Embed Size (px)
Citation preview

EFFECT OF POLYMERIC RELEASE MODULATORS ON DRUG
RELEASE FROM SOLID LIPID MICROPARTICLES
By
Asma Abdel-Raouf Khaled Fakhoury
Supervisor
Dr. Hatim Alkhatib
Co-supervisor
Dr. Mohammad Khalil
This thesis was submitted in Partial Fulfillment of the Requirements for
the Master Degree in Pharmaceutical Sciences
Faculty of Graduate Studies
The University of Jordan
May, 2010

ii
This Thesis/Dissertation (Effect of Polymeric Release Modulators on
Drug Release from Solid Lipid Microparticles) was successfully
defended and approved on……………..
Examination Committee Signature
Dr. Hatim Alkhatib, Supervisor
Assistant Professor of Pharmaceutics --------------------
Dr. Mohammad Khalil, Co-supervisor
Assistant Professor of Pharmaceutics --------------------
Dr. Bassam Amro, Member
Associate Professor of Pharmaceutics --------------------
Dr. Basher Alkhaldi, Member
Assistant Professor of Pharmaceutical Technology --------------------
Dr Ayman Khdair, Member
Assistant Professor of Pharmaceutics --------------------
Applied Science University

iii
Dedication
To my mother and my father who stand with me all the
time and give me all the care and love until the end
You have been the wind beneath my wings until I
completed this work.

iv
Acknowledgment
I am heartily thankful to my supervisor, Dr. Hatim Al-khatib for his
encouragement, guidance and support throughout the course of this
thesis
I am also thankful to Dr. mohammad khalil for his support.
Special thanks for Shorouq for her support and patience.
I offer my regards and blessings to all of those who supported me in any
respect during the completion of the thesis.

v
List of Contents
Subject Page
Committee Decision…………………………………………………………..
Dedication……………………………………………………………………..
Acknowledgment………...……………………………………………………
List of contents…………………………………………………………..……
List of Tables…………………………………………………………...……...
List of Figures………………………………………………………..………..
List of Abbreviations………………………………………………..………...
Abstract……………………………………………………………..…………
Introduction………………………………………………………..………….
1. Epilepsy……………………………………………………………………...
1.1 carbamazepine……………………………………………………………..
2. Solid lipid microparticles……………………………………………………
2.1 Preparation Techniques of SLM ……………………………………….….
2.2 Characterization of Solid Lipid Microparticles…………………………….
2.2.1 Particle Size Analysis……………………………………………………..
2.2.2Particle Morphology………………………………………….……………
2.2.3 Solid State Analysis of Solid Lipid Microparticles ……………………...
2.2.4 Drug loading and entrapment efficiency ………………………………...
2.2.5 Dissolution Test………………………………………………………….
2.2.6 Rheological behavior of the suspension…………………………………
3- Related studies ………………………………….…………………………...
4- Objective, hypothesis and specific aims of the study.…..…………………..
4.1 Objective ………………………………………………………………….
4.2 Hypothesis ………………………………………………………………...
ii
iii
vi
v
viii
ix
xi
xii
1
1
2
5
8
11
11
13
13
15
16
17
18
20
20
20

vi
4.3 Specific aims ………………………………………………..…………….
Experimental Part…………………………….……………..………………...
1. Materials and Suppliers:………………………………..…………………...
2. Equipments…………………………………………………………………..
3. Methods………………………………………………………………………
3.1 Preparation of carbamazepine- loaded solid lipid microparticles…………..
3.2 Characterization of carbamazepine-loaded SLMs………………………….
3.2.1 Determination of drug content……………………………………………
3.2.2 Thermal analysis…………………………………………………………
3.2.3 In-vitro drug release (dissolution)….…………………………………….
3.2.4 Stability ………………………………………………………..………...
3.3 Preparation of suspension ………………………………………….………
3.4 Characterization of suspension…………………………………………….
3.4.1 Separation fraction Experimental Part…………..……………….………
3.4.2 Rheological behavior ……………………………………………………
3.4.3 In-vitro drug release (dissolution)………………………………..………
3.4.4 Stability …………………………………………………………...……..
3.6 Experimental design……………………………………………………….
3.6.2Effect of initial drug amount on the properties of carbamazepine-loaded
SLMs …………………………………………………………………………..
3.6.2 Effect of release modulator type on the properties of carbamazepine-loaded
SLMs ………………………………………………………………….
3.6.2 Effect of release modulator amount on the properties of carbamazepine-
loaded SLMs …………………….…………………………………………….
3.6.3 Effect of method of addition of PVP as a release modulator……………
Results and discussion………………………………………………………
1. Characterization of carbamazepine-loaded SLMs…………………………
1.1 total drug loading………………………………….………………………
20
21
21
24
25
25
26
26
26
26
27
27
28
28
28
28
29
29
29
30
31
32
33
33
33

