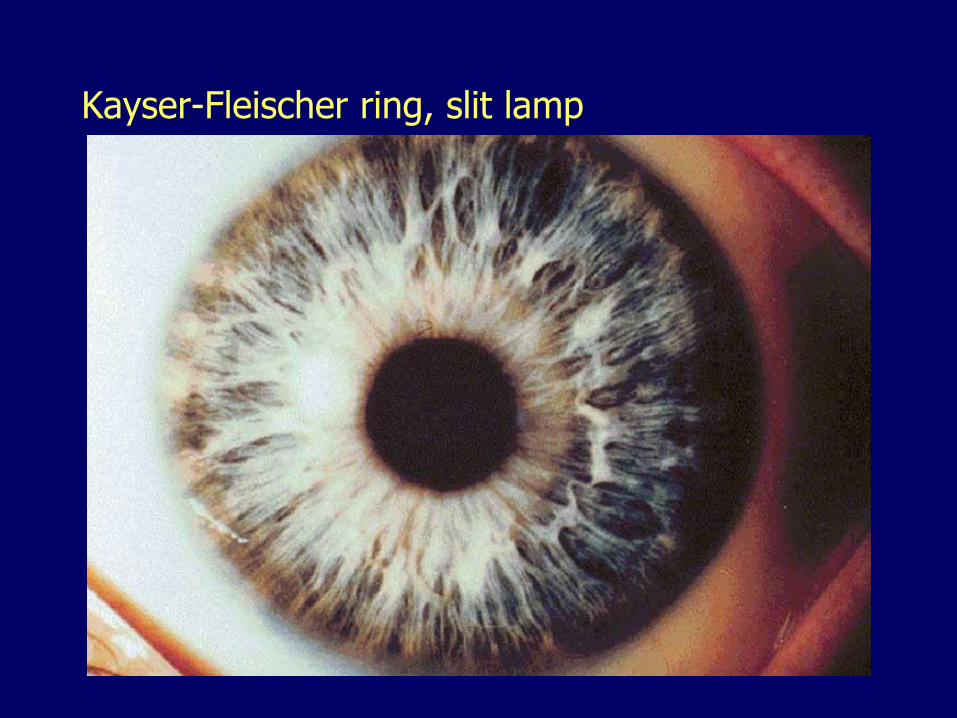
Kayser-Fleischer ring, slit lamp

Wilson disease
In hepatocytes copper is:
– Transported into the golgi apparatus where it is incorporated into apo-ceruloplasmin to form ceruloplasmin which is SECRETED into circulation (90-95% of plasma copper)
– Transported into bile (canaliculi) to be EXCRETED
ATP7B gene codes for a copper-transporting ATPase required for both functions
Defect in the ATP7B gene interferes with both secretion and excretion

ATP7B-mediated copper export in the hepatocyte
Copper entrance: copper transporter CTR1. Copper is delivered to ATP7B in the golgi apparatus by the copper chaperone ATOX1. In the golgi apparatus copper is incorporated in various cuproenzymes including ceruloplasmin. When copper levels rise, ATP7B redistributes to a vesicular compartment and participates in copper excretion in the bile via an unknown mechanism that probably involves COMMD1

Wilson disease
Diagnosis– LOW serum ceruloplasmin
– HIGH non-ceruloplasmin serum copper
– HIGH urinary copper
– HIGH hepatic copper
– Ophthalmologic evaluation
– Genetic testing is now available• Sequencing of the ATP7B gene, more than 500
different mutations identified

Alpha1-antitrypsin deficiency
Autosomal recessive disorder with codominant expression
The most common genetic cause of liver disease in children
The most common genetic cause of emphysema in adults

Alpha1-antitrypsin deficiency A1AT is a serine protease inhibitor (Pi, SERPINA1)
– Inhibition of neutrophil elastase and other proteases
SERPINA1 gene, has more than 100 allelic variants
Each gene allele provides half of the enzyme level
The normal gene product is PiM
Most common deficiency alleles are:
– PiS (50-60% enzyme levels)
– PiZ (10-20% enzyme levels)
Most common carrier genotypes: PiMS, PiMZ
Most common deficiency genotypes: PiSS, PiSZ, PiZZ

Alpha-1-antitrypsin deficiency
A single amino acid substitution
Alpha-1 antitrypsin abnormally folded
Cannot move from ER
Form large aggregates that resist normal degradation including autophagy
Appear as PAS-positive cytoplasmic globes

Alpha1-antitrypsin deficiency, biopsy, PAS stain

Alpha-1-antitrypsin deficiency
Defective degradation of the misfolded protein appears to induce apoptosis, inflammation
Only 10% of PiZZ individuals develop clinical disease
Other genetic/environmental factors must play a role

Alpha-1-antitrypsin deficiency
Liver manifestations
– Neonatal (giant cell) hepatitis
– Chronic hepatitis (adolescents)
– Cirrhosis (adults)
– Hepatocellular carcinoma
Lung manifestations:
– Emphysema

Alpha-1-antitrypsin deficiency
Diagnosis
– Serum levels of A1AT, used for screening
– Protein electrophoresis (phenotype)
– DNA analysis (genotype)
– Liver biopsy
Underdiagnosed
– PiZZ, present in 1/1800 live births

Hemochromatosis

Iron metabolism Absorbed in duodenum and proximal jejunum
No natural mechanism of excretion
Amount absorbed must balance out losses: epithelial exfoliation, minor hemorrhages, menstruation
Ferroportin, an iron channel, controls the release of iron into the circulation from: enterocytes, macrophages, hepatocytes
Hepcidin, produced in the liver, binds ferroportin which is then internalized and degraded preventing the release of iron

Hepcidin, ferroportin
Low iron, low hepcidin: ferroportin exports iron into the circulation
High iron, high hepcidin: hepcidin binds to ferroportin inducing its internalization and degradation; iron remains in the cells

Iron transport and storage Iron is transported in blood by TRANSFERRIN
Iron is stored as:– FERRITIN (circulation, readily available)
– HEMOSIDERIN (macrophages, tissues, poorly available)
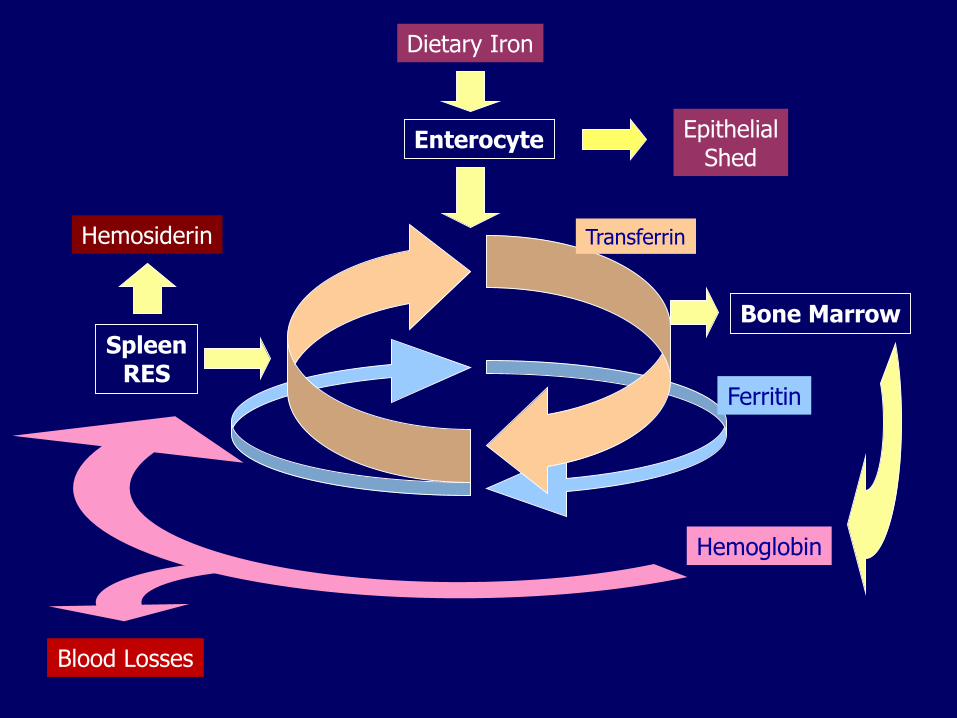
Dietary Iron
Enterocyte
Hemoglobin
SpleenRES
Ferritin
Hemosiderin
Bone Marrow
Blood Losses
EpithelialShed
Transferrin

Hemosiderosis, hemochromatosis
HEMOSIDEROSIS: generic term, excessive accumulation of iron
HEMOCHROMATOSIS: the disease state caused by the generation of free radicals and oxidative injury

Hemochromatosis Secondary, acquired
– Ineffective erythropoiesis (the most common cause)
• Thalassemia
• Sideroblastic anemia
• Pyruvate kinase deficiency
• Other
– Parenteral administration• Transfusions, injections
– Increased oral intake• Bantu siderosis
Primary, hereditary– Excessive intestinal iron absorption

Hereditary hemochromatosis Adult form Autosomal recessive Mutations in proteins that regulate hepcidin
synthesis in response to iron levels– HFE (the most common)– Transferrin receptor 2 (TfR-2)– Hemojuvelin (HJV)
These mutations result in:– Lower hepcidin levels– Greater ferroportin levels– Greater transport of iron into the circulation

Hereditary hemochromatosis HFE mutations cause most cases of adult
hemochromatosis Two mutations: C282Y, H63D, account for
more than 90% of HH cases C282Y is the most common Common among whites (Northern European)
– Homozygotes, 1 of every 200– Heterozygotes, 1 of every 8 (accumulate less iron)

