Embed Size (px)
Citation preview

Drug-excipient behavior in polymeric amorphous soliddispersions.
Surikutchi Bhanu Tejaa, Shashank Pralhad Patila, Ganesh Shetea, Sarsvatkumar Patelb andArvind Kumar Bansala,*
aDepartment of Pharmaceutics, National Institute of Pharmaceutical Education and Research (NIPER), S.A.S. Nagar, Punjab 160 062, IndiabCollege of Pharmacy, Long Island University, 75 Dekalb Avenue, Brooklyn, New York, 11201-8423, USA
Received: May 6, 2013; Accepted: May 31, 2013 Review Paper
ABSTRACT
Amorphous drug delivery systems are increasingly utilized to enhance aqueous solubility and oralbioavailability. However, they lack physical and/or chemical stability. One of the most common ways ofstabilizing an amorphous form is by formulating it as an amorphous solid dispersion. This review focuses onpolymeric amorphous solid dispersions wherein polymers are used as excipients to stabilize the amorphousform. A brief introduction to the basic concepts of amorphous systems such as glass transition temperatureand the solubility advantage of amorphous systems is provided. Additionally, information on types ofpolymers used for the development of amorphous solid dispersions, their structural attributes andmechanisms of stabilization are presented here. Molecular aspects of drug-polymer miscibility and drug-polymer interactions are also discussed.
KEY WORDS: Amorphous, solid dispersion, miscibility, polymeric excipients, amorphous form stabilization,mechanism of stabilization
INTRODUCTION
Poor aqueous solubility of drugs has emergedas one of the major challenges in drug delivery.It has been reported that about 70 % of newchemical entities have aqueous solubilityproblems and consequently poor oralbioavailability and delivery problems (1).Formulation strategies, such as particle size
reduction (2, 3), amorphous solid dispersions(ASDs) (4), co-crystal formation (5, 6),complexation employing cyclodextrins (7), co-solvents (8) and lipid formulations (9) havebeen used to improve aqueous solubility.
The crystalline form of a drug offers advantagesin terms of high purity and physical/chemicalstability compared to ASDs. However,constraints contributed by lattice energy mustbe overcome to allow solute molecules todissolve. The amorphous state exhibits adisordered structure in comparison to thecrystalline state and possesses higher free
*Corresponding author: Department of Pharmaceutics, National Institute of Pharmaceutical Education and Research, S.A.S. Nagar, Sector-67, Mohali, Punjab (INDIA)–160 062, Tel: +91-172- 2214682 Ext: 2126, Fax: +91-172- 2214692, E-mail: [email protected];[email protected]
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 70

Review Paper
Figure 1 Classification of solid dispersions. (Reproducedwith modification and permission from Reference 13).
energy. Thus it offers enhanced apparentsolubility, dissolution rate and oralbioavailability (10). Pure amorphous drugs arerarely developed alone into medicinal productsbecause of their inherent physical/chemicalinstability. This has encouraged investigation ofthe stability of ASDs where advances have beenmade in recent years. ASDs are defined as solidsystems consisting of a drug and carrierexcipient(s) prepared using thermal and/orsolvent based methods. A variety of excipientssuch as polymers and surfactants may be usedto prepare and stabilize the amorphous form ofa drug (11).
In the late 1960's, solid dispersions (SDs)emerged as a formulation strategy to improveapparent solubility by stabilizing amorphousforms of drugs (12). Solid dispersions havebeen classified based on the distribution ofsolute molecules within the carrier matrix aseutectic mixtures, solid solutions and microfinecrystalline dispersions. Figure 1 shows acomprehensive classification system for SDs(13).
A eutectic system typically consists of twocompounds which, when mixed in a particular
proportion, forms a composition which has asingle melting point that is lower than themelting points for the individual components.
Solid solutions are further classified intosubstitutional crystalline solid, interstitial solidand amorphous solid solutions. In subs-titutional crystalline solid solutions, a moleculeof carrier/solvent is replaced by a solutemolecule whereas an interstitial solid solutionconsists of dissolved solute moleculesoccupying interstices within the carrier matrix.Amorphous solid solutions consist of the drugmolecularly, but irregularly, dispersed withinthe amorphous carrier. Microfine crystallinedispersions consists of a molecular dispersionof a crystalline drug in a carrier.
A wide range of pharmaceutical excipients havebeen used in the preparation of ASDs.Materials such as lipids, carbohydrates, proteinsand surfactants have been used to kineticallystabilize an amorphous form of a drug (14).Small molecules such as meglumine (15), urea(16), sugars (17), amino acids (18) and organicacids (19) have also been investigated in thestabilizion of the amorphous form. PolymericASDs (PASDs) have shown considerablepromise in the stabilization of amorphousforms providing significant increase in solubilityand dissolution rate. Several products using thismethod have been commercialized to date, e.g.,Novir®, Kaletra® and Sporana®.
This paper reviews the theoretical and technicalaspects of PASDs and briefly describes thepotential advantages of polymers as inertpharmaceutical excipients in ASDs. This reviewalso discusses the molecular aspects of thegeneration and performance of PASDs.
Amorphous form
The amorphous form is ubiquitous in natureand plays an important role in material,biological and more recently pharmaceuticalsciences. Amorphous forms are also known asglass, vitrified, disordered or frustrated systems(20).
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 71
Review Paper
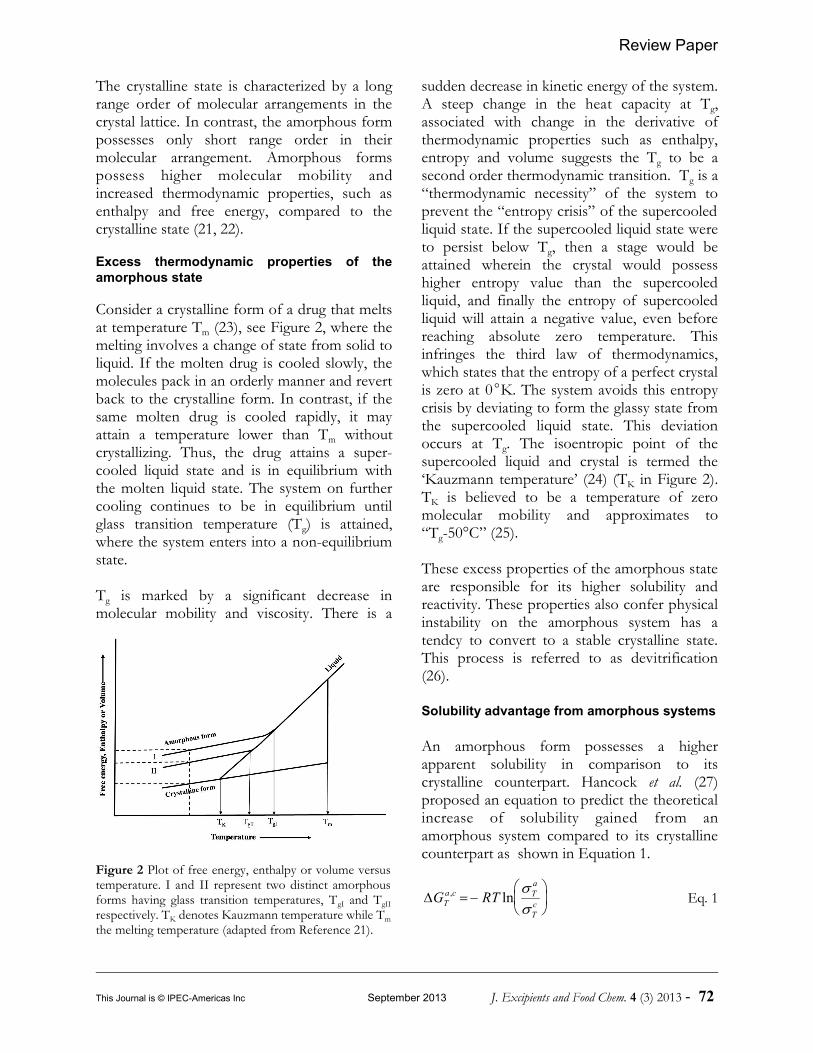
Figure 2 Plot of free energy, enthalpy or volume versustemperature. I and II represent two distinct amorphousforms having glass transition temperatures, TgI and TgIIrespectively. TK denotes Kauzmann temperature while Tmthe melting temperature (adapted from Reference 21).
The crystalline state is characterized by a longrange order of molecular arrangements in thecrystal lattice. In contrast, the amorphous formpossesses only short range order in theirmolecular arrangement. Amorphous formspossess higher molecular mobility andincreased thermodynamic properties, such asenthalpy and free energy, compared to thecrystalline state (21, 22).
Excess thermodynamic properties of theamorphous state
Consider a crystalline form of a drug that meltsat temperature Tm (23), see Figure 2, where themelting involves a change of state from solid toliquid. If the molten drug is cooled slowly, themolecules pack in an orderly manner and revertback to the crystalline form. In contrast, if thesame molten drug is cooled rapidly, it mayattain a temperature lower than Tm withoutcrystallizing. Thus, the drug attains a super-cooled liquid state and is in equilibrium withthe molten liquid state. The system on furthercooling continues to be in equilibrium untilglass transition temperature (Tg) is attained,where the system enters into a non-equilibriumstate.
Tg is marked by a significant decrease inmolecular mobility and viscosity. There is a
sudden decrease in kinetic energy of the system.A steep change in the heat capacity at Tg,associated with change in the derivative ofthermodynamic properties such as enthalpy,entropy and volume suggests the Tg to be asecond order thermodynamic transition. Tg is a“thermodynamic necessity” of the system toprevent the “entropy crisis” of the supercooledliquid state. If the supercooled liquid state wereto persist below Tg, then a stage would beattained wherein the crystal would possesshigher entropy value than the supercooledliquid, and finally the entropy of supercooledliquid will attain a negative value, even beforereaching absolute zero temperature. Thisinfringes the third law of thermodynamics,which states that the entropy of a perfect crystalis zero at 0K. The system avoids this entropycrisis by deviating to form the glassy state fromthe supercooled liquid state. This deviationoccurs at Tg. The isoentropic point of thesupercooled liquid and crystal is termed the‘Kauzmann temperature’ (24) (TK in Figure 2).TK is believed to be a temperature of zeromolecular mobility and approximates to“Tg-50°C” (25).
These excess properties of the amorphous stateare responsible for its higher solubility andreactivity. These properties also confer physicalinstability on the amorphous system has atendcy to convert to a stable crystalline state.This process is referred to as devitrification(26).
Solubility advantage from amorphous systems
An amorphous form possesses a higherapparent solubility in comparison to itscrystalline counterpart. Hancock et al. (27)proposed an equation to predict the theoreticalincrease of solubility gained from anamorphous system compared to its crystallinecounterpart as shown in Equation 1.
Eq. 1G RTTa c T
a
Tc
, ln
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 72

