Embed Size (px)
Citation preview

Bioseparation 10: 73–85, 2001.© 2001 Kluwer Academic Publishers. Printed in the Netherlands.
73
Direct process integration of cell disruption and fluidised bed adsorptionin the recovery of labile microbial enzymes
Horst Bierau1, Roger J. Hinton2 & Andrew Lyddiatt1,∗1Biochemical Recovery Group, Centre for Bioprocess Engineering, School of Chemical Engineering, University ofBirmingham, Edgbaston, Birmingham, B15 2TT, UK. 2CAMR, Porton Down, Salisbury, Wiltshire, SP4 0JG, UK(∗corresponding author)
Received 3 July 2000; accepted 4 December 2000
Key words: bead mill, cell debris, cell disruption, fluidised bed, intracellular proteins, process integration
Abstract
The practical feasibility and generic applicability of the direct integration of cell disruption by bead milling with thecapture of intracellular products by fluidised bed adsorption has been demonstrated. Pilot-scale purification of theenzyme L-asparaginase from unclarified Erwinia chrysanthemi disruptates exploiting this novel approach yieldedan interim product which rivalled or bettered that produced by the current commercial process employing discreteoperations of alkaline lysis, centrifugal clarification and batch adsorption. In addition to improved yield and qualityof product, the process time during primary stages of purification was greatly diminished. Two cation exchangeadsorbents, CM HyperD LS (Biosepra/Life Technologies) and SP UpFront (custom made SP form of a prototypestainless steel/agarose matrix, UpFront Chromatography) were physically and biochemically evaluated for suchdirect product sequestration. Differences in performance with regard to product capacity and adsorption/desorptionkinetics were demonstrated and are discussed with respect to the design of adsorbents for specific applications.In any purification of L-asparaginase (pI = 8.6), product-debris interactions commonly diminish the recovery ofavailable product. It was demonstrated herein, that immediate disruptate exposure to a fluidised bed adsorbentpromoted concomitant reduction of product in the liquid phase, which clearly counter-acted the product-debrisinteractions to the benefit of product yield.
Introduction
Adsorption in expanded or fluidised beds is nowwidely adopted for the direct recovery of proteinproducts from particulate feedstocks. The circum-vention of process bottlenecks encountered with thesolid liquid separation required upstream of fixed bedadsorption, whilst achieving considerable concentra-tion and primary purification of products, has beena major driver in process selection (Chase, 1994;Hjorth, 1997). However, such technology still exploitsdiscrete upstream operations of fermentation or celldisruption, and is commonly characterised by poten-tially detrimental hold-up periods whilst batches offeedstock are accumulated and/or conditioned prior tofluidised bed processing. Hold-up risks product modi-fication, inactivation or degradation by system antag-
onists (proteases, carbohydrases and drifting physicalconditions of temperature, pH and ionic strength;Kaufmann, 1997; Grodberg and Dunn, 1988) andproduct losses promoted by protein-debris interac-tions. However, a logical physical coupling of flu-idised bed adsorption with upstream operations offermentation or cell disruption might be predicted togain additional benefits of product yield and qualityby virtue of the immediate and direct sequestration ofproducts from process feedstocks at source (Hamiltonet al., 1999; Bierau et al., 1999; Hamilton et al.,2000). The present study was designed to identifyperformance advantages in the direct exploitation ofthe output of bacterial bead milling as the input anddriving force for fluidised bed adsorption of a target in-tracellular enzyme contained in cell disruptates. Thus,cell disruption and primary product capture are made

74
possible in a common time frame dependent upon thechosen feedstock throughput for the combined opera-tion. Provided that similar or better purification factorsare achieved, such an arrangement has clear advant-ages of process efficiency over discrete and sequentialoperation of cell disruption, disruptate clarificationand fixed bed chromatography commonly adopted inconventional downstream processing.
Cell disruption by mechanical or (bio)chemicalmeans is the first requirement for the purification of in-tracellular microbial enzymes but commonly initiatescellular and molecular degradation processes analog-ous to those of natural cell death and lysis (Schütteand Kula, 1987; Middelberg, 1995). In addition, thegeneration of fine cell debris may promote electro-static and/or hydrophobic product-debris interactions.Such adverse effects will compromise the yield andmolecular fidelity of protein products, particularlywhen feedstocks are accumulated and processed intime-dependent batch operations. Direct product se-questration at cell disruption (achieved in a commontime frame) should minimise such degradation and en-hance the yield and quality of even the most labileproducts.
Mechanical cell breakage in a bead-mill has manyattractive process characteristics including high dis-ruption efficiency, high throughput and biomass load-ing, good temperature control and unlimited scale-upfor most bioprocesses. For example bead mills, ori-ginally developed for colloid mixing, are availablewith chamber volumes of 275 litres, whilst processingof microbial disruption may be intensified by op-erating with biomass concentration up to 50% wetweight/volume and flow rates of up to 150 chambervolumes of feedstock per hour (discussed in White andMarcus, 1988; Bierau et al., 1999). In addition, the op-erational characteristic of single-pass and continuousdisruption of a single batch of cells recommends thebead-mill as the ideal feedstock generator for directsequestration of released products in a fluidised bedoperated in a common time frame. We report here acase study of the pilot-scale purification of the enzymeL-asparaginase (EC 3.5.1.1) sourced as an intracellu-lar product in Erwinia chrysanthemi. The commercialmanufacture of this enzyme currently exploits con-ventional unit operations of chemical lysis, proteinprecipitation, centrifugation, filtration and batch andfixed bed chromatography. However, the developmentof an integrated bead-milling/fluidised bed adsorptionprocess has been shown to improve the yield and qual-
ity of the product and diminish the processing timeduring the primary stages of purification.
Materials and methods
Mechanical cell disruption
Cell disruption was performed by bead milling in aDyno Mill KDL-I (Willi A. Bachofen AG, Switzer-land) consisting of a silicon-carbide lined stainlesssteel chamber (600 ml) cooled by re-circulating icedwater (0 ◦C). The chamber was loaded with zirconiabeads (0.3 mm) to 83% settled volume occupancy. Theagitator speed was 3200 rpm corresponding to a peri-pheral tip speed of the agitating discs of 10.5 m s−1.Frozen cells were thawed and resuspended in equilib-ration buffer (buffer A, 20 mM citric acid / tri-sodiumcitrate, pH 5.5). The cell suspension was adjustedto a pH of 5.5 (20 mM citric acid) and a biomassconcentration of 15% wet weight per volume (ww/v).
Adsorbents
Two different cation exchangers were exploited forthis study: (i) CM HyperD LS, 100–300 µm, 1.4 gml−1 (kindly supplied by BioSepra/Life Technolo-gies) and (ii) a novel, prototype material, comprisedof agarose-coated stainless steel beads, 151–323 µm,2.35 g ml−1 (kindly supplied by UpFront Chromato-graphy). The latter had been derivatised in order toobtain a cation exchange matrix in SP form (referredto as SP UpFront). Here, activation of the base mat-rix was achieved by reaction with allyl glycidyletherunder alkaline conditions followed by sulphonationusing metabisulphite and sodium sulphite.
