Embed Size (px)
Citation preview

J. Cell Sd. 67, 1-23 (1984)Printed in Great Britain © The Company of Biologists Limited 1984
EFFECT OF ALTERED OLIGOSACCHARIDESTRUCTURE ON THE CELL SURFACE NUMBER,DISTRIBUTION AND TURNOVER OF THE HIGHMOLECULAR WEIGHT ACIDIC GLYCOPROTEINS OFCHO CELLS
L. A. FITZGERALD*, J. B. DENNYf, G. A. BAUMBACH, C. M.KETCHAM AND R. M. ROBERTS}Department of Biochemistry and Molecular Biology, College of Medicine, University ofFlorida, Gainesville, FL 32610, U.SA.
SUMMARY
The influence of altered carbohydrate structure on the surface number, distribution and turnoverof plasma membrane glycoproteins has been studied in Chinese hamster ovary (CHO) cells bycomparing three lines that are resistant to the cytotoxic effects of wheat germ agglutinin (WG A) withparental CHO cells. The glycoproteins investigated were members of a group of high molecularweight acidic glycoproteins (HMWAG). On parental cells these represent the major surface com-ponents that become labelled by lactoperoxidase-catalysed iodination. They are the only plasmamembrane glycoproteins that bind WGA. The mutant lines also possess iodinatable surfacepolypeptides of high molecular weight, but these were less acidic and electrophoretically less diffusethan those from parental cells. These polypeptides in general did not bind [I25I]WGA when two-dimensional polyacrylamide gels were overlaid with iodinated lectin. Mutant cells treated withfluorescein-conjugated WGA showed low surface fluorescence. However, the nuclear envelope anda small region in the perinuclear zone fluoresced strongly. Together, these results confirm that thesurface glycoproteins of mutant cells had altered carbohydrate structure. Mouse antiserum preparedagainst the HMWAG, however, bound equally effectively to the mutant lines as to the parental lines.Indirect immunofluorescence experiments showed that the HMWAG had a fairly uniformdistribution over the surface, and that internalization induced by second antibody occurred at asimilar rate and in a similar manner in all lines, including the mutants. Electron microscopicobservations using immunoperoxidase procedures confirmed the similarities in glycoproteindistribution on mutant and parental cells. Two mouse monoclonal antibodies raised against theHMWAG also revealed no difference in the number or topography of surface glycoproteins. Finally,the half-lives of several HMWAG in a parental and a mutant line (15B) maintained on low-serummedium were compared by means of a I/1311 double-label technique. Half-lives of HMWAG fromthe former averaged 12 h and from the latter 11 h. It is concluded that the lack of complex terminion oligosaccharides of this particular group of CHO plasma membrane glycoproteins has no effecton their number, distribution or turnover.
INTRODUCTION
The rates at which cytoplasmic proteins are turned over are influenced by a numberof features, including their size (Dice & Goldberg, 1975a), isoelectric point (Dice &
•Present address: The Gladstone Foundation Laboratories, San Francisco, CA, U.S.A.f Present address: Rockefeller University, New York, U.S.A.| Author for correspondence.

2 L. A. Fitzgerald and others
Goldberg, 19756), susceptibility to proteolytic attack and ease of denaturation (Gold-berg & St John, 1976; Goldberg & Dice, 1974; Segal, Winkler & Miyagi, 1974;Knowles & Ballard, 1976). However, previous studies have indicated that size andisoelectric point may be poor predictors of turnover rates of membrane-associatedpolypeptides (Horst & Roberts, 1979). One potential factor that may influence thehalf-lives of membrane components is the structure of their carbohydrate groups, ifpresent. For example, the transport of the envelope proteins of Sindbis virus and somestrains of vesicular stomatitis virus to the plasma membrane of the host cell is inhibitedin the presence of tunicamycin. The loss of carbohydrate apparently causes a fall inthe solubility of the proteins within the endoplasmic reticulum where they becamedenatured (Leavitt, Schlesinger&Kornfeld, 1977). Prives& Olden (1980) found thatin embryonic muscle cells treated with tunicamycin the acetylcholine receptor turnedover at a rate three to four times faster than normal, and it was concluded that this wasdue to its increased susceptibility to proteolysis. Similarly the non-glycosylated orpartially glycosylated T-25 cell surface glycoprotein of murine T-cell lymphoma cells(Trowbridge, Hyman & Mazouskas, 1978) and non-glycosylated fibronectinsynthesized by chick fibroblast (Olden, Pratt & Yamada, 1978) turn over much fasterthan their normal counterparts. In all three cases there was also a decrease in theamount of non-glycosylated protein transported to the surface.
The effect of smaller alterations in carbohydrate structure on membraneglycoprotein residence and turnover is less clear. It is well recognized that the circulat-ing half-lives of glycoproteins in plasma, can be reduced from days to minutes byspecific alterations in their carbohydrate groups (see Ashwell & Harford, 1982). Withmembrane glycoproteins the situation is less clear. Enzymic removal of terminalcarbohydrate groups from surface glycoproteins did not appear to increase turnoverrates (Buck & Warren, 1976; Baumann, Hou & Jahreis, 1983), with the possibleexception of the asialoglycoprotein receptor itself (Pricer & Ashwell, 1976). Inaddition to effects on turnover, it has been suggested that saccharide groups might actas sorting signals for placement of glycoproteins into specific organelles or membranesystems and, therefore, influence not only the location of glycoproteins in the cell buttheir movement to and from the surface during such processes as endocytosis andmembrane recycling (see Olden, Parent & White, 1982).
One approach to studying the influence of such changes in carbohydrate structureon membrane glycoprotein dynamics is to use mutant cell lines that have been selectedfor their resistance to the cytotoxic effect of plant lectins and have altered oligo-saccharide chains on their surfaces. The best-described series of mutants have beenderived from Chinese hamster ovary (CHO) cells (see Stanley, 1980). The wild-typeparents of such mutant CHO lines possess a group of iodinatable, high molecularweight acidic glycoproteins (HMWAG) on their surface, which are the only plasmamembrane glycoproteins that bind the lectin wheat germ agglutinin (WGA) (Horst,Baumbach, Olympio & Roberts, 1980b; Fitzgerald et al. 1981). Mutants resistant toWGA, therefore, might be expected to lack these molecules or to possess some withaltered carbohydrate groups (Gottleib, Skinner & Kornfeld, 1974; Li & Kornfeld,1978; Briles, Li & Kornfeld, 1977; Stanley, Sudo & Carver, 1980). The aims of the

