Embed Size (px)
Citation preview

Rm
GCa
Ab
c
a
ARRA
KCSMID
1
1
tteoehc
iiT
h1
Drug Resistance Updates 26 (2016) 10–27
Contents lists available at ScienceDirect
Drug Resistance Updates
jo ur nal homep age: www.elsev ier .com/ locate /drup
epositioning of drugs for intervention in tumor progression andetastasis: Old drugs for new targets
iridhar Muddulurua,b, Wolfgang Walthera, Dennis Kobelta, Mathias Dahlmanna,hristoph Treesea, Yehuda G. Assaraf c, Ulrike Steina,b,∗
Experimental and Clinical Research Center, Charité Universitätsmedizin, Berlin and Max-Delbrück-Center for Molecular Medicine in the Helmholtzssociation, Robert-Rössle-Straße 10, 13125 Berlin, GermanyGerman Cancer Consortium, GermanyThe Fred Wyszkowski Cancer Research Laboratory, Department of Biology, Technion – Israel Institute of Technology, Haifa 32000, Israel
r t i c l e i n f o
rticle history:eceived 2 December 2015eceived in revised form 14 March 2016ccepted 18 March 2016
eywords:ancerolid tumorsetastasis
nhibitorsrug development
a b s t r a c t
The increasing unraveling of the molecular basis of cancer offers manifold novel options for interventionstrategies. However, the discovery and development of new drugs for potential clinical applicationsis a tremendously time-consuming and costly process. Translating a novel lead candidate compoundinto an approved clinical drug takes often more than a decade, and the success rate is very low due toversatile efforts including defining its pharmacokinetics, pharmacodynamics, side effects as well as lack ofsufficient efficacy. Thus, strategies are needed to minimize time and costs, while maximizing success rates.A very attractive strategy for novel cancer therapeutic options is the repositioning of already approveddrugs. These medicines, approved for the treatment of non-malignant disorders, have already passedsome early costs and time, have been tested in humans and are ready for clinical trials as anti-cancerdrugs. Here we discuss the repositioning of nonsteroidal anti-inflammatory drugs (NSAID), statins, anti-
psychotic drugs, anti-helminthic drugs and vitamin D as anti-tumor agents. We focus on their novelactions and potential for inhibition of cancer growth and metastasis by interfering with target moleculesand pathways, which drive these malignant processes. Furthermore, important pre-clinical and clinicaldata are reviewed herein, which elucidate their therapeutic mechanisms which enable their repositioningfor cancer therapy and disruption of metastasis.© 2016 Elsevier Ltd. All rights reserved.
. Introduction
.1. Drug development: where we stand
For decades a plethora of drugs have been developed for thereatment of different malignant and non-malignant disorders. Inhis effort, natural or synthetic compounds were isolated or gen-rated and extensively tested to finally use them for the treatmentf a particular disease in the clinic. The process of drug discov-
ry usually starts with the identification of a compound, whichas the potential to become an active drug. Such a compound thenan enter the very time-consuming and costly process to achieve a∗ Corresponding author at: Experimental and Clinical Research Center, Char-té Universitätsmedizin, Berlin and Max-Delbrück-Center for Molecular Medicinen the Helmholtz Association, Robert-Rössle-Str. 10, 13125 Berlin, Germanyel.: +49 3094063432; fax: +49 3094062780.
E-mail address: [email protected] (U. Stein).
ttp://dx.doi.org/10.1016/j.drup.2016.03.002368-7646/© 2016 Elsevier Ltd. All rights reserved.
Downloaded for Anonymous User (n/a) at Guilin Medical College from ClinFor personal use only. No other uses without permission. Copyright ©2
validation of the preclinical therapeutic efficacy of a given com-pound and to perform pharmacokinetics, pharmacodynamics, andtoxicity studies before clinical application is anticipated (Fig. 1).Due to the immensely high costs of pre-clinical as well as clinicalevaluation over many years, introduction of new drugs for a partic-ular disease including cancer has become a challenging endeavor.
Analyses regarding scientific, clinical, and financial efforts andthe respective establishment of a novel drug for routine clinicalapplication revealed a significant decrease in the number of drugsintroduced to the clinic (Mullard, 2014; Pammolli et al., 2011;Scannell et al., 2012). The number of novel drugs has halved everynine years during the past 40 years of drug development per billionUS dollars invested into research and development. The averagetime for the development of a novel drug has also increased consid-erably over time. During the 1990s the average time required from
drug discovery to its use in the clinic (market launch) was 9.7 years.This has then increased to almost 14 years from 2000 onwards – andstill increases (Pammolli et al., 2011). In addition to this dilemma,the Tufts Center for Study of Drug Development stated that costsicalKey.com by Elsevier on April 03, 2019.019. Elsevier Inc. All rights reserved.

G. Mudduluru et al. / Drug Resistance Updates 26 (2016) 10–27 11
F discovp finanw s like c
fcfiia
1
bmi6etaeoea2
tabwuantto
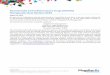
ig. 1. Schematic representation of the flow and timely efforts for classical drug
rocedure for novel drug discovery requires more time in association with higherhich is revealed, could contribute to an accelerated clinical use for other disorder
or market launch of a new drug in 2014 has increased 1.7-foldompared to the costs required in 2003. These facts reflect the inef-ciency of the current drug development process in the developed
ndustrial countries as well as the need of repositioning of alreadypproved drugs.
.2. Drug development for cancer treatment
The general hurdles in drug development also apply to theurning need to develop novel anti-cancer drugs. Here the esti-ated time required for drug discovery until the drug is approved
s ∼8 years, in association with a very low success rate of only.7% in phase I clinical trials (Kaitin and DiMasi, 2011; Pantziarkat al., 2014). This is in spite of the tremendous progress made forhe identification of novel molecular targets for cancer treatmentnd the molecular deciphering of signaling pathways, which aressential for tumor development, metastasis, immune surveillancef tumors, drug response or drug resistance mechanisms (Fishert al., 2013; Gonen and Assaraf, 2012; Huang et al., 2014; Livneynd Assaraf, 2013; Niewerth et al., 2015; Zhitomirsky and Assaraf,016).
The revolution in computational bioanalyses does further con-ribute to the acceleration in this field due to improved data miningnd correlation analyses, such as correlation of drug response andiomarker expression or of mutations of particular genes. Oneould expect that with such progress in understanding the molec-lar basis of cancer biology, drug development of novel anti-cancergents should be easier, faster and will also promote the entry of
ovel drugs into clinical application. However, this is hindered byhe immense time and financial investment required for introduc-ion of novel drugs into the clinic. In fact, the expected and alreadybserved increase in cancer incidence in association with agingDownloaded for Anonymous User (n/a) at Guilin Medical College from ClinicalFor personal use only. No other uses without permission. Copyright ©2019
ery in comparison to drug repositioning. The scheme indicates that the classicalcial efforts. Therefore, repositioning of already approved drugs, the mechanism ofancer.
populations in developed countries clearly calls for the develop-ment of novel, more targeted, effective, and affordable drugs in thecoming years (Vineis and Wild, 2014). This will be essential to betterfulfill the modern needs for a more individualized cancer therapy.
1.3. Repositioning of drugs for cancer therapy
Taking into account the various hurdles in the development ofnovel anti-cancer therapeutics, which aims at new druggable tar-gets but also at already known ones, the use of known compoundsdoes represent an attractive alternative. To achieve this goal, drugrepositioning or repurposing came into focus. This approach isbased on identification of new indications from already existingapproved drugs. More importantly these drugs could be used for thetreatment of diseases other than the original indication of the drug.This approach is certainly of particular interest in the search for newdrugs for cancer treatment. To achieve this goal more efficiently,the Repurposing Drugs in Oncology (ReDO) project was initiated, toidentify candidate drugs with the best potential of clinical use andto bring such drugs to better attention of the basic research and clin-ical community (Pantziarka et al., 2015, 2014). The ReDO project hasdefined key criteria, by which such drugs can be efficiently selectedas high-potential candidates for further re-evaluation:
1. The compound should be well known for many years and widelyused in the clinic. Generics of the drug are of advantage.
2. The drug should have low toxicity (good toxicology profile), evenupon long-term administration.
3. A putative mechanism or defined target of action should beknown for its use in cancer therapy.
4. High-level evidence of anti-cancer activity in vitro as well asin vivo should be shown. In this respect, data generated in
Key.com by Elsevier on April 03, 2019.. Elsevier Inc. All rights reserved.

1 sistan
5
6
uoraitJikbiturs
t((Nu
tdiartt
1d
caomtfsSai
d
1
2
3
4
2 G. Mudduluru et al. / Drug Re
syngeneic, orthotopic transplantable models or in geneticallyengineered mice is of highest rating.
. Anti-cancer activity should be demonstrated at standard dosingat low toxicity.
. The drug of interest with anti-cancer activity should currentlynot be widely pursued as an active compound in cancer therapy.
Such drugs can be identified via high-throughput screenssing small molecule libraries, which incorporate approved drugsf different clinical uses; other than cancer therapy. This isecently supported by various open-access libraries of approvednd investigational compounds enabling better drug reposition-ng, as provided by the National Clinical Guidance Center (NCGC),he Pharmaceutical Collection (NPC) for virtual screening, or theohns Hopkins Drug Library (JHDL) as another assembly of exist-ng drugs (Huang et al., 2011; Shim and Liu, 2014). Furthermore,nowledge of the molecular structure of target proteins could alsoe indicative (in silico analysis) that particular compounds might
nterfere with important functional domains of such target proteinso specifically inhibit their activities. This approach is currentlynder intense evaluation, called “drug-protein interaction-basedepurposing (DPIR)”, analyzing drug-protein interactions for inilico prediction of promising drug candidates (Liu et al., 2014).
The first six compounds the ReDO project has selected for fur-her evaluation are mebendazole (anti-helminthic), nitroglycerinvasodilator), cimetidine (H2-receptor antagonist), clarithromycinantibiotic), diclofenac (nonsteroidal anti-inflammatory drug,SAID), and itraconazole (anti-fungal), drugs which were not inse for cancer therapy (Pantziarka et al., 2014).
As discussed in the context of drug repositioning, the key ques-ions remain, at which point clinical testing should start for suchrugs, if pre-clinical results clearly demonstrate anti-cancer activ-
ty? Should the entry level be at clinical phase II or even phase II/IIIt reduced size of the respective patient numbers required for theespective patient groups in the trial? To maintain the initial advan-age of repositioning of drugs during the phases for clinical testing,he enhanced and widely accepted clinical testing is essential.
.4. Cancer progression and metastasis: rewarding targets for oldrugs
In this review not only the search for drug repositioning to treatancer in general is in focus, but also the immense potential of thispproach for identification of drugs, which can effectively preventr inhibit metastasis. This is of particular interest, since for manyalignancies patients die of their cancer metastases rather due
o the primary tumor. Therefore, drugs which are able to inter-ere with tumor progression and also with metastasis-associatedignaling pathways, are attractive candidates for intense testing.uch drugs could then be used as inhibitors of tumor progressionnd metastasis, or even as tumor and metastasis preventing drugsn a long-term treatment of defined patient groups.
For such drugs, similar criteria for their selection will apply asefined by the ReDO project:
. The compound should be well known and widely used in theclinic, with a good toxicology profile, particularly for the long-term medication in metastasis prevention settings.
. A putative mechanism or defined target of tumor and metastasisinhibition/prevention should be known.
. High-level in vitro evidence of anti-tumor and anti-metastaticactivity as well as in vivo validation should be demonstrated at
standard dosing.Furthermore:. The proposed drug candidate might deliberately have multi-
target promiscuous properties (polypharmacology), to hit at
Downloaded for Anonymous User (n/a) at Guilin Medical College from ClinFor personal use only. No other uses without permission. Copyright ©2
ce Updates 26 (2016) 10–27
multiple sites of tumor progression and metastatic signalingpathways.
In the following chapter, different classes of clinically useddrugs are introduced and discussed for their application in anti-tumor and anti-metastatic treatments, including their mechanismsof action.
2. Old drugs for new targets
2.1. NSAIDs in cancer therapy
NSAIDs do represent the most widely used group of compoundsfor many decades. These drugs interfere with the prostanoid syn-thesis (Vane, 1971). They represent an alternative to the steroidalanti-inflammatory drugs (cortisone, hydrocortisone, dexametha-sone), which are related to the glucocorticoids, acting at the levelof arachidonic acid release to prevent eicosanoid (prostanoids andleukotrienes) synthesis. In contrast, the NSAIDs target, e.g. thecyclooxygenase (COX) subunit of the prostaglandin endoperox-ide synthase (PTGS1 and PTGS2), which led to their simplifiednomenclature as COX-1/COX-2 inhibitors with anti-inflammatory,anti-pyretic and analgesic activities (Vane et al., 1998). Since theirdiscovery it was long assumed, that NSAIDs solely hit this targeteither as irreversible inhibitors acting via covalent modification ofCOX-1 and COX-2 isoenzymes (aspirin), or as low- or high-affinitycompetitive inhibitors of arachidonic acid binding (e.g. ibupro-fen, diclofenac, indomethacin). However, the current view aboutthis group of compounds expanded significantly by the observa-tion of a link between inflammatory processes, COX-2 expressionand activity, and cancer development. Thus, in the late 1980s, firstideas emerged to use NSAIDs as anti-cancer agents (Kune et al.,1988). In this context, clinical observations comparing NSAID-userswith non-users revealed that NSAIDs could have an impact ontumor development and tumor growth (Méric et al., 2006). Par-ticularly this observation was initially made in patients sufferingfrom colon adenomas, as well as in colon cancer patients (Ruderet al., 2011). Meanwhile, numerous meta-analyses suggest thatNSAIDs might also exert anti-tumor and chemopreventive effectsin other malignancies (Jacobs et al., 2007). This therapeutic con-cept is currently further developed by combining NSAID therapieswith conventional therapies, such as chemo- or radiotherapy. Theanti-tumor effects observed for NSAIDs are either based on thedirect inhibition of COX-2, which is known to be overexpressedin cancer due to stimulation by, e.g. rat sarcoma (Ras) signaling,protein kinase C (PKC), human epidermal growth factor receptor 2(HER2), or by off-target effects of NSAIDs affecting other oncogenicproteins and signaling pathways (Dannenberg et al., 2001; Shaoet al., 2000). In the following chapter, the major NSAID representa-tives are reviewed regarding their anti-cancer and anti-metastaticpotential, and their use in the context of drug repurposing.
2.1.1. AspirinAspirin is known since the 1880s as an effective anti-pyretic,
anti-inflammatory drug and is therefore the first NSAID generatedby chemical synthesis (1897, by Felix Hoffmann). Its mechanism ofaction is based on the inhibition of COX-1 and COX-2 by irreversibleacetylation of serine in the catalytic site of the enzyme. In the lastdecade however, this old drug has gained new attention due tomultiple observations that aspirin might prevent cancer. Large-cohort and case-control studies, covering observation periods of
up to 10 years, demonstrated a strong correlation between low-dose aspirin intake, based on daily or at least regular use, andcancer prevention. The reduction of cancer risk and cancer-relatedmortality in colon, lung, esophageal, and prostate cancer, definesicalKey.com by Elsevier on April 03, 2019.019. Elsevier Inc. All rights reserved.