vii
1.2 thermal analysis…………………..………….…………………………….
1.3 in vitro release……………………………………………………………..
1.3.1 Effect of initial drug amount on the in-vitro drug release of carbamazepine-
loaded SLMs…………………………………………………………………..
1.3.2 Effect of release modulator type used on the in-vitro drug release of
carbamazepine-loaded SLMs………………………………………………….
1.3.3 Effect of release modulator amount on the in-vitro drug release of
carbamazepine-loaded SLMs …………..…………………………………..…
1.3.4 Effect of method of addition of PVP on the in-vitro drug release of
carbamazepine-loaded SLMs …………….……………………………………
1.4 Stability Study …………….………………………………………………
2. Characterization of suspension prepared from carbamazepine loaded SLMs
2.1 Rheological behavior………………………………………………………..
2.2 Separation Fraction………………………………………………………….
2.3 in vitro drug release…………………………………………………………
2.4 stability study………………………………………………………………..
3- Mechanism of carbamazepine release………………………………………..
Conclusion………………………………………………………………………
References………………………………………………………………………
Appendices……………………………………………………………………...
Abstract in Arabic……………………………………..……………………….
34
37
38
40
45
50
51
55
55
56
57
59
61
64
65
69
76

viii
List of Tables
Number Title Page
1 Formulas prepared to investigate the effect of initial drug amount on
carbamazepine-loaded SLMs. 29
2 Formulas prepared to investigate the effect of type of release modulator
used on carbamazepine-loaded SLMs. 30
3 Formulas prepared to investigate the effect of release modulator amount
on carbamazepine-loaded SLMs. 31
4 List of formulas prepared with their total drug content. 33
5 F2 test value of average % release of prepared formulas compared to no
surfactant formula. 43
6 List of values of F2 test using different amounts of PVP 48
7 Parameter values for model fitting to the release data of the investigated
formulas. 62
8 Parameter values for model fitting to the release data of the prepared
SLMs before and after storage in ambient conditions for 2 weeks. 63
9 Parameter values for model fitting to the release data of the prepared
SLMs suspensions before and after storage in ambient conditions for 1
month.
63
ix
List of Figures
Number Title Page
1 Molecular structure of carbamazepine 2
2 DSC thermograms of bulk compritol and carbamazepine and the
thermogram of formula used no release modulator
35
3 DSC thermograms of the release modulator used to prepare SLMs 35
4 Effect of initial drug amount on the in-vitro drug release of carbamazepine-loaded SLMs using PVA-L and Pluronic-F 127 as release modulators
38
5 DSC thermograms of formulas containing different initial drug
amount with pluronic-F 127 as a release modulator and PL-
127/PVA formula
39
6 DSC thermograms of formulas containing different initial drug
amount with PVA as a release modulator and different PVA-L
content.
40
7 Thermogram of PVP-carbamazepine mixture after second heating. 43
8 Effect of release modulator type used on the in-vitro drug release of
carbamazepine-loaded SLMs.
44
9 Effect of GMS amount on the in-vitro drug release of
carbamazepine-loaded SLMs.
46
10 DSC thermograms of formulas containing different GMS content 46
11 Effect of PVP amount on the in-vitro drug release of
carbamazepine-loaded SLMs.
47
12 DSC thermograms of formulas containing different PVP amount
and PVP-2/water formula.
48
13 Effect of PVA-L amount on the in-vitro drug release of
carbamazepine-loaded SLMs.
49
14 Effect of method of addition of PVP on the in-vitro drug release of
carbamazepine-loaded SLMs.
50
15 Effect of storage conditions (40°C and 75% RH) on the release
profile of CBZ-loaded SLMs containing GMS as a release
modulator.
53
16 Effect of storage conditions (40°C and 75% RH) on the
release profile of CBZ-loaded SLMs containing PVP as a
release modulator
54