Review Paper
Figure 3 Comparison of dissolution profile of crystallineand amorphous curcumin. (Reproduced with modificationand permission from Reference 29).
Where; G is the difference in free energy, R is agas constant, T is the given temperature, and(σa/ σc) is the solubility ratio of the amorphousand the crystalline form, respectively.
The increase in solubility of the amorphousdrug, determined experimentally, remains lesserthan theoretically predicted values in mostcases. When an amorphous drug is added to theaqueous media, it shows a rapid increase insolubility, and appears as a ‘peak’ in thesolubility curve. Subsequently, a decrease in thesolubility is observed as the amorphous formdevitrifies to the crystalline form. Theappearance of a peak and subsequent decreasein the solubility curve of an amorphous systemis known as the ‘spring and parachute effect’,and has been reported by several researchers(28, 29). The decrease in solubility can beexplained by a devitrification mediated processwhich is caused by two simultaneouslyoccurring phenomena: (i) crystallization of thedrug due to supersaturation, and (ii)plasticization of undissolved amorphous drugby water and its subsequent devitrification. Thelatter phenomenon initiates from the surface ofthe particles and percolates into the bulk of theamorphous sample. Together these twophenomena manifest as a downward trend inthe solubility curve for the amorphous drug.
The solubility profile of amorphous andcrystalline curcumin is shown in Figure 3wherein the amorphous form of curcuminshows the ‘spring and parachute effect’.
Although the initial apparent solubility of theamorphous form is noticeably higher, theconversion of the amorphous material to itscrystalline counterpart creates considerablechallenges during dissolution. Strategies havebeen devised to kinetically ‘stabilize’ theamorphous form so that the advantage ofincreasing solubility of these systems can beretained.
Strategies for stabilizing amorphous forms
An amorphous form can be stabilized primarilyby minimizing its molecular mobility which inturn prevents crystallization. The simplestapproach is to store an amorphous form belowits Tg. An empirical rule known as the ‘Tg-50 °Crule recommends storage of the amorphousform at temperature 50C below its Tg. Thisretards the molecular mobility of theamorphous state making it sufficiently stable toprovide a practical shelf life (30). However, thisstrategy has limited applications as it affectsstability during shelf life only. It has no effecton subsequent events during dissolution.
A more practical approach towards stabilizingthe amorphous form is formulating an ASDwherein the amorphous drug is incorporatedinto a matrix of excipient(s). Various methodshave been reported for achieving this includingmilling (31), fusion (32), hot melt extrusion(33), freeze drying (34), spray drying (35) andsupercritical fluid precipitation (36). Formationof an ASD provides stability to the amorphousform both during shelf life and dissolution.
Polymers as stabilizers
Polymers consist chemically of repetitivestructural units called monomers. Eachmonomer is covalently linked to anothermonomer forming an extended structuralframework. They can be classified based on
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 73

Review Paper
their origin: a) natural polymers, e.g., cellulosederivatives, starch derivatives, gums, etc., andb ) s y n t h e t i c p o l y m e r s , e . g . ,poly(vinlypyrollidone) and its copolymers,poly(ethyleneglycol) and poly(acrylate) (14).
A polymer that consists of only one type ofmonomer to form a long chain is known as ahomopolymer or simply a polymer. If the chainis composed of two different types ofmonomer units then it is known as acopolymer. Both homopolymers (e.g.,methylcellulose, hypromellose) and copolymers(e.g., copovidone) are widely used aspharmaceutical excipients. From a structuralperspective, polymers can be classified asamorphous, semi-crystalline or crystalline. Bothpolymer strength and stiffness increases with anincrease in the degree of crystallinity as a resultof greater intermolecular interactions (37).
Owing to their complex structural properties,polymers form extensive inter- and intra-chaincross links, ultimately forming a network likestructure. Incorporation of any heterogeneousmolecule (amorphous drug) into these networkshinders the molecular mobility of the latter.
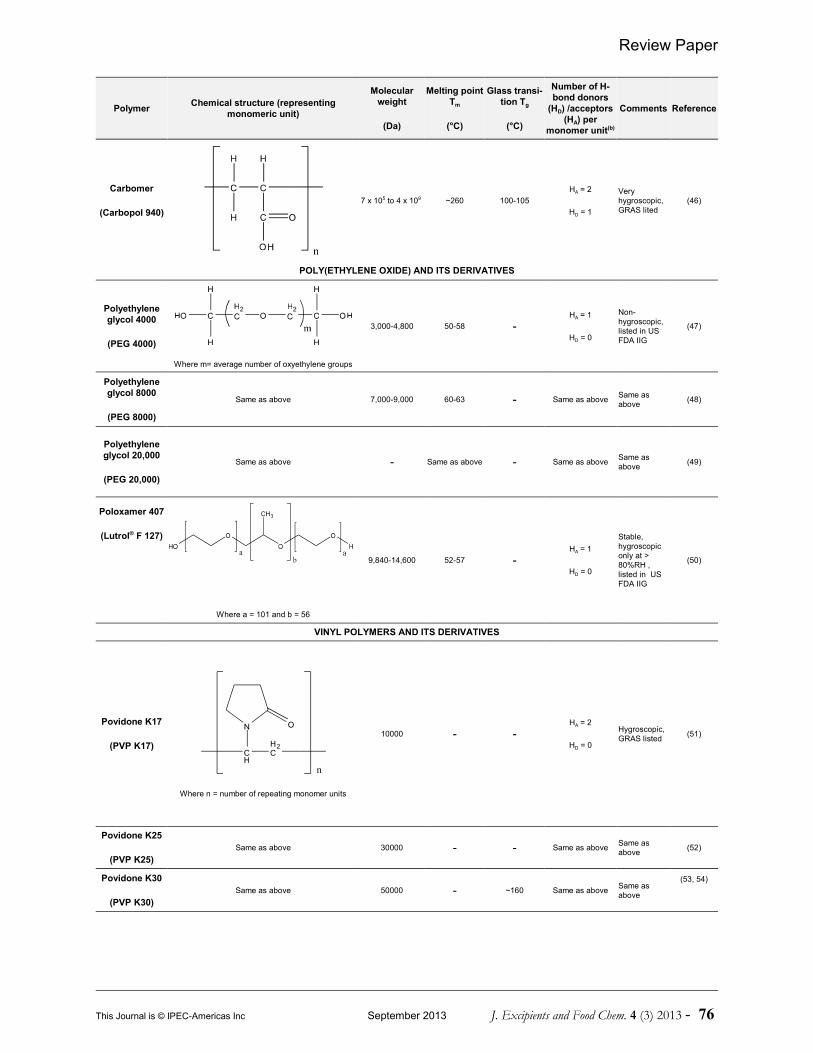
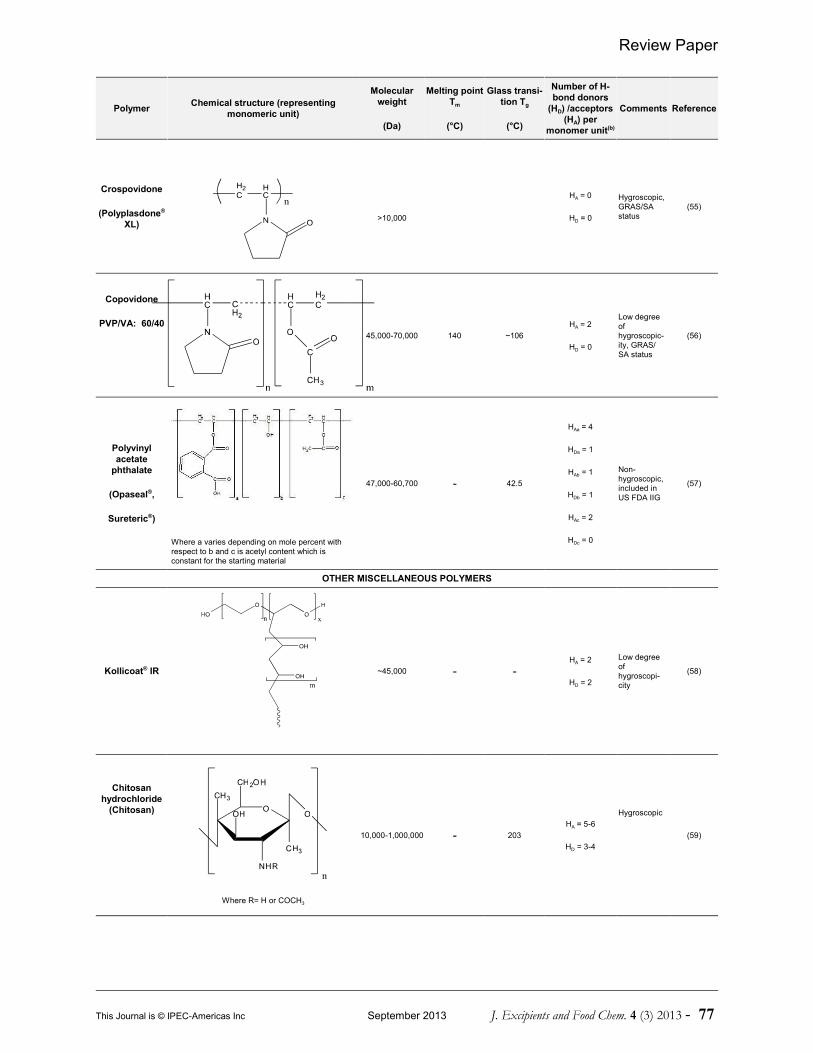
As a result, polymer chains act as crystallizationinhibitors, thereby preserving the viability ofthe amorphous form intact (38, 39). Variouspolymers have been investigated incorporatingthe amorphous drug into matrices in order todevelop PSADs. A comprehensive list of thesepolymers is given in Table 1. Table 1 includesthe polymer name, chemical structure, averagemolecular weight, Tm/Tg and number ofhydrogen bond (H-bond) donors andacceptors.
M E C H A N I S M S F O R S T A B I L I Z I N GAMORPHOUS DRUG IN PASDs
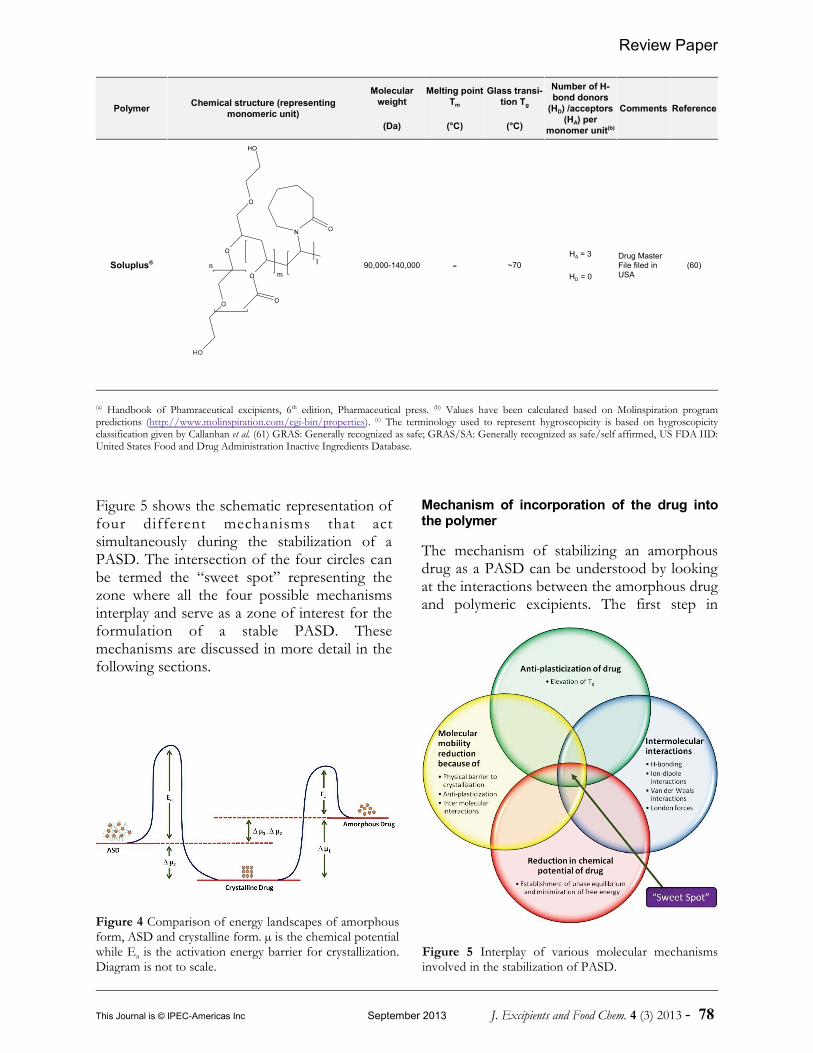
The amorphous form, because of itsthermodynamic properties, less stable and isable undergo phase transformation to a stablecrystalline form. As mentioned previously, theamorphous form can be stabilized byformulating it as a PASD. Figure 4 shows theenergy landscape of a crystalline form, anamorphous form and a PASD. Formulation ofan amorphous drug into a PASD lowers thefree energy of the amorphous form closer tothe energy level of crystalline form (62).
Table 1 Examples of various polymeric carriers employed in the formulation of PASDs(a)
Polymer Chemical structure (representingmonomeric unit)
Molecularweight
(Da)
Melting pointTm
(°C)
Glass transi-tion Tg
(°C)
Number of H-bond donors
(HD) /acceptors(HA) per
monomer unit(b)
Comments Reference
CELLULOSE DERIVATIVES
Methylcellulose 10,000-220,000 290-305 -HA = 5
HD = 0
Slightlyhygroscopic(c)
GRAS listed(31)
Hypromellose
[Hydroxypropyl-methylcellulose
(HPMC)] Where R = H, CH3 or CH2CH(OH)CH2
10,000-1,500,000 190-220 170-180HA = 10 - 17
HD = 0 - 7
Hygroscopicbut stable,soluble incold waterandpracticallyinsoluble inhot water,GRAS listed
(40)
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 74

