Embed Size (px)
Citation preview

68 Research Article
IntroductionCellular senescence limits the proliferation (growth) of damagedcells that are at risk for neoplastic transformation by imposing anessentially irreversible growth arrest. Cells senesce in response tomany potentially oncogenic stressors, including dysfunctionaltelomeres, DNA damage, chromatin alterations and strongmitogenic signals such as those delivered by some oncogenes(Ben-Porath and Weinberg, 2004; Campisi and d’Adda di Fagagna,2007). The senescence response depends crucially on the cellulartumor antigen p53 (also known as tumor suppressor TP53) and theretinoblastoma-associated protein (pRb) tumor suppressor pathwaysand is now accepted as a potent cell-autonomous mechanism forsuppressing the development of cancer (Braig and Schmitt, 2006;Campisi, 2005; Dimri, 2005; Prieur and Peeper, 2008). Accordingly,loss of the senescence response increases the incidence of cancerin humans and mice.
Unlike apoptotic cells, which rapidly disintegrate, senescentcells remain viable in culture for long intervals and are found withincreasing frequency in aged tissues and at sites of age-relatedpathology, including preneoplastic lesions (Collado et al., 2005;Dimri et al., 1995; Erusalimsky and Kurz, 2005; Jeyapalan et al.,
2007; Price et al., 2002). In addition, they develop a senescence-associated secretory phenotype (SASP) with potent autocrine andparacrine activities. The SASP includes numerous cytokines, growthfactors and proteases, and develops several days after cells receivea senescence stimulus and cease growth (Coppe et al., 2010; Coppeet al., 2008; Rodier et al., 2009). Some SASP components reinforcethe growth arrest (Acosta et al., 2008; Kuilman et al., 2008;Wajapeyee et al., 2008). Others disrupt epithelial differentiation(Parrinello et al., 2005) or promote cancer cell growth and invasionin culture and in vivo (Bavik et al., 2006; Coppe et al., 2008;Krtolica et al., 2001; Liu and Hornsby, 2007). Because senescentcells can strongly influence nearby cells, it is important tounderstand how the SASP develops.
Several signaling cascades are associated with the establishmentand maintenance of senescence-associated phenotypes, includinggrowth arrest and SASP (Campisi and d’Adda di Fagagna, 2007;Kuilman and Peeper, 2009). Many senescence-inducing stimuligenerate a persistent DNA damage response (DDR), normallyassociated with DNA double-strand breaks (DSBs) (d’Adda diFagagna, 2008). Recent findings show that DDR signaling isessential for establishing and maintaining senescent phenotypes.
Accepted 23 August 2010Journal of Cell Science 124, 68-81 © 2011. Published by The Company of Biologists Ltddoi:10.1242/jcs.071340
SummaryDNA damage can induce a tumor suppressive response termed cellular senescence. Damaged senescent cells permanently arrestgrowth, secrete inflammatory cytokines and other proteins and harbor persistent nuclear foci that contain DNA damage response(DDR) proteins. To understand how persistent damage foci differ from transient foci that mark repairable DNA lesions, we identifysequential events that differentiate transient foci from persistent foci, which we term ‘DNA segments with chromatin alterationsreinforcing senescence’ (DNA-SCARS). Unlike transient foci, DNA-SCARS associate with PML nuclear bodies, lack the DNA repairproteins RPA and RAD51, lack single-stranded DNA and DNA synthesis and accumulate activated forms of the DDR mediators CHK2and p53. DNA-SCARS form independently of p53, pRB and several other checkpoint and repair proteins but require p53 and pRb totrigger the senescence growth arrest. Importantly, depletion of the DNA-SCARS-stabilizing component histone H2AX did not deplete53BP1 from DNA-SCARS but diminished the presence of MDC1 and activated CHK2. Furthermore, depletion of H2AX reduced boththe p53-dependent senescence growth arrest and p53-independent cytokine secretion. DNA-SCARS were also observed followingsevere damage to multiple human cell types and mouse tissues, suggesting that they can be used in combination with other markersto identify senescent cells. Thus, DNA-SCARS are dynamically formed distinct structures that functionally regulate multiple aspectsof the senescent phenotype.
Key words: Aging, Cancer, Cellular senescence, DNA repair, Homologous recombination, Interleukin 6 (IL6), Promyelocytic leukemia protein (PML)
DNA-SCARS: distinct nuclear structures that sustaindamage-induced senescence growth arrest andinflammatory cytokine secretionFrancis Rodier1,2,*, Denise P. Muñoz1,2, Robert Teachenor1,‡, Victoria Chu1, Oanh Le3, Dipa Bhaumik2,Jean-Philippe Coppé1,2, Eric Campeau4, Christian M. Beauséjour3, Sahn-Ho Kim1,§, Albert R. Davalos1,2 andJudith Campisi1,2,¶
1Lawrence Berkeley National Laboratory, One Cyclotron Road, Berkeley, CA 94720, USA2The Buck Institute for Age Research, 8001 Redwood Boulevard, Novato, CA 94945, USA3CHU Ste-Justine, Département de Pharmacologie, Université de Montréal, 3175 Cote Ste-Catherine, Montreal, QC H3T 1C5, Canada4Program in Gene Function and Expression, University of Massachusetts Medical School, 364 Plantation Street, Worcester, MA 01605, USA*Present address: CRCHUM/Institut du cancer de Montréal, Department of Radiology, Radio-Oncology and Nuclear Medicine, Université de Montréal, Montreal, QC H2L 4M1, Canada‡Present address: University of California San Diego, Division of Biological Sciences, 9500 Gilman Drive, La Jolla, CA 92037, USA§Present address: Henry Ford Health System, Department of Urology, One Ford Place, Detroit, MI 48202, USA¶Author for correspondence ([email protected])
Jour
nal o
f Cel
l Sci
ence

Thus, loss of DDR checkpoint kinases such as ATM or theserine/threonine-protein kinase CHK2, which phosphorylate andactivate p53, not only prevents the p53-dependent senescencegrowth arrest (Bartkova et al., 2006; Beausejour et al., 2003; DiMicco et al., 2006; Gire et al., 2004; Herbig et al., 2004) but alsoprevents the p53-independent inflammatory cytokine secretion thatcomprises the SASP (Rodier et al., 2009).
DDR signaling is initiated at DSBs by sensor proteins such asthe phosphoinositide 3-kinase-like kinases (PIKKs) ATM and ATR,and amplified by the MRN (MRE11–RAD50–NBS1) complex.These proteins help recruit and further activate PIKKs, andparticipate in DNA repair. PIKKs promote local chromatinremodeling, which spreads for megabases surrounding the DSBand facilitates repair. PIKKs also transduce the DDR signal todownstream mediators, such as CHK2 and p53, which integratethe signal with cellular physiology and coordinate DNA repairwith cell cycle checkpoints (Bartek and Lukas, 2007; Berkovich etal., 2007; Rodier et al., 2007). Many of these DDR signaling andrepair proteins assemble rapidly (within minutes) around DSBsand can be detected in the nuclei of fixed or living cells as focalaggregates termed DNA damage foci. Two components are typicallyused to detect these foci by fluorescence microscopy: the PIKK-phosphorylated form of the histone variant H2A.x (H2AX), andthe adaptor protein tumor suppressor p53-binding protein 1 (53BP1)(Celeste et al., 2003; Huyen et al., 2004; Lobrich et al., 2010;Meier et al., 2007; Rogakou et al., 1999).
When DNA lesions are repairable, DNA damage foci aretransient. They typically resolve within 24 hours, during whichtime cells transiently arrest growth, presumably to allow time forrepair. However, severe or irreparable DNA damage, such ascomplex breaks or uncapped telomeres, causes many cells tosenesce with persistent DNA damage foci, constitutive DDRsignaling and chronic p53 activation. These persistent changesprecede establishment of senescence-associated phenotypes,including growth arrest (Beausejour et al., 2003; d’Adda di Fagagnaet al., 2003; Herbig et al., 2004) and SASP (Coppe et al., 2008;Rodier et al., 2009).
Much is known about the hierarchy and temporal recruitmentof DDR signaling and repair proteins that aggregate immediatelyafter DNA damage and disaggregate as transient damage fociresolve (Bekker-Jensen et al., 2006; Lisby et al., 2004; Paull etal., 2000). Many proteins that associate with transient foci alsooccur in the persistent foci that mark senescent cells (d’Adda diFagagna et al., 2003; Herbig et al., 2004; Kim et al., 2004; Takaiet al., 2003). However, DNA damage foci per se might not beaccurate senescence markers. For example, telomere dysfunction-induced foci (TIF) – H2AX or 53BP1 foci localized to telomeres– can identify senescent cells in culture and tissues (Herbig et al.,2006; Herbig et al., 2004; Takai et al., 2003), but TIF can alsooccur in pre-senescent cells (Beliveau et al., 2007; Verdun et al.,2005) and the damage foci in senescent cells are not alwaystelomere associated (Nakamura et al., 2008; Passos et al., 2010;Wang et al., 2009). Furthermore, it is not known whether thepersistent foci associated with senescence contain componentsthat are distinct from those in transient foci. In addition, whilethe persistent foci clearly contain active DDR components, it isnot known whether these structures maintain the DDR activitythat is required for the senescence-associated growth arrest orcytokine secretion.
Here, we investigate the spatiotemporal dynamics of persistentDNA damage foci and the impact these structures have on
69DNA-SCARS sustain senescence phenotypes
establishing and maintaining senescence-associated phenotypes.We use X-irradiation to initiate damage-induced senescencesynchronously in normal human cells, follow DNA damage fociover time and identify characteristics that distinguish transientfrom persistent foci. Because markers of persistent foci are recruitedto locally modified chromatin, we termed these persistent foci‘DNA segments with chromatin alterations reinforcing senescence’or DNA-SCARS. Finally, we show that DNA-SCARS are importantfor maintaining the senescence-associated growth arrest andsecretion of IL-6, an important SASP component.
