Embed Size (px)
Citation preview

Doublecortin-Expressing Cells in theIschemic Penumbra of aSmall-Vessel Stroke
Rui Hua,1 Ron Doucette,2 and Wolfgang Walz1*1Department of Physiology, University of Saskatchewan, Saskatoon, Saskatchewan, Canada2Department of Anatomy and Cell Biology, University of Saskatchewan, Saskatoon, Saskatchewan, Canada
A cortical lesion was induced by disrupting the me-dium-size pial vessels, which led to a cone-shapedcortical lesion and turned into a fluid-filled cavity sur-rounded by a glial acidic fibrillary protein-positive(GFAP1) glia limitans 21 days after injury. Therefore, itmimics conditions of lacunar infarctions, one of themost frequent human stroke pathologies. Doublecortin(DCX)-positive cells were present in the neocortex andcorpus callosum at the base of the lesion. The numberof DCX-positive cells in the corpus callosum was signif-icantly increased from day 5 to day 14 compared withthe control group. In contrast, there were no DCX-posi-tive cells in neocortex of control animals; the DCX-posi-tive cells appeared in the neocortex after lesioning andwere maintained until 14 days postlesioning. Some ofthe DCX-positive cells were also immunoreactive forbIII-tubulin, another marker of immature neurons. Theydid not stain positively for markers of glia cells. Thepresence of these DCX-positive cells near the lesionmight indicate a migratory pathway for developing neu-roblasts from the subventricular zone (SVZ) through thecorpus callosum to the lesion. SVZ cells were labeledwith a lipophilic molecule, 5- (and 6-) carboxyfluores-cein diacetate succinimidyl ester (CFSE) stereotaxicalinjections. Although rostral migratory stream and olfac-tory bulb were intensely labeled, no CFSE-containingcells were found in the cortex beneath the lesion.These results do not support the idea that the DCX-positive cells were originating from neural precursors ofthe SVZ, but they might be generated from local pro-genitor cells. VVC 2007 Wiley-Liss, Inc.
Key words: 5- (and 6-) carboxyfluorescein diacetatesuccinimidyl ester; focal ischemia; migration; neuroblasts;neurogenesis
Adult neurogenesis is a phenomenon that hasreceived much attention over the last decade (Perettoet al., 1999; Hallbergson et al., 2003; Alvarez-Buylla andLim, 2004; Lindsey and Tropepe, 2006; Lindvall andKokaia, 2006). Adult neurogenesis in the mammalianCNS is a robust and reproducible occurrence, but fourqualifications have to be considered. 1) Adult neurogen-esis is species dependent, with what appears to be a
gradual restriction during evolution (Bhardwaj et al.,2006). 2) Although there is no doubt that this phenom-enon occurs in the subventricular zone, olfactory bulb,and dentate gyrus of many species, especially rodents, itsoccurrence in other brain areas such as cerebral cortex iscontroversial in rodents and nonexistent in humans(Bhardwaj et al., 2006). 3) Adult neurogenesis can beenhanced and induced by factors in the environmentsuch as behavior patterns associated with learning as wellas by injury (e.g., ischemia, hemorrhage) or drug treat-ment (Lindsey and Tropepe, 2006). 4) It has also to bekept in mind that many newly generated neurons mighthave a transient appearance and might never turn intomature cells, much less into full-fledged participants inneural circuits (Lledo et al., 2006).
Stroke seems to be the injury that is most apt topromote increased neurogenesis, especially in rodents(Lichtenwalner and Parent, 2006; Taupin, 2006). Most ofthe studies on poststroke neurogenesis have used strokemodels involving large-vessel occlusion, resulting in mas-sive lesions, such as MCAO (Zhang et al., 2005). Wangand Walz (2003) introduced a modified pial-vessel class IIdisruption (PVDII) model. It is a model of small-vesselocclusion and creates a small, reproducible lesion, shapedlike an inverted cone, with the tip reaching into layers IVand V of the cerebral cortex. The tip of the lesion neverreaches deep enough to touch the corpus callosum. After2 weeks, cavitation develops at the site of the PVDIIlesion (Wang and Walz, 2003; Hua and Walz, 2006a).This stroke model therefore has features resembling lacu-nar infarcts as observed in humans as a result of small-ves-sel occlusion and silent strokes. Reactive astrocytes sur-round the lesion, and those in the space between the tipof the lesion and the corpus callosum were found to be
Contract grant sponsor: Heart and Stroke Foundation of Saskatchewan
(to W.W.).
*Correspondence to: Dr. Wolfgang Walz, Department of Physiology,
University of Saskatchewan, 107 Wiggins Road, Saskatoon, Saskatchewan
S7N 5E5, Canada. E-mail: [email protected]
Received 4 June 2007; Revised 3 August 2007; Accepted 9 August 2007
Published online 25 October 2007 in Wiley InterScience (www.
interscience.wiley.com). DOI: 10.1002/jnr.21546
Journal of Neuroscience Research 86:883–893 (2008)
' 2007 Wiley-Liss, Inc.

highly reactive, expressing a high density of vimentin1
cells (Wang et al., 2004). This is in contrast to the reac-tive astrocytes surrounding the remainder of the lesion.We were not able to detect a higher proliferation rate ofcells in this area of high reactivity. This area containingvimentin1 reactive astrocytes is strategically situated alonga hypothetical axis that includes subventricular zone(SVZ)—rostral migratory stream (RMS)—corpus callosum(CC)—lesion. One function of these vimentin1 reactiveastrocytes could be to serve as a guiderail for the migra-tion of neuroblasts or other newly created precursor cells.Indeed, we show here the appearance of doublecortin-positive (DCX1) cells with the morphology of migratingneuroblasts. Although these DCX1 cells were seen bothin the portion of the ischemic penumbra containingvimentin1 astrocytes and in the adjacent part of the cor-pus callosum, we were not able to confirm that these cellsarose from the SVZ—RMS pathway.
MATERIALS AND METHODS
Modified Cortical Devascularizing Lesion(PVDII model)
Male adult Wistar rats (Charles River Inc., St. Constant,Quebec, Canada) with a weight of 270–330 g were used. Afterarrival, the animals were kept for at least 1 week before beingprepared for surgery. This model of focal cerebral ischemia wasinduced as described by Wang and Walz (2003) by disruptingmedium-sized (class II) vessels on the cortical surface of adultrats. Rats were anesthetized with an intraperitoneal injection ofketamine (125 mg/kg) and xylazine (7 mg/kg) cocktail. A cra-niotomy was performed with a 5-mm-diameter trephine posi-tioned on the right and rostral side of the bregma adjacent tothe coronal and saggital sutures. Cool sterile saline was appliedintermittently to prevent overheating from the high-speed dril-ling. After removal of the dura, the cortical surface and theoverlying pial vessels were exposed. Medium-size (class II) pialvessels (Bar, 1980) were disrupted by fine-tipped forceps. Thepiece of bone was replaced, and the scalp was then closed witha wound clip, and buprenorphine (dosage 0.035 mg/kg,) wasinjected subcutaneously for pain management. The body tem-perature was monitored throughout the surgery by a rectalprobe and maintained at 378C by a heating pad. Animals werekept in a cage separately under a warm lamp during the recov-ery from anesthesia. Thereafter, the animals were returned totheir cages and sacrificed at different survival times by intracar-diac perfusion. All studies were performed following a protocol(20020024) approved by the Animal Care Review Committeeof the University of Saskatchewan.
