Embed Size (px)
Citation preview

Published OnlineFirst September 24, 2010; DOI: 10.1158/1541-7786.MCR-10-0011
DNA Damage and Cellular Stress Responses Molecular
CancerResearch
DNA Damage–Induced Modulation of GLUT3 Expression IsMediated through p53-Independent Extracellular Signal-Regulated Kinase Signaling in HeLa Cells
Masaru Watanabe, Hiroaki Naraba, Tomoko Sakyo, and Takayuki Kitagawa
Abstract
Authors'Pathology,Iwate, Japa
Note:SuppResearch O
Corresponand Molecu2-1-1 Nishi651-5111;
doi: 10.115
©2010 Am
www.aacr
D
Many cancer cells exhibit increased rates of uptake and metabolism of glucose compared with normalcells. Glucose uptake in mammalian cells is mediated by the glucose transporter (GLUT) family. Here,we report that DNA-damaging anticancer agents such as Adriamycin and etoposide suppressed the expres-sion of GLUT3, but not GLUT1, in HeLa cells and a tumorigenic HeLa cell hybrid. Suppression ofGLUT3 expression determined by the real-time PCR was also evident with another DNA-damaging agent,camptothecin, which reduced the promoter's activity as determined with a luciferase-linked assay. The sup-pression by these agents seemed to be induced independently of p53, and it was evident when wild-typep53 was overproduced in these cells. In contrast, the mitogen-activated protein kinase/extracellular signalregulated kinase (MAPK/ERK) kinase (MEK) inhibitor U0126 (but not the phosphoinositide 3-kinaseinhibitor LY294002) prevented the drug-induced suppression as determined by reverse transcription-PCRand promoter assays. Furthermore, overexpression of GLUT3 in HeLa cell hybrids increased resistance tothese drugs, whereas depletion of the gene by small interfering RNA rendered the cells more sensitive to thedrugs, decreasing glucose consumption. The results suggest that DNA-damaging agents reduce GLUT3expression in cancer cells through activation of the MEK–ERK pathway independently of p53, leadingto cell death or apoptosis. The findings may contribute to the development of new chemotherapeutic drugsbased on the GLUT3-dependent metabolism of glucose. Mol Cancer Res; 8(11); 1547–57. ©2010 AACR.
Introduction
Many cancer cells use nutrients such as glucose as asource of energy for growth and survival (1-3). Cancer cellsprefer to metabolize glucose by aerobic glycolysis instead ofoxidative phosphorylation, a phenomenon termed as theWarburg effect (4). Increased glucose uptake in cancer cellscan be measured clinically by [18F]deoxyglucose positronemission tomography (5, 6).The glucose transporter (GLUT) mediates facilitated up-
take of D-glucose in mammalian cells. The GLUT familyhas >10 members, all expressed in a tissue-specific manner(7). GLUT1 is widely expressed in all proliferating cells,whereas GLUT3 is predominantly expressed in neuronal
Affiliation: Department of Cell Biology and MolecularIwate Medical University, School of Pharmacy, Yahaba,n
lementary data for this article are available at Molecular Cancernline (http://mcr.aacrjournals.org/).
ding Author: Takayuki Kitagawa, Department of Cell Biologylar Pathology, Iwate Medical University, School of Pharmacy,tokuta, Yahaba, Shiwa, Iwate 028-3694, Japan. Phone: 81-19-Fax: 81-19-698-1844. E-mail: [email protected]
8/1541-7786.MCR-10-0011
erican Association for Cancer Research.
journals.org
Researchon February 11mcr.aacrjournals.org ownloaded from
cells (7). Both isoforms show high affinity for D-glucoseunder normal physiologic conditions (8, 9). An upregula-tion of GLUT1 or GLUT3 expression is observed in manytumors, such as gliomas, non–small cell lung carcinomas,gastroenterologic tumors, and ovarian carcinomas (10-16).The increased glucose uptake correlates with the level ofexpression of GLUTs in the plasma membrane (17, 18).Because expression of the GLUT family often shows astrong association with malignancy (14), the componentscontrolling their expression could be important as targetsfor cancer therapy as well as for the early diagnosis of tu-mors and as molecular markers of malignancy (1).We have previously reported a tumor-associated alter-
ation of GLUT1 or GLUT3 in human cell hybrids derivedfrom cervical carcinoma HeLa cells and normal fibroblasts(19-21). The tumorigenic hybrid (CGL4) expressed bothGLUT1 and GLUT3, whereas the tumor-suppressed hy-brid (CGL1) expressed GLUT1 alone (21). This tumor-associated GLUT3 expression is regulated at the level oftranscription at least (21).The regulation of GLUT3 transcription is associated
with several signaling molecules such as the nuclear fac-tors Sp1 and cyclic AMP in rodent cells. In rat neurons,Sp1 and Sp3 regulated GLUT3 expression and bound tocis elements in the promoter (22, 23). In mouse musclecells, insulin-like growth factor-I played a role in maintaining
1547
. , 2018. © 2010 American Association for Cancer

Watanabe et al.
1548
Published OnlineFirst September 24, 2010; DOI: 10.1158/1541-7786.MCR-10-0011
GLUT3 expression via Sp1 (24). The mouse GLUT3 pro-moter also responded to cyclic AMP in a breast cancercell line (25). However, human GLUT3 expression dur-ing tumorigenesis has not been extensively studied.It is also known that genotoxic stress-induced signals
regulate GLUT3 expression. Multiple genotoxic stimulisuch as Adriamycin (ADR), etoposide (ETOP), cisplatin,UV, and γ-radiation resulted in a suppression of GLUT3expression and glucose metabolism (26) and also initiatedcell death pathways (27). However, the role of GLUT3 incell survival remains unclear.A well-known tumor suppressor, p53, plays an impor-
tant role in the response to DNA damage (28). The p53protein is mainly degraded by Mdm2-mediated ubiquitin-dependent proteolysis and is kept at low levels in vivo (29).In response to DNA damage, stabilization of p53 is medi-ated by the ATM and ATR protein kinases (27, 28). Thestabilized p53 promotes cell cycle arrest to allow repair orthe initiation of apoptosis (27, 28).Recent studies have highlighted a link between p53-
mediated surveillance of damaged DNA and the metab-olism of glucose. Kawauchi et al. (30), using p53-nullmouse embryonic fibroblasts (MEF), showed that theexpression of GLUT3 is regulated through NF-κB ina p53-dependent manner. Although the p53 proteinplays a critical role in responses to DNA damage,p53-independent responses to DNA damage have alsobeen reported (31-34). Damage to DNA activates extra-cellular signal-regulated kinase (ERK) pathways in paral-lel with p53 in an ATM-dependent manner. The ERKactivation associated with DNA damage mediates cellcycle arrest and apoptosis (33).Here, we report that DNA-damaging agents such as ADR
and camptothecin (CPT) suppressed the expression ofGLUT3 at a transcriptional level in HeLa cells and humancell hybrids. The levels of mRNA were quantitatively andstatistically determined by the real-time PCR method. Wealso show that this suppression occurred independently ofp53 and mainly involved the activation of ERK. We showthat the level of GLUT3 expression was linked to DNAdamage–induced cell death. Our data indicate that GLUT3plays a key role in the surveillance of DNA damage inhuman tumor cells.