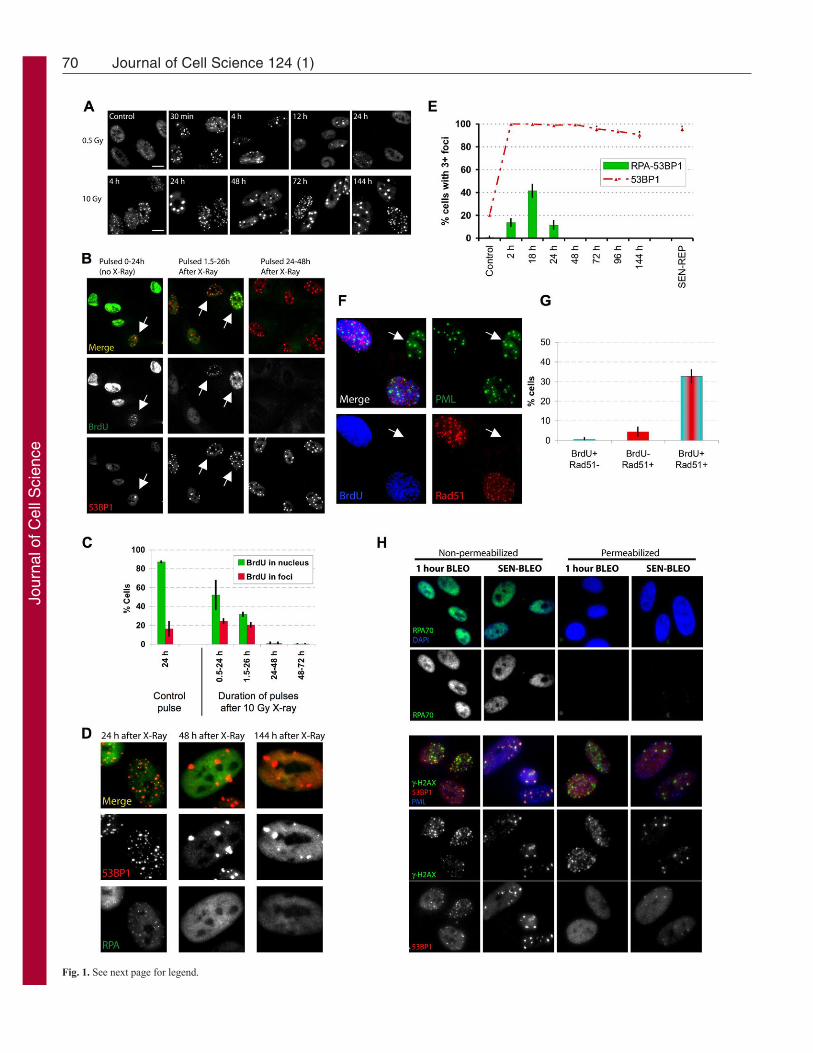
ResultsKinetics of transient versus persistent DNA damage fociTo investigate the kinetics of damage foci formation and resolution,we exposed proliferating normal human fibroblasts (strain HCA2)to 0.5 Gy ionizing radiation (IR; X-ray), which causes a transientgrowth arrest that reverses within 24 hours; alternatively, we used10 Gy, which causes a (permanent) senescence arrest (Rodier et al.,2009). We monitored damage foci by 53BP1 immunofluorescence.As expected, 0.5 Gy rapidly produced many small foci, whichresolved almost completely within 12 hours; only a few residualfoci remained 24 hours later (Fig. 1A; supplementary material Fig.S1A). 10 Gy also rapidly produced many small foci, but asubfraction failed to resolve, became enlarged and persisted fordays (Fig. 1A,E) and months (data not shown). These IR-inducedpersistent DNA damage foci contained many proteins that are alsopresent in transient foci or foci that mark dysfunctional telomeres(d’Adda di Fagagna et al., 2003; Herbig et al., 2004; Lisby et al.,2004; Meier et al., 2007; Rogakou et al., 1999; Takai et al., 2003).Such proteins included the modified chromatin component H2AX(see Fig. 1H), the repair or adaptor proteins MDC1 (see below),NBS1 and MRE11 (data not shown), the activated DDR signalingprotein ATM-pSer-1981 (data not shown) and proteins containingthe ATM/ATR-pSer/Thr substrate motif (supplementary materialFig. S1B). Virtually all the 10-Gy-irradiated proliferating cellsdeveloped persistent foci, demonstrating that the cell cycle phaseat the time of irradiation did not influence their formation. Asenescence-inducing dose of bleomycin, a radiomimetic used incancer therapy (Regulus et al., 2007), also produced both smalltransient and large persistent foci (Fig. 1H, nonpermeabilizedpanels). Not surprising, the higher IR and bleomycin dosesproduced more small transient foci, and possibly some larger earlyfoci (although difficult to distinguish from coincident foci), thanlower doses, but, in all cases, the foci largely resolved after lowdoses, whereas large foci persisted after the higher doses.
Persistent damage foci lack evidence of the active DNArepair that occurs in transient fociTo determine whether transient and/or persistent foci are sites ofactive DNA synthesis, indicative of DNA replication or repair, weexposed proliferating cells to 10 Gy IR, pulsed them withbromodeoxyuridine (BrdU) for 24 hours at varying times after IRand analyzed single cells.
In unirradiated cells, >80% showed uniform nuclear staining,consistent with passage through S phase. ~15% of unirradiatedcells contained BrdU-positive foci, indicating DNA synthesis andpresumably repair of spontaneous damage at these sites (Fig.1B,C).
As expected (Rodier et al., 2009), 10 Gy IR arrested cell cycleprogression within 24 hours. Thus, between 0.5 and 24 hours afterIR, only ~50% of cells showed uniform BrdU labeling; this fraction
Jour
nal o
f Cel
l Sci
ence
70 Journal of Cell Science 124 (1)
Fig. 1. See next page for legend.
Jour
nal o
f Cel
l Sci
ence

declined to <30% 1.5–26 hours after IR, and <1% 24–48 hoursafter IR (Fig. 1B,C). DNA synthesis was also evident in foci, butonly those that were pulsed early after IR. Thus, 20–25% of cellshad BrdU-positive foci when pulsed 0.5–24 hours or 1.5–26 hoursafter IR. However, 48 hours after IR, few if any cells had BrdU-positive foci, despite numerous persistent foci (Fig. 1B,C). Evenlong BrdU pulses, from 3–8 days, failed to label persistent foci inirradiated senescent cells; the same was true for replicativelysenescent cells, which contain persistent damage foci localized todysfunctional telomeres (TIF) (data not shown). We conclude thatpersistent damage foci are not sites of replicative or repair DNAsynthesis.
Because we irradiated proliferating cells, we hypothesized thatcells irradiated during S phase incorporated BrdU into foci owingto homologous recombination repair (HRR), the preferred mode ofDSB repair during S phase. Indeed, immunostaining revealed thepresence of RPA70, a single-strand DNA (ssDNA) binding andHRR protein, in some 53BP1 foci. A fraction of RPA70 alsoremained nucleoplasmic. However, focal coincidence betweenRPA70 and 53BP1 was limited to 24 hours after IR, with a peak~18 hours after IR (Fig. 1D,E). RPA70 and 53BP1 coincided atfrequencies similar to the number of cells in S phase (compare Fig.1C and 1E), but foci that persisted for >48 hours were devoid ofRPA70. RPA70 was also absent from 53BP1 foci in replicativelysenescent cells (Fig. 1E), suggesting that persistent foci, regardlessof origin, lack ssDNA. We also exposed proliferating cells to 10Gy IR, allowed recovery for 3 hours, pulsed with BrdU for 4hours, then stained for BrdU and RAD51, another ssDNA bindingor HRR protein. Cells with RAD51 foci were almost exclusivelypositive for BrdU (Fig. 1F,G). Thus, in contrast to early transientfoci, the persistent foci that remain 24–48 hours after senescence-inducing damage lack evidence of DNA synthesis, ssDNA andHRR.
71DNA-SCARS sustain senescence phenotypes
All proliferating cells exposed to 10 Gy developed persistentfoci, suggesting that S phase (or repair associated with S phase) isnot required for their formation or maintenance. To test this idea,we arrested cell proliferation using a lentivirus to overexpress thecyclin-dependent kinase inhibitor 2A p16INK4A. As expected, cellsarrested with a G1 DNA content ~24 hours after infection. Fortyeight hours after infection, we irradiated the cells with 10 Gy. Dualimmunostaining showed that the p16INK4A-arrested cells resolvedearly transient foci in a manner similar to that of pre-senescentcells. Thus, at least for cells induced to senesce by p16INK4A, earlyrepair is not strongly affected, although other senescent cells mightrepair DSBs less efficiently (Gorbunova et al., 2007). Importantly,5 days later, most irradiated p16INK4A-positive cells harboredpersistent 53BP1 foci, whereas few unirradiated p16INK4A-positivecells had foci (supplementary material Fig. S1C,D). Becausepersistent foci do not incorporate BrdU or harbor certain repairproteins (RPA70, RAD51) found in early foci, we suggest that theymight not be sites of damaged DNA per se but instead are stablechromatin alterations resulting from damage. Indeed, live-cellimaging of damage foci labeled with MDC1–eGFP showed that asignificant fraction of the persistent foci were stable during at least6 hours (supplementary material Fig. S1E), a period that waslargely sufficient to resolve most of the transient foci induced by0.5 Gy IR (Fig. 1A; supplementary material Fig. S1A). Similarobservations were recently made by others using a 53BP1–eGFPfusion protein (Passos et al., 2010). For this and the reasonsexplained below, we refer to persistent foci as DNA-SCARS.
53BP1 is less soluble in DNA-SCARSTo evaluate how proteins associate with transient foci and DNA-SCARS, we treated cells with bleomycin and either fixed beforepermeabilization to detect both soluble and non-soluble proteins orpermeabilized before fixation to extract soluble proteins and detectonly non-soluble proteins. Pre-permeabilization completelyremoved RPA70 from the nucleus (Fig. 1H, upper panels),indicating that RPA70 is readily soluble, regardless of whether itis nucleoplasmic or in early foci. By contrast, pre-permeabilizationfailed to remove H2AX (lower panels), regardless of whether itwas in early foci or DNA-SCARS (Fig. 1H), as expected for a corehistone tightly integrated into chromatin. 53BP1, however, extracteddifferentially, depending on the type of foci. Pre-permeabilizationremoved 53BP1 from foci that formed early after damage butfailed to remove 53BP1 from DNA-SCARS. This difference inextractability could be due to the greater amount of 53BP1 inDNA-SCARS (Fig. 1H) but nonetheless identifies a notabledifference between early transient foci and DNA-SCARS. Thisfinding supports the idea that early foci differ from DNA-SCARS,one characteristic being decreased 53BP1 solubility.
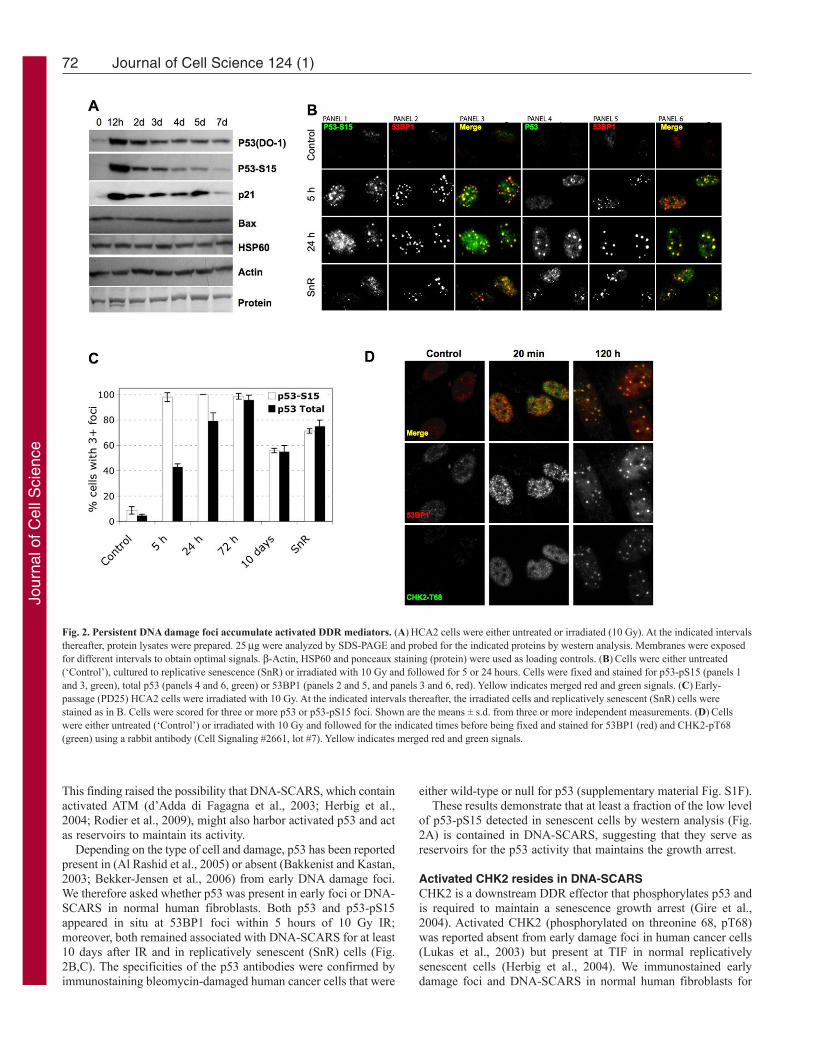
Activated p53 resides in DNA-SCARSIn many human cells, active p53 is required for both establishmentand maintenance of the senescence growth arrest (Beausejour etal., 2003; Gire and Wynford-Thomas, 1998; Rodier et al., 2009),suggesting that there is a reservoir of active p53 in senescent cells.p53 activation occurs through stabilization and posttranslationalmodifications, particularly Ser15 phosphorylation (p53-pS15) byDDR kinases such as ATM (Appella and Anderson, 2001). After10 Gy IR, HCA2 cells showed the expected rapid rise in p53 andp53-pS15 levels, followed by a decline, as detected by westernanalysis (Fig. 2A). However, p53 and p53-pS15 typically declinedto levels slightly above those of pre-senescent controls (Fig. 2A).