Fluidised bed contactors
Fluidised bed contactors were operated and studied atthree different scales. A 1 cm diameter column (Up-Front Chromatography) employing a magnetic stirrerfor flow distribution was used in scouting experimentsfor both adsorbents. In addition, two custom builtcontactors, referred to as BRG contactors (Lan et al.,1999), were used for large scale development stud-ies. These comprised glass columns (2.5 and 4.5 cminternal diameter) with a hemispherical inlet wherea 2 cm bed of glass beads (710–1180 µm) servedas a flow distributor. All contactors were fitted withan adjustable top adaptor in order to minimise head-space above the fluidised bed. Adsorbent matrices

75
were equilibrated and washed using buffer A (citricacid/tri-sodium citrate, 20 mM, pH 5.5). Prior to theapplication of feedstock all beds were fluidised at thesuperficial flow velocity planned for particular adsorp-tion experiments. Elution was routinely performedin fluidised bed mode using buffer B (20 mM citricacid/tri-sodium citrate, pH 5.5, 1 M NaCl).
Batch binding experiments (debris adsorption study)
Batch binding experiments were performed by incub-ating 1 ml settled volume of the cation exchanger CMHyperD LS (equilibrated in buffer A) with differentvolumes (5/10/15/20/30/40 ml) of freshly generatedErwinia disruptate (bead mill feed rate 5 l h−1, 15%original biomass wet w/v at pH 5.5). The reactiontubes were placed on a roller incubator and at giventime intervals tubes were removed from the roller and,after the matrix was allowed to settle (approx. 10–15s), a small sample (200 µl) of the supernatant, i.e.the particulate disruptate was taken. Cell debris in thesample was eliminated by centrifugation (7600 g, 10min) and the clear supernatant removed, quantifiedvolumetrically, and assayed for asparaginase activityto yield the variation with time of enzyme in the li-quid phase. The cell debris was re-suspended in a50 mM borate buffer (pH 8.5) in order to desorb as-paraginase bound to sedimented cell debris. After a2 h incubation time, samples were again clarified bycentrifugation (as above), and the supernatant assayedfor free L-asparaginase activity.
Matrix adsorption kinetics
Batch binding experiments were performed byincubating both adsorbents at defined volumes(0.25/0.5/1.0/1.5/2.0/3.0 ml) with purified L-aspara-ginase (supplied by CAMR, Porton Down) in buffer A(2 mg ml−1, 25 ml total volume). The reaction tubeswere placed on a roller incubator and at given timeintervals the tubes were removed from the roller and,after the matrix was allowed to settle (approx. 5–10s), a small sample of the supernatant was removed andassayed for protein concentration.
Results and discussion
Process intensification by process integration
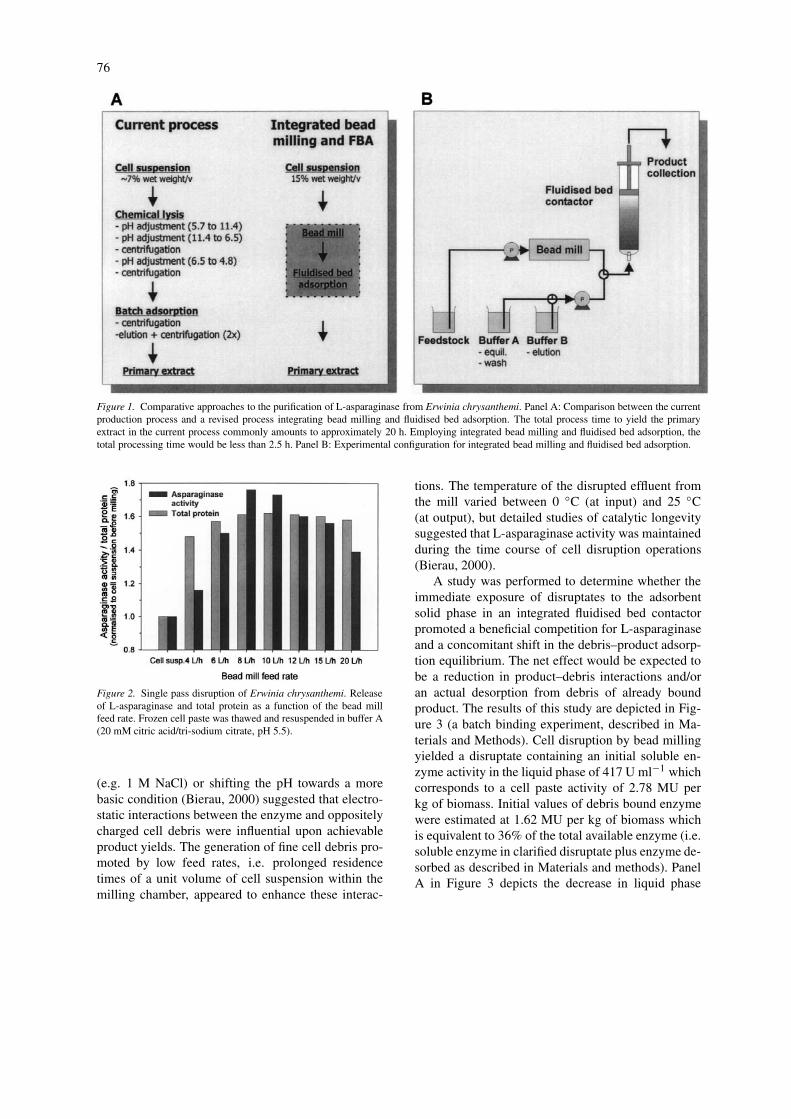
Figure 1 depicts a comparison between the current anda potential revised process of L-asparaginase product
release for Erwinia cell paste and its primary purifica-tion. The current commercial process exploits alkalinelysis of the cells and an adsorption step performedin batch mode following centrifugal clarification. Celllysis is achieved by adjustment of the pH of a cell sus-pension to 11.4. The pH is then incrementally loweredto 6.5 and 4.8. At both pH values the broth is clarifiedby centrifugation. The pH is not directly adjusted to4.8 in a single step in order to minimise loss of enzymedue to adsorption to cell debris (Goward et al., 1992).Batch adsorption of the product is then performed ina stirred tank using a cation exchange CM cellulosematrix. The matrix is separated from the broth bycentrifugation and eluted in batch mode with addedsalts. Commonly, the total process time to yield theprimary extract amounts to approximately 20 h (referto Figure 1).