sistan
aewabmpa((t(ebsrcbbmbcfca2d(o(
efsn�mtocapoaasoac
2
kdefia
aaddlncd
G. Mudduluru et al. / Drug Re
spirin as a chemopreventive drug (Cook et al., 2005; Hochmutht al., 2015; Huang et al., 2015; Rothwell et al., 2011). These studiesere supported by analyses of the molecular mechanisms medi-
ting the anti-tumor effects of aspirin. Most of these mechanisticases were focused on the interplay of COX-2 overexpression inany cancers and the resulting deregulation of cellular signaling
athways. Here, reduced synthesis of prostaglandin E2 (PGE2) isssociated with inhibition of Ras, mitogen activated protein kinaseMAPK) and phosphatidylinositide 3-kinase (PI3K)/Ak-thymomaAkt) signaling or normalization of epidermal growth factor recep-or (EGFR) signaling by reducing EGFR expression in cancer cellsDovizio et al., 2012; Li et al., 2015; Pozzi et al., 2004; Uddint al., 2010; Wang et al., 2004). Of particular interest is the linketween PGE2 and glycogen synthase kinase 3� (GSK3�)/�-cateninignaling, in which aspirin can inhibit �-catenin activation viaeduced PGE2 synthesis, leading to increased GSK3� activity for �-atenin phosphorylation and degradation. In addition, an interplayetween aspirin action on COX-1 and its anti-cancer activity haseen shown. Apart from this, it was shown that low-dose aspirinediated inhibition of COX-1 in platelets results in reduced throm-
oxane A2 levels. This was correlated with reduced masking ofirculating tumors cells (CTC) by thrombocytes, which is importantor CTC’s evasion from immune surveillance, promoting survival ofolorectal cancer (CRC) cells. More importantly, this inhibition byspirin, in turn, leads to reduced metastasis by CTC (Ogawa et al.,014; Rothwell et al., 2012). As alternative mechanisms of low-ose aspirin, the drug action on vascular endothelial growth factorVEGF) signaling is discussed, which also contributes to inhibitionf metastasis by aspirin via promotion of anti-angiogenic signalingHolmes et al., 2013).
Regarding COX-independent activities of aspirin, its anti-tumorffects have also been linked to its ability to interfere with nuclearactor ‘kappa-light-chain-enhancer’ of activated B-cells (NF-�B)ignaling. It has been shown, that high-dose aspirin preventsuclear translocation of NF-�B and inhibits transcription of NF-B target genes, which contribute to cancer cell proliferation,igration and metastasis (D. Liao et al., 2015; Yin et al., 1998). Fur-
hermore, aspirin has the ability to increase the steady-state levelf mismatch repair (MMR) enzymes, which are impaired in cancerells. Thus, aspirin treatment prevents accumulation of mutationsnd stimulates activity of DNA repair mechanisms acting as cancerreventing drug (Goel et al., 2003). In conclusion, all these activitiesf aspirin highlight the great and wide spectrum (COX-dependentnd -independent) of mechanisms by which aspirin can act either as
chemopreventive or as anti-cancer and anti-metastatic drug. Suchtudies further reveal the importance of both the dose and durationf aspirin treatment, which determines the molecular mechanismsnd direction of the mode of action for this drug in clinical use forancer patient therapies.
.1.2. SulindacChemoprevention of CRC by NSAIDs such as sulindac is long
nown and well documented (Clevers, 2006; Szabo, 2006). Sulin-ac, introduced in the 1970s, is a prodrug that is converted by livernzymes to the active drug: by reversible reduction to sulindac sul-de, and by irreversible oxidation to sulindac sulfone. The biologicalctive compound is thought to be the sulfide metabolite.
Sulindac has pleiotropic activities both as a COX inhibitors well as an inhibitor of polyamine biosynthesis. This latterctivity provided the rationale for its combination with �-ifluoromethylornithine (DFMO), a reversible inhibitor of ornithineecarboxylase, the key enzyme in polyamine biosynthesis, in a
arge scale trial for chemoprevention of sporadic colorectal ade-omas (Meyskens et al., 2008; Sporn and Hong, 2008). Thisombination trial produced a striking preventive effect in the sulin-ac/DMFO arm.
Downloaded for Anonymous User (n/a) at Guilin Medical College from ClinicalFor personal use only. No other uses without permission. Copyright ©2019
ce Updates 26 (2016) 10–27 13
The use of NSAIDs such as sulindac is linked to decreased can-cer incidence, regression of precancerous lesions, delayed tumorprogression, and reduced risk of cancer-related mortality. Despitenumerous reports describing these effects via its action as a COXinhibitor, important COX-independent mechanisms of sulindacwere currently unveiled (Gurpinar et al., 2014). For example, a fur-ther clinical trial employed sulindac in combination with etadolac,which resulted in eradication of aberrant crypt foci and preventedsporadic colorectal polyps. Etadolac is an NSAID able to inhibit �-catenin signaling (Takayama et al., 2011).
It is less well known, that pharmacological manipulation withsulindac negatively regulates Wingless-type MMTV integration site(Wnt)/�-catenin signaling, a pathway activated in more than 90%of all CRC. NSAIDs have been repeatedly evaluated as potentialWnt/�-catenin pathway therapeutics (Kundu et al., 2006). Experi-mental studies suggest that apoptosis induction and suppression of�-catenin-dependent transcription are important aspects of theiranti-neoplastic activity mechanisms independent of COX (Gurpinaret al., 2014). Sulindac acts as a pharmacological inhibitor of �-catenin by inhibiting �-catenin expression in CRC cells and inpatients with hereditary nonpolyposis CRC and familial adeno-matous polyposis (FAP) (Gardner et al., 2004; Han et al., 2008;Koornstra et al., 2005; Tai et al., 2014). Furthermore, sulindacinduces proteasome-dependent degradation of �-catenin, therebysuppressing tumorigenesis in vivo by downregulating �-cateninsignaling (Orner et al., 2003; Rice et al., 2003). It inhibits the nuclearaccumulation of �-catenin in CRC cell lines and in adenomas ofpatients with FAP, leading to reduced downstream signaling (Boonet al., 2004). Sulindac, in combination with celecoxib (NSAID, COX-2inhibitor), regulated angiogenesis during the early neoplasm of thecolon via the PI3K/phosphatase and tensin homologue (PTEN)/Aktpathway to the canonical Wnt/�-catenin signaling (Vaish andSanyal, 2012). Consequently, �-catenin target genes like c-Met, c-myc, and cyclin D1, are transcriptionally downregulated followingsulindac treatment.
Apart from these known �-catenin target genes, we newly iden-tified the metastasis inducer S100A4 as a transcriptional targetof Wnt/�-catenin signaling (Stein et al., 2006). S100A4, a mem-ber of the S100 family of calcium-binding proteins, allows theearly identification of patients at high risk for distant metasta-sis. S100A4 is overexpressed in many different types of cancersincluding CRC, and represents a dismal prognosis and poor patientsurvival (Liu et al., 2013). Moreover, we developed a non-invasivepatient plasma-based assay for quantification of circulating S100A4transcripts, which are of prognostic value for the disease course ofthe patients (Stein et al., 2011b). Consequently, although sulindacwas evaluated in a large body of studies for chemoprevention of pri-mary CRC, arising in inflammatory bowel disease, FAP, and Lynchsyndrome patients (Fajardo and Piazza, 2015; Lang and Gasche,2015), we probed for the impact of sulindac on prevention ofmetastasis in CRC (Stein et al., 2011a). We reported the expressionknock-down of �-catenin by sulindac, leading to its reduced nuclearaccumulation. The binding of �-catenin to T-cell factor (TCF) wasdecreased, resulting in reduced S100A4 promoter activity. Expres-sion of S100A4 at the mRNA and protein levels was inhibited bysulindac. This correlated well with inhibition of cell migration andinvasion, which could be rescued by ectopic S100A4 expression.In mice, sulindac treatment resulted in reduced tumor growthand significantly decreased formation of liver metastasis in humanCRC xenografts. Tumors and liver metastases of sulindac-treatedmice showed lowered �-catenin and S100A4 levels. These resultssuggest that modulators of �-catenin signaling, such as sulindac,
have a potential as anti-metastatic agents by preventing S100A4expression. Based on our findings, we hypothesize that sulindacadministration will abrogate Wnt/�-catenin-mediated signalingand thus decrease S100A4 activity in patients with metastatic CRC.Key.com by Elsevier on April 03, 2019.. Elsevier Inc. All rights reserved.

1 sistan
Wti
sciorsagc
iirea2ebsmt(aisfir
btRm
2
oaapaawHTtriifug
tiiriiepdi
4 G. Mudduluru et al. / Drug Re
e postulate that there may be a clinical benefit in treatment ofhese patients following resection to mitigate disease progressionn this high-risk population.
Apart from sulindac itself, novel sulindac derivatives such asulindac benzylamine, have been developed, that also inhibit colonancer cell growth by suppressing �-catenin transcriptional activ-ty (Whitt et al., 2012). Another derivative, NOSH-sulindac, a nitricxide- and hydrogen sulfide-releasing hybrid, has been reportedecently (Kashfi et al., 2015). This derivative is gastrointestinallyafe, and maintains the anti-inflammatory, analgesic, anti-pyretic,nd anti-platelet properties of its parent compound sulindac, withrowth inhibitory activity against a wide variety of human cancerells.
Besides its impact on CRC tumorigenesis and metastasis byntervening with COX and Wnt/�-catenin signaling, novel sulindac-nduced COX and Wnt signaling-independent pathways have beeneported in recent years, such as sulindac-induced cleavage ofpithelial cell adhesion molecule protein (Liggett et al., 2014),ctivation of NF-�B signaling (Li et al., 2012; Mladenova et al.,013), or by modulating secreted protein expression (Greeningt al., 2013). Further regulatory mechanisms of sulindac have alsoeen identified, such as its anti-fibrotic effects by suppressingignal transducer and activator of transcription (STAT3)-relatediR-21 (Zhou et al., 2015), its inhibitory effects of aldose reduc-
ase and aldo-keto reductase family 1, member B10 (AKR1B10)Cousido-Siah et al., 2015), its synergistic effect with simvastatin,
3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductasenhibitor, on apoptosis through Akt-dependent downregulation ofurvivin (Kim et al., 2015), and the involvement of myeloid zincnger 1 mediating the sulindac-induced upregulation of deatheceptor 5 (DR5) (Horinaka et al., 2014).
Taken together, the NSAID sulindac or derivatives thereof haveeen shown to act for chemoprevention of CRC and other cancerypes via COX-dependent as well as -independent mechanisms.epositioning of sulindac for chemoprevention of CRC metastasisight provide clinical benefit for CRC patients at high risk.
.1.3. IbuprofenAs all other NSAIDs, ibuprofen, a phenylpropanoic acid, is also
riginally used for COX-inhibition as analgesic, anti-phlogistic andnti-pyretic drug. Besides its anti-inflammatory effects, ibuprofennd derivatives thereof came into focus as potential thera-eutic drugs for cancer treatment. Particularly for CRC, suchnti-tumor activity was observed, since ibuprofen showed alsonti-proliferative effects in human HCT15 and HCT116 CRC cells,hich did not express the intended target, COX (Elder et al., 1997;anif et al., 1996; Janssen et al., 2006; Kashfi and Rigas, 2005).herefore, alternative modes of action for this drug were inves-igated in numerous studies and indeed, this concentrated effortevealed other therapy-relevant “off target” activities, which havempact on cancer cell proliferation, cell cycle regulation, apoptosisnduction, and potentially on metastasis and chemoprevention. Inact, this was additionally fueled by the observation that regularse of NSAIDs, including ibuprofen, might decrease cancer risk ineneral or improve survival of cancer patients.
The Wnt/�-catenin and NF-�B pathways bear great impor-ance in promoting tumor growth and metastasis. Interestingly,buprofen was found to inhibit both of these pathways by reduc-ng �-catenin translocation to the nucleus in association witheduced cyclin D1 expression. This downregulation of cyclin D1n association with c-Jun N-terminal kinase (JNK) activation bybuprofen resulted in G1 cell cycle arrest in CRC cell lines (Grösch
t al., 2003). Regarding its effects on NF-�B signaling, ibuprofenromoted NF-�B enhancer in B-cells inhibitor, alpha (I�B�) degra-ation but induced NF-�B nuclear translocation. More importantly,buprofen reduced the expression of B-cell lymphoma 2 (Bcl-2) and
Downloaded for Anonymous User (n/a) at Guilin Medical College from ClinFor personal use only. No other uses without permission. Copyright ©2
ce Updates 26 (2016) 10–27
survivin expression, induced inhibitory phosphorylation of GSK3�,which in turn suppressed NF-�B signaling in human SW480 CRCcells (Greenspan et al., 2011). This indicates that ibuprofen has thepotential to target these two important signaling pathways, act-ing in a cancer preventive manner. Another target for ibuprofenaction, peroxisome proliferator-activated receptor gamma (PPAR�)was identified, which is a pro-apoptotic transcription factor. Here,ibuprofen was shown to promote PPAR� nuclear activity and todownregulate NF-�B to prevent tumor formation in chemicallyinduced rat colon cancer models (Vaish et al., 2010). Regardingibuprofen-mediated induction of apoptosis, another study revealedthat ibuprofen was able to upregulate DR5 to promote tumornecrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in human HCT116 CRC cells (Todo et al., 2013).It was further shown that apoptosis induction by ibuprofen istightly associated with intact tumor suppressor protein 53 (p53)signaling in HCT116 cells, indicating p53 selectivity of ibuprofenaction (Janssen et al., 2008). Taken together, ibuprofen exerts manynotable “off-target” and COX-independent activities, interferingwith vital pathways of tumor cell proliferation, migration and apo-ptosis regulation. Therefore, this drug harbors great potential asanti-cancer, anti-metastatic and also as a chemopreventive agent.
2.1.4. DiclofenacThe phenylacetic acid diclofenac is routinely used as an
anti-inflammatory and analgesic drug, reducing inflammationsymptoms and pain, which like all other NSAIDs, is mainly basedon COX-2 inhibition. In addition, this drug has also inhibitorypotential on lipoxygenase and phospholipase A2, and thereby caninterfere with leukotriene and prostaglandin synthesis. As withibuprofen, diclofenac also came into focus as an NSAID with poten-tial for anti-cancer activities. In this regard, over the last decade,numerous new target proteins as well as target signaling pathwayswere identified for diclofenac-mediated anti-tumor effects. Amongthose, Wnt/�-catenin signaling is of great interest, since the expres-sion of numerous Wnt/�-catenin target genes was influenced bydiclofenac treatment. In this regard it has been demonstrated thatdiclofenac reduced Wnt/�-catenin signaling in association withreduction in translocation of �-catenin from the cytoplasm intothe nucleus in glioblastoma cells. These effects of the drug led todownregulation of �-catenin target genes, such as axin-like protein2 (Axin-2), cyclin D1 and c-myc, causing inhibition of cell prolifer-ation, colony formation and cell migration (Sareddy et al., 2013),supporting its anti-tumor and anti-metastatic potential. Similarly,diclofenac was shown to inhibit 1,2-dimethylhydrazine (DMH)-mediated activation of Wnt, �-catenin, PI3K and Akt, but increasedexpression of GSK3� as well as the pro-apoptotic proteins Bcl-2-associated X (Bax) and Bcl-2-associated agonist of cell death (Bad)in an experimental rat colon cancer model (Kaur and Sanyal, 2010;Rana et al., 2015a). This activity restored apoptosis induction, whichstrongly points to the chemopreventive and anti-tumor potentialof diclofenac. This is further supported by another study in whichdiclofenac suppressed DMH-induced tumorigenesis by repressingvascular endothelial growth factor (VEGF), monocyte chemoattrac-tant protein-1 (MCP-1) and macrophage inflammatory protein-1(MIP-1) in a rat colon cancer model (Kaur and Sanyal, 2011). In thesame DMH-induced colon cancer model, diclofenac led to down-regulation of telomerase activity in conjunction with cell cyclearrest and induction of apoptosis (Rana et al., 2015b). As furthertargets of diclofenac action, the E2F1 transcription factor and c-mycwere identified (Gottfried et al., 2013; Valle et al., 2013); diclofenacwas able to downregulate E2F1 expression, causing growth inhi-
bition in human ovarian cancer cells in vitro and in vivo. Further,the diclofenac-mediated inhibition of c-myc reduced tumor cellproliferation in Mel Im human melanoma, U937 human histio-cytic leukemia, and in PC3 prostate carcinoma cells. In addition,icalKey.com by Elsevier on April 03, 2019.019. Elsevier Inc. All rights reserved.

G. Mudduluru et al. / Drug Resistance Updates 26 (2016) 10–27 15
PI3K /Akt
Gene Regul a�on
VEGF
e.g., Cyclin D, E2F1, S100A4, Bcl-2, Bcl-xl,miR-21, Su rvivi n ...e tc
NSAIDs induc e cel l cycle ar rest an d inh ibi t cel l mig ra�on, invasion, EMT …. etc
: Ac� va�on or inhibi �on off unc� on by drug : End effect of NSAIDs: Transcri p�onal regul a�on
(Aspirin, Sulindac, Ibup rofen, Diclo fena c )
NSAI Ds
COX-1 /2
PGE2/Thromb oxan e etc
Ras MAPK GSK3ββ STAT3NF-kB Wnt/ββ-c at MMRPPARγ
COX-depende nt COX-independe nt
F Ds. Thf s inhib
toet(aolmat
(a
dptutbdmu
2
erSs
ig. 2. Schematic overview of COX-dependent and -independent inhibition by NSAIunction of one or more molecules or signaling pathways in parallel. Indeed, NSAID
his particular study revealed the impact of diclofenac treatmentn glucose metabolism in these tumor cells, showing decreasedxpression of the glucose transporter protein 1 (GLUT-1), lac-ase dehydrogenase A (LDHA) and monocarboxylate transporter 1MCT1). By this activity, diclofenac diminished glucose uptake inssociation with reduced tumor cell respiration and accumulationf intracellular lactate. In fact, this effect of diclofenac could be uti-ized for novel therapeutic concepts in cancer treatment, combining
etabolic interference and conventional chemotherapy to inducend to enforce apoptotic signaling in cancer cells for more effectiveumor cell eradication.
Comprehensive molecular or signaling axis inhibition of NSAIDsdiscussed in this subsection) and their mediated anti-cancer andnti-metastasis evidences are summarized in Fig. 2.
In conclusion for the above discussed drugs ibuprofen andiclofenac, their anti-cancer activity is a valuable feature with greatotential to treat solid cancers of the gastrointestinal tract and alsohe skin. This opens new avenues for adjuvant therapy regimenssing these drugs to intervene with tumor progression and metas-asis formation, or to prevent recurrent disease. In this context,oth the COX-dependent and COX-independent effects of theserugs seem to effectively contribute to their anti-cancer and anti-etastatic activities, rendering them of particular value for clinical
se.
.2. Statins
Statins are widely used to lower the blood cholesterol lev-
ls in patients with hypercholesterinemia, thereby reducing theisk of cardiovascular and cerebrovascular incidents (Endo, 2010).tatins are produced by fungi as secondary metabolites. All statinshare a hexahydro-naphtalene system and a �-hydroxylactone asDownloaded for Anonymous User (n/a) at Guilin Medical College from ClinicalFor personal use only. No other uses without permission. Copyright ©2019
e discussed drugs aspirin, sulindac, ibuprofen and diclofenac inhibit or activate theit cellular proliferation, survival, migration, invasion, EMT and metastasis.
common structural features; differences are due to their side chains(Manzoni et al., 1998). The first statin, mevastatin (ML-236B, com-pactin), was isolated and described in Japan during the late 1970s(Endo, 1992). The discovery was driven by the idea to isolate a nat-urally occurring compound by screening the inhibitory effects ofsecondary metabolites on the biosynthesis of cholesterol. Althougheffective, it never reached the markets. This was mainly due to tox-icity testing in dogs, where doses as high as 1000-fold as comparedto human dosage were applied (Hata et al., 1980). Today, thereare several statins available: lovastatin (natural), pravastatin, sim-vastatin (semisynthetic), atorvastatin, fluvastatin, pitavastatin, androsuvastatin (synthetic). The most severe but rare side effects aremyositis, elevated creatine kinase and rhabdomyolysis. Overall, theuse of statins in the past three decades is known as generally safe,even after prolonged treatment.
Statins act as potent inhibitors of HMG-CoA reductase. Thisenzyme catalyzes the rate-limiting step in the biosyntheticsequence from acetyl-CoA to mevalonate. The inhibition of themevalonate pathway results in reduced low density lipopro-tein, triglycerides, and total cholesterol. In addition, high densitylipoprotein might be increased. The block of HMG-CoA reductase ismediated by competitive binding of statins instead of the naturalsubstrate. The interference with the mevalonate pathway does notonly lead to reduced cholesterol synthesis, but also inhibits synthe-sis of downstream molecules like geranylgeranyl-pyrophosphate(gpp) and farnesyl-pyrophosphate (fpp) (Likus et al., 2016).
Following the reduction of intracellular gpp and fpp, levels ofrespective post-translational modifications of cellular proteins is
reduced. This was shown for Ras, which requires farnesylationfor activation and for Ras homologue (Rho) GTP-binding proteins,which are activated by geranylgeranylation. The loss of this post-translational modification will result in inhibition of proliferationKey.com by Elsevier on April 03, 2019.. Elsevier Inc. All rights reserved.