Materials and Methods
Antibodies, reagents, and RNA interferenceThe mouse monoclonal antibody to β-actin was
purchased from Sigma, whereas those to human p21,phosphorylated ERK1/2 and ERK1, were from BD Bio-sciences and human p53 was from Santa Cruz Biotechnol-ogy. An enhanced chemiluminescence kit was obtainedfrom GE Healthcare. Lipofectamine 2000 reagent andsmall interfering RNAs (siRNA) to p53 (HSS186390),ERK1 (VHS40318), and GLUT3 (HSS143955) were pur-chased from Invitrogen. ADR, ETOP, CPT, vinblastine,paclitaxel, U0126, and LY294002 were obtained from
Mol Cancer Res; 8(11) November 2010
Researchon February 11mcr.aacrjournals.org Downloaded from
Calbiochem. Nutlin-3 and cisplatin were obtained fromSigma.
Cell culture and transfectionHeLa-S3 and Caco-2 were obtained from the American
Type Culture Collection. CGL4 was previously estab-lished as a tumorigenic derivative of the parental tumor-suppressed cell hybrid ESH5 from HeLa D98/AH2 cellsand normal human fibroblasts (19-21, 35-37). HeLa-S3and CGL4 were cultured in DMEM (Invitrogen) contain-ing 5% fetal bovine serum (MBL), and Caco-2 was cul-tured in DMEM containing 10% fetal bovine serum,supplemented with penicillin (100 units/mL) and strepto-mycin (100 μg/mL) under humidified 5% CO2/95% airat 37°C, as described previously (21). These cells were freefrom mycoplasmic contamination. One day before thetransfection, the cells were trypsinized and seeded ontosix-well plastic culture plates (Asahi Glass). On the follow-ing day, transfection procedures were done using 6 μL ofLipofectamine 2000 diluted in 250 μL of DMEM (Sigma)and 1 μg of DNA diluted in 250 μL of supplementaryDMEM in the six-well plates. Cells were incubated inthe presence of the lipofectamine/DNA complex for24 hours at 37°C in 5% CO2. At 24 hours posttransfec-tion, cells were used for luciferase assays or immunoblottingas described below.
Reverse transcription-PCRTotal RNA was extracted using TRIzol (Invitrogen).
cDNA was prepared using oligonucleotides (dTs;Invitrogen) and Superscript III (Invitrogen). Reversetranscription-PCR (RT-PCR) analyses of GLUT1, GLUT3,p21, and p53 or β-actin as a control were carried out usingthe following primer pairs: GLUT1, 5′-AAGTCCT-TTGAGATGCTGATCCT-3′ forward and 5′-AA-GATGGCCACGATGCTCAGATA-3′ reverse; GLUT3,5′-AACCAGCTGGGCATCGTTGTTGG-3′ forwardand 5′-GCCACAATAAACCAGGGAATGGG-3′ reverse;p21, 5′-GGATGTCCGTCAGAACCCAT-3′ forwardand 5′-CCAGCACTCTTAGGAACCTCT-3′ reverse;p53, 5′-CCACCATGAGCGCTGCTCA-3′ forwardand 5′-GCAGGGGAGGGAGAGATG-3′ reverse; β-actin,5′-TTAAGGAGAAGCTGTGCTACGTC-3′ forwardand 5′-AGGAGCAATGATCTTGATCTTCA-3′ reverse.β-Actin was used as a loading control.
Quantitative real-time PCR analysisThis was done using a TaqMan Universal Master Mix
II (Applied Biosystems) under the following conditions:15 minutes at 95°C followed by 40 cycles of 95°C for15 seconds and 60°C for 1 minute using a 7500 real-time PCR system (Applied Biosystems). The predesignedprimer and probe sets for human GLUT1, GLUT3, andβ-actin are commercially available (Applied Biosystems;GLUT1, Hs00892681_m1; GLUT3, Hs00359840_m1;β-actin, Hs99999903_m1). Threshold cycle (Ct) valueswere automatically calculated for each replicate and usedto determine the relative expression of the gene of interest
Molecular Cancer Research
. , 2018. © 2010 American Association for Cancer

GLUT3 Mediates DNA Damage-Induced Cell Death
Published OnlineFirst September 24, 2010; DOI: 10.1158/1541-7786.MCR-10-0011
relative to reference genes for both treated and untreatedsamples by the 2−ΔΔCt method.
PlasmidsThe full-length wild-type (wt) human p53was cloned into
a pCDNA3.1+ vector (Invitrogen). A p53 mutant (L14Q/F19S), unable to bind DNA (38), was generated by two-stepmutagenesis (Stratagene) using pCDNA-p53 wt as atemplate. PCR products were sequenced to confirm that se-quences were correct.Mutant p53 cDNAwas subcloned intopCDNA3.1+ to generate pCDNA3-p53 (L14Q/F19S).Full-length human GLUT1 and GLUT3 promoter-reporterconstructs (GLUT1-P-Full and GLUT3-P-Full) were gener-ated by subcloning of the upstream 5′ region of the humanGLUT1 (−3,057 to +1) orGLUT3 (−2,788 to +1) gene intopGL4.10 (Promega) upstream of the firefly luciferase gene.Sequential 5′ deletion mutants of the GLUT1 and GLUT3promoter were generated by the digestion of GLUT1-P-Fullor GLUT3-P-Full with suitable restriction endonucleases,followed by ligation. The control plasmid pGL4.74 (Renillaluciferase reporter) was also obtained from Promega. TheNH2 terminal EGFP-GLUT3 (GFP-G3) cDNA constructwas described previously (39).
ImmunoblottingCells were cultured in DMEM plus 5% (v/v) fetal bovine
serum (MBL) overnight. After transfection or the additionof appropriate inhibitors, the cells were incubated foranother 24 hours. They were then harvested and lysed inlysis buffer [20 mmol/L Tris-HCl (pH 7.5), 150 mmol/LNaCl, 1 mmol/L EDTA, 1.0 mmol/L dithiothreitol, 20mmol/L glycerophosphate, 2 mmol/L Na3VO4, 1%NP40, and 1 mmol/L phenylmethylsulfonyl fluoride].The whole-cell lysate was electrophoresed on a 10%SDS-PAGE gel, transferred to polyvinylidene difluoridemembranes, and immunoblotted with the antibody. β-Actin was used as a loading control. The intensity of bandswas quantified with ImageJ (Wayne Rasband, NIH).
Transcriptional reporter assaysFor theGLUT promoter-luciferase assay, CGL4 cells were
cotransfected with either GLUT promoter-Luc or vectorplasmids (pGL4.10) and pGL4.74 as an internal controlfor the transfection rate. A dual-luciferase assay kit (Prome-ga) was used according to themanufacturer's directions. Theactivity levels were expressed relative to a vector control.
Cell viability analysisCell viability was determined with Cell Counting Kit-8 -
(Dojindo) as described previously (40). Briefly, cells (2.5 × 103
for CGL4 and CGL4/gt3) were plated onto 96-well plates andtreated with the appropriate inhibitor as indicated in figurelegends. After incubation at 37°C in 5% CO2/95% air for 72hours, cell viability was calculated relative to theDMSOcontrol.
Measurements of glucose consumptionCells were seeded in six-well dishes, and the medium
changed after an overnight culture. Cells (∼90% confluent)
www.aacrjournals.org
Researchon February 11mcr.aacrjournals.org Downloaded from
were incubated for 24 hours, and then the culture mediumwas collected for measurements of glucose concentrationsusing a Glucose (GO) Assay kit (Sigma). Glucose con-sumption was calculated from a standard curve.
Statistical analysisThe statistical significance of differences in data was de-
termined using the unpaired Student's t test. A P value of<0.01 or <0.05 was considered statistically significant.