Hereditary hemochromatosis Men predominate 5:1
Occurs earlier in men (40’s) than in women (50’s)
Classic triad:– Cirrhosis (micronodular)
– Diabetes mellitus (pancreatic fibrosis, atrophy)
– Bronze skin pigmentation
Other manifestations:– Joint pain (arthropathy), the most common presenting
complaint, may precipitate pseudogout
– Cardiomyopathy: arrhythmia, heart failure
– Thyroid and adrenal insufficiency
– Hypogonadism: impotence, amenorrhea, early menopause
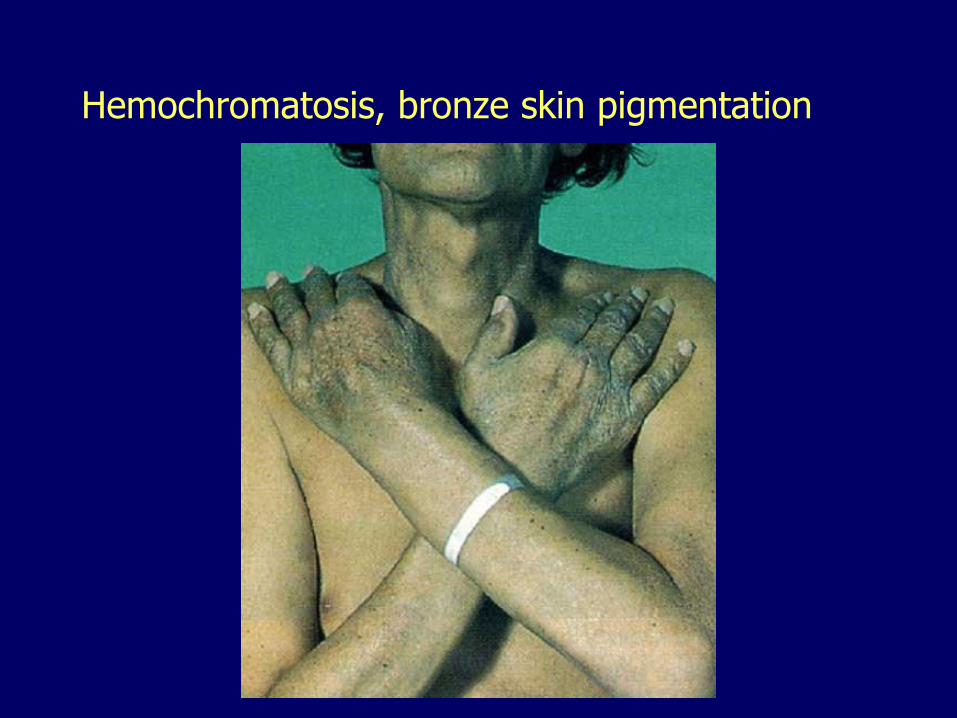
Hemochromatosis, bronze skin pigmentation
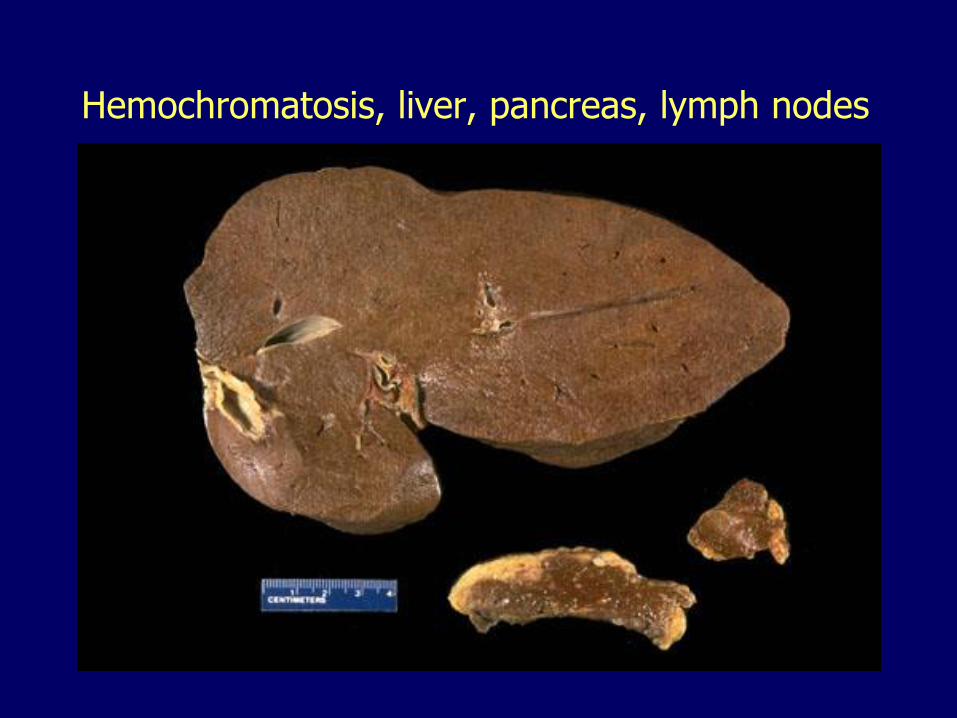
Hemochromatosis, liver, pancreas, lymph nodes
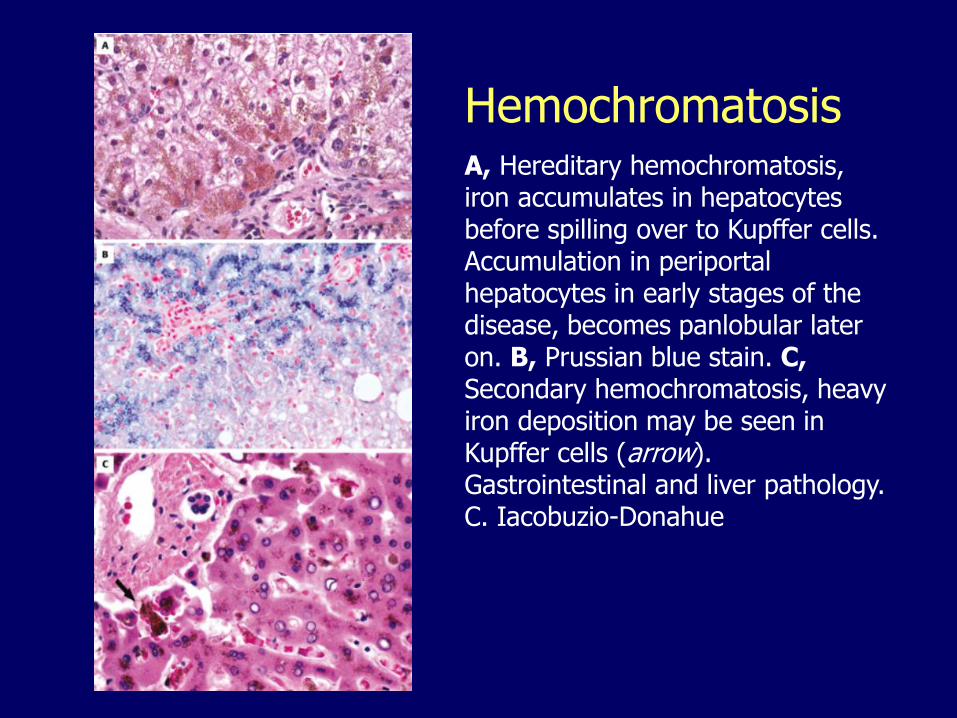
HemochromatosisA, Hereditary hemochromatosis, iron accumulates in hepatocytes before spilling over to Kupffer cells. Accumulation in periportal hepatocytes in early stages of the disease, becomes panlobular later on. B, Prussian blue stain. C,Secondary hemochromatosis, heavy iron deposition may be seen in Kupffer cells (arrow). Gastrointestinal and liver pathology. C. Iacobuzio-Donahue

Hemochromatosis
Diagnosis
– Elevated serum iron
– Increased transferrin saturation
– Elevated serum ferritin
– Increased iron concentration in liver
– Genetic analysis (HFE gene)
Hepatocellular carcinoma 200-fold risk
The liver III11. Inborn errors of metabolism
12. Intrahepatic biliary tract diseases
– Congenital abnormalities
– Primary and secondary biliary cirrhosis
– Primary sclerosing cholangitis13. Circulatory disorders
11. Hepatic disease in pregnancy
12. Nodular hyperplasias
13. Benign neoplasms
14. Malignant neoplasms

Congenital Abnormalities of the Intrahepatic Biliary Tree
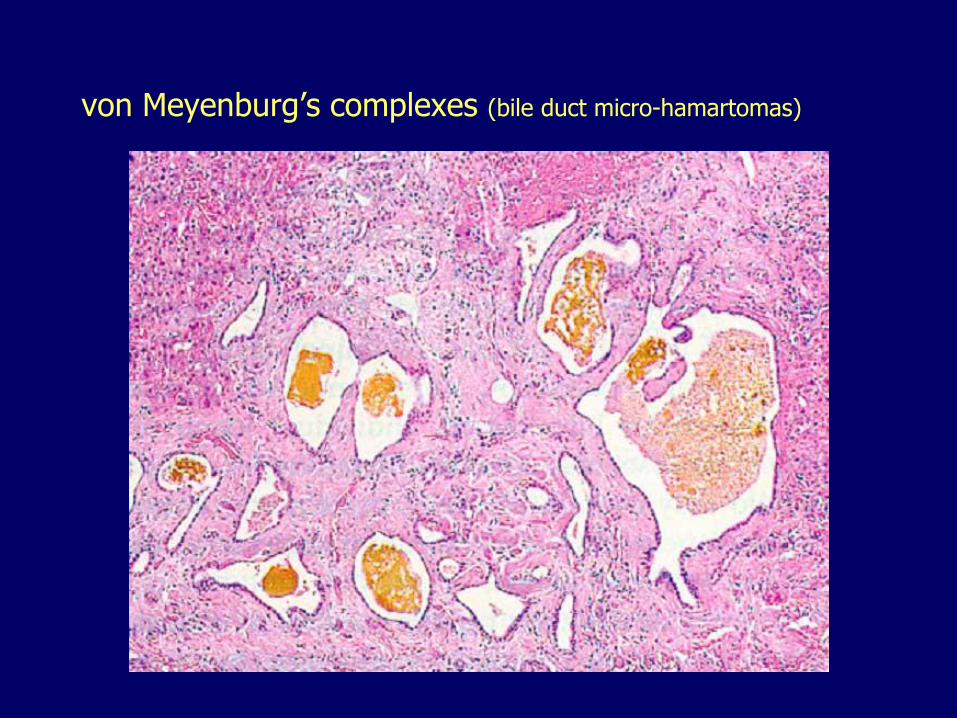
von Meyenburg’s complexes (bile duct micro-hamartomas)
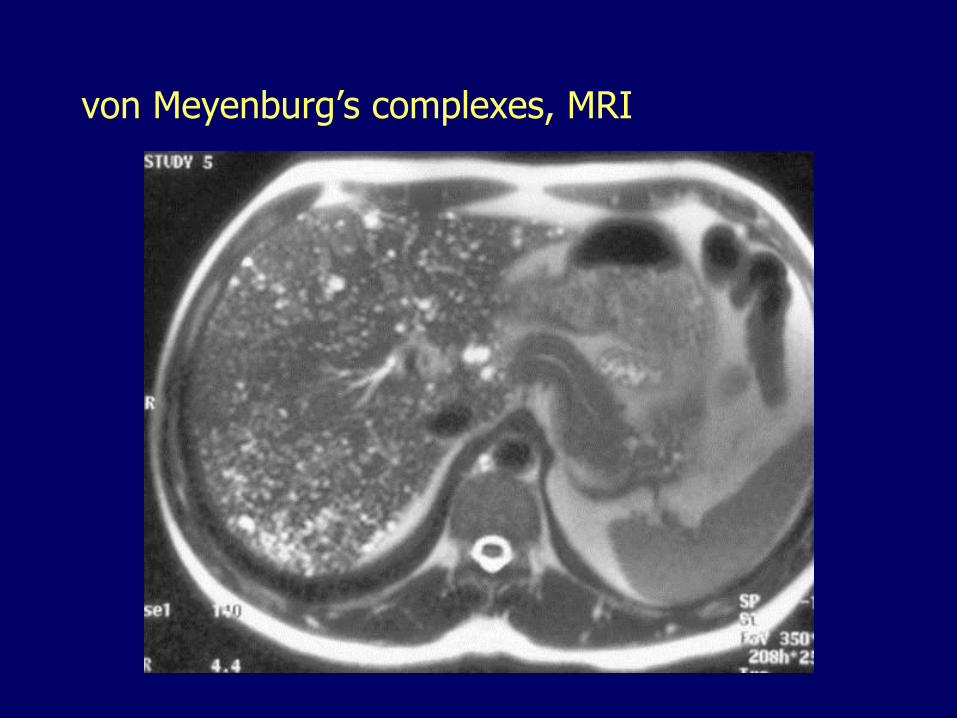
von Meyenburg’s complexes, MRI
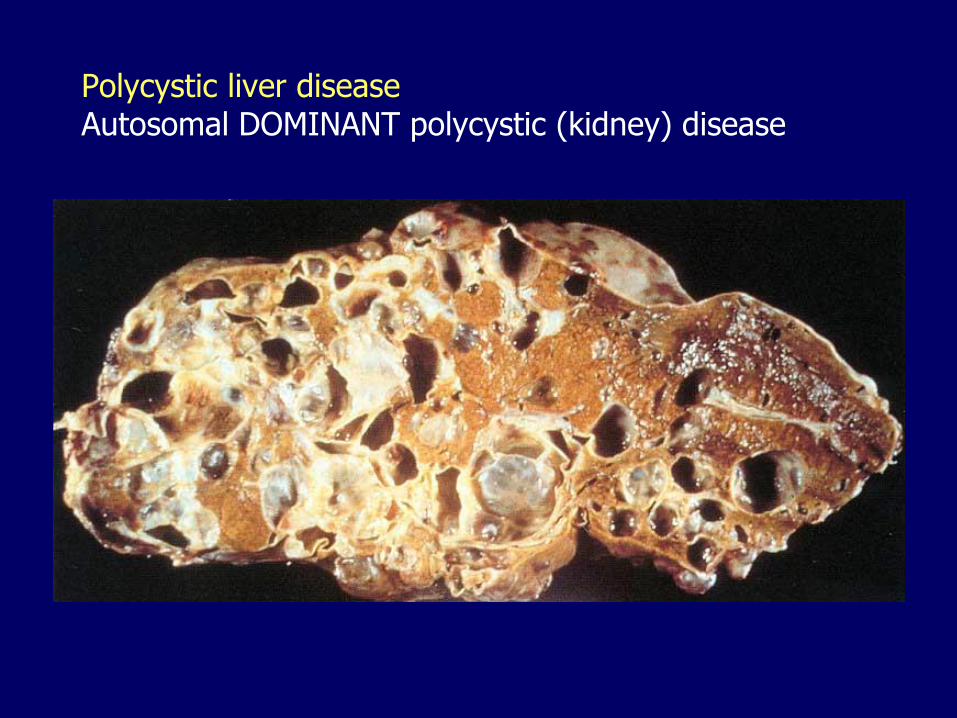
Polycystic liver diseaseAutosomal DOMINANT polycystic (kidney) disease
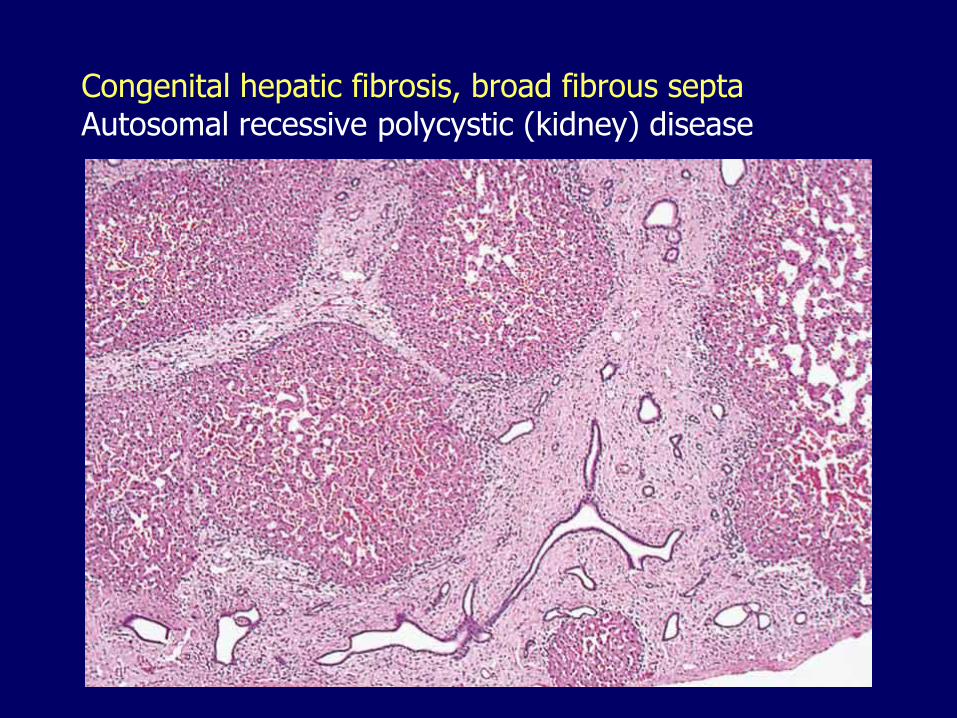
Congenital hepatic fibrosis, broad fibrous septa Autosomal recessive polycystic (kidney) disease