In Vivo Labeling of the SVZ Cells With CFSE
To undertake in vivo labeling of SVZ cells of adult rats,the cell tracker carboxyfluorescein diacetate succinimidyl ester(CFSE; Molecular Probes, Eugene, OR) was injected into theanterior part of SVZ (SVZa). CFSE is a lipophilic dye, whichis nonfluorescent until it is transported into the cells and itsacetate group is cleaved by an intracellular esterase. This willyield a highly fluorescent, amine-reactive carboxyfluoresceinsuccinimidyl ester. The covalent reaction of the succinimidyl
ester group with intracellular amines means that CFSE is wellretained in the intracellular space throughout the fixation andstaining procedures. The fluorescent dye is inherited bydaughter cells after cell division and is not transferred to adja-cent cells in a population (Hu et al., 2003; Li et al., 2003).Because of its instability, CFSE solution was made up freshlybefore the in vivo injection.
Stereotaxic Surgery and Tracer Injection
For the study of neuroblast migration after the ischemicinjury, CFSE was injected either right before the induction ofthe PVDII or 2, 3, or 5 days after lesioning. Adult animalswere anesthetized as described above, placed in stereotaxicframes, and stabilized with ear bars to obtain a flat skull (Har-vard Apparatus, ASI Instruments). The rats were maintained at378C with aid of a heating pad during anesthesia. A dremelwith a 1-mm drill bit was used to drill a burr hole at 1.4 mmlateral to the bregma and was irrigated continuously with coolsaline to prevent overheating. Two hundred nanoliters of10 mM CFSE (500 lg CFSE in 90 ll dimetheylsulfoxide) wasinjected into SVZa (AP 5 0 mm, ML 5 1.4 mm, DV 54.7 mm from bregma skull) at a speed of 0.1 ll/min. This wasdone by means of a 5-ll Hamilton syringe with a 32-gaugeneedle attached to a microinjector. Three minutes after theneedle was placed into the final position, the injection processbegan and lasted for 2 min. The syringe was left in place for anadditional 3 min before slow withdrawal was initiated to pre-vent any backflow. The scalp incision was closed with suture.
Tissue Preparation and Immunohistochemistry
The rats were anesthetized 1, 3, 5, 7, 10, 14, and 21days after lesioning with a ketamine (125 mg/kg) and xylazine(7 mg/kg) mixture and intracardially perfused with PBS, fol-lowed by cold freshly depolymerized 4% formaldehyde (FA)in 0.1 M PBS (pH 7.4). The brains were removed, postfixedin 4% FA overnight, then moved into 30% sucrose solutionand stored at 48C before sectioning. The brains were embed-ded in Tissue-Tek optimal cutting compound (OCT; SakuraFinetek, Torrance, CA), frozen in liquid nitrogen-cooled iso-pentane, and sectioned at 40 lm on a cryostat.
The sections were blocked in PBS containing 5% don-key serum and 0.3% Triton X-100 and incubated with pri-mary antibodies overnight at 48C and with secondary antibod-ies for 2 hr at room temperature in blocking solution withoutTriton X-100. The following primary antibody dilutions wereused (with the quotation for antibody specificity given): anti-doublecortin (DCX; guinea pig polyclonal IgG, 1:100–1:300;Chemicon, Temecula, CA; catalog No. AB5910; Lee et al.,2003); antineuronal class bIII-tubulin (TuJ1 clone, mousemonoclonal IgG, 1:500; Covance, Berkeley, CA; catalog No.MMS-435P; Jepsen et al., 2000); anti-NG2 chondroitin sulfateproteoglycans (mouse monoclonal IgG1, 1:100; Chemicon;catalog No. MAB5384; Ughrin et al., 2003); anti-Ki67 (rabbitpolyclonal IgG, 1:200; Novocastra; Key et al., 1993); antivi-mentin (mouse monoclonal IgG1, 1:200; Santa Cruz Biotech-nology, Santa Cruz, CA; catalog No. SC6260); and antiglialfibrillary acidic protein (GFAP, rabbit polyclonal IgG, 1:500;Dako, Carpinteria, CA; catalog No. Z0334; mouse monoclo-
884 Hua et al.
Journal of Neuroscience Research

nal IgG1; Sigma, St. Louis, MO; catalog No. G3893). Nega-tive controls omitting the primary antibodies were carried outin all experiments for all the secondary antibodies used. Afterwashing in PBS, sections were incubated with secondary anti-bodies conjugated with fluorescence dyes (TRITC or FITC)at room temperature for 2 hr in blocking solution withoutTriton X-100. The secondary antibodies included donkeyanti-guinea pig, donkey anti-mouse IgG, donkey anti-rabbit,and goat anti-mouse. All secondary antibodies were used at1:100 or 1:200 dilutions and were species cross-adsorbed fromJackson Immunoresearch (West Grove, PA).
Heat-induced antigen retrieval (AR) technique was usedin DCX/Ki67 immunostaining for retrieval of antigens thathad been masked by formalin fixation (Boenisch, 2001). Slidesfor DCX and Ki67 immunostaining were treated in citratebuffer (pH 6.0) at 908C about 45 min before blocking toachieve a stronger intensity of immunohistochemical staining.
The fluorescein filter set was used to visualize CFSE-la-beled cells. Nuclei were stained with Hoechst 33243 to distin-guish the cortical lesion, neocortex, corpus callosum, andother brain structures.
Quantification and Imaging
The images of stained specimens were captured by aphotometrics CoolSnap fx CCD camera mounted on a micro-scope (Olympus IX71) and analyzed in Image Pro Plus (ver-sion 4.5). Cellular colocalization was confirmed via confocalmicroscopy (Zeiss LSM-410 or Olympus FV300). Singleconfocal images were taken as 0.5-lm optical sections from40-lm cryostat sections.
For quantification of DCX1 cells, three to six parasaggitalsections through the lesion (1.4–2.4 mm lateral to the bregma)were examined. DCX1 cells in the cortex surrounding theinjury and between the injury and the corpus callosum werequantified, and only positive cells with nuclei were counted.DCX1 cells (TRITC filter) were localized at 3 20 magnifica-tion; then, the filter was switched to examine whether the cellwas positive for Hoechst 33243. Higher magnification (340)was used to confirm that DCX and nucleus belonged to thesame cell by focusing up and down through several planes offocus and ascertaining that DCX immunoreactivity completelysurrounded the nuclear immunoreactivity. The average numberof DCX1 nuclei per 40-lm cryostat section between the lesionand the RMS is indicated in the figures.
Values were expressed as the mean 6 SE. One-wayANOVA was used to compare the number of DCX1 cells inexperimental rats to control rats, with days postischemia beingthe independent variable; the numbers of DCX1 cells in corpuscallosum and neocortex were analyzed separately for statisticalsignificance. Multiple post hoc comparisons among the differentgroups were performed with Bonferonni’s multiple-compari-sons posttest, with the significance level set at P < 0.05.