Fig. 1. Persistent DNA damage foci lack evidence of ssDNA and DNAsynthesis. (A)HCA2 [population doubling 25 (PD25)] cells were irradiatedwith 0.5 or 10 Gy X-rays and followed for the indicated intervals before beingfixed and stained for 53BP1. Scale bars: 10m. (B)Cells, either untreated orirradiated with 10 Gy, were pulsed with BrdU for the indicated intervals afterirradiation before being fixed and stained for 53BP1 (red) and BrdU (green).Arrows indicate cells with BrdU-positive 53BP1 foci. (C)Cell populations inB were analyzed for the percentage of cells synthesizing DNA (BrdU positivethroughout the nucleus) and the percentage that harbor BrdU-positive 53BP1foci (BrdU in foci). Shown are the means ± s.d. from three or moreindependent measurements. (D)Cells were irradiated with 10 Gy and followedfor the indicated intervals before being fixed and stained for 53BP1(red) andRPA70 (RPA, green). Note the RPA70 foci 24 hours after irradiation. (E)Cellstreated and stained in D were analyzed for the percentage harboring three ormore 53BP1 foci and the percentage with three or more foci positive for bothRPA and 53BP1. Shown are the means ± s.d. from three or more independentmeasurements. (F)Cells were irradiated with 10 Gy. Three hours later, theywere pulsed for 4 hours with BrdU, then fixed and stained for Rad51 (red),PML (green) and BrdU (blue). The arrow indicates a BrdU-Rad51-negativecell. (G)Single cells in the cell populations in F were analyzed simultaneouslyfor the presence of nuclear RAD51 foci and nucleoplasmic BrdU. Shown arethe means ± s.d. from three or more independent measurements. (H)Cellswere given 10g/ml bleomycin (BLEO) for 30 minutes and processed 30minutes later (1 hour BLEO), or induced to senesce by an exposure for 2 hoursto 20g/ml BLEO and processed 12 days later (SEN-BLEO). Cells wereeither fixed (‘Non-permeabilized’) or permeabilized before fixation(‘Permeabilized’), then stained for RPA70 (green) and DNA (DAPI, blue) inthe top panels or 53BP1 (red), -H2AX (green) and PML (blue) in the lowerpanels.
Jour
nal o
f Cel
l Sci
ence
This finding raised the possibility that DNA-SCARS, which containactivated ATM (d’Adda di Fagagna et al., 2003; Herbig et al.,2004; Rodier et al., 2009), might also harbor activated p53 and actas reservoirs to maintain its activity.
Depending on the type of cell and damage, p53 has been reportedpresent in (Al Rashid et al., 2005) or absent (Bakkenist and Kastan,2003; Bekker-Jensen et al., 2006) from early DNA damage foci.We therefore asked whether p53 was present in early foci or DNA-SCARS in normal human fibroblasts. Both p53 and p53-pS15appeared in situ at 53BP1 foci within 5 hours of 10 Gy IR;moreover, both remained associated with DNA-SCARS for at least10 days after IR and in replicatively senescent (SnR) cells (Fig.2B,C). The specificities of the p53 antibodies were confirmed byimmunostaining bleomycin-damaged human cancer cells that were
72 Journal of Cell Science 124 (1)
either wild-type or null for p53 (supplementary material Fig. S1F).These results demonstrate that at least a fraction of the low level
of p53-pS15 detected in senescent cells by western analysis (Fig.2A) is contained in DNA-SCARS, suggesting that they serve asreservoirs for the p53 activity that maintains the growth arrest.
Activated CHK2 resides in DNA-SCARSCHK2 is a downstream DDR effector that phosphorylates p53 andis required to maintain a senescence growth arrest (Gire et al.,2004). Activated CHK2 (phosphorylated on threonine 68, pT68)was reported absent from early damage foci in human cancer cells(Lukas et al., 2003) but present at TIF in normal replicativelysenescent cells (Herbig et al., 2004). We immunostained earlydamage foci and DNA-SCARS in normal human fibroblasts for
Fig. 2. Persistent DNA damage foci accumulate activated DDR mediators. (A)HCA2 cells were either untreated or irradiated (10 Gy). At the indicated intervalsthereafter, protein lysates were prepared. 25g were analyzed by SDS-PAGE and probed for the indicated proteins by western analysis. Membranes were exposedfor different intervals to obtain optimal signals. -Actin, HSP60 and ponceaux staining (protein) were used as loading controls. (B)Cells were either untreated(‘Control’), cultured to replicative senescence (SnR) or irradiated with 10 Gy and followed for 5 or 24 hours. Cells were fixed and stained for p53-pS15 (panels 1and 3, green), total p53 (panels 4 and 6, green) or 53BP1 (panels 2 and 5, and panels 3 and 6, red). Yellow indicates merged red and green signals. (C)Early-passage (PD25) HCA2 cells were irradiated with 10 Gy. At the indicated intervals thereafter, the irradiated cells and replicatively senescent (SnR) cells werestained as in B. Cells were scored for three or more p53 or p53-pS15 foci. Shown are the means ± s.d. from three or more independent measurements. (D)Cellswere either untreated (‘Control’) or irradiated with 10 Gy and followed for the indicated times before being fixed and stained for 53BP1 (red) and CHK2-pT68(green) using a rabbit antibody (Cell Signaling #2661, lot #7). Yellow indicates merged red and green signals.
Jour
nal o
f Cel
l Sci
ence
CHK2-pT68. Minutes after 10 Gy IR, CHK2-pT68 stainingintensity increased, but it did so throughout the nucleus and withoutdiscernible enrichment at foci (Fig. 2D). This nucleoplasmicstaining generally declined within 24 hours, after which focal
73DNA-SCARS sustain senescence phenotypes
staining began to appear. Many hours later, CHK2-pT68 clearlylocalized to DNA-SCARS (Fig. 2D). The specificity of thephospho-CHK2 antibody was confirmed by immunostainingirradiated wild-type and CHK2-null cancer cells (supplementarymaterial Fig. S1G) and using a different antibody against phospho-CHK2 (supplementary material Fig. S1H). We also detected CHK2-pT68 in the DNA-SCARS (TIF) of replicatively senescent cells(data not shown). These results reconcile earlier results (Herbig etal., 2004; Lukas et al., 2003) and suggest that, although activatedCHK2 diffuses through the nucleus immediately after damage, afraction accumulates at DNA-SCARS in senescent cells. They alsosuggest that TIF are selectively localized DNA-SCARS.
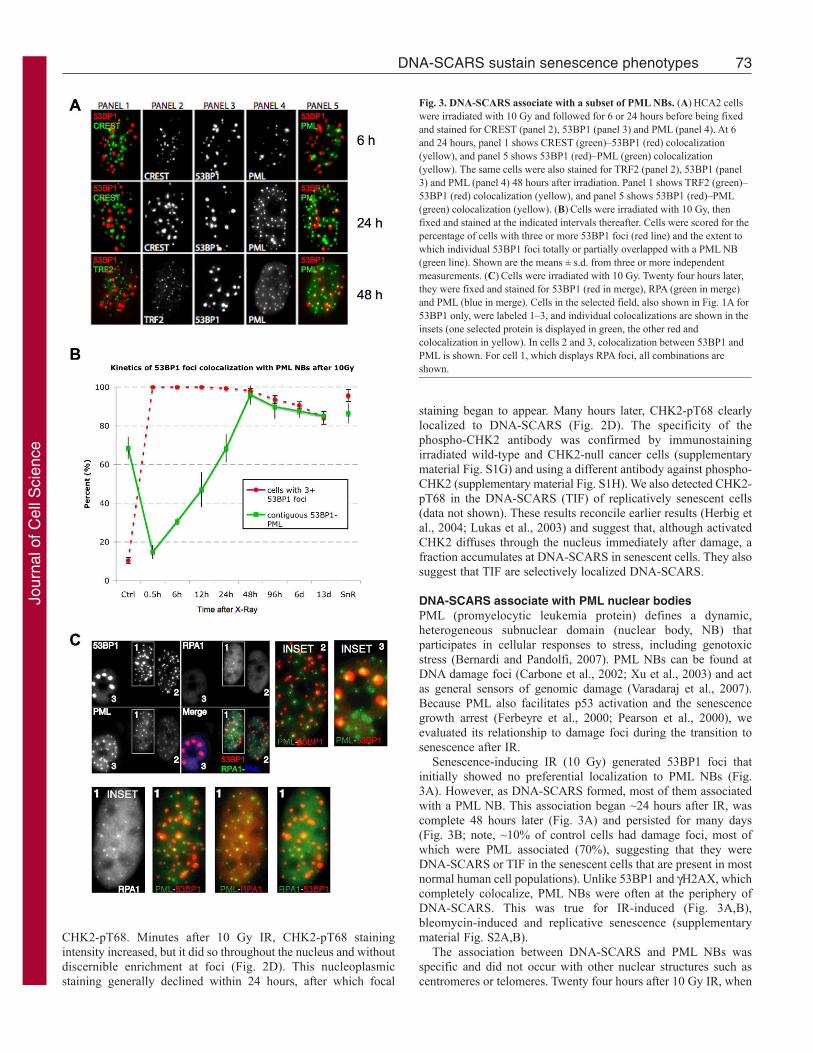
DNA-SCARS associate with PML nuclear bodiesPML (promyelocytic leukemia protein) defines a dynamic,heterogeneous subnuclear domain (nuclear body, NB) thatparticipates in cellular responses to stress, including genotoxicstress (Bernardi and Pandolfi, 2007). PML NBs can be found atDNA damage foci (Carbone et al., 2002; Xu et al., 2003) and actas general sensors of genomic damage (Varadaraj et al., 2007).Because PML also facilitates p53 activation and the senescencegrowth arrest (Ferbeyre et al., 2000; Pearson et al., 2000), weevaluated its relationship to damage foci during the transition tosenescence after IR.
Senescence-inducing IR (10 Gy) generated 53BP1 foci thatinitially showed no preferential localization to PML NBs (Fig.3A). However, as DNA-SCARS formed, most of them associatedwith a PML NB. This association began ~24 hours after IR, wascomplete 48 hours later (Fig. 3A) and persisted for many days(Fig. 3B; note, ~10% of control cells had damage foci, most ofwhich were PML associated (70%), suggesting that they wereDNA-SCARS or TIF in the senescent cells that are present in mostnormal human cell populations). Unlike 53BP1 and H2AX, whichcompletely colocalize, PML NBs were often at the periphery ofDNA-SCARS. This was true for IR-induced (Fig. 3A,B),bleomycin-induced and replicative senescence (supplementarymaterial Fig. S2A,B).