Caroli diseaseSegmental cystic dilatation of bile ducts
RadioGraphics, 2006, 26:715-731

Caroli disease, cystically dilated bile ducts
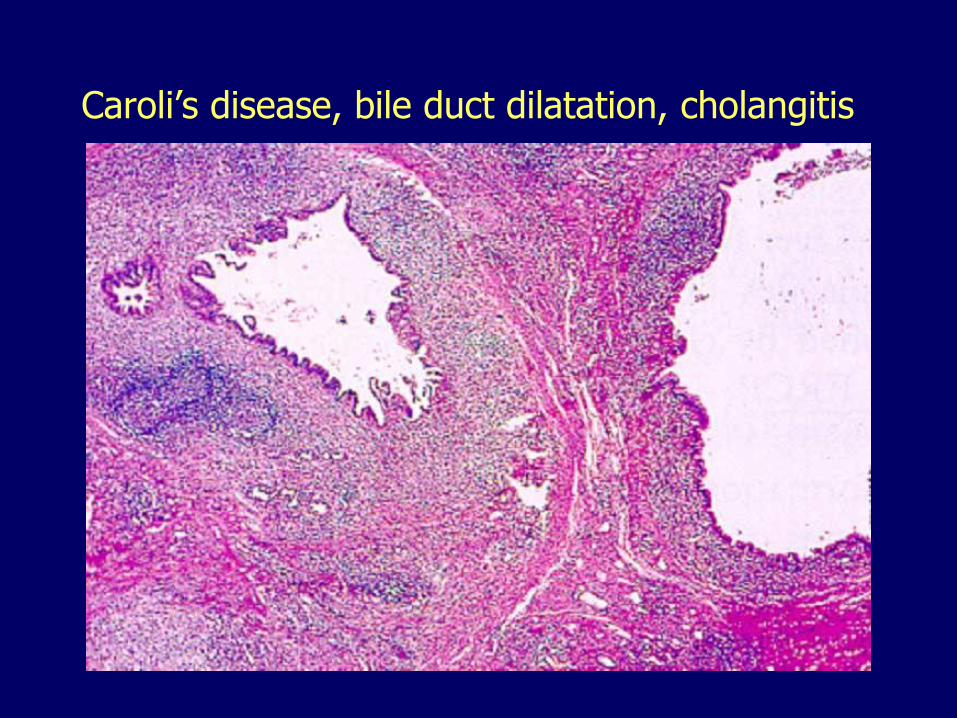
Caroli’s disease, bile duct dilatation, cholangitis
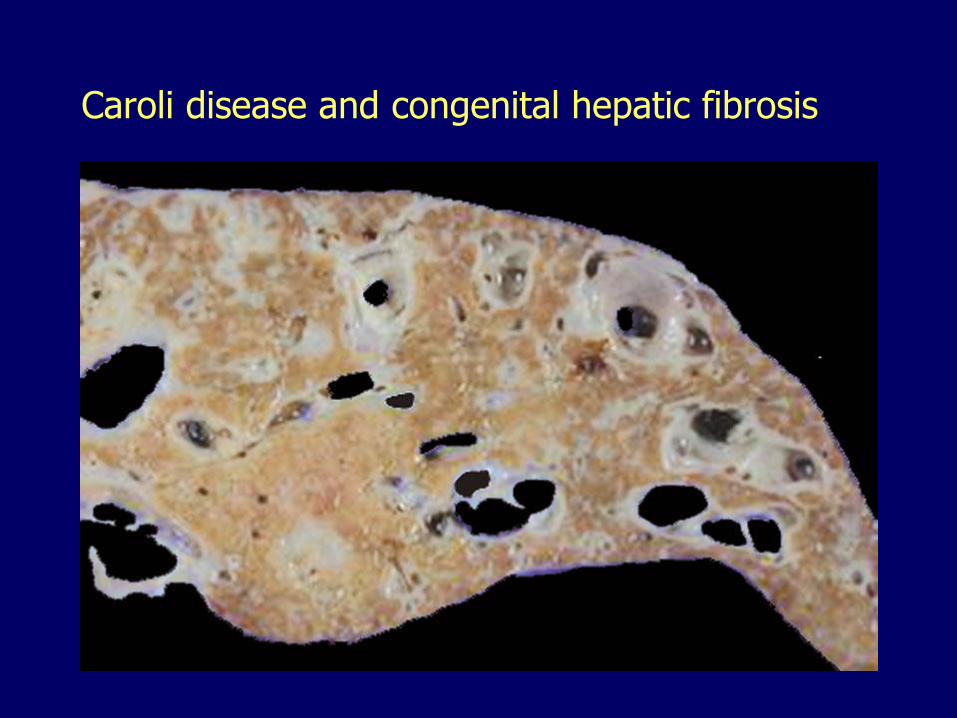
Caroli disease and congenital hepatic fibrosis

Primary Biliary Cirrhosis Autoimmune
– Destruction of small and medium-size intrahepatic bile ducts
– Chronic hepatitis
– Biliary cirrhosis
Antimitochondrial antibodies– E2 component of pyruvate dehydrogenase
Association with other autoimmune conditions
Female predominance > 6:1
Peak incidence, 40-50 years
Family and geographic clustering
Symptoms: those associated with cholestasis and portal hypertension
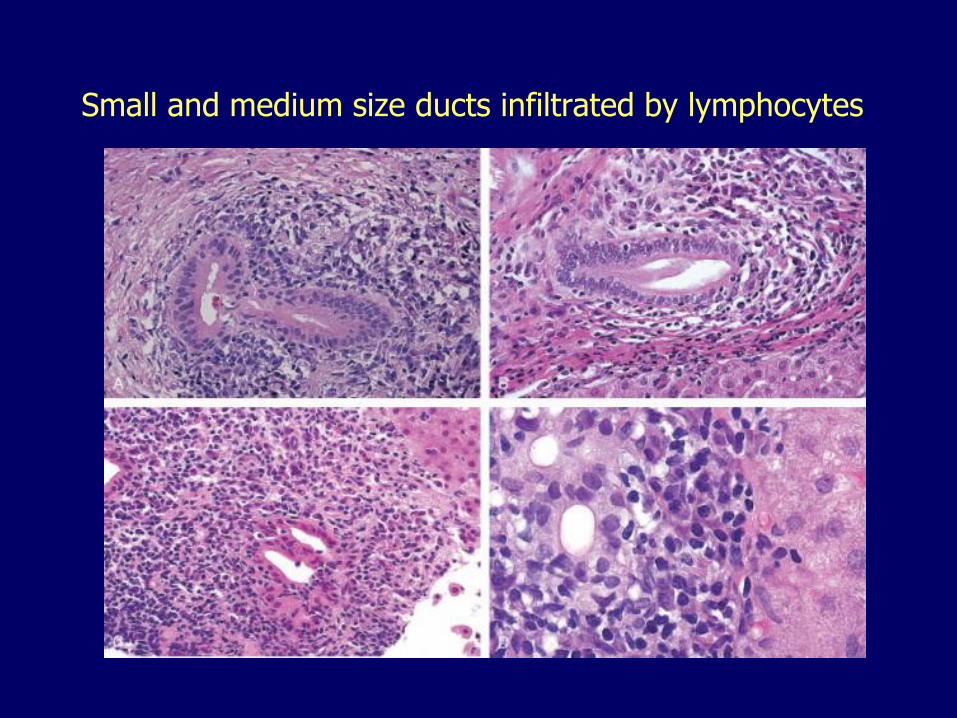
Small and medium size ducts infiltrated by lymphocytes