16 G. Mudduluru et al. / Drug Resistan
Sta�ns
HMG-CoA reductase
Ras
VEGF
Sta�ns inhibits cel l me tabolism, angiogenesis an d cancer progression
: Inhibi �on off unc� on by sta�ns
: End effect of sta�n s on cellula r func�ons
Rho
AMP K
ERK1/2
Proteasome
Signaling th rough
Geranylgeranyl-, farnesyl-pyrophosph ate
Direct effectsGene exp ression
: Increase of synthesis / gene regula�o n / signalin g axis
Fig. 3. Schematic overview of statin-mediated effects. Statins inhibit the enzymeHMG-CoA reductase, thereby leading to reduced mevalonate production aswell as its downstream products like geranylgeranyl-pyrophosphate or farnesyl-pyrophosphate. The latter are crucial for post-translational modifications of varioussignaling molecules. Thus, different pathways are altered in their activity, which isttV
(tiaag
ttsimis
mractiTW(t1scl
he basis underlying the anti-tumor and anti-metastatic activity of statins. In addi-ion, proteasome function and gene expression of angiogenesis-inducing genes likeEGF is inhibited.
Dai et al., 2007). The inhibition of farnesylation, geranylgeranyla-ion and the proteasome system by statins can lead to autophagyn different in vitro models. This is caused by activation of the AMP-ctivated protein kinase (AMPK) pathway, the accumulation of p21nd endoplasmic reticulum stress. Autophagy can then limit tumorrowth and metastasis (Mathew et al., 2007) (Fig. 3).
Cellular metabolism relies on proper protein quality control andight regulation of protein synthesis and degradation, mediated byhe ubiquitin proteasome system. Statins can inhibit the protea-ome, leading to accumulation of cyclin-dependent kinase (CDK)nhibitors p21 and p27. The prolonged appearance of these two
olecules results in G1 arrest of the affected cells. This proteasomenhibition seems to be mediated by the inactive, not metabolizedtatin variant (Rao et al., 1999).
Apoptosis is the process of controlled cell death aimed toaintain cellular homeostasis. In many cancer types, cells do not
espond properly to apoptotic stimuli. Therefore, restoration ofpoptotic response and activation of apoptosis is a major field inancer treatment. Agarwal et al., have shown as early as 1999 thathe combination of sulindac with lovastatin leads to an increasedn vitro cell death due to apoptosis in colon cancer cell models.his was confirmed in an in vivo rat model (Agarwal et al., 1999).u et al., have shown in a model of acute myeloid leukemia
AML), that statin treatment can alter several pathways; in 2004hey presented a combination approach with a MAPK/ERK kinase
(MEK1) inhibitor resulting in increased apoptosis compared totatin use alone (Wu et al., 2004). Thus, besides their intendedholesterol lowering properties, statins can inhibit cellular pro-iferation, immune modulation, activation of apoptosis, and have
Downloaded for Anonymous User (n/a) at Guilin Medical College from ClinFor personal use only. No other uses without permission. Copyright ©2
ce Updates 26 (2016) 10–27
effects on the proteasome system as important mechanism for theiranti-tumor action (Cafforio et al., 2005; Rao et al., 1999). Moreover,angiogenesis is a prerequisite for solid tumors which have reachedcertain size and require proper vascularization. The inhibition ofthis key process leads to nutrient deprivation. The anti-angiogenicproperties of statins are not fully established, but in a pre-clinicalmodel, lovastatin reduced angiogenesis synergistically in combi-nation with TNF�. This was at least partially due to reduced VEGFexpression. This activity might be dose-dependent, since very lowstatin concentrations were shown to increase angiogenesis (Weiset al., 2002).
Metastasis is the major cause of patient death for many cancertypes, and epithelial to mesenchymal transition (EMT) is associ-ated with cancer progression and metastasis (Kalluri and Weinberg,2009). During EMT, markers like E-cadherin are downregulated;vimentin and N-cadherin are upregulated. Warita et al., haveshown that the loss of E-cadherin expression (i.e. a more mes-enchymal phenotype) can be linked to statin sensitivity. Ectopicoverexpression of E-cadherin in different in vitro cancer modelsled to increased resistance towards statin application (Warita et al.,2014). These observations led to the notion that statin treatmentmight be employed for cancer prevention and could be used for can-cer and metastasis intervention. Recently, it was debated, whetheror not statins can prevent cancer (Ravnskov et al., 2015). How-ever, there is currently still a controversy for statin effects, sincestatins can also increase the chance to develop cancer, which hasbeen linked to low density lipoprotein (LDL) as a vital player inthe immune system in general. This counteraction is especiallytrue for prolonged statin application without further therapeuticneed, e.g. hypercholesterolemia. Several studies documented thatthe use of statins is associated with an increased risk of differentcancer types, as discussed by Ravnskov et al. In contrast, Farwellet al., showed that statin (Simvastatin) treatment reduced the can-cer incidences (Farwell et al., 2008). However, in this regard othertrials have shown that the incidence can increase upon treatmentwith statins for certain cancer types, e.g. non-melanoma skin cancer(Platz, 2008). This controversy highlights the need for a thoroughevaluation of the clinical context, in which statins were applied tothe patients.
Despite this opposite action, statins still remain potential drugsto treat patients with already established malignant disease andmight reduce the risk for metastasis. Several observational trialshave shown, at least in part, an association of statin use and reducedcancer risk or a therapeutic value (Cardwell et al., 2014; Katz et al.,2005; Kawata et al., 2001; Knox et al., 2005; Konings et al., 2010;Lersch et al., 2004; López-Aguilar et al., 1999; Mace et al., 2013;Minden et al., 2001; Vitols et al., 1997; Zanders et al., 2015). Analyz-ing the Danish population, Nielsen et al., concluded that statin usecan reduce cancer-related mortality in 13 different cancer entities(Nielsen et al., 2012). However, these trials lack proper controls andrandomization. In fact, in 2013 Klop et al., argued that observationaltrials can lead to biased results compared to randomized controlledstudies (Klop et al., 2013). One of these studies is the meta-analysisof Emberson et al., in which they analyzed 27 controlled trials anddid not observe an effect of statin use on cancer incidence or mor-tality in any cancer type (Cholesterol Treatment Trialists’ (CTT)Collaboration et al., 2012). Recently, Hoffmeister et al., analyzedsurvival of 2697 CRC patients after statin treatment. They did notfind an association of statin use and overall survival in their patientcohort. CRC-specific survival was not associated with statin appli-cation either. A subgroup comprising stage I and II patients showedan increase in recurrence-free survival (Hoffmeister et al., 2015).
Statins are valuable tools in the treatment of hypercholes-terolemia. Their use for decades shows their good safety profilewith regard to otherwise severe conditions. Statins can alter differ-ent cellular properties, leading to tumor formation and metastasis,
icalKey.com by Elsevier on April 03, 2019.019. Elsevier Inc. All rights reserved.

sistan
acaturuatamtcin
2
msqf(oiaAf2au2
i2fidPflodrihmitmac5
omenan
2
gveu
G. Mudduluru et al. / Drug Re
s shown in numerous experimental settings. This leads to the con-lusion that statins will be valuable tools in the treatment of cancernd metastasis only under certain conditions. To date, many clinicalrials and analyses have shown just a limited effect, if any, of statinse with regard to cancer. If high-risk patients can be stratified withegard to target molecules responding to statin treatment, this pop-lation might benefit from statin application. It is necessary to usepplication schemes that avoid additional cancer development dueo chronic non-physiologic reduction of LDL. The combination withdditional treatment options or drugs might improve the statin-ediated anti-cancer effects. In this case, patients with established
umors might benefit from a statin treatment. For this, more clini-al trials exploring the effects of combined treatment with statinsn stratified patient cohorts with regard to target molecules areeeded.
.3. Anti-psychotic drugs
Anti-psychotics, the main treatment option for patients withental disorders, like schizophrenia, bipolar disorder, and depres-
ion, are mainly used in cancer treatment to improve patients’uality of life. Following diagnosis, cancer patients often encountereelings of despair and anxiety, leading to severe depressionFitzgerald et al., 2015; Walker et al., 2013), while the integrationf mental health care in cancer treatment significantly reduces itsncidence and grade, shown in the second SMaRT (Symptom Man-gement Research Trial in Oncology-2) study (Sharpe et al., 2014).
high fraction of patients undergoing chemotherapy is sufferingrom chemotherapy-induced nausea and vomiting (Haiderali et al.,011), while anti-emetic drugs are given to prevent this side effectnd, in turn, to improve quality of life during therapy cycles. These and effect of these medicines was recently reviewed (Natale,015).
Anti-psychotic agents can be classified into two groups: typ-cal and atypical anti-psychotics (reviewed in Kapur and Mamo,003). Most of the typical anti-psychotics are derived from therst compound used to effectively treat neuropsychiatric disor-ers in the 1950s – the tricyclic phenothiazine chlorpromazine.rominent members of the phenothiazine family are thioredazine,uphenazine, and trifluoperazine. Structurally different membersf the typical anti-psychotics are the butyrophenones haloperi-ol and penfluridol. All anti-psychotic drugs affect dopamine D2eceptors, lessening the effect of abnormal dopaminergic functionn the brain. Treatment with these ‘first generation’ drugs withigh dopamine receptor affinity caused extrapyramidal side effects,anifested in Parkinson-like symptoms in these patients. The atyp-
cal anti-psychotics originating from clozapine, were designed inhe 1960s to improve the effect of chlorpromazine. Prominent
embers of this ‘second generation’ are risperidone, olanzapine,nd quetiapine. Blocking of dopamine receptors is reduced in thislass of anti-psychotics; they mainly act by antagonizing serotonin-HT2A receptors.
Further modification of chlorpromazine gave rise to the classf tricyclic anti-depressants (TCA), with imipramine as the firstember. TCAs interfere with the reuptake of serotonin or nor-
pinephrine, or both, and prominent members are clomipramine,ortriptyline, and amitriptyline. Here we focus on the reportednti-cancer potential of anti-psychotic drugs, in particular phe-othiazines and the tricyclic anti-depressant clomipramine.
.3.1. Typical anti-psychotics: phenothiazinesMore than 10 years ago, phenothiazines were shown to inhibit
rowth and induce apoptosis in leukemic cells at clinically rele-ant concentrations, in contrast to normal lymphocytes (Zhelevt al., 2004). A recent study of altered gene expression in tumorspon treatment with phenothiazines predicts common modes of
Downloaded for Anonymous User (n/a) at Guilin Medical College from ClinicalFor personal use only. No other uses without permission. Copyright ©2019
ce Updates 26 (2016) 10–27 17
action of these drugs (Qi and Ding, 2013). The research team pos-tulated a regulatory effect of phenothiazines on cell proliferationand cellular metabolism via CDK2, GSK3�, insulin-like growth fac-tor 1 receptor (IGF1R), fibroblast growth factor receptor 2 (FGFR2),and MAPK10, which are regulated by altered activity of MAPK8and PPAR�. Experimental evidence of common effects of phe-nothiazines that might be used in cancer therapy was reported(Daniel et al., 2015). Clathrin-mediated endocytosis is effectivelyblocked by these drugs at vesicle fission due to the inhibition ofthe dynamin GTPase activity. Besides affecting tumor growth, tri-fluoperazine was also found to inhibit cancer cell motility in a3D cell invasion assay (Pulkoski-Gross et al., 2015). Interestingly,the anti-metastatic potential of trifluoperazine was postulated viablocking of dopamine D2 receptors in cells of several cancer types,including prostate cancer and fibrosarcoma, but not in the respec-tive normal tissues. Chlorpromazine was recently found to inhibittumor growth of CRC cells by JNK-mediated sirtuin 1 (SIRT1) degra-dation, leading to enhanced p53-acetylation and hence apoptosisof tumor cells (Lee et al., 2015). Taken together, phenothiazinesmight contribute to new therapeutic options in cancer treatment,especially those with a broader range of cellular targets. As deter-mined by Kastrinsky et al., refining the molecule structures mightlead to a new class of drugs that are less anti-psychotic and moreanti-metastatic (Kastrinsky et al., 2015). Similar studies like the lat-ter would further enhance the development of effective drugs forcancer.
2.3.2. Tricyclic anti-depressants: clomipramineClomipramine (CMI) is a highly selective inhibitor of serotonin
reuptake, due to its high affinity to the serotonin receptor. More-over, the active metabolite N-desmethylclomipramine additionallyshows a high affinity to the norepinephrine transporter. A poten-tial role for CMI in cancer treatment has been assumed since the1970s, when CMI was shown to induce cell cycle arrest and celldeath in human skin fibroblasts for the first time, depending ontheir malignancy. CMI blocked the uptake of oxygen in the cells,leading to reduced energy metabolism (Wilkie and Delhanty, 1970).Later, CMI was rediscovered as anti-cancer treatment option asit inhibited cell growth and reduced cellular Ca2+ influx (Sauter,1989; Yamaji et al., 1996). Cytotoxicity upon CMI treatment wasalso reported for melanoma cell lines and primary cell cultures frommetastatic cutaneous melanoma (Parker et al., 2012).
Human acute myeloid leukemia cells responded to CMI treat-ment with elevated generation of intracellular reactive oxygenspecies, causing caspase-3 activation and thus apoptosis (Xia et al.,1999). The mechanism of reduced oxygen consumption and cyto-toxicity in human glioma cells at CMI treatment was later revealedas an inhibition of mitochondrial complex III, which triggerscaspase-3 activity in these cells (Daley et al., 2005).
N-desmethylclomipramine was recently discovered to inhibitautophagic flux at lower concentrations than reported for cytotox-icity (Rossi et al., 2009). This effect has been confirmed in cancerstem cells (CSC) of lung tumors, without elevating apoptotic signals,where it also reduced the stemness of these cells by inhibition oftheir self-renewal and cell proliferation (Bongiorno-Borbone et al.,2015).
Besides the induction of apoptosis and the reduction ofautophagic flux, CMI treatment can be used to overcome theresistance to several common anti-cancer drugs (actinomycin D,vincristine, doxorubicin, cisplatin, gemcitabine, paclitaxel). How-ever, the clear molecular mechanism is not yet elucidated inthis regard (Bongiorno-Borbone et al., 2015; Merry et al., 1991;
Pommerenke and Volm, 1995). Another cellular target of CMI is theHECT domain-containing E3 ligase ITCH (Rossi et al., 2014), whichis involved in the regulation of several important cancer-relatedprocesses, including apoptosis, cell growth, and inflammationKey.com by Elsevier on April 03, 2019.. Elsevier Inc. All rights reserved.

1 sistan
(a2r
g(ssisilrC(mu
2
daacs
2
t(1ta1cafstTtoLceatbc2
mewasivi�slsb
8 G. Mudduluru et al. / Drug Re
Bernassola et al., 2008; Melino et al., 2008). As inhibition of ITCHlso sensitized the cells to chemotherapeutic agents (Rossi et al.,014), this mechanism might be the cause for the decreased drugesistance mentioned before.
The identification of CMI as a functional inhibitor of acid sphin-omyelinase (FIASMA) also points to a possible anti-cancer functionKornhuber et al., 2011). Acid sphingomyelinase (ASM) hydrolyzesphingomyelin into ceramide and phosphorylcholine, thereby pos-ibly re-modulating the local membrane composition. ASM isnvolved in several cellular processes, like stress response, apopto-is and cancer cell resistance (Savic and Schuchman, 2013). Thenhibition of ASM by FIASMAs, in turn, leads to cancer-specificysosomal cell death, reduced tumor growth, and reversal of drugesistance (Petersen et al., 2013). Taken together, the effect ofMI in cellular processes that are crucial for cancer progressionautophagy, energy metabolism, CSC) or effective anti-cancer treat-
ent (drug resistance) renders this drug highly interesting for these in cancer-related clinical trials.
.4. Anti-helminthics
Since decades, pre- and post-clinical studies resulted in theevelopment of efficient drugs to treat helminthic parasites. Thesenti-helminthic drugs niclosamide, mebendazole and flubendazolere discussed in the following section as repositioning drugs forancer therapy with the emphasis on inhibition of tumor progres-ion and metastasis.
.4.1. NiclosamideNiclosamide is a teniacide, included in the list of the most essen-
ial drugs needed for basic health by the World Health OrganizationWHO) (18th edition of Essential Medicines). Since the mid of the960s, niclosamide is used as an anti-helminthic drug, which func-ionally inhibits glucose uptake, oxidative phosphorylation andnaerobic metabolism in its target cells (Weinbach and Garbus,969). In the early 1980s, the US National Cancer Institute (NCI)reated a drug screening panel of 60 human cell lines, which is
useful platform to screen the effects of newly developed drugsor their anti-cancer properties. Recently, Li et al., showed, using aimilar approach, that niclosamide could inhibit the cell prolifera-ion below an IC50 of 1 �M in all tested cell lines (Li et al., 2014).o date, it has been shown that niclosamide functions as an anti-umor agent in numerous cancers including CRC (Sack et al., 2011),steosarcoma (Z. Liao et al., 2015), breast cancer (Balgi et al., 2009;ondono-Joshi et al., 2014), ovarian cancer (Yo et al., 2012), prostateancer (Lu et al., 2011), non-small cell lung cancer (NSCLC) (M. Lit al., 2013), glioblastoma (Wieland et al., 2013), multiple myeloma,nd leukemia (Jin et al., 2010; Khanim et al., 2011). From a func-ional perspective, it has been described that niclosamide is able tolock multiple signaling pathways, which play a major role in can-er initiation, progression and metastasis (Li et al., 2014; Pan et al.,012).
The calcium-binding protein S100A4 is known to induceetastasis in different experimental and animal models. Its over-
xpression correlates with poor patients’ prognosis. In our study,e found that niclosamide inhibits S100A4 gene expression and,
s a consequence, the S100A4-mediated cell migration and inva-ion. Furthermore, we showed in mouse models, that niclosamidenhibited liver metastasis of CRC cell lines and increased overall sur-ival (Sack et al., 2011). In this experimental setting, niclosamidenterfered with the Wnt signaling pathway by abrogating binding of-catenin/TCF with the S100A4 gene promoter. The Wnt/�-catenin
ignaling pathway regulates major aspects of cancer progressionike cancer initiation, growth, cell differentiation and metasta-is (Clevers and Nusse, 2012). More than 90% of CRC patientsear mutations in the Wnt/�-catenin signaling pathway moleculesDownloaded for Anonymous User (n/a) at Guilin Medical College from ClinFor personal use only. No other uses without permission. Copyright ©2
ce Updates 26 (2016) 10–27
which are known to have an active status. S100A4 is transcrip-tionally activated and regulated by the Wnt/�-catenin signalingpathway in CRC (Bienz and Clevers, 2000; Sack and Stein, 2009; Sacket al., 2011; Stein et al., 2006). This evidence shows that niclosamideinhibits Wnt/�-catenin signaling, one of the key pathways inCRC and also other cancers like osteosarcoma (Chen et al., 2009;Stein et al., 2006). Further reports demonstrated that niclosamidealso interferes with the Wnt signaling pathway via inhibition ofWnt/Frizzled 1 signaling or by inducing the degradation of theWnt co-receptor LDL receptor-related protein 6 (LRP6) (Chen et al.,2009; Lu et al., 2011; Osada et al., 2011).
Like the Wnt/�-catenin signaling pathway, the mammalian tar-get of rapamycin complex 1 (mTORC1) mediated pathway plays akey role in the translation of a subset of mRNAs that are criticalfor cell growth and metabolism (Dancey, 2010). Activation of theAkt pathway, the ERK pathway or inhibition of the AMPK path-way leads to activation of mTORC signaling (Dancey, 2010; Guertinand Sabatini, 2007). Recent studies demonstrated that niclosamideinhibits the mTORC1 activity by lysosomal dysfunction in differentcancer types (Balgi et al., 2009; Fonseca et al., 2012; M. Li et al.,2013).
STAT3 also plays a key role in the transcriptional activationof different key genes which control cell proliferation, apoptosisand other cell functions (Zhong et al., 1994). STAT3 enhances cellsurvival by activating the anti-apoptotic genes Bcl-2 and Bcl-xl.Acquired resistance to erlotinib treatment activates the STAT3/Bcl-2/Bcl-xl pathway, which could be reversed by niclosamide throughinhibiting STAT3 in head and neck cancer cells, and NSCLC cells (R.Li et al., 2013a, 2013b). Niclosamide further blocked STAT3 phos-phorylation and its translocation into the nucleus in prostate cancercells (Ren et al., 2010). Niclosamide also inhibited the NF-�B andNotch pathways (Jin et al., 2010; Wieland et al., 2013), which arealso important in cancer initiation and progression.
Very recently, we initiated a first phase II clinical trial based onour previous findings (Sack et al., 2011; Stein et al., 2011b, 2006)evaluating the safety and efficacy of orally applied niclosamidein patients who progressed with metachronous or synchronousmetastases of CRC after the previous therapy (ClinicalTrials.gov;NCT02519582) All patients in this monocentric open-label clinicaltrial receive 2 g niclosamide per day. The primary outcome param-eter is progression-free survival (PFS) at 4 months. The secondaryoutcome parameters are overall survival (defined as the time fromthe date of randomization until the date of death, assessed upto 2 years, or defined as the time from patient inclusion to thedate of death or date of last follow-up news, censured data), timeto progression (defined as the time from the date of randomiza-tion until the date of first documented progression, assessed upto 2 years; progression according to RECIST = Response Evalua-tion Criteria In Solid Tumor criteria), disease control rate (definedas the time from the date of randomization, assessed up to 2years, remission + partial remission + stable disease), number ofadverse events > grade 2 toxicities according to NCI Common Tox-icity Criteria for Adverse Effects v 4.03 (time frame from the dateof randomization, assessed up to 1 month after end of therapy),and the number of serious adverse events (time frame from thedate of randomization, assessed up to 1 month after end of ther-apy). Thirty seven patients will be enrolled in this interventionaltrial. A first evaluation with PFS at 4 months as primary endpointwill be performed after treatment of the first 17 patients. Theestimated duration of the entire trial is until February 2018. Mean-while, based on the promising findings obtained with niclosamide,derivatives thereof have been tested. For these compounds with
the niclosamide chemotype it has been shown in cell culture, inmice as well as in patient samples, that they act by interven-ing in the Wnt/�-catenin signaling pathway (Mook et al., 2015;Walters Haygood et al., 2015). However, their clinical usefulness foricalKey.com by Elsevier on April 03, 2019.019. Elsevier Inc. All rights reserved.