Effects of altered oligosaccharide structure 3
experiments described here were to determine whether these glycoproteins wereabsent or present in reduced numbers on the mutant lines, whether the surfacedistribution of the molecules was changed and whether the glycoproteins had alteredturnover rates.
MATERIALS AND METHODS
Chemicals and reagents
The origins of most of the reagents used in this study have been described (Fitzgerald et al. 1981)Biotinyl-WGA and avidin coupled to horseradish peroxidase, mouse immunoglobulin G and otheranitbodies were purchased from Miles. Carrier-free Na125I (1-7 X104 Ci/g) was purchased from theAmersham Corp.
Cell cultures, membrane isolation and HMWAG purification
Cell lines Pro"5 and Pro"5 WGARI were gifts from Dr Pamela Stanley, Albert Einstein Collegeof Medicine, Bronx, New York. The lectin-resistant lines 15B and 13 and the parental line (P), fromwhich the mutant lines were derived, were gifts from Dr S. Kornfeld, Washington UniversitySchool of Medicine, St Louis, Missouri. The latter, like Pro"5, are prolme-requiring auxotrophsand originated from the original proline-negative clone Kl of Kao & Puck (1968). Line 13 wasselected for resistance to the cytotoxic effects of WGA (Brilesef al. 1977) Line 15B was selected forresistance to ricin agglutinin I (Gottlieb et al. 1974) but is also resistant to WGA.
CHO cells were grown as described previously (Horst & Roberts, 1979) except the medium wassupplemented with 10 % (V/v) colostrum-free newborn calf serum rather than 7'5 % (v/v) foetalcalf serum.
Plasma membranes were isolated from cells broken by means of a fluid pump according to themethods of Horst et al. (19806) and Baumbach et al. (1981). They were solubilized and theHMWAG were purified by affinity chromatography on WGA-Sepharose by methods described indetail by Fitzgerald et al. (1981).
Lactoperoxidase-catalysed, surface iodination of cells in monolayer was performed according tostandard procedures (see Denny & Roberts, 1982).
Two-dimensional electrophoresis and WGA staining of gelsThis was performed according to Horst et al. (1980a). Samples were separated in the first
dimension by isoelectric focusing using a mixture of ampholytes to give a pH gradient from 3-6 to8-9. The second dimension consisted of electrophoresis in 10% (w/v) polyacrylamide gels in thepresence of sodium dodecyl sulphate.
Gels were fixed, stained and dried. Autoradiography was performed using Kodak XRP-1 filmwith intensifying screens and with exposures of up to 1 week at — 80°C (Roberts et al. 1983). Thetechnique of [ I]WGA 'staining' of two-dimensional gels has been described by Horst et al.(1980a).
Preparation of sheep anti-mouse IgGA sheep was immunized at multiple sites on its back with mouse immunoglubulin G (IgG) (1 mg
in Freund's complete adjuvant). After 1 month it was reimmunized using 1 mg of protein inFreund's incomplete adjuvant. At 60 days the sheep was bled from its jugular vein. The whole serum(500 ml) was then frozen in small lots. Antibody was purified by passing samples of serum throughan affinity column (mouse IgG coupled to Sepharose 4B; 5 mg IgG per 3 ml of gel) and eluted with0-1 M-glycine buffer (pH2-2). The pH was quickly adjusted to 7-2, and the antibody was dialysedagainst phosphate-buffered saline (PBS: 014M-NaCl, 1-5 mM-KH2PO4, 8mM-Na2HPO5,3mM-KCl, 0-5mM-MgCl2, lmM-CaCl2, pH 74) .

4 L. A. Fitzgerald and others
Preparation of monoclonal antibodiesBalb/c mice (females, 6-10 weeks old) were immunized with HMWAG as described previously
(Fitzgerald et al. 1981). Fusion with mouse myeloma P3-X63Ag8 (P3) cells (Kohler & Milstein,1975) or SP2/0-Ag(SP2/0) cells (Schulman, Wilder & Kohler, 1978) was performed 5 days follow-ing the final injection by standard procedures (Galfr6 & Milstein, 1981). A mixture of 108 spleencells and 2x 10 myeloma cells was used for each fusion. Selection for hybridoma cells was perfor-med using hypoxanthine (10~4M)/aminopterin (4xlO~7M)/thymidine ( 1 - 6 X 1 0 ~ 5 M ) medium(Littlefield, 1964). Colonies showing good growth were screened for antibody production using thesolid-phase plate assay (Fitzgerald et al. 1981). Positive colonies were expanded into 24-well platesand cells were cloned by limiting dilution in 96-well plates at a density of 0-5 to 2 cells/well. Cloneswere selected from those groups in which less than 33 % of the wells exhibited growth, and in whichthere was no evidence of multiple clones per well. These positive clones were then expanded,screened for antibody production and then recloned. Ascites fluids were prepared by injecting 1X107
cloned cells into 4 to 8-month-old Balb/c mice that had been injected 10 days previously with 0-5 mlPristane (2, 6, 10, 14-tetramethylpentadecane).
ImmunoprecipitationCells (107) were surface-labelled with 125I, washed three times with calcium/magnesium-free PBS
(CMF-PBS: 014M-NaCl, 1-5 mM-KH2PC>4, 8 mM-Na2HPO4, 3 mM-KCl, pH 74) solubilized at4°C in 2 ml CMF-PBS containing 0-05 % (v/v) Nonidet P-40 and 0-1 mM-phenylmethanesulphonylfluoride. Nuclei were removed by centrifugation at 1500 gfor 5 min and the supernatant fraction wascentrifuged at 10000#for 15 min. The resulting supernatant fraction was dialysed (M, cut-off 3000)against the above buffer overnight. Growth medium from hybridoma clones and from myeloma cellswere concentrated 10-fold using a Millipore immersible CX ultrafilter. Immunoprecipitation fromsolubilized cell extracts (0'2 ml) was initiated using 0-2 ml of concentrated hybridoma cell medium.After 4 h at 4°C sheep anti-mouse antiserum (0-6 ml) was added, the mixture was incubated at 4 °Covernight and the immunoprecipitate was collected by centrifugation at 10000#. After washingthree times in buffered detergent solution the immunoprecipitate was analysed by two-dimensionalelectrophoresis. Controls in these experiments included the use of 10 times concentrated P3 andSP2/0 growth media and non-immune mouse antiserum. Immunoprecipitations have also beencarried out using 0-025 ml of ascites fluid rather than concentrated culture medium, with similarresults.
Cell labelling for light microscopyWheat germ agglutinin (WGA) was conjugated to fluorescein isothiocyanate (FITC) by the
method of Rinderknecht (1962). CHO cells grown on 22 mm2 glass coverslips were labelled using35mm diameter culture dishes as a chamber. The basic protocol was to: (1) wash the cells threetimes using Hanks' buffered saline solution (HBSS); (2) place the dishes on ice for 5-10 min toequilibrate to 4°C; (3) incubate with either lOOg/ml of FITC-WGA or a mouse anti-HMWAGantiserum diluted 1 to 20 in HBSS for 15-30min; (4) wash with cold HBSS three times; (5)incubate with FITC-rabbit anti-mouse IgG (diluted 1 to 50) for 30 min at 4°C followed by a washin HBSS; and (6) fix using formaldehyde generated from 3 % (w/v) paraformaldehyde in CMF-PBS at 4°C for at least 30 min. In some experiments, labelled, unfixed cells were wanned at 37 CCby replacing the labelling medium with warm HBSS and incubating the cells in a CO2-containingtissue-culture incubator for up to 2 h before fixation. In control experiments, either lOOmM-JV-acetyl-D-glucosamine along with the FITC-WGA or non-immune mouse serum were used for theprimary incubations. Cells were fixed and mounted in glycerol/CMF-PBS (1: 9, v/v).
Permeabilization of cells was by the method of Laurila, Virtanen, Wartiovaara & Stenman (1978).In this procedure cells were fixed as above. They were then permeabilized in 0-05 % (v/v) NonidetP-40 in CMF-PBS and subsequently labelled with either FITC-WGA or HMWAG-antisera asdescribed above. In these experiments, all incubations following treatment with Nonidet P-40included 1 mg/ml bovine serum albumin to reduce non-specific labelling of cytoplasmic com-ponents.
Fluorescence microscopy was performed on a Zeiss universal microscope equipped with an