Results
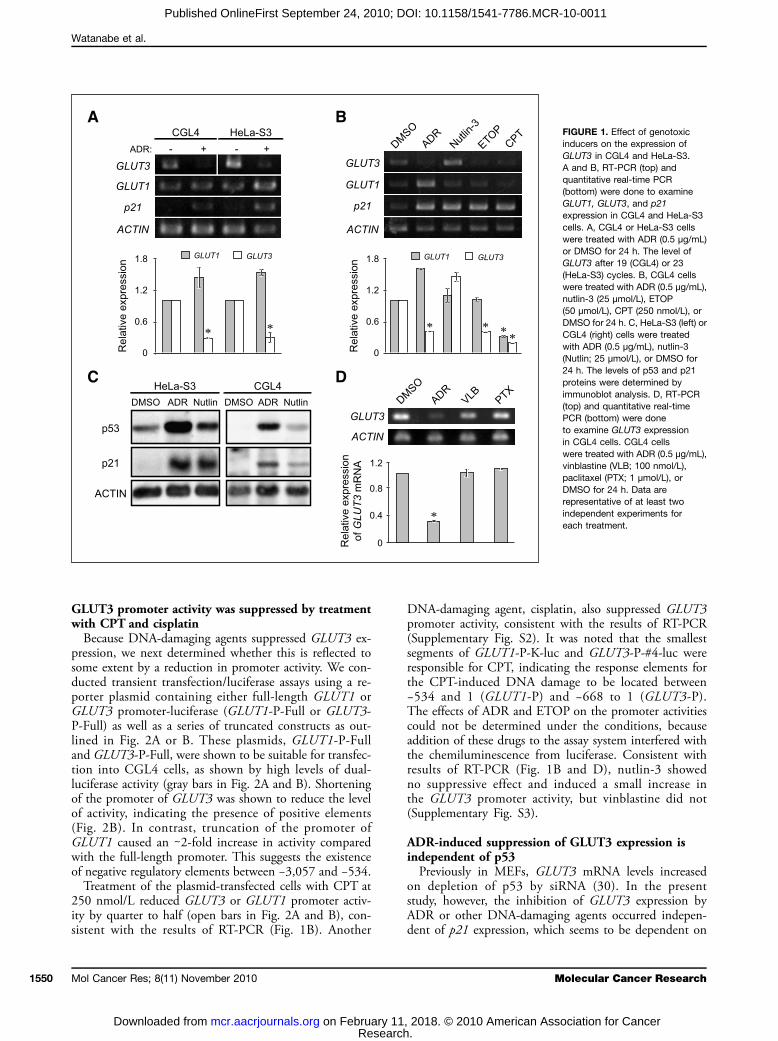
DNA-damaging agents suppress GLUT3 mRNAaccumulation in HeLa cellsPreviously, we have reported that the tumorigenic HeLa
cell hybrid CGL4 expressed GLUT1 and GLUT3, whereasa tumor-suppressed hybrid, CGL1, expressed GLUT1alone (21, 35). The increased glucose uptake by thetumorigenic cells was well associated with the level ofGLUT3 expression (21). To search for drugs to suppressGLUT3 expression in CGL4 cells, we analyzed the effectof ADR in HeLa and CGL4 cells by RT-PCR (Fig. 1A).Inhibition was obtained in a dose-dependent manner, con-sistent with the inhibition of cell growth (SupplementaryFig. S1; Fig. 5D). The accumulation of GLUT3 mRNAwas further quantitated by the real-time PCR methodand was shown to be reduced by ∼30% by treatment withADR (0.5 μg/mL) for 24 hours. In contrast, the expressionof GLUT1 was not suppressed by vehicle (−) or ADR (+)treatment (Fig. 1A), and a small increase in GLUT1 ex-pression with ADR was observed, suggesting the effectsof ADR on GLUTs to be more selective between GLUT3and GLUT1.We also tested other DNA-damaging agents such as
ETOP and CPT. As expected, the expression of p21 wasupregulated after treatment with each of these agents(Fig. 1B). ETOP (50 μmol/L) or CPT (250 nmol/L) likeADR (0.5 μg/mL) reduced the amount of GLUT3 mRNAaccumulated (Fig. 1B). These effects were also dose depen-dent (Supplementary Fig. S1). Although ETOP and ADRdid not affect GLUT1 expression, CPT inhibited it(Fig. 1B). A small increase in GLUT3 expression was ob-served on exposure to nutlin-3 (25 μmol/L), a small mo-lecular antagonist for the p53-binding protein MDM-2,despite that nutlin-3 upregulated p21 expression and accu-mulation of p53 to almost the same extent as the otherDNA-damaging agents (Fig. 1C). These results suggestthat the HeLa and CGL4 cell lines are capable of respond-ing to nutlin-3 to accumulate p53 protein and upregulatep21 expression.The suppressive effect on GLUT3 expression of these
DNA-damaging agents was preserved because it wasnot evident on treatment with non–DNA-damagingand microtubule-attacking agents such as vinblastine(100 nmol/L) or paclitaxel (1 μmol/L; Fig. 1D), whichinhibited cell growth (Supplementary Table S1; datanot shown).
Mol Cancer Res; 8(11) November 2010 1549
. , 2018. © 2010 American Association for Cancer
Watanabe et al.
1550
Published OnlineFirst September 24, 2010; DOI: 10.1158/1541-7786.MCR-10-0011
GLUT3 promoter activity was suppressed by treatmentwith CPT and cisplatinBecause DNA-damaging agents suppressed GLUT3 ex-
pression, we next determined whether this is reflected tosome extent by a reduction in promoter activity. We con-ducted transient transfection/luciferase assays using a re-porter plasmid containing either full-length GLUT1 orGLUT3 promoter-luciferase (GLUT1-P-Full or GLUT3-P-Full) as well as a series of truncated constructs as out-lined in Fig. 2A or B. These plasmids, GLUT1-P-Fulland GLUT3-P-Full, were shown to be suitable for transfec-tion into CGL4 cells, as shown by high levels of dual-luciferase activity (gray bars in Fig. 2A and B). Shorteningof the promoter of GLUT3 was shown to reduce the levelof activity, indicating the presence of positive elements(Fig. 2B). In contrast, truncation of the promoter ofGLUT1 caused an ∼2-fold increase in activity comparedwith the full-length promoter. This suggests the existenceof negative regulatory elements between −3,057 and −534.Treatment of the plasmid-transfected cells with CPT at
250 nmol/L reduced GLUT3 or GLUT1 promoter activ-ity by quarter to half (open bars in Fig. 2A and B), con-sistent with the results of RT-PCR (Fig. 1B). Another
Mol Cancer Res; 8(11) November 2010
Researchon February 11mcr.aacrjournals.org Downloaded from
DNA-damaging agent, cisplatin, also suppressed GLUT3promoter activity, consistent with the results of RT-PCR(Supplementary Fig. S2). It was noted that the smallestsegments of GLUT1-P-K-luc and GLUT3-P-#4-luc wereresponsible for CPT, indicating the response elements forthe CPT-induced DNA damage to be located between−534 and 1 (GLUT1-P) and −668 to 1 (GLUT3-P).The effects of ADR and ETOP on the promoter activitiescould not be determined under the conditions, becauseaddition of these drugs to the assay system interfered withthe chemiluminescence from luciferase. Consistent withresults of RT-PCR (Fig. 1B and D), nutlin-3 showedno suppressive effect and induced a small increase inthe GLUT3 promoter activity, but vinblastine did not(Supplementary Fig. S3).