sistan
rctft
2
dwp1ac1slezmFcosceaeid
mlecatlaFm2oe(
aptpLMbet2i8ppcsrct(
G. Mudduluru et al. / Drug Re
estriction or even prevention of tumor growth and metastasis inancer patients has to be demonstrated in clinical trials. Takenogether, these in vitro and in vivo anti-tumor and anti-metastaticunctions and the ongoing phase II clinical trial of niclosamide showhe potential as an anti-cancer drug.
.4.2. MebendazoleAnother anti-helminthic drug, mebendazole, is a highly effective
rug to treat nematode infections like pinworm, roundworm, hook-orm, whipworm and threadworm at the initial states of infectionsrior to its spread beyond the digestive track. In the mid of the980s, mebendazole was used as a post-operative treatment as anntibiotic for the prevention of infectious episodes of ovarian car-inoma, where the patients showed a better survival (Mousa et al.,987). However, Mukhopadhyay et al., published the first reporttating that mebendazole has a potent anti-tumor effect on humanung cancer cell lines both in vitro and in vivo (Mukhopadhyayt al., 2002). In this study the authors demonstrated that mebenda-ole treatment increased apoptosis in a dose- and time-dependentanner, and also increased the cell cycle arrest at G2/M phase.
urthermore, it showed less toxicity to normal cells and signifi-antly inhibited tumor growth and metastatic lesions in the lungsf nude mice. Moreover, they noticed reduced blood vessel den-ities in mebendazole-treated mice, in comparison to untreatedontrols (Mukhopadhyay et al., 2002). A similar study by Sasakit al., showed that mebendazole treatment induced mitotic arrestnd apoptosis by depolymerizing tubulin in NSCLC cells (Sasakit al., 2002). These two initial comprehensive mebendazole stud-es, both in vitro and in vivo, demonstrated that this anti-helminthicrug shows anti-tumor properties.
The primary main mode of action of mebendazoles isicrotubule-disruption, which hinders the polymerization of tubu-
in in the gut of helminths hence inducing parasites death (Laclettet al., 1980). Tubulin polymerization and structuring is vital forell division. This basic microtubule cell function is a target for
number of widely used anti-cancer drugs. Mebendazole bindso the colchicine-binding domain of tubulin and blocks tubu-in polymerization, which was first confirmed in glioblastomand melanoma models (Bai et al., 2011; Doudican et al., 2008;riedman and Platzer, 1980). It was further demonstrated that theebendazole-initiated microtubule disruption is mediated by Bcl-
phosphorylation and also showed a decreased protein amountf X-linked inhibitor of apoptosis (XIAP) in melanoma cells. How-ver, this effect could not be shown in non-melanoma cell linesDoudican et al., 2013).
Apart from the initial evidences showing mebendazole as annti-cancer drug and its mode of action by inhibiting microtubuleolymerization, other studies for example in CRC, medulloblas-oma, and mammary adenocarcinoma demonstrated anti-tumorroperties of mebendazoles (Bai et al., 2015; Coyne et al., 2014;arsen et al., 2015; Nygren and Larsson, 2014; Nygren et al., 2013).ebendazole-induced apoptosis appears to be effective through
oth p53-dependent and -independent manner. Mukhopadhyayt al., showed that after mebendazole treatment, the stabilization ofhe p53 protein indeed increased the p21 and mouse double minute
homologue (MDM2) levels, whereas p53 null-cells showed anncreased accumulation of cytochrome c and activation of caspases-
and -9, and cleavage of Poly (ADP-Ribose) Polymerase (PARP) androcaspase-3 (Mukhopadhyay et al., 2002). Dakshanamurthy et al.,redicted in silico, and experimentally proved, that mebendazolean bind to VEGF receptor 2, which indeed inhibits angiogene-is (Dakshanamurthy et al., 2012). Another recent and interesting
eport by Larsen et al., shows, that the hedgehog pathway,ommonly activated in most cancers, is inhibited in medulloblas-oma cells at clinically attainable concentrations of mebendazoleLarsen et al., 2015). Mebendazole treatment of a mammaryDownloaded for Anonymous User (n/a) at Guilin Medical College from ClinicalFor personal use only. No other uses without permission. Copyright ©2019
ce Updates 26 (2016) 10–27 19
adenocarcinoma cell line (which is resistant to chemotherapy)complemented the potency and efficacy of gemcitabine and epiru-bicin (Coyne et al., 2014), and revealed that it can also be used asa combination drug in drug resistant breast cancers to achieve abetter outcome.
Thus far, two active clinical trials with this drug are ongoing inbrain tumors. One is a phase I study with open label, at Sidney Kim-mel Comprehensive Cancer Center, Johns Hopkins Hospital, USA,of mebendazole in newly diagnosed high-grade glioma patientsreceiving temozolomide (ClinicalTrials.gov; NCT01729260). Thepurpose of this study is to find out the highest dose of mebendazolethat can be used safely in patients with malignant brain tumors incombination with the current temozolomide treatment, withoutsevere side effects. The second trial is a phase I and II study withopen label, at the North Shore Long Island Jewish Health System,USA, of mebendazole in combination with vincristine, carboplatin,and temozolomide (ClinicalTrials.gov; NCT01837862). The purposeof this study is to determine the safety and efficacy of the drugwhen used in combination with standard chemotherapeutic drugsfor the treatment of pediatric brain tumors. Both of these clinicaltrials are ongoing. In conclusion, the above in vitro and in vivo pre-clinical and clinical studies strongly suggest that mebendazole is apotential drug to inhibit cancer progression and metastasis.
2.4.3. FlubendazoleFlubendazole is a safe and effective drug widely used in
the treatment of gastrointestinal parasites (Yangco et al., 1981).Flubendazole is a member of the benzimidazole family and hasa typical benzimidazole moiety with an additional fluorine atom,which makes it different from the other benzimidazoles (Ceballoset al., 2011). Like mebendazole, flubendazole also inhibits tubu-lin polymerization by binding to tubulin (Katiyar et al., 1994;Spagnuolo et al., 2010). Very few studies have been performed toassess the anti-cancer activity of flubendazole when compared withmebendazole.
As a first evidence, treatment with flubendazole at nanomo-lar concentrations induced cell death in a panel of leukemia andmyeloma cell lines. Secondly, flubendazole inhibited the clono-genic growth of primary acute myeloid leukemia cells derivedfrom patients. Mice xenograft models showed a significant delayedtumor growth in leukemia and myeloma cells after flubendazoletreatment (Spagnuolo et al., 2010). Another study with fluben-dazole treatment of CRC cells showed a significant time- anddose-dependent inhibition of the cell cycle with an arrest at theG2/M phase (Králová et al., 2013). Combination treatment of fluben-dazole and paclitaxel showed a more effective inhibition of cellproliferation than paclitaxel treatment alone (Králová et al., 2013).
Michaelis et al., studied flubendazole sensitivity of 321 differ-ent cancer cell lines from 26 cancer lineages. Among the panelof tumor cell lines screened for flubendazole sensitivity, onlyleukemia, myeloma and neuroblastoma cell lines showed highersensitivity, compared to other cell lines. Moreover, flubendazoleshowed reduced cell viability in primary neuroblastoma cells andalso in a panel of 140 cell lines which acquired drug resistance forvarious anti-cancer drugs at lower concentrations. A chorioallan-toic membrane (CAM) assay demonstrated the in vivo relevanceof flubendazole by inhibiting the blood vessel formation andneuroblastoma tumor growth. Short interfering (si)RNA-mediateddepletion of p21, BAX or PUMA, molecules downstream of p53,reduced the neuroblastoma cell sensitivity, hence demonstratingthat the p53 pathway plays a major role in flubendazole-mediatedanti-tumor activity (Michaelis et al., 2015).
Breast cancer stem-like cells with increased resistance to ther-apy, induced EMT, metastasis and relapse, are known to have amolecular profile of CD44+/CD24− and elevated aldehyde dehy-drogenase 1 (ALDH1) expression (Al-Hajj et al., 2003; Ginestier
Key.com by Elsevier on April 03, 2019.. Elsevier Inc. All rights reserved.

20 G. Mudduluru et al. / Drug Resistance Updates 26 (2016) 10–27
External s�muli(ex., TGF, EGF, IGF1)
Gli
Axin /APC/GSK3ββ/Dvl
ββ-cateni n
Notch
NICD
NICD
JAK/STAT3 PI3K/AktMAPK
NF-kBAP-1mTORC1
Gene Regul a�on
STAT3
e.g., S100A4, Bcl-2, Bcl-xl, c- myc ...e tc
Cell death
p53
Bax
Caspase 3
An�-helminthic s Inh ibi t mig ra�on, invasion, EM T,metastasis, tumor growt h and angiogenesis
: Ac� va�o n of p53 induced apop tosi s by an�-helmenthi c drugs : Ac� ve molecul e translo ca�o n to nucleous
: Induce d gene expressio n inhibit s cel l de ath
Niclosamide
Mebendazole
Flubendazole
: Inhibi�on of respec� ve path ways by an�-helme nthi c dru gs
2 3
1 1
1
3
2
1
2
3
11
1
: End effect of an�-helmenthic drugs : Transcri p�onal regul a�on
LRP5/6
WntFrizzled
Fig. 4. Schematic representation of anti-helminthic drugs mediating signaling pathways. Drugs include: (1) Niclosamide (STAT3, Akt, MAPK, NF-�B, Notch and Wnt/�-c athwai -medid
eTn(imcYtevsbc(tTacsematadvt
atenin signaling pathways), (2) Mebendazole (Hedgehog and activating the p53 pnhibits tumor progression and metastasis, induces apoptosis and tumor suppressoromain).
t al., 2007; Li et al., 2008; Mani et al., 2008; Phillips et al., 2006).hese cells also showed active hedgehog, Wnt/�-catenin, andotch signaling pathways; all of them contributed to self-renewalNicolini et al., 2011). Treatment of these cells with flubendazolenhibited cell proliferation and delayed tumor growth in a xenograft
odel. Furthermore, flubendazole reduced the self-renewal genes-myc, octamer binding protein 4 (Oct4), sex determining region-box 2 (Sox2), Nanog and cyclin D1, thus reducing the subpopula-ion of CD44+/CD24− cells. Additionally, it inhibited cell migration,xpression of mesenchymal markers �-catenin, N-cadherin andimentin and induced the epithelial marker keratin 18. In con-istence with other results, flubendazole treatment also arrestedreast cancer cells at the G2/M phase and sensitized cells foronventional therapeutic drugs as 5-fluorouracil and doxorubicinHou et al., 2015). This evidence shows that apart from its anti-umor activity, it also suppressed breast cancer stem-cell like cells.hese cumulative evidences suggest that flubendazole could be
potential drug to treat at least neuroblastoma and breast can-er, by inhibiting cell proliferation, migration, and EMT while alsoensitizing drug resistant cells to chemotherapy. Comprehensivevidence of anti-helminthic drugs discussed here (niclosamide,ebendazole and flubendazole), proteins and signaling pathways
re represented in Fig. 4. In conclusion, along with the tradi-ional chemotherapeutic drugs, these anti-helminthic drugs could
lso function as anti-cancer drugs either alone or in combination,epending on the cancer type and molecular signature of the indi-idual patients. However, more basic research has to be performedo delineate the particular mechanism of these anti-helminthicDownloaded for Anonymous User (n/a) at Guilin Medical College from ClinFor personal use only. No other uses without permission. Copyright ©2
y), and (3) Flubendazole (Wnt/�-catenin and activates the p53 signaling pathway)ated signaling axis. (SMO: Smoothened; Dvl: Dishevelled; NICD: Notch intracellular
drugs in either activating or inhibiting the multiple importantsignaling pathways and their secondary mediator molecules, whichwill certainly enhance the development of efficient cancer treat-ment strategies.
2.5. Vitamin D
Vitamin D, originally described as a lipid-soluble vitamin is nowconsidered a secosteroid prehormone. The major subtypes are vita-min D3 (cholecalciferol) and vitamin D2 (ergocalciferol). From abiological perspective, vitamin D3 is the most active derivativeand is synthesized non-enzymatically upon exposure to ultravioletrays in sunlight from 7-dehydrocholesterol to cholecalciferol in theepidermis and dermis.
Severe vitamin D deficiency (serum level below 15 nmol/l)leads to rickets in children or osteomalacia in adults and children.Whereas severe hypovitaminosis D is rare, a moderate deficiency(defined as 25(OH)D3 serum level below 25 nmol/l) is very com-mon (8% of adults in the US) (Looker et al., 2011) and is known tocontribute to the development of osteoporosis. Recent data suggestthat vitamin D deficiency is associated with a higher incidence ofcardiovascular diseases, diabetes and cancer (Ryan et al., 2015).
As mentioned before, vitamin D3 is a prehormone which hasto be activated by double hydroxylation to form 25(OH)D3 in the
liver and further to 1,25(OH)2D3 in the kidney, which is the maincontributor to vitamin D activity. The latter exerts two differentmodes of action. The main (slow) process is the transcriptional reg-ulation of target genes. Vitamin D3 binds to the nuclear vitamin DicalKey.com by Elsevier on April 03, 2019.019. Elsevier Inc. All rights reserved.

sistan
rttpt
m(Pb(mT
cGecso1sTvmH
FDah
G. Mudduluru et al. / Drug Re
eceptor (VDR), which is a member of the steroid hormone recep-or superfamily. The activated VDR specifically binds along withhe retinoic X receptor, to the vitamin D response elements in theromoter region of target genes and regulates the transcription ofhese multiple (∼50) genes (Deeb et al., 2007).
As the non-genomic (rapid) activity of vitamin D3, binding to aembranous VDR with activation of store-operated Ca2+ channels
SOC) is assumed. The increased intracellular Ca2+ level activatesKC. A subsequent activation of the MEK/ERK-pathway by PKC haseen described for normal colon cells and skeletal muscle cellsDeeb et al., 2007), in contrast to malignant cell lines, where vita-
in D3 leads to a decreased activity of Erk1 (Narayanan et al., 2004).hese pathways are not fully elucidated yet.
The main cancer-related research focus of vitamin D3 is its can-er preventive potential. In an epidemiologic analysis Garland andarland (1980) showed that sunlight exposure and vitamin D lev-ls were inversely correlated with the incidence of CRC. This resultould be confirmed in many other studies (Song et al., 2015). Severaltudies analyzed the effect of vitamin D supplementation on vari-us types of cancer. A Cochrane meta-analysis from 2014, including8 trials with 50,623 participants, analyzed the effects of vitamin Dupplementation on prevention of several solid cancers in adults.
he authors showed a significant positive overall effect on pre-ention of mortality (7.5% vs. 8.0%; p = 0.009) and cancer-relatedortality (2.5% vs. 2.8%; p = 0.02) upon vitamin D supplementation.owever, this positive effect could not be confirmed in the trialVDR
RXR -
Vita
Vitamin D3
HIF1α VEGF
Bax
Bak Bad
Bcl-2
RAS
RAF
MEK
ERK
Vitamin D3
Angiogenesis
Apop tosis
VDRE
: Inhibi�on : Transc
ig. 5. Schematic representation of vitamin D3-mediated signaling pathways: Vitamin D33 complex binds directly to �-catenin and blocks the formation of LEF-1/TCF complex. Fund expression of DKK1 that inhibits LRP5/6, which in turns deactivates the Wnt receptorence sensitizing the cells to apoptosis by inducing the Bax, Bcl-2-antagonist/killer 1 (Bak
Downloaded for Anonymous User (n/a) at Guilin Medical College from ClinicalFor personal use only. No other uses without permission. Copyright ©2019
ce Updates 26 (2016) 10–27 21
sequential analysis, which indicated a possible intervention effecton cancer-related mortality. In conclusion, the authors did not findevidence for cancer prevention by vitamin D (Bjelakovic et al.,2014).
Despite the lack of effect in cancer prevention, vitamin D wasshown to play a role in cancer signaling. Several in vitro and invivo studies showed anti-tumor effect of vitamin D3 by sensitizingcells for apoptosis or inhibiting EMT, proliferation and angiogenesis(Deeb et al., 2007) (Fig. 5).
Resistance to cell death is a major characteristic of malig-nant tissues. In breast cancer cell lines, vitamin D3 activates theintrinsic pathway of apoptosis by inducing the expression of Bax,Bcl-2-antagonist/killer (Bak) and Bad and decrease the expressionof anti-apoptotic proteins like Bcl-2 (Kubis and Piwowar, 2015;Narvaez and Welsh, 2001). It is known that the activity of vitaminD3 on the MEK/ERK pathway differs between malignant and normaltissues without knowledge of the exact mechanisms. An analy-sis in squamous cancer cell lines could show a caspase-mediatedcleavage of MEK following treatment with vitamin D3. After MEKcleavage, the following anti-apoptotic ERK signaling is interrupted(McGuire et al., 2001).
Vitamin D3 treatment inhibited the Wnt signaling induced
EMT in colon and breast cancer cells via different mechanismsin vitro (Larriba et al., 2013). The VDR-vitamin D3 complex bindsdirectly to �-catenin and avoids the formation of the trans-criptional �-catenin/TCF complex (Shah et al., 2006). Thus, theFrizzled
LEF-1/TCF
LRP5/6
Wnt
ββ-catenin
DKK1
E-cadherin
DKK1
Canonical Wnttarg et gens
p21
p27
min D3
EMT
Cell cycle
rip�ona l regul a�on
blocks EMT via inhibition of Wnt signaling by different mechanisms: VDR-vitaminrthermore, it induces the expression of E-cadherin, leading to �-catenin inhibition. Cell cycle is inhibited by vitamin D3 by increasing the expression of p21 and p27,) and Bad, and decreasing the expression of Bcl-2.
Key.com by Elsevier on April 03, 2019.. Elsevier Inc. All rights reserved.