The association between DNA-SCARS and PML NBs wasspecific and did not occur with other nuclear structures such ascentromeres or telomeres. Twenty four hours after 10 Gy IR, when
Fig. 3. DNA-SCARS associate with a subset of PML NBs. (A)HCA2 cellswere irradiated with 10 Gy and followed for 6 or 24 hours before being fixedand stained for CREST (panel 2), 53BP1 (panel 3) and PML (panel 4). At 6and 24 hours, panel 1 shows CREST (green)–53BP1 (red) colocalization(yellow), and panel 5 shows 53BP1 (red)–PML (green) colocalization(yellow). The same cells were also stained for TRF2 (panel 2), 53BP1 (panel3) and PML (panel 4) 48 hours after irradiation. Panel 1 shows TRF2 (green)–53BP1 (red) colocalization (yellow), and panel 5 shows 53BP1 (red)–PML(green) colocalization (yellow). (B)Cells were irradiated with 10 Gy, thenfixed and stained at the indicated intervals thereafter. Cells were scored for thepercentage of cells with three or more 53BP1 foci (red line) and the extent towhich individual 53BP1 foci totally or partially overlapped with a PML NB(green line). Shown are the means ± s.d. from three or more independentmeasurements. (C)Cells were irradiated with 10 Gy. Twenty four hours later,they were fixed and stained for 53BP1 (red in merge), RPA (green in merge)and PML (blue in merge). Cells in the selected field, also shown in Fig. 1A for53BP1 only, were labeled 1–3, and individual colocalizations are shown in theinsets (one selected protein is displayed in green, the other red andcolocalization in yellow). In cells 2 and 3, colocalization between 53BP1 andPML is shown. For cell 1, which displays RPA foci, all combinations areshown.
Jour
nal o
f Cel
l Sci
ence
many 53BP1 foci (DNA-SCARS) associated with PML NBs, theywere largely excluded from centromeres, as determined by stainingfor CREST (Fig. 3A), and telomeres, as determined by staining forTRF2 (Fig. 3A). Because PML NB association began ~24 hoursafter IR, we asked whether RPA70, which showed a peak ofassociation with damage foci ~18 hours after IR, was excludedfrom PML-associated foci. Triple immunofluorescence showedthat, 24 hours after IR, before RPA70 foci completely declined, itwas possible to detect foci containing RPA70, 53BP1 and PML(Fig. 3C). Thus, ssDNA-binding proteins do not precludeassociation of DNA-SCARS with PML NBs.
74 Journal of Cell Science 124 (1)
DNA-SCARS do not depend on several DNA repair orsignaling proteins or PML organizationThe formation and resolution of damage foci are linked to damagesignaling and repair pathways. We therefore asked whetherimportant components of these pathways altered DNA-SCARformation, as determined by association with PML NBs.
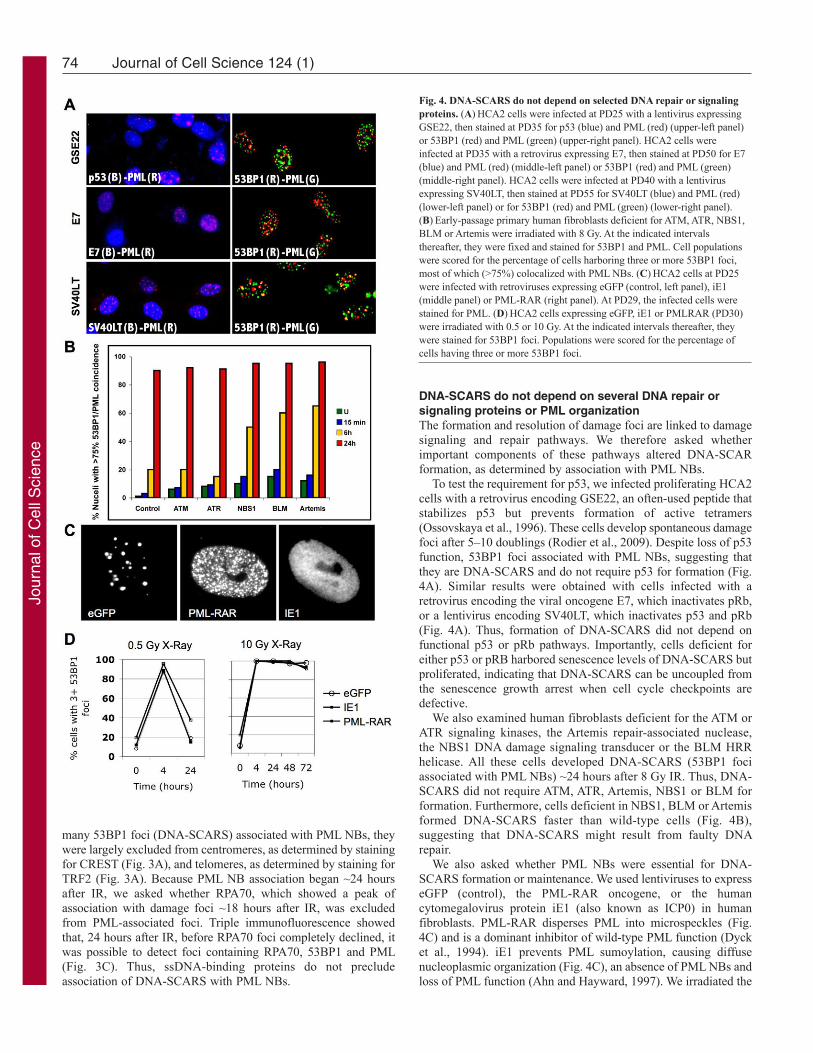
To test the requirement for p53, we infected proliferating HCA2cells with a retrovirus encoding GSE22, an often-used peptide thatstabilizes p53 but prevents formation of active tetramers(Ossovskaya et al., 1996). These cells develop spontaneous damagefoci after 5–10 doublings (Rodier et al., 2009). Despite loss of p53function, 53BP1 foci associated with PML NBs, suggesting thatthey are DNA-SCARS and do not require p53 for formation (Fig.4A). Similar results were obtained with cells infected with aretrovirus encoding the viral oncogene E7, which inactivates pRb,or a lentivirus encoding SV40LT, which inactivates p53 and pRb(Fig. 4A). Thus, formation of DNA-SCARS did not depend onfunctional p53 or pRb pathways. Importantly, cells deficient foreither p53 or pRB harbored senescence levels of DNA-SCARS butproliferated, indicating that DNA-SCARS can be uncoupled fromthe senescence growth arrest when cell cycle checkpoints aredefective.
We also examined human fibroblasts deficient for the ATM orATR signaling kinases, the Artemis repair-associated nuclease,the NBS1 DNA damage signaling transducer or the BLM HRRhelicase. All these cells developed DNA-SCARS (53BP1 fociassociated with PML NBs) ~24 hours after 8 Gy IR. Thus, DNA-SCARS did not require ATM, ATR, Artemis, NBS1 or BLM forformation. Furthermore, cells deficient in NBS1, BLM or Artemisformed DNA-SCARS faster than wild-type cells (Fig. 4B),suggesting that DNA-SCARS might result from faulty DNArepair.
We also asked whether PML NBs were essential for DNA-SCARS formation or maintenance. We used lentiviruses to expresseGFP (control), the PML-RAR oncogene, or the humancytomegalovirus protein iE1 (also known as ICP0) in humanfibroblasts. PML-RAR disperses PML into microspeckles (Fig.4C) and is a dominant inhibitor of wild-type PML function (Dycket al., 1994). iE1 prevents PML sumoylation, causing diffusenucleoplasmic organization (Fig. 4C), an absence of PML NBs andloss of PML function (Ahn and Hayward, 1997). We irradiated the
Fig. 4. DNA-SCARS do not depend on selected DNA repair or signalingproteins. (A)HCA2 cells were infected at PD25 with a lentivirus expressingGSE22, then stained at PD35 for p53 (blue) and PML (red) (upper-left panel)or 53BP1 (red) and PML (green) (upper-right panel). HCA2 cells wereinfected at PD35 with a retrovirus expressing E7, then stained at PD50 for E7(blue) and PML (red) (middle-left panel) or 53BP1 (red) and PML (green)(middle-right panel). HCA2 cells were infected at PD40 with a lentivirusexpressing SV40LT, then stained at PD55 for SV40LT (blue) and PML (red)(lower-left panel) or for 53BP1 (red) and PML (green) (lower-right panel).(B)Early-passage primary human fibroblasts deficient for ATM, ATR, NBS1,BLM or Artemis were irradiated with 8 Gy. At the indicated intervalsthereafter, they were fixed and stained for 53BP1 and PML. Cell populationswere scored for the percentage of cells harboring three or more 53BP1 foci,most of which (>75%) colocalized with PML NBs. (C)HCA2 cells at PD25were infected with retroviruses expressing eGFP (control, left panel), iE1(middle panel) or PML-RAR (right panel). At PD29, the infected cells werestained for PML. (D)HCA2 cells expressing eGFP, iE1 or PMLRAR (PD30)were irradiated with 0.5 or 10 Gy. At the indicated intervals thereafter, theywere stained for 53BP1 foci. Populations were scored for the percentage ofcells having three or more 53BP1 foci.
Jour
nal o
f Cel
l Sci
ence

infected cells and followed the appearance, resolution and retentionof 53BP1 foci over time. The rapid appearance and resolution ofearly 53BP1 foci after 0.5 or 10 Gy IR were unaffected by PML-RAR or iE1 (Fig. 4D). Likewise, the appearance of DNA-SCARSwas unaffected by PML-RAR or iE1 (Fig. 4D). Thus, althoughDNA-SCARS associate with PML-NBs, PML function is notrequired for formation of DNA-SCARS.
DNA-SCARS integrity regulates senescence-associatedphenotypesWe previously showed that persistent damage foci correlate withthe DDR signaling that is essential for maintaining the senescence-associated growth arrest and inflammatory cytokine secretion(Rodier et al., 2009), consistent with our finding here that DNA-SCARS contain active CHK2 and p53 (Fig. 2). Histone H2AX isdispensable for the initial formation of DNA damage foci but isrequired for their stabilization (Celeste et al., 2003). We thereforetested the idea that depletion of H2AX might disrupt DNA-SCARSand hence senescence-associated phenotypes.
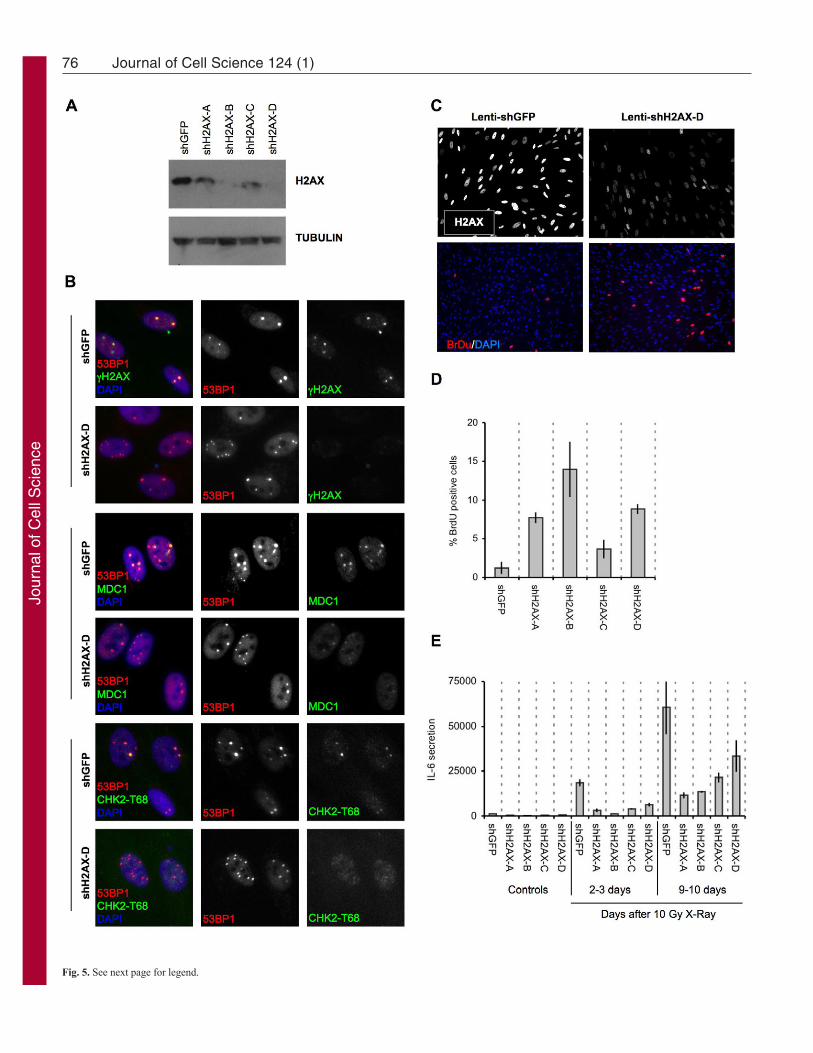
We depleted H2AX by RNA interference (RNAi) usinglentiviruses encoding short-hairpin (sh) RNAs against eGFP(control) or H2AX. Western blotting confirmed the decrease inlevels of H2AX (Fig. 5A). We then followed the formation of53BP1 foci after wild-type and H2AX-deficient cells wereirradiated with 10 Gy. Forty eight hours after IR, 53BP1 efficientlyformed persistent foci in both cell types. However, the DDRadaptor protein MDC1 and CHK2-pT68 were either absent orpresent at much reduced levels in H2AX-depleted 53BP1 foci(Fig. 5B). Thus, H2AX deficiency interfered with the efficientassembly of some DDR proteins into DNA-SCARS.