Ducts, granulomatous inflammation
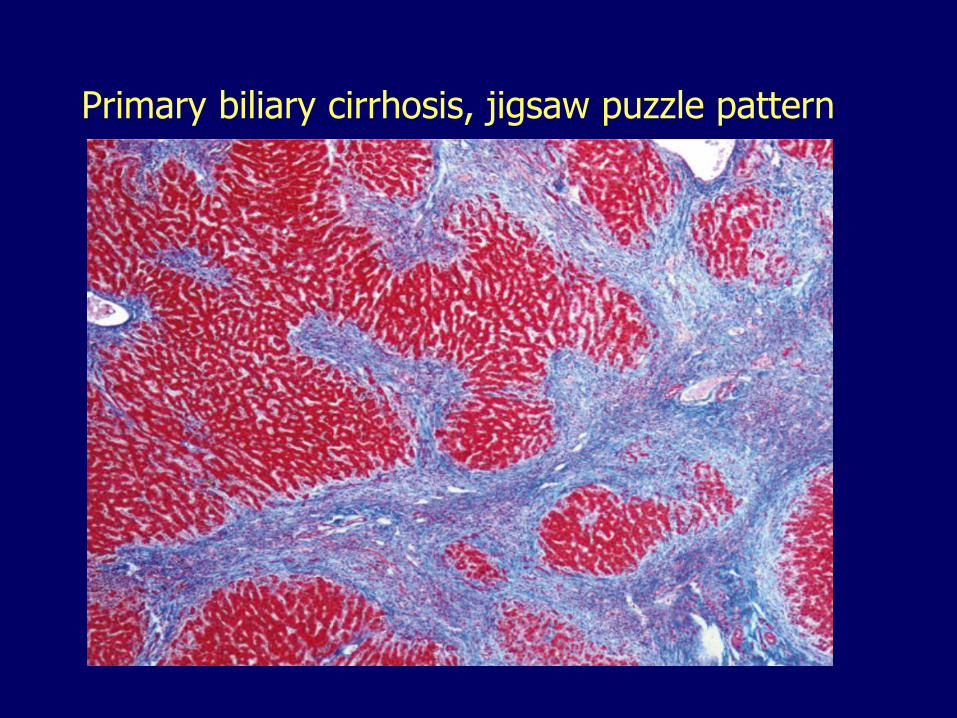
Primary biliary cirrhosis, jigsaw puzzle pattern
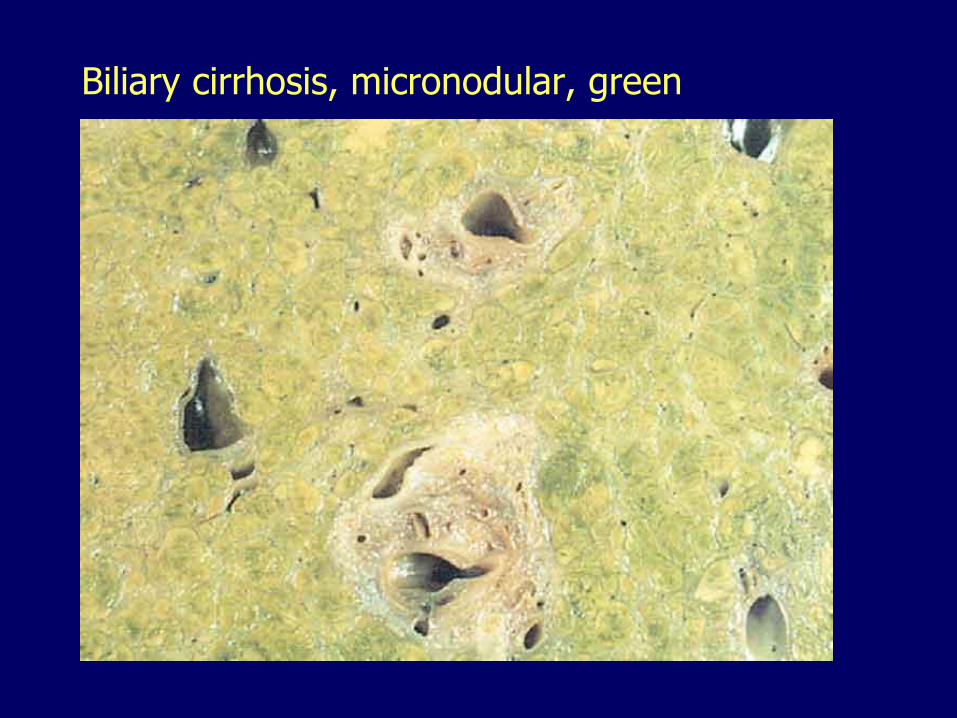
Biliary cirrhosis, micronodular, green

Secondary biliary cirrhosis
In adults:
– Choledocholithiasis
– Malignant neoplasms (head of the pancreas, biliary tree)
– Biliary strictures from surgical procedures
In children:
– Biliary atresia
– Choledochal cysts
– Cystic fibrosis

Primary Sclerosing Cholangitis Immune-mediated SEGMENTAL inflammation, fibrosis
and obliteration of LARGE intrahepatic and extrahepatic bile ducts, and pancreatic ducts
Males predominate 2:1
Third through fifth decades of life
Ulcerative colitis, 70% of patients
p-ANCA antibodies
Toxins released by inflamed gut may cause injury in the biliary tree
T cells and antibodies cross-reactivity between intestinal or bacterial antigens and bile ducts
Genetic predisposition

Primary sclerosing cholangitis, onion-skin fibrosis

Primary sclerosing cholangitis, fibrous obliteration
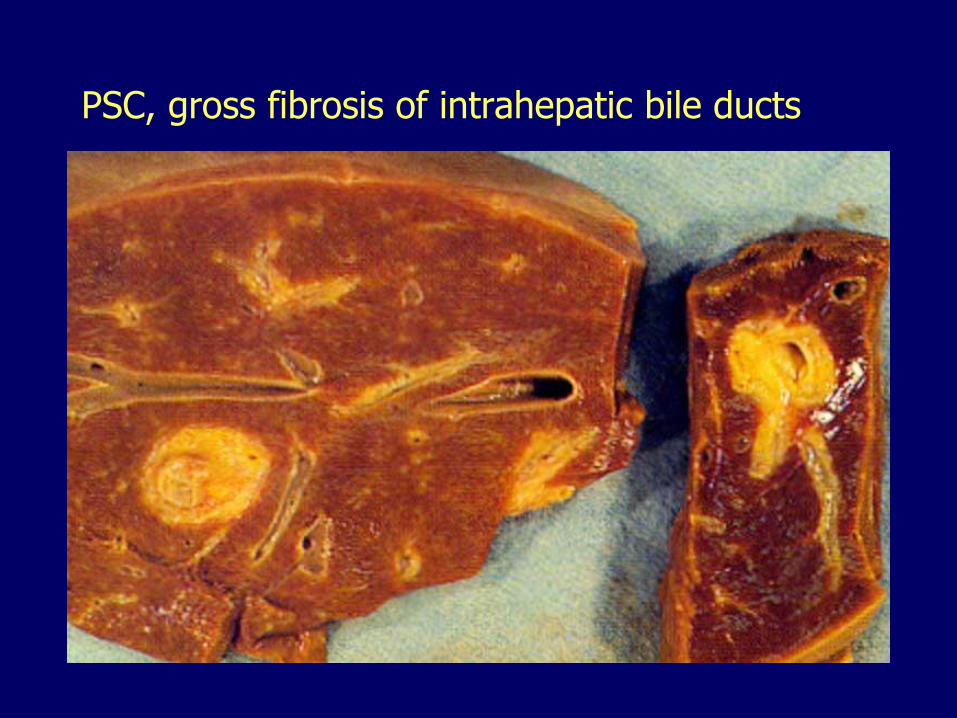
PSC, gross fibrosis of intrahepatic bile ducts

Primary sclerosing cholangitis, chain of lakes

Primary Sclerosing Cholangitis Inflammation (cholangitis) and stone
formation (hepatolithiasis) in dilated ducts
May involve gallbladder (cholecystitis) and pancreatic ducts (pancreatitis)
Risk for cholangiocarcinoma
Many progress to cirrhosis and liver failure and need liver transplantation
May recur after transplantation

The liver III11. Inborn errors of metabolism
12. Intrahepatic biliary tract diseases
13. Circulatory disorders
– Passive congestion
– Infarcts
– Portal hypertension
– Budd-Chiari syndrome
– Veno-occlusive disease14. Hepatic disease in pregnancy
15. Nodular hyperplasias
16. Benign neoplasms
17. Malignant neoplasms
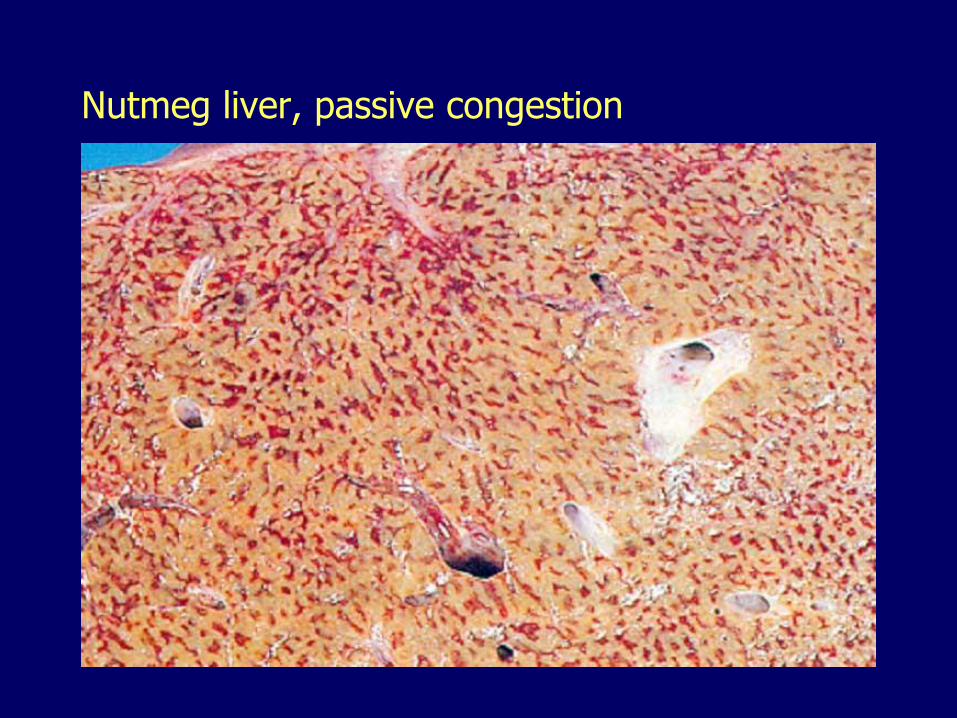
Nutmeg liver, passive congestion
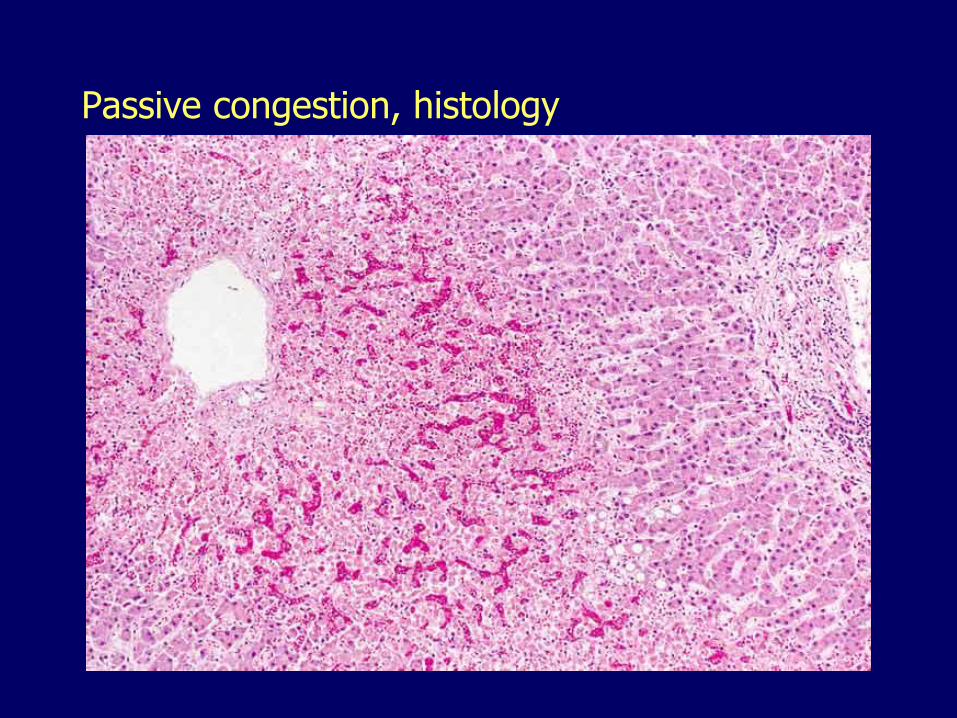
Passive congestion, histology
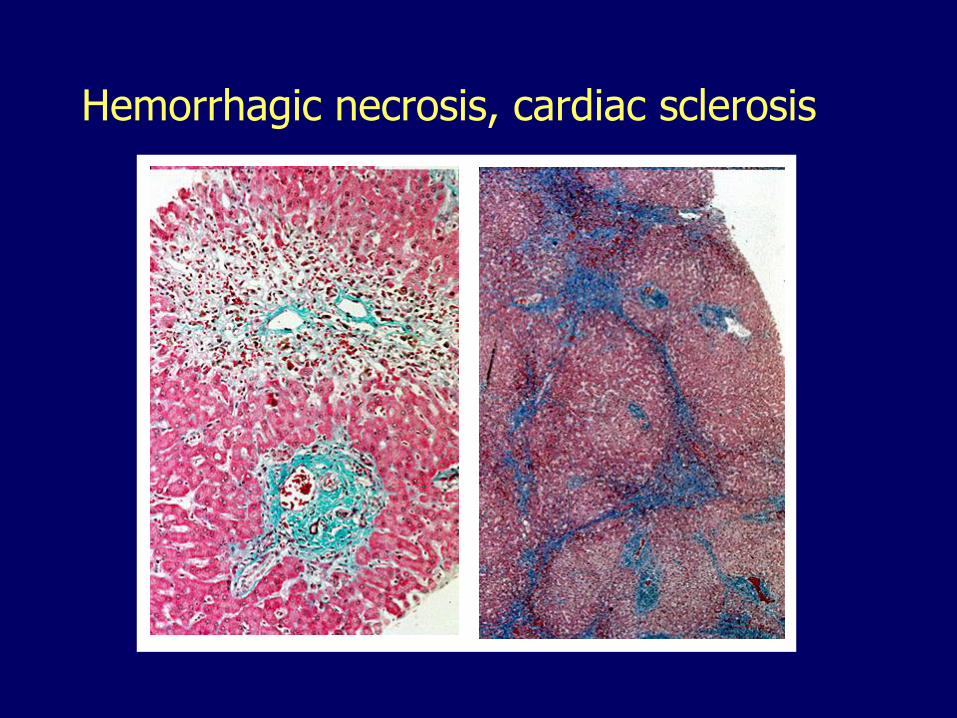
Hemorrhagic necrosis, cardiac sclerosis