x
17 Rheogram for the suspension prepared from SLMs using PVP
and GMS as a release modulator
55
18 Separation fraction of the suspensions prepared from SLMs
containing PVP or GMS as a release modulator over one week.
57
19 Release profile of CBZ-loaded SLMs as suspension and powder
using PVP as release modulator compared to the immediate release
of Tegretol suspension.
58
20 Release profile of CBZ-loaded SLMs as suspension and powder
using GMS as release modulator compared to the immediate release
of Tegretol suspension.
58
21 Release profile of PVP-2 suspension after 1 month storage in
ambient conditions compared to its dissolution after immediate
preparation.
59
22 Release profile of GMS-2 suspension after 1 month storage in
ambient conditions compared to its dissolution after immediate
preparation.
60

xi
List Of Abbreviations
1 SLMs Solid lipid microparticles
2 O/W Oil in water
3 B.P British pharmacopoeia
4 HCl Hydrochloric acid
5 KCl Potassium chloride
6 HLB Hydrophilic lipophilic balance
7 SLS Sodium lauryl sulfate
8 CMC Carboxymethyl cellulose
9 µm micrometer
10 mL milliliter
11 DSC Differential scanning calorimetry
12 RH Relative humidity
13 F2 Similarity factor
14 Pl-127 Pluronic F 127
15 PVP polyvinyl pyrrolidone
16 GMS Glyceryl monostearate
17 PVA-l Polyvinyl alcohol with low molecular weight
18 PVA-M Polyvinyl alcohol with medium molecular weight
19 CBZ carbamazepine
20 SEM Scanning electron microscopy
21 FT-IR Fourier Transform Infrared
22 HSM Hot stage microscopy
23 XRD x- ray diffraction
24 LD Laser diffractometry
25 W/O/W Water in oil in water
26 AEDs Antiepileptic drugs
27 PHT phynetoin
xii
28 VPA valproate
29 t.i.d Three timed daily
30 q.i.d Four times daily
31 CMS Solid lipid nanoparticles microparticles
32 PEITC Phenyl isothiocyanate

xiii
EFFECT OF POLYMERIC RELEASE MODULATORS ON
DRUG RELEASE FROM SOLID LIPID MICROPARTICLES
By
Asma abdel-raouf khaled fakhoury
Supervisor
Dr. Hatim Alkhatib
Co-supervisor
Dr. mohammad khalil
Abstract
Melt emulsification technique was used to prepare different carbamazepine-loaded
Compritol solid lipid microparticles (SLMs) formulations using different release modulators in an
attempt to study the effect of their incorporation on the physical and controlled release properties
of these SLMs. The effect of increasing drug or release modulator amount and the effect of
method addition of release modulator were also evaluated.
Suspensions prepared from selected formulas were evaluated in terms of rheology,
separation fraction and in-vitro drug release. SLMs and their corresponding suspensions were
subjected to accelerated stability conditions and evaluated in term of in-vitro drug release.
Formulas used pluronic-F 127, glyceryl monostearate and polyvinyl pyrrolidone showed
an enhanced in vitro drug release and stability.
Optimum formula of those used polyvinyl pyrrolidone was the one that contains 2 grams,
further increase in polyvinyl pyrrolidone amount decreased in vitro release of carbamazepine
Increasing glyceryl monostearate amount was with no effect on the release rate of
carbamazepine.
Both suspensions of PVP and GMS stabilized formulas showed increase in release
profile compared to their corresponding powder. This increase was significant with formula used
PVP as a release modulator.
Stability studies showed that both formulas containing PVP or GMS as stabilizer were
stable over two weeks and one month, GMS suspension showed instability after one month
storage in ambient condition, while PVP suspension was stable under the same conditions.
In conclusion, solid lipid microparticles containing polyvinyl pyrrolidone as a release
modulator give promising results to be used as a sustained release suspension with appreciable in
vitro drug release and good stability.