To determine the functional consequences of H2AX deficiency,we irradiated (10 Gy) control and H2AX-deficient cells. After 7days, we pulsed the cells for 24 hours with BrdU to assess DNAsynthesis. H2AX deficiency increased BrdU incorporation by cellsthat would normally remain growth arrested (Fig. 5C,D). All fourH2AX shRNAs tested gave similar results. We also asked whetherH2AX-deficient cells secreted a SASP cytokine in response tosenescence-inducing DNA damage (Rodier et al., 2009). H2AXdeficiency suppressed IL-6 secretion 2–3 days after IR, whencells just begin secreting, and 9–10 days later, when secretion ismore robust (Fig. 5E). Thus, H2AX depletion reduced certainDDR signaling proteins in DNA-SCARS and suppressed theDDR-dependent senescence-associated growth arrest and IL-6secretion.
DNA-SCARS – multiple senescence inducers, multiple celltypes and formation in vivoWe tested the occurrence of DNA-SCARS, determined byassociation of 53BP1 foci with PML NBs, in HCA2 cells inducedto senesce by means other than IR: expression of an activatedoncogene (RAS) (Serrano et al., 1997), expression of a dominant-negative telomeric protein (DN-TIN2) (Kim et al., 2004) ortreatment with hydrogen peroxide (H2O2). Each manipulationinduced DNA-SCARS within 48 hours (Fig. 6A). Furthermore, weobserved DNA-SCARS in four different human fibroblast strains(WI-38, IMR-90, BJ, HCA2) induced to senesce by replicativeexhaustion or IR (data not shown) and in IR-induced senescenthuman mammary epithelial cells (HMECs), human umbilical veinendothelial cells (HUVECs) and human aortic endothelial cells(HAECs) (Fig. 6B). These findings suggest that DNA-SCARS area general feature of damaged senescent cells.
75DNA-SCARS sustain senescence phenotypes
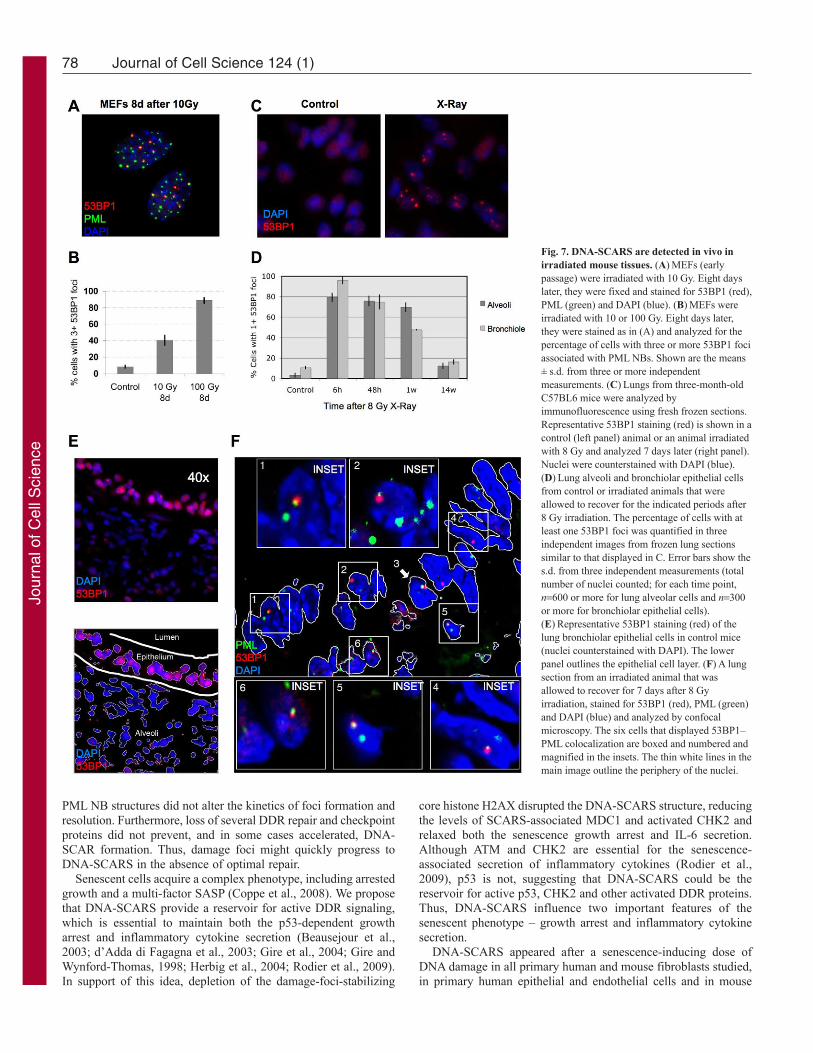
To determine whether DNA-SCARS form in vivo, we examinedmouse cells and tissues. First, we exposed primary mouseembryonic fibroblasts (MEFs) to 10–100 Gy IR and, 8 days later,immunostained for 53BP1 and PML. Coincident staining wasreadily apparent, although higher IR doses were needed relative tothose in human cells (Fig. 7A,B). Thus, DNA-SCARS form inboth human and mouse cells. We then subjected mice to whole-body non-lethal IR (8 Gy) and prepared frozen tissue sections forimmunostaining at varying times thereafter. 53BP1 foci werereadily detected in both the alveoli and bronchioles of lungs 6hours, 48 hours and 7 days after IR (Fig. 7C,D; supplementarymaterial Figs S3, S4). Nearly 100% of cells harbored foci 6 hoursafter IR. Many foci resolved after 48 hours, especially in thebronchioles, but a fraction of cells (45–70%) maintained at leastone 53BP1 focus 1 week after IR (Fig. 7D; see supplementarymaterial Fig. S5 for similar results in liver). After 14 weeks,persistent 53BP1 foci diminished further, but remained two- tofour-fold higher than the low level in matched unirradiated controls(Fig. 7D). This delayed decline might reflect damage repair,clearance of damaged cells by the immune system (Ventura et al.,2007; Xue et al., 2007) and/or cell turnover (Le et al., 2010).
To determine whether the 53BP1 foci that persisted 1 week afterIR were DNA-SCARS, we asked whether they associated withPML NBs. Fig. 7E illustrates how bronchiolar layers were selectedand single cells analyzed for coincidental 53BP1 and PML staining.As shown in the Fig. 7F insets, most bronchiolar epithelial cellnuclei showed an association between 53BP1 and PML NBs (cells1–4). Additionally, non-epithelial cells (underlying the airwayepithelial border) also showed associated 53BP1 foci and PMLNBs (cells 5–6). Similar results were obtained in liver and lungalveolar cells, although only 30–50% of 53BP1 foci associatedwith PML NBs in these compartments (supplementary materialFigs S4, S5). These results suggest that association between PMLNBs and 53BP1 can be used to distinguish early transient DNAdamage foci from DNA-SCARS in vivo.
DiscussionConstitutive DDR signaling is an important component of theintegrated tumor suppressor network that establishes and maintainsthe senescence phenotypes of growth arrest and the SASP (d’Addadi Fagagna et al., 2003; Herbig et al., 2004; Rodier et al., 2009).Here, we show that senescence-associated DNA damage foci, orDNA-SCARS, are relatively stable structures that are distinct fromtransient damage foci and functionally important for both thegrowth arrest and IL-6 secretion, an important SASP feature.
DNA damage foci can be detected by immunofluorescencebecause chromatin modifications generated by the DDR at DSBsspread over megabases (Meier et al., 2007; Rogakou et al., 1999).Whether these foci form early and transiently after DNA damageor are the persistent foci associated with senescence, they reflectelevated concentrations of modified histones (e.g. H2AX) andother proteins that are recruited to remodeled chromatin. Weidentified several molecular events that differentiate transient frompersistent foci, which we followed after X-irradiation to generateDSBs (among other lesions) synchronously.
In transient foci, lesions are likely to be repaired by both non-homologous end joining (NHEJ) and HRR, depending on the cellcycle phase in which the damage was inflicted. For cells damagedin S phase, we detected the ssDNA-binding proteins RAD51 andRPA70, which bind the resected DNA that is generated duringHRR; these cells also incorporated BrdU, indicative of repair.
Jour
nal o
f Cel
l Sci
ence
76 Journal of Cell Science 124 (1)
Fig. 5. See next page for legend.
Jour
nal o
f Cel
l Sci
ence

H2AX can be found at ssDNA, in addition to its predominantassociation with DNA DSBs (Lobrich et al., 2010). Nonetheless,both ssDNA-binding proteins and DNA synthesis were absentfrom persistent foci. Furthermore, individual persistent foci werestable for several hours in living cells, and, in fixed senescent cells,were detected for days and weeks after their formation. Thus, HRRand NHEJ are likely either inactive or incapable of resolving thesefoci after ~48 hours. Formation of DNA-SCARS was acceleratedin cells that are deficient in certain DNA repair proteins (Fig. 4B),supporting the idea that ineffective or defective repair initiates theformation of these structures. We propose that, by ~48 hours aftersenescence-inducing DNA damage, a general repair phase endsand the remaining foci contain either unresolved lesions or stablechromatin modifications resulting from the damage. Alternatively,repair mechanisms that do not require DNA synthesis, HRR orNHEJ could be actively attempting to repair lesions in DNA-SCARS, but these (presumably abortive) repair processes were notdetected in our study. Because these foci persist, and theirdestabilization relaxed the senescence growth arrest and IL-6secretion, we term them ‘DNA segments with chromatin alterationsreinforcing senescence’ or DNA-SCARS.