Hepatic artery obstruction, infarcts
Liver infarcts are rare
Double blood supply
– Hepatic artery 30 -40%
– Portal vein 60 -70%
Interruption of the main hepatic artery can be tolerated
– Except in transplanted liver: thrombosis of the hepatic artery a major cause of graft loss
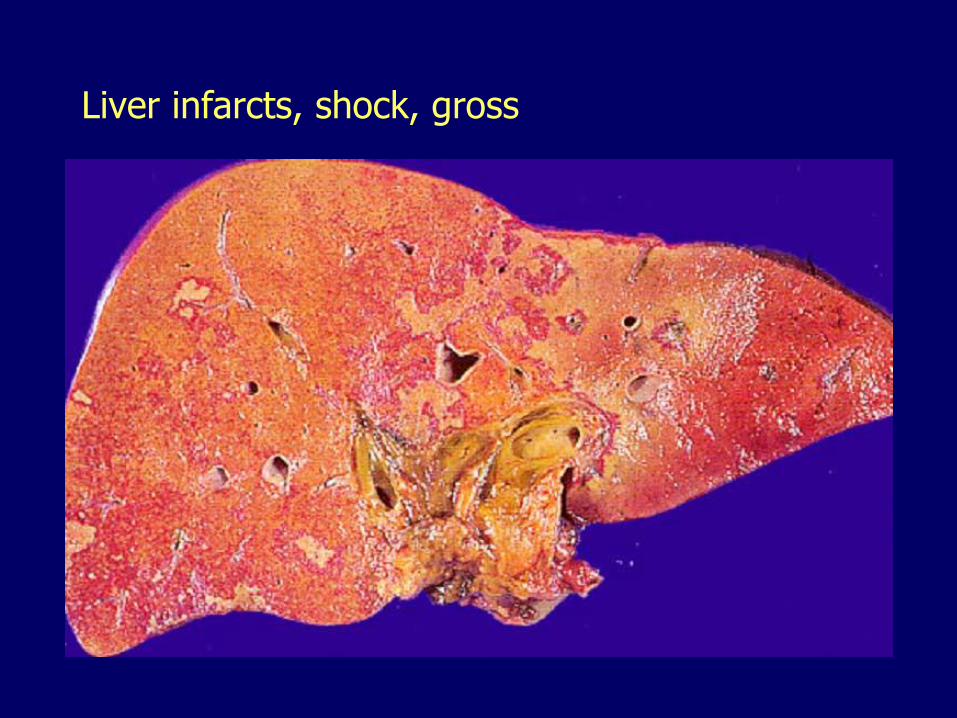
Liver infarcts, shock, gross

Infarct of Zahn
Thrombosis of a portal vein radicle
Results in:
– Ischemia
– Dilatation and congestion of sinusoids
– Flattening of the surrounding hepatocytes
– No necrosis (pseudoinfarct)
Grossly visible as sharply demarcated area of congestion
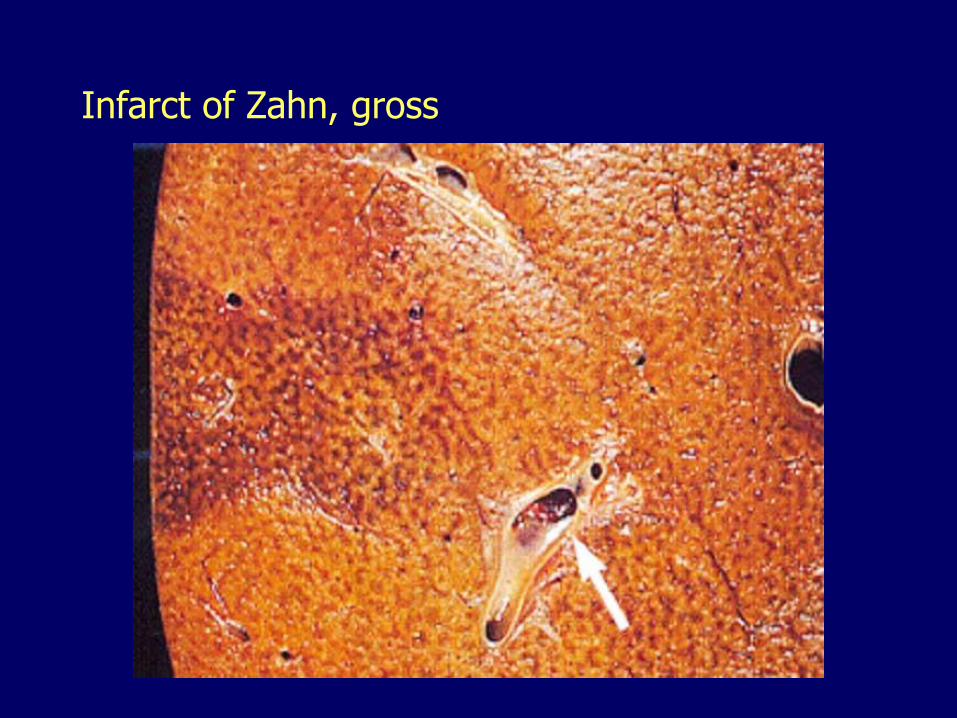
Infarct of Zahn, gross

Posthepatic– Right-sided heart failure
– Hepatic veins thrombosis• Budd-Chiari syndrome
Intrahepatic– Cirrhosis (the most common cause)
– Nodular regenerative hyperplasia
– Sinusoidal obstruction syndrome
Prehepatic– Portal vein obstruction
• Pylephlebitis (portal thrombophlebitis)
Portal Hypertension, causes:
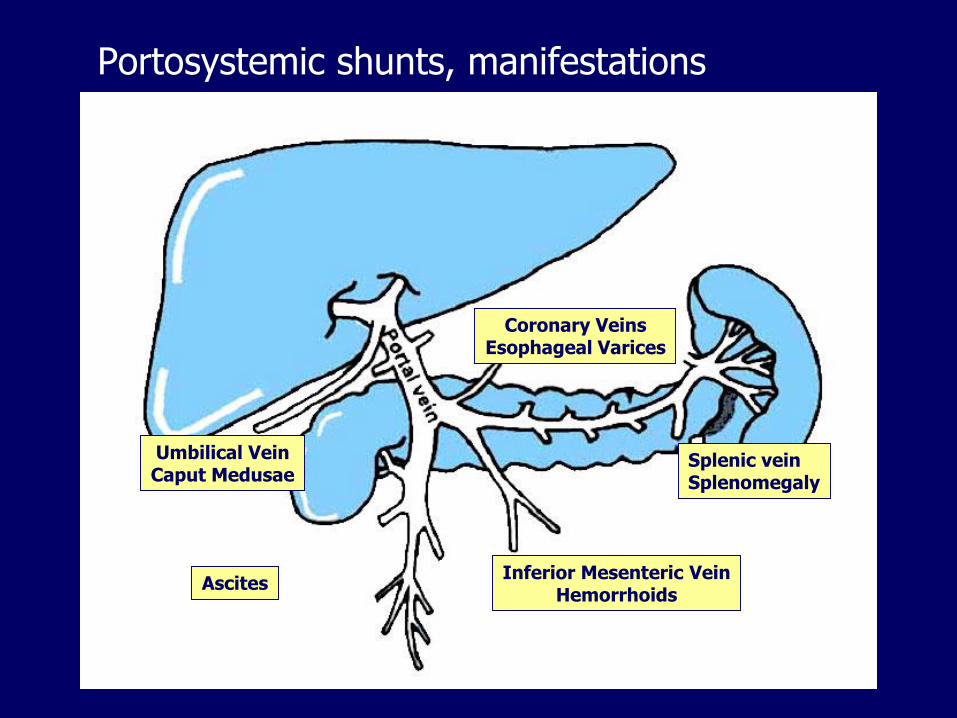
Portosystemic shunts, manifestations
Coronary VeinsEsophageal Varices
Splenic veinSplenomegaly
Inferior Mesenteric VeinHemorrhoids
Umbilical VeinCaput Medusae
Ascites
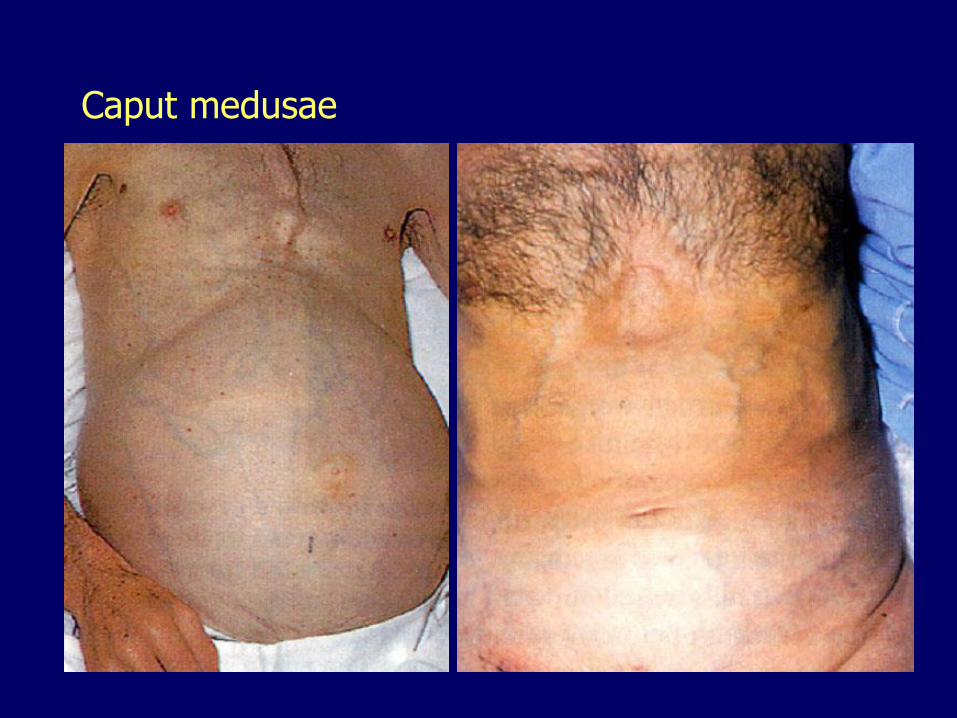
Caput medusae