RESULTS
There Are No DCX-Positive Cells in theNeocortex of Control Rats
Control rats include normal rats that had no sur-gery and sham rats that underwent the whole procedure
except for the disruption of the medium-size pial vessels.As expected, the strongest DCX immunoreactivity innormal and sham rats was found in the lateral and dorsalSVZ of the lateral ventricle, the RMS, the accessory ol-factory bulb, and the dentate gyrus of the hippocampus(data not shown). DCX1 cells in the RMS assembledinto chains and showed the characteristic morphology ofmigrating neurons: an ovoid cell body with a leadingprocess (data not shown). Isolated or clusters of DCX1
cells were also seen in the corpus callosum (data notshown) and in the striatum of these sham and normalrats. Some DCX1 cells were also seen in the dorsal cor-pus callosum, their morphology resembling that ofmigrating neurons. The characteristic morphology ofDCX1 cells in the striatum appeared more like differen-tiated neurons, with long, branched processes. Numer-ous DCX1 cells in the RMS coexpressed were double-labeled with Ki67 (data not shown), which is a markerfor dividing cells (Lu et al., 2005). The expression ofKi67 by the DCX1 cells indicates that they are prolifer-ating as they migrate along the RMS, as shown previ-ously by Komitova et al. (2005).
DCX immunostaining of parasagittal sections ofboth normal rats (n 5 3) and sham rats (10 days afterlesioning, n 5 3) did not reveal any DCX1 cells in theneocortex. In addition, there were no DCX1 cells inthe cortex of the contralateral hemisphere of rats thatunderwent PVDII at any time period we investigated inthis study.
DCX-Positive Cells in Neocortex and CorpusCallosum After PVDII Lesioning
We showed previously that disruption of the me-dium-sized (class II) pial vessels consistently induced asmall cortical lesion in the somatosensory cerebral cortexwithout damaging the corpus callosum underneath(Wang and Walz, 2003; Hua and Walz, 2006a). Thecortical infarct was formed by day 1, with a distinct bor-der forming by day 6 and a cystic cavity by day 21(Wang and Walz, 2003; Hua and Walz, 2006a). Thiscystic cavity resembles lacunar infarctions.
In the present study, adult rats were sacrificed 1(n 5 5), 3 (n 5 3), 5 (n 5 5), 10 (n 5 5), and 14 (n 5 4)days after pial vessel disruption. As pointed out above,neither normal rats nor sham rats 10 days after injuryhad any DCX1 cells in the neocortex. However, in thePVDII-lesioned rats, DCX1 cells were found in theneocortex between the area of the lesion and the CC.The distribution in the different time periods after thestart of the lesion was as follows: DCX1 cells werefound in one of five rats in the day 1 postinjury group,two of three rats in the day 3 postinjury group, two offive rats in the day 5 postinjury group, four of five ratsin the day 10 postinjury group, and three of four rats inthe day 14 post injury group. These DCX1 cells in theneocortex had a morphology resembling that of differen-tiated postmigratory neurons, with long, branched pro-cesses (Fig. 1C–E). Larger numbers of DCX1 cells were
Neuroblasts in the Ischemic Penumbra 885
Journal of Neuroscience Research
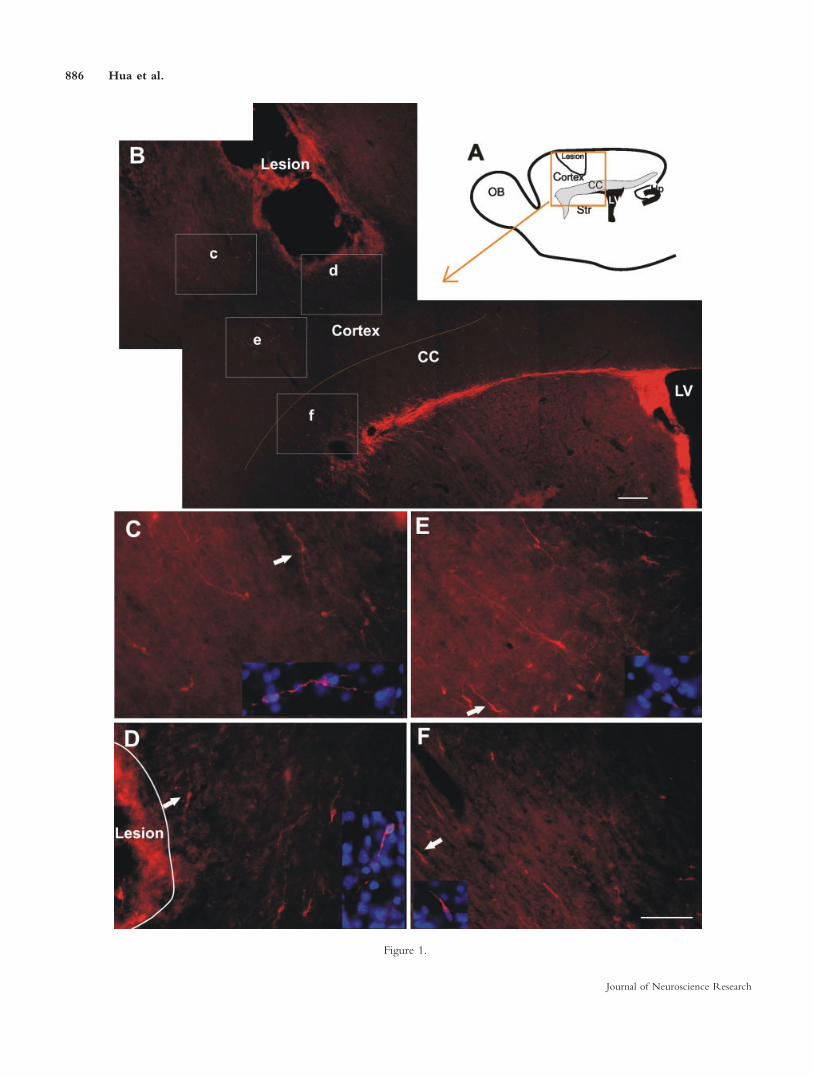
Figure 1.
886 Hua et al.
Journal of Neuroscience Research

observed in the CC of PVDII-lesioned rats, and thesecells were more likely to exhibit a migratory morphol-ogy, with leading and trailing processes (Fig. 1F).
The number of DCX1 cells in the neocortex andCC beneath the PVDII lesion was quantified in allgroups. The one-way ANOVA revealed a significantincrease in the number of DCX1 cells in the corpus cal-losum on days 5, 10, and 14 compared with the numberin normal (n 5 3) and day 1 postinjury rats. The one-way ANOVA did not revealed a significant effect of the
time postinfarct for the number of DCX1 cells in thenerocortex (Fig. 2). However, in contrast, there were noDCX1 cells in neocortex of normal rats and sham rats(see above); the DCX1 cells appeared only in the neo-cortex after PVDII and were maintained until 14 daysafter PVDII lesion (Fig. 2). By 21 days, the DCX1 cellswere still present in the ischemic penumbra of the neo-cortex in one of three animals. However, by 21 dayspostlesion, the numbers of DCX1 cells in both corpuscallosum and neocortex appeared to decrease.
Further Characterization of the DCX1 Cells
DCX, a microtubule-associated protein, is requiredfor normal neuronal migration in the developing CNS,and it is expressed in both radially and tangentiallymigrating neuroblasts (Brown et al., 2003; Koizumiet al., 2006). In contrast to the frequent colocalization ofDCX and Ki67 in SVZ and RMS cells, the DCX1 cellsunderlying the lesion were not colabeled with Ki67,which suggests that they were postmitotic (Fig. 3). Nota single DCX1 cell was observed that coexpressedmarkers of glia precursors or mature glia, NG-2, vimen-tin, and GFAP. However, 70% of the DCX1 cellsstained for bIII-tubulin were positive for both markers(Fig. 3). The expression of bIII-tubulin, which isanother marker of immature neurons, further confirmedtheir neuronal lineage. Thus, the appearance of DCXcan be confidently taken as a sign of immature, migrat-ing cells of the neuronal lineage.