Review Paper
Polymer Chemical structure (representingmonomeric unit)
Molecularweight
(Da)
Melting pointTm
(°C)
Glass transi-tion Tg
(°C)
Number of H-bond donors
(HD) /acceptors(HA) per
monomer unit(b)
Comments Reference
Hypromelloseacetate
succinate
(HPMCAS)
Where R = H, CH3, CH3CH(OH)CH2, CH3CO, orsuccinoyl
55,000-90,000 - 113±2HA = 10 - 28
HD = 0 – 6
Hygroscopicandsusceptible tohydrolysisuponexposure tomoisture,listed in USFDA IIG
(41)
HPC (L-HPC)
Chemical structure same as HPMC
Substitutions:
Where R = H or CH3 or CH2CH(OH)CH3
50,000-125,000 130-260 -HA = 10 - 16
HD = 0 - 6
Hygroscopicbut stable,GRAS listed
(42)
Hypromellosephthalate(HPMCP)
Where R = H, CH3, CH2CH(OH)CH3,
or
80,000-130,000 150
137 (for HP-50)
133 (for HP-55)
HA = 28
HD = 18
Hygroscopic,listed in USFDA IIG
(43)
POLYACRYLATES AND METHACRYLATES
Ammoniomethacrylatecopolymer
(Eudragit® E)Where R1, R3 = CH3
R2 = CH2CH2N(CH3)2
R4 = CH3, C4H9
- - -HA = 10
HD = 0- (44)
Ammoniomethacrylatecopolymer,
Type A
(Eudragit® RL)
Chemical formula same as above
Substitutions:
Where R1 = H, CH3
R2 = CH3, C2H5
R3 = CH3
R4 = CH2CH2N(CH3)3+ Cl-
- - -HA = 10
HD = 0- (45)
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 75
Review Paper
Polymer Chemical structure (representingmonomeric unit)
Molecularweight
(Da)
Melting pointTm
(°C)
Glass transi-tion Tg
(°C)
Number of H-bond donors
(HD) /acceptors(HA) per
monomer unit(b)
Comments Reference
Carbomer
(Carbopol 940)7 x 105 to 4 x 109 ~260 100-105
HA = 2
HD = 1
Veryhygroscopic,GRAS lited
(46)
POLY(ETHYLENE OXIDE) AND ITS DERIVATIVES
Polyethyleneglycol 4000
(PEG 4000)
Where m= average number of oxyethylene groups
3,000-4,800 50-58 -HA = 1
HD = 0
Non-hygroscopic,listed in USFDA IIG
(47)
Polyethyleneglycol 8000
(PEG 8000)
Same as above 7,000-9,000 60-63 - Same as above Same asabove (48)
Polyethyleneglycol 20,000
(PEG 20,000)
Same as above - Same as above - Same as above Same asabove (49)
Poloxamer 407
(Lutrol® F 127)
Where a = 101 and b = 56
9,840-14,600 52-57 -HA = 1
HD = 0
Stable,hygroscopiconly at >80%RH ,listed in USFDA IIG
(50)
VINYL POLYMERS AND ITS DERIVATIVES
Povidone K17
(PVP K17)
Where n = number of repeating monomer units
10000 - -HA = 2
HD = 0
Hygroscopic,GRAS listed (51)
Povidone K25
(PVP K25)Same as above 30000 - - Same as above Same as
above (52)
Povidone K30
(PVP K30)Same as above 50000 - ~160 Same as above Same as
above
(53, 54)
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 76
Review Paper
Polymer Chemical structure (representingmonomeric unit)
Molecularweight
(Da)
Melting pointTm
(°C)
Glass transi-tion Tg
(°C)
Number of H-bond donors
(HD) /acceptors(HA) per
monomer unit(b)
Comments Reference
Crospovidone
(Polyplasdone®
XL)>10,000
HA = 0
HD = 0
Hygroscopic,GRAS/SAstatus
(55)
Copovidone
PVP/VA: 60/4045,000-70,000 140 ~106
HA = 2
HD = 0
Low degreeofhygroscopic-ity, GRAS/SA status
(56)
Polyvinylacetate
phthalate
(Opaseal®,
Sureteric®)
Where a varies depending on mole percent withrespect to b and c is acetyl content which isconstant for the starting material
47,000-60,700 - 42.5
HAa = 4
HDa = 1
HAb = 1
HDb = 1
HAc = 2
HDc = 0
Non-hygroscopic,included inUS FDA IIG
(57)
OTHER MISCELLANEOUS POLYMERS
Kollicoat® IR ~45,000 - -HA = 2
HD = 2
Low degreeofhygroscopi-city
(58)
Chitosanhydrochloride
(Chitosan)
Where R= H or COCH3
10,000-1,000,000 - 203HA = 5-6
HD = 3-4
Hygroscopic
(59)
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 77
Review Paper
Polymer Chemical structure (representingmonomeric unit)
Molecularweight
(Da)
Melting pointTm
(°C)
Glass transi-tion Tg
(°C)
Number of H-bond donors
(HD) /acceptors(HA) per
monomer unit(b)
Comments Reference
Figure 5 Interplay of various molecular mechanismsinvolved in the stabilization of PASD.
Figure 4 Comparison of energy landscapes of amorphousform, ASD and crystalline form. µ is the chemical potentialwhile Ea is the activation energy barrier for crystallization.Diagram is not to scale.
Soluplus® 90,000-140,000 - ~70HA = 3
HD = 0
Drug MasterFile filed inUSA
(60)
(a) Handbook of Phamraceutical excipients, 6th edition, Pharmaceutical press. (b) Values have been calculated based on Molinspiration programpredictions (http://www.molinspiration.com/cgi-bin/properties). (c) The terminology used to represent hygroscopicity is based on hygroscopicityclassification given by Callanhan et al. (61) GRAS: Generally recognized as safe; GRAS/SA: Generally recognized as safe/self affirmed, US FDA IID:United States Food and Drug Administration Inactive Ingredients Database.
Figure 5 shows the schematic representation offour different mechanisms that actsimultaneously during the stabilization of aPASD. The intersection of the four circles canbe termed the “sweet spot” representing thezone where all the four possible mechanismsinterplay and serve as a zone of interest for theformulation of a stable PASD. Thesemechanisms are discussed in more detail in thefollowing sections.
Mechanism of incorporation of the drug intothe polymer
The mechanism of stabilizing an amorphousdrug as a PASD can be understood by lookingat the interactions between the amorphous drugand polymeric excipients. The first step in
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 78

Review Paper
Figure 6 Schematic representation of formation of aPASD through hot melt extrusion and spray dryingprocesses.
designing PASD is incorporating an amorphousdrug into the matrix of a polymeric excipient(s).The incorporation of an amorphous drug in apolymer matrix results in intimate contactbetween the drug and the polymer. Drugmolecules occupy void spaces between thepolymer chains, thus making the polymerchains relatively flexible (63). The incorporationof a drug into a polymer matrix followsdifferent steps, depending on how the PASD isformed. Figure 6 shows the schematicrepresentation of the incorporation of a druginto the polymer using hot melt extrusion andspray drying, both commonly used processes inthe pharmaceutical industry. Hot meltextrusion, also known as melt cooling, involvesloosening the polymer chains by heat, followedby the incorporation of the drug molecules.The presence of the polymer usually lowers themelting point of the crystalline drug, known as‘melting point depression’, discussed in detaillater. The molten drug undergoes molecularassociation with polymer chains, governed bycomplementary chemical domains in the drugand the polymer. The drug molecules becomeincorporated into the polymer matrix, thusforming a PASD.
For spray drying, a solution of drug andpolymer in a suitable solvent or solvent mixtureis prepared. The solution of the polymer in the
solvent loosens the cohesive inter- and intra-molecular interactions of the polymer chainsand resulting in the formation of solvent-polymer interactions. Drug molecules that aredissolved in the solvent are incorporated intothe loosened polymer chains. The solutioncontaining polymer and drug is then spray driedto remove the solvent, thus forming the PASD.
PASDs prepared by different methods do notnecessarily exhibit the same physical properties.Dong et al. (64), performed a comparativeevaluation of PASDs of Compound A inHPMCAS, prepared by hot-melt extrusion andsolvent co-precipitation process. Powder X-raydiffractometry, thermal analysis and watervapor sorption analysis indicated that therewere no significant differences between thePASDs prepared by either method. However,the PASD prepared by co-precipitation wasmore porous and had a larger specific surfacearea than the product prepared by hot-meltextrusion. Dissolution studies showed that theco-precipitated product had a faster dissolutionprofile, but slower, intrinsic dissolution rate,than the hot-melt extruded product. Both theproducts showed acceptable physical stabilityafter storage at 40°C/75% RH for 3 months.The hot-melt extruded product was found to bemore stable in an aqueous suspension.
Anti-plasticization
Anti-plasticization is widely used in theliterature term for when the mechanicalproperties of a substance change into stiff andbrittle when another substance is added (65).From a thermodynamic perspective, anti-plasticization can be explained as an increase inthe Tg of the system. When a low Tg compoundis mixed with a high Tg compound, the Tg ofcorresponding mixture would fall somewhere inbetween the Tg’s of both components.
Mixing of an amorphous drug that has a low Tgwith a high Tg polymer at the molecular level ina PASD leads to the development of a systemwith Tg intermediate to these two components.Hence, the Tg of the drug increases incomparison to its native amorphous state.Thus, the amorphous drug undergoes anti-
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 79