Effects of altered oligosaccharide structure 5
epifluorescence light source. Kodak Tri-X film was used and developed in Eduwal FG-7 developer.Within each experiment identical exposure times were used to show relative fluorescence intensity.
Electron microscopyCHO cells were grown to confluence on 60 mm dishes, washed three times using HBSS and
equilibrated to 4 °C on ice. For labelling the HMWAG, the cells were labelled using a 1: SO dilutionof mouse anti-HMWAG antiserum followed after washing by a 1:20 dilution of rabbit, peroxidase-conjugated, anti-mouse IgG. In parallel control experiments, non-immune serum was used at a 1: 50dilution in an identical manner to that for immune serum. In some experiments cells were wannedat 37 °C for various times. The labelled cells were fixed for 30-60 min using 3 % (w/v) glutaral-dehyde in 0-1 M-sodium cacodylate (pH 7-4) at 4°C. The fixed cells were washed several times inthe cacodylate buffer followed by 0-1 M-Tris-HCl (pH7-6). The cell surface or internalizedperoxidase was revealed by using the 3-3'-diaminobenzidine method of Graham & Karnovsky(1966). The 3-3 '-diaminobenzidine was used at a concentration of 0-05 % (w/v) and H2O2 at 0-06 %(v/v) in 0-1 M-Tris'HCl (pH7-6). The reaction was performed for 10-15 min at room temperature.The labelled cells were washed for several hours in 0-1 M-Tris-HCl (pH7-6) followed by 0-1 M-cacodylate. Finally, the cells were scraped off the plastic by means of a rubber policeman and a pelletwas formed by mixing 0'2 ml of the cell suspension with 0-2 ml of 2 % (w/v) OsG^ in 0-1 M-sodiumcacodylate in a Microfuge tube. The preparation was centrifuged for 5 min using a Beckman modelB microfuge. The pellets were dehydrated in a graded series of ethanol, followed by propylene oxideand then embedded in a mixture of Epon-Araldite (Mollenhauer, 1964). Light gold sections werepost-stained with uranyl acetate and lead citrate and examined in an Hitachi HU-11E electronmicroscope.
Labelling techniques with monoclonal antibodiesTo investigate the distribution of monoclonal antibody binding sites on CHO cells, the super-
natant fraction from cultured hybridoma cells was diluted 1: 5 using HBSS and incubated with cellsgrown on 22mm2 glass coverslips for 30 min at 4°C. After three washes, the cells were incubatedusing a 1: 30 dilution of FITC-rabbit anti-mouse IgG and examined. In this study only two super-natants c226'l 1 (a P3 hybrid line) and 1-2 (an SP2/0 hybrid line) were used.
The distributions of hybridoma antibody bound to CHO cell surface were also investigated at theelectron-microscopic level by culturing CHO cells in 60 mm diameter dishes. The cells were in-cubated in the presence of hybridoma culture media or myeloma supernatant at a 1: 3 dilution for30 min at 4°C. Following three washes, the dishes were incubated in a 1: 20 dilution of peroxidase-conjugated anti-mouse IgG for 30 min at 4°C. Some dishes were warmed to 37 °C for 30 min andothers were fixed, dehydrated and embedded according to the method described in the previoussection.
Turnover of iodinated surface polypeptidesThe general scheme for all of the turnover experiments reported here was to label cells initially
with l2iI and then incubate the cells for various periods of time at 37 °C in culture medium containingmaintenance levels only (0-1 %, v/v) of serum. During this time, cell division rates were significant-ly reduced and degradation of the '"1-labelled proteins occurred (Horst & Roberts, 1979; Denny& Roberts, 1984). At the end of the incubation period, the lziI-labelled cells were mixed with anequivalent amount of 131I-labelled cells that had just been surface labelled, but had been maintainedon low-serum medium for the same length of time as the l25I-labelled cells. This mixture is referredto as the 'experimental' cells. Control cells were prepared by mixing equivalent amounts of125I-labelled and 131I-labelled cells, neither of which had been allowed to undergo protein turnoverafter labelling. The control and experimental cells were then lysed and analysed by two-dimensionalelectrophoresis and autoradiography. Each major radioactive spot in both the experimental andcontrol gels was cut out using the autoradiograms as templates, and the gel plugs were counted ina gamma counter. The ratio of 125I to 131I for each protein was then determined after correction wasmade for isotope spillover and radioactive decay. By means of a first-order rate equation (see Horst& Roberts, 1979; Denny & Roberts, 1984), the rate constant for degradation and corresponding half-life for each radioactive protein can be computed from the decrease in 125I/13II ratio with time. This

6 L. A. Fitzgerald and others
ratio is decreased for each protein in the experimental gel when compared with the ratio of thecorresponding protein in the control gel due to the turnover of the lzsI-labelled proteins by theexperimental cells.
An initial set of experiments was performed in order to determine the correct time interval for theturnover experiments. Eight 75 cm2 flasks of cells were grown to 80 % confluency in monolayer. Themedium was then replaced with McCoy's 5A containing 0-1 % (v/v) heat-inactivated newborn calfserum (low-serum medium), followed by incubation of the flasks at 37°Cfor 24 h. Three flasks wereiodinated with I25I and placed at 37°C for either 31, 55 or 79 h. At the end of each of these timeintervals, one flask of cells was iodinated with '31I and was mixed with the l25I-labelled cells andfrozen. Finally, control cells were prepared by mixing equivalent numbers of I25I- and l31I-labelledcells that had not been allowed to undergo turnover after labelling. The above procedures resultedin four groups of double-labelled cells. Each group was suspended in 02ml of lOmin-Tris-HCl(pH7-4) that contained 5 mM MgCb. and pancreatic ribonuclease (0-05 jlg/ml). The cells werebroken by five freeze-thaw cycles in an acetone/solid CO2 bath, and each lysate then received 10 /dof a 1 mg/ml solution of deoxyribonuclease I in lOniM-Tris-HCI (pH 7-4). After lOmin on ice, thematerial was solubilized (Horst et al. 1980a) and analysed by two-dimensional electrophoresis.
In the experiment involving the comparison of the parental and ricin-resistant line 15B, the cellswere grown in monolayer to 80 % confluency in four 75 cm2 tissue-culture flasks. The flasks wereplaced on low-serum medium, and the iodination was carried out using a time interval of 24 hbetween the I2SI and 131I labelling. The flasks were washed three times with 10ml CMF-PBS(pH7-4), and were incubated for 15min at room temperature with 10ml of CMF-PBS (pH7-4)containing 005 % (w/v) Na2EDTA and 5 min-glucose. The detached cells were collected bycentrifugation, and each flask of l25I-labelled cells was mixed with a corresponding flask of l :"l-labelled cells. The resulting four cell pellets (a control and experimental pellet for each of the twocell lines) were lysed by the addition of 1 ml of CMF-PBS (pH 7-4) containing 0-5 % (w/v) NP-40and 1 mM-phenylmethanesulphonyl fluoride (freshly added from a 0-1 M stock solution in absoluteethanol), followed by trituration with a Pasteur pipette. The lysed cells were placed on ice for20min, and nuclei were then removed by centrifugation (1500|f, 5min). The supernatants weredialysed for 16 h at 4 °C against 1 litre of 0-5 % (w/v) Nonidet P-40 in distilled water. The retentateswere then lyophilized and redissolved in 400/il of a solution containing 9-3 M-urea, 5 mM-KzCC^ ,2% (w/v) Nonidet P-40, and 5 mg/ml dithiothreitol. Samples (0-2 ml) were then analysed by two-dimensional electrophoresis and autoradiography (12 h) and the radioactive content of differentpolypeptides was analysed as above.
RESULTS
Iodinatable surface components on CHO lines
Those components that incorporated 12SI after lactoperoxidase-catalysed iodinationof intact CHO cells were identified on the two 'wild-type' parental lines and the threelectin-resistant mutant lines by means of two-dimensional polyacrylamide gelelectrophoresis followed by autoradiography of the dried gels (Fig. 1A-E). Theparental lines (Pro"5 and P in A and c, respectively) contained a similar but notidentical group of iodinatable components, the HMWAG, which were located in theupper, acidic corner of the gels. Most of these formed rather diffuse, slanting lines of
Fig. 1. Autoradiograms of two-dimensional gels of plasma membrane fractions isolatedfrom CHO cells that had been iodinated with I using the lactoperoxidase technique. Celllines were: A, Pro"5; B, Pro"5 WGARI; c, P; D, 13; E, 15B. Approximately 200/ig ofprotein and 20000disints/min were loaded on each gel and exposure was for 1 week. Amolecular weight scale and pH gradient are indicated. The regions arrowed in A corres-pond to radioactive spots (I, II and III) analysed for exponential decay of 12SI duringturnover experiments (see Table 2 and Fig. 7).