1
Introduction
1-Epilepsy:
Epilepsy is a biomedical disturbance that results in abnormal episodic bursts of electrical
activity in certain neurons. These bursts of electrical activity may spread to the entire brain. And
such abnormal neuronal activity may have significant impact on the normal cognitive processes
and behavior of the affected individuals. (Motamedi and Meador, 2003) The recurrent
unprovoked seizures that characterize epilepsy can be successfully treated and controlled in most
patients with mono or polytherapy. (Zeng et al, 2009)
Epilepsy is considered as one of the most common neurological disorders, affecting about
50 million people worldwide. (Shehata et al, 2009; Zeng et al, 2009) Antiepileptic drugs (AEDs)
are commonly prescribed to control epileptic seizures. Traditional AEDs, carbamazepine (CBZ),
phenytoin (PHT) and valproate (VPA) are considered to be the first-line treatments for epilepsy
in Europe and the United States. (Shehata et al, 2009)
Carbamazepine and PHT are considered to be the first-line treatments for partial-onset
seizures (simple partial, complex partial and secondarily generalized tonic–clonic seizures).
(Backman et al, 1987; Zeng et al, 2009)
Carbamazepine was introduced in the early sixties, and has become the most frequently
prescribed drug for the treatment of several forms of epilepsy. It is also used in the treatment of
neuropathic pain and in psychiatric disorders. (Ambrósio et al, 2001)
Carbamazepine acts by inhibiting sodium channel activity, and this may be the main
mechanism of its anticonvulsant effect. It also antagonizes the A1 adenosine receptor, increases
dopaminergic transmission and potentiated voltage-gated potassium channels, CBZ increases
extracellular serotonin concentration, interact with peripheral-type benzodiazepine receptors, and
2
decreases basal and stimulated level of cAMP. Those effects may also be of importance for the
anticonvulsant action of CBZ (Ambrósio et al, 2001)
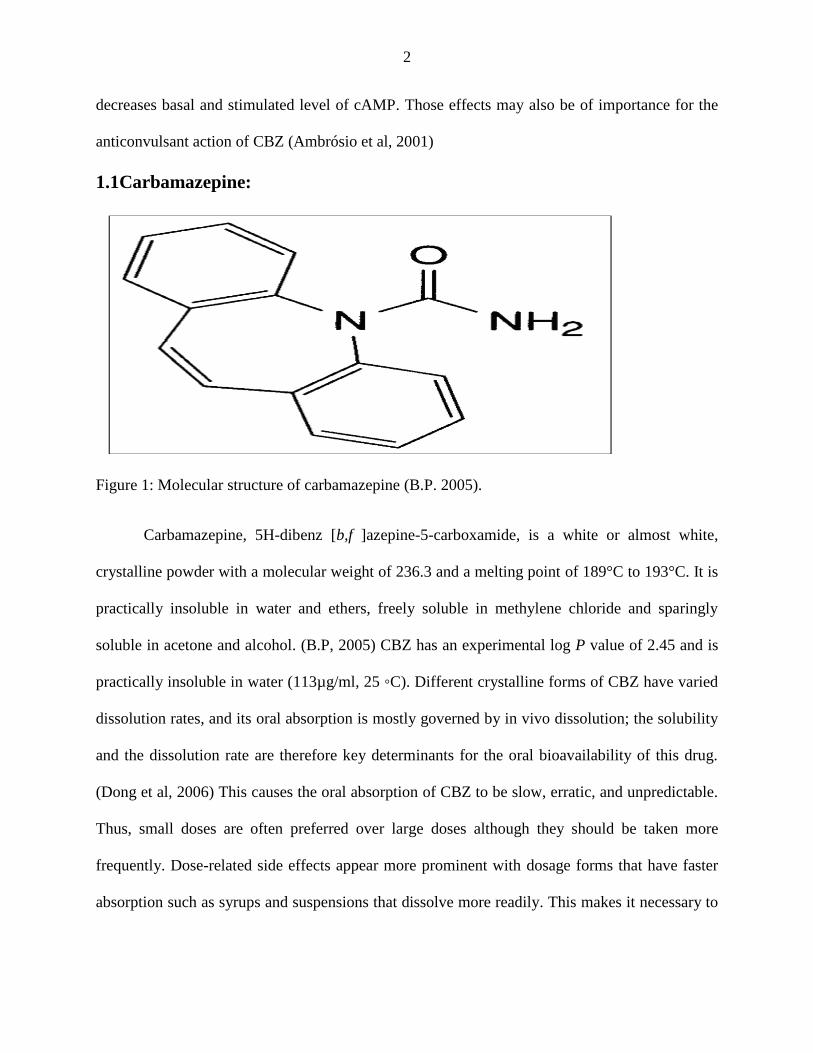
1.1Carbamazepine:
Figure 1: Molecular structure of carbamazepine (B.P. 2005).
Carbamazepine, 5H-dibenz [b,f ]azepine-5-carboxamide, is a white or almost white,
crystalline powder with a molecular weight of 236.3 and a melting point of 189°C to 193°C. It is
practically insoluble in water and ethers, freely soluble in methylene chloride and sparingly
soluble in acetone and alcohol. (B.P, 2005) CBZ has an experimental log P value of 2.45 and is
practically insoluble in water (113µg/ml, 25 ◦C). Different crystalline forms of CBZ have varied
dissolution rates, and its oral absorption is mostly governed by in vivo dissolution; the solubility
and the dissolution rate are therefore key determinants for the oral bioavailability of this drug.
(Dong et al, 2006) This causes the oral absorption of CBZ to be slow, erratic, and unpredictable.
Thus, small doses are often preferred over large doses although they should be taken more
frequently. Dose-related side effects appear more prominent with dosage forms that have faster
absorption such as syrups and suspensions that dissolve more readily. This makes it necessary to