DNA-SCARS include TIF, which occur at dysfunctionaltelomeres (Herbig et al., 2004; Takai et al., 2003). They alsoinclude foci generated by senescence-inducing DNA damageinflicted by H2O2 or oncogenic RAS. However, as with othersenescence markers, DNA-SCARS are associated with, but notexclusive to, senescent cells. In particular, p53-deficient cellsspontaneously develop DNA-SCARS, yet they proliferate. Suchcells resemble those in pre-cancerous lesions in which DDRsignaling is detected before full transformation (Gorgoulis et al.,2005). Furthermore, PML NBs can harbor damaged telomeres intelomerase-deficient cancer cells (Nabetani et al., 2004). In theseALT (‘alternative lengthening of telomeres’) cells, markers oftelomeres, DSBs and PML colocalize in a structure reminiscent ofDNA-SCARS. These results suggest that, although DNA-SCARSare novel markers of senescence in normal cells, the associationcan be uncoupled in preneoplastic or cancer cells. Thus, DNA-SCARS or TIF can be used as senescence markers, but only whencombined with other markers, such as senescence-associated
77DNA-SCARS sustain senescence phenotypes
beta-galactosidase (SA-Bgal) (Dimri et al., 1995), p16INK4A
(Krishnamurthy et al., 2004) or senescence-associatedheterochromatic foci (Narita et al., 2003). Notably, only the growtharrest, not inflammatory cytokine secretion, was uncoupled fromDNA-SCARS in cells expressing viral oncogenes that inactivatethe p53 and/or pRB tumor-suppressor pathways. Therefore, in thecontext of cancer, DNA-SCARS could mark locally increasedinflammatory cytokine secretion, which is associated with persistentDDR signaling (Rodier et al., 2009).
The association between DNA-SCARS and PML NBs, wheremany repair proteins localize, might occur to allow furtherprocessing of lesions. In addition, persistent foci might associatewith PML NBs, where many chromatin-modifying proteins alsolocalize, to promote senescence-associated gene expression changessuch as the SASP. Alternatively, the presence of repair proteins inPML NBs could simply reflect their normal localization when theimmediate repair phase ends. In support of this idea, disruption of
Fig. 6. DNA-SCARS are caused by multiple senescence inducers and inseveral cell types. (A)HCA2 cells were stained for 53BP1 foci (panel 1) andPML NBs (panel 2). Colocalization is shown in yellow (panel 3). Untreatedcells were fixed at PD23 (‘Control’), and PD25 cells were infected with alentivirus encoding either oncogenic Rasv12 (Ras) or a dominant-negativeTIN2 protein (DN-Tin2). After a recovery period of 3–4 days, the cells werefixed and stained. Cells were also treated for 1 hour with 100M H2O2
(H2O2); 5 days later, they were fixed and stained. (B)Primary HMECs,HUVECs and HAECs were fixed and stained as in A 8 or 10 days, asindicated, after receiving 10 Gy irradiation.
Fig. 5. Histone H2AX is required for senescence-associated phenotypes.(A)HCA2 cells were infected with lentiviruses expressing short hairpin RNAsdirected against eGFP (control, shGFP) or H2AX (shH2AX-A to shH2AX-D).Forty eight hours later, cells were selected for 4 days and allowed to recoverfor 3 days. Whole-cell lysates were analyzed by western blotting for theindicated proteins. (B)HCA2 cells were infected with lentiviruses expressingshGFP (control) or shH2AX-A–shH2AX-D, as in A. Infected cells wereirradiated with 8 Gy. Two days later, they were stained for the indicated DDRproteins. (C)HCA2 cells were infected with lentiviruses expressing shGFP orshH2AX-A–shH2AX-D. Infected cells were irradiated with 10 Gy. 7 dayslater, BrdU was added for 24 hours. Top panels: cells were fixed and stainedfor H2AX (grayscale). Lower panels: cells were fixed and stained for DNAsynthesis (BrdU incorporation, red) and DNA (DAPI, blue). (D)Cells in Awere treated as in B and analyzed for DNA synthesis (BrdU positive). Shownare the means ± s.d. from three or more independent measurements. (E)HCA2cells were infected as in A and either untreated or irradiated with 10 Gy. After2 days, conditioned media were collected over an interval of 24 hours fromuntreated cells (‘Controls’) and irradiated cells 2 days after irradiation (2–3days) and 9 days after irradiation (9–10 days). Conditioned media wereassessed for IL-6 by ELISA. IL-6 secretion is reported as 10–6 pg per cell perday. Shown are the means ± s.d. from three or more independentmeasurements.
Jour
nal o
f Cel
l Sci
ence
PML NB structures did not alter the kinetics of foci formation andresolution. Furthermore, loss of several DDR repair and checkpointproteins did not prevent, and in some cases accelerated, DNA-SCAR formation. Thus, damage foci might quickly progress toDNA-SCARS in the absence of optimal repair.
Senescent cells acquire a complex phenotype, including arrestedgrowth and a multi-factor SASP (Coppe et al., 2008). We proposethat DNA-SCARS provide a reservoir for active DDR signaling,which is essential to maintain both the p53-dependent growtharrest and inflammatory cytokine secretion (Beausejour et al.,2003; d’Adda di Fagagna et al., 2003; Gire et al., 2004; Gire andWynford-Thomas, 1998; Herbig et al., 2004; Rodier et al., 2009).In support of this idea, depletion of the damage-foci-stabilizing
78 Journal of Cell Science 124 (1)
core histone H2AX disrupted the DNA-SCARS structure, reducingthe levels of SCARS-associated MDC1 and activated CHK2 andrelaxed both the senescence growth arrest and IL-6 secretion.Although ATM and CHK2 are essential for the senescence-associated secretion of inflammatory cytokines (Rodier et al.,2009), p53 is not, suggesting that DNA-SCARS could be thereservoir for active p53, CHK2 and other activated DDR proteins.Thus, DNA-SCARS influence two important features of thesenescent phenotype – growth arrest and inflammatory cytokinesecretion.
DNA-SCARS appeared after a senescence-inducing dose ofDNA damage in all primary human and mouse fibroblasts studied,in primary human epithelial and endothelial cells and in mouse
Fig. 7. DNA-SCARS are detected in vivo inirradiated mouse tissues. (A)MEFs (earlypassage) were irradiated with 10 Gy. Eight dayslater, they were fixed and stained for 53BP1 (red),PML (green) and DAPI (blue). (B)MEFs wereirradiated with 10 or 100 Gy. Eight days later,they were stained as in (A) and analyzed for thepercentage of cells with three or more 53BP1 fociassociated with PML NBs. Shown are the means± s.d. from three or more independentmeasurements. (C)Lungs from three-month-oldC57BL6 mice were analyzed byimmunofluorescence using fresh frozen sections.Representative 53BP1 staining (red) is shown in acontrol (left panel) animal or an animal irradiatedwith 8 Gy and analyzed 7 days later (right panel).Nuclei were counterstained with DAPI (blue).(D)Lung alveoli and bronchiolar epithelial cellsfrom control or irradiated animals that wereallowed to recover for the indicated periods after8 Gy irradiation. The percentage of cells with atleast one 53BP1 foci was quantified in threeindependent images from frozen lung sectionssimilar to that displayed in C. Error bars show thes.d. from three independent measurements (totalnumber of nuclei counted; for each time point,n600 or more for lung alveolar cells and n300or more for bronchiolar epithelial cells).(E)Representative 53BP1 staining (red) of thelung bronchiolar epithelial cells in control mice(nuclei counterstained with DAPI). The lowerpanel outlines the epithelial cell layer. (F)A lungsection from an irradiated animal that wasallowed to recover for 7 days after 8 Gyirradiation, stained for 53BP1 (red), PML (green)and DAPI (blue) and analyzed by confocalmicroscopy. The six cells that displayed 53BP1–PML colocalization are boxed and numbered andmagnified in the insets. The thin white lines in themain image outline the periphery of the nuclei.
Jour
nal o
f Cel
l Sci
ence

79DNA-SCARS sustain senescence phenotypes
tissues. This finding indicates that formation of DNA-SCARS is arobust phenomenon that is conserved between mouse and human,as are many other features of cellular senescence (Coppe et al.,2010), and that it occurs in culture and in vivo. A deeperunderstanding of how these structures assemble and function willprobably enrich our insights into the mechanisms that link DNAdamage, inflammation, cancer and aging. Furthermore, it mightsuggest novel strategies for ameliorating the effects of DNAdamage, whether encountered during normal aging, environmentalexposure or certain therapies, such as those used to treat manycancers.
Materials and MethodsCellsIn general, cells were cultured at 37°C in 10% CO2, DMEM, 10% fetal bovine serumand 100 U/ml streptomycin–penicillin. Early-passage cells had 24 hours BrdUlabeling indices of >75%, and <10% expressed SA-Bgal (Dimri et al., 1995). Cellpopulations were considered senescent when they had labeling indices of <5% and>75% expressed SA-Bgal. HCA2 cells were from J. Smith (University of Texas, SanAntonio, TX). Artemis-deficient cells were from S. Yannone, ATM-deficient cellsfrom E. Blakeley and human mammary epithelial cells from M. Stampfer, all fromLawrence Berkeley National Laboratory. Fibroblasts deficient in ATR (GM09812),NBS1 (GM07166A) or BLM (GM02085) were from the Coriell Institute. Humanumbilical vein endothelial cells (HUVECs) and aortic endothelial cells (HAECs)were from CloneTics.
Viruses and infectionsViruses were produced as described previously (Beausejour et al., 2003) and titersadjusted when necessary to achieve ~90% infectivity. Retroviruses encoding eGFP(Rodier et al., 2009), GSE22 (Itahana et al., 2002) or E7 (Itahana et al., 2003) havebeen described previously. iE1 and PML-RAR were cloned into the pBABE-puroretroviral backbone. Lentiviruses encoding dominant-negative TIN2 (TIN2-15C)(Kim et al., 2004), eGFP (Rodier et al., 2009), SV40LT (Beausejour et al., 2003) andoncogenic RASV12 (Rodier et al., 2009) have been described previously. The full-length coding sequence for human MDC1 was fused with eGFP (entry vector 770-7) and inserted in the lentiviral backbone 775-1 to generate the pLenti CMV/TOGFP–MDC1 expression vector (779-2) (Campeau et al., 2009). Lentiviruses encodingshRNAs against eGFP and H2AX were purchased from Open Biosystems (shGFP3:#RHS4459; shH2AX-A: #TRCN0000073278; shH2AX-B: #TRCN0000073280;shH2AX-C: #TRCN0000073279; and shH2AX-D: #TRCN0000073281).
Irradiation and bleomycinCells were X-irradiated at rates equal to or above 0.75 Gy/minute using a Pantak X-ray generator (320 kV/10 mA with 0.5 mm copper filtration). Cells were treated withbleomycin, either 10 g/ml for 30 minutes with a recovery period of 30 minutes or20 g/ml for 2 hours with a recovery period of 10–12 days.
Frozen tissue sectionsC57BL6 mice were sacrificed and excised organs were dipped in OCT (Tissue-Tek#4583), flash-frozen in liquid nitrogen and preserved at –80°C. Organs were sectioned(6–10 m thickness) at –20°C and slices were placed on superfrost plus microslides(VWR #48311-703). Slices were air-dried and stored at –80°C.
Pre-permeabilization extractionSoluble proteins were extracted as described previously (He et al., 1990). Briefly,cells in chamber-slides were washed on ice with phosphate-buffered saline (PBS)and soluble proteins extracted by an incubation for 10 minutes with ice-coldpermeabilization buffer containing inhibitors of RNAses (New England Biolabs),phosphatases (Sigma) and proteases (Complete-Mini Roche). Cells were washed incold permeabilization buffer and PBS before being fixed in formalin (roomtemperature) and processed for immunofluorescence (below).
ImmunofluorescenceFrozen tissue-section slides were brought to room temperature for 10 minutes, orcells in four-well chamber-slides were washed in PBS. Frozen sections or cells werefixed in formalin (10%, neutral buffered) for 10 minutes at room temperature,washed in PBS, permeabilized in PBS plus 0.5% Triton X-100 for 10 minutes andwashed again. Slides were blocked for 1 hour in PBS plus 1% BSA and 4% serumcorresponding to the secondary antibody (goat or donkey) and incubated withprimary antibodies diluted in blocking buffer overnight at 4°C. The slides werewashed with PBS and incubated with secondary antibodies for 1 hour at roomtemperature, washed and mounted with slow-fade gold (Molecular Probes). Whennoted, DAPI was added to secondary antibodies.