In the integrated process the disruptate is directlyfed from the mill into the fluidised adsorbent bed.Due to the direct coupling of the two unit operations,temporary storage of the disruptate is eliminated andexposure of the product to potential antagonists (debrisadsorbents, degrading enzymes) is minimised to thetime required for loading the adsorbent to an appro-priate saturation and for washing the bed from debrisand unbound contaminants. In addition, no clarific-ation steps are required to process the broth whichconsiderably reduces the number of unit operationsand thus shortens the overall process time. Here, thetotal process time (cell disruption/loading, washingand elution) to yield an equivalent primary extractwould be less than 2.5 h when employing an appro-priately up-scaled fluidised bed (e.g. column diameter48 cm, settled bed volume 28 l).
Cell disruption by bead milling
Product release from Erwinia cell suspension in buf-fer A by bead milling in single pass mode is depictedin Figure 2. Both enzyme and total protein releasereached a maximum at a feed rate of 8 l h−1. Higherfeed rates resulted in a decrease in enzyme activitywhich indicated incomplete disruption. However, feedrates less than 8 l h−1 were also characterised bya decrease in measurable enzyme activity. Prelimin-ary work has demonstrated that L-asparaginase (pI =8.6) adsorbed to negatively charged cell debris in apH and ionic strength dependent manner similar tothe behaviour observed with a cation exchange mat-rix. The fact that levels of asparaginase activity indisruptates could be increased by the addition of salt
76
Figure 1. Comparative approaches to the purification of L-asparaginase from Erwinia chrysanthemi. Panel A: Comparison between the currentproduction process and a revised process integrating bead milling and fluidised bed adsorption. The total process time to yield the primaryextract in the current process commonly amounts to approximately 20 h. Employing integrated bead milling and fluidised bed adsorption, thetotal processing time would be less than 2.5 h. Panel B: Experimental configuration for integrated bead milling and fluidised bed adsorption.
Figure 2. Single pass disruption of Erwinia chrysanthemi. Releaseof L-asparaginase and total protein as a function of the bead millfeed rate. Frozen cell paste was thawed and resuspended in buffer A(20 mM citric acid/tri-sodium citrate, pH 5.5).
(e.g. 1 M NaCl) or shifting the pH towards a morebasic condition (Bierau, 2000) suggested that electro-static interactions between the enzyme and oppositelycharged cell debris were influential upon achievableproduct yields. The generation of fine cell debris pro-moted by low feed rates, i.e. prolonged residencetimes of a unit volume of cell suspension within themilling chamber, appeared to enhance these interac-
tions. The temperature of the disrupted effluent fromthe mill varied between 0 ◦C (at input) and 25 ◦C(at output), but detailed studies of catalytic longevitysuggested that L-asparaginase activity was maintainedduring the time course of cell disruption operations(Bierau, 2000).
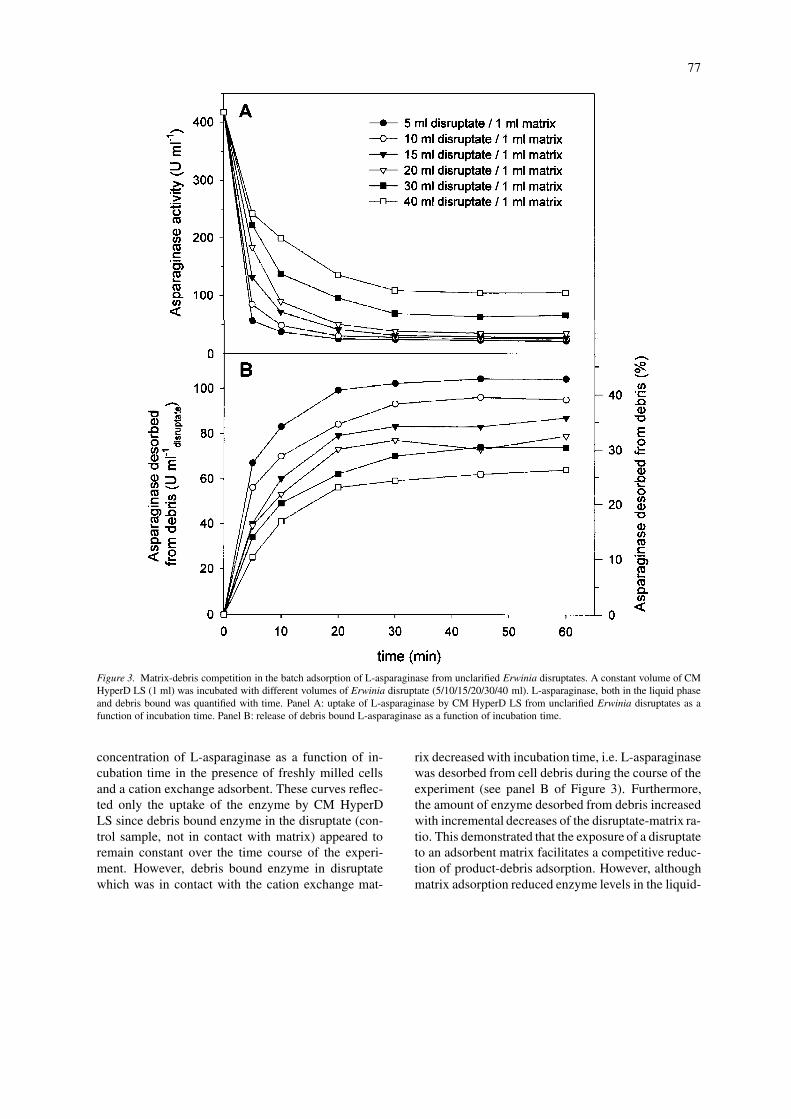
A study was performed to determine whether theimmediate exposure of disruptates to the adsorbentsolid phase in an integrated fluidised bed contactorpromoted a beneficial competition for L-asparaginaseand a concomitant shift in the debris–product adsorp-tion equilibrium. The net effect would be expected tobe a reduction in product–debris interactions and/oran actual desorption from debris of already boundproduct. The results of this study are depicted in Fig-ure 3 (a batch binding experiment, described in Ma-terials and Methods). Cell disruption by bead millingyielded a disruptate containing an initial soluble en-zyme activity in the liquid phase of 417 U ml−1 whichcorresponds to a cell paste activity of 2.78 MU perkg of biomass. Initial values of debris bound enzymewere estimated at 1.62 MU per kg of biomass whichis equivalent to 36% of the total available enzyme (i.e.soluble enzyme in clarified disruptate plus enzyme de-sorbed as described in Materials and methods). PanelA in Figure 3 depicts the decrease in liquid phase
77
Figure 3. Matrix-debris competition in the batch adsorption of L-asparaginase from unclarified Erwinia disruptates. A constant volume of CMHyperD LS (1 ml) was incubated with different volumes of Erwinia disruptate (5/10/15/20/30/40 ml). L-asparaginase, both in the liquid phaseand debris bound was quantified with time. Panel A: uptake of L-asparaginase by CM HyperD LS from unclarified Erwinia disruptates as afunction of incubation time. Panel B: release of debris bound L-asparaginase as a function of incubation time.
concentration of L-asparaginase as a function of in-cubation time in the presence of freshly milled cellsand a cation exchange adsorbent. These curves reflec-ted only the uptake of the enzyme by CM HyperDLS since debris bound enzyme in the disruptate (con-trol sample, not in contact with matrix) appeared toremain constant over the time course of the experi-ment. However, debris bound enzyme in disruptatewhich was in contact with the cation exchange mat-
rix decreased with incubation time, i.e. L-asparaginasewas desorbed from cell debris during the course of theexperiment (see panel B of Figure 3). Furthermore,the amount of enzyme desorbed from debris increasedwith incremental decreases of the disruptate-matrix ra-tio. This demonstrated that the exposure of a disruptateto an adsorbent matrix facilitates a competitive reduc-tion of product-debris adsorption. However, althoughmatrix adsorption reduced enzyme levels in the liquid-

78
phase by approximately 95% (panel A in Figure 3, 5ml disruptate per ml of matrix) the amount of debrisbound enzyme was only reduced by 43% (panel B,Figure 3).