3
administer the syrup and suspension formulations in smaller, more frequent doses in order to
reduce these adverse effects. (Winkler et al, 1996)
Carbamazepine is primarily metabolized in the liver. The major metabolite is
carbamazepine 10,11- epoxide, which is pharmacologically active and contributes to both the
efficacy and toxicity observed with carbamazepine therapy. (Altafullah et al, 1988; Winkler et al,
1996) CBZ also induces the enzymes responsible for its own metabolism (autoinduction), as well
as the liver metabolism of other drugs (heteroinduction). Autoinduction begins after 3–5 days of
starting therapy and takes 3–4 weeks to complete, which increases drug clearance over time.
Thus, chronic therapy of CBZ results in lower concentrations than that during the first days of
therapy. (Winkler et al, 1996)
Patients taking CBZ often require dosing 2–4 times per day due to the short terminal half-
life of CBZ after the autoinduction process. In cases of polytherapy, heteroinduction caused by
other agents may further shorten the circulating half-life and increase daily dosing requirements.
(Winkler et al, 1996)
The pediatric suspension formulations of CBZ is recommended in the treatment of pediatric
epilepsy to be given for 6-12 years of age at an initial dose of 200 mg/day divided in fuor doses
followed by a dose increase at weekly intervals by adding up to 100 mg/day using t.i.d. or q.i.d.
regimen until the optimal response is obtained. For children under 6 years of age an initial
recommended dose of 10-20 mg/kg/day q.i.d. is recommended with a weekly escalation to
achieve optimal clinical response with a dose administered t.i.d. or q.i.d. (Jarrar and Buchhalter,
2003).
Frequent dosing of the pediatric suspension is a risk to the success of the management of
epilepsy in pediatric patients in particular. This risk is linked to the fact that the suspension form

4
of CBZ provides higher peak levels and lower trough levels than those obtained from the
conventional tablet for the same dosage regimen making those patients at higher risks for
therapeutic failures or concentration dependent adverse reactions. (Jarrar and Buchhalter, 2003).
These problems can be potentially overcome by developing a controlled release
suspension that reduces both dosing frequency and plasma concentration fluctuations.

5
2-Solid Lipid Microparticles:
Extended-release products are designed to prolong the absorption of drugs that suffers
from short half-lives, and thus allow longer dosing intervals and minimize fluctuations in serum
drug levels. Extended-release formulations have been shown to be particularly valuable for
carbamazepine, in the prolongation of the dosing interval. (Zhang et al, 2008)
During the last few decades, increasing attention has been paid to the extended release
formulations of various drugs. Extended release is usually accomplished using a membrane or
matrix formulations. Matrix type formulations are prepared from either swellable hydrophilic
polymers or non-swellable hydrophobic excipients, like waxes and fats. (Savolainen et al, 2002)
Solid dispersion techniques offer an interesting formulation approach for enhancing drug
solubility and controlling the release rate of the drug. Solid dispersion controlled-release
formulations offer the advantage of forming a matrix type system with the probability of
avoiding the risk of dose dumping. Both polymers and lipids have been used as matrix forming
materials. (Savolainen et al, 2003) However, constraints of solubility, potential interaction of the
drug with excipients, and physical stability Limitations have restricted the use of hydrophilic
polymers. (Jaeghere et al, 2001) Encapsulating the drug in solid lipid microparticles is a
common approach to control drug release. Drug release is controlled at a proper rate over
prolonged time, which improves its efficacy and reduces its side effect. In addition, the physical
stability and dissolution properties of drugs with short half-life and low solubility are improved
being embedded in micro-size drug carriers, which enhance their therapeutic effect. (Long et al,
2006)