In Vivo Labeling of DCX-Positive Cells in theSVZ: Control Rats
The appearance of DCX1 cells in the neocortex isan indication of neurogenesis in response to injury, ashas been shown previously using other stroke models(Jin et al., 2003), we hypothesized that these DCX1
cells were recruited from the SVZ and the RMS inresponse to the ischemic. To test this hypothesis, we la-beled SVZ cells of adult rats by stereotaxic injection ofCFSE into the SVZa.
One day after CFSE injection, labeled cells werefound mainly in the SVZ and RMS, but not in the coreof the OB. Three days after injection, labeled cells weredetected not only in the RMS but also in the entry ofthe OB, which is in accordance with previouslydescribed reports in which neuroblast migration fromSVZ to OB occurs within 2–9 days (Petreanu andAlvarez-Buylla, 2002). CFSE-labeled cells in the RMS
Fig. 2. Quantification of DCX1 cells in the neocortex and CCbeneath the lesion per 40-lm-thick section at different time pointsafter PVDII. The one-way ANOVA was applied to the data, withthe data for the CC and neocortex analyzed separately for statisticalsignificance. Multiple post hoc comparisons were performed withBonferonni’s multiple-comparisons posttest. The one-way ANOVAshowed a significant main effect of the time postinfarct for the num-ber of DCX1 cells in the CC, F(5,19) 5 5.622, P < 0.002. Themain effect of the time postinfarct was nonsignificant for the numberof DCX1 cells in the neocortex, F(5,19) 5 1.064, P 5 0.41. In con-trast, there were no DCX1 cells in neocortex of normal rats andsham rats; the DCX1 cells appeared only in the neocortex afterPVDII and were maintained until 14 days after PVDII lesion. Bon-feronni’s posttest revealed that the number of DCX1 cells in the cor-pus callosum was significantly higher (yP < 0.05) in the postinjurydays 5, 10, and 14 rats than in either postinfarct day 1 or normal ani-mals. CC, corpus callosum; DCX, doublecortin.
Fig. 1. Distribution of DCX1 cells 14 days after PVDII. A: Sche-matic drawing of a representative parasaggital plane in an adult ratbrain with the highest density of DCX1 cells. B: DCX1 cells locatedbetween the RMS and the lesion in both neocortex and CC. Thearea, which is represented by this overview picture is given in the or-ange box in the drawing in A. C–F: Higher magnification of therectangular areas c–f in B. They represent white matter (f) and theneocortex surrounding the lesion (c–e). Inset pictures are theenlarged view of DCX1 cell (white arrow) merged with nuclei label-
ing (Hoechst, blue). The DCX1 cells in the neocortex exhibit amorphology more like that of differentiated postmigratory neurons(arrows in C–E), and DCX1 cells underlying white matter exhibit amigratory morphology with leading and trailing processes (arrow inF). CC, corpus callosum; OB, olfactory bulb; LV, lateral ventricle;Str, striatum; Hp, hippocampus; DCX, doublecortin. Scale bars 5200 lm in B; 100 lm in F (applies to C–F).
3
Neuroblasts in the Ischemic Penumbra 887
Journal of Neuroscience Research

showed the typical morphology of migrating neuroblasts.This consists of an elongated cell body and a leadingprocess usually pointing toward the OB (Fig. 4). By days5–7, CFSE-labeled cells were observed along the RMSand in the core of the OB. From 7 days onward, labeledcells started to appear in the granule cell layer (GCL) ofthe OB. By days 10–14, more labeled cells wereobserved in the GCL, and some labeled cells were alsoseen located in the more superficial layers of OB, suchas the glomerular layer. At day 14, the fluorescence in-tensity was decreased, and the fluorescence distributionbecame uneven, but the labeling was still detectable. Inaddition, cells in the SVZ were always strongly labeledat any time examined.
Both ipsilateral SVZ cells and ependymal cells werelabeled with CFSE, whereas contralateral ependymalcells were rarely labeled, and no CFSE-labeled cells werefound on the pial surface. These findings suggest that,
with this injection method, CFSE was not transportedwithin the bulk flow of cerebrospinal fluid and did notlabel the contralateral lateral ventricle. Outside theSVZ-RMS-OB axis, some CFSE-labeled cells wereobserved in the striatum at all time points. Other thanthese cells in the striatum, very few CFSE-labeled cellswere observed outside of the SVZ-RMS-OB axis. Todetermine the identity of the labeled cells, immuno-staining was performed: some DCX1 cells in the SVZand the RMS were also labeled with CFSE (Fig. 4F,G).This indicates that considerable numbers of migratingneuroblasts were labeled by the CFSE injection intothe SVZa. CFSE was contained and retained in theSVZ migrating cells and their progeny for at least 14days. Therefore, this method could be a useful tool toaddress the hypothesis of whether DCX1 cells weerecruited from SVZ and RMS in response to the ische-mic injury.
Fig. 3. Characterization of the DCX1 cells. A–C: High-power confocal images showing a subsetof DCX1 cells expressing bIII-tubulin. Cells doubly labeled (arrow) for DCX (green, A), bIII-tubulin (red, B), and overlap (orange, C) in the neocortex surrounding the lesion 5 days afterPVDII. D–F: Conventional fluorescent images showing that DCX1 cells (green, D) do not coloc-alize with Ki671 cells (red, E) at the base of the lesion 10 days post-PVDII. DCX, doublecortin.Scale bars 5 20 lm in A–C; 40 lm in D–F.
Fig. 4. CFSE-labeled cells in the adult rat brain. A: Schematic draw-ing with the injection site on the parasaggtial brain section. B:Enlargement of the orange box in A, giving an overview of CFSE-labeled cells 7 days after stereotaxic injection into SVZa. C: CFSE-labeled cells in the RMS; enlarged view of the right square outlinedarea in B. D: CFSE-labeled cells in the GCL of OB; enlarged viewof the left square outlined area in B. E: GFAP immunostainingshows that CFSE-labeled cells in the RMS do not express GFAP.These CFSE-labeled cells migrate through a tube-like structure,
which is formed by cells expressing GFAP. F: DCX immnostainingshows that all CFSE-labeled cells also express DCX in the RMS. G:Same section viewed with the aid of a confocal microscope, whichconfirmed the colocalization of DCX and CFSE (white arrows).SVZa, anterior of subventricular zone; RMS, rostral migratorystream; OB, olfactory bulb; LV, lateral ventricle; GCL, granule celllayer; OB, olfactory bulb; DCX, doublecortin; CFSE, 5- (and 6-)carboxyfluorescein diacetate succinimidyl ester; GFAP, glial acidicfibrillary protein. Scale bars 5 200 lm in B–D; 50 lm in E–G.