BrdU stainingCells in chamber slides were pulsed with BrdU before fixation and permeabilization,as described above. After two PBS washes and a wash in enzyme reaction buffer[0.75X EXO III reaction buffer (Promega) plus 10 mM MgCl2], DNA was denaturedby incubation for 30 minutes at 37°C in enzyme reaction buffer plus 5 U/ml DNASEI (Roche) and 200 U/ml EXOIII (Promega). Cells were washed with PBS andprocessed for immunofluorescence.
MicroscopyImages were acquired on an Olympus BX60 fluorescence microscope with thespotfire 3.2.4 software (Diagnostics Instruments) or a Nikon PCM-2000 laserconfocal scanning microscope and prepared for figure creation using Photoshop CS2(Adobe).
Western blotsCells were lysed in 50 mM Tris–HCl pH 6.8, 100 mM DTT, 2% SDS and 10%glycerol. The mixture was sonicated (30 seconds), boiled (10 minutes) and spun.Samples were separated by electrophoresis on 5% or 4–15% gradient Tris–glycineSDS-polyacrylamide gels. Proteins were electroblotted overnight onto PVDFmembranes and probed overnight at 4°C in PBS with 5% milk. Secondary antibodiesconjugated to horseradish peroxidase were used at 1:15,000 (JacksonImmunoResearch). Blots were developed using the supersignal pico reagent(PIERCE).
AntibodiesThe antibodies used were: 53BP1, Bethyl, BL182 or Upstate, BP13 (against human)or Novus, 100-305 (against mouse); H2AX, Upstate, JBW301; PML, Santa CruzBiotechnology, N-19 (against human) or Upstate, 36.1-104 (against mouse); p53,Oncogene Research Products, DO-1; phospho-p53 serine 15, Cell Signaling, #9286;p21, BD Biosciences, 556430; p16INK4A, Neomarkers, JC8; actin, Chemicon,MAB3128; tubulin, Sigma, T5168; phospho-ATM/ATR STK substrates, CellSignaling #2851; TRF2, Imgenex, IMG-124; CREST, Antibodies Incorporated, 15-235-F; phospho-CHK2 threonine 68, Cell Signaling #2661 (lots #1 and #7) or therabbit monoclonal #2197; BrdU; Abcam, ab1893 or Roche, BMC 9318; Rad51,Santa Cruz Biotechnology, H-92; RPA70, Calbiochem, NA13; SV40LT, Santa CruzBiotechnology, Pab 101; E7, Neomarkers, TVG701Y; HSP60, Santa CruzBiotechnology, N-20; MDC1, rabbit polyclonal (Campeau et al., 2009); donkeysecondary antibodies conjugated to Alexa Fluor dyes were from Molecular Probes(Alexa 350, 488 and 594).
ELISAConditioned media (CM) were prepared by washing cells three times in PBS andincubating in serum-free DMEM plus antibiotics for 24 hours. CM were filtered andstored at –80°C. Cell number was determined for every sample. ELISAs wereperformed using kits and procedures from R&D (IL-6 #D06050). The data werenormalized to cell number and reported as 10–6 pg secreted protein per cell per day.
We thank Patrick Kaminker for valuable assistance with the confocalmicroscope, Dolf Beems for comments on mouse histology and PierreDesprez for critically reading the manuscript. This work was supportedby grants from the Canadian Institutes of Health Research MPO79317to C.B., US NIH AG09909 and AG17242 to J.C. and AG025708 to theBuck Institute, and DOE contract AC03-76SF00098. Deposited inPMC for release after 12 months.
Supplementary material available online athttp://jcs.biologists.org/cgi/content/full/124/1/68/DC1
ReferencesAcosta, J. C., O’Loghlen, A., Banito, A., Guijarro, M. V., Augert, A., Raguz, S.,
Furnagalli, M., DaCosta, M., Brown, C., Popov, N. et al. (2008). Chemokine signalingvia the CXCR2 receptor reinforces senescence. Cell 133, 1006-1018.
Ahn, J. H. and Hayward, G. S. (1997). The major immediate-early proteins IE1 and IE2of human cytomegalovirus colocalize with and disrupt PML-associated nuclear bodiesat very early times in infected permissive cells. J. Virol. 71, 4599-4613.
Al Rashid, S. T., Dellaire, G., Cuddihy, A., Jalali, F., Vaid, M., Coackley, C., Folkard,M., Xu, Y., Chen, B. P., Chen, D. J. et al. (2005). Evidence for the direct binding ofphosphorylated p53 to sites of DNA breaks in vivo. Cancer Res. 65, 10810-10821.
Appella, E. and Anderson, C. W. (2001). Post-translational modifications and activationof p53 by genotoxic stresses. Eur. J. Biochem. 268, 2764-2772.
Bakkenist, C. J. and Kastan, M. B. (2003). DNA damage activates ATM throughintermolecular autophosphorylation and dimer dissociation. Nature 421, 499-506.
Bartek, J. and Lukas, J. (2007). DNA damage checkpoints: from initiation to recoveryor adaptation. Curr. Opin. Cell Biol. 19, 238-245.
Bartkova, J., Rezaei, N., Liontos, M., Karakaidos, P., Kletsas, D., Issaeva, N., Vassiliou,L. V., Kolettas, E., Niforou, K., Zoumpourlis, V. C. et al. (2006). Oncogene-inducedsenescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints.Nature 444, 633-637.
Jour
nal o
f Cel
l Sci
ence

80 Journal of Cell Science 124 (1)
Bavik, C., Coleman, I., Dean, J. P., Knudsen, B., Plymate, S. and Nelson, P. S. (2006).The gene expression program of prostate fibroblast senescence modulates neoplasticepithelial cell proliferation through paracrine mechanisms. Cancer Res. 66, 794-802.
Beausejour, C. M., Krtolica, A., Galimi, F., Narita, M., Lowe, S. W., Yaswen, P. andCampisi, J. (2003). Reversal of human cellular senescence: roles of the p53 and p16pathways. EMBO J. 22, 4212-4222.
Bekker-Jensen, S., Lukas, C., Kitagawa, R., Melander, F., Kastan, M. B., Bartek, J.and Lukas, J. (2006). Spatial organization of the mammalian genome surveillancemachinery in response to DNA strand breaks. J. Cell Biol. 173, 195-206.
Beliveau, A., Bassett, E., Lo, A. T., Garbe, J., Rubio, M. A., Bissell, M. J., Campisi, J.and Yaswen, P. (2007). p53-dependent integration of telomere and growth factordeprivation signals. Proc. Natl. Acad. Sci. USA 104, 4431-4436.
Ben-Porath, I. and Weinberg, R. A. (2004). When cells get stressed: an integrative viewof cellular senescence. J. Clin. Invest. 113, 8-13.
Berkovich, E., Monnat, R. J. and Kastan, M. B. (2007). Roles of ATM and NBS1 inchromatin structure modulation and DNA double-strand break repair. Nat. Cell Biol. 9,683-690.
Bernardi, R. and Pandolfi, P. P. (2007). Structure, dynamics and functions ofpromyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 8, 1006-1016.
Braig, M. and Schmitt, C. A. (2006). Oncogene-induced senescence: putting the brakeson tumor development. Cancer Res. 66, 2881-2884.
Campeau, E., Ruhl, V. E., Rodier, F., Smith, C. L., Rahmberg, B. L., Fuss, J. O.,Campisi, J., Yaswen, P., Cooper, P. K. and Kaufman, P. D. (2009). A versatile viralsystem for expression and depletion of proteins in mammalian cells. PLoS ONE 4,e6529.
Campisi, J. (2005). Suppressing cancer: The importance of being senescent. Science 309,886-887.
Campisi, J. and d’Adda di Fagagna, F. (2007). Cellular senescence: when bad thingshappen to good cells. Nat. Rev. Mol. Cell Biol. 8, 729-740.
Carbone, R., Pearson, M., Minucci, S. and Pelicci, P. G. (2002). PML NBs associatewith the hMre11 complex and p53 at sites of irradiation induced DNA damage.Oncogene 21, 1633-1640.
Celeste, A., Fernandez-Capetillo, O., Kruhlak, M. J., Pilch, D. R., Staudt, D. W., Lee,A., Bonner, R. F., Bonner, W. M. and Nussenzweig, A. (2003). Histone H2AXphosphorylation is dispensable for the initial recognition of DNA breaks. Nat. Cell Biol.5, 675-679.
Collado, M., Gil, J., Efeyan, A., Guerra, C., Schuhmacher, A. J., Barradas, M.,Benguria, A., Zaballos, A., Flores, J. M., Barbacid, M. et al. (2005). Tumor biology:senescence in premalignant tumours. Nature 436, 642.
Coppe, J. P., Patil, C. K., Rodier, F., Sun, Y., Munoz, D., Goldstein, J., Nelson, P. S.,Desprez, P. Y. and Campisi, J. (2008). Senescence-associated secretory phenotypesreveal cell non-automous functions of oncogenic RAS and the p53 tumor suppressor.PLoS Biol. 6, 2853-2868.
Coppe, J. P., Patil, C. K., Rodier, F., Krtolica, A., Beausejour, C. M., Parrinello, S.,Hodgson, J. G., Chin, K., Desprez, P. Y. and Campisi, J. (2010). A human-likesenescence-associated secretory phenotype is conserved in mouse cells dependent onphysiological oxygen. PLoS ONE 5, e9188.
d’Adda di Fagagna, F. (2008). Living on a break: cellular senescence as a DNA-damageresponse. Nat. Rev. Cancer 8, 512-522.
d’Adda di Fagagna, F., Reaper, P. M., Clay-Farrace, L., Fiegler, H., Carr, P., VonZglinicki, T., Saretzki, G., Carter, N. P. and Jackson, S. P. (2003). A DNA damagecheckpoint response in telomere-initiated senescence. Nature 426, 194-198.
Di Micco, R., Fumagalli, M., Cicalese, A., Piccinin, S., Gasparini, P., Luise, C.,Schurra, C., Garre, M., Nuciforo, P. G., Bensimon, A. et al. (2006). Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication.Nature 444, 638-642.
Dimri, G. P. (2005). What has senescence got to do with cancer? Cancer Cell 7, 505-512.Dimri, G. P., Lee, X., Basile, G., Acosta, M., Scott, G., Roskelley, C., Medrano, E. E.,
Linskens, M., Rubelj, I., Pereira-Smith, O. M. et al. (1995). A novel biomarkeridentifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad.Sci. USA 92, 9363-9367.
Dyck, J. A., Maul, G. G., Miller, W. H., Chen, J. D., Kakizuka, A. and Evans, R. M.(1994). A novel macromolecular structure is a target of the promyelocyte-retinoic acidreceptor oncoprotein. Cell 76, 333-343.
Erusalimsky, J. D. and Kurz, D. J. (2005). Cellular senescence in vivo: its relevance inageing and cardiovascular disease. Exp. Gerontol. 40, 634-642.
Ferbeyre, G., de Stanchina, E., Querido, E., Baptiste, N., Prives, C. and Lowe, S. W.(2000). PML is induced by oncogenic ras and promotes premature senescence. GenesDev. 14, 2015-2027.
Gire, V. and Wynford-Thomas, D. (1998). Reinitiation of DNA synthesis and celldivision in senescent human fibroblasts by microinjection of anti-p53 antibodies. Mol.Cell. Biol. 18, 1611-1621.
Gire, V., Roux, P., Wynford-Thomas, D., Brondello, J. M. and Dulic, V. (2004). DNAdamage checkpoint kinase Chk2 triggers replicative senescence. EMBO J. 23, 2554-2563.
Gorbunova, V., Seluanov, A., Mao, Z. and Hine, C. (2007). Changes in DNA repairduring aging. Nucleic Acids Res. 35, 7466-7474.