2 sistan
tpeecabLcptDaie
tV1astM
cd(2nvds0ccpdteca5amAdppewloEb
3
gdmemta
2 G. Mudduluru et al. / Drug Re
ranscription of several target genes including c-myc, TCF1, lym-hoid enhancer-binding factor 1 (LEF1), AXIN2, PPAR�, CD44 andctoderm-neuronal cortex protein 1 (ENC1) is inhibited (Pálmert al., 2001). Furthermore, vitamin D3 induced the expression of E-adherin and Dickkopf1 (DKK1). DKK1 binds to the LRP5/6 receptornd inhibits the Wnt signaling pathway by blocking the interactionetween LRP5/6 and the frizzled receptor (Aguilera et al., 2007;arriba et al., 2013). Cell cycle progression is positively regulated byyclins and CDKs and negatively controlled by CDK-Inhibitors (e.g.21, p27 and p53). Vitamin D3 inhibits proliferation by increasinghe expression of both CDK-inhibitors, by binding to the vitamin
response elements in the gene promoters of p21 and growthrrest and DNA damage-inducible protein 45 � (GADD45A), whichn turn induces p27 expression (Goeman et al., 2014; Verlindent al., 1998).
An important factor for tumor proliferation and metastasis is theumor-associated vascularization by angiogenesis. In this processEGFA, thrombospondin-1 (TBS-1) and the hypoxia inducing factor
� (HIF1�) play an important role. In vitro analyses in prostate, lungnd CRC cell lines and in vivo analysis in a breast cancer xenografthowed an inhibition of angiogenesis by vitamin D3 by decreasinghe expression of VEGFA and HIF1� (Ben-Shoshan et al., 2007;
antell et al., 2000).Further, in vitro and in vivo studies showed an enhanced
ytotoxic effect of anti-tumor agents like cisplatin, carboplatin,ocetaxel or paclitaxel, when supplemented with vitamin D3Hershberger et al., 2002; Moffatt et al., 1999; Wang et al.,000). A recently published retrospective analysis in patients withon-metastatic breast cancer showed, that patients receiving aitamin D supplementation because of a vitamin D deficiencyuring adjuvant chemotherapy have an improved disease-freeurvival compared to unsupplemented patients (HR 0.36 (95%-CI.15–0.88); p = 0.026) (Zeichner et al., 2015). In combination withytotoxic drugs, vitamin D supplementation was effective in severallinical studies. Blanke et al., showed in 25 patients a longer time torogression (3.6 vs. 1.5 months mono-docetaxel) after weekly, highose calcitriol supplementation (0.5 �g/kg), combined with doce-axel in advanced pancreatic cancer (Blanke et al., 2009). Medionit al., tested the additional effect of the vitamin D analogue inecal-itol over the standard docetaxel therapy in prostate cancer. Theuthors could show that the combination therapy leads to a faster0% reduction of the serum tumor marker PSA (prostate specificntigen) than the standard docetaxel monotherapy (2.9 vs. 5.3onths) (Medioni et al., 2014). In contrast to these findings, theSCENT 2 trial, a phase II trial comparing docetaxel plus high-ose calcitriol to docetaxel plus prednisone in castration-resistantrostate cancer, showed a lower survival for calcitriol treatedatients (17.8 vs. 20.2 months median survival; p = 0.002) (Schert al., 2011). In conclusion, vitamin D either alone or in combinationith other cytotoxic drugs effectively inhibited cancer hallmarks
ike cell proliferation, invasion, migration, and angiogenesis. More-ver, it sensitized cancer patients for traditional cytotoxic drugs.specially patients with proven vitamin D deficiency had survivalenefits from vitamin D supplementation during chemotherapy.
. Conclusions
Repositioning of established “old” drugs for new targets bearsreat promise in cancer therapy. Targeting certain proteins andisrupting molecular cascades driving tumor progression andetastasis by exploiting already approved drugs will certainly
nhance cancer treatment options. In the era of personalizededicine, the molecular classification of patient sub-cohorts,
he identification of high-risk patients, and patient stratificationccording to molecular biomarkers, the repositioning of known
Downloaded for Anonymous User (n/a) at Guilin Medical College from ClinFor personal use only. No other uses without permission. Copyright ©2
ce Updates 26 (2016) 10–27
drugs could offer great benefit for the patients. Is this optimismjustified? Drugs constituting the class of NSAIDs such as aspirin,sulindac, ibuprofen and diclofenac demonstrate, apart from theirchemopreventive properties, also anti-cancer and anti-metastaticactivities. Statins applied for treatment of hypercholesterolemiamight be used for anti-cancer and anti-metastatic purposes aswell. Anti-psychotic drugs like phenothiazines and clomipramineharbor potential to interfere with cancer development and metas-tasis. Anti-helminthics including niclosamide and mebendazole,demonstrate anti-tumorigenic and anti-metastatic activities. Fur-thermore, vitamin D as a supplement during chemotherapy isimproving cancer patient prognosis.
Thus, are there drawbacks to this repositioning strategy?Although our knowledge on the molecular basis of cancer is rapidlyincreasing, we are still far away from comprehensively understand-ing all the pathways, their cross talks and interactions, buildingthe potential targets for intervention by using the drugs supposedto be repositioned. Not to mention tumor heterogeneity, genomicinstability, immune surveillance, and mechanisms of multidrugresistance. However, programs like the ReDO initiative are excel-lent examples for a systematic investigation of the repositoriesof old drugs, based on well-defined criteria to select promising,high-potential candidates. First drugs were already re-evaluatedand identified for novel anti-cancer treatments. Further optionsin maximizing the effects of the old drugs are their combinatorialuse with further treatment modalities depending on cancer typeand molecular signature of the individual patients. Taken together,repositioning of old drugs for novel cancer targets is an indispens-able approach exploiting an inestimable source, in order to meetthe great need for improving cancer therapy and to accelerate theclinical use of these drugs.
Acknowledgements
This work was supported by the German Cancer Consortium(DKTK) (to GM and US). CT is a participant in the Charité ClinicalScientist Program funded by the Charité Universitätsmedizin Berlinand the Berlin Institute of Health.
References
Agarwal, B., Rao, C.V., Bhendwal, S., Ramey, W.R., Shirin, H., Reddy, B.S., Holt, P.R.,1999. Lovastatin augments sulindac-induced apoptosis in colon cancer cellsand potentiates chemopreventive effects of sulindac. Gastroenterology 117,838–847.
Aguilera, O., Pena, C., García, J.M., Larriba, M.J., Ordónez-Morán, P., Navarro, D., Bar-báchano, A., López de Silanes, I., Ballestar, E., Fraga, M.F., Esteller, M., Gamallo,C., Bonilla, F., González-Sancho, J.M., Munoz, A., 2007. The Wnt antagonistDICKKOPF-1 gene is induced by 1alpha, 25-dihydroxyvitamin D3 associated tothe differentiation of human colon cancer cells. Carcinogenesis 28, 1877–1884.
Al-Hajj, M., Wicha, M.S., Benito-Hernandez, A., Morrison, S.J., Clarke, M.F., 2003.Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad.Sci. U. S. A. 100, 3983–3988.
Bai, R.-Y., Staedtke, V., Aprhys, C.M., Gallia, G.L., Riggins, G.J., 2011. Antiparasiticmebendazole shows survival benefit in 2 preclinical models of glioblastomamultiforme. Neuro Oncol. 13, 974–982.
Bai, R.-Y., Staedtke, V., Rudin, C.M., Bunz, F., Riggins, G.J., 2015. Effective treatmentof diverse medulloblastoma models with mebendazole and its impact on tumorangiogenesis. Neuro Oncol. 17, 545–554.
Balgi, A.D., Fonseca, B.D., Donohue, E., Tsang, T.C.F., Lajoie, P., Proud, C.G., Nabi, I.R.,Roberge, M., 2009. Screen for chemical modulators of autophagy reveals noveltherapeutic inhibitors of mTORC1 signaling. PLoS ONE 4, e7124.
Ben-Shoshan, M., Amir, S., Dang, D.T., Dang, L.H., Weisman, Y., Mabjeesh, N.J., 2007.1alpha, 25-dihydroxyvitamin D3 (Calcitriol) inhibits hypoxia-inducible factor-1/vascular endothelial growth factor pathway in human cancer cells. Mol. CancerTher. 6, 1433–1439.
Bernassola, F., Karin, M., Ciechanover, A., Melino, G., 2008. The HECT family of E3ubiquitin ligases: multiple players in cancer development. Cancer Cell 14, 10–21.
Bienz, M., Clevers, H., 2000. Linking colorectal cancer to Wnt signaling. Cell 103,311–320.
Bjelakovic, G., Gluud Lise, L., Nikolova, D., Whitfield, K., Krstic, G., Wetterslev, J.,Gluud, C., 2014. Vitamin D supplementation for prevention of cancer in adults.Cochrane Database Syst. Rev. 6, CD007469.
icalKey.com by Elsevier on April 03, 2019.019. Elsevier Inc. All rights reserved.

sistan
B
B
B
C
C
C
C
C
C
C
C
C
C
D
D
D
D
D
D
D
D
D
D
E
E
E
F
G. Mudduluru et al. / Drug Re
lanke, C.D., Beer, T.M., Todd, K., Mori, M., Stone, M., Lopez, C., 2009. Phase II study ofcalcitriol-enhanced docetaxel in patients with previously untreated metastaticor locally advanced pancreatic cancer. Invest. New Drugs 27, 374–378.
ongiorno-Borbone, L., Giacobbe, A., Compagnone, M., Eramo, A., De Maria, R.,Peschiaroli, A., Melino, G., 2015. Anti-tumoral effect of desmethylclomipraminein lung cancer stem cells. Oncotarget 6, 16926–16938.
oon, E.M.J., Keller, J.J., Wormhoudt, T., a, M., Giardiello, F.M., Offerhaus, G.J.a., van derNeut, R., Pals, S.T., 2004. Sulindac targets nuclear beta-catenin accumulation andWnt signalling in adenomas of patients with familial adenomatous polyposisand in human colorectal cancer cell lines. Br. J. Cancer 90, 224–229.
afforio, P., Dammacco, F., Gernone, A., Silvestris, F., 2005. Statins activate the mito-chondrial pathway of apoptosis in human lymphoblasts and myeloma cells.Carcinogenesis 26, 883–891.
ardwell, C.R., Hicks, B.M., Hughes, C., Murray, L.J., 2014. Statin use after colorectalcancer diagnosis and survival: a population-based cohort study. J. Clin. Oncol.32, 3177–3183.
eballos, L., Elissondo, C., Sánchez Bruni, S., Denegri, G., Lanusse, C., Alvarez, L., 2011.Comparative performances of flubendazole and albendazole in cystic echinococ-cosis: ex vivo activity, plasma/cyst disposition, and efficacy in infected mice.Antimicrob. Agents Chemother. 55, 5861–5867.
hen, M., Wang, J., Lu, J., Bond, M.C., Ren, X.-R., Lyerly, H.K., Barak, L.S., Chen, W., 2009.The anti-helminthic niclosamide inhibits Wnt/Frizzled1 signaling. Biochemistry48, 10267–10274.
holesterol Treatment Trialists’ (CTT) CollaborationEmberson, J.R., Kearney, P.M.,Blackwell, L., Newman, C., Reith, C., Bhala, N., Holland, L., Peto, R., Keech, A.,Collins, R., Simes, J., Baigent, C., 2012. Lack of effect of lowering LDL cholesterol oncancer: meta-analysis of individual data from 175,000 people in 27 randomisedtrials of statin therapy. PLoS ONE 7, e29849.
levers, H., 2006. Colon cancer – understanding how NSAIDs work. N. Engl. J. Med.354, 761–763.
levers, H., Nusse, R., 2012. Wnt/�-catenin signaling and disease. Cell 149,1192–1205.
ook, N.R., Lee, I.-M., Gaziano, J.M., Gordon, D., Ridker, P.M., Manson, J.E., Hen-nekens, C.H., Buring, J.E., 2005. Low-dose aspirin in the primary prevention ofcancer: the Women’s Health Study: a randomized controlled trial. JAMA 294,47–55.
ousido-Siah, A., Ruiz, F.X., Crespo, I., Porté, S., Mitschler, A., Parés, X., Podjarny, A.,Farrés, J., 2015. Structural analysis of sulindac as an inhibitor of aldose reductaseand AKR1B10. Chem. Biol. Interact. 234, 290–296.
oyne, C.P., Jones, T., Bear, R., 2014. Anti-neoplastic cytotoxicity of gemcitabine-(C4-amide)-[anti-EGFR] in dual-combination with epirubicin-(C3-amide)-[anti-HER2/neu] against chemotherapeutic-resistant mammary adenocarcinoma(SKBr-3) and the complementary effect of mebendazole. J. Cancer Res. Ther.Oncol. 2, 203.
ai, Y., Khanna, P., Chen, S., Pei, X.-Y., Dent, P., Grant, S., 2007. Statins synergisti-cally potentiate 7-hydroxystaurosporine (UCN-01) lethality in human leukemiaand myeloma cells by disrupting Ras farnesylation and activation. Blood 109,4415–4423.
akshanamurthy, S., Issa, N.T., Assefnia, S., Seshasayee, A., Peters, O.J., Madha-van, S., Uren, A., Brown, M.L., Byers, S.W., 2012. Predicting new indicationsfor approved drugs using a proteochemometric method. J. Med. Chem. 55,6832–6848.
aley, E., Wilkie, D., Loesch, A., Hargreaves, I.P., Kendall, D.A., Pilkington, G.J., Bates,T.E., 2005. Chlorimipramine: a novel anticancer agent with a mitochondrialtarget. Biochem. Biophys. Res. Commun. 328, 623–632.
ancey, J., 2010. mTOR signaling and drug development in cancer. Nat. Rev. Clin.Oncol. 7, 209–219.
aniel, J.A., Chau, N., Abdel-Hamid, M.K., Hu, L., von Kleist, L., Whiting, A., Krishnan,S., Maamary, P., Joseph, S.R., Simpson, F., Haucke, V., McCluskey, A., Robinson, P.J.,2015. Phenothiazine-derived antipsychotic drugs inhibit dynamin and clathrin-mediated endocytosis. Traffic 16, 635–654.
annenberg, A.J., Altorki, N.K., Boyle, J.O., Dang, C., Howe, L.R., Weksler, B.B., Subbara-maiah, K., 2001. Cyclo-oxygenase 2: a pharmacological target for the preventionof cancer. Lancet. Oncol. 2, 544–551.
eeb, K.K., Trump, D.L., Johnson, C.S., 2007. Vitamin D signalling pathways in cancer:potential for anticancer therapeutics. Nat. Rev. Cancer 7, 684–700.
oudican, N.A., Rodriguez, A., Osman, I., Orlow, S.J., 2008. Mebendazole induces apo-ptosis via Bcl-2 inactivation in chemoresistant melanoma cells. Mol. Cancer Res.6, 1308–1315.
oudican, N.A., Byron, S.A., Pollock, P.M., Orlow, S.J., 2013. XIAP downregulationaccompanies mebendazole growth inhibition in melanoma xenografts. Anti-cancer Drugs 24, 181–188.
ovizio, M., Tacconelli, S., Sostres, C., Ricciotti, E., Patrignani, P., 2012. Mechanisticand pharmacological issues of aspirin as an anticancer agent. Pharmaceuticals(Basel) 5, 1346–1371.
lder, D.J., Halton, D.E., Hague, A., Paraskeva, C., 1997. Induction of apoptotic celldeath in human colorectal carcinoma cell lines by a cyclooxygenase-2 (COX-2)-selective nonsteroidal anti-inflammatory drug: independence from COX-2protein expression. Clin. Cancer Res. 3, 1679–1683.
ndo, A., 2010. A historical perspective on the discovery of statins. Proc. Jpn. Acad.Ser. B: Phys. Biol. Sci. 86, 484–493.
ndo, A., 1992. The discovery and development of HMG-CoA reductase inhibitors. J.
Lipid Res. 33, 1569–1582.ajardo, A.M., Piazza, G.A., 2015. Chemoprevention in gastrointestinal physiologyand disease. Anti-inflammatory approaches for colorectal cancer chemo-prevention. Am. J. Physiol. Gastrointest. Liver Physiol. 309, G59–G70.
Downloaded for Anonymous User (n/a) at Guilin Medical College from ClinicalFor personal use only. No other uses without permission. Copyright ©2019
ce Updates 26 (2016) 10–27 23
Farwell, W.R., Scranton, R.E., Lawler, E.V., Lew, R.A., Brophy, M.T., Fiore, L.D., Gaziano,J.M., 2008. The association between statins and cancer incidence in a veteranspopulation. J. Natl. Cancer Inst. 100, 134–139.
Fisher, R., Pusztai, L., Swanton, C., 2013. Cancer heterogeneity: implications for tar-geted therapeutics. Br. J. Cancer 108, 479–485.
Fitzgerald, P., Lo, C., Li, M., Gagliese, L., Zimmermann, C., Rodin, G., 2015. The rela-tionship between depression and physical symptom burden in advanced cancer.BMJ Support. Palliat. Care 5, 381–388.
Fonseca, B.D., Diering, G.H., Bidinosti, M.A., Dalal, K., Alain, T., Balgi, A.D., Forestieri,R., Nodwell, M., Rajadurai, C.V., Gunaratnam, C., Tee, A.R., Duong, F., Andersen,R.J., Orlowski, J., Numata, M., Sonenberg, N., Roberge, M., 2012. Structure-activityanalysis of niclosamide reveals potential role for cytoplasmic pH in control ofmammalian target of rapamycin complex 1 (mTORC1) signaling. J. Biol. Chem.287, 17530–17545.
Friedman, P.A., Platzer, E.G., 1980. Interaction of anthelmintic benzimidazoles withAscaris suum embryonic tubulin. Biochim. Biophys. Acta 630, 271–278.
Gardner, S.H., Hawcroft, G., Hull, M.A., 2004. Effect of nonsteroidal anti-inflammatory drugs on beta-catenin protein levels and catenin-relatedtranscription in human colorectal cancer cells. Br. J. Cancer 91, 153–163.
Garland, C.F., Garland, F.C., 1980. Do sunlight and vitamin D reduce the liklihood ofcolon cancer? Int. J. Epidemiol. 9, 227–231.
Ginestier, C., Hur, M.H., Charafe-Jauffret, E., Monville, F., Dutcher, J., Brown, M.,Jacquemier, J., Viens, P., Kleer, C.G., Liu, S., Schott, A., Hayes, D., Birnbaum, D.,Wicha, M.S., Dontu, G., 2007. ALDH1 is a marker of normal and malignant humanmammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 1,555–567.
Goel, A., Chang, D.K., Ricciardiello, L., Gasche, C., Boland, C.R., 2003. A novel mecha-nism for aspirin-mediated growth inhibition of human colon cancer cells. Clin.Cancer Res. 9, 383–390.
Goeman, F., De Nicola, F., De Meo, P.D.O., Pallocca, M., Elmi, B., Castrignanò, T., Pesole,G., Strano, S., Blandino, G., Fanciulli, M., Muti, P., 2014. VDR primary targetsby genome-wide transcriptional profiling. J. Steroid Biochem. Mol. Biol. 143,348–356.
Gonen, N., Assaraf, Y.G., 2012. Antifolates in cancer therapy: structure, activity andmechanisms of drug resistance. Drug Resist. Updat. 15, 183–210.
Gottfried, E., Lang, S.A., Renner, K., Bosserhoff, A., Gronwald, W., Rehli, M., Einhell,S., Gedig, I., Singer, K., Seilbeck, A., Mackensen, A., Grauer, O., Hau, P., Dettmer,K., Andreesen, R., Oefner, P.J., Kreutz, M., 2013. New aspects of an old drug –diclofenac targets MYC and glucose metabolism in tumor cells. PLOS ONE 8,e66987.
Greening, D.W., Ji, H., Kapp, E.a., Simpson, R.J., 2013. Sulindac modulates secretedprotein expression from LIM1215 colon carcinoma cells prior to apoptosis.Biochim. Biophys. Acta 1834, 2293–2307.
Greenspan, E.J., Madigan, J.P., Boardman, L.A., Rosenberg, D.W., 2011. Ibupro-fen inhibits activation of nuclear {beta}-catenin in human colon adenomasand induces the phosphorylation of GSK-3{beta}. Cancer Prev. Res. (Phila.) 4,161–171.
Grösch, S., Tegeder, I., Schilling, K., Maier, T.J., Niederberger, E., Geisslinger, G., 2003.Activation of c-Jun-N-terminal-kinase is crucial for the induction of cell cyclearrest in human colon carcinoma cells caused by flurbiprofen enantiomers.FASEB J. 17, 1316–1318.
Guertin, D.A., Sabatini, D.M., 2007. Defining the role of mTOR in cancer. Cancer Cell12, 9–22.
Gurpinar, E., Grizzle, W.E., Piazza, G.a., 2014. NSAIDs inhibit tumorigenesis, but how?Clin. Cancer Res. 20, 1104–1113.
Haiderali, A., Menditto, L., Good, M., Teitelbaum, A., Wegner, J., 2011. Impact on dailyfunctioning and indirect/direct costs associated with chemotherapy-inducednausea and vomiting (CINV) in a U.S. population. Support. Care Cancer 19,843–851.
Han, A., Song, Z., Tong, C., Hu, D., Bi, X., Augenlicht, L.H., Yang, W., 2008. Sulindacsuppresses beta-catenin expression in human cancer cells. Eur. J. Pharmacol.583, 26–31.
Hanif, R., Pittas, A., Feng, Y., Koutsos, M.I., Qiao, L., Staiano-Coico, L., Shiff, S.I., Rigas,B., 1996. Effects of nonsteroidal anti-inflammatory drugs on proliferation andon induction of apoptosis in colon cancer cells by a prostaglandin-independentpathway. Biochem. Pharmacol. 52, 237–245.
Hata, Y., Shigematsu, H., Oikawa, T., Yamamoto, M., Yamauchi, Y., Goto, Y., 1980.Treatment of hypercholesterolemia with an HMG-CoA reductase inhibitor(CS-500). II. Determination of unit weight effect and daily doses by an inte-gration method and observation of safety in initial stage. Geriatr. Med. 18,104–112.
Hershberger, P.a., McGuire, T.F., Yu, W.-D., Zuhowski, E.G., Schellens, J.H.M.,Egorin, M.J., Trump, D.L., Johnson, C.S., 2002. Cisplatin potentiates 1,25-dihydroxyvitamin D3-induced apoptosis in association with increased mitogen-activated protein kinase kinase kinase 1 (MEKK-1) expression. Mol. Cancer Ther.1, 821–829.
Hochmuth, F., Jochem, M., Schlattmann, P., 2015. Meta-analysis of aspirin use andrisk of lung cancer shows notable results. Eur. J. Cancer Prev. [Epub ahead ofprint].
Hoffmeister, M., Jansen, L., Rudolph, A., Toth, C., Kloor, M., Roth, W., Bläker, H., Chang-Claude, J., Brenner, H., 2015. Statin use and survival after colorectal cancer: theimportance of comprehensive confounder adjustment. J. Natl. Cancer Inst. 107,
djv045.Holmes, C.E., Jasielec, J., Levis, J.E., Skelly, J., Muss, H.B., 2013. Initiation of aspirin ther-apy modulates angiogenic protein levels in women with breast cancer receivingtamoxifen therapy. Clin. Transl. Sci. 6, 386–390.
Key.com by Elsevier on April 03, 2019.. Elsevier Inc. All rights reserved.