"
888 Hua et al.
Journal of Neuroscience Research
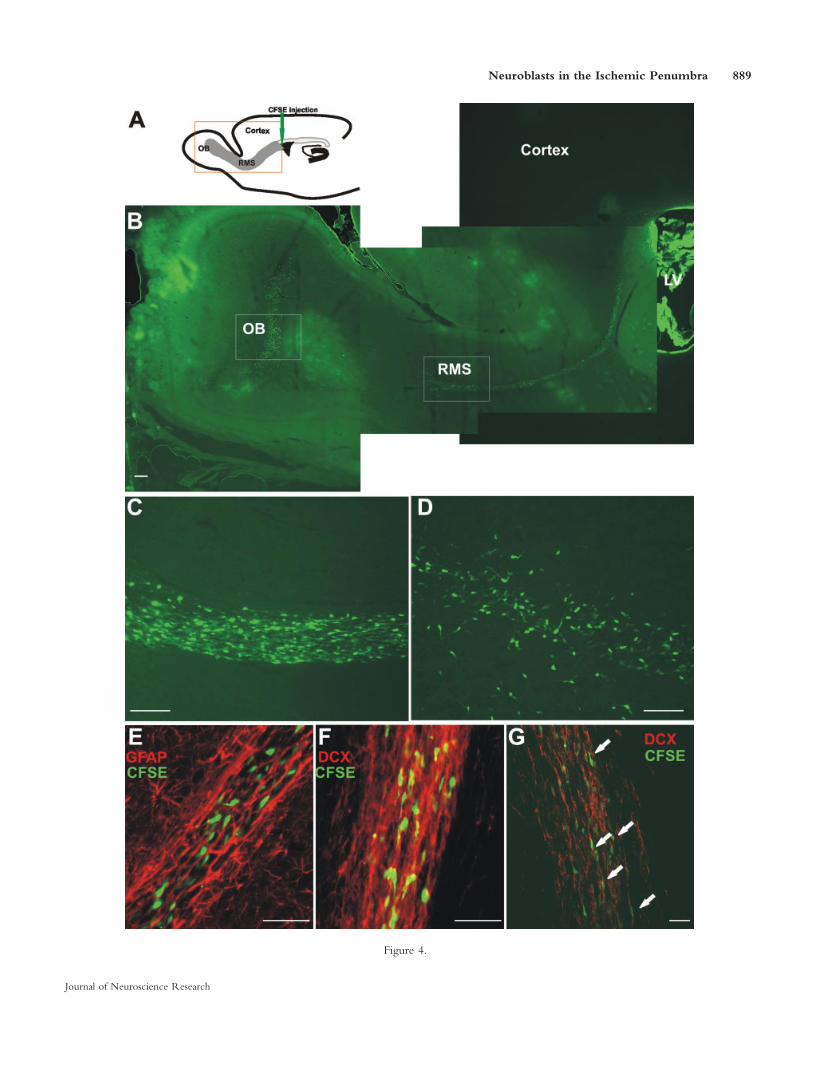
Figure 4.
Neuroblasts in the Ischemic Penumbra 889
Journal of Neuroscience Research

In Vivo Labeling of DCX-Positive Cells in theSVZ: PVDII-Induced Lesion
The fluorescent cell tracker CFSE was injectedunilaterally into the SVZa before induction of the corti-cal ischemic lesion by disrupting the medium-sized pialvessels. Rats were sacrificed 1 (n 5 6), 3 (n 5 6), 5(n 5 7), 7 (n 5 5), 10 (n 5 6), and 14 (n 5 8) daysafter the surgery. The successful labeling of SVZ cells ineach animal was confirmed by the strong labeling ofCFSE- labeled cells in the SVZ, in the RMS, and in theOB (data not shown). Although many CFSE-labeledcells were observed in the RMS and OB, there were noCFSE-labeled cells either in the CC or in the ischemicpenumbra.
To test the possibility that the neuroblasts born af-ter the PVDII-induced injury were the ones preferen-tially being recruited to migrate toward the lesion, CFSEwas injected into the SVZa 2, 3, and 5 days after pialvessel disruption, and the animals were sacrificed at 10days (n 5 3) or 14 days (n 5 3) after making the lesion.There were still no CFSE-labeled cells in either the is-chemic penumbra or the CC beneath the lesion.
DISCUSSION
PVDII Is a Model of Human Small-Vessel Disease
The PVD model of focal ischemic stroke, which isoften also called pial stripping, results in a large lesionthat reaches far into the CC and creates hemorrhage(Farr and Whishaw, 2002; Gonzalez and Kolb, 2003).This massive injury is due to the disruption of the largeclass I vessels. It resembles, therefore, more a situationcaused by neurotrauma rather than by ischemia. Tomake a lesion more representative of small-vessel stroke,we used a modification of the PVD model that disruptsonly medium-sized (class II) vessels, creates a muchsmaller lesion, does not extend as deep as the CC, and ishighly reproducible. This PVDII model fills a void inanimal ischemia models by reproducing pertinent fea-tures of small-vessel disease. This disease accounts forabout one-third of all human strokes (Greenberg, 2006).The small size of the ischemic injury (one to severalcubic millimeters) and the presence of a fluid-filled cav-ity are hallmarks of human small-vessel strokes and silentinfarcts (Wardlaw, 2004).
However, the ischemic lesion in the PVDII modelis in the neocortex, whereas most small-vessel strokes inhumans occur in subcortical areas. There are, however,other small-vessel diseases within the human neocortex(Chester et al., 1978), with many of these causing vascu-lar dementia (Greenberg, 2006). In addition, the PVDIImodel uses an irreversible interruption of blood flowand therefore does not allow for reperfusion of the is-chemic tissue. However, about 80% of reperfusion inhuman stroke is not through clot lysis but through col-lateralization (Carmichael, 2005), which is not affectedin the PVDII model. Thus, the cone-shaped lesion witha volume of approximately 1-mm3 that turns into afluid-filled cavity by the third week postischemia (Wang
and Walz, 2003) is a realistic approximation of humansmall-vessel disease (Hua and Walz, 2006b).
Neurogenesis in the Ischemic Penumbra asEvidenced by the Appearanceof DCX-Positive Cells
In response to the PVDII-induced ischemic injury,there were DCX-expressing cells in the ischemic pe-numbra and in the CC beneath the lesion during thefirst 2 weeks after injury. In this study, DCX was usedas the principle marker of migrating neuroblasts andimmature neurons. DCX is a microtubule-associatedprotein that is required for both radial and tangentialneuronal migration in the developing CNS (Gleesonet al., 1999; Ocbina et al., 2006). In the adult brain,DCX expression is retained mainly within the two areasof continuous neurogenesis, the SVZ—RMS—OB axisand the dentate gyrus of hippocampus (Nacher et al.,2001). DCX is suggested to be a reliable and specificmarker for adult neurogenesis (Brown et al., 2003;Couillard-Despres et al., 2005). During adult neurogene-sis, the expression of DCX starts as the generation ofneuroblasts and is down-regulated with the maturation(Couillard-Despres et al., 2005; Plumpe et al., 2006).
DCX1 cells in white matter displayed a morphol-ogy typical of migrating neuroblasts and in the neocortexof differentiated neuroblasts (Nacher et al., 2001; Brownet al., 2003) but do not express markers of either astro-cytes or oligodendrocytes. Some of the DCX1 cells alsoexpress bIII-tubulin (TUJ1), another marker proteintransiently expressed by immature neurons during theirmaturation (Lledo et al., 2006). Therefore, based onboth morphological and immunohistochemical charac-teristics, the appearance of DCX1 cells is an attempt bythe brain to replace neurons lost as a result of the ische-mic injury.