After cell disruption the disruptate was stored for60 min during preparations for the batch binding ex-periment depicted in Figure 3. During this period,the product was exposed to cell debris and some ad-sorption inevitably took place. It would have beenbeneficial to expose the disruptate immediately aftercell disruption to an adsorbent matrix such that contacttimes between product and cell debris are minimisedand gross product losses by adsorption to debris re-duced overall. Such a facilitation has been proposedearlier by the integration of cell disruption by beadmilling and the primary product capture by fluidisedbed adsorption (Bierau et al., 1999). It would be ex-pected to favour further benefits of decreased productdegradation, shortened process times and lower oper-ational costs.
Method development (fluidised bed adsorption)
CM HyperD and SP UpFront represent two differenttypes of adsorbent solid phases. The former is charac-terised by a dense ceramic shell filled with a porouscontinuum whilst the latter has a more limited pel-licular structure, i.e. a porous agarose coating of anon-porous steel core). As a result, their inherent ad-sorption/desorption characteristics may be expected todiffer significantly. Depth of the porous agarose layerof the pellicular SP UpFront (bead size range 151–323 µm) was estimated as 40% of the bead radius(calculation based on the densities of the core andcoating material and the composite bead). Diffusiondistances within such an assembly are thus shorter incomparison with CM HyperD (bead size range 100–300 µm) and therefore rates of adsorption/desorptionmay be higher. On the other hand, the equilibrium ca-pacity of pellicular matrices is expected to be reducedin comparison with an equivalently sized macropor-ous continuum by virtue of a reduced volume foradsorption.
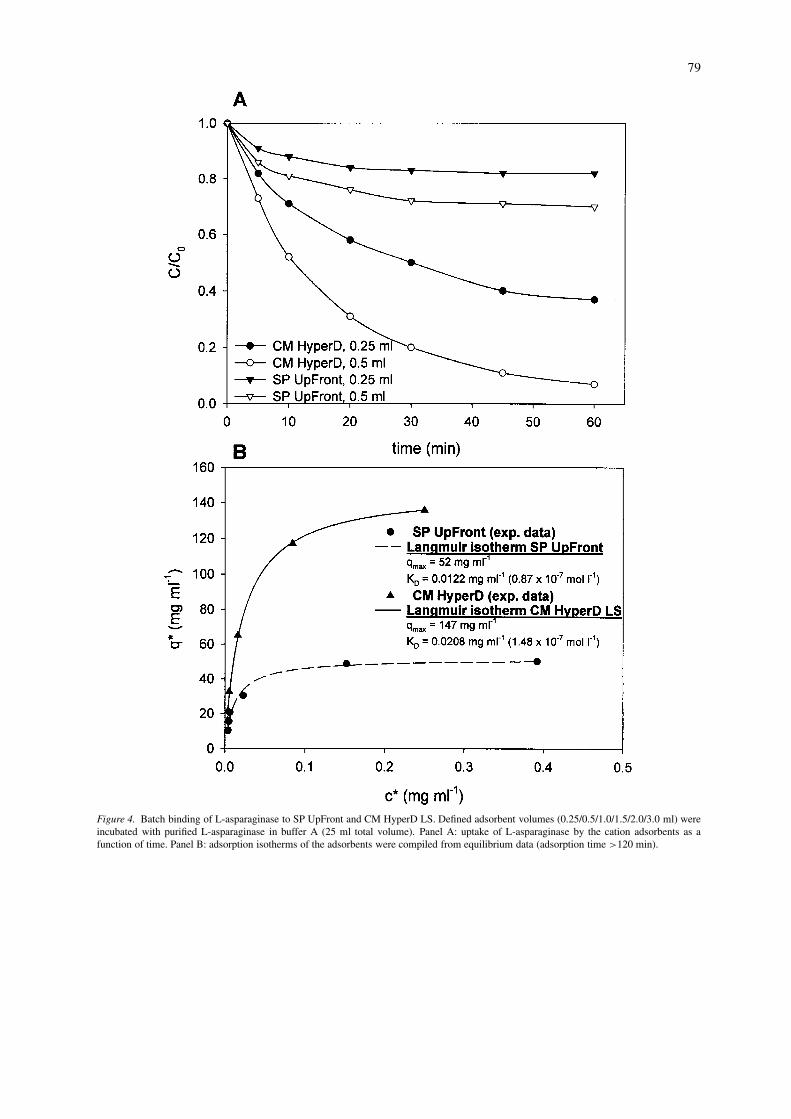
In order to investigate and address comparativeaspects of adsorption kinetics, a series of batch bind-ing experiments were designed and performed as de-scribed in the Materials and methods section. Uptakeof L-asparaginase as a function of time for the twoadsorbents is depicted in panel A of Figure 4 and ad-sorption isotherms are given in panel B of Figure 4. Itcan be seen from panel A that enzyme adsorption by
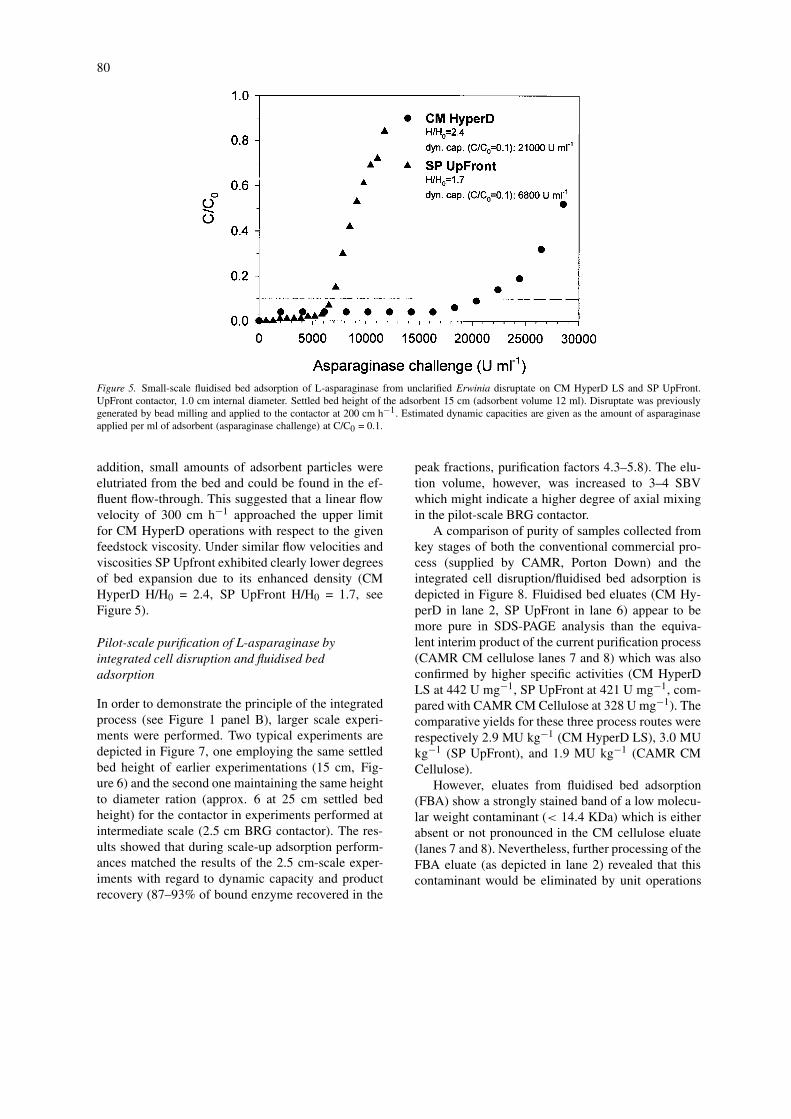