Review Paper
KCCCKp
p
1
2
plasticization, whereas the Tg of the polymerdecreases and it undergoes plasticization. Anti-plasticization decreases the molecular mobilityof the drug, stabilizing the amorphous state. Interms of free volume, the energy required forthe amorphous drug to reach the critical freevolume increases, leading to its ‘stabilization’.The Tg of the drug-polymer mixture can bepredicted according to Fox (66) (Equation 2),Gordon-Taylor (67) (Equation 3), Couchman-Karasz (68) (Equation 4) or Kwei (24)(Equation 5).
Eq. 21 1
1
2
2TwT
wTg g g
Eq. 3Tw T K w Tw K wgg GT g
GT
1 1 2 2
1 2
Eq. 4Tw T K w Tw K wgg CK g
CK
1 1 2 2
1 2
Eq. 5T w T w T qw wg g g 1 1 2 2 1 2
Where; Tg1 and Tg2 are the glass transitionstemperatures of drug and polymer, respectively,and w1 and w2 indicate their weight fractions.KGT, KCK and q are the constants whichindicate a measure of interaction between twocomponents. KGT and KCK may be expressedmathematically as shown in Equation 6 and 7.
Eq. 6KTTGTg
g
1 1
2 2
Where; ρ1 and ρ2 are the densities of two com-ponents;
Eq. 7
Where; Cp1 and Cp2 are the specific heatcapacities of two components.
These equations can be used for the predictionof Tg for drug-polymer mixture and are usefultools for designing PASDs. It has beenobserved that experimental Tg values of PASDsdeviate from theoretically predicted Tg values.
The reasons for these deviations can beascribed to ‘volume non-additivity’ and ‘non-ideality in mixing’ of drug and polymer systems(68).
When the drug is mixed with a polymer in aPASD, they interact with each other. Thisinteraction can be represented as following:
(1) D-D + E-E > 2 (D-E)(2) D-D + E-E < 2 (D-E)(3) D-D + E-E = 2 (D-E)
Where; D and E indicate drug and excipient,respectively.
In the first case, the interaction between theindividual components, i.e., D-D and E-E isstronger than the interaction between the drugand excipient (D-E). Thus, when a PASD isformed, there would be a net contraction in thevolume. In the second case, D-D and E-E areweaker than D-E and thus a net expansion isobtained. The third case indicates the idealsituation wherein there is no net increase ordecrease in volume and perfect volumeadditivity is found (26, 68).
Non-ideality in mixing is another reason fordeviations between experimental and predictedTg values. It can be predicted by determiningexcess enthalpy, entropy and free energy ofmixing using Equation 8.
Eq. 8
H H H
w C dt w C dT
EmG
mSCL
p pT
T
T
T
g
g
g
g
1 1 2 2
21
expexp
Where; ΔHE is the enthalpy of mixing of amixture, whose Tg has deviated from idealbehavior assuming that the mixing is non-thermal. ΔHm
G and ΔHmSCL are the enthalpies
of mixing of glass and supercooled liquid states,respectively. ΔCp1 and ΔCp2 are specific heatcapacity changes at absolute Tg for drug andpolymer, respectively. w1 and w1 are the weightfractions of drug and polymer, respectively.
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 80

Review Paper
Figure 7 Deviation of experimental Tg from predicted Tgfor amorphous drug/PVP dispersions (a) IMC/PVPsystem, (b) UDCA/PVP system and (c) IDP/PVP system.Solid lines represent predicted Tg values using theCouchman-Karasz version of the Gordon-Taylor equation(reproduced with permission from Reference 70).
Tg-exp is the experimentally observed Tg of themixture.
When the entropy changes on mixing are inexcess of those corresponding to a regularsolution (i.e., the entropy change is purelycombinatorial) there is an excess entropy ofmixing which is calculated according toEquation 9.
Eq. 9
S S S
w C d T w C d T
EmG
mSCL
p pT
T
T
T
g
g
g
g
1 1 2 2
21
ln lnexpexp
The excess entropy of mixing (ΔSE) is related tothe difference in the entropy of mixing in theglass (ΔSm
G) and the supercooled liquid states(ΔSm
SCL) and is the difference between entropyof the mixture at Tg-exp and the theoreticalentropy of the mixture at a temperature thatrepresents the theoretically predicted Tg of arandomly mixed system. The relativecontribution of ΔHE and ΔSE to the excess freeenergy of mixing, ΔGE can be calculated byfollowing Equation 10.
Eq. 10 G H T SE Eg
E exp
Crowley et al. (70) reported the non-ideality ofmixing in some PASDs including indomethacin(IMC), ursodeoxycholic acid (UDCA) andindapamide (IDP), prepared using PVP (70).Negative deviations were observed inIMC/PVP and UDCA/PVP PASDs, where theTg of the binary mixture was found to be lowerthan predicted values at PVP concentrationsgreater than 50%. Similarly, positive deviationwas observed in IDP/PVP PASDs, where theTg of the binary mixture was found to be higherthan the predicted Tg in intermediate dispersioncompositions of 30-80% w/w PVP. Figure 7shows the positive and negative deviations ofthese three dispersion systems. Thesedeviations from predictions were explainedbased on the relative extent of hetero-molecularto homo-molecular interactions. IMC/PVP andUDCA/PVP dispersions experienced negativedeviation at high PVP compositions, as thelevel of drug-PVP interaction was less than thesum of drug-drug and PVP-PVP interactions at
these compositions. However, stronger IDP-PVP interactions relative to the homo-molecular interactions were responsible forpositive deviation from predicted Tg values.
Inter-molecular interactions
Besides anti-plasticization, drug-excipientinteractions also contribute towards‘stabilization’ of a PASD. Several studies haveshown the formation of ion-dipole interactionsand intermolecular H-bonding between drugsand polymers.
Makiko et al. (71) reported the role of specificinteractions in stabilizing solid dispersions.They examined the effects of differentsubstituents on benzodiazepines (nitrazepam,nimetazepam, diazepam and medazepam) inASDs with phosphatidylcholine. It was foundthat nitrazepam was the only one of the fourwhich remained stable at the limit of miscibilityafter one year of storage. Nitrazepam was alsothe only compound with an H-bond donorgroup. Interactions between nitrazepam andphosphatidylcholine were verified usinginfrared spectroscopy (IR). Specific interactionswere not observed with the other threebenzodiazepines. It was concluded that someinteraction between a drug and a carrier wasnecessary for a solid dispersion to stabilize.
Another interesting study was carried out byTaylor et al. (72) which highlighted theimportance of chemical interactions betweenamorphous IMC and PVP. ASDs, of IMC withPVP were prepared by a solvent evaporationtechnique and studied using vibrational
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 81

Review Paper
Figure 9 Stereoview of intermolecular associationbetween CEL and PVP, the monomeric unit of PVP. TheH-bonding is represented by dotted lines between theinteracting groups of CEL and PVP (reproduced withpermission from Reference 75).
Figure 9 Stereoview of intermolecular associationbetween CEL and PVP, the monomeric unit of PVP. TheH-bonding is represented by dotted lines between theinteracting groups of CEL and PVP (reproduced withpermission from Reference 75).
Figure 8 chemical structures of (a) celecoxib and (b) PVP.(c) FTIR spectra indicating the molecular interactions ofCEL and PVP (reproduced with permission fromReference 75).
spectroscopy. The investigation confirmed theexistence of drug-polymer interactions even atlower concentrations of PVP where anti-plasticization had little effect on stabilization.This study highlighted the changes in stretchingvibrations of IMC and PVP. The authorssuggested that interactions between PVP andIMC were mainly H- bonding interactions. PVPcan act as a proton acceptor (through either theO or N atoms of the pyrrole ring) and IMC hasonly one proton donor site, the OH group ofthe carboxyl acid function. IR spectra of thecarbonyl region for IMC, PVP and soliddispersions, confirmed the presence of H-bonding interactions between IMC and PVP inASDs.
These conclusions were in accordance with thework carried out by Yoshioka et al. (72), whoconcluded that the inhibition could not beexplained solely by the antiplasticizing effect ofthe polymer. Moreover, in solution PVP wasfound to interact with numerous organicmolecules and it was reported that themechanism of crystallization inhibition isrelated to the extent of interaction betweendrug and polymer.
The contribution of drug-polymer interactionhas further been highlighted in a study byKhougaz and Clas (74). They demonstratedthat the onset of crystallization relative toamorphous MK-0591 increased in all SDs evenwhere Tg of the polymer blend (PVP K-12 and
PVP/VA) was less than Tg of drug. Thiscounter-intuitive finding led to the conclusionthat anti-plasticization alone cannot explaincrystallization inhibition. They demonstrated anion-dipole interaction between PVP and theCOO-Na+ moiety of MK-0591 based on anincrease in the intensity of the carbonylstretching in the amide group of PVP observedin the IR spectrum. This contributed to thestabilization of the ASD.
Another study highlighting the role ofmolecular interaction in the stability ofcelecoxib(CEL)-PVP amorphous systems wascarried out by Gupta et al. (75). Amorphoussolid dispersions of celecoxib in PVP wereprepared using a solvent evaporation method.The authors demonstrated that molecularinteractions play an important role in thestabilization of amorphous forms. This wasconfirmed by computational simulation,Differential Scanning Calorimetric (DSC) andFourier Transform Infrared (FTIR)spectroscopy. DSC analysis showed that the Tgvalues for CEL-PVP binary systems of varyingPVP composition exhibited a positive deviationfrom predicted Tg values based on the Gordon-Taylor/Kelley-Bueche equation. The positivedeviation from expected Tg values wasattributed to intermolecular interactionsbetween CEL and PVP. This was furtherconfirmed by FTIR analysis.
FTIR studies confirmed the formation of H-bonding between -NH2 groups of CEL and
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 82