Budd-Chiari syndrome Thrombosis of 2 or more major hepatic veins
Massive enlargement of the liver
Severe portal hypertension
Severe ascites
Etiology:– Hypercoagulable states
• Pregnancy, oral contraceptives
• Postoperative, postpartum states
• Deficiencies in antithrombin III, protein S or protein C
• Polycythemia vera, sickle cell disease
– Hepatocellular carcinoma
– Idiopathic, 10%

Budd-Chiari, hepatic vein thrombosis
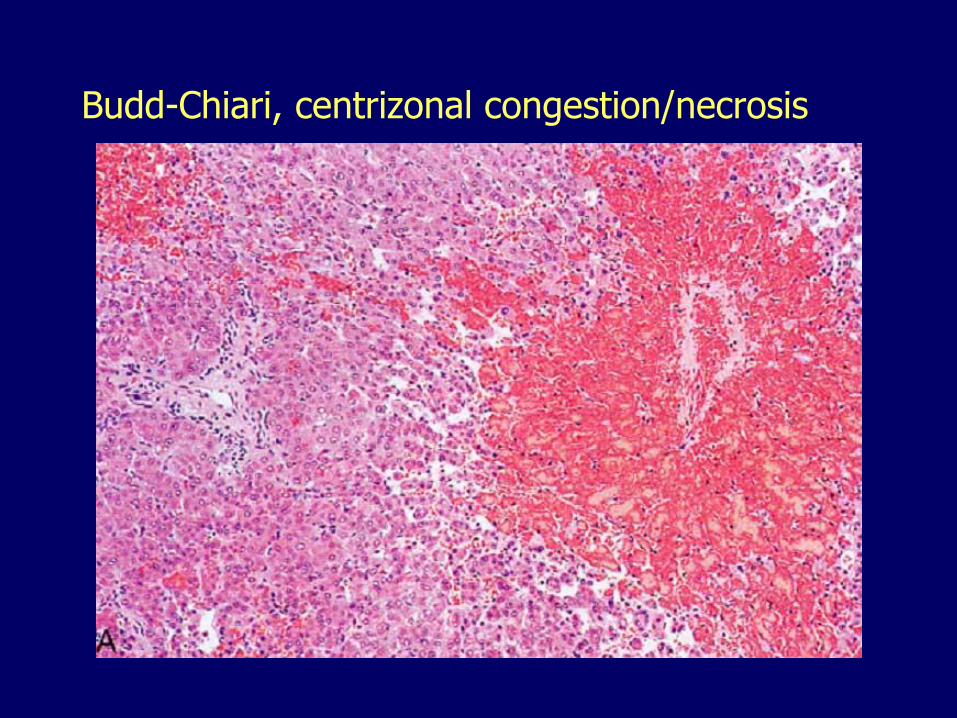
Budd-Chiari, centrizonal congestion/necrosis

Sinusoidal obstruction syndrome
Also called veno-occlusive disease– Toxic damage of sinusoidal endothelial cells
– Desquamation, embolization into the terminal hepatic venules
– Extravasation of red blood cells
– Marked centrizonal congestion, necrosis, fibrosis
Etiology:– Acute: After bone marrow transplantation
– Subacute or chronic: Chemotherapy, herbal remedies (Jamaican bush tea, pyrrolizidine alkaloids found in many plants)

Veno-occlusive disease, centrizonal congestion/necrosis

The liver III11. Inborn errors of metabolism
12. Intrahepatic biliary tract diseases
13. Circulatory disorders
14. Liver disease in pregnancy– Preeclampsia-Eclampsia
– Acute fatty liver of pregnancy
– Intrahepatic cholestasis of pregnancy
15. Nodular hyperplasias
16. Benign neoplasms
17. Malignant neoplasms

Preeclampsia-Eclampsia
Hypertension, proteinuria, edema, coagulation disorders
Hyperreflexia, seizures
HELLP syndrome
– Hemolysis, elevated liver enzymes, low platelets
Unknown etiology
Periportal fibrin deposits
Periportal coagulative necrosis
Parenchymal hemorrhage and infarcts
Subcapsular hematoma, catastrophic rupture
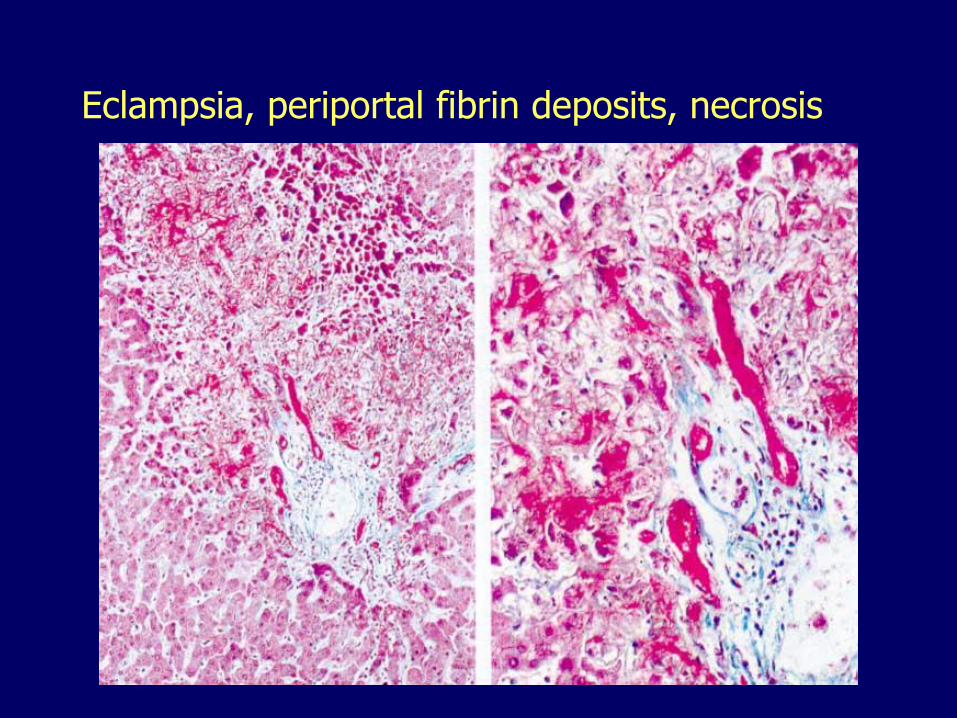
Eclampsia, periportal fibrin deposits, necrosis

Eclampsia, subcapsular hematoma, CT scan

Eclampsia, subcapsular hematoma

Acute Fatty Liver of Pregnancy
Third trimester
Jaundice
Acute liver failure
Microvesicular steatosis
Parental/fetal defect in mitochondrial oxidation of long-chain fatty acids that accumulate and cause liver toxicity
20-40% coexistent preeclampsia
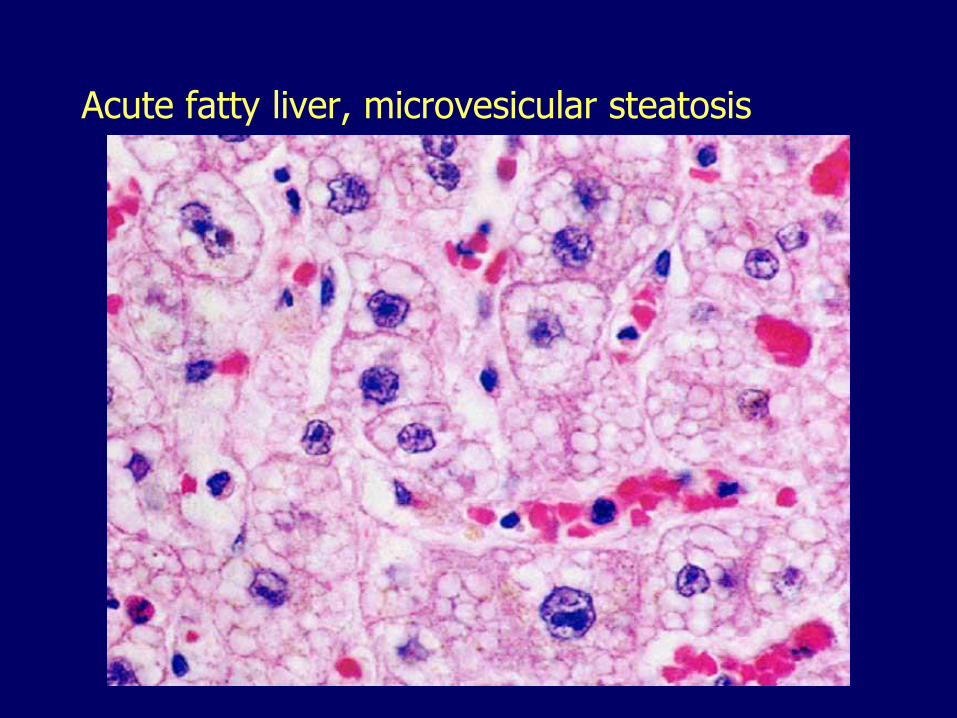
Acute fatty liver, microvesicular steatosis

Intrahepatic Cholestasis of Pregnancy
Conjugated hyperbilirubinemia, < 5 mg/dl Pruritus and jaundice Third trimester More common in multiple pregnancies,
Scandinavia, Chile Resolve rapidly after delivery, may recur Increased incidence of fetal distress,
stillbirths, prematurity Pathogenesis not entirely clear:
– Estrogen/progesterone inhibit bile secretion– Mutation in genes encoding biliary transport
proteins

Intrahepatic cholestasis of pregnancy

The liver III11. Inborn errors of metabolism
12. Intrahepatic biliary tract diseases
13. Circulatory disorders
14. Liver disease in pregnancy
15. Nodular hyperplasias
– Focal nodular hyperplasia
– Nodular regenerative hyperplasia16. Benign neoplasms
17. Malignant neoplasms

Focal nodular hyperplasiaHyperplastic nodule with central stellate scar

Focal nodular hyperplasiaFibrous scar with misshaped vessels and bile ducts, hyperplastic lobular parenchyma
Robbins, Cotran

Focal nodular hyperplasia
Hyperplastic response to focal increased blood flood (vascular malformations)
More common in females
Questionable association with oral contraceptives
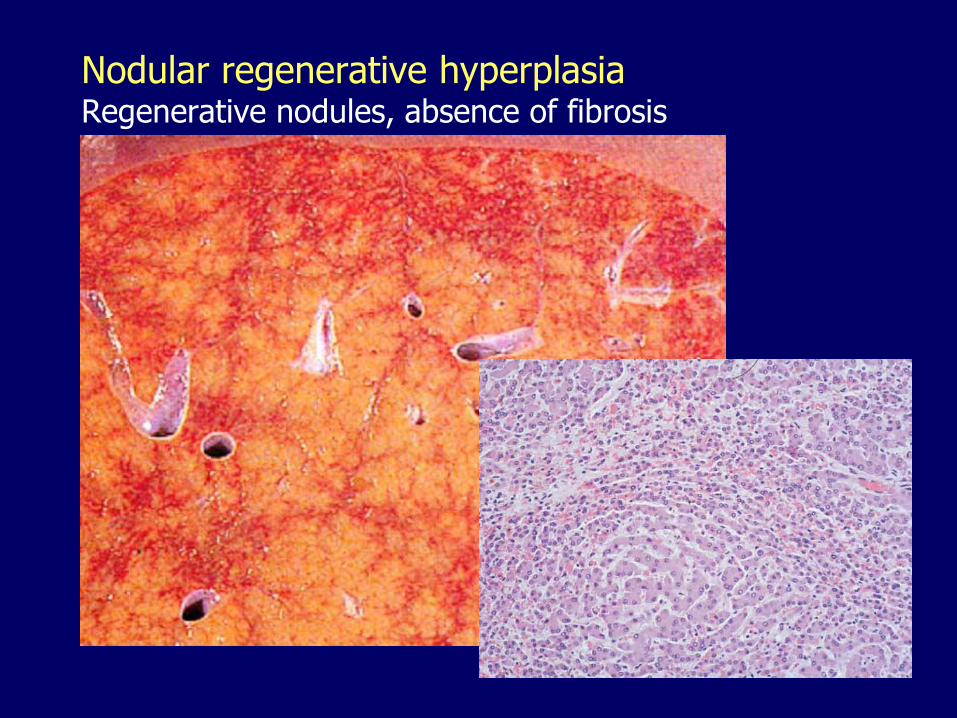
Nodular regenerative hyperplasiaRegenerative nodules, absence of fibrosis