SP UpFront approached a steady state value at about20 min of incubation time whereas in the case of CMHyperD LS significant adsorption was still detectableat times up to 60 min. A higher rate of adsorptionfor the pellicular adsorbent may also be indicated bya lower apparent KD value (see adsorption isotherms,panel B in Figure 4). On the other hand, adsorptionisotherms show a threefold higher equilibrium capa-city for CM HyperD LS. These results confirmed thepredictions made from the structural differences ofthe two adsorbents, i.e. adsorption takes place morerapidly on the pellicular adsorbent whilst its bindingcapacity is considerably lower. A threefold differencein capacity, i.e. CM HyperD LS 21000 U ml−1 and SPUpFront 6800 U ml−1, was also confirmed by small-scale fluidised bed adsorption experiments (UpFrontcontactor, 1.0 cm) employing unclarified Erwinia dis-ruptate (see Figure 5). In the light of advantages withregard to binding capacity, CM HyperD LS was expec-ted to perform best in integrated recoveries and wasused exclusively for further experimentation.
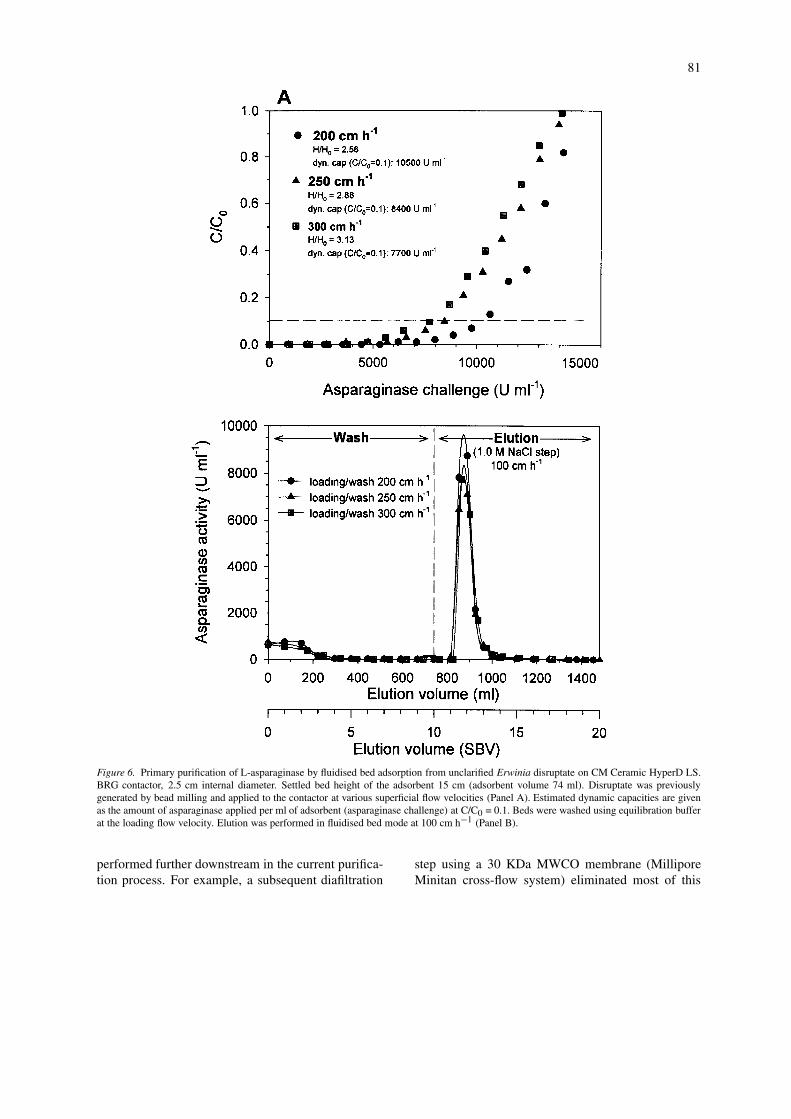
Fluidised bed adsorption of L-asparaginase fromunclarified Erwinia disruptates on CM HyperD LSin a 2.5 cm BRG contactor is depicted in Figure 6.Here, previously generated feedstock was applied tothe contactor at various superficial flow velocities.All experiments exhibited an extended period of neg-ligible breakthrough. However, dynamic capacities(estimated from the asparaginase challenge requiredto achieve 10% breakthrough, i.e. C/C0 = 0.1) werelower in comparison with the experiments performedin the 1.0 cm UpFront contactor (Figure 5) which sug-gested a higher degree of axial mixing in the BRGcontactor (the performance of such a contactor hasbeen characterised by Lan et al., 1999). The achieveddynamic capacities also decreased to some extent withincreasing superficial flow velocities. A wash volumeof 5 settled bed volumes was generally sufficient toreduce the concentration of contaminating proteins inthe effluent of the fluidised bed by more than 90%.Step elution in 1 M NaCl resulted in a sharp peak ofenzyme which could be collected in about 2 settledbed volumes (SBV). This was equivalent to resultsobtained from scouting experiments with fixed bedelution (data not shown). Between 76 and 82% ofthe adsorbed enzyme was recovered in the peak frac-tions and purification factors between 4.7 and 5.0were achieved. During loading at higher flow rates,i.e. 300 cm h−1, the top of the fluidised bed becameincreasingly diffuse which aggravated the determina-tion of the true value of the expanded bed height. In
79
Figure 4. Batch binding of L-asparaginase to SP UpFront and CM HyperD LS. Defined adsorbent volumes (0.25/0.5/1.0/1.5/2.0/3.0 ml) wereincubated with purified L-asparaginase in buffer A (25 ml total volume). Panel A: uptake of L-asparaginase by the cation adsorbents as afunction of time. Panel B: adsorption isotherms of the adsorbents were compiled from equilibrium data (adsorption time >120 min).