Review Paper
Figure 10 Events taking place during glass formation. Localmotions in glass leading to structural relaxation.
–C=O groups of PVP. Figure 8 shows thechemical structure of PVP and CEL and theFTIR spectra of the corresponding PASD. Theappearance of a band at 1662 cm-1 foramorphous CEL was attributed to thealteration of intermolecular H-bonding of CELin the crystalline and amorphous forms.Spectral shifts from 1662-1655 cm-1 at low PVPcontent (0.25% w/w to 1.00% w/w) impliedthat CEL-PVP binary amorphous systemsconsisted of CEL-CEL as well as CEL-PVP inH-bonded states. Computer simulations ofCEL and N-vinyl-2-pyrrolidone (NVP), themonomeric unit of PVP, also favoredinteractions between –C=O group of NVP and–NH2 group of CEL, as shown in Figure 9.
In some cases, studies of H-bondinginteractions are not possible because of thechemistry of the system. Vippagunta et al. (76)concluded that fenofibrate did not exhibitspecific interactions with polyethylene glycol(PEG), irrespective of the number of H-bonddonating groups present. The absence ofspecific chemical interactions betweenketoconazole and PVP K25 was reported byVan den Mooter et al. (77). Nevertheless, themajority of the drugs contain H-bonding sitesand thus exhibit specific directional H-bondinginteractions that contribute to their stabilizationin ASDs.
Alteration of chemical potential of a drug
Chemical potential, also known as partial molarfree energy, is a form of potential energy thatcan be absorbed or released during a chemicalreaction (79). All systems tend to reduce theirchemical potential to the state of lowestpotential energy. A drug in its nativeamorphous state has a higher chemicalpotential compared to its crystalline state whichacts as a driving force for crystallization. Thechemical potential of the amorphous drug canbe lowered by mixing it with a polymer. In abinary blend, thermodynamic criterion for twophases at equilibrium is that the chemicalpotentials of the components should be equalin two coexisting phases represented inEquation 11.
Eq. 11 1 1a b 2 2
a b
Where; 1 and 2 represent the two components,and a and b represent the phases.
If the drug and polymer form a miscibleamorphous system, the chemical potential ofthe drug in such a system is less than the pureamorphous drug. The reduction in chemicalpotential results from the increased entropy in amixture. Additionally, if the strength and/orextent of enthalpic interactions are greater inthe mixture than the sum of the interactions inthe pure components, the drug chemicalpotential is further reduced. Hence, an idealsolid dispersion will be in chemical equilibriumor phase equilibrium such that the total sum ofchemical potentials is zero and the free energyof the system is at minimum (80).
Reduction in molecular motions of drug
The stabilization of the amorphous drug inPASDs can also be explained in terms ofmolecular mobility. Amorphous systems byvirtue of their short range order possess excessproperties such as enthalpy, entropy and freeenergy relative to the crystalline state (81). Dueto their thermodynamic instability they tend toapproach equilibrium over extended periods oftime when stored at a temperature close to Tg.The excess enthalpy and entropy of amorphousforms present in entrapped frozen molecules islost gradually on storage at temperatures closeto Tg for a specified period of time. As a resultthere is a reduction in the molecular mobility,enthalpy, entropy, and free volume as a
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 83
Review Paper
Figure 11 Enthalpy recoveries of CEL and CELdispersions. Key: (Ib) is CEL + PVP K30, (II) is CEL +PVP K17, (III) is CEL + HPMC and (IV) is CEL +Trehalose. (reproduced with permission from Reference85).
function of storage time. This phenomenon isknown as structural relaxation (82). Figure 10represents a sequence of events in terms ofmolecular mobility that happens during glassformation.
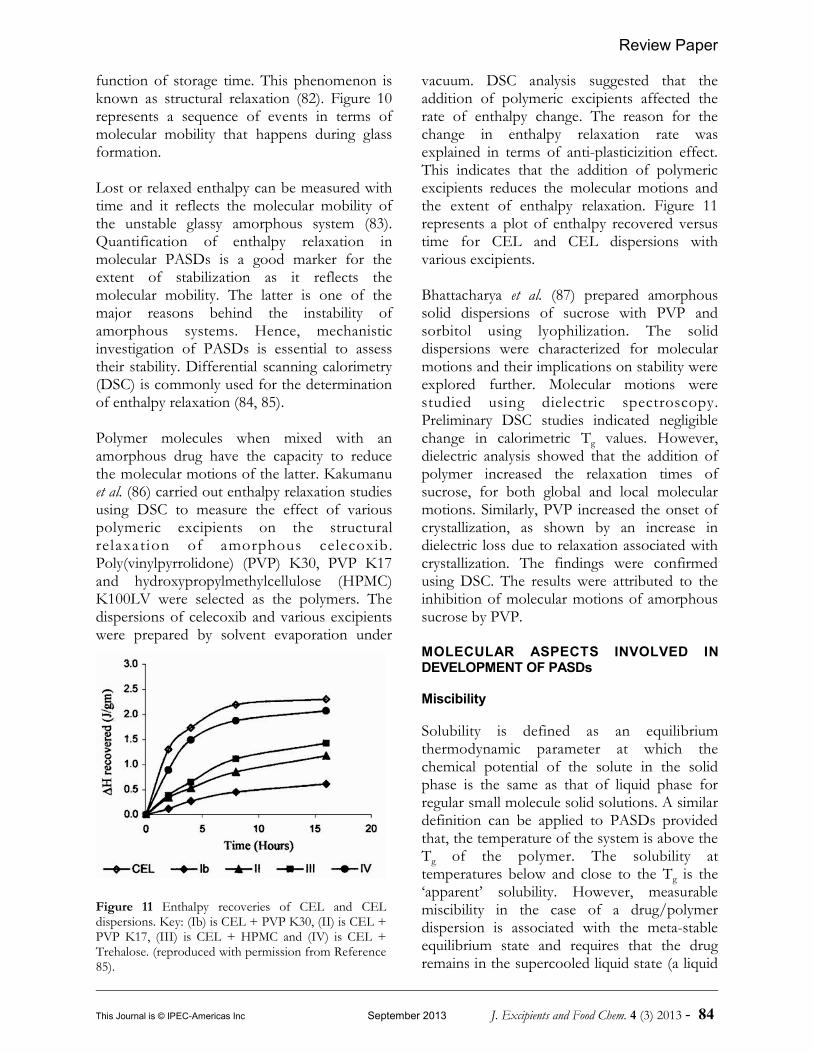
Lost or relaxed enthalpy can be measured withtime and it reflects the molecular mobility ofthe unstable glassy amorphous system (83).Quantification of enthalpy relaxation inmolecular PASDs is a good marker for theextent of stabilization as it reflects themolecular mobility. The latter is one of themajor reasons behind the instability ofamorphous systems. Hence, mechanisticinvestigation of PASDs is essential to assesstheir stability. Differential scanning calorimetry(DSC) is commonly used for the determinationof enthalpy relaxation (84, 85).
Polymer molecules when mixed with anamorphous drug have the capacity to reducethe molecular motions of the latter. Kakumanuet al. (86) carried out enthalpy relaxation studiesusing DSC to measure the effect of variouspolymeric excipients on the structuralrelaxat ion of amorphous celecoxib.Poly(vinylpyrrolidone) (PVP) K30, PVP K17and hydroxypropylmethylcellulose (HPMC)K100LV were selected as the polymers. Thedispersions of celecoxib and various excipientswere prepared by solvent evaporation under
vacuum. DSC analysis suggested that theaddition of polymeric excipients affected therate of enthalpy change. The reason for thechange in enthalpy relaxation rate wasexplained in terms of anti-plasticizition effect.This indicates that the addition of polymericexcipients reduces the molecular motions andthe extent of enthalpy relaxation. Figure 11represents a plot of enthalpy recovered versustime for CEL and CEL dispersions withvarious excipients.
Bhattacharya et al. (87) prepared amorphoussolid dispersions of sucrose with PVP andsorbitol using lyophilization. The soliddispersions were characterized for molecularmotions and their implications on stability wereexplored further. Molecular motions werestudied using dielectric spectroscopy.Preliminary DSC studies indicated negligiblechange in calorimetric Tg values. However,dielectric analysis showed that the addition ofpolymer increased the relaxation times ofsucrose, for both global and local molecularmotions. Similarly, PVP increased the onset ofcrystallization, as shown by an increase indielectric loss due to relaxation associated withcrystallization. The findings were confirmedusing DSC. The results were attributed to theinhibition of molecular motions of amorphoussucrose by PVP.
MOLECULAR ASPECTS INVOLVED INDEVELOPMENT OF PASDs
Miscibility
Solubility is defined as an equilibriumthermodynamic parameter at which thechemical potential of the solute in the solidphase is the same as that of liquid phase forregular small molecule solid solutions. A similardefinition can be applied to PASDs providedthat, the temperature of the system is above theTg of the polymer. The solubility attemperatures below and close to the Tg is the‘apparent’ solubility. However, measurablemiscibility in the case of a drug/polymerdispersion is associated with the meta-stableequilibrium state and requires that the drugremains in the supercooled liquid state (a liquid
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 84

Review Paper
Figure 12 Schematic illustration of Flory-Huggins theory.
at a temperature above Tg and below Tm),without crystallization within the experimentaltime frame. This can only be achieved whenboth of the components of the binary mixtureform a single amorphous phase. In order toform a single phase, the two liquids should bethermodynamically miscible. The system isperturbed during the preparation stage and thesystem must re-equilibrate at the post-processing conditions. The system may remainas a single phase or become metastable/unstable. Thermodynamics dictates thatmetastable/unstable systems tend to phaseseparate. However, owing to slow dynamics,the dispersion may be sufficiently kineticallystable for its intended use. Formation of asingle amorphous phase of the drug with theexcipient is most commonly assessed from theformation of a single Tg in DSC. However,systematic study using advanced techniquesshow the homogeneity of PASD (88-90).
The extent of mixing is also governed by thenature and magnitude of the interactionsbetween the drug and the polymer. Themagnitude of enthalpic interactions in each ofthe pure amorphous components (cohesive orhomo-molecular interactions) relative to theenthalpic interactions in the blend (adhesive orhetero-molecular interactions) essentiallydetermines the miscibility of components (91).The relative strength of these interactionsdepends upon the chemistry of the drug andthe polymer.
Tools to assess miscibility of components
Solubility parameter as a predictor ofmiscibility
The use of solubility parameters to predictmiscibility can be applied to low molecularweight materials and polymers. Hildebrand (92)proposed the solubility parameter as a squareroot of the cohesive energy density (CED).CED represents the total attractive forceswithin a condensed state material and can bedefined as the amount of energy required tocompletely remove a unit volume of moleculesfrom their neighbours to infinite separation.Further, CED can be used to predict the
solubility of one material into another material.Similar values of CED for two materialsindicate the likelihood of miscibility, as bothmaterials possess similar interactions. Thus, theoverall energy needed to facilitate mixingshould be small, as the energy required to breakthe interactions within the components shouldbe compensated by the energy released due tothe interactions between unlike molecules.
Greenhalgh et al. (93) reported such types ofinteractions and possible incompatibilities insolid dispersions of hydrophobic drugs withhydrophilic carriers. They used the 'Hildebrandsolubility parameters' as a means to interpretsuch interactions. A trend between thedifferences in drug/excipient solubilityparameters and immiscibility was reported.Incompatibilities were evident when largesolubility parameter differences existed betweenthe drug and the carrier. Table 2 representsvarious drug/excipient systems, their miscibilityand their corresponding solubility parametervalues.
Despite their ease of application, using the'Hildebrand solubility parameters' to accu-ratelypredict the phase diagram, and the specific levelof interaction between drugs and excipients,remains limited. They only provide informationabout the overall cohesive energy in materials,but little information on relative strengths ofvarious types of forces viz dispersion, polar andH-bonding.
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 85

Review Paper
Table 2 Solubility parameters as indicator of miscibilitybetween drug and excipient (reproduced with permissionfrom Reference 93).
DRUG/CARRIERSYSTEM DESCRIPTION δ (MPa)0.5 δ
Ibuprofen/PVP Forms a 1:1 drug: polymercomplex in the solid state 20.9/22.5 1.6
Ibuprofen/LutrolF68
Forms a eutectic system. Eutecticcomposition contained 30-35%w/w Ibuprofen
20.9/19 1.9
Ibuprofen/Maltose Immiscible when both componentsare molten at 1% and 99% w/w 20.9/38.9 18
Ibuprofen/Sorbitol Immiscible when both componentsare molten at 1% and 99% w/w 20.9/38.2 17.3
Ibuprofen/Xylitol
Immiscible when both componentsare molten at 25% w/w, 50% w/w,75% w/w and 99.9% w/w,Ibuprofen
20.9/37.1 16.2
δ (MPa)0.5 represents the solubility parameter (Mega Pascals)
δ represents the difference in solubility parameter between the drugand excipient
Improved predictive qualities can be obtainedusing 'Hansen partial solubility parameters' (94).These can be predicted by various groupcontribution methods tha t involveconsideration of contribution of eachfunctional group to the cohesive energy of themolecule. Methods such as, ‘Hoftyzer and Van-Krevelen’, ‘Hoy’ and ‘Fedors’ (95) providespecific numerical values to each functionalgroup that provides theoretical estimates ofsolubility parameters (96, 97). One practicalhurdle of calculating Hansen parameters usinggroup contribution methods is the limited dataavailable for different structural groups whichlimits its application to complex molecules suchas polymers.
Flory-Huggins theory
Solution models involving small molecules andsolvents are inadequate to describe how smallmolecules mix into the polymers. The Flory-Huggins lattice theory provides an explanationof the thermodynamics of polymer solutions(98, 99). In the case of polymer solutions, thefree energy of the mixture is more accuratelydescribed in terms of volume fraction of thematerial rather than mole fraction. This isbecause entropy of mixing for large molecularweight materials is significantly reduced due tothe limited number of possible configurationsof two components of the binary mixture (100,101). The Flory-Huggins lattice theory takesmolecular size into account when predicting theentropy of mixing.
Consider a large molecular weight polymermixed with a small molecular weight solvent.Flory-Huggins theory defines a hypothetical“lattice” in the space (see Figure 12). The sizeof each position in the lattice may be describedby the molecular volume of a solvent moleculeor any other convenient volume. Eachcomponent of the mixture will occupy severaladjacent positions in the lattice and the numberof lattice positions required to accommodateeach component is equal to the ratio ofmolecular volume of each component to thatof the lattice cell.
The Flory-Huggins model can be applied todescribe the thermodynamics of drug-polymersystems by considering an amorphous drug assimilar to a solvent. Following this rationale,and after the addition of the Flory-Hugginsinteraction parameter, χ to account for theenthalpy of mixing, the free energy of mixing ofa drug-polymer system, GM can be describedusing Equation 12.
Eq. 12
GRT
n n
n x
Mdrug drug polymer polymer
drug polymer
ln ln
Where; ndrug and npolymer denote the number ofmoles of drug and polymer respectively. Φdrugand Φpolymer denote the volume fraction of thedrug and polymer, respectively. R denotes gasconstant and T denotes absolute temperature ofthe system. The application of above equationto a drug-polymer system enables theevaluation of thermodynamic parametersrelated to mixing of drug and polymer.
Most of the experimental techniques forprediction of interaction parameters, such asvapour pressure reduction, inverse gaschromatography and osmotic pressuredepression are not applicable to drug-polymerblends because of their viscous and non-volatilecharacter. Methods applicable to drug-polymersystems are: 1) a priori estimates using solubilityparameters and 2) melting point depression(102).
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 86