The small number of DCX1 cells that we found,however, suggests that the recruitment of neuroblasts tothe ischemic penumbra of the PVDII lesion was modestat best (Lichtenwalner and Parent, 2006). To date, post-stroke neurogenesis has been studied mostly in the mas-sive ischemic injury model induced by occlusion of theMCA. Several reports indicate that focal ischemic injuryin the striatum (Parent et al., 2002; Arvidsson et al.,2002; Jin et al., 2003; Zhang et al., 2004; Nygren et al.,2006; Thored et al., 2006; Yamashita et al., 2006) orneocortex (Gu et al., 2000; Jiang et al., 2001; Jin et al.,2003; Ohab et al., 2006) can also induce neurogenesis inthe ischemic penumbra. Within the first 2 weeks afterinjury, newborn immature neurons as indicated by theexpression of neuronal lineage cell markers such as DCXor bIII-tubulin were observed in tissue surrounding theinfarct area (Parent et al., 2002; Arvidsson et al., 2002;Jin et al., 2003; Zhang et al., 2004; Nygren et al., 2006;Yamashita et al., 2006). A few weeks later, some ofthese newly generated cells (BrdU1) expressed themarkers for mature neurons, such as NeuN, and region-specific mature neuronal markers, such as DARPP-42
890 Hua et al.
Journal of Neuroscience Research

and calbindin (Arvidsson et al., 2002; Parent et al., 2002;Yamashita et al., 2006). Interestingly, studies using theMCAO model, which mimics focal ischemia and indu-ces an excessive infarct in both neocortex and striatum,found poststroke neurogenesis in the neocortical ische-mic penumbra (Jiang et al., 2001; Jin et al., 2003). How-ever, others failed to detect cortical neurogensis(Arvidsson et al., 2002; Parent et al., 2002; Zhang et al.,2004; Yamashita et al., 2006). In contrast, all of thesestudies found poststroke neurogenesis in the striatum.
A recent study has shown substantial cortical neu-rogenesis in the periinfarct cortex following permanentocclusion in mice of the distal middle cerebral artery andtransient occlusion (bilaterally) of the common carotidartery (Ohab et al., 2006). In that study, with stereologi-cal quantification, Ohab et al. found about 2,000 DCX1
cells in periinfarct cortex per animal at day 3 after thestroke, with the number increasing to more than 10,000DCX1 cells per animal at day 7. The number of DCX1
cells post-PVDII was much lower (approximately 150DCX1 cells in the neocortical ischemic penumbra peranimal) compared with the number of DCX1 cellsreported by Ohab et al. (2006), and we did not observecorical DCX1 cells in all post-PVDII animals. A similarvariation of DCX1 cells among different animals (withsome animals expressing no DCX at all) was alsoobserved by Ramaswamy et al. (2005) in a cortical neu-rotrauma model. As mentioned above, the PVDII modelinduces a smaller cortical ischemic injury than othercortical ischemic models, and the PVDII-induced lesionis located mainly in the primary motor cortex. It is to beexpected that the nature, location, and size of the ische-mic injury will all influence the magnitude of the sig-nal(s) necessary to recruit migrating neuroblasts and toinduce their diffentiation into neurons. Indeed, withrespect to ischemic injury of the striatum, it has beenshown that the numbers of immature neurons and ofnewly generated neurons are both positively correlatedwith the volume of the injury (Thored et al., 2006).
Source of DCX-Positive Cells That Appear in theIschemic Penumbra
The appearance of DCX1 cells in the ischemic pe-numbra of the neocortex and in the CC of adult rats af-ter a cortical ischemic injury raises the question of thecellular origin of these cells. Do they arise locally fromparenchymal precursors or at a distance from neurogenicregions such as SVZ or the bone marrow and thus beinduced to migrate long distances toward the ischemicinjury? By directly labeling SVZ cells with DiI or a viralvector, neuroblasts of the SVZ have been shown tomigrate into the damaged striatum and/or neocortex inresponse to ischemic (Jin et al., 2003; Ohab et al., 2006;Yamashita et al., 2006) or traumatic (Ramaswamy et al.,2005) injury. Recent studies, using transgenic targetingstrategies (Cre-loxP recombination system) to label SVZcells specifically, have also provided direct evidence thatstroke can induce the migration of neuroblasts from the
SVZ into the damaged striatum (Yamashita et al., 2006)or neocortex (Ohab et al., 2006).
With the PVDII model, the appearance of DCX1
cells showed that the ischemic injury first induced a peakof DCX1 cells in the subcortical white matter, followedby a second peak in the periinfarct cortex. Although thesedata are consistent with the DCX1 cells migrating fromthe CC into the injured area of neocortex, they do notaddress whether the lesion might have induced them tomigrate away from the SVZ. To determine whether thePVDII ischemic injury induced neuroblasts of the SVZ tomigrate away from the RMS, through the CC, and intothe neocortical ischemic penumbra, we performed in vivolabeling of SVZ cells with CFSE. No CFSE-labeled cellswere seen outside of the SVZ—RMS—OB pathway, sothe ischemic injury itself does not appear to be inducingDCX1 cells to migrate away from the SVZ of the lateralventricle. It is important to note, however, that theapproach used in this study to trace the migratory pathtaken by SVZ cells has detection limits. As a lipophilicdye, CFSE may be diluted over time by cell divisions. Inaddition, the one-time injection of CFSE into the SVZmight label only a portion of the SVZ cells. Therefore,we cannot rule out the possibility of a very small butsteady stream of neuroblasts emigrating out of the RMSin response to cortical ischemic injury. If these DCX1
cells are indeed derived by migration from the SVZ, thetracing of such a low number of cells can be achievedonly by genetically tagging all the SVZ neural progeni-tors, such as using the Cre-loxP transgenic mice used byYamashita et al. (2006) and Ohab et al. (2006).
Thus, it is possible that the precursor cells giving riseto the DCX1 cells might reside locally within the CC,and it is also possible that they could derive from multipo-tential precursors that possess gliogenic as well as neuro-genic potentials. Resident glial precursors isolated fromhuman subcortical white matter appear to be multipoten-tial cells that are capable of undergoing not only gliogene-sis but also neurogenesis in vitro and following transplanta-tion in vivo (Nunes et al., 2003). Oligodencrocyte pro-genitor cells (OPCs) could make up at least a portion ofthese multipotential precursors, insofar as they appear topossess a wider differentiation potential than previouslyassumed and thus could be considered to be multipotential(Kondo and Raff, 2000; Belachew et al., 2003). Aftertransplantation into the adult rat hippocampus, substantialnumbers of transplanted oligodendrocyte progenitor cellsdifferentiate into cells expressing DCX (Gaughwin et al.,2006). In this study, although DCX1 cells did not coex-press the NG2 antigenic epitope, which is a marker foroligodendrocyte progenitor cells, it could very well be thatthese OPCs lost the expression of the NG2 epitope oncethey began to differentiate into DCX1 cells.
Another possible source of DCX1 cells in thePVDII model is bone marrow-derived cells (BMDCs).For example, migrating neuroblasts cluster in the periin-farct area in the vicinity of blood vessels (Zhang et al.,2001; Thored et al., 2006; Yamashita et al., 2006). Thisclose association with the vasculature is consistent with
Neuroblasts in the Ischemic Penumbra 891
Journal of Neuroscience Research

the DCX1 cells either being recruited from local multi-potent precursor cells or due to infiltration of blood-borne cells (Lledo et al., 2006). Bone marrow contains avide variety of stem and progenitor cells, such as hema-topoietic stem cells, that can and do invade ischemicsites (Rafii and Lyden, 2003). After cerebral ischemia,BMDCs rapidly infiltrate the brain, where they give riseprimarily to microglia and endothelial cells, althoughsome cells do express neuronal markers (NeuN; Hesset al., 2002, 2004; Cogle et al., 2004).