ADR-induced suppression of GLUT3 expression isindependent of p53Previously in MEFs, GLUT3 mRNA levels increased
on depletion of p53 by siRNA (30). In the presentstudy, however, the inhibition of GLUT3 expression byADR or other DNA-damaging agents occurred indepen-dent of p21 expression, which seems to be dependent on
M
. , 2018. © 2010 American Ass
FIGURE 1. Effect of genotoxicinducers on the expression ofGLUT3 in CGL4 and HeLa-S3.A and B, RT-PCR (top) andquantitative real-time PCR(bottom) were done to examineGLUT1, GLUT3, and p21expression in CGL4 and HeLa-S3cells. A, CGL4 or HeLa-S3 cellswere treated with ADR (0.5 μg/mL)or DMSO for 24 h. The level ofGLUT3 after 19 (CGL4) or 23(HeLa-S3) cycles. B, CGL4 cellswere treated with ADR (0.5 μg/mL),nutlin-3 (25 μmol/L), ETOP(50 μmol/L), CPT (250 nmol/L), orDMSO for 24 h. C, HeLa-S3 (left) orCGL4 (right) cells were treatedwith ADR (0.5 μg/mL), nutlin-3(Nutlin; 25 μmol/L), or DMSO for24 h. The levels of p53 and p21proteins were determined byimmunoblot analysis. D, RT-PCR(top) and quantitative real-timePCR (bottom) were doneto examine GLUT3 expressionin CGL4 cells. CGL4 cellswere treated with ADR (0.5 μg/mL),vinblastine (VLB; 100 nmol/L),paclitaxel (PTX; 1 μmol/L), orDMSO for 24 h. Data arerepresentative of at least twoindependent experiments foreach treatment.
olecular Cancer Research
ociation for Cancer

GLUT3 Mediates DNA Damage-Induced Cell Death
Published OnlineFirst September 24, 2010; DOI: 10.1158/1541-7786.MCR-10-0011
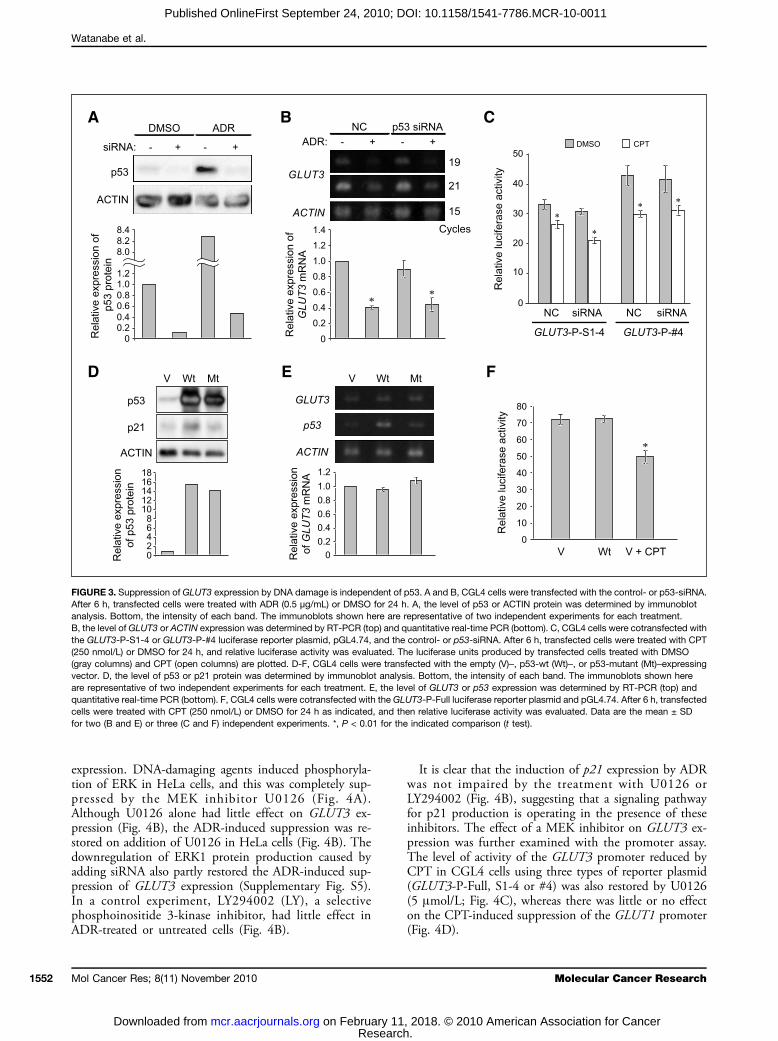
p53 (Fig. 1B and C). We next examined whether or notknockdown of p53 affects the ADR-induced suppressionof GLUT3 expression in CGL4 cells. p53 protein wastemporarily depleted in CGL4 cells by adding siRNA,whose concentration was also enough to lower the levelsof p53 protein raised by ADR treatment (Fig. 3A). Un-der these conditions, the GLUT3 expression in CGL4cells was not significantly affected by p53-siRNA(Fig. 3B). In addition, it was suppressed by ADR inthe p53-depleted CGL4 cells (Fig. 3B). In accordancewith this result, the basal level of activity of the GLUT3promoter was not greatly affected by the depletion andwas suppressed by CPT in the p53-depleted cells (Fig.3C). The results further support the idea that DNA-damaging agents suppress GLUT3 expression indepen-dently of p53.Because it has been reported that p53 downregulates
GLUT1, GLUT3, and GLUT4 expression in SaOS-2,RD, and C2C12 cells and MEFs (30, 41), we next ex-amined the direct effect of p53 on GLUT3 expression inthe CGL4 cell lines. CGL4 cells were transfected witheither the wt-p53 cDNA (Wt) or the p53-inactivated mu-tant cDNA (Mt). A lower level of p53 protein was de-
www.aacrjournals.org
Researchon February 11mcr.aacrjournals.org Downloaded from
tected in the control vector–transfected cells (Fig. 3D).Significantly, higher levels of p53 protein and p21 proteinwere observed in wt-p53 transfected cells, consistent withchanges in the mRNA (Fig. 3D and E). However, nooverproduction of p21 protein or mRNA was obvious inthe cells transfected with mt-p53 (Fig. 3D). Under theseconditions, GLUT3 expression at the levels of mRNA(Fig. 3E) and protein (data not shown) was unaffected.Thus, these results indicate that wt-p53 does not directlycontrol GLUT3 expression in CGL4 cell lines. Transfec-tion of CGL4 cells with wt-p53 did not affect the promot-er activity detected by GLUT3-P-Full, and a reduction dueto CPT was observed (Fig. 3F). In addition, the suppres-sion of GLUT3 expression by treatment with ADR wasobserved in Caco-2 cells (Supplementary Fig. S4), whichis reported as a p53-null colonic carcinoma (42).
MEK inhibitor, U0126, blocks ADR-inducedsuppression of GLUT3 expressionBecause damage to DNA induces activation of ERK
in several cell lines in a p53-independent manner (33),we investigated whether the activation plays a role inthe DNA damage–induced suppression of GLUT3
FIGURE 2. Dissection of human GLUT1 or GLUT3 promoter response elements. Left, a promoter map of the GLUT1 (A) or GLUT3 (B) gene and sequentialdeletions of promoter regions. A, “Full” contains the full-length promoter, whereas “SE” and “K” contain a series of deleted GLUT1 promoters. B, “Full”contains the full-length promoter, whereas “S1-4,” “SX,” and “#4” contain a series of deleted GLUT3 promoters. “Luc” represents the luciferase reportergene. The transcription start site is indicated by +1. The locations of the 5′ ends of the promoters are indicated by the negative numbering of nucleotidesrelative to the start site. The graphs to the left display luciferase activity. Transient transfection and luciferase assays of GLUT promoter reporterplasmids were done. CGL4 cells were cotransfected with the luciferase reporter plasmid and pGL4.74. At 6 h after transfection, cells were treated with CPT(250 nmol/L) or DMSO for 24 h. The luciferase units produced by transfected cells treated with DMSO (gray columns) and CPT (open columns) areplotted. Data are the mean ± SD for three independent experiments. *, P < 0.01 for the indicated comparison (t test).