80
Figure 5. Small-scale fluidised bed adsorption of L-asparaginase from unclarified Erwinia disruptate on CM HyperD LS and SP UpFront.UpFront contactor, 1.0 cm internal diameter. Settled bed height of the adsorbent 15 cm (adsorbent volume 12 ml). Disruptate was previouslygenerated by bead milling and applied to the contactor at 200 cm h−1. Estimated dynamic capacities are given as the amount of asparaginaseapplied per ml of adsorbent (asparaginase challenge) at C/C0 = 0.1.
addition, small amounts of adsorbent particles wereelutriated from the bed and could be found in the ef-fluent flow-through. This suggested that a linear flowvelocity of 300 cm h−1 approached the upper limitfor CM HyperD operations with respect to the givenfeedstock viscosity. Under similar flow velocities andviscosities SP Upfront exhibited clearly lower degreesof bed expansion due to its enhanced density (CMHyperD H/H0 = 2.4, SP UpFront H/H0 = 1.7, seeFigure 5).
Pilot-scale purification of L-asparaginase byintegrated cell disruption and fluidised bedadsorption
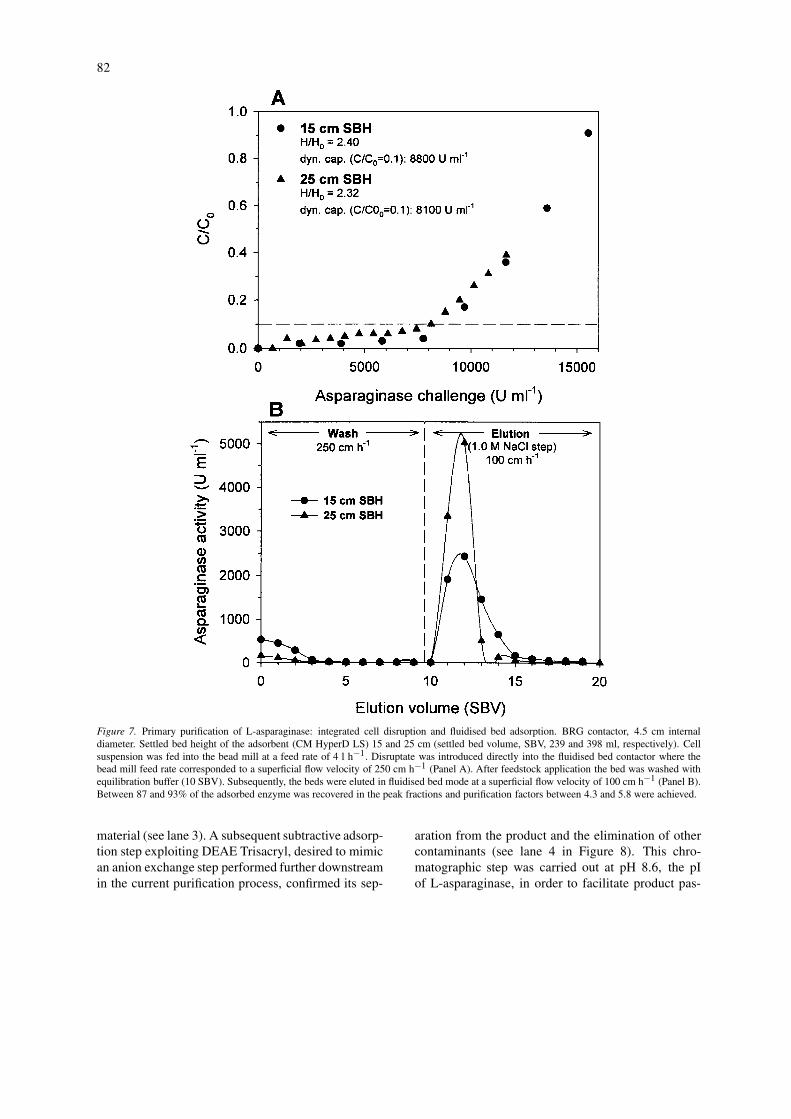
In order to demonstrate the principle of the integratedprocess (see Figure 1 panel B), larger scale experi-ments were performed. Two typical experiments aredepicted in Figure 7, one employing the same settledbed height of earlier experimentations (15 cm, Fig-ure 6) and the second one maintaining the same heightto diameter ration (approx. 6 at 25 cm settled bedheight) for the contactor in experiments performed atintermediate scale (2.5 cm BRG contactor). The res-ults showed that during scale-up adsorption perform-ances matched the results of the 2.5 cm-scale exper-iments with regard to dynamic capacity and productrecovery (87–93% of bound enzyme recovered in the
peak fractions, purification factors 4.3–5.8). The elu-tion volume, however, was increased to 3–4 SBVwhich might indicate a higher degree of axial mixingin the pilot-scale BRG contactor.