Nodular regenerative hyperplasia, reticulumNodules surrounded by rims of compressed cords

Nodular regenerative hyperplasia
May be subclinical, no liver dysfunction
Presents with portal hypertension
Uncertain etiology
Associated with : toxic/drug vascular injury, autoimmune, infectious, hematologic/thrombotic conditions.
Obstructive vasculopathy with global decrease of the (low-pressure) venous portal blood supply and compensatory increase of the (high pressure) arterial blood supply
The liver III11. Inborn errors of metabolism
12. Intrahepatic biliary tract diseases
13. Circulatory disorders
14. Liver disease in pregnancy
15. Nodular hyperplasias
16. Benign neoplasms
– Hemangiomas
– Adenomas17. Malignant neoplasms

Cavernous hemangiomas
The most common liver neoplasm
Red-blue soft nodules
Usually less than 2 cm
Often directly beneath the capsule
Significance:
– Mistaken for metastatic tumors
– Percutaneous biopsies may cause profuse bleeding

Cavernous hemangiomas

Hepatic adenoma Well-demarcated proliferation of hepatocytes
– No bile ducts
Often subcapsular
Young women
Related to the use of oral contraceptives and anabolic steroids
May regress on discontinuance
Significance:– Mistaken for hepatocellular carcinoma
– Rupture, bleeding
– May undergo malignant transformation
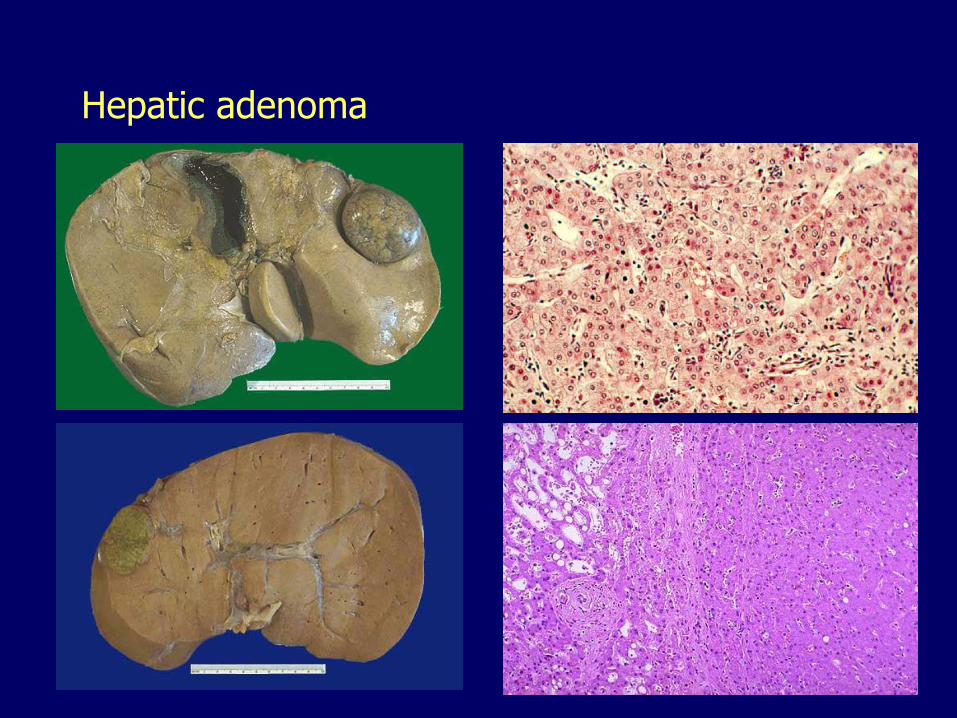
Hepatic adenoma

Adenoma subtypes
HNF1α mutation, fatty, almost no malignant transformation
β-catenin mutation, cytologic atypia, highest-risk for malignant transformation
Inflammatory, associated with non-alcoholic fatty liver disease, small risk of malignant transformation
The liver III11. Inborn errors of metabolism
12. Intrahepatic biliary tract diseases
13. Circulatory disorders
14. Liver disease in pregnancy
15. Nodular hyperplasias
16. Benign neoplasms
17. Malignant neoplasms
– Hepatoblastoma
– Angiosarcoma
– Hepatocellular carcinoma
– Cholangiocarcinoma
– Metastases

Hepatoblastoma
Infants
More common in boys
Immature hepatocytes
Some have mesenchymal elements (cartilage, bone, striated muscle)
Survival has improved
Only chance for cure: total resection

Hepatoblastoma, fetal-like or embryonic-like hepatocytes
Odze & Goldblum Surgical Pathology of the GI Tract, Liver, Biliary Tract and Pancreas
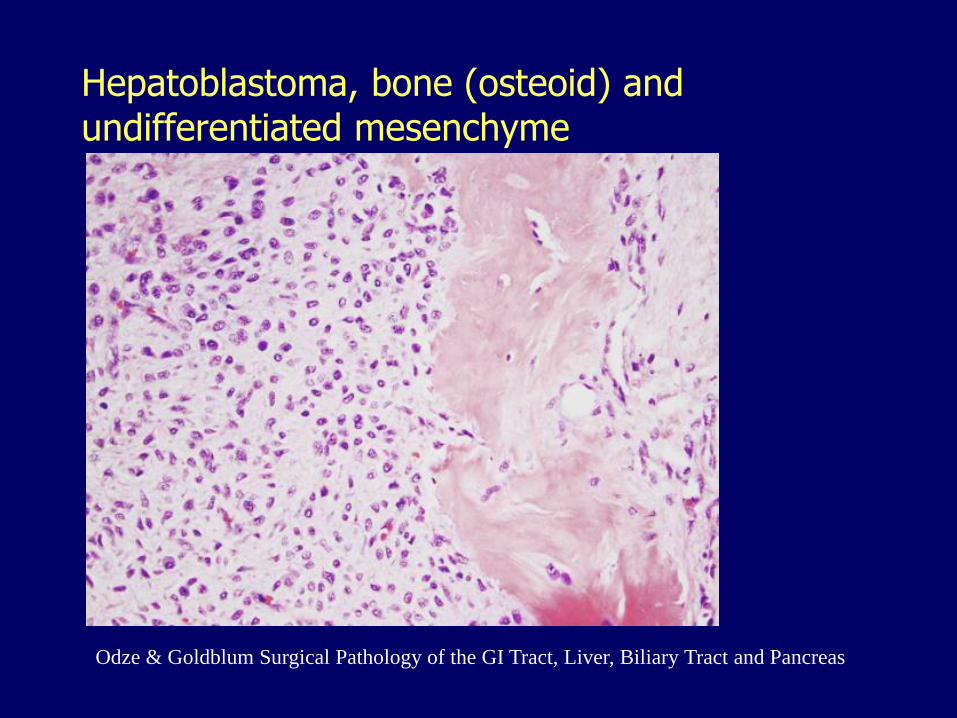
Hepatoblastoma, bone (osteoid) and undifferentiated mesenchyme
Odze & Goldblum Surgical Pathology of the GI Tract, Liver, Biliary Tract and Pancreas

Angiosarcoma
Exposure to
– Poly-vinyl chloride (PVC) industry
– Arsenic
– Thorotrast
The latency period several decades
Highly aggressive, metastasize widely

Angiosarcoma

Liver carcinomas, mayor types
Hepatocellular carcinoma (hepatoma)
– 90% of all primary cancers
Cholangiocarcinoma
Mixed forms

Liver and intrahepatic bile duct cancer incidence rates

Hepatocellular carcinoma
Uncommon in the US
Cirrhosis present in > 85%
Male predominance 3:1
Rare before age 60

Hepatocellular carcinoma
HBV endemic regions
20-40% of all cancers
Vertical transmission (200-fold risk)
Cirrhosis may not be present
Male predominance 8:1
Younger age (20-40 years)

Risk factors
Cirrhosis
HBV
HCV
Alcohol, NASH
Primary hemochromatosis, A1AT deficiency, Wilson disease
Hereditary tyrosinemia (40%)
Aflatoxins– Food spoilage molds (Aspergillus flavus)
– Activated in hepatocytes

Hepatocellular carcinoma
Background cirrhosis and chronic hepatitis
May be preceded by hepatocellular dysplasia
Unifocal, multifocal or diffusely infiltrative
Strong propensity for vascular invasion
– Extensive intrahepatic metastases
– Extension to portal vein, inferior vena cava
– Hematogenous metastases to other organs tend to occur late
Poor prognosis
Elevated alpha-fetoprotein, 60-75%
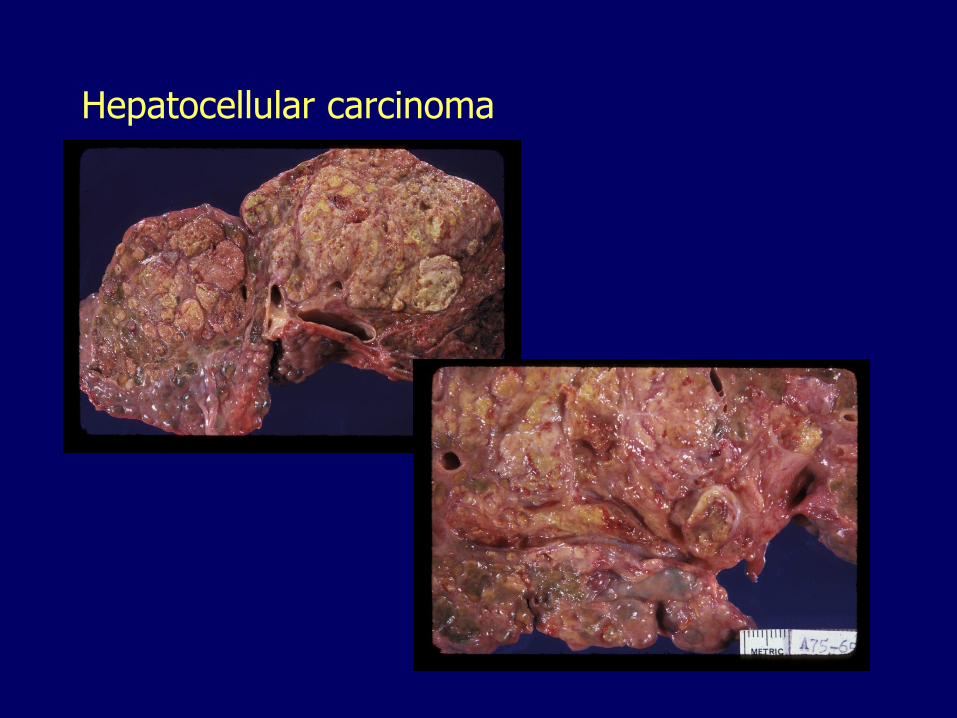
Hepatocellular carcinoma