Review Paper
Figure 13 Schematic representation of the phenomenonoccurring during dissolution of encapsulated amorphouscelecoxib (A-CLB) and amorphous celecoxib soliddispersion (ACSD) (reproduced with permission fromReference 107).
Estimation of Flory-Huggins interactionparameter using solubility parameter
Solubility parameter differences have beenproposed as a means to predict miscibility inpharmaceutical systems. The interactionparameter (χ) can be estimated from solubilityparameters, δ as shown in Equation 13.
Eq. 13 x VRTsite
drug polymer 2
Where; Vsite denotes the volume of thehypothetical lattice.
The relationship shown in Equation 13 assumesthat enthalpic interactions between unlikespecies are equal to like species. Thisassumption is reasonable for systemscontaining van der Waals type interactions, butnot for systems with specific directionalinteractions. Many drugs and pharmaceuticalpolymers are known to involve specificinteractions and hence solubility parameterestimates of the interaction parameter can beinaccurate for such systems.
Estimation of Flory-Huggins interactionparameters using melting point depression
When a mixture of a drug and a polymer issubjected to thermal analysis such as DSC, themelting point of the drug system is generallyobserved at temperatures lower than themelting point of the drug alone, provided thetwo components are miscible. Thisphenomenon has been introduced previously inthis paper and is known as ‘melting pointdepression’.
Melting point depression is a manifestation ofthe reduced thermodynamic activity of thedrug, in the presence of the polymer relative tothe activity of the pure drug. Systematicexplanation of the reason behind melting pointdepression can be explained by the reduction invapour pressure of the drug and increasedentropy (S) after mixing. The melting point isthe temperature at which vapour pressure ofsolid equals the vapour pressure of liquid andaddition of non-volatile substance decreases thevapour pressure of solid and as a result the
melting point decreases. Moreover, it is obviousthat polymer molecules do not interfere withmolecular interactions of drug in the crystallinestate, but in turn they increase the entropy ofthe liquid state after melting. Hence, ΔSincreases for a solid, melting into an impureliquid (polymer molecules in molten drug)compared to a pure liquid. At the melting point(Tm), the change in free energy oftransformation from solid to liquid (ΔG) equalszero. Hence, solving the equation ΔG = ΔH -TΔS and assuming the value of ΔG=0 at Tm,yields T = ΔH/ΔS. If ΔS increases, meltingtemperature should decrease (63).
Melting point depression arises because thechemical potential of the drug in solid andliquid phase must be identical at the meltingpoint. The drug in amorphous molecular soliddispersion possesses a chemical potential equalto that of the crystalline drug at a temperaturelower than the fusion temperature of the puredrug, thus resulting in depression of the meltingpoint. This approach has been reported to beuseful to estimate the interaction parameter indrug-polymer systems. The prediction of theinteraction parameters between drug andpolymer using melting point depression data isshown in Equation 14.
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 87

Review Paper
Eq. 14
1 1
1 12
T T
RH m
x
Mmix
Mpure
fusdrug polymer polymer
ln
Where; TMmix is the melting temperature of the
drug in the presence of the polymer, TMpure is
the melting temperature of the drug in theabsence of the polymer, ΔHfus is the heat offusion of the pure drug, and m is the ratio ofthe volume of the polymer to that of the latticesite (defined here by the volume of the drug).
The slope of a linear plot, (1/TMmix- 1/TM
pure) *(ΔHfus/-R) – ln(Φdrug) – (1 – 1/m)Φpolymer vs.[Φpolymer ]2, gives the interaction parameter, χ.Marsac et al. (102) determined the miscibility ofnifedipine in PVP K12 using melting pointdepression data obtained from DSCmeasurements. For these systems, melting pointdepression is usually kinetically favourablebecause of the low melting point of thepolymer being around 60C. At the meltingtemperature of the drug, the polymer is molten,and therefore the drug can easily interact andequilibrate with the polymer in the liquid state.The interaction parameter determined usingEquation 14 was found to be -3.8 for nifedipinein the presence of PVP K12 which indicatedmutual miscibility of the two components. Thisobservation was consistent with experimentalobservations.
ROLE OF DRUG-CARRIER INTERACTIONSDURING PRODUCT PERFORMANCE
Amorphous solid dispersions are expected tomaintain sustained supersaturation levels in theaqueous environment of gastrointestinal tractduring the dissolution process. However, thedissolution of drug from a PASD is influencedby parameters such as the particle size of thePASD (103), type of polymer (104), drug load,interaction strength of the drug/polymercomplex, compaction pressure during tabletting(105), aqueous solubility of components, drugmiscibility in the polymer and the drugrecrystallization tendency. Furthermore, in adosage form, solid-liquid interfacial phenomenacan become the rate controlling step for drugrelease. These multiple factors simultaneouslyinfluence drug release from a PASD (106).
Literature reports indicate that some PASDbased drug products exhibited poor dissolutionperformance, thus limiting their commercialapplication. This deviation from the desiredperformance can be attributed to any of theabove mentioned factors.
In a study carried out by Puri et al. (107), releaseprofiles of CEL from its ASD comprising ofamorphous celecoxib, PVP and meglumine(7:2:1, w/w) were compared with crystallineCEL, in powder and capsule form. Although,the powder displayed 28- to 50-fold higherdissolution efficiency at 60 minutes (DE60), theDE60 of the encapsulated powder wasdrastically reduced due to the formation of anon-dispersible plug. Rapid hydration followedby leaching out of meglumine and formation ofwater mediated H-bond interlinks in the ASDmatrix were found to be the reasons behindincreased interparticle cohesivity. Figure 13represents the schematic representation of thephenomena occurring during dissolution ofencapsulated amorphous CEL (A-CLB) and theASD of amorphous CEL with PVP andmeglumine (ACSD). These conclusions shouldbe considered when formulating an ASD.
CONTRIBUTION OF POLYMERIC CARRIERSTO PROCESSIBILITY
PASDs must be developed into convenientdosage forms such as capsules or tablets forclinical use and successful commercialization.However, manufacturing of PASDs offersnumerous challenges with respect to polymerproperties such as hygroscopicity, tackiness andaging (108). As a result of advances inmanufacturing technologies, many productsbased on ASD platform have beencommercialized (shown in Table 3).
Some of the technologies now in use are hotmelt extrusion, spray drying and supercriticalfluid technology. Hot melt extrusion is a fastcontinuous manufacturing process whichallows the processing of amorphous drugs andhas the advantage of short thermal exposure.Spray drying, either using a conventional spraydryer or a fluidized bed spray dryer hasfacilitated development of PASDs.
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 88

Review Paper
Table 3 List of commercial amorphous solid dispersions
PRODUCT DRUG DISPERSIONPOLYMER MANUFACTURER
Cesamet® Nabilone PVP Eli Lilly and Company
Certican® Everolimus HPMC Novartis
Gris-PEG® Griseofulvin PEG 6000 Pedinol/ValeantPharmacueticals
Intelence® Etravirine HPMC Tibotec/ Johnson &Johnson
Isoptin SR-E® Verapamil HPC/HPMC Abbott
Kaletra®Lopinavir
RitonavirPVP/VA Abbott
Nivadil® Nivaldipine HPC/HPMC Fujisawa PharmaceuticalsCo., Ltd.
Prograf® Tacrolimus HPMC Fujisawa PharmaceuticalsCo., Ltd.
Rezulin®* Troglitazone PVP Pfizer, Inc.
Sporanox® Itraconazole HPMC Janssen Pharmaceuticals,Inc.
*Withdrawn in 2000 due to adverse drug reactions
The polymeric carriers that are used inmanufacturing of PASDs largely contribute totheir processing. Suitability of a manufacturingprocess is greatly influenced by the type ofpolymer used for the preparation of the PASD.
Work carried out by Chokshi et al. (109). Is agood example of a systematic experimentalprotocol for the selection of polymers and theassessment of their suitability for use in the hotmelt extrusion manufacture of a PASD.Indomethacin was used as a model drugtogether with polymers such as Eudragit® EPO,PVP-VA, PVP K30 and Poloxamer 188. Thesedrug-polymer systems were characterized fortheir thermal and rheological properties as afunction of drug concentration. Solubilityparameters were used as initial screening toolsfor the selection of the most useful polymers.Further evaluation of various binary mixturesusing DSC provided an estimate of Tg and Tm.The extrusion temperatures were set at 10 to20C above the Tg or Tm to assure propermaterial flow in the extruder. Rheologicalevaluation was performed to obtain theparameters: zero rate viscosity (viscosities atlow shear rates, whose values are generallyconstant) and activation energy (energyrequired to initiate the flow) which are useful inevaluating the extrudability and predicting themiscibility of the drug/polymer blends. Theunderstanding of thermal and rheologicalproperties of the various drug/polymer
mixtures assisted in establishing the processingconditions for hot melt extrusion, as well as,providing insights into the properties of thefinal extrudates.
During high speed continuous manufacturingoperations, various properties of the materialsuch as Tg, moisture content, hygroscopicityand mechanical properties play a significantrole. An understanding of the contribution ofpolymer carriers to these properties would helpin efficient large scale product development. Anunderstanding of how polymer carrierscontribute to these properties could help indeveloping efficient large scale productmanufacturing. Research shows that during thepreparation of PASDs, the influence ofvariables such as moisture, temperature anddrug loading varies from polymer to polymer(110, 111). Therefore judicious selection offormulation components, especially the choiceof polymers and manufacturing technology, canaid the efficient development andmanufacturing of PASDs.
CONCLUSION
PASDs provide a potential advantage instabilizing the amorphous form of a drug.Polymers play an important role in stabilizationthrough various mechanisms including anti-plastic ization, specific intermolecularinteractions, alteration of chemical potentialand reduction in molecular mobility of theamorphous drug. Molecular aspects ofmiscibility and interaction between drug andpolymer should be considered whenformulating PASDs. Proper selection of thepolymer, composition of the PASD, and themethod of preparation can aid in formulating astable amorphous system. However, challengessuch as poor stability as a result ofhygroscopicity, deviations from expecteddissolution behavior, and poor processabilitymay limit their utility as a viable formulationstrategy. Careful control of these parameterscan help in harnessing the benefits of theamorphous state by converting them tosuccessful commercial products.
REFERENCES
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 89