Mol Cancer Res; 8(11) November 2010 1551
. , 2018. © 2010 American Association for Cancer
Watanabe et al.
1552
Published OnlineFirst September 24, 2010; DOI: 10.1158/1541-7786.MCR-10-0011
expression. DNA-damaging agents induced phosphoryla-tion of ERK in HeLa cells, and this was completely sup-pressed by the MEK inhibitor U0126 (Fig. 4A).Although U0126 alone had little effect on GLUT3 ex-pression (Fig. 4B), the ADR-induced suppression was re-stored on addition of U0126 in HeLa cells (Fig. 4B). Thedownregulation of ERK1 protein production caused byadding siRNA also partly restored the ADR-induced sup-pression of GLUT3 expression (Supplementary Fig. S5).In a control experiment, LY294002 (LY), a selectivephosphoinositide 3-kinase inhibitor, had little effect inADR-treated or untreated cells (Fig. 4B).
Mol Cancer Res; 8(11) November 2010
Researchon February 11mcr.aacrjournals.org Downloaded from
It is clear that the induction of p21 expression by ADRwas not impaired by the treatment with U0126 orLY294002 (Fig. 4B), suggesting that a signaling pathwayfor p21 production is operating in the presence of theseinhibitors. The effect of a MEK inhibitor on GLUT3 ex-pression was further examined with the promoter assay.The level of activity of the GLUT3 promoter reduced byCPT in CGL4 cells using three types of reporter plasmid(GLUT3-P-Full, S1-4 or #4) was also restored by U0126(5 μmol/L; Fig. 4C), whereas there was little or no effecton the CPT-induced suppression of the GLUT1 promoter(Fig. 4D).
FIGURE 3. Suppression of GLUT3 expression by DNA damage is independent of p53. A and B, CGL4 cells were transfected with the control- or p53-siRNA.After 6 h, transfected cells were treated with ADR (0.5 μg/mL) or DMSO for 24 h. A, the level of p53 or ACTIN protein was determined by immunoblotanalysis. Bottom, the intensity of each band. The immunoblots shown here are representative of two independent experiments for each treatment.B, the level of GLUT3 or ACTIN expression was determined by RT-PCR (top) and quantitative real-time PCR (bottom). C, CGL4 cells were cotransfected withthe GLUT3-P-S1-4 or GLUT3-P-#4 luciferase reporter plasmid, pGL4.74, and the control- or p53-siRNA. After 6 h, transfected cells were treated with CPT(250 nmol/L) or DMSO for 24 h, and relative luciferase activity was evaluated. The luciferase units produced by transfected cells treated with DMSO(gray columns) and CPT (open columns) are plotted. D-F, CGL4 cells were transfected with the empty (V)–, p53-wt (Wt)–, or p53-mutant (Mt)–expressingvector. D, the level of p53 or p21 protein was determined by immunoblot analysis. Bottom, the intensity of each band. The immunoblots shown hereare representative of two independent experiments for each treatment. E, the level of GLUT3 or p53 expression was determined by RT-PCR (top) andquantitative real-time PCR (bottom). F, CGL4 cells were cotransfected with the GLUT3-P-Full luciferase reporter plasmid and pGL4.74. After 6 h, transfectedcells were treated with CPT (250 nmol/L) or DMSO for 24 h as indicated, and then relative luciferase activity was evaluated. Data are the mean ± SDfor two (B and E) or three (C and F) independent experiments. *, P < 0.01 for the indicated comparison (t test).
Molecular Cancer Research
. , 2018. © 2010 American Association for Cancer

GLUT3 Mediates DNA Damage-Induced Cell Death
Published OnlineFirst September 24, 2010; DOI: 10.1158/1541-7786.MCR-10-0011
GLUT3 contributes to resistance againstgenotoxic stressBecause a signaling pathway via MEK-ERK is thought
to be involved in ADR-induced cell death, we next as-sessed the role of GLUT3 in cell death induced byDNA-damaging agents in CGL4. We have previouslyconstructed transfectants (CGL4/gt3) that stably over-expressed GLUT3 in CGL4 cells (21). Although GLUT3mRNA levels declined during storage in liquid nitrogen,they remained ∼5-fold higher than those in the parentalCGL4 cells (Fig. 5A and B). In contrast, the expressionof GLUT1 was comparable between these cell lines(Fig. 5B). Greater consumption of glucose was also notedin CGL4/gt3 compared with CGL4 (Fig. 5C). We havecompared the effect of ADR on the growth of these cellsin vitro by treating them with various amounts of ADR for
www.aacrjournals.org
Researchon February 11mcr.aacrjournals.org Downloaded from
3 days. As shown in Fig. 5D, ADR had a dose-dependenteffect on cell survival. The IC50 value of ADR for CGL4cells was 9.8 ± 1.3 ng/mL, but CGL4/gt3 cells weremore resistant with IC50s of 17.9 ± 1.0 ng/mL (Supple-mentary Table S1). Similar drug resistance amongGLUT3-expressing cells was seen for CPT but not vin-blastine (Supplementary Table S1). We also found thatoverexpression of GLUT3 gene in CGL4 cells increasedglucose consumption, rendering the cells more resistantto ADR-induced cell death (Supplementary Fig. S6).To further examine the relationship between drug resis-
tance and GLUT3 expression, we determined howknockdown of the GLUT3 gene affects sensitivity toADR in CGL4 or CGL4/gt3 cells. GLUT3 mRNA wastemporarily depleted in CGL4 and CGL4/gt3 cells byadding siRNA (Fig. 6A). Incorporation of GLUT3 siRNA
FIGURE 4. ERK contributed to DNA damage–induced GLUT3 gene suppression. A, HeLa-S3 cells were exposed to ADR (0.5 μg/mL), ETOP (50 μmol/L), orCPT (250 nmol/L) with or without U0126 (5 μmol/L) for 24 h. The proteins were resolved by SDS-PAGE and immunoblotted using indicated antibodies.Bottom, the normalized intensity of phosphorylated ERK1/2. B, HeLa-S3 cells were exposed to ADR (0.5 μg/mL) or ETOP (50 μmol/L) with or withoutLY294002 (5 μmol/L) or U0126 (5 μmol/L) for 24 h as indicated. The level of GLUT3 or p21 expression was determined by RT-PCR (top) and quantitativereal-time PCR (bottom). C and D, CGL4 cells were transfected with the GLUT3 (C) or GLUT1 (D) promoter luciferase reporter plasmid and pGL4.74.After 6 h, transfected cells were treated with CPT (250 nmol/L) or DMSO with or without U0126 (5 μmol/L) for 24 h as indicated, and relative luciferaseactivity was evaluated. The luciferase units produced by transfected cells treated with DMSO (gray columns), CPT (open columns), and CPT with U0126(border columns) are plotted. Data are the mean ± SD for three independent experiments. *, P < 0.01 for the indicated comparison (t test).