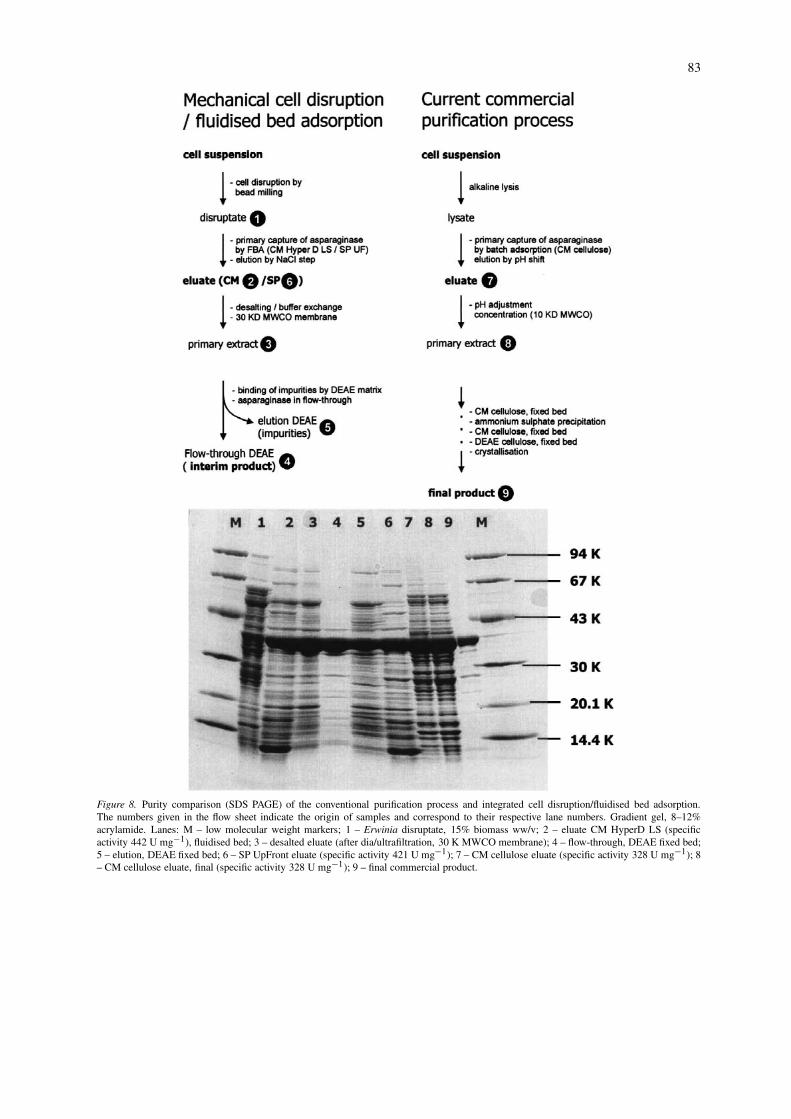
A comparison of purity of samples collected fromkey stages of both the conventional commercial pro-cess (supplied by CAMR, Porton Down) and theintegrated cell disruption/fluidised bed adsorption isdepicted in Figure 8. Fluidised bed eluates (CM Hy-perD in lane 2, SP UpFront in lane 6) appear to bemore pure in SDS-PAGE analysis than the equiva-lent interim product of the current purification process(CAMR CM cellulose lanes 7 and 8) which was alsoconfirmed by higher specific activities (CM HyperDLS at 442 U mg−1, SP UpFront at 421 U mg−1, com-pared with CAMR CM Cellulose at 328 U mg−1). Thecomparative yields for these three process routes wererespectively 2.9 MU kg−1 (CM HyperD LS), 3.0 MUkg−1 (SP UpFront), and 1.9 MU kg−1 (CAMR CMCellulose).
However, eluates from fluidised bed adsorption(FBA) show a strongly stained band of a low molecu-lar weight contaminant (< 14.4 KDa) which is eitherabsent or not pronounced in the CM cellulose eluate(lanes 7 and 8). Nevertheless, further processing of theFBA eluate (as depicted in lane 2) revealed that thiscontaminant would be eliminated by unit operations
81
Figure 6. Primary purification of L-asparaginase by fluidised bed adsorption from unclarified Erwinia disruptate on CM Ceramic HyperD LS.BRG contactor, 2.5 cm internal diameter. Settled bed height of the adsorbent 15 cm (adsorbent volume 74 ml). Disruptate was previouslygenerated by bead milling and applied to the contactor at various superficial flow velocities (Panel A). Estimated dynamic capacities are givenas the amount of asparaginase applied per ml of adsorbent (asparaginase challenge) at C/C0 = 0.1. Beds were washed using equilibration bufferat the loading flow velocity. Elution was performed in fluidised bed mode at 100 cm h−1 (Panel B).
performed further downstream in the current purifica-tion process. For example, a subsequent diafiltration
step using a 30 KDa MWCO membrane (MilliporeMinitan cross-flow system) eliminated most of this
82
Figure 7. Primary purification of L-asparaginase: integrated cell disruption and fluidised bed adsorption. BRG contactor, 4.5 cm internaldiameter. Settled bed height of the adsorbent (CM HyperD LS) 15 and 25 cm (settled bed volume, SBV, 239 and 398 ml, respectively). Cellsuspension was fed into the bead mill at a feed rate of 4 l h−1. Disruptate was introduced directly into the fluidised bed contactor where thebead mill feed rate corresponded to a superficial flow velocity of 250 cm h−1 (Panel A). After feedstock application the bed was washed withequilibration buffer (10 SBV). Subsequently, the beds were eluted in fluidised bed mode at a superficial flow velocity of 100 cm h−1 (Panel B).Between 87 and 93% of the adsorbed enzyme was recovered in the peak fractions and purification factors between 4.3 and 5.8 were achieved.
material (see lane 3). A subsequent subtractive adsorp-tion step exploiting DEAE Trisacryl, desired to mimican anion exchange step performed further downstreamin the current purification process, confirmed its sep-
aration from the product and the elimination of othercontaminants (see lane 4 in Figure 8). This chro-matographic step was carried out at pH 8.6, the pIof L-asparaginase, in order to facilitate product pas-
83
Figure 8. Purity comparison (SDS PAGE) of the conventional purification process and integrated cell disruption/fluidised bed adsorption.The numbers given in the flow sheet indicate the origin of samples and correspond to their respective lane numbers. Gradient gel, 8–12%acrylamide. Lanes: M – low molecular weight markers; 1 – Erwinia disruptate, 15% biomass ww/v; 2 – eluate CM HyperD LS (specificactivity 442 U mg−1), fluidised bed; 3 – desalted eluate (after dia/ultrafiltration, 30 K MWCO membrane); 4 – flow-through, DEAE fixed bed;5 – elution, DEAE fixed bed; 6 – SP UpFront eluate (specific activity 421 U mg−1); 7 – CM cellulose eluate (specific activity 328 U mg−1); 8– CM cellulose eluate, final (specific activity 328 U mg−1); 9 – final commercial product.

84
sage through the adsorbent bed and to retain moreacidic contaminants (lane 5 in Figure 8). In prelimin-ary studies, about 40% of the applied L-asparaginasealso adsorbed to the anion exchanger (refer to lane 5).Clearly, this step needs further optimisation in order tominimise adsorptive losses of asparaginase. However,the purity of the enzyme product after 3 operationalsteps of integrated bead milling/FBA, diafiltration andfixed bed adsorption is highly encouraging.
Conclusions
Bead milling as a means for cell disruption
Bead milling proved to be an efficient means for dis-rupting Erwinia chrysanthemi in single pass mode. Inthe preliminary experimentation discussed herein, theproduct yield per unit of biomass (i.e. resuspended cellpaste) was slightly lower in comparison with the oneachieved by alkaline lysis employed for the same batchof cell paste (e.g. bead milling 3.8 MU kg−1, alkalinelysis 4.6 MU kg−1). This may be attributed to a com-bination of process intensification (up to 15% biomassww/v for bead milling; see Figure 1) and back mixingphenomena in the disruption chamber. This perform-ance was judged acceptable in view of the process ad-vantages of single-pass disruption, but requires furtheroptimisation. Within the range of feed rates tested, thedisruptate temperature did not exceed 25 ◦C and heatdenaturation of the product was shown to be insigni-ficant. Under disruption conditions suited for fluidisedbed adsorption of L-asparaginase on cation exchangeadsorbents, the enzyme exhibited detrimental adsorpt-ive interactions with cell debris. A partial eliminationof debris bound losses (> 40%) could be promotedby the immediate exposure of disruptates to adsorp-tion by establishing competition between the cationexchange adsorbent and debris. The observation thatthe release of debris bound enzyme in batch bind-ing experiments was increased at lower volume ratiosof disruptate to adsorbent (panel B, Figure 3) mightindicate that within a fluidised bed L-asparaginase re-lease from debris is enhanced at low degrees of bedvoidage, i.e. low degrees of bed expansion.