Hepatocellular carcinoma

Fibrolamellar carcinoma
Young adults (20 to 40 years of age)
Equal male and female incidence
No association with underlying liver disease or cirrhosis
No elevation of alpha-fetoprotein
Better prognosis, 76% alive at 5 years
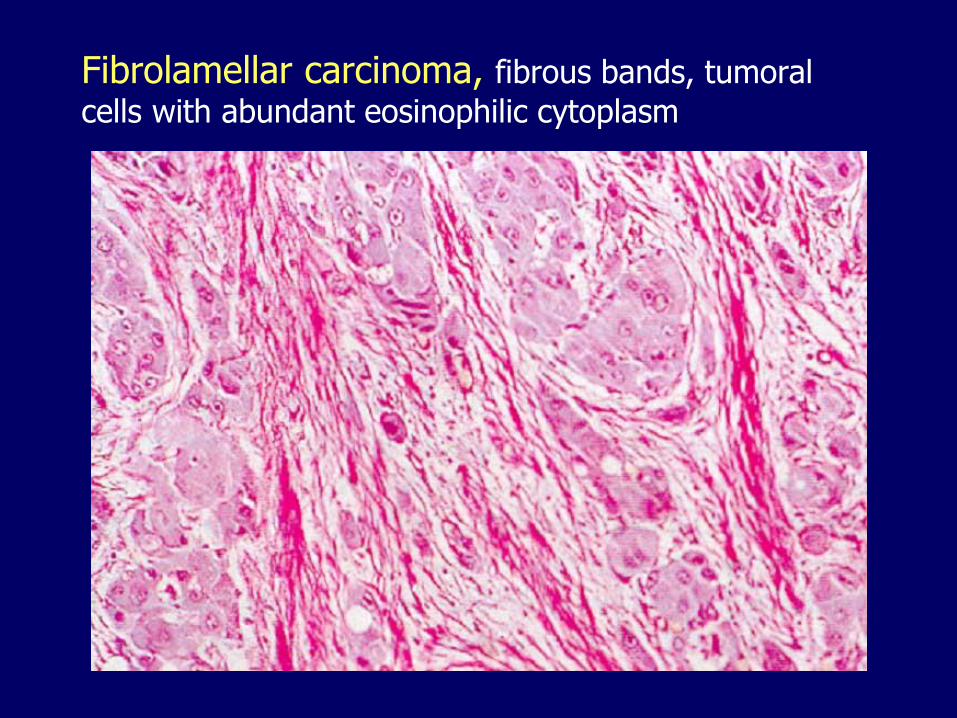
Fibrolamellar carcinoma, fibrous bands, tumoral
cells with abundant eosinophilic cytoplasm

Cholangiocarcinomas
Resemble adenocarcinomas arising in the pancreas
Characteristically desmoplastic (fibrous)
Not commonly associated with cirrhosis
Preceded by biliary intraepithelial neoplasia
Usually detected late
Death within 6 months
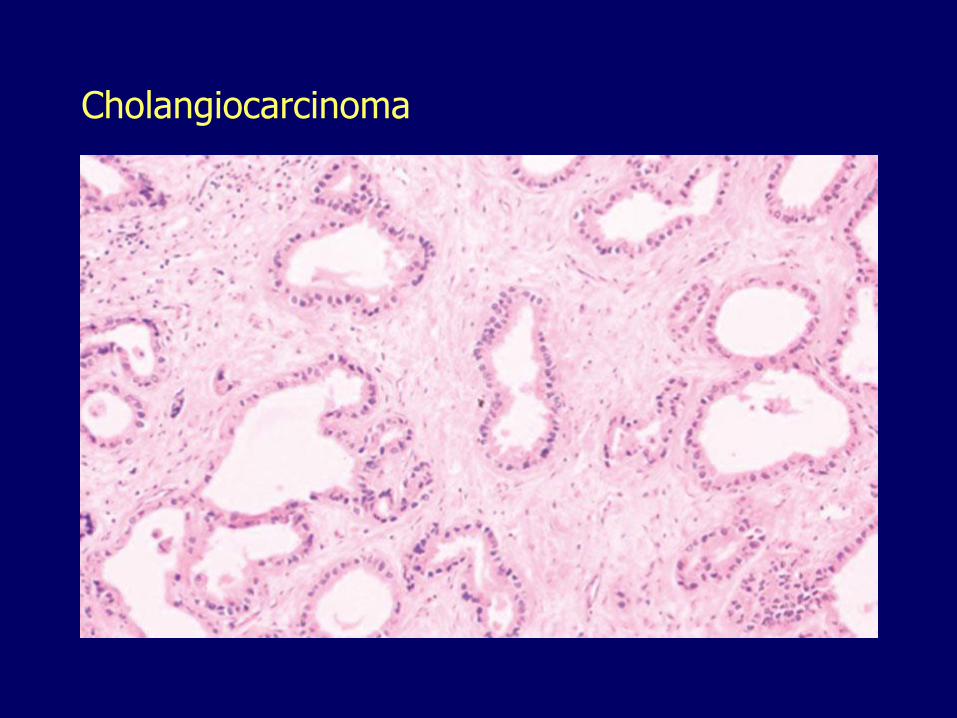
Cholangiocarcinoma
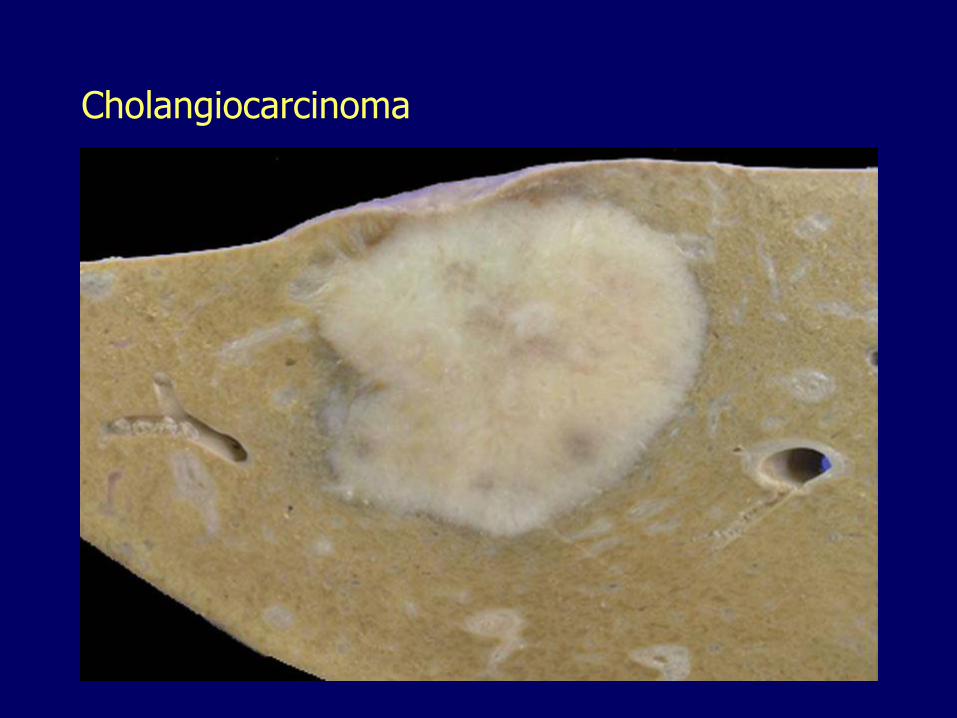
Cholangiocarcinoma
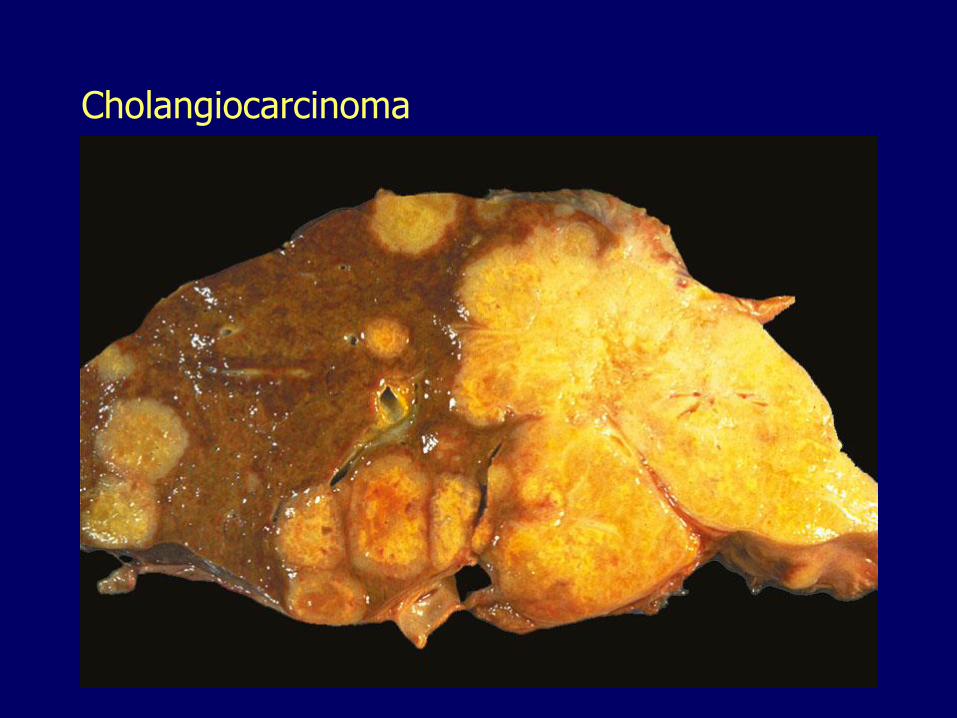
Cholangiocarcinoma

Cholangiocarcinoma, risk factors
Primary sclerosing cholangitis
Congenital hepatic fibrosis
Caroli disease
Choledochal cysts
Hepatolithiasis, liver flukes
Thorotrast
HCV

Metastatic tumors Far more common than primary neoplasia Most common:
– Breast– Lung– Colon– Pancreas
Multiple implants– Cannon balls with central necrosis
Striking hepatomegaly Nodularity palpable on the free edge Extensive involvement may be silent
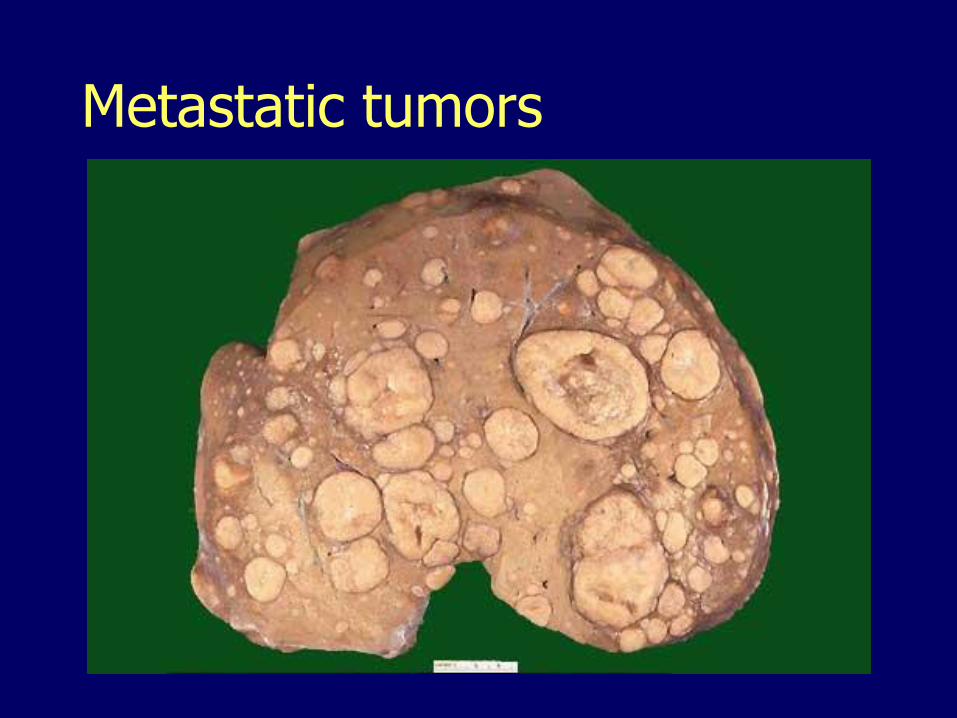
Metastatic tumors





![Biliary cystadenoma – A rare cystic neoplasm of livercystic lesions of liver with majority of the cases being intrahepatic (80-85%) [5,6]. These lesions constitute less than 5 %](https://img.pdfslide.net/doc/110x75/5f10af037e708231d44a5012/biliary-cystadenoma-a-a-rare-cystic-neoplasm-of-liver-cystic-lesions-of-liver.jpg)