2 sistan
H
H
H
H
H
J
J
J
J
K
K
K
K
K
K
K
K
K
K
K
K
K
K
K
K
K
4 G. Mudduluru et al. / Drug Re
orinaka, M., Yoshida, T., Tomosugi, M., Yasuda, S., Sowa, Y., Sakai, T., 2014. Myeloidzinc finger 1 mediates sulindac sulfide-induced upregulation of death receptor5 of human colon cancer cells. Sci. Rep. 4, 6000.
ou, Z.-J., Luo, X., Zhang, W., Peng, F., Cui, B., Wu, S.-J., Zheng, F.-M., Xu, J., Xu,L.-Z., Long, Z.-J., Wang, X.-T., Li, G.-H., Wan, X.-Y., Yang, Y.-L., Liu, Q., 2015.Flubendazole, FDA-approved anthelmintic, targets breast cancer stem-like cells.Oncotarget 6, 6326–6340.
uang, M., Shen, A., Ding, J., Geng, M., 2014. Molecularly targeted cancer therapy:some lessons from the past decade. Trends Pharmacol. Sci. 35, 41–50.
uang, R., Southall, N., Wang, Y., Yasgar, A., Shinn, P., Jadhav, A., Nguyen, D.-T., Austin,C.P., 2011. The NCGC pharmaceutical collection: a comprehensive resource ofclinically approved drugs enabling repurposing and chemical genomics. Sci.Transl. Med. 3, 80ps16.
uang, W.-K., Tu, H.-T., See, L.-C., 2015. Aspirin use on incidence and mortality ofgastrointestinal cancers: current state of epidemiological evidence. Curr. Pharm.Des. 21, 5108–5115.
acobs, E.J., Thun, M.J., Bain, E.B., Rodriguez, C., Henley, S.J., Calle, E.E., 2007. A largecohort study of long-term daily use of adult-strength aspirin and cancer inci-dence. J. Natl. Cancer Inst. 99, 608–615.
anssen, A., Maier, T.J., Schiffmann, S., Coste, O., Seegel, M., Geisslinger, G., Grösch,S., 2006. Evidence of COX-2 independent induction of apoptosis and cell cycleblock in human colon carcinoma cells after S- or R-ibuprofen treatment. Eur. J.Pharmacol. 540, 24–33.
anssen, A., Schiffmann, S., Birod, K., Maier, T.J., Wobst, I., Geisslinger, G., Grösch,S., 2008. p53 is important for the anti-proliferative effect of ibuprofen in coloncarcinoma cells. Biochem. Biophys. Res. Commun. 365, 698–703.
in, Y., Lu, Z., Ding, K., Li, J., Du, X., Chen, C., Sun, X., Wu, Y., Zhou, J., Pan, J., 2010.Antineoplastic mechanisms of niclosamide in acute myelogenous leukemia stemcells: inactivation of the NF-kappaB pathway and generation of reactive oxygenspecies. Cancer Res. 70, 2516–2527.
aitin, K.I., DiMasi, J.A., 2011. Pharmaceutical innovation in the 21st century:new drug approvals in the first decade, 2000–2009. Clin. Pharmacol. Ther. 89,183–188.
alluri, R., Weinberg, R.A., 2009. The basics of epithelial-mesenchymal transition. J.Clin. Invest. 119, 1420–1428.
apur, S., Mamo, D., 2003. Half a century of antipsychotics and still a central rolefor dopamine D2 receptors. Prog. Neuropsychopharmacol. Biol. Psychiatry 27,1081–1090.
ashfi, K., Chattopadhyay, M., Kodela, R., 2015. NOSH-sulindac (AVT-18A) is a novelnitric oxide- and hydrogen sulfide-releasing hybrid that is gastrointestinal safeand has potent anti-inflammatory, analgesic, antipyretic, anti-platelet, and anti-cancer properties. Redox Biol. 6, 287–296.
ashfi, K., Rigas, B., 2005. Non-COX-2 targets and cancer: expanding the moleculartarget repertoire of chemoprevention. Biochem. Pharmacol. 70, 969–986.
astrinsky, D.B., Sangodkar, J., Zaware, N., Izadmehr, S., Dhawan, N.S., Narla, G.,Ohlmeyer, M., 2015. Reengineered tricyclic anti-cancer agents. Bioorg. Med.Chem. 23, 6528–6534.
atiyar, S.K., Gordon, V.R., McLaughlin, G.L., Edlind, T.D., 1994. Antiprotozoal activi-ties of benzimidazoles and correlations with beta-tubulin sequence. Antimicrob.Agents Chemother. 38, 2086–2090.
atz, M.S., Minsky, B.D., Saltz, L.B., Riedel, E., Chessin, D.B., Guillem, J.G., 2005.Association of statin use with a pathologic complete response to neoadjuvantchemoradiation for rectal cancer. Int. J. Radiat. Oncol. Biol. Phys. 62, 1363–1370.
aur, J., Sanyal, S.N., 2011. Diclofenac, a selective COX-2 inhibitor, inhibits DMH-induced colon tumorigenesis through suppression of MCP-1, MIP-1� and VEGF.Mol. Carcinog. 50, 707–718.
aur, J., Sanyal, S.N., 2010. PI3-kinase/Wnt association mediates COX-2/PGE(2)pathway to inhibit apoptosis in early stages of colon carcinogenesis: chemo-prevention by diclofenac. Tumour Biol. 31, 623–631.
awata, S., Yamasaki, E., Nagase, T., Inui, Y., Ito, N., Matsuda, Y., Inada, M., Tamura,S., Noda, S., Imai, Y., Matsuzawa, Y., 2001. Effect of pravastatin on survival inpatients with advanced hepatocellular carcinoma. A randomized controlled trial.Br. J. Cancer 84, 886–891.
hanim, F.L., Merrick, B.A.M.E., Giles, H.V., Jankute, M., Jackson, J.B., Giles, L.J.,Birtwistle, J., Bunce, C.M., Drayson, M.T., 2011. Redeployment-based drugscreening identifies the anti-helminthic niclosamide as anti-myeloma therapythat also reduces free light chain production. Blood Cancer J. 1, e39.
im, Y.-S., Seol, C.-H., Jung, J.-W., Oh, S.-J., Hwang, K.-E., Kim, H.-J., Jeong, E.-T., Kim,H.-R., 2015. Synergistic effect of sulindac and simvastatin on apoptosis in lungcancer A549 cells through AKT-dependent downregulation of survivin. CancerRes. Treat. 47, 90–100.
lop, C., Driessen, J.H.M., de Vries, F., 2013. Statin use and reduced cancer-relatedmortality. N. Engl. J. Med. 368, 574.
nox, J.J., Siu, L.L., Chen, E., Dimitroulakos, J., Kamel-Reid, S., Moore, M.J., Chin,S., Irish, J., LaFramboise, S., Oza, A.M., 2005. A Phase I trial of prolongedadministration of lovastatin in patients with recurrent or metastatic squa-mous cell carcinoma of the head and neck or of the cervix. Eur. J. Cancer 41,523–530.
onings, I.R.H.M., van der Gaast, A., van der Wijk, L.J., de Jongh, F.E., Eskens, F.A.L.M.,Sleijfer, S., 2010. The addition of pravastatin to chemotherapy in advanced gas-tric carcinoma: a randomised phase II trial. Eur. J. Cancer 46, 3200–3204.
oornstra, J.J., Rijcken, F.E.M., Oldenhuis, C.N.A.M., Zwart, N., van der Sluis, T.,
Hollema, H., DeVries, E.G.E., Keller, J.J., Offerhaus, J.A., Giardiello, F.M., Kleibeuker,J.H., 2005. Sulindac inhibits beta-catenin expression in normal-appearing colonof hereditary nonpolyposis colorectal cancer and familial adenomatous polypo-sis patients. Cancer Epidemiol. Biomarkers Prev. 14, 1608–1612.Downloaded for Anonymous User (n/a) at Guilin Medical College from ClinFor personal use only. No other uses without permission. Copyright ©2
ce Updates 26 (2016) 10–27
Kornhuber, J., Muehlbacher, M., Trapp, S., Pechmann, S., Friedl, A., Reichel, M., Mühle,C., Terfloth, L., Groemer, T.W., Spitzer, G.M., Liedl, K.R., Gulbins, E., Tripal, P., 2011.Identification of novel functional inhibitors of acid sphingomyelinase. PLoS ONE6, e23852.
Králová, V., Hanusová, V., Stanková, P., Knoppová, K., Cánová, K., Skálová, L., 2013.Antiproliferative effect of benzimidazole anthelmintics albendazole, ricoben-dazole, and flubendazole in intestinal cancer cell lines. Anticancer. Drugs 24,911–919.
Kubis, M.A., Piwowar, A., 2015. New insights into the regulatory role of vitaminD3 in metabolic pathways involved in carcinogenesis and neurodegenerativediseases. Ageing Res. Rev. 24, 126–137.
Kundu, J.K., Choi, K.-Y., Surh, Y.-J., 2006. beta-Catenin-mediated signaling: a novelmolecular target for chemoprevention with anti-inflammatory substances.Biochim. Biophys. Acta 1765, 14–24.
Kune, G.A., Kune, S., Watson, L.F., 1988. Colorectal cancer risk, chronic illnesses,operations, and medications: case control results from the Melbourne ColorectalCancer Study. Cancer Res. 48, 4399–4404.
Laclette, J.P., Guerra, G., Zetina, C., 1980. Inhibition of tubulin polymerization bymebendazole. Biochem. Biophys. Res. Commun. 92, 417–423.
Lang, M., Gasche, C., 2015. Chemoprevention of colorectal cancer. Dig. Dis. 33,58–67.
Larriba, M.J., González-Sancho, J.M., Barbáchano, A., Niell, N., Ferrer-Mayorga, G.,Munoz, A., 2013. Vitamin D is a multilevel repressor of Wnt/b-catenin signalingin cancer cells. Cancers (Basel) 5, 1242–1260.
Larsen, A.R., Bai, R.-Y., Chung, J.H., Borodovsky, A., Rudin, C.M., Riggins, G.J., Bunz,F., 2015. Repurposing the antihelmintic mebendazole as a hedgehog inhibitor.Mol. Cancer Ther. 14, 3–13.
Lee, W.-Y., Lee, W.-T., Cheng, C.-H., Chen, K.-C., Chou, C.-M., Chung, C.-H., Sun, M.-S.,Cheng, H.-W., Ho, M.-N., Lin, C.-W., 2015. Repositioning antipsychotic chlor-promazine for treating colorectal cancer by inhibiting sirtuin 1. Oncotarget 6,27580–27595.
Lersch, C., Schmelz, R., Erdmann, J., Hollweck, R., Schulte-Frohlinde, E., Eckel, F.,Nader, M., Schusdziarra, V., 2004. Treatment of HCC with pravastatin, octreotide,or gemcitabine – a critical evaluation. Hepatogastroenterology 51, 1099–1103.
Li, H., Zhu, F., Boardman, L.a., Wang, L., Oi, N., Liu, K., Li, X., Fu, Y., Limburg, P.J., Bode,A.M., Dong, Z., 2015. Aspirin prevents colorectal cancer by normalizing EGFRexpression. EBioMedicine 2, 447–455.
Li, M., Khambu, B., Zhang, H., Kang, J.-H., Chen, X., Chen, D., Vollmer, L., Liu, P.-Q.,Vogt, A., Yin, X.-M., 2013. Suppression of lysosome function induces autophagyvia a feedback down-regulation of MTOR complex 1 (MTORC1) activity. J. Biol.Chem. 288, 35769–35780.
Li, R., Hu, Z., Sun, S.-Y., Chen, Z.G., Owonikoko, T.K., Sica, G.L., Ramalingam, S.S., Cur-ran, W.J., Khuri, F.R., Deng, X., 2013a. Niclosamide overcomes acquired resistanceto erlotinib through suppression of STAT3 in non-small cell lung cancer. Mol.Cancer Ther. 12, 2200–2212.
Li, R., You, S., Hu, Z., Chen, Z.G., Sica, G.L., Khuri, F.R., Curran, W.J., Shin, D.M., Deng,X., 2013b. Inhibition of STAT3 by niclosamide synergizes with erlotinib againsthead and neck cancer. PLOS ONE 8, e74670.
Li, X., Gao, L., Cui, Q., Gary, B.D., Dyess, D.L., Taylor, W., Shevde, L.a., Samant, R.S.,Dean-Colomb, W., Piazza, G.a., Xi, Y., 2012. Sulindac inhibits tumor cell inva-sion by suppressing NF-�B-mediated transcription of microRNAs. Oncogene 31,4979–4986.
Li, X., Lewis, M.T., Huang, J., Gutierrez, C., Osborne, C.K., Wu, M.-F., Hilsenbeck, S.G.,Pavlick, A., Zhang, X., Chamness, G.C., Wong, H., Rosen, J., Chang, J.C., 2008. Intrin-sic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl. CancerInst. 100, 672–679.
Li, Y., Li, P.-K., Roberts, M.J., Arend, R.C., Samant, R.S., Buchsbaum, D.J., 2014. Multi-targeted therapy of cancer by niclosamide: a new application for an old drug.Cancer Lett. 349, 8–14.
Liao, D., Zhong, L., Duan, T., Zhang, R.-H., Wang, X., Wang, G., Hu, K., Lv, X., Kang, T.,2015. Aspirin suppresses the growth and metastasis of osteosarcoma throughthe NF-�B pathway. Clin. Cancer Res. 21, 5349–5359.
Liao, Z., Nan, G., Yan, Z., Zeng, L., Deng, Y., Ye, J., Zhang, Z., Qiao, M., Li, R., Denduluri, S.,Wang, J., Wei, Q., Geng, N., Zhao, L., Lu, S., Wang, X., Zhou, G., Luu, H.H., Haydon,R.C., He, T.-C., Wang, Z., 2015. The anthelmintic drug niclosamide inhibits theproliferative activity of human osteosarcoma cells by targeting multiple signalpathways. Curr. Cancer Drug Targets 15, 726–738.
Liggett, J.L., Min, K.-W., Smolensky, D., Baek, S.J., 2014. A novel COX-independentmechanism of sulindac sulfide involves cleavage of epithelial cell adhesionmolecule protein. Exp. Cell Res. 326, 1–9.
Likus, W., Siemianowicz, K., Bienk, K., Pakuła, M., Pathak, H., Dutta, C., Wang, Q.,Shojaei, S., Assaraf, Y.G., Ghavami, S., Cieslar-Pobuda, A., Łos, M.J., 2016. Coulddrugs inhibiting the mevalonate pathway also target cancer stem cells? DrugResist. Updat. 25, 13–25.
Liu, R., Singh, N., Tawa, G.J., Wallqvist, A., Reifman, J., 2014. Exploiting large-scaledrug–protein interaction information for computational drug repurposing. BMCBioinformatics 15, 210.
Liu, Y., Tang, W., Wang, J., Xie, L., Li, T., He, Y., Qin, X., Li, S., 2013. Clinicopathologicaland prognostic significance of S100A4 overexpression in colorectal cancer: ameta-analysis. Diagn. Pathol. 8, 181.
Livney, Y.D., Assaraf, Y.G., 2013. Rationally designed nanovehicles to overcome can-cer chemoresistance. Adv. Drug Deliv. Rev. 65, 1716–1730.
Londono-Joshi, A.I., Arend, R.C., Aristizabal, L., Lu, W., Samant, R.S., Metge, B.J.,Hidalgo, B., Grizzle, W.E., Conner, M., Forero-Torres, A., Lobuglio, A.F., Li, Y.,Buchsbaum, D.J., 2014. Effect of niclosamide on basal-like breast cancers. Mol.Cancer Ther. 13, 800–811.
icalKey.com by Elsevier on April 03, 2019.019. Elsevier Inc. All rights reserved.