Effects of altered oligosaccharide structure
86 8Mr • •103)
2 0 0 -1 0 0 -
5 0 -4 0 -3 0 -
2 0 -3
J^^r
Fig. 1

I L. A. Fitzgerald and others
protein spots. When the mutant lines were examined several labelled polypeptideswere also seen to be present in the upper acidic quadrant, but the spots were in generalless diffuse and of somewhat higher pi than those of the parental lines. The mutantpatterns were sufficiently dissimilar from parental patterns for it to be impossible todecide unequivocally which mutant polypeptide was related to which parental com-ponent. However, there appeared to be some strong similarities in the iodinationpatterns of the three lectin-resistant lines, although each line was distinctive, and evenapparently corresponding polypeptides appeared to have relatively different inten-sities.
Identification of WGA-binding polypeptides on mutant plasma membranes
We have shown (Horst et al. 19806; Fitzgerald et al. 1981) that the major WGA-binding glycoproteins of CHO cells consist of the group of iodinatable components,
-2A B_
IFig. 2. Binding of 12sI-labelled WGA to surface glycoproteins following two-dimensionalgel electrophoresis of plasma membrane fractions isolated from unlabelled CHO cells. Celllines were: A, Pro"5; B, Pro"5 WGARI; c, P; D, 13. [1Z5I]WGA did not bind to anycomponents of 1SB as determined by autoradiography. Approximately 200 /.ig of mem-brane protein was loaded onto each gel. The autoradiograms for A and c were exposed forabout 12 h; those for B and D for 1 week.

Effects of altered oligosacchande structure 9
Fig. 3. FITC-WGA labelling of CHO cells that had been fixed and then perfneabilizedprior to addition of fluorescent lectin. A, Pro"5; B, Pro"5 WGARI; c, 13.; D, 15B. Otherwild-type lines such as P show a pattern of labelling identical to A. Note the intensefluorescence of both cell surface and perinuelear region in Pro""5 (A). In contrast, thelectin-resistant lines (B, C, D) exhibit negligible cell surface fluorescence. However, eachmutant line shows a compact region of fluorescence within the perinuelear zone. Note alsothat the nuclear envelope of each line binds FITC-WGA. In control incubations, using0-1 M-iV-acetyl-D-glucosamine as a competing hapten, only faint cytoplasmic fluorescencewas observed (data not shown). X1500.
which are located in the upper acidic quadrants of Fig. 1A and c. When two-dimensional electrophoretic gels prepared from the lectin-resistant lines were overlaidwith 125I-labelled WGA, the patterns obtained differed markedly from those seen withthe parental lines (Fig. 2). For example, there appeared to be no binding to anycomponent of 15B even after exposure of the autoradiograph for 1 week (result notshown). With WGARI, however, there was binding to an acidic macromolecule ofMr ~ 70 000 and to another component of higher molecular weight, which failed toenter the second-dimensional gel (Fig. 2B). By contrast, binding of 125I-labelled WGA

10 L. A. Fitzgerald and others
to plasma membrane polypeptides of line 13 was very weak. However, after relativelylong exposures a number of diffuse streaks could be detected on the autoradiograms.These results confirm the fact that all three mutant lines show a reduced number ofcomponents that can bind WGA. The question therefore arises as to whether themutant lines lacked a number of parental-type components completely or whether thelow WGA binding was due simply to loss of terminal carbohydrate groups on other-wise identical polypeptides.
Binding ofFITC-WGA
FITC-WGA binding to the three mutant lines was relatively weak as compared towild-type lines when unpermeabilized cells at 4 °C were used (results not shown), thusconfirming the earlier experiments with 125I-labelled WGA. This lack of surfacefluorescence was also evident when the cells were first permeabilized with detergentprior to adding the fluorescent lectin (Fig. 3). With lines Pro"5 and P (the latter notshown) a surface rim of fluorescence, lacking in the mutants, was clearly evident (Fig.3A). The nuclear envelopes of both parental and lectin-resistant lines bound WGA,however. On the other hand, while Pro"5 demonstrated a large, diffuse patch ofinternal fluorescence in the perinuclear region, the mutants each possessed a discrete,more compact band of WGA-binding adjacent to the nucleus.
Binding of Hti'IWAG antiserum to cell lines
Antibodies generated against the HMWAG of Pro"5 cells bound fairly uniformlyover the surface of all CHO lines tested (Fig. 4A, C, E, G), including the three mutants.As observed previously, the pattern of labelling was somewhat punctate and theantibody appeared to be concentrated most intensely on surface protuberances andmicrovilli (see Fig. 5A, B). When cells that had been surface-labelled with mouse anti-HMWAG antiserum followed by fluorescent second antibody were warmed for30min at 37 °C, fluorescent label was rapidly removed from the surface and enteredcytoplasmic vesicles in the cortical cytoplasm (Figs 4B, D, F, H and 5F). All linesappeared to internalize bound antibody in a qualitatively similar manner. Internaliza-tion involved the formation of surface patches and clustering of label in pits, par-ticularly at the base of microvilli (Fig. 5D, E). The internal vacuoles were generallyconnected to the surface by neck-like structures during the earliest periods of warming(Fig. 5D, E). Stained material in regions of cell-to-cell contact were not internalizedafter warming for 30min (Fig. 5F).
These results suggest that the lectin-resistant lines possess the HMWAG inamounts comparable to those in the parental cells, that these molecules are not
Fig. 4. Immunofluorescence micrographs of CHO cell lines labelled using HMWAG anti-serum followed by FITC-labelled anti-mouse IgG. A, C, E, G . Cells labelled and then fixed at4°C. B, D, F, H. Cells labelled at 4 °C and subsequently warmed to 37 °C for 30 min. The celllinesarePro~5(A1B);Pro~5WGARI(c1D);13(E,F);andl5B(G,H).Theotherwild-typelineP labelled identically to Pro"5. In control incubations using non-immune serum only a faintfluorescence was observed (data not shown, but see Fitzgerald et al. 1981, fig. 3c). X800.