Mol Cancer Res; 8(11) November 2010 1553
. , 2018. © 2010 American Association for Cancer

Watanabe et al.
1554
Published OnlineFirst September 24, 2010; DOI: 10.1158/1541-7786.MCR-10-0011
had no significant effect on the viability of CGL4and CGL4/gt3 cells (Fig. 6B and C). In both types ofcell, knockdown of GLUT3 decreased glucose consump-tion and sensitized the cells to ADR-induced death(Fig. 6B-D), indicating that the level of GLUT3 expres-sion is directly linked to cell damage induced by DNA-damaging agents.
Discussion
Different types of tumor cells show an increase in glu-cose uptake, which is often associated with elevated levelsof GLUT family members such as GLUT1 and GLUT3(14-16). This characteristic has been exploited clinicallyfor the detection of tumors by [18F]deoxyglucose positronemission tomography (5, 6). The downregulation of theexpression of genes involved in glucose metabolism, in-cluding genes for GLUTs, may lead to a functional deficitin glucose consumption and cell growth. Hence, variousantitumor drugs induce the death of tumor cells by activat-ing apoptotic signaling pathways (27). It has been recentlyreported that some antitumor agents that damage DNAthrough a variety of targets suppress the expression ofGLUT genes (GLUT1 or GLUT3) and glucose uptake(26). However, the mechanism by which these agents af-fect the gene expression of the GLUT family in human tu-mor cells and its role in cell death are largely unknown.
Mol Cancer Res; 8(11) November 2010
Researchon February 11mcr.aacrjournals.org Downloaded from
In this paper, we report novel findings on the responseto DNA damage in cancer cells. Previous reports showedthat treatment of cells with ADR, ETOP, or cisplatin ledto GLUT3 suppression (26). Our new observations resultfrom detailed characterization of the mechanisms andconsequences of GLUT3 suppression after ADR-inducedDNA damage. We have mapped the activation of ERK toa point upstream of GLUT3 expression in response toDNA damage independent of p53 (Figs. 3 and 4). To-gether with p53, such GLUT3 expression contributedto resistance against DNA damage–induced cell death(Figs. 5 and 6).Because suppression of GLUT3 expression was found
with ADR and ETOP (topoisomerase II inhibitor) as wellas CPT (topoisomerase I inhibitor; Fig. 1B), the damage toDNA by these inhibitors might be involved (26). It hasbeen proved that the molecular responses to these inhibi-tors by topoisomerases I and II are different (43, 44). In-deed, ADR or ETOP was unable to suppress GLUT1expression in HeLa or CGL4 cells, whereas CPT inhibitedboth GLUT1 and GLUT3 expression (Fig. 1B). The mo-lecular basis for the difference in suppression of GLUT1and GLUT3 by these drugs seems to be an important sub-ject for future study.p53 has a role in the regulation of glycolysis through in-
hibition of the expression of the GLUTs directly as well asindirectly (30, 45). In addition, p53 protein is also a majoreffector in the response to DNA damage (27, 28). There-fore, it is essential to investigate any relationship betweenp53 and the inhibition of GLUT3. However, we found thatthe inhibition occurred independently of p53 in HeLa orHeLa cell hybrids in the case of the DNA damage response.This is based on the MDM2 inhibitor, nutlin-3, sustainedp53 protein levels, and activated p21 expression but did notmodulate GLUT3 expression (Fig. 1B; SupplementaryFig. S1); the maximum level of GLUT3 suppression byADR or CPT was not attenuated in p53-depleted cells(Fig. 3A-C), and overexpression of wt-p53 did not preventGLUT3 expression with activation of p21 expression(Fig. 3D-E). These findings strongly suggest that a p53-independent pathway is involved in the GLUT3 gene's sup-pression by DNA-damaging agents in these cell lines.However, the present results are different from a previous
report showing a link between GLUT3 expression and p53.Kawauchi et al. (30) showed that overexpression of wt-p53led to GLUT3 suppression. Although there is a discrepancyamong these results, this seems to be partly due to differentcell types and signaling pathways. Kawauchi et al. studiedMEFs, and we have used human-derived epithelial cancercell lines (30).Although in various types of cells p53 protein plays an
essential role in the response to DNA damage and apo-ptosis caused by DNA-damaging agents (27, 28, 46),p53-deficient cells are not completely unable to undergoapoptosis (47). Severe DNA damage could activate p53-independent apoptosis via backup systems. Because p53 isfrequently mutated in tumors (48), this backup systemwould clearly be important. In this study, we found the
FIGURE 5. DNA damage–induced cell death is suppressed by theexpression of GLUT3. A and B, RT-PCR (A) and quantitative real-timePCR (B) were done to examine GLUT1 and GLUT3 expression in CGL4 orCGL4/gt3 cells. C, glucose consumption in CGL4 or CGL4/gt3 cells.Values were calculated from a standard curve. D, survival of CGL4 orCGL4/gt3 cells treated with various concentrations of ADR. Cells weretreated with ADR or DMSO for 3 d (CGL4 and CGL4/gt3). Cell numberswere counted as described in Materials and Methods. Cell survivalrelative to the DMSO control (100% value) is plotted.
Molecular Cancer Research
. , 2018. © 2010 American Association for Cancer

GLUT3 Mediates DNA Damage-Induced Cell Death
Published OnlineFirst September 24, 2010; DOI: 10.1158/1541-7786.MCR-10-0011
existence of a p53-independent signaling pathway to reg-ulate the expression of GLUT3 in human carcinomaHeLa cells. This novel pathway may be involved in theapoptosis caused by anticancer agents in p53-mutatedtumor cells. Our finding may provide another way tofind anticancer drugs to trigger p53-independent DNAdamage–induced apoptosis.Although this p53-independent pathway remains largely
unknown, we also found that the activation of ERK con-tributes to GLUT3 expression dependent on DNA dam-age, because the MEK inhibitor U0126 or ERK-siRNAprevented the ADR- or CPT-induced suppression ofGLUT3 expression, and this coincided with the effects ofCPT and U0126 on the promoter activity (Fig. 4). Theseresults are in line with the findings that DNA damage–induced apoptosis in NIH3T3 cells leads to activation ofthe MEK-ERK pathway in a p53-independent manner(33). The major cellular response to intense DNA damageis apoptosis (49). DNA damage activates p53, which theninduces apoptosis. Alternatively, DNA damage induced by
www.aacrjournals.org
Researchon February 11mcr.aacrjournals.org Downloaded from
genotoxic stress activates the MEK-ERK signaling pathway(33), resulting in suppression of GLUT3 expression. Theexpression of GLUT3 is involved in resistance to cell deaththrough an unknown mechanism.It should also be noted that a scatter factor, a pleiotrophic
cytokine, accumulates in tumors and prevents ADR-induced cell death through NF-κB activity (50) regulatedby p53 (30). These results may indicate that DNA damageactivates both MEK-ERK and p53-NF-κB pathways lead-ing to apoptosis. We have shown that GLUT3 expressionmakes important contributions to cell survival after DNAdamage. Depletion ofGLUT3 by siRNA enhanced sensitiv-ity to ADR (Fig. 6). Conversely, enforced expressionof GLUT3 increased resistance to ADR in CGL4 cells(Fig. 5; Supplementary Fig. S6). Thus, activation of theMEK-ERK signaling pathway may require suppression ofGLUT3 expression, reducing glycolysis. It would be inter-esting to examine how GLUT3 controls the uptake andconsumption of glucose and survival in tumor cells. In con-nection with this, it should be remembered that GLUT3
FIGURE 6. Effect of depletion ofGLUT3 using siRNA on DNAdamage–induced cell death inCGL4 cells. A, GLUT3 expressionin CGL4 or CGL4/gt3 cellstransfected with control- orGLUT3-siRNA determined byRT-PCR (top) and quantitativereal-time PCR (bottom).B and C, cultures of CGL4 orCGL4/gt3 were transfected withcontrol- or GLUT3-siRNA. Cellnumbers were quantified using ahematometer after treatment withADR (62.5, 125, or 250 ng/mL) for24 h. Cell numbers relative to theDMSO control (100% value)are plotted (left). D, glucoseconsumption in CGL4 or CGL4/gt3cells transfected with control- orGLUT3-siRNA was measured(right). *, P < 0.01; **, P < 0.05, forthe indicated comparison (t test).