CONCLUSIONS
Stroke is a leading cause of the death and disability,and it is a diverse disease in terms of causes, manifesta-tions, severity, and anatomic sites. In response to modi-fied PVDII, a model mimicking small-vessel stroke,DCX1 neuroblasts appear in the neocortex and CC ofadult rats until up to 14 days postinjury. These resultsdemonstrate that endogenous neural precursors can beinduced to differentiate into neocortical neuronal lineagecells in the neocortex, a nonneurogenic region of theadult rodent brain. Regeneration of the lost neurons iscritical for functional improvement. Our results suggestthat, although this neurogenic response might be lowand transient, progenitor cells of the rodent adult braincan be triggered to produce new cortical neurons inresponse to small-vessel stroke. This opens up the possi-bility of a therapeutic approach to enhance the genera-tion and survival of these new neurons and their incor-poration into reorganizing neural circuits.
This study did not attempt to determine the long-term survival of these DCX-expressing neuroblasts.Therefore, the ultimate fate of these DCX1 cells isunclear. Long-term survival studies suggest that the ma-jority of the newly generated neuroblasts in the damagedarea undergo programmed cell death, and a small portionsurvive for many months, differentiate into neuronsexpressing mature neuron marker and region-specificneuron markers (Arvidsson et al., 2002; Ohab et al.,2006; Thored et al., 2006), and form synapses in thedamaged area (Yamashita et al., 2006). This is an indica-tion that these cells may be functional mature neurons.However, the ultimate test is functional analysis, such asusing electrophysiology to determine the neuronal prop-erties of these newborn neurons.
ACKNOWLEDGMENTS
We thank Lyndon Osman and Viktor Skihar for theirhelp with some of the techniques involved in this study.
REFERENCES
Alvarez-Buylla A, Lim DA. 2004. For the long run: maintaining germinal
niches in the adult brain. Neuron 41:683–686.
Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O. 2002. Neuronal
replacement from endogenous precursors in the adult brain after stroke.
Nat Med 8:963–970.
Bar T. 1980. The vascular system of the cerebral cortex. Adv Anat
Embryol Cell Biol 59:I–VI, 1–62.
Belachew S, Chittajallu R, Aguirre AA, Yuan X, Kirby M, Anderson S,
Gallo V. 2003. Postnatal NG2 proteoglycan-expressing progenitor cells
are intrinsically multipotent and generate functional neurons. J Cell
Biol 161:169–186.
Bhardwaj RD, Curtis MA, Spalding KL, Buchholz BA, Fink D, Bjork-
Eriksson T, Nordborg C, Gage FH, Druid H, Eriksson PS, Frisen J.
2006. Neocortical neurogenesis in humans is restricted to development.
Proc Natl Acad Sci U S A 103:12564–12568.
Boenisch T. 2001. Formalin-fixed and heat-retrieved tissue antigens: a
comparison of their immunoreactivity in experimental antibody
diluents. Appl Immunohistochem Mol Morphol 9:176–179.
Brown JP, Couillard-Despres S, Cooper-Kuhn CM, Winkler J, Aigner
L, Kuhn HG. 2003. Transient expression of doublecortin during adult
neurogenesis. J Comp Neurol 467:1–10.
Carmichael ST. 2005. Rodent models of focal stroke: size, mechanism,
and purpose. NeuroRx 2:396–409.
Chester EM, Agamanolis DP, Banker BQ, Victor M. 1978. Hypertensive
encephalopathy: a clinicopathologic study of 20 cases. Neurology
28:928–939.
Cogle CR, Yachnis AT, Laywell ED, Zander DS, Wingard JR, Steindler
DA, Scott EW. 2004. Bone marrow transdifferentiation in brain after
transplantation: a retrospective study. Lancet 363:1432–1437.
Couillard-Despres S, Winner B, Schaubeck S, Aigner R, Vroemen M,
Weidner N, Bogdahn U, Winkler J, Kuhn HG, Aigner L. 2005. Dou-
blecortin expression levels in adult brain reflect neurogenesis. Eur J
Neurosci 21:1–14.
Farr TD, Whishaw IQ. 2002. Quantitative and qualitative impairments
in skilled reaching in the mouse (Mus musculus) after a focal motor
cortex stroke. Stroke 33:1869–1875.
Gaughwin PM, Caldwell MA, Anderson JM, Schwiening CJ, Fawcett
JW, Compston DA, Chandran S. 2006. Astrocytes promote neurogene-
sis from oligodendrocyte precursor cells. Eur J Neurosci 23:945–956.
Gleeson JG, Lin PT, Flanagan LA, Walsh CA. 1999. Doublecortin is a
microtubule-associated protein and is expressed widely by migrating
neurons. Neuron 23:257–271.
Gonzalez CL, Kolb B. 2003. A comparison of different models of stroke
on behaviour and brain morphology. Eur J Neurosci 18:1950–1962.
Greenberg SM. 2006. Small vessels, big problems. N Engl J Med
354:1451–1453.
Gu W, Brannstrom T, Wester P. 2000. Cortical neurogenesis in adult
rats after reversible photothrombotic stroke. J Cereb Blood Flow Metab
20:1166–1173.
Hallbergson AF, Gnatenco C, Peterson DA. 2003. Neurogenesis and
brain injury: managing a renewable resource for repair. J Clin Invest
112:1128–1133.
Hess DC, Hill WD, Martin-Studdard A, Carothers J, Brailer J, Carroll J.
2002. Blood into brain after stroke. Trends Mol Med 8:452–453.
Hess DC, Abe T, Hill WD, Studdard AM, Carothers J, Masuya M,
Fleming PA, Drake CJ, Ogawa M. 2004. Hematopoietic origin of
microglial and perivascular cells in brain. Exp Neurol 186:134–144.
Hu H, Dong Y, Feng P, Fechner J, Hamawy M, Knechtle SJ. 2003.
Effect of immunosuppressants on T-cell subsets observed in vivo using
carboxy-fluorescein diacetate succinimidyl ester labeling. Transplanta-
tion 75:1075–1077.
Hua R, Walz W. 2006a. Minocycline treatment prevents cavitation in
rats after a cortical devascularizing lesion. Brain Res 1090:172–181.
Hua R, Walz W. 2006b. The need for animal models in small-vessel
brain disease. Crit Rev Neurobiol (in press).
Jepsen K, Hermanson O, Onami TM, Gleiberman AS, Lunyak V, McE-
villy RJ, Kurokawa R, Kumar V, Liu F, Seto E, Hedrick SM, Mandel
G, Glass CK, Rose DW, Rosenfeld MG. 2000. Combinatorial roles of
the nuclear receptor corepressor in transcription and development. Cell
102:753–763.
892 Hua et al.
Journal of Neuroscience Research

Jiang W, Gu W, Brannstrom T, Rosqvist R, Wester P. 2001. Cortical
neurogenesis in adult rats after transient middle cerebral artery occlu-
sion. Stroke 32:1201–1207.
Jin K, Sun Y, Xie L, Peel A, Mao XO, Batteur S, Greenberg DA. 2003.
Directed migration of neuronal precursors into the ischemic cerebral
cortex and striatum. Mol Cell Neurosci 24:171.
Key G, Becker MH, Baron B, Duchrow M, Schluter C, Flad HD,
Gerdes J. 1993. New Ki-67-equivalent murine monoclonal antibodies
(MIB 1–3) generated against bacterially expressed parts of the Ki-67
cDNA containing three 62 base pair repetitive elements encoding for
the Ki-67 epitope. Lab Invest 68:629–636.