Effects of altered oligosaccharide structure 11
Fig. 4

12 L. A. Fitzgerald and others
Fig. 5. Immunoperoxidase localization of HMWAG on CHO Pro 5 cells. Cells weretreated either with anti-HMWAG serum (A, B, D, E, F) or with non-immune serum (c),each followed by peroxidase-conjugated anti-mouse IgG. The product of reaction of thebound peroxidase with 3-3'-diaminobenzidine stains black, A. Cells labelled at 4°C; B,same as A but higher magnification; c, control incubation at 4°C using non-immuneserum; D, cells labelled at 4°C and warmed to 37 °C for 2min; E, cell surface after 5 minof wanning at 37°C; F, cells labelled at 4°C and wanned to 37°C for 30 min. Magnifica-tion: A, c, D, F, X8000; B, X48000; E, X67000.
Effects of altered oligosaccharide structure 13
abnormally distributed on the cell surface and that the pattern of internalizationinduced by a constant amount of anti-HMWAG antiserum followed by a constantamount of second antibody occurs in a similar manner in all of the lines.
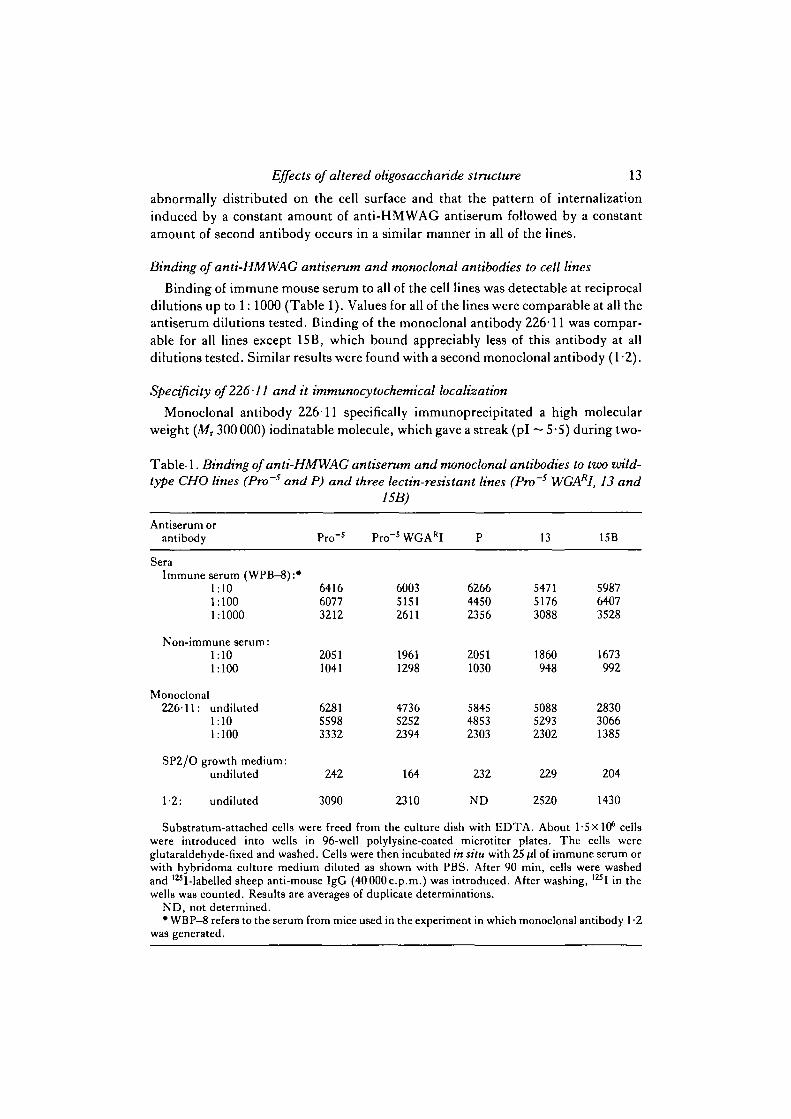
Binding of anti-HMWAG antiserum and monoclonal antibodies to cell lines
Binding of immune mouse serum to all of the cell lines was detectable at reciprocaldilutions up to 1: 1000 (Table 1). Values for all of the lines were comparable at all theantiserum dilutions tested. Binding of the monoclonal antibody 226-11 was compar-able for all lines except 15B, which bound appreciably less of this antibody at alldilutions tested. Similar results were found with a second monoclonal antibody (1 -2).
Specificity of 226-11 and it immunocytochemical localization
Monoclonal antibody 226-11 specifically immunoprecipitated a high molecular
weight (Mr 300000) iodinatable molecule, which gave a streak (pi ~5-5) during two-
Table- 1. Binding of anti-HMWAG antiserum and monoclonal antibodies to tvx> ivild-
type CHO lines (Pro~s and P) and three lectin-resistant lines (Pro~s WGARI, 13 and15B)
Antiserum orantibody
SeraImmune serum (WPB-8):*
1:101:1001:1000
Non-immune serum:1:101:100
Monoclonal22611: undiluted
1:101:100
SP2/O growth medium:undiluted
1-2: undiluted
Pro"5
641660773212
20511041
628155983332
242
3090
Pro"5 WGARI
600351512611
19611298
473652522394
164
2310
P
626644502356
20511030
584548532303
232
ND
13
547151763088
1860948
508852932302
229
2520
15B
598764073528
1673992
283030661385
204
1430
Substratum-attached cells were freed from the culture dish with EDTA. About 1-5X106 cellswere introduced into wells in 96-well polylysine-coated microtiter plates. The cells wereglutaraldehyde-fixed and washed. Cells were then incubated in situ with 25 fA of immune serum orwith hybridoma culture medium diluted as shown with PBS. After 90 min, cells were washedand l25I-labelled sheep anti-mouse IgG (40 000 c.p.m.) was introduced. After washing, l z i I in thewells was counted. Results are averages of duplicate determinations.
ND, not determined.• WBP—8 refers to the serum from mice used in the experiment in which monoclonal antibody 1 -2
was generated.

14 L. A. Fitzgerald and others
dimensional electrophoresis (Fig. 6A). It corresponds to component 3 in Fig. 8A. Arelatively high proportion of the radioactivity failed to enter the second-dimension geland existed as a long streak at the stacking gel-running gel interface. The antibody alsoprecipitated a similar high molecular weight component from line 13 (Fig. 6B).
The distribution of the antigen bound by 226-11 was investigated by immuno-fluorescence and immunoperoxidase techniques at the level of the light and electronmicroscope, respectively. The patter of surface labelling was indistinguishable fromthat seen with anti-HMWAG immune mouse serum (results not shown).
Labelling with monoclonal 1-2 also occurred relatively uniformly over the entirecell and, like 226-11 and whole antiserum, the antibody bound to membrane surfaces
6A
B
Fig. 6. Two-dimensional polyacrylamide gel electrophoresis of product immuno-precipitated from solubilized plasma membrane preparations of iodinated CHO cells bymonoclonal antibody 226-11. CHO cells (A, line P; B, line 13) were iodinated by thelactoperoxidase procedure, plasma membrane rnaterial isolated and solubilized, and anti-gen cross-reacting with 226-11 was isolated by immunoprecipitation. The precipitate wasanalysed by two-dimensional electrophoresis and autoradiography. Approximately1000 disints/min of radioactivity was loaded on the gels. Exposure of the autoradiogramswas for 3 weeks. Controls using P-3 medium and non-immune mouse serum were blank.