Review Paper
1 Che E, Zheng X, Sun C, Chang D, Jiang T, Wang S.Drug nanocrystals: a state of art formulation strategyfor preparing the poorly water-soluble drugs. Asian JPharm Sci, 7(2): 85-95, 2012.
2 Elamin AA, Ahlneck C, Alderborn G, Nyström C.Increased metastable solubility of milled griseofulvin,depending on the formation of a disordered surfacestructure. Int J Pharm, 111(2): 159-170, 1994.
3 Jinno J, Kamada N, Miyake M, Yamada K, Mukai T,Odomi M, Toguchi H, Liversidge GG, Higaki K,Kimura T. Effect of particle size reduction ondissolution and oral absorption of a poorly water-soluble drug, cilostazol, in beagle dogs. J ControlledRelease, 111(1): 56-64, 2006.
4 Vasconcelos T, Sarmento B, Costa P. Soliddispersions as strategy to improve oral bioavailabilityof poor water soluble drugs. Drug Discovery Today,12(23): 1068-1075, 2007.
5 Smith AJ, Kavuru P, Wojtas L, Zaworotko MJ,Shytle RD. Cocrystals of quercetin with improvedsolubility and oral bioavailability. Mol Pharm, 8(5):1867-1876, 2011.
6 Aakeroy CB, Forbes S, Desper J. Using cocrystalsto systematically modulate aqueous solubility andmelting behavior of an anticancer drug. J Am ChemSoc, 131(47): 17048-17049, 2009.
7 Kimura K, Hirayama F, Arima H, Uekama K.Effects of aging on crystallization, dissolution andabsorption characteristics of amorphoustolbutamide-2-hydroxypropyl-beta-cyclodextrincomplex. Chem Pharm Bull, 48(5): 646-650, 2000.
8 Yalkowsky SH, Rubino JT. Solubilization bycosolvents I: Organic solutes in propylene glycol-water mixtures. J Pharm Sci, 74(4): 416-421, 1985.
9 Porter CJ, Trevaskis NL, Charman WN. Lipids andlipid-based formulations: optimizing the oral deliveryof lipophilic drugs. Nature Rev Drug Discovery,6(3): 231-248, 2007.
10 Zhang M, Li H, Lang B, O’Donnell K, Zhang H,Wang Z, Dong Y, Wu C, Williams III RO.Formulation and Delivery of Improved AmorphousFenofibrate Solid Dispersions Prepared by ThinFilm Freezing. Eur J Pharm Biopharm, 2012.
11 Broman E, Khoo C, Taylor LS. A comparison ofalternative polymer excipients and processingmethods for making solid dispersions of a poorlywater soluble drug. Int J Pharm, 222(1): 139-151,2001.
12 Chiou WL, Riegelman S. Pharmaceutical applicationsof solid dispersion systems. J Pharm Sci, 60(9): 1281-1302, 1971.
13 Qiu Y, Chen Y, Geoff G, Zhang Z. DevelopingSolid Oral Dosage Forms - Pharmaceutical Theoryand Practice. Academic Press, 2009.
14 Leuner C, Dressman J. Improving drug solubility fororal delivery using solid dispersions. Eur J PharmBiopharm, 50(1): 47-60, 2000.
15 Gupta P, Bansal AK. Spray drying for generation ofa ternary amorphous system of celecoxib, PVP, andmeglumine. Pharm Dev Technol, 10(2): 273-281,2005.
16 Goldberg AH, Gibaldi M, Kanig JL, Mayersohn M.Increasing dissolution rates and gastrointestinalabsorption of drugs via solid solutions and eutecticmixtures IV: Chloramphenicol—urea system. JPharm Sci, 55(6): 581-583, 1966.
17 Van Drooge D, Hinrichs W, Frijlink H. Anomalousdissolution behavior of tablets prepared from sugarglass-based solid dispersions. J Controlled Release,97(3): 441-452, 2004.
18 Löbmann K, Grohganz H, Laitinen R, Strachan C,Rades T. Amino acids as co-amorphous stabilisersfor poorly water soluble drugs-Part 1: Preparation,stability and dissolution enhancement. Eur J PharmBiopharm, 2013.
19 Goldberg AH, Gibaldi M, Kanig JL. Increasingdissolution rates and gastrointestinal absorption ofdrugs via solid solutions and eutectic mixtures III:Experimental evaluation of griseofulvin-succinic acidsolid solution. J Pharm Sci, 55(5): 487-492, 1966.
20 Kaushal AM, Gupta P, Bansal AK. Amorphous drugdelivery systems: molecular aspects, design, andperformance. Crit Rev Ther Drug, 21(3): 133, 2004.
21 Shah B, Kakumanu VK, Bansal AK. Analyticalt e c h n i q u e s f o r q u a n t i f i c a t i o n o famorphous/crystalline phases in pharmaceuticalsolids. J Pharm Sci, 95(8): 1641-1665, 2006.
22 Cui Y. A material science perspective ofpharmaceutical solids. Int J Pharm, 339(1): 3-18,2007.
23 Ediger M, Angell C, Nagel SR. Supercooled liquidsand glasses. J Phys Chem, 100(31): 13200-13212,1996.
24 Kwei T. The effect of hydrogen bonding on theglass transition temperatures of polymer mixtures. JPolym Sci: Polym Lett Ed, 22(6): 307-313, 2003.
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 90

Review Paper
25 Torquato S. Glass transition: Hard knock forthermodynamics. Nature, 405(6786): 521-523, 2000.
26 Gupta P, Bansal AK. Devitrification of amorphouscelecoxib. AAPS PharmSciTech, 6(2): E223-E230,2005.
27 Hancock BC, Parks M. What is the true solubilityadvantage for amorphous pharmaceuticals? PharmRes, 17(4): 397-404, 2000.
28 Babu NJ, Nangia A. Solubility advantage ofamorphous drugs and pharmaceutical cocrystals.Cryst. Growth Des., 11: 2662-2679, 2011.
29 Pawar YB, Shete G, Popat D, Bansal AK. Phasebehavior and oral bioavailability of amorphouscurcumin. Eur J Pharm Sci, 47: 56-64, 2012.
30 Hancock BC, Zografi G. Characteristics andsignificance of the amorphous state inpharmaceutical systems. J Pharm Sci, 86(1): 1-12,1997.
31 Onoue S, Sato H, Kawabata Y, Mizumoto T,Hashimoto N, Yamada S. In vitro and in vivocharacterization on amorphous solid dispersion ofcyclosporine A for inhalation therapy. J ControlledRelease, 138(1): 16-23, 2009.
32 Law D, Krill SL, Schmitt EA, Fort JJ, Qiu Y, WangW, Porter WR. Physicochemical considerations inthe preparation of amorphous ritonavir-poly(ethylene glycol) 8000 solid dispersions. J Pharm Sci,90(8): 1015-1025, 2001.
33 Lakshman JP, Cao Y, Kowalski J, Serajuddin AT.Application of melt extrusion in the development ofa physically and chemically stable high-energyamorphous solid dispersion of a poorly water-soluble drug. Mol Pharm, 5(6): 994-1002, 2008.
34 Yang W, Tam J, Miller DA, Zhou J, McConville JT,Johnston KP, Williams RO. High bioavailabilityfrom nebulized itraconazole nanoparticle dispersionswith biocompatible stabilizers. Int J Pharm, 361(1):177-188, 2008.
35 Ambike AA, Mahadik K, Paradkar A. Spray-driedamorphous solid dispersions of simvastatin, a low Tg drug: in vitro and in vivo evaluations. Pharm Res,22(6): 990-998, 2005.
36 Won DH, Kim MS, Lee S, Park JS, Hwang SJ.Improved physicochemical characteristics offelodipine solid dispersion particles by supercriticalanti-solvent precipitation process. Int J Pharm,301(1): 199-208, 2005.
37 Patrick J. Martin's physical pharmacy andpharmaceutical sciences. Lippincott Williams andWilkins, 2010.
38 Konno H, Handa T, Alonzo DE, Taylor LS. Effectof polymer type on the dissolution profile ofamorphous solid dispersions containing felodipine.Eur J Pharm Biopharm, 70(2): 493-499, 2008.
39 Rawlinson CF, Williams AC, Timmins P, Grimsey I.Polymer-mediated disruption of drug crystallinity.Int J Pharm, 336(1): 42-48, 2007.
40 Miyazaki T, Aso Y, Yoshioka S, Kawanishi T.Differences in crystallization rate of nitrendipineenantiomers in amorphous solid dispersions withHPMC and HPMCP. Int J Pharm, 407(1): 111-118,2011.
41 Kennedy M, Hu J, Gao P, Li L, Ali-Reynolds A,Chal B, Gupta V, Ma C, Mahajan N, Akrami A.Enhanced bioavailability of a poorly soluble VR1antagonist using an amorphous solid dispersionapproach: a case study. Mol Pharm, 5(6): 981-993,2008.
42 Ambike AA, Mahadik K, Paradkar A. Stability studyof amorphous valdecoxib. Int J Pharm, 282(1): 151-162, 2004.
43 Wang L, Cui FD, Hayase T, Sunada H. Preparationand evaluation of solid dispersion for nitrendipine-carbopol and nitrendipine-HPMCP systems using atwin screw extruder. Chem Pharm Bull, 53(10):1240-1245, 2005.
44 Liu H, Wang P, Zhang X, Shen F, Gogos CG.Effects of extrusion process parameters on thedissolution behavior of indomethacin in Eudragit® EPO solid dispersions. Int J Pharm, 383(1): 161-169,2010.
45 Huang J, Li Y, Wigent RJ, Malick WA, Sandhu HK,Singhal D, Shah NH. Interplay of formulation andprocess methodology on the extent of nifedipinemolecular dispersion in polymers. Int J Pharm,420(1): 59-67, 2011.
46 Jachowicz R, Czech A. Preparation and evaluation ofpiroxicam-HPMCAS solid dispersions for ocularuse. Pharm Dev Technol, 13(6): 495-504, 2008.
47 Xie Y, Xie P, Song X, Tang X, Song H. Preparationof esomeprazole zinc solid dispersion and study onits pharmacokinetics. Int J Pharm, 360(1): 53-57,2008.
48 Law D, Schmitt EA, Marsh KC, Everitt EA, WangW, Fort JJ, Krill SL, Qiu Y. Ritonavir–PEG 8000amorphous solid dispersions: In vitro and in vivoevaluations. J Pharm Sci, 93(3): 563-570, 2004.
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 91