sistan
L
L
L
M
M
M
M
M
M
M
M
M
M
M
M
M
M
M
M
M
M
MN
N
N
N
N
G. Mudduluru et al. / Drug Re
ooker, A.C., Ph, D., Johnson, C.L., Lacher, D.A., Pfeiffer, C.M., Ph, D., Schleicher, R.L.,Ph, D., Sempos, C.T., Ph, D., 2011. Vitamin D status: United States, 2001–2006.NCHS Data Briefs 59, 1–8.
ópez-Aguilar, E., Sepúlveda-Vildósola, A.C., Rivera-Márquez, H., Cerecedo-Diaz, F.,Valdez-Sánchez, M., Villasis-Keever, M.A., 1999. Security and maximal tolerateddoses of fluvastatin in pediatric cancer patients. Arch. Med. Res. 30, 128–131.
u, W., Lin, C., Roberts, M.J., Waud, W.R., Piazza, G.A., Li, Y., 2011. Niclosamide sup-presses cancer cell growth by inducing Wnt co-receptor LRP6 degradation andinhibiting the Wnt/�-catenin pathway. PLoS ONE 6, e29290.
ace, A.G., Gantt, G.A., Skacel, M., Pai, R., Hammel, J.P., Kalady, M.F., 2013. Statintherapy is associated with improved pathologic response to neoadjuvantchemoradiation in rectal cancer. Dis. Colon Rectum 56, 1217–1227.
ani, S.A., Guo, W., Liao, M.-J., Eaton, E.N., Ayyanan, A., Zhou, A.Y., Brooks, M., Rein-hard, F., Zhang, C.C., Shipitsin, M., Campbell, L.L., Polyak, K., Brisken, C., Yang,J., Weinberg, R.A., 2008. The epithelial-mesenchymal transition generates cellswith properties of stem cells. Cell 133, 704–715.
antell, D.J., Owens, P.E., Bundred, N.J., Mawer, E.B., Canfield, A.E., 2000. 1 alpha,25-dihydroxyvitamin D(3) inhibits angiogenesis in vitro and in vivo. Circ. Res.87, 214–220.
anzoni, M., Rollini, M., Bergomi, S., Cavazzoni, V., 1998. Production and purificationof statins from Aspergillus terreus strains. Biotechnol. Tech. 12, 529–532.
athew, R., Karantza-Wadsworth, V., White, E., 2007. Role of autophagy in cancer.Nat. Rev. Cancer 7, 961–967.
cGuire, T.F., Trump, D.L., Johnson, C.S., 2001. Vitamin D(3)-induced apoptosisof murine squamous cell carcinoma cells. Selective induction of caspase-dependent MEK cleavage and up-regulation of MEKK-1. J. Biol. Chem. 276,26365–26373.
edioni, J., Deplanque, G., Ferrero, J.M., Maurina, T., Rodier, J.M.P., Raymond, E.,Allyon, J., Maruani, G., Houillier, P., MacKenzie, S., Renaux, S., Dufour Lamartinie,J.F., Elaidi, R., Le Rest, C., Oudard, S.M., 2014. Phase I Safety and Pharmacodynamicof inecalcitol, a novel VDR agonist with docetaxel in metastatic castration-resistant prostate cancer patients. Clin. Cancer Res. 20, 4471–4478.
elino, G., Gallagher, E., Aqeilan, R.I., Knight, R., Peschiaroli, A., Rossi, M., Scialpi, F.,Malatesta, M., Zocchi, L., Browne, G., Ciechanover, A., Bernassola, F., 2008. Itch:a HECT-type E3 ligase regulating immunity, skin and cancer. Cell Death Differ.15, 1103–1112.
éric, J.-B., Rottey, S., Olaussen, K., Soria, J.-C., Khayat, D., Rixe, O., Spano, J.-P., 2006.Cyclooxygenase-2 as a target for anticancer drug development. Crit. Rev. Oncol.Hematol. 59, 51–64.
erry, S., Hamilton, T.G., Flanigan, P., Freshney, R.I., Kaye, S.B., 1991. Circumventionof pleiotropic drug resistance in subcutaneous tumours in vivo with verapamiland clomipramine. Eur. J. Cancer 27, 31–34.
eyskens, F.L., McLaren, C.E., Pelot, D., Fujikawa-Brooks, S., Carpenter, P.M., Hawk, E.,Kelloff, G., Lawson, M.J., Kidao, J., McCracken, J., Albers, C.G., Ahnen, D.J., Turgeon,D.K., Goldschmid, S., Lance, P., Hagedorn, C.H., Gillen, D.L., Gerner, E.W., 2008.Difluoromethylornithine plus sulindac for the prevention of sporadic colorectaladenomas: a randomized placebo-controlled, double-blind trial. Cancer Prev.Res. (Phila.) 1, 32–38.
ichaelis, M., Agha, B., Rothweiler, F., Löschmann, N., Voges, Y., Mittelbronn, M.,Starzetz, T., Harter, P.N., Abhari, B.A., Fulda, S., Westermann, F., Riecken, K., Spek,S., Langer, K., Wiese, M., Dirks, W.G., Zehner, R., Cinatl, J., Wass, M.N., Cinatl, J.,2015. Identification of flubendazole as potential anti-neuroblastoma compoundin a large cell line screen. Sci. Rep. 5, 8202.
inden, M.D., Dimitroulakos, J., Nohynek, D., Penn, L.Z., 2001. Lovastatin inducedcontrol of blast cell growth in an elderly patient with acute myeloblasticleukemia. Leuk. Lymphoma 40, 659–662.
ladenova, D., Pangon, L., Currey, N., Ng, I., Musgrove, E.a., Grey, S.T., Kohonen-Corish, M.R.J., 2013. Sulindac activates NF-�B signaling in colon cancer cells.Cell Commun. Signal. 11, 73.
offatt, K.A., Johannes, W.U., Miller, G.J., 1999. 1Alpha, 25dihydroxyvitamin D3 andplatinum drugs act synergistically to inhibit the growth of prostate cancer celllines. Clin. Cancer Res. 5, 695–703.
ook, R.A., Wang, J., Ren, X.-R., Chen, M., Spasojevic, I., Barak, L.S., Lyerly, H.K.,Chen, W., 2015. Structure-activity studies of Wnt/�-catenin inhibition in theNiclosamide chemotype: identification of derivatives with improved drug expo-sure. Bioorg. Med. Chem. 23, 5829–5838.
ousa, A.M., Mehrez, M.G., Muhtaseb, S.A., Al-Mudalal, D.S., Kamel, S.M., 1987.Disseminated pelvic hydatidosis presenting as ovarian carcinomatosis: success-ful post-operative treatment with mebendazole. Int. J. Gynaecol. Obstet. 25,473–478.
ukhopadhyay, T., Sasaki, J., Ramesh, R., Roth, J.A., 2002. Mebendazole elicits apotent antitumor effect on human cancer cell lines both in vitro and in vivo.Clin. Cancer Res. 8, 2963–2969.
ullard, A., 2014. 2013 FDA drug approvals. Nat. Rev. Drug Discov. 13, 85–89.arayanan, R., Sepulveda, V.a.T., Falzon, M., Weigel, N.L., 2004. The functional con-
sequences of cross-talk between the vitamin D receptor and ERK signalingpathways are cell-specific. J. Biol. Chem. 279, 47298–47310.
arvaez, C.J., Welsh, J., 2001. Role of mitochondria and caspases in vitamin D-mediated apoptosis of MCF-7 breast cancer cells. J. Biol. Chem. 276, 9101–9107.
atale, J.J., 2015. Reviewing current and emerging antiemetics for chemotherapy-induced nausea and vomiting prophylaxis. Hosp. Pract. (1995) 43, 226–234.
icolini, A., Ferrari, P., Fini, M., Borsari, V., Fallahi, P., Antonelli, A., Carpi, A., Miccoli,P., 2011. Cancer stem cells: perspectives of new therapeutical approaches forbreast cancer. Front. Biosci. (Schol. Ed.) 3, 1486–1499.
ielsen, S.F., Nordestgaard, B.G., Bojesen, S.E., 2012. Statin use and reduced cancer-related mortality. N. Engl. J. Med. 367, 1792–1802.
Downloaded for Anonymous User (n/a) at Guilin Medical College from ClinicalFor personal use only. No other uses without permission. Copyright ©2019
ce Updates 26 (2016) 10–27 25
Niewerth, D., Jansen, G., Assaraf, Y.G., Zweegman, S., Kaspers, G.J.L., Cloos, J., 2015.Molecular basis of resistance to proteasome inhibitors in hematological malig-nancies. Drug Resist. Updat. 18, 18–35.
Nygren, P., Fryknäs, M., Agerup, B., Larsson, R., 2013. Repositioning of theanthelmintic drug mebendazole for the treatment for colon cancer. J. CancerRes. Clin. Oncol. 139, 2133–2140.
Nygren, P., Larsson, R., 2014. Drug repositioning from bench to bedside: tumourremission by the antihelmintic drug mebendazole in refractory metastatic coloncancer. Acta Oncol. 53, 427–428.
Ogawa, F., Amano, H., Ito, Y., Matsui, Y., Hosono, K., Kitasato, H., Satoh, Y., Majima, M.,2014. Aspirin reduces lung cancer metastasis to regional lymph nodes. Biomed.Pharmacother. 68, 79–86.
Orner, G.a, Dashwood, W.-M., Blum, C.a., Díaz, G.D., Li, Q., Dashwood, R.H., 2003.Suppression of tumorigenesis in the Apc(min) mouse: down-regulation of beta-catenin signaling by a combination of tea plus sulindac. Carcinogenesis 24,263–267.
Osada, T., Chen, M., Yang, X.Y., Spasojevic, I., Vandeusen, J.B., Hsu, D., Clary, B.M.,Clay, T.M., Chen, W., Morse, M.a., Lyerly, H.K., 2011. Antihelminth compoundniclosamide downregulates Wnt signaling and elicits antitumor responses intumors with activating APC mutations. Cancer Res. 71, 4172–4182.
Pálmer, H.G., González-Sancho, J.M., Espada, J., Berciano, M.T., Puig, I., Baulida, J.,Quintanilla, M., Cano, A., de Herreros, A.G., Lafarga, M., Munoz, A., 2001. VitaminD(3) promotes the differentiation of colon carcinoma cells by the induction ofE-cadherin and the inhibition of beta-catenin signaling. J. Cell Biol. 154, 369–387.
Pammolli, F., Magazzini, L., Riccaboni, M., 2011. The productivity crisis in pharma-ceutical R&D. Nat. Rev. Drug Discov. 10, 428–438.
Pan, J.-X., Ding, K., Wang, C.-Y., 2012. Niclosamide, an old antihelminthic agent,demonstrates antitumor activity by blocking multiple signaling pathways ofcancer stem cells. Chin. J. Cancer 31, 178–184.
Pantziarka, P., Bouche, G., Meheus, L., Sukhatme, V., Sukhatme, V.P., 2015. Repurpos-ing drugs in your medicine cabinet: untapped opportunities for cancer therapy?Future Oncol. 11, 181–184.
Pantziarka, P., Bouche, G., Meheus, L., Sukhatme, V., Sukhatme, V.P., 2014. Repur-posing drugs in oncology (ReDO)-mebendazole as an anti-cancer agent.Ecancermedicalscience 8, 443.
Parker, K.A., Glaysher, S., Hurren, J., Knight, L.A., McCormick, D., Suovouri, A.,Amberger-Murphy, V., Pilkington, G.J., Cree, I.A., 2012. The effect of tricyclicantidepressants on cutaneous melanoma cell lines and primary cell cultures.Anticancer Drugs 23, 65–69.
Petersen, N.H.T., Olsen, O.D., Groth-Pedersen, L., Ellegaard, A.-M., Bilgin, M., Redmer,S., Ostenfeld, M.S., Ulanet, D., Dovmark, T.H., Lønborg, A., Vindeløv, S.D., Hanahan,D., Arenz, C., Ejsing, C.S., Kirkegaard, T., Rohde, M., Nylandsted, J., Jäättelä, M.,2013. Transformation-associated changes in sphingolipid metabolism sensitizecells to lysosomal cell death induced by inhibitors of acid sphingomyelinase.Cancer Cell 24, 379–393.
Phillips, T.M., McBride, W.H., Pajonk, F., 2006. The response of CD24(−/low)/CD44+breast cancer-initiating cells to radiation. J. Natl. Cancer Inst. 98, 1777–1785.
Platz, E.A., 2008. Re: The association between statins and cancer incidence in aveterans population. J. Natl. Cancer Inst. 100, 972.
Pommerenke, E.W., Volm, M., 1995. Reversal of doxorubicin-resistance in solidtumors by clomipramine. In Vivo 9, 99–101.
Pozzi, A., Yan, X., Macias-Perez, I., Wei, S., Hata, A.N., Breyer, R.M., Morrow,J.D., Capdevila, J.H., 2004. Colon carcinoma cell growth is associated withprostaglandin E2/EP4 receptor-evoked ERK activation. J. Biol. Chem. 279,29797–29804.
Pulkoski-Gross, A., Li, J., Zheng, C., Li, Y., Ouyang, N., Rigas, B., Zucker, S., Cao, J., 2015.Repurposing the antipsychotic trifluoperazine as an antimetastasis agent. Mol.Pharmacol. 87, 501–512.
Qi, L., Ding, Y., 2013. Potential antitumor mechanisms of phenothiazine drugs. Sci.China Life Sci. 56, 1020–1027.
Rana, C., Piplani, H., Vaish, V., Nehru, B., Sanyal, S.N., 2015a. Downregulation ofPI3-K/Akt/PTEN pathway and activation of mitochondrial intrinsic apopto-sis by Diclofenac and Curcumin in colon cancer. Mol. Cell. Biochem. 402,225–241.
Rana, C., Piplani, H., Vaish, V., Nehru, B., Sanyal, S.N., 2015b. Downregulation of tel-omerase activity by diclofenac and curcumin is associated with cell cycle arrestand induction of apoptosis in colon cancer. Tumour Biol. 36, 5999–6010.
Rao, S., Porter, D.C., Chen, X., Herliczek, T., Lowe, M., Keyomarsi, K., 1999. Lovastatin-mediated G1 arrest is through inhibition of the proteasome, independentof hydroxymethyl glutaryl-CoA reductase. Proc. Natl. Acad. Sci. U. S. A. 96,7797–7802.
Ravnskov, U., Rosch, P.J., McCully, K.S., 2015. Statins do not protect against cancer:quite the opposite. J. Clin. Oncol. 33, 810–811.
Ren, X., Duan, L., He, Q., Zhang, Z., Zhou, Y., Wu, D., Pan, J., Pei, D., Ding, K., 2010.Identification of niclosamide as a new small-molecule inhibitor of the STAT3signaling pathway. ACS Med. Chem. Lett. 1, 454–459.
Rice, P.L., Kelloff, J., Sullivan, H., Driggers, L.J., Beard, K.S., Kuwada, S., Piazza,G., Ahnen, D.J., 2003. Sulindac metabolites induce caspase- and proteasome-dependent degradation of beta-catenin protein in human colon cancer cells.Mol. Cancer Ther. 2, 885–892.
Rossi, M., Munarriz, E.R., Bartesaghi, S., Milanese, M., Dinsdale, D., Guerra-Martin,M.A., Bampton, E.T.W., Glynn, P., Bonanno, G., Knight, R.A., Nicotera, P., Melino,
G., 2009. Desmethylclomipramine induces the accumulation of autophagymarkers by blocking autophagic flux. J. Cell Sci. 122, 3330–3339.Rossi, M., Rotblat, B., Ansell, K., Amelio, I., Caraglia, M., Misso, G., Bernassola, F.,Cavasotto, C.N., Knight, R.A., Ciechanover, A., Melino, G., 2014. High throughput
Key.com by Elsevier on April 03, 2019.. Elsevier Inc. All rights reserved.