Adsorbent characteristics
CM HyperD LS, representing a macroporous con-tinuum, distinguished itself by its high capacity forL-asparaginase but was restricted by a moderate dens-ity. The pellicular SP UpFront, on the other hand,
is ideal by virtue of its elevated density for applic-ations with high flow velocities and/or high viscousfeedstocks. The mass transfer characteristics of thepellicular structure also encourage novel routes tothe design of an adsorptive step in downstream pro-cessing. Thus, the use of a manifold of appropriatelydown-scaled fluidised beds of such adsorbents wouldbe characterised by short cycle times of adsorption,desorption and cleaning/re-equilibration (Morton andLyddiatt, 1994), see Figure 9. Advantages of such amulti-bed system would be a reduced adsorbent in-ventory as well as short residence times for feedstocks(discussed in Hamilton et al., 1999). Dense, pellicu-lar adsorbents, operated at high fluid velocities andviscosities with low bed expansions and rapid adsorp-tion/desorption rates appeared to be ideal for suchapplications.
Integration of cell disruption and product capture byfluidised bed adsorption
The feasibility of directly integrating conventionallydiscrete operations of cell disruption by bead millingand the product capture by fluidised bed adsorptionin a common time frame has been demonstrated. Ina side-by-side comparison with the established puri-fication process for a therapeutical enzyme, the novelprocess proved to be more efficient by yielding apurer interim product using either CM HyperD or SPUpFront as an adsorbent (refer to Figure 8). Yieldswere routinely in excess of 2.8 MU kg−1 of cellpaste with specific activities exceeding 400 U mg−1
of protein which bettered the performance of the com-mercial process at an equivalent stage. Furthermore,operational sequences were considerably compressedwithout the need of disruptate clarification and/or stor-age. With the given system configuration operated atpilot-scale, the throughput of biomass was only lim-ited by the dimensions of the fluidised bed contactorsince optimal product release was achieved at a beadmill feed rate of 8 l h−1 (corresponding to a superficialflow velocity of 500 cm h−1 in the column, a twofoldincrease in comparison with flow velocities used inthis study). Process intensification, i.e. increased bio-mass throughput, could be achieved by increasing thecontactor diameter and/or by the use of an adsorbentwith enhanced density. Such material, provided thatcapacity and mass transfer characteristics were appro-priate, would facilitate the use of higher superficialflow velocities and/or higher biomass concentrationsin the feedstock.

85
Figure 9. Principle of a multi-fluidised bed system.
Acknowledgement
This work was supported by a studentship fun-ded by CAMR (The Centre for Applied Microbio-logy and Research, Porton Down, Salisbury, Wilt-shire, SP4 0JG). Adsorbents were kindly supplied byBiosepra/Life Technologies (CM HyperD LS) andUpFront Chromatography (SP stainless steel, pellicu-lar prototype).
References
Bierau H (2001) Process integration of cell disruption and fluid-ised bed adsorption of microbial enzymes: application to theretro-design of the purification of L-asparaginase. Phothesis,University of Birmingham.
Bierau H, Zhang Z and Lyddiatt A (1999) Direct process integrationof cell disruption and fluidised bed adsorption for the recovery ofintracellular proteins. J. Chem. Tech. Biotechnol. 74: 208–212.
Chase HA (1994) Purification of proteins by adsorption chromato-graphy in expanded beds. Trends Biotechnol. 12: 296–303.
Goward CR, Stevens GB, Tattersall R and Atkinson T (1992) Rapidlarge-scale preparation of recombinant Erwinia chrysanthemi L-asparaginase. Bioseparation 2: 335–341.
Grodberg J and Dunn JJ (1988) ompT encodes the Escherichiacoli outer membrane protease that cleaves T7 RNA polymeraseduring purification. J. Bacteriol. 170: 1245–1253.
Hamilton GE, Morton PH, Young TW and Lyddiatt A (1999) Pro-
cess intensification by direct product sequestration from batchfermentations: Application of a fluidised bed, multi-bed externalloop contactor. Biotechnol. Bioeng. 64: 310–321.
Hamilton GE, Luechau F, Burton SC and Lyddiatt A (2000) De-velopment of a mixed mode adsorption process for the directproduct sequestration of an extracellular protease from microbialbatch cultures. J. Biotechnol. 79: 103–115.
Hjorth R (1997) Expanded-bed adsorption in industrial biopro-cessing: Recent developments. Trends Biotechnol. 15: 230–235.
Kaufmann M (1997) Unstable proteins: how to subject themto chromatographic separations for purification procedures. J.Chromatogr. B 699: 347–369.
Lan JC-W, Hamilton GE and Lyddiatt A (1999) Physical andbiochemical characterization of a simple intermediate betweenfluidised and expanded bed contactors. Bioseparation 8: 43–51.
Middelberg APJ (1995) Process-scale disruption of microorgan-isms. Biotechnol. Adv. 13: 491–551.
Morton P and Lyddiatt A (1994) Direct integration of protein recov-ery with productive fermentations. In: Pyle DL (ed.) Separationsfor Biotechnology 3 (pp. 329–335). Royal Society of Chemistry.
Schütte H and Kula M-R (1987) Purification of proteins and the celldisruption of microbial cells. Biotechnol. Progress 3: 31–42.
White MD and Marcus D (1988) Disintegration of Microorgan-isms. In: Mizrahi A [ed.], Downstream Processes: Equipmentand Techniques, Vol. 8 (pp. 53–90. Alan R. Liss Inc., New York.
Address for correspondence: Prof. A. Lyddiatt, Universityof Birmingham, Chemical Engineering Department, Edgbaston,Birmingham B15 2TT, UK (Tel./Fax: 0121 414 5278; E-mail:[email protected])












![nÃÛÖ] äËm†˙ å‚nÏ - Khalifatullah Mehdikhalifatullahmehdi.info/books/urdu/Majmua-Rasayel-BandagiMiyan-RZ.pdfï ï ï ï ï ï ï ï ï ï ï ï ï ï ï ï ï ï ï ï](https://img.pdfslide.net/doc/110x75/5e752ae12f0d2b679431cf1d/nf-ma-an-khalifatullah-meh-.jpg)
