Review Paper
49 Janssens S, Denivelle S, Rombaut P, Van denMooter G. Influence of polyethylene glycol chainlength on compatibility and release characteristics ofternary solid dispersions of itraconazole inpolyethylene glycol/hydroxypropylmethylcellulose2910 E5 blends. Eur J Pharm Sci, 35(3): 203-210,2008.
50 Nepal PR, Han HK, Choi HK. Enhancement ofsolubility and dissolution of Coenzyme Q10 usingsolid dispersion formulation. Int J Pharm, 383(1):147-153, 2010.
51 Li S, Liu Y, Liu T, Zhao L, Zhao J, Feng N.Development and in-vivo assessment of thebioavailability of oridonin solid dispersions by thegas anti-solvent technique. Int J Pharm, 411(1): 172-177, 2011.
52 Ayenew Z, Paudel A, Van den Mooter G. Cancompression induce demixing in amorphous soliddispersions? A case study of naproxen-PVP K25.Eur J Pharm Biopharm, 2012.
53 Balani P, Wong S, Ng W, Widjaja E, Tan R, Chan S.Influence of polymer content on stabilizing milledamorphous salbutamol sulphate. Int J Pharm, 391(1):125-136, 2010.
54 Muela S, Escalera B, Peña M, Bustamante P.Influence of temperature on the solubilization ofthiabendazole by combined action of soliddispersions and co-solvents. Int J Pharm, 384(1): 93-99, 2010.
55 Shibata Y, Fujii M, Sugamura Y, Yoshikawa R,Fujimoto S, Nakanishi S, Motosugi Y, Koizumi N,Yamada M, Ouchi K. The preparation of a soliddispersion powder of indomethacin withcrospovidone using a twin-screw extruder orkneader. Int J Pharm, 365(1): 53-60, 2009.
56 Moneghini M, Bellich B, Baxa P, Princivalle F.Microwave generated solid dispersions containingibuprofen. Int J Pharm, 361(1): 125-130, 2008.
57 Newman A, Knipp G, Zografi G. Assessing theperformance of amorphous solid dispersions. JPharm Sci, 2012.
58 Janssens S, Anné M, Rombaut P, Van den MooterG. Spray drying from complex solvent systemsbroadens the applicability of Kollicoat® IR as acarrier in the formulation of solid dispersions.European Journal of Pharmaceutical Sciences, 37(3):241-248, 2009.
59 Portero A, Remunan-Lopez C, Vila-Jato J. Effect ofchitosan and chitosan glutamate enhancing thedissolution properties of the poorly water solubledrug nifedipine. Int J Pharm, 175(1): 75-84, 1998.
60 Liu X, Lu M, Guo Z, Huang L, Feng X, Wu C.Improving the chemical stability of amorphous soliddispersion with cocrystal technique by hot meltextrusion. Pharm Res, 29(3): 806-817, 2012.
61 Callahan J, Cleary G, Elefant M, Kaplan G, KenslerT, Nash R. Equilibrium moisture content ofpharmaceutical excipients. Drug Dev Ind Pharm,8(3): 355-369, 1982.
62 Qian F, Huang J, Hussain MA. Drug–polymersolubility and miscibility: Stability consideration andpractical challenges in amorphous solid dispersiondevelopment. J Pharm Sci, 99(7): 2941-2947, 2010.
63 Stukalin EB, Douglas JF, Freed KF. Plasticizationand antiplasticization of polymer melts diluted bylow molar mass species. J Chem Phys, 132: 1-11,2010.
64 Dong Z, Chatterji A, Sandhu H, Choi DS, ChokshiH, Shah N. Evaluation of solid state properties ofsolid dispersions prepared by hot-melt extrusion andsolvent co-precipitation. Int J Pharm, 355(1–2): 141-149, 2008.
65 Anderson S, Grulke E, DeLassus P, Smith P,Kocher C, Landes B. A model for antiplasticizationin polystyrene. Macromol, 28(8): 2944-2954, 1995.
66 Fox GT., Bull Am Phys Soc, 1:123, 1956.
67 Gordon M, Taylor JS. Ideal copolymers and thesecond-order transitions of synthetic rubbers. i. non-crystalline copolymers. J Appl Chem, 2(9): 493-500,2007.
68 Couchman P, Karasz F. A classical thermodynamicdiscussion of the effect of composition on glass-transition temperatures. Macromol, 11(1): 117-119,1978.
69 Shamblin SL, Taylor LS, Zografi G. Mixing behaviorof colyophilized binary systems. J Pharm Sci, 87(6):694-701, 1998.
70 Crowley KJ, Zografi G. Water vapor absorption intoa m o r p h o u s h y d r o p h o b i c d r u g / p o l y(vinylpyrrolidone) dispersions. J Pharm Sci, 91(10):2150-2165, 2002.
71 Makiko F, Junko H, Hideaki K, Mitsuo M. The solidd i s p e r s i o n o f b e n z o d i a z e p i n s w i t hphosphatidylcholine. The effect of substituents ofbenzodiazepins on the formation of soliddispersions. Chem Pharm Bull, 39(11): 3013-3017,1991.
72 Taylor LS, Zografi G. Spectroscopic characterizationof interactions between PVP and indomethacin in
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 92

Review Paper
amorphous molecular dispersions. Pharm Res,14(12): 1691-1698, 1997.
73 Sekikawa H, Arita T, Nakano M. Dissolutionbehavior and gastrointestinal absorption ofphenytoin-polyvinylpyrolidone and dicumarol-beta-cyclodextrin. Chem Pharm Bull, 26: 118-126, 1978.
74 Khougaz K, Clas SD. Crystallization inhibition insolid dispersions of MK-0591 and poly(vinylpyrrolidone) polymers. J Pharm Sci, 89(10):1325-1334, 2000.
75 Gupta P, Thilagavathi R, Chakraborti AK, BansalAK. Role of molecular interaction in stability ofcelecoxib-PVP amorphous systems. Mol Pharm,2(5): 384-391, 2005.
76 Vippagunta SR, Wang Z, Hornung S, Krill SL.Factors affecting the formation of eutectic soliddispersions and their dissolution behavior. J PharmSci, 96(2): 294-304, 2006.
77 Van den Mooter G, Wuyts M, Blaton N, Busson R,Grobet P, Augustijns P, Kinget R. Physicalstabilisation of amorphous ketoconazole in soliddispersions with polyvinylpyrrolidone K25. Eur JPharm Sci, 12(3): 261-269, 2001.
78 Gunawan L, Johari G, Shanker RM. Structuralrelaxation of acetaminophen glass. Pharm Res, 23(5):967-979, 2006.
79 Job G, Herrmann F. Chemical potential-a quantity insearch of recognition. Eur J Phys, 27(2): 353, 2006.
80 Marsac PJ, Shamblin SL, Taylor LS. Theoretical andpractical approaches for prediction of drug-polymermiscibility and solubility. Pharm Res, 23(10): 2417-2426, 2006.
81 Westrum E, McCullough J. Thermodynamics ofcrystals. Phys Chem Org Solid State, 1: 89-102, 1963.
82 Bansal SS, Kaushal AM, Bansal AK. Enthalpyrelaxation studies of two structurally relatedamorphous drugs and their binary dispersions. DrugDev Ind Pharm, 36(11): 1271-1280, 2010.
83 Surana R, Pyne A, Rani M, Suryanarayanan R.Measurement of enthalpic relaxation by differentialscanning calorimetry-effect of experimentalconditions. Thermochim Acta, 433(1): 173-182,2005.
84 Hancock BC, Shamblin SL, Zografi G. Molecularmobility of amorphous pharmaceutical solids belowtheir glass transition temperatures. Pharm Res, 12(6):799-806, 1995.
85 Craig DQ. A review of thermal methods used forthe analysis of the crystal form, solutionthermodynamics and glass transition behavior ofpolyethylene glycols. Thermochim Acta, 248: 189-203, 1995.
86 Kakumanu VK, Bansal AK. Enthalpy relaxationstudies of celecoxib amorphous mixtures. PharmRes, 19(12): 1873-1878, 2002.
87 Bhattacharya S, Suryanarayanan R. MolecularMotions in Sucrose-PVP and Sucrose-SorbitolDispersions: I. Implications of Global and LocalMobility on Stability. Pharm Res, 28(9): 2191-2203,2011.
88 Rumondor AC, Ivanisevic I, Bates S, Alonzo DE,Taylor LS. Evaluation of drug-polymer miscibility inamorphous solid dispersion systems. Pharm Res,26(11): 2523-2534, 2009.
89 Ivanisevic I, Bates S, Chen P. Novel methods for theassessment of miscibility of amorphous drug-polymer dispersions. J Pharm Sci, 98(9): 3373-3386,2009.
90 Qian F, Huang J, Zhu Q, Haddadin R, Gawel J,Garmise R, Hussain M. Is a distinctive single Tg areliable indicator for the homogeneity of amorphoussolid dispersion? Int J Pharm, 395(1): 232-235, 2010.
91 Painter PC, Graf JF, Coleman MM. Effect ofhydrogen bonding on the enthalpy of mixing and thecomposition dependence of the glass transitiontemperature in polymer blends. Macromol, 24(20):5630-5638, 1991.
92 Hildebrand JH. Solubility. J Am Chem Soc, 38(8):1452-1473, 1916.
93 Greenhalgh DJ, Williams AC, Timmins P, York P.Solubility parameters as predictors of miscibility insolid dispersions. J Pharm Sci, 88(11): 1182-1190,1999.
94 Hansen CM. Hansen solubility parameters: a user'shandbook. CRC, 2007.
95 Fedors RF. A method for estimating both thesolubility parameters and molar volumes of liquids.Polym Eng Sci, 14(2): 147-154, 2004.
96 Krevelen DWv, Nijenhuis Kt. Properties ofpolymers. Elsevier, 2009.
97 Barton AF. Handbook of solubility parameters andother cohesion parameters. CRC, 1991.
98 Flory PJ. Thermodynamics of high polymersolutions. J Chem Phys, 9: 660-661, 1941.
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 93

Review Paper
99 Young RJ, Lovell PA. Introduction to polymers.CRC, 2009.
100 Flory PJ. Principles of polymer chemistry. CornellUniversity Press, 1953.
101 Huggins ML. Thermodynamic Properties ofSolutions of Long-chain Compounds. Ann NY AcadSci, 43(1): 1-32, 1942.
102 Marsac PJ, Li T, Taylor LS. Estimation of drug-polymer miscibility and solubility in amorphous soliddispersions using experimentally determinedinteraction parameters. Pharm Res, 26(1): 139-151,2009.
103 Ford JL, Elliott PN. The effect of particle size onsome in vitro and in vivo properties of indomethacin-polyethylene glycol 6000 solid dispersions. DrugDev Ind Pharm, 11(2-3): 537-549, 1985.
104 Akbuga J, Gürsoy A, Yetimoglu F. Preparation andproperties of tablets prepared from furosemide-PVPsolid dispersion systems. Drug Dev Ind Pharm,14(15-17): 2091-2108, 1988.
105 Sjökvist E, Nyström C. Physicochemical aspects ofdrug release. XI. Tableting properties of soliddispersions, using Xylitol as carrier material. Int JPharm, 67(2): 139-153, 1991.
106 Hideshi S, Hisakazu S. Some factors influencing thedissolution of solid dispersions with nicotinamideand hydroxypropylmethylcellulose as combinedcarriers. Chem Pharm Bull, 46(6): 1015-1020, 1998.
107 Puri V, Dantuluri AK, Bansal AK. Investigation ofatypical dissolution behavior of an encapsulatedamorphous solid dispersion. J Pharm Sci, 100(6):2460-2468, 2011.
108 Serajuddin A. Solid dispersion of poorly water-soluble drugs: early promises, subsequent problems,and recent breakthroughs. J Pharm Sci, 88(10): 1058-1066, 1999.
109 Chokshi RJ, Sandhu HK, Iyer RM, Shah NH, MalickAW, Zia H. Characterization of physico-mechanicalproperties of indomethacin and polymers to assesstheir suitability for hot-melt extrusion processs as ameans to manufacture solid dispersion/solution. JPharm Sci, 94(11): 2463-2474, 2005.
110 Lamm MS, Simpson A, McNevin M, Frankenfeld C,Nay R, Variankaval N. Probing the Effect of DrugLoading and Humidity on the Mechanical Propertiesof Solid Dispersions with Nanoindentation:Antiplasticization of a Polymer by a Drug Molecule.Mol Pharm, 9(11): 3396-3402, 2012.
111 Marsac PJ, Rumondor ACF, Nivens DE, Kestur US,Stanciu L, Taylor LS. Effect of temperature andmoisture on the miscibility of amorphousdispersions of felodipine and poly(vinyl pyrrolidone).J Pharm Sci, 99(1): 169-185, 2010.
This Journal is © IPEC-Americas Inc September 2013 J. Excipients and Food Chem. 4 (3) 2013 - 94





