Effects of altered oligosaccharide structure 15
20 40
Time (h)
60 80
Fig. 7. I25I to I31I ratios of three selected polypeptides as a function of time. Double-labelanalysis was carried out using experimental intervals of 0, 31, 55 and 79 h between theinitial l25I-labelling and the final I-labelling. Two-dimensional electrophoresis was per-formed each of the four sets of cells. Labelled spots were detected by autoradiography,punched from the dried gels, and their 125I and 131I contents were determined. The ratiosfor each spot were plotted on a semilogarithmic plot as a function of hours betweenlabelling. ( • ) Protein region I; (A) protein region II; (O) protein region III, in Fig. 1A.
between cells as well as to regions of no contact (results not shown). The pattern ofinternalization following addition of second antibody was similar to that observedwith whole antiserum, and the label rapidly became concentrated in endocytoticvesicles in the cortical cytoplasm.
Turnover of iodinatable surface components on parental and mutant linesTurnover of the HMWAG was assessed using a double-label, 125I/131I procedure.
In initial experiments it was established that the loss of radioactivity from three majorgroups of iodinatable components occurred via a process that appeared to be first-order. Parental cells were first labelled with 1Z5I and then at intervals of 31, 55 and 79 hlater were labelled with 13II. After two-dimensional polyacrylamide by overnightautoradiography. Spots were punched and counted from three major regions of thegel, corresponding to regions I, II and III of Fig. 1A. When the 125I/131I ratios of these


Effects of altered oligosaccharide structure 17
spots were plotted versus time on a semilogarithmic scale, a straight line relationshipwas obtained for all three components, indicating that the loss of 125I followedapproximate first-order kinetics (Fig. 7). In addition, it can be seen that the amountsof 125I in each of the three regions decreased to about half their initial values in about19 h. By 79 h the levels of 1Z5I present were very low (Table 2) and any calculatedvalues for KD made at this stage would, therefore, be very unreliable compared tothose obtained at earlier times.
In the experiments in which the turnover of surface iodinatable components of themutant line 15B was compared with that of its parental line, a single time interval of24 h between 12SI and 131I labellings was used. Fig. 8A and B shows the seven majorpolypeptides, from the upper right-hand quadrant of the gel, that were analysed indetail in this experiment.
Table 3 summarizes the labelling data for the parental line. The 125I/131I ratios ofa total of 19 proteins from all regions of the gel were examined (data not shown). Thecontrol ratios for these 19 components (i.e. the values obtained after the two groupsof cells were labelled simultaneously with 12SI and 131I, mixed and then analysed) was2-55 ± 0*34. The control values for the seven HMWAG fell very close to this averageand none was more than one standard deviation from it. Table 3 lists the 13SI and 131Icounts and the isotope ratios for each of these proteins when a 24 h period separatedthe two iodinations. The half-lives calculated from these data were all fairly similarand averaged about 12 h.
With clone 15B, the average control ratios for 16 iodinated components punchedfrom gel was 2-51 ± 0-73. The control ratios for the seven HMWAG were very closeto this average (2-45 ± 0-35). However, the major polypeptide marked with an arrow
V
8 B
Fig. 8. Radioactive polypeptides analysed for l2Sl/1311 ratios in the experiments describedin Tables 3 and 4. Cells labelled with 12SI, and 24h later with 13II, were analysed by two-dimensional polyacrylamide gel electrophoresis. Labelled proteins were detected byautoradiography. The photographs are of autoradiographs prepared from line P (A) andline 15B (B). Only the region of the gel showing the HMWAG is shown (i.e. the upperright-hand portion). Regions from components numbered l ' - 7 ' were punched from thedried gel using the overlying autoradiogram as a template, and their IZSI and 13II contentswere measured. The spot marked with an arrow was also punched and gave anomalousl 2 i I / m I ratios even in the control experiment.

18 L. A. Fitzgerald and others
Table 3. / 2 5 / / » ' / double-label analysis of turnover of HMWAG in CHO line P
Polypeptide*
1234567
Controlratiof
2-432-302-392-842-782-532-71
125J
(c.p.m.)
3382481325479617325
1793
131,
(c.p.m.)
5616789631754
1140520
2390
Experimentalratioj
0-6020-6030-5150-6350-5420-6250-750
(h )
0-0600-0600-0670-0580-0650-0590-051
(b)
12121012111214
• Polypeptide number refers to designations in Fig. 8A.•f Control ratios are the 125I/131I values of control cells that were labelled simultaneously
with 125I and 131I.X Experimental ratios are the 125I/1311 values of cells in which the labelling with m I occurred 24 h
after the labelling with 125I.§ A'D is the rate constant of degradation; ti the calculated half-life.
Table 4. 12SI/>3II double-label analysis of turnover of HMWAG in CHO line 15B
Control I25I I31I Experimental KD tiPolypeptide* ratiof (c.p.m.) (c.p.m.) ratioj (h~5) (h)
r2'3'4'5'6'7'
2-522-542-50l-86§2-812-122-82
754178194188229238440
2133271298278430278857
0-3530-6580-6520-6790-5320-8550-514
00830-0570-05700560-0660-0460-067
8121212111510
•Polypeptide number refers to designations in Fig. 8B.f Control ratios are the 125I/131I values of control cells that were labelled simultaneously
with ml and 131I.X Experimental ratios are the ]2s\/nil values of cells in which the labelling with 131I occurred 24 h
after the labelling with 1ZSI.§ Value here may be unreliable since only a low recovery of redioactivity (12SI, 114 c.p.m.; 131I,
61 c.p.m.) occurred. It is possible that the spot was not punched accurately.
in Fig. 8B exhibited a control ratio of 1-35 and was not considered further in this studysince it fell significantly outside the average range. Possibly, it is a secreted proteinrecovered in varying amounts from cell preparation to cell preparation. Of the seven(l '-7 ') HMWAG listed in Table 4, the average half-life was 11 ±2h , and did notdiffer significantly from the value obtained with the parental line. However, somevariability was evident. Components 1' and 6' had half-lives of 8 and 15 h, respectively.
DISCUSSION
In this study we have attempted to determine whether alterations in the carbo-hydrate structures of membrane glycoproteins influence the amount of these