6
Solid lipid microparticles (SLM) are microsized drug carriers made of solid lipids and of
particle size between 0.2 and 100 µm. (Saraf et al, 2005; Zhang et al, 2008). SLM are made up of
solid hydrophobic core in which the bioactive compound is dissolved or dispersed. (Esposito et
al, 2005; Jasper et al, 2005) SLMs also contain surfactants and water .SLM are relatively more
stable than liposomes at room temperature. (Jaspert et al, 2005; Saraf et al, 2005; Long et al,
2006) The solid matrix is made from solid lipid, such as fatty acid, glyceride, fatty alcohol and
solid wax. (Jaspert et al, 2005; Long et al, 2006; Saupe and Rades, 2006) Drugs encapsulated in
SLM are released mainly due to gradual degradation of the lipid by lipase found in the small
intestines in human body when taken by oral route. (Kukizaki, 2009)
Because of their large range of particle size, SLM can be administered by different routes
such as orally, subcutaneously, intramuscularly, or topically, and they have been used for the
controlled delivery of various types of drugs, such as vasodilator and antiplatelet drugs, anti-
inflammatory drugs, local anesthetics, antibiotics, and anticancer agents.In addition they have
been also investigated as carriers of vaccines and adjuvants. (Esposito et al, 2005)
Solid lipid microparticles offer many advantages compared to other sustained release structures;
including:
1- Low drug mobility, due to incorporating solid lipids instead of liquid oils.(Jaspert et al,
2005)
2- Higher bio-compatibility and biodegradability than polymeric microparticles because of
using lipids as a matrix which have lower toxicitis. (Erni et al, 2002; Sanna et al, 2004;
Esposito et al, 2005; Jaspert et al, 2005)

7
3- Production of SLMs is relatively easy and can be scaled up with low raw materials and
production cost.( Esposito et al, 2005; Jaspert et al, 2005; Saupe and Rades, 2006;
Dalpiaz et al, 2008)
4- Improved physical stability and dissolution properties, which enhance therapeutic
efficacy of the drug. (Savolainen et al; 2002; Esposito et al, 2005; Jaspert et al, 2005;
Saupe and Rades, 2006; Dalpiaz et al, 2008)
5- High entrapment efficiency of hydrophobic drugs and, hence, higher bioavailability. This
overcomes the limitations of previous approaches such as salt formation, solubilization
by cosolvents and particle size reduction.( Savolainen et al, 2002; Esposito et al, 2005;
Jaspert et al, 2005;)
6- Control and sustain drug release effectively.( Sanna et al, 2004; Jaspert et al, 2005; Zhang
et al, 2007)
7- Reduction of the risk of dose dumping. (Savolainen et al, 2003)
Although SLM have those attractive properties, they have some disadvantages, for
example, drugs that are preferable to be incorporated are the lipophilic ones to ensure high
encapsulation efficiency (Trotta et al, 2004; Jasper et al, 2005), whereas hydrophilic drugs are
poorly incorporated in the lipid particles. In order to solve this problem, complex production
procedures are constructed or hydrophobic derivatives of the target drug are prepared prior to its
encapsulation. (Dalpiaz et al, 2008) In addition, in most cases, hydrophilic drugS show burst
release when incorporated in SLM. (Trotta et al, 2004) further, some SLMs were associated with
progressive loss of activity or the formation of aggregates. These problems were encountered at
higher incubation temperatures suggesting an instability mechanism related to the conditions
during in vitro dissolution testing. (Koennings et al, 2007) SLM may also suffer from expulsion

8
of core material during storage, which happens due to the polymorphic changes of many lipid
materials during the solidification and crystallization process with a reduction in the amorphous
regions of the carrier matrix (Chambi et al, 2007)
2.1 Preparation Techniques of SLM:
Several techniques can be used to prepare SLM, these techniques offer the drug to be
dissolved or dispersed into the solid matrix:
1- Solvent evaporation technique:
This approach is used with the aim of reducing the exposure to the high temperatures of
thermo-labile compounds, such as proteins and nucleic acids. (Jaspert et al, 2005) where the
lipid matrix is dissolved in an organic solvent and then emulsified with an external aqueous
phase containing the surfactant. The resulting O/W emulsion is stirred for several hours
under ambient conditions, allowing the solvent to evaporate. Another way to obtain the
SLMs is to pour the emulsion in ice bath with stirring until SLMs are produced. By either
ways, SLMs are then collected by filtration, (Erni et al, 2002; Trotta et al, 2004; Jasper et al,
2005) or the drug lipid mixture that is dissolved in an organic solvent can be directly sprayed
to get SLMs. (Jaspert et al, 2005)
In this method, if we use water immiscible solvent we will get high encapsulation
efficiency for hydrophilic drugs, but more hydrophilic solvents will cause the encapsulation
efficiency to be reduced. (Trotta et al, 2004)
2- Melt dispersion technique(melt emulsification technique):
In this method the lipid and the lipophilic drug are melted at a degree above the melting point of
the lipid and then emulsified with an external previously heated aqueous phase to a temperature above
the melting point of the lipid containing a suitable surfactant. (Sanna et al, 2004; Trotta et al, 2004;
Jasper et al, 2005; Chunxia et al, 2006; Zhang et al, 2007)