2 sistan
R
R
R
R
S
S
S
S
S
S
S
S
S
S
S
S
S
S
S
S
S
S
S
T
T
6 G. Mudduluru et al. / Drug Re
screening for inhibitors of the HECT ubiquitin E3 ligase ITCH identifies antide-pressant drugs as regulators of autophagy. Cell Death Dis. 5, e1203.
othwell, P.M., Fowkes, F.G.R., Belch, J.F.F., Ogawa, H., Warlow, C.P., Meade, T.W.,2011. Effect of daily aspirin on long-term risk of death due to cancer: analysis ofindividual patient data from randomised trials. Lancet (Lond., Engl.) 377, 31–41.
othwell, P.M., Wilson, M., Price, J.F., Belch, J.F.F., Meade, T.W., Mehta, Z., 2012. Effectof daily aspirin on risk of cancer metastasis: a study of incident cancers duringrandomised controlled trials. Lancet (Lond., Engl.) 379, 1591–1601.
uder, E.H., Laiyemo, A.O., Graubard, B.I., Hollenbeck, A.R., Schatzkin, A., Cross, A.J.,2011. Non-steroidal anti-inflammatory drugs and colorectal cancer risk in alarge, prospective cohort. Am. J. Gastroenterol. 106, 1340–1350.
yan, J.W., Anderson, P.H., Morris, H.A., 2015. Pleiotropic activities of vitamin Dreceptors – adequate activation for multiple health outcomes. Clin. Biochem.Rev. 36, 53–61.
ack, U., Stein, U., 2009. Wnt up your mind – intervention strategies for S100A4-induced metastasis in colon cancer. Gen. Physiol. Biophys. 28, F55–F64.
ack, U., Walther, W., Scudiero, D., Selby, M., Kobelt, D., Lemm, M., Fichtner, I., Schlag,P.M., Shoemaker, R.H., Stein, U., 2011. Novel effect of antihelminthic niclosamideon s100a4-mediated metastatic progression in colon cancer. J. Natl. Cancer Inst.103, 1018–1036.
areddy, G.R., Kesanakurti, D., Kirti, P.B., Babu, P.P., 2013. Nonsteroidal anti-inflammatory drugs diclofenac and celecoxib attenuates Wnt/�-catenin/Tcfsignaling pathway in human glioblastoma cells. Neurochem. Res. 38,2313–2322.
asaki, J., Ramesh, R., Chada, S., Gomyo, Y., Roth, J.A., Mukhopadhyay, T., 2002.The anthelmintic drug mebendazole induces mitotic arrest and apoptosis bydepolymerizing tubulin in non-small cell lung cancer cells. Mol. Cancer Ther. 1,1201–1209.
auter, C., 1989. Cytostatic activity of commonly used tricyclic antidepressants.Oncology 46, 155–157.
avic, R., Schuchman, E.H., 2013. Use of acid sphingomyelinase for cancer therapy.Adv. Cancer Res. 117, 91–115.
cannell, J.W., Blanckley, A., Boldon, H., Warrington, B., 2012. Diagnosing the declinein pharmaceutical R&D efficiency. Nat. Rev. Drug Discov. 11, 191–200.
cher, H.I., Jia, X., Chi, K., De Wit, R., Berry, W.R., Albers, P., Henick, B., Waterhouse, D.,Ruether, D.J., Rosen, P.J., Meluch, A.a., Nordquist, L.T., Venner, P.M., Heidenreich,A., Chu, L., Heller, G., 2011. Randomized, open-label phase III trial of docetaxelplus high-dose calcitriol versus docetaxel plus prednisone for patients withcastration-resistant prostate cancer. J. Clin. Oncol. 29, 2191–2198.
hah, S., Islam, M.N., Dakshanamurthy, S., Rizvi, I., Rao, M., Herrell, R., Zinser, G.,Valrance, M., Aranda, A., Moras, D., Norman, A., Welsh, J., Byers, S.W., 2006. Themolecular basis of vitamin D receptor and �-catenin crossregulation. Mol. Cell21, 799–809.
hao, J., Sheng, H., Inoue, H., Morrow, J.D., DuBois, R.N., 2000. Regulation of consti-tutive cyclooxygenase-2 expression in colon carcinoma cells. J. Biol. Chem. 275,33951–33956.
harpe, M., Walker, J., Holm Hansen, C., Martin, P., Symeonides, S., Gourley,C., Wall, L., Weller, D., Murray, G., SMaRT (Symptom Management ResearchTrials) Oncology-2 Team, 2014. Integrated collaborative care for comorbidmajor depression in patients with cancer (SMaRT Oncology-2): a multi-centre randomised controlled effectiveness trial. Lancet (Lond., Engl.) 384,1099–1108.
him, J.S., Liu, J.O., 2014. Recent advances in drug repositioning for the discovery ofnew anticancer drugs. Int. J. Biol. Sci. 10, 654–663.
ong, M., Garrett, W.S., Chan, A.T., 2015. Nutrients, foods, and colorectal cancerprevention. Gastroenterology 148, 1244–1260.
pagnuolo, P.A., Hu, J., Hurren, R., Wang, X., Gronda, M., Sukhai, M.A., DiMeo, A., Boss, J., Ashali, I., Beheshti Zavareh, R., Fine, N., Simpson, C.D.,Sharmeen, S., Rottapel, R., Schimmer, A.D., 2010. The antihelmintic flubenda-zole inhibits microtubule function through a mechanism distinct from Vincaalkaloids and displays preclinical activity in leukemia and myeloma. Blood 115,4824–4833.
porn, M.B., Hong, W.K., 2008. Concomitant DFMO and sulindac chemopreventionof colorectal adenomas: a major clinical advance. Nat. Clin. Pract. Oncol. 5,628–629.
tein, U., Arlt, F., Smith, J., Sack, U., Herrmann, P., Walther, W., Lemm, M., Ficht-ner, I., Shoemaker, R.H., Schlag, P.M., 2011a. Intervening in �-catenin signalingby sulindac inhibits S100A4-dependent colon cancer metastasis. Neoplasia 13,131–144.
tein, U., Arlt, F., Walther, W., Smith, J., Waldman, T., Harris, E.D., Mertins, S.D., Heiz-mann, C.W., Allard, D., Birchmeier, W., Schlag, P.M., Shoemaker, R.H., 2006. Themetastasis-associated gene S100A4 is a novel target of �-catenin/T-cell factorsignaling in colon cancer. Gastroenterology 131, 1486–1500.
tein, U., Burock, S., Herrmann, P., Wendler, I., Niederstrasser, M., Wernecke, K.-D., Schlag, P.M., 2011b. Diagnostic and prognostic value of metastasis inducerS100A4 transcripts in plasma of colon, rectal, and gastric cancer patients. J. Mol.Diagn. 13, 189–198.
zabo, E., 2006. Selecting targets for cancer prevention: where do we go from here?Nat. Rev. Cancer 6, 867–874.
ai, W.-P., Hu, P.-J., Wu, J., Lin, X.-C., 2014. The inhibition of Wnt/�-catenin signalingpathway in human colon cancer cells by sulindac. Tumori 100, 97–101.
akayama, T., Nagashima, H., Maeda, M., Nojiri, S., Hirayama, M., Nakano, Y., Taka-
hashi, Y., Sato, Y., Sekikawa, H., Mori, M., Sonoda, T., Kimura, T., Kato, J., Niitsu,Y., 2011. Randomized double-blind trial of sulindac and etodolac to eradicateaberrant crypt foci and to prevent sporadic colorectal polyps. Clin. Cancer Res.17, 3803–3811.Downloaded for Anonymous User (n/a) at Guilin Medical College from ClinFor personal use only. No other uses without permission. Copyright ©2
ce Updates 26 (2016) 10–27
Todo, M., Horinaka, M., Tomosugi, M., Tanaka, R., Ikawa, H., Sowa, Y., Ishikawa,H., Fujiwara, H., Otsuji, E., Sakai, T., 2013. Ibuprofen enhances TRAIL-inducedapoptosis through DR5 upregulation. Oncol. Rep. 30, 2379–2384.
Uddin, S., Ahmed, M., Hussain, A., Assad, L., Al-Dayel, F., Bavi, P., Al-Kuraya, K.S.,Munkarah, A., 2010. Cyclooxygenase-2 inhibition inhibits PI3K/AKT kinase activ-ity in epithelial ovarian cancer. Int. J. Cancer 126, 382–394.
Vaish, V., Sanyal, S.N., 2012. Role of Sulindac and Celecoxib in the regulation of angio-genesis during the early neoplasm of colon: exploring PI3-K/PTEN/Akt pathwayto the canonical Wnt/�-catenin signaling. Biomed. Pharmacother. 66, 354–367.
Vaish, V., Tanwar, L., Sanyal, S.N., 2010. The role of NF-�B and PPAR� in experi-mentally induced colorectal cancer and chemoprevention by cyclooxygenase-2inhibitors. Tumour Biol. 31, 427–436.
Valle, B.L., D’Souza, T., Becker, K.G., Wood, W.H., Zhang, Y., Wersto, R.P., Morin,P.J., 2013. Non-steroidal anti-inflammatory drugs decrease E2F1 expression andinhibit cell growth in ovarian cancer cells. PLOS ONE 8, e61836.
Vane, J.R., 1971. Inhibition of prostaglandin synthesis as a mechanism of action foraspirin-like drugs. Nat. N. Biol. 231, 232–235.
Vane, J.R., Bakhle, Y.S., Botting, R.M., 1998. Cyclooxygenases 1 and 2. Annu. Rev.Pharmacol. Toxicol. 38, 97–120.
Verlinden, L., Verstuyf, A., Convents, R., Marcelis, S., Van Camp, M., Bouillon, R., 1998.Action of 1,25(OH)2D3 on the cell cycle genes, cyclin D1, p21 and p27 in MCF-7cells. Mol. Cell. Endocrinol. 142, 57–65.
Vineis, P., Wild, C.P., 2014. Global cancer patterns: causes and prevention. Lancet(Lond., Engl.) 383, 549–557.
Vitols, S., Angelin, B., Juliusson, G., 1997. Simvastatin impairs mitogen-inducedproliferation of malignant B-lymphocytes from humans – in vitro and in vivostudies. Lipids 32, 255–262.
Walker, J., Holm Hansen, C., Martin, P., Sawhney, A., Thekkumpurath, P., Beale, C.,Symeonides, S., Wall, L., Murray, G., Sharpe, M., 2013. Prevalence of depressionin adults with cancer: a systematic review. Ann. Oncol. 24, 895–900.
Walters Haygood, C.L., Arend, R.C., Gangrade, A., Chettiar, S., Regan, N., Hassmann,C.J., Li, P.-K., Hidalgo, B., Straughn, J.M., Buchsbaum, D.J., 2015. Niclosamideanalogs for treatment of ovarian cancer. Int. J. Gynecol. Cancer 25, 1377–1385.
Wang, D., Wang, H., Shi, Q., Katkuri, S., Walhi, W., Desvergne, B., Das, S.K., Dey, S.K.,DuBois, R.N., 2004. Prostaglandin E(2) promotes colorectal adenoma growth viatransactivation of the nuclear peroxisome proliferator-activated receptor delta.Cancer Cell 6, 285–295.
Wang, Q., Yang, W., Uytingco, M.S., Christakos, S., Wieder, R., 2000. 1,25-Dihydroxyvitamin D3 and all-trans-retinoic acid sensitize breast cancer cellsto chemotherapy-induced cell death. Cancer Res. 60, 2040–2048.
Warita, K., Warita, T., Beckwitt, C.H., Schurdak, M.E., Vazquez, A., Wells, A., Olt-vai, Z.N., 2014. Statin-induced mevalonate pathway inhibition attenuates thegrowth of mesenchymal-like cancer cells that lack functional E-cadherin medi-ated cell cohesion. Sci. Rep. 4, 7593.
Weinbach, E.C., Garbus, J., 1969. Mechanism of action of reagents that uncoupleoxidative phosphorylation. Nature 221, 1016–1018.
Weis, M., Heeschen, C., Glassford, A.J., Cooke, J.P., 2002. Statins have biphasic effectson angiogenesis. Circulation 105, 739–745.
Whitt, J.D., Li, N., Tinsley, H.N., Chen, X., Zhang, W., Li, Y., Gary, B.D., Keeton, A.B.,Xi, Y., Abadi, A.H., Grizzle, W.E., Piazza, G.A., 2012. A novel sulindac derivativethat potently suppresses colon tumor cell growth by inhibiting cGMP phospho-diesterase and �-catenin transcriptional activity. Cancer Prev. Res. (Phila.) 5,822–833.
Wieland, A., Trageser, D., Gogolok, S., Reinartz, R., Höfer, H., Keller, M., Leinhaas,A., Schelle, R., Normann, S., Klaas, L., Waha, A., Koch, P., Fimmers, R., Pietsch,T., Yachnis, A.T., Pincus, D.W., Steindler, D.A., Brüstle, O., Simon, M., Glas, M.,Scheffler, B., 2013. Anticancer effects of niclosamide in human glioblastoma.Clin. Cancer Res. 19, 4124–4136.
Wilkie, D., Delhanty, J., 1970. Effects of chlorimipramine on human cells in tissueculture. Br. J. Exp. Pathol. 51, 507–511.
Wu, J., Wong, W.W.-L., Khosravi, F., Minden, M.D., Penn, L.Z., 2004. Blockingthe Raf/MEK/ERK pathway sensitizes acute myelogenous leukemia cells tolovastatin-induced apoptosis. Cancer Res. 64, 6461–6468.
Xia, Z., Bergstrand, A., DePierre, J.W., Nässberger, L., 1999. The antidepressantsimipramine, clomipramine, and citalopram induce apoptosis in human acutemyeloid leukemia HL-60 cells via caspase-3 activation. J. Biochem. Mol. Toxicol.13, 338–347.
Yamaji, T., Kagaya, A., Okamoto, Y., Hayashi, T., Motohashi, N., Yamawaki, S.,1996. Effects of clomipramine and verapamil on 5-HT-induced intracellularcalcium changes in individual C6 rat glioma cells. Neuropsychobiology 33,55–59.
Yangco, B.G., Klein, T.W., Deresinski, S.C., Vickery, A.C., Craig, C.P., 1981. Flubendazoleand mebendazole in the treatment of trichuriasis and other helminthiases. Clin.Ther. 4, 285–290.
Yin, M.J., Yamamoto, Y., Gaynor, R.B., 1998. The anti-inflammatory agents aspirinand salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 396, 77–80.
Yo, Y.-T., Lin, Y.-W., Wang, Y.-C., Balch, C., Huang, R.-L., Chan, M.W.Y., Sytwu, H.-K., Chen, C.-K., Chang, C.-C., Nephew, K.P., Huang, T., Yu, M.-H., Lai, H.-C., 2012.Growth inhibition of ovarian tumor-initiating cells by niclosamide. Mol. CancerTher. 11, 1703–1712.
Zanders, M.M.J., van Herk-Sukel, M.P.P., Vissers, P.A.J., Herings, R.M.C., Haak, H.R., vande Poll-Franse, L.V., 2015. Are metformin, statin and aspirin use still associated
with overall mortality among colorectal cancer patients with diabetes if adjustedfor one another? Br. J. Cancer 113, 403–410.Zeichner, S.B., Koru-Sengul, T., Shah, N., Liu, Q., Markward, N.J., Montero, A.J.,Glück, S., Silva, O., Ahn, E.R., 2015. Improved clinical outcomes associated with
icalKey.com by Elsevier on April 03, 2019.019. Elsevier Inc. All rights reserved.

sistan
Z
Z
sine phosphorylation in response to epidermal growth factor and interleukin-6.Science 264, 95–98.
G. Mudduluru et al. / Drug Re
vitamin d supplementation during adjuvant chemotherapy in patients withHER2+ nonmetastatic breast cancer. Clin. Breast Cancer 15, e1–e11.
helev, Z., Ohba, H., Bakalova, R., Hadjimitova, V., Ishikawa, M., Shinohara, Y., Baba,
Y., 2004. Phenothiazines suppress proliferation and induce apoptosis in culturedleukemic cells without any influence on the viability of normal lymphocytes.Phenothiazines and leukemia. Cancer Chemother. Pharmacol. 53, 267–275.hitomirsky, B., Assaraf, Y.G., 2016. Lysosomes as mediators of drug resistance incancer. Drug Resist. Updat. 24, 23–33.
Downloaded for Anonymous User (n/a) at Guilin Medical College from ClinicalFor personal use only. No other uses without permission. Copyright ©2019
ce Updates 26 (2016) 10–27 27
Zhong, Z., Wen, Z., Darnell, J.E., 1994. Stat3: a STAT family member activated by tyro-
Zhou, X., Li, Y.-J., Gao, S.-Y., Wang, X.-Z., Wang, P.-Y., Yan, Y.-F., Xie, S.-Y., Lv, C.-J., 2015. Sulindac has strong antifibrotic effects by suppressing STAT3-relatedmiR-21. J. Cell. Mol. Med. 19, 1103–1113.
Key.com by Elsevier on April 03, 2019.. Elsevier Inc. All rights reserved.