Effects of altered oligosaccharide structure 19
molecules present on the cell surface, their distribution on the plasma membrane andtheir turnover rates. To accomplish these goals we have used, three cell lines selectedfor their resistance to plant lectins. The first of these, clone 1SB, is deficient in uridinediphosphate-iV-acetylglucosamine : glycoprotein ./V-acetylglucosaminyl transferase(Li & Kornfeld, 1978) and is resistant to the cytotoxic lectin, ricin I. Its carbohydratechains lack the outer tier of sialic acid, galactose and VV-acetylglucosamine residues,characteristic of many fully 'processed' asparaginyl-linked oligosaccharides, and ter-minate in a-D-mannosyl residues. Clone 13 was selected for WGA resistance (Brileset al. 1977). Its membrane glycoproteins are low in sialic acid and galactose but appearto bear a normal complement of ./V-acetylglucosamine and mannose residues, althoughthe nature of the enzymic defect remains unclear. CHO cell mutant WGARI, unlike15B was selected for WGA rather than ricin resistance, but like 15B appears to possessdeficient /V-acetylglucosaminyl transferase I activity (Stanley et al. 1980). Like 15B,it presumably lacks the outer saccharide units characteristic of complex, fullyprocessed glycoproteins. Indeed, our studies show that 15B and WGARI possessedsimilar surface-labelling patterns after lactoperoxidase-catalysed iodination of intactcells.
Although all three mutant lines had few surface glycoproteins that bound WGA,the nuclear envelope and a small compact region of membrane in the perinuclear zonefluoresced intensely after exposure to FITC-WGA, suggesting that the oligo-saccharide chains on these structures had a biosynthetic origin different from thoseon the plasma membrane. Possibly, the fluorescent region within the perinuclear zonecontains types of glycoprotein identical to those in the nuclear envelope and playssome role in the elaboration of that structure.
Studies using mouse antiserum against the entire group of HMWAG have shownthat the mutant lines bind these antibodies in comparable amounts to the parentallines. The binding of monoclonal antibodies c226-ll and 1-2 was also generally un-affected by lectin resistance. Although clone 15B showed a somewhat reduced bindingof 226-11, WGARI, which possess similar types of carbohydrate chain to 15B, bound226-11 in similar amounts to those observed with the parental lines.
All lines showed a similar distribution of the glycoprotein antigens on their surface.In general, the molecules were distributed fairly uniformly over the entire plasmamembrane, although the pattern was somewhat punctate over the upper surfaces andedges of the cell, and appeared to be concentrated relatively strongly on microvilli andcell projections. The glycoproteins were also found in regions of cell-to-cell contact.Internalization promoted by successive addition of anti-HMWAG antibodies and asecond anti-mouse immunoglobulin followed by a period of warming occurred in asimilar manner in both mutant and parental lines. When carried out at a fixed con-centration of serological reagents internalization, except at regions of cell-to-cell con-tact, occurred quickly into endocytotic vacuoles in the cortical cytoplasm. Together,these results suggest that the mutant lines possessed a relatively unchanged numberof HMWAG on their surfaces and that the majority of these glycoproteins were notdistributed in an unusual manner.
The monoclonal antibodies used in this study (226-11 and 1-2) were both specific

20 L. A. Fitzgerald and others
for plasma membrane antigens that were distributed over the total cell surface. How-ever, it is possible that some of the individual HMWAG do not have such a generaldistribution and may be more strongly localized in regions such as coated pits and celljunctions, as has been noted for plasma membrane glycoproteins of mouse fibroblasts(Murphy, Decker & August, 1983). Conceivably, alterations in the carbohydratestructures of such glycoproteins could influence their topographical distribution onthe cell surface. The use of monoclonal antibodies directed against a wider range ofindividual HMWAG in the mutant cells should help resolve this issue.
The results of the experiments in which the turnover rates of the HMWAG weremeasured also indicated little difference between the parental and mutant cell lines,although at least one component (1' in Fig. 8B) appeared to have a shorter half-lifethan its partner in Fig. 8A. However, there was no indication that this was a generalphenomenon. Moreover, it was not possible to match components in the mutant andparental lines with any degree of confidence; therefore, it is possible that 1 and 1' werenot homologous. The high molecular weight glycoprotein recognized by monoclonalantibody 226-11 (3 and 3' in Fig. 8) had apparent half-lives of 10 and 12 h, respectively,in the parent and mutant line, a difference that was not significant.
It is important to stress that these turnover experiments were carried out using cellsplaced in low-serum medium prior to the experiment. Such a treatment reduces therates of cell division and is necessary in experiments with CHO cells in order to obtainmeasurable rates of membrane turnover. Actively proliferating CHO cells, whichhave a population doubling time of about 12-18 h, show very slow rates of turnoverof their surface iodinatable components, with half-lives exceeding 100 h (Roberts &Yuan, 1974). Double-label experiments of the type described here cannot, therefore,be done since there would be at least a thirtyfold dilution of the initial label before thepolypeptides had even undergone a single half-life. Nevertheless, low-serum con-ditions might lead to destruction of surface molecules by processes different fromthose occurring under normal growth conditions. The half-lives we reported in Tables3 and 4 were unexpectedly low. In previous experiments, using smaller amounts ofradioactivity, half-lives in the 14— 20h range were noted for the HMWAG (J. B.Denny & R. M. Roberts, unpublished results). Other membrane-associated polypep-tides, including components that were probably part of the cell cytoskeleton, havebeen estimated to have half-lives averaging 54 ± 18 h (Horst & Roberts, 1979). How-ever, the latter experiments were carried out using reutilizable precursors (L-[3 5S]-
and L-[3H]methionine) and the HMWAG themselves were not investigated. Thereason for the high rate of turnover of the HMWAG in the present experiment isunclear. Nevertheless, there was still no evidence to suggest that the abnormalglycoproteins of the mutant cells were degraded at faster rates than their structurallymore normal counterparts.
In summary, therefore, we have found no evidence that truncation of the carbo-hydrate chains and exposure of normally subterminal saccharides on membraneglycoprotein molecules of CHO cells lead to reduced numbers of these molecules onthe surface, or to increased turnover rates, or to an abnormal distribution of thesemolecules on the plasma membrane. This is not to suggest, however, that these

Effects of altered oligosaccharide structure 21
alterations in carbohydrate structure do not have other, more subtle influences on cellbehaviour and physiology.
We thank Dr Henry Aldrich for access to the electron microscope. This work was supported bygrant PCM 8104071 from the National Science Foundation. J. B. Denny and G. A. Baumbach weresupported by predoctoral fellowships on a Training grant (CA 09126) from the National CancerInstitute. We thank Drs P. Stanley and S. Kornfeld for supplying us with the lectin-resistant linesused in this study and Mr John Berceann for typing the manuscript.
REFERENCES
ASHWELL, G. &HARFORD, J. (1982). Carbohydrate-specific receptors of the liver. A. Rev. Biochem.51, 531-554.
BAUMANN, H., HOU, E. & JAHREIS, G. P. (1983). Preferential degradation of the terminalcarbohydrate moiety of membrane glycoproteins in rat hepatoma cells and after transfer to themembranes of mouse fibroblasts. J. Cell Biol. 96, 139-150.
BAUMBACH, G. A., HORST, M. N., OLYMPIO, M. A., ALDRICH, H. C. & ROBERTS, R. M. (1981).
Isolation and characterization of plasma membrane from cultured cancer cells in Cancer CellOrganelles: Methodological Surveys in Biochemistry, vol. II (ed. E. Reid, G. M. Cook & D. J.Morr6e), pp. 351-370. Chichester: Ellis-Horwood Ltd.
BRILES, E. B., L I , E. & KORNFELD, S. (1977). Isolation of wheat germ agglutinin-resistant clonesof Chinese Hamster Ovary cells deficient in membrane sialic acid and galactose. J. biol. Chem.252, 1107-1116.
BUCK, C. A. & WARREN, L. (1976). The repair of the surface structure of animal cells. J. cell.Physiol. 89, 187-200.
DENNY, J. B.& ROBERTS, R. M. (1982). Anew immunoreactive probe for the isolation and analysisof plasma membrane polypeptides. Synthesis and propertiesofisethionyl3-(iV-2,4-dinitrophenyl)-aminopropioimidate. J. biol. Chem. 257, 2460—2468.
DENNY, J. B. & ROBERTS, R. M. (1984). Turnover of plasma membrane polypeptides in trans-formed and normal cells using 1 2 5I /" ' I double-label techniques. In Cancer Cell Organelles:Methodological Surveys in Biochemistry, vol. II (ed. E. Reid, G. M. W. Cook & D. J. Morr6).Chichester: Ellis-Horwood (in press).
DICE, J. F. & GOLDBERG, A. L. (1975a). A statistical analysis of the relationship betweendegradative rates and molecular weights of proteins. Archs Biochem. Biophys, 170, 213-219.
DICE, J. F. & GOLDBERG, A. F. (19756). Relationship between in vivo degradative rates and theisoelectric points of proteins. Proc. natn. Acad. Sci. U.SA. 72, 3893-3897.
FITZGERALD, L. A., BAUMBACH, G. A., HORST, M. N., NOONAN, K. D. & ROBERTS, R. M.
(1981). The purification and immunocytochemical localization of the major iodinatable cellsurface glycoproteins of Chinese hamster ovary cells. J. Cell Sci. 52, 405—424.
GALFRE\ G. & MILSTEIN, C. (1981). Preparation of monoclonal antibodies: Strategies andprocedures. In Methods in Enzymology, vol.73B (ed. J. J. Langone&H. VanVanakis), pp. 3—46.New York: Academic Press.
GOLDBERG, A. L. & DICE, J. F. (1974). Intracellular protein degradation in mammalian andbacterial cells. I. A. Rev. Biochem. 43, 835-869.
GOLDBERG, A. L. & S T JOHN, A. C. (1976). Intracellular protein degradation in mammalian andbacterial cells. II. A. Rev. Biochem. 45, 747-803.
GOTTLIEB, C , SKINNER, A. M. & KORNFELD, S. (1974). Isolation of a clone of Chinese hamsterovary cells deficient in plant lectin-binding sites. Proc. natn. Acad. Sci. U.SA. 71, 1078-1082.
GRAHAM, R. C. & KARNOVSKY, M. J. (1966). The early stages of adsorption of horseradishperoxidase into the proximal tubules of mouse kidney: Ultrastructural cytochemistry by a newtechnique. J . Histochem. Cytochem. 14, 291-297.
HORST, M. N., BASHA, S. M. M., BAUMBACH, G. A., MANSFIELD, E. H. & ROBERTS, R. M.(1980a). Alkaline urea solubilization, two-dimensional electrophoresis and lectin staining ofmammalian cell plasma membrane and plant seed proteins. Analyt. Biochem. 102, 399-408.