Mol Cancer Res; 8(11) November 2010 1555
. , 2018. © 2010 American Association for Cancer

Watanabe et al.
1556
Published OnlineFirst September 24, 2010; DOI: 10.1158/1541-7786.MCR-10-0011
and GLUT1 distribute to different microdomains in theplasma membrane, GLUT3 in a fluid domain and GLUT1in raft-like domains (35, 39).Cancer cells show higher rates of glucose transport and
glycolysis, and recent studies indicate the biological sig-nificance of this adaptation. Vaughn et al. showed thatcancer cells inhibit cytochrome c–mediated apoptosis bya mechanism dependent on glucose metabolism (51).Schafer et al. also showed that detachment of mammaryepithelial cells from the extracellular matrix causes reac-tive oxygen species–mediated apoptosis due to loss of glu-cose transport (52). In addition, in human colon cancercell lines, the upregulation of glucose uptake induced byendogenous KRAS or BRAF mutations was essential tosurvival under cytotoxic conditions (53). These findingsare consistent with our observation that the depletionof GLUT3 expression and glucose uptake cooperate toprotect against genotoxic stress induced by ADR. Theseresults may contribute to new developments for anti-cancer agents based on glucose metabolism.
Mol Cancer Res; 8(11) November 2010
Researchon February 11mcr.aacrjournals.org Downloaded from
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the Screening Committee of Anticancer Drugs (Dr. Takao Yamori,Japanese Foundation for Cancer Research) for providing the SCADS inhibitor kitsupported by grants-in-aid for Scientific Research on the Priority Area “Cancer”from the Ministry of Education, Culture, Sports, Science, and Technology,Japan.
Grant Support
Ministry of Education, Culture, Sports, Science, and Technology of Japan grants-in-aid (H. Naraba and M. Watanabe).
The costs of publication of this article were defrayed in part by the payment ofpage charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received 01/07/2010; revised 09/13/2010; accepted 09/14/2010; publishedOnlineFirst 09/24/2010.
References
1. Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond.Cell 2008;134:703–7.2. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biol-
ogy of cancer: metabolic reprogramming fuels cell growth and pro-liferation. Cell Metab 2008;7:11–20.
3. Vander Heiden MG, Cantley LC, Thompson CB. Understanding theWarburg effect: the metabolic requirements of cell proliferation.Science 2009;324:1029–33.
4. Warburg O. On respiratory impairment in cancer cells. Science 1956;124:269–70.
5. Pauwels EK, Sturm EJ, Bombardieri E, Cleton FJ, Stokkel MP.Positron-emission tomography with [18F]fluorodeoxyglucose: Part I.Biochemical uptake mechanism and its implication for clinical stud-ies. J Cancer Res Clin Oncol 1999;126:549–59.
6. Ak I, Stokkel MP, Pauwels EK. Positron emission tomography with2-[18F]fluoro-2-deoxy-D-glucose in oncology: Part II. The clinicalvalue in detecting and staging primary tumours. J Cancer Res ClinOncol 2000;126:560–74.
7. Zhao FQ, Keating AF. Functional properties and genomics of glu-cose transporters. Curr Genomics 2007;8:113–28.
8. Keller K, Strube M, Mueckler M. Functional expression of the humanHepG2 and rat adipocyte glucose transporters in Xenopus oocytes.Comparison of kinetic parameters. J Biol Chem 1989;264:18884–9.
9. Burant CF, Bell GI. Mammalian facilitative glucose transporters:Evidence for similar substrate recognition sites in functionally mono-meric proteins. Biochemistry 1992;31:10414–20.
10. Boado RJ, Black KL, Pardridge WN. Gene expression of GLUT3 andGLUT1 glucose transporters in human brain tumors. Brain Res MolBrain Res 1994;27:51–7.
11. Younes M, Lechago LV, Somoano JR, Mosharaf M, Lechago J. Wideexpression of the human erythrocyte glucose transporter GLUT1 inhuman cancers. Cancer Res 1996;56:1164–7.
12. Younes M, Lechago LV, Somoano JR, Mosharaf M, Lechago J.Immunohistochemical detection of GLUT3 in human tumors and nor-mal tissues. Anticancer Res 1997;17:2747–50.
13. Younes M, Brown RW, Stephenson M, Gondo M, Cagle PT. Over-expression of GLUT1 and GLUT3 in stage I nonsmall cell lung carci-noma is associated with poor survival. Cancer 1997;80:1046–51.
14. Macheda ML, Rogers S, Best JD. Molecular and cellular regulation ofglucose transporter (GLUT) proteins in cancer. J Cell Physiol 2005;202:654–62.
15. Kim SJ, Lee HW, Kim DC, Rha SH, Hong SH, Jeong JS. Significance
of GLUT1 expression in adenocarcinoma and adenoma of the am-pulla of Vater. Pathol Int 2008;58:233–8.
16. Tsukioka M, Matsumoto Y, Noriyuki M, et al. Expression of glucosetransporters in epithelial ovarian carcinoma: correlation with clinicalcharacteristics and tumor angiogenesis. Oncol Rep 2007;18:361–7.
17. Flier JS, Mueckler MM, Usher P, Lodish HF. Elevated levels ofglucose transport and transporter mRNA are induced by ras or srconcogenes. Science 1987;235:1492–5.
18. Kitagawa T, Tanaka M, Akamatsu Y. Regulation of glucose transportactivity and expression of glucose transporter mRNA by serum,growth factors and phorbol ester in quiescent mouse fibroblasts.Biochim Biophys Acta 1989;980:100–8.
19. Kitagawa T, Tsuruhara Y, Hayashi M, Endo T, Stanbridge EJ.A tumor-associated glycosylation change in the glucose transporterGLUT1 controlled by tumor suppressor function in human cellhybrids. J Cell Sci 1995;108:3735–43.
20. Noto Y, Iwazaki A, Nagao J, et al. Altered N-glycosylation of glucosetransporter-1 associated with radiation-induced tumorigenesis of hu-man cell hybrids. Biochem Biophys Res Commun 1997;240:395–8.
21. Suzuki T, Iwazaki A, Katagiri H, et al. Enhanced expression of glu-cose transporter GLUT3 in tumorigenic HeLa cell hybrids associ-ated with tumor suppressor dysfunction. Eur J Biochem 1999;262:534–40.
22. Rajakumar RA, Thamotharan S, Menon RK, Devaskar SU. Sp1 andSp3 regulate transcriptional activity of the facilitative glucose trans-porter isoform-3 gene in mammalian neuroblasts and trophoblasts.J Biol Chem 1998;273:27474–83.
23. Rajakumar RA, Thamotharan S, Raychaudhuri N, Menon RK,Devaskar SU. Trans-activators regulating neuronal glucose trans-porter isoform-3 gene expression in mammalian neurons. J BiolChem 2004;279:26768–79.