9
Homogenization is carried out at a temperature above the melting point of the lipid and
SLMs are formed by the subsequent cooling of the emulsion to room temperature or below.
(Trotta et al, 2004; Jasper et al, 2005)
While melt dispersion of miscible components results in amorphous solid solution formation,
melt dispersion of immiscible components leads to amorphous drug dispersed in crystalline
excipient. (Karanth et al, 2006)
3- Spray congealing method (spray chilling):
In this method the drug is dispersed or dissolved in the molten lipid. The molten mixture
is then atomized with a pneumatic nozzle into a vessel stored in CO2 ice bath. SLMs are then
dried at room temperature in vacuum. (Savolainen et al, 2002; Jaspert et al, 2005; Chambi et al,
2007)
The advantage of spray congealing is that no additional manufacturing step is needed to
pulverize the solid dispersion. (Savolainen, et al, 2002)
4- W/O melt dispersion technique:
This method is suitable for hydrophilic drugs, where no water is used to avoid excessive
drug solubility. (Jaspert et al, 2005)
In this method the drug and the surfactant are dispersed in the molten lipid. This melt is
then mixed with a hot non-aqueous continuous phase. And the mixture is rapidly cooled by
adding cold oil and immersion in ice bath. SLMs are obtained by centrifugation. (Jaspert et al,
2005)
5- W/O/W multiple emulsion technique:
The hydrophilic drug is dissolved in an aqueous phase and emulsified with the molten
lipid. A W/O/W emulsion is formed by adding the W/O emulsion to another aqueous phase with

10
stirring to get W/O/W emulsion. SLMs are obtained after cooling the emulsion at R.T or in ice
bath. (Jaspert et al, 2005)
6- High pressure homogenization:
The drug- molten lipid mixture is mixed with a hot aqueous surfactant solution using a
high shear device. The mixture is then homogenized with a previously heated high pressure
homogenizer and mixed once or several times, the mixture is then allowed to cool at room
temperature. (Jaspert et al, 2005)
7- Micro-channel emulsifying technique:
The prepared emulsion is forced into a continuous phase through silicon micro-channel
plate. SLM suspension is obtained after cooling at room temperature. (Jaspert et al, 2005;
Kukizaki, 2009)
The advantage of this method is that the resulting droplet size is controlled by the choice
of the membrane and not by the generation of turbulent droplet break-up. The technique is highly
attractive given its simplicity, potentially lower energy demands in comparison with
conventional methods, need for less surfactant, and resulting narrow droplet size distributions.
(Jaspert et al, 2005; D’oria et al, 2009; Kukizaki, 2009)
8- Cryogenic micronization:
The drug-molten lipid mixture is dissolved in a solvent and stored at -80ºC and then
micronized in a customized apparatus supplying liquid nitrogen during the process. The
apparatus contains an automatic sieving apparatus where the powder produced is then sieved.
(Jaspert et al, 2005)

11
2.2 Characterization of Solid Lipid Microparticles:
Colloidal dispersions of solid lipids are extremely complex systems. Several types of
particles may coexist in dispersions, and the solid state of the particles generally allows more
complicated processes to occur (e, g, polymorphism and variations in particle shape and size).
(Bunjes, 2005) SLMs microstructures, such as drug distribution in the lipid matrix, the crystal
structure of matrices, Particle size and morphology, surfactant distribution on particle surface,
and the structure of the surfactant layer, strongly affect the properties of SLMs, and thus the
characteristics of the dispersions have to be known in details. (Zhang et al, 2008).Most important
characteristics that should be scanned for SLM are listed below:
2.2.1 Particle Size Analysis:
Particle size is one of the most important characterization parameters for SLMs, Particle size
determinations are predominantly performed to ensure that the desired colloidal size range has
been obtained during preparation and that it is retained during storage or upon further processing.
(Bunjes, 2005)
Laser diffractometry (LD; also called laser light scattering) is probably a good choice for
getting an impression of the particle size distribution. Broad, narrow or monomodal distributions
can all be characterized by LD. (Bunjes, 2005, Jaspert et al, 2005) A laser diffractometer
determines particle size based on the principle that particles of a given size diffract light in a
specific angle.( Bunjes, 2005, Jaspert et al, 2005) Large particles scatter light predominantly in a
forward direction, whereas very small particles emit a more sphere-like “cloud” of scattered
light.( Bunjes, 2005) Analysis of the angular intensity distribution of the scattered light thus
gives information about the particle size. The advantage of using this technique is that it gives
an estimation of the particle size distribution and characteristic particle size values (e.g., mean,