22 L. A. Fitzgerald and others
HORST, M. N., BAUMBACH, G. A., OLYMPIO, M. A. & ROBERTS, R. M. (19806). Isolation of a
domain of the plasma membrane in Chinese hamster ovary cells which contains iodinatable, acidicglycoproteins of high molecular weight. Biochim. biophys. Ada 600, 48—61.
HORST, M. N. & ROBERTS, R. M. (1979). Analysis of polypeptide turnover rates in Chinese hamsterovary cell plasma membranes using two-dimensional electrophoresis. J. biol. Chem. 254,5000-5007.
KAO, F. T. & PUCK, T. T. (1968). Genetics of somatic mammalian cells. VII. Induction andisolation of nutritional mutants in Chinese hamster cells. Proc. natn. Acad. Set. U.SA. 60,1275-1281.
KOHLER, G. & MILSTEIN, C. (1975). Continuous culture of fused cells secreting antibody ofpredefined specificity. Nature, Land. 2S6, 495-497.
KNOWLES, S. E. & BALLARD, F. J. (1976). Selective control of the degradation of normal andaberrant proteins in Reuber H35 hepatoma cells. Biochem.J. 156, 609-617.
LAURILA, P., VIRTANEN, I., WARTIOVAARA, J. & STENMAN, S. (1978). Fluorescent antibodiesand lectins stain intracellular structures in fixed cells treated with nonionic detergent. J.Histochem. Cytochem. 26, 251-257.
LEAVITT, R., SCHLESINGER, S. & KORNFELD, S. (1977). Impaired intracellular migration andaltered solubility of nonglycosylated glycoproteins of Vesicular Stomatitis virus and Sindbisvirus. J . biol. Chem. 252, 9018-9023.
Li, E. & KORNFELD, S. (1978). Structure of the altered oligosaccharide present in glycoproteinsfrom a clone of Chinese hamster ovary cells deficient in N-acetylglucosaminyltransferase activity.J. biol. Chem. 253, 6426-6431.
LITTLEFIELD, J. W. (1964). Selection of hybrids from mating of fibroblasts in vitro and theirpresumed recombinants. Science, N.Y. 145, 709.
MOLLENHAUER, H. H. (1964). Plastic embedding mixtures for use in electron microscopy. StainTechnol. 39, 111-114.
MURPHY, T. L., DECKER, G. & AUGUST, J. T. (1983). Glycoproteins of coated pits, cell junctionsand the entire cell surface revealed by monoclonal antibodies and immunoelectron microscopy.J. Cell Biol. 97, 533-541.
OLDEN, K., PARENT, J. B. & WHITE, R. L. (1982). Carbohydrate moieties of glycoproteins: A re-evaluation of their function. Biochim. biophys. Ada 650, 209—232.
OLDEN, K., PRATT, R. M. & YAMADA, K. M. (1978). Role of carbohydrates in protein secretionand turnover: Effect of tunicamycin on the major cell surface glycoprotein of chick embryofibroblasts. Cell 13, 461-473.
PRICER, W. E. & ASHWELL, G. (1976). Subcellular distribution of a mammalian hepatic bindingprotein specific for asialoglycoproteins. J. biol. Chem. 251, 7539-7544.
PRIVES, J. M. & OLDEN, K. (1980). Carbohydrate requirements for expression and stability ofacetylcholine receptor on the surface of embryonic muscle cells in culture. Proc. natn. Acad. Sci.U.SA. 77, 5263-5267.
RINDERKNECHT, H. (1962). Ultra-rapid fluorescent labelling of proteins. Nature, Land. 193,167-168.
ROBERTS, R. M., BAUMBACH, G. A., BUHI, W. C , DENNY, J. B., FITZGERALD, L. A., BABELYN,
S. F. & HORST, M. N. (1984). Analysis of membrane polypeptides by two-dimensionalpolyacrylamide gel electrophoresis. In Receptor Biochemistry and Methodology, vol. I l l (ed. J.C. Venter & L. Harrison). New York: Alan R. Liss (in press).
ROBERTS, R. M. & YUAN, B. O.-C. (1974). Chemical modification of the plasma membrane poly-peptides of cultured mammalian cells as an aid to studying turnover. Biochemistry 13, 4846—4855.
SEGAL, H. I., WINKLER, J. R. & MIYAGI, M. P. (1974). Relationship between degradation rates ofproteins in vivo and their susceptibility to lysosomal proteases. .7. biol. Chem. 249, 6364—6365.
SCHULMAN, M., WILDER, C. D. & KOHLER, G. (1978). A better cell line for making hybridomassecreting specific antibodies. Nature, Land. 276, 269-270.
STANLEY, P. (1980). Surface carbohydrate alterations of mutant mammalian cells selected forresistance to plant lectins. In The Biochemistry of Glycoproteins and Proteoglycans (ed. W. J.Lennarz), pp. 161-189. New York: Plenum Press.
STANLEY, P., SUDO, T. & CARVER, J. P. (1980). Differential involvement of cell surface sialic acidresidues in wheat germ agglutinin binding to parental and wheat germ agglutinin-resistantChinese Hamster Ovary cells. J . Cell Biol. 85, 60-69.

Effects of altered oligosaccharide structure 23
TROWBRIDGE, I. S., HYMAN, R. & MAZOUSKAS, C. (1978). The synthesis and properties of T25glycoprotein in Thy-1-negative mutant lymphoma cells. Cell 14, 21-32.
(Received 4 October 1983-Accepted 2 November 1983)






