24. Copland JA, Pardini AW, Wood TG, et al. IGF-I controls GLUT3 ex-pression in muscle via the transcriptional factor Sp1. Biochim Bio-phys Acta 2007;1769:631–40.
25. Meneses AM, Medina RA, Kato S, et al. Regulation of GLUT3 andglucose uptake by the cAMP signalling pathway in the breast cancercell line ZR-75. J Cell Physiol 2008;214:110–6.
26. Zhou R, Vander Heiden MG, Rudin CM. Genotoxic exposure is as-sociated with alterations in glucose uptake and metabolism. CancerRes 2002;62:3515–20.
27. Roos WP, Kaina B. DNA damage-induced cell death by apoptosis.Trends Mol Med 2006;12:440–50.
Molecular Cancer Research
. , 2018. © 2010 American Association for Cancer

GLUT3 Mediates DNA Damage-Induced Cell Death
Published OnlineFirst September 24, 2010; DOI: 10.1158/1541-7786.MCR-10-0011
28. Meek DW. Tumour suppression by p53: a role for the DNA damageresponse? Nat Rev Cancer 2009;9:714–23.
29. Ringshausen I, O'Shea CC, Finch AJ, Swigart LB, Evan GI. Mdm2 iscritically and continuously required to suppress lethal p53 activityin vivo. Cancer Cell 2006;10:501–14.
30. Kawauchi K, Araki K, Tobiume K, Tanaka N. p53 regulates glucosemetabolism through an IKK-NF-κB pathway and inhibits cell transfor-mation. Nat Cell Biol 2008;10:611–8.
31. Strasser A, Harris AW, Jacks T, Cory S. DNA damage can induceapoptosis in proliferating lymphoid cells via p53-independent mech-anisms inhibitable by Bcl-2. Cell 1994;79:329–39.
32. Huang L, Sowa Y, Sakai T, Pardee AB. Activation of the p21WAF1/CIP1 promoter independent of p53 by the histone deacetylase inhib-itor suberoylanilide hydroxamic acid (SAHA) through the Sp1 sites.Oncogene 2000;19:5712–9.
33. Tang D, Wu D, Hirao A, et al. ERK activation mediates cell cyclearrest and apoptosis after DNA damage independently of p53. J BiolChem 2002;277:12710–7.
34. Wu CY, Gómez-Curet I, Funanage VL, Scavina M, Wang W.Increased susceptibility of spinal muscular atrophy fibroblasts tocamptothecin is p53-independent. BMC Cell Biol 2009;16:10–40.
35. Sakyo T, Kitagawa T. Differential localization of glucose transporterisoforms in non-polarized cells: distribution of GLUT1 but not GLUT3to detergent-resistant membrane domain. Biochim Biophys Acta2002;1567:165–75.
36. Stanbridge EJ, Flandermeyer RR, Daniels DW, Nelson-Rees WA.Specific chromosome loss associated with the expression of tumor-igenicity in human cell hybrids. Somatic Cell Genet 1981;7:699–712.
37. Stanbridge EJ, Der CJ, Doersen CJ, et al. Human cell hybrids: anal-ysis of transformation and tumorigenicity. Science 1982;215:252–9.
38. Lin J, Chen J, Elenbaas B, Levine AJ. Several hydrophobic aminoacids in the p53 amino-terminal domain are required for transcrip-tional activation, binding to mdm-2 and the adenovirus 5 E1B55-kD protein. Genes Dev 1994;8:1235–46.
39. Sakyo T, Naraba H, Teraoka H, Kitagawa T. The intrinsic structure ofglucose transporter isoforms GLUT1 and GLUT3 regulates their dif-ferential distribution to detergent-resistant membrane domains innonpolarized mammalian cells. FEBS J 2007;274:2843–53.
40. Watanabe M, Fiji HD, Guo L, et al. Inhibitors of protein geranylgera-
www.aacrjournals.org
Researchon February 11mcr.aacrjournals.org Downloaded from
nyltransferase I and Rab geranylgeranyltransferase identified from alibrary of allenoate-derived compounds. J Biol Chem 2008;283:9571–9.
41. Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumorsuppressor p53 down-regulates glucose transporters GLUT1 andGLUT4 gene expression. Cancer Res 2004;64:2627–33.
42. Djelloul S, Forgue-Lafitte ME, Hermelin B, Mareel M, Bruyneel E,Baldi A, Giordano A, Chastre E, Gespach C. Enterocyte differentia-tion is compatible with SV40 large T expression and loss of p53 func-tion in human colonic Caco-2 cells. Status of the pRb1 and pRb2tumor suppressor gene products. FEBS Lett 1997;406:234–42.
43. Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond.Nat Rev Cancer 2006;6:789–802.
44. Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy.Nat Rev Cancer 2009;9:338–50.
45. Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer 2009;9:691–700.
46. Cotter TG. Apoptosis and cancer: the genesis of a research field. NatRev Cancer 2009;9:501–7.
47. Roos WP, Batista LF, Naumann SC, et al. Apoptosis in malignantglioma cells triggered by the temozolomide-induced DNA lesionO6-methylguanine. Oncogene 2006;26:186–97.
48. Hollstein M, Hergenhahn M, Yang Q, Bartsch H, Wang ZQ, Hainaut P.New approaches to understanding p53 gene tumor mutation spectra.Mutat Res 1999;431:199–209.
49. Vousden KH, Prives C. Blinded by the light: the growing complexityof p53. Cell 2009;137:413–31.
50. Fan S, Meng Q, Laterra JJ, Rosen EM. Role of Src signal transduc-tion pathways in scatter factor-mediated cellular protection. J BiolChem 2009;284:7561–77.
51. Vaughn AE, Deshmukh M. Glucose metabolism inhibits apoptosis inneurons and cancer cells by redox inactivation of cytochrome c. NatCell Biol 2008;10:1477–83.
52. Schafer ZT, Grassian AR, Song L, et al. Antioxidant and oncogenerescue of metabolic defects caused by loss of matrix attachment.Nature 2009;461:109–13.
53. Yun J, Rago C, Cheong I, et al. Glucose deprivation contributes tothe development of KRAS pathway mutations in tumor cells. Science2009;325:1555–9.
Mol Cancer Res; 8(11) November 2010 1557
. , 2018. © 2010 American Association for Cancer

2010;8:1547-1557. Published OnlineFirst September 24, 2010.Mol Cancer Res Masaru Watanabe, Hiroaki Naraba, Tomoko Sakyo, et al. Signal-Regulated Kinase Signaling in HeLa CellsMediated through p53-Independent Extracellular
Expression IsGLUT3Induced Modulation of −DNA Damage
Updated version
10.1158/1541-7786.MCR-10-0011doi:
Access the most recent version of this article at:
Cited articles
http://mcr.aacrjournals.org/content/8/11/1547.full#ref-list-1
This article cites 53 articles, 15 of which you can access for free at:
Citing articles
http://mcr.aacrjournals.org/content/8/11/1547.full#related-urls
This article has been cited by 4 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
SubscriptionsReprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://mcr.aacrjournals.org/content/8/11/1547To request permission to re-use all or part of this article, use this link
Research. on February 11, 2018. © 2010 American Association for Cancermcr.aacrjournals.org Downloaded from
Published OnlineFirst September 24, 2010; DOI: 10.1158/1541-7786.MCR-10-0011





