12
mode, median diameter, and standard percentiles of the diameter of the distribution) in a
comparatively short time. (Bunjes, 2005)
As a drawback, the models assume the particles to be spherical, which lead to uncertainty
in the results when non-spherical lipid particles are under investigation. And using this technique
for the characterization of particles with nano or micrometer size ranges gives additional
uncertainties in the results. (Bunjes, 2005)
Scanning electron microscopy (SEM) and optical microscopy are also used for the
determination of particle size of SLMs; they are used also to determine particles morphology and
surface characteristics of the microparticles.(Bunjes, 2005, Jaspert et al, 2005) In SEM
microparticles are usually dried, and their surface is coated with a conductive layer, usually gold,
and the specimen is scanned point by point with an electron beam, and secondary electrons
emitted by the particles surface are detected. (Bunjes, 2005)
The disadvantage of these two techniques is that they examine only a small number of
particles, which makes them slow and tedious, since a sufficient amount of particles should be
examined (300-500) to give a good estimation of the particle size distribution of the sample.
(Jaspert et al, 2005)In addition, optical microscopy does not give estimation of the thickness of
the particles, only the length and the breadth. (Jaspert et al, 2005)
Two other methods can be used to determine the particle size distribution, electrical zone
sensing method and image analysis system. Electrical zone sensing method depends on
measuring electric resistance of the particles suspended in a conducting liquid when gets in a
small orifice on either sides of which are electrodes. The increase in resistance is proportional to
the particle size. (Jaspert et al, 2005)

13
Image analysis system can be used to determine particles shape and size in the range of
0.7-2000 µm. this method is increasingly used owing to its precision and sensitivity and being
carried in a real time or within few minutes although it still remains rather expensive. (Jaspert et
al, 2005)
2.2.2Particle Morphology:
Particle morphology is an important characteristic of SLMs. Particle shape may influence
drug loading and release characteristics of solid lipid particles and particles with a spherical
shape give the highest potential for protecting and controlling the release of the incorporated
drugs, as they provide minimum contact with the aqueous environment, and the longest diffusion
pathways. (Bunjes, 2005)
Morphology of SLMs can be determined using SEM, optical microscopy or image analysis
technology. (Bunjes, 2005, Jaspert et al, 2005)
2.2.3 Solid State Analysis of Solid Lipid Microparticles:
Monitoring of the crystalline status is an important point in the characterization of SLMs
in order to detect any possible modifications in the physicochemical properties of the
incorporated drug and the excipients. Although raw materials used in preparing SLMs are of
crystalline form, the preparation technique, lipid matrix composition, the stabilizer composition
or the presence of a drug can affect the solid state of the microparticles, and thus lead to
amorphous or partially crystallized metastable systems. (Jaspert et al, 2005; Zhang et al, 2008)
Polymorphic transformations may occur also during the preparation of dosage forms or upon
storage. (Jaspert et al, 2005) and this polymorphic transformations may cause changes in drugs

14
solubilities in the matrices and hence their entrapment efficiencies and release rates (Jaspert et al,
2005; Zhang et al, 2008)
Differential scanning Calorimetry (DSC) and X-ray diffraction (XRD) are the techniques
most widely used for the characterization of crystallinity and polymorphism of solid lipid
particles. (Bunjes, 2005; Jaspert et al, 2005) DSC can determine purity, stability and
polymorphism of a compound based on the principle that solid state modifications have different
melting points and melting enthalpies. Crystallization, melting and polymorphic transitions can
all be evaluated with regard to phase transition temperatures and transition enthalpy. (Jaspert et
al, 2005)
X-ray diffraction investigates