Koizumi H, Higginbotham H, Poon T, Tanaka T, Brinkman BC, Glee-
son JG. 2006. Doublecortin maintains bipolar shape and nuclear translo-
cation during migration in the adult forebrain. Nat Neurosci 9:779–
786.
Komitova M, Zhao LR, Gido G, Johansson BB, Eriksson P. 2005. Post-
ischemic exercise attenuates whereas enriched environment has certain
enhancing effects on lesion-induced subventricular zone activation in
the adult rat. Eur J Neurosci 21:2397–2405.
Kondo T, Raff M. 2000. Oligodendrocyte precursor cells reprogrammed
to become multipotential CNS stem cells. Science 289:1754–1757.
Lee EJ, Kim IB, Lee E, Kwon SO, Oh SJ, Chun MH. 2003. Differential
expression and cellular localization of doublecortin in the developing
rat retina. Eur J Neurosci 17:1542–1548.
Li X, Dancausse H, Grijalva I, Oliveira M, Levi AD. 2003. Labeling
Schwann cells with CFSE—an in vitro and in vivo study. J Neurosci
Methods 125:83–91.
Lichtenwalner RJ, Parent JM. 2006. Adult neurogenesis and the ischemic
forebrain. J Cereb Blood Flow Metab 26:1–20.
Lindsey BW, Tropepe V. 2006. A comparative framework for under-
standing the biological principles of adult neurogenesis. Prog Neurobiol
80:281–307.
Lindvall O, Kokaia Z. 2006. Stem cells for the treatment of neurological
disorders. Nature 441:1094–1096.
Lledo PM, Alonso M, Grubb MS. 2006. Adult neurogenesis and func-
tional plasticity in neuronal circuits. Nat Rev Neurosci 7:179–193.
Lu G, Wai SM, Poon WS, Yew DT. 2005. Ki67 and doublecortin posi-
tive cells in the human prefrontal cortices of normal aging and vascular
dementia. Microsc Res Techniq 68:255–257.
Nacher J, Crespo C, McEwen BS. 2001. Doublecortin expression in the
adult rat telencephalon. Eur J Neurosci 14:629.
Nunes MC, Roy NS, Keyoung HM, Goodman RR, McKhann G 2nd,
Jiang L, Kang J, Nedergaard M, Goldman SA. 2003. Identification and
isolation of multipotential neural progenitor cells from the subcortical
white matter of the adult human brain. Nat Med 9:439–447.
Nygren J, Wieloch T, Pesic J, Brundin P, Deierborg T. 2006. Enriched
environment attenuates cell genesis in subventricular zone after focal is-
chemia in mice and decreases migration of newborn cells to the stria-
tum. Stroke 37:2824–2829.
Ocbina PJ, Dizon ML, Shin L, Szele FG. 2006. Doublecortin is necessary
for the migration of adult subventricular zone cells from neurospheres.
Mol Cell Neurosci 33:126–135.
Ohab JJ, Fleming S, Blesch A, Carmichael ST. 2006. A neurovascular
niche for neurogenesis after stroke. J Neurosci 26:13007–13016.
Parent JM, Vexler ZS, Gong C, Derugin N, Ferriero DM. 2002. Rat
forebrain neurogenesis and striatal neuron replacement after focal stroke.
Ann Neurol 52:802–813.
Peretto P, Merighi A, Fasolo A, Bonfanti L. 1999. The subependymal
layer in rodents: a site of structural plasticity and cell migration in the
adult mammalian brain. Brain Res Bull 49:221–243.
Petreanu L, Alvarez-Buylla A. 2002. Maturation and death of adult-born
olfactory bulb granule neurons: role of olfaction. J Neurosci 22:6106–
6113.
Plumpe T, Ehninger D, Steiner B, Klempin F, Jessberger S, Brandt M,
Romer B, Rodriguez GR, Kronenberg G, Kempermann G. 2006. Var-
iability of doublecortin-associated dendrite maturation in adult hippo-
campal neurogenesis is independent of the regulation of precursor cell
proliferation. BMC Neurosci 7:77.
Rafii S, Lyden D. 2003. Therapeutic stem and progenitor cell transplan-
tation for organ vascularization and regeneration. Nat Med 9:702–712.
Ramaswamy S, Goings GE, Soderstrom KE, Szele FG, Kozlowski DA.
2005. Cellular proliferation and migration following a controlled corti-
cal impact in the mouse. Brain Res 1053:38–53.
Taupin P. 2006. Stroke-induced neurogenesis: physiopathology and
mechanisms. Curr Neurovasc Res 3:67–72.
Thored P, Arvidsson A, Cacci E, Ahlenius H, Kallur T, Darsalia V,
Ekdahl CT, Kokaia Z, Lindvall O. 2006. Persistent production of neu-
rons from adult brain stem cells during recovery after stroke. Stem Cells
24:739–747.
Ughrin YM, Chen ZJ, Levine JM. 2003. Multiple regions of the NG2
proteoglycan inhibit neurite growth and induce growth cone collapse.
J Neurosci 23:175–186.
Wang K, Walz W. 2003. Unusual topographical pattern of proximal
astrogliosis around a cortical devascularizing lesion. J Neurosci Res
73:497–506.
Wang K, Bekar LK, Furber K, Walz W. 2004. Vimentin-expressing
proximal reactive astrocytes correlate with migration rather than prolif-
eration following focal brain injury. Brain Res 1024:193.
Wardlaw J. 2004. ACCESS: the acute cerebral CT evaluation stroke
study. Emerg Med J 21:666.
Yamashita T, Ninomiya M, Hernandez Acosta P, Garcia-Verdugo JM,
Sunabori T, Sakaguchi M, Adachi K, Kojima T, Hirota Y, Kawase T,
Araki N, Abe K, Okano H, Sawamoto K. 2006. Subventricular zone-
derived neuroblasts migrate and differentiate into mature neurons in the
post-stroke adult striatum. J Neurosci 26:6627–6636.
Zhang RL, Zhang ZG, Zhang L, Chopp M. 2001. Proliferation and dif-
ferentiation of progenitor cells in the cortex and the subventricular
zone in the adult rat after focal cerebral ischemia. Neuroscience
105:33–41.
Zhang R, Zhang Z, Wang L, Wang Y, Gousev A, Zhang L, Ho KL,
Morshead C, Chopp M. 2004. Activated neural stem cells contribute to
stroke-induced neurogenesis and neuroblast migration toward the
infarct boundary in adult rats. J Cereb Blood Flow Metab 24:441–448.
Zhang RL, Zhang ZG, Chopp M. 2005. Neurogenesis in the adult is-
chemic brain: generation, migration, survival, and restorative therapy.
Neuroscientist 11:408–416.
Neuroblasts in the Ischemic Penumbra 893
Journal of Neuroscience Research





![Realistic Soft Shadows by Penumbra-Wedges Blending · Penumbra-wedges X + Specular & diffuse Visibility buffer Modulated spec+diff Ambient Final image. Penumbra-wedges [3/4] Penumbra-wedges](https://img.pdfslide.net/doc/110x75/5f543a4c0135c76e2b226697/realistic-soft-shadows-by-penumbra-wedges-penumbra-wedges-x-specular-diffuse.jpg)






