Gorgoulis, V. G., Vassiliou, L. V., Karakaidos, P., Zacharatos, P., Kotsinas, A., Liloglou,T., Venere, M., Ditullio, R. A., Kastrinakis, N. G., Levy, B. et al. (2005). Activationof the DNA damage checkpoint and genomic instability in human precancerous lesions.Nature 434, 907-913.
He, D. C., Nickerson, J. A. and Penman, S. (1990). Core filaments of the nuclear matrix.J. Cell Biol. 110, 569-580.
Herbig, U., Jobling, W. A., Chen, B. P., Chen, D. J. and Sedivy, J. (2004). Telomereshortening triggers senescence of human cells through a pathway involving ATM, p53,and p21(CIP1), but not p16(INK4a). Mol. Cell 14, 501-513.
Herbig, U., Ferreira, M., Condel, L., Carey, D. and Sedivy, J. M. (2006). Cellularsenescence in aging primates. Science 311, 1257.
Huyen, Y., Zgheib, O., Ditullio, R. A., Gorgoulis, V. G., Zacharatos, P., Petty, T. J.,Sheston, E. A., Mellert, H. S., Stavridi, E. S. and Halazonetis, T. D. (2004).Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature432, 406-411.
Itahana, K., Dimri, G. P., Hara, E., Itahana, Y., Zou, Y., Desprez, P. Y. and Campisi,J. (2002). A role for p53 in maintaining and establishing the quiescence growth arrestin human cells. J. Biol. Chem. 277, 18206-18214.
Itahana, K., Zou, Y., Itahana, Y., Martinez, J. L., Beausejour, C., Jacobs, J. J., VanLohuizen, M., Band, V., Campisi, J. and Dimri, G. P. (2003). Control of the replicativelife span of human fibroblasts by p16 and the polycomb protein Bmi-1. Mol. Cell. Biol.23, 389-401.
Jallepalli, P. V., Lengauer, C., Vogelstein, B. and Bunz, F. (2003). The Chk2 tumorsuppressor is not required for p53 responses in human cancer cells. J. Biol. Chem. 278,20475–20479.
Jeyapalan, J. C., Ferreira, M., Sedivy, J. M. and Herbig, U. (2007). Accumulation ofsenescent cells in mitotic tissue of aging primates. Mech. Ageing Dev. 128, 36-44.
Kim, S. H., Beausejour, C., Davalos, A. R., Kaminker, P., Heo, S. J. and Campisi, J.(2004). TIN2 mediates the functions of TRF2 at telomeres. J. Biol. Chem. 279, 43799-43804.
Krishnamurthy, J., Torrice, C., Ramsey, M. R., Kovalev, G. I., Al-Regaiey, K., Su, L.and Sharpless, N. E. (2004). Ink4a/Arf expression is a biomarker of aging. J. Clin.Invest. 114, 1299-1307.
Krtolica, A., Parrinello, S., Lockett, S., Desprez, P. and Campisi, J. (2001). Senescentfibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer andaging. Proc. Natl. Acad. Sci. USA 98, 12072-12077.
Kuilman, T. and Peeper, D. S. (2009). Senescence-messaging secretome: SMS-ingcellular stress. Nat. Rev. Cancer 9, 81-94.
Kuilman, T., Michaloglou, C., Vredeveld, L. C. W., Douma, S., van Doorn, R., Desmet,C. J., Aarden, A. L., Mooi, W. J. and Peeper, D. S. (2008). Oncogene-inducedsenescence relayed by an interleukin-dependent inflammatory network. Cell 133, 1019-1031.
Le O. N., Rodier, F., Fontaine, F., Coppe, J. P., Campisi, J., DeGregori, J., Laverdiere,C., Kokta, V., Haddad, E. and Beausejour, C. M. (2010). Ionizing radiation-inducedlong-term expression of senescence markers in mice is independent of p53 and immunestatus. Aging Cell 9, 398-409.
Lisby, M., Barlow, J. H., Burgess, R. C. and Rothstein, R. (2004). Choreography of theDNA damage response: spatiotemporal relationships among checkpoint and repairproteins. Cell 118, 699-713.
Liu, D. and Hornsby, P. J. (2007). Senescent human fibroblasts increase the early growthof xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 67, 3117-3126.
Lobrich, M., Shibata, A., Beucher, A., Fisher, A., Ensminger, M., Goodarzi, A. A.,Barton, O. and Jeggo, P. A. (2010). gammaH2AX foci analysis for monitoring DNAdouble-strand break repair: strengths, limitations and optimization. Cell Cycle 9, 662-669.
Lukas, C., Falck, J., Bartkova, J., Bartek, J. and Lukas, J. (2003). Distinctspatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage.Nat. Cell Biol. 5, 255-260.
Meier, A., Fiegler, H., Muñoz, P., Ellis, P., Rigler, D., Langford, C., Blasco, M. A.,Carter, N. and Jackson, S. P. (2007). Spreading of mammalian DNA-damage responsefactors studied by ChIP-chip at damaged telomeres. EMBO J. 26, 2707-2718.
Nabetani, A., Yokoyama, O. and Ishikawa, F. (2004). Localization of hRad9, hHus1,hRad1, and hRad17 and caffeine-sensitive DNA replication at the alternative lengtheningof telomeres-associated promyelocytic leukemia body. J. Biol. Chem. 279, 25849-25857.
Nakamura, A. J., Chiang, Y. J., Hathcock, K. S., Horikawa, I., Sedelnikova, O. A.,Hodes, R. J. and Bonner, W. M. (2008). Both telomeric and non-telomeric DNAdamage are determinants of mammalian cellular senescence. Epigenetics Chromatin 1,6.
Narita, M., Nunez, S., Heard, E., Narita, M., Lin, A. W., Hearn, S. A., Spector, D. L.,Hannon, G. J. and Lowe, S. W. (2003). Rb-mediated heterochromatin formation andsilencing of E2F target genes during cellular senescence. Cell 113, 703-716.
Ossovskaya, V. S., Mazo, I. A., Chernov, M. V., Chernova, O. B., Strezoska, Z.,Kondratov, R., Stark, G. R., Chumakov, P. M. and Gudkov, A. V. (1996). Use ofgenetic suppressor elements to dissect distinct biological effects of separate p53 domains.Proc. Natl. Acad. Sci. USA 93, 10309-10314.
Parrinello, S., Coppe, J. P., Krtolica, A. and Campisi, J. (2005). Stromal-epithelialinteractions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation.J. Cell Sci. 118, 485-496.
Passos, J. F., Nelson, G., Wang, C., Richter, T., Simillion, C., Proctor, C. J., Miwa, S.,Olijslagers, S., Hallinan, J., Wipat, A. et al. (2010). Feedback between p21 andreactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 6, e347.
Paull, T. T., Rogakou, E. P., Yamazaki, V., Kirchgessner, C. U., Gellert, M. andBonner, W. M. (2000). A critical role for histone H2AX in recruitment of repair factorsto nuclear foci after DNA damage. Curr. Biol. 10, 886-895.
Pearson, M., Carbone, R., Sebastiani, C., Cioce, M., Fagioli, M., Saito, S., Higashimoto,Y., Appella, E., Minucci, S., Pandolfi, P. P. et al. (2000). PML regulates p53 acetylationand premature senescence induced by oncogenic Ras. Nature 406, 207-210.
Price, J. S., Waters, J., Darrah, C., Pennington, C., Edwards, D. R., Donell, S. T. andClark, I. M. (2002). The role of chondrocyte senescence in osteoarthritis. Aging Cell1, 57-65.
Jour
nal o
f Cel
l Sci
ence

81DNA-SCARS sustain senescence phenotypes
Prieur, A. and Peeper, D. S. (2008). Cellular senescence in vivo: a barrier to tumorigenesis.Curr. Opin. Cell Biol. 20, 150-155.
Regulus, P., Duroux, B., Bayle, P. A., Favier, A., Cadet, J. and Ravanat, J. L. (2007).Oxidation of the sugar moiety of DNA by ionizing radiation or bleomycin could inducethe formation of a cluster DNA lesion. Proc. Natl. Acad. Sci. USA 104, 14032-14037.
Rodier, F., Campisi, J. and Bhaumik, D. (2007). Two faces of p53: aging and tumorsuppression. Nucleic Acids Res. 35, 7475-7584.
Rodier, F., Coppe, J. P., Patil, C. K., Hoeijmakers, W. A. M., Munoz, D., Raza, S. R.,Freund, A., Campeau, E., Davalos, A. R. and Campisi, J. (2009). Persistent DNAdamage signaling triggers senescence-associated inflammatory cytokine secretion. Nat.Cell Biol. 11, 973-979.
Rogakou, E. P., Boon, C., Redon, C. and Bonner, W. M. (1999). Megabase chromatindomains involved in DNA double-strand breaks in vivo. J. Cell Biol. 146, 905-916.
Serrano, M., Lin, A. W., McCurrach, M. E., Beach, D. and Lowe, S. W. (1997).Oncogenic ras provokes premature cell senescence associated with accumulation of p53and p16INK4a. Cell 88, 593-602.
Takai, H., Smogorzewska, A. and de Lange, T. (2003). DNA damage foci at dysfunctionaltelomeres. Curr. Biol. 13, 1549-1556.
Varadaraj, A., Dovey, C. L., Laredj, L., Ferguson, B., Alexander, C. E., Lubben, N.,Wyllie, A. H. and Rich, T. (2007). Evidence for the receipt of DNA damage stimuli byPML nuclear domains. J. Pathol. 211, 471-480.
Ventura, A., Kirsch, D. G., McLaughlin, M. E., Tuveson, D. A., Grimm, J., Lintault,L., Newman, J., Reczek, E. E., Weissleder, R. and Jacks, T. (2007). Restoration ofp53 function leads to tumour regression in vivo. Nature 445, 661-665.
Verdun, R. E., Crabbe, L., Haggblom, C. and Karlseder, J. (2005). Functional humantelomeres are recognized as DNA damage in G2 of the cell cycle. Mol. Cell 20, 551-561.
Wajapeyee, N., Serra, R. W., Zhu, X., Mahalingam, M. and Green, M. R. (2008).Oncogenic BRAF induces senescence and apoptosis through pathways mediated by thesecreted protein IGFBP7. Cell 132, 363-374.
Wang, C., Jurk, D., Maddick, M., Nelson, G., Martin-Ruiz, C. and von Zglinicki, T.(2009). DNA damage response and cellular senescence in tissues of aging mice. AgingCell 8, 311-323.
Wang, Y., Blandino, G. and Givol, D. (1999). Induced p21waf expression in H1299 cellline promotes cell senescence and protects against cytotoxic effect of radiation anddoxorubicin. Oncogene 18, 2643–2649.
Xu, Z. X., Timanova-Atanasova, A., Zhao, R. X. and Chang, K. S. (2003). PMLcolocalizes with and stabilizes the DNA damage response protein TopBP1. Mol. Cell.Biol. 23, 4247-4256.
Xue, W., Zender, L., Miething, C., Dickins, R. A., Hernando, E., Krizhanovsky, V.,Cordon-Cardo, C. and Lowe, S. W. (2007). Senescence and tumour clearance istriggered by p53 restoration in murine liver carcinomas. Nature 445, 656-660.
Jour
nal o
f Cel
l Sci
ence

![Topological Scars - pks.mpg.de · Scar characteristics Non-integrable model [Turner et. al, Nature Physics (2018)] Low (sub-volume law) entanglement entropy states No disorder (distinct](https://img.pdfslide.net/doc/110x75/5e067ba8a705735abc119809/topological-scars-pksmpgde-scar-characteristics-non-integrable-model-turner.jpg)










![[DDD] Microservice scars](https://img.pdfslide.net/doc/110x75/587756241a28ab84388b74a9/ddd-microservice-scars.jpg)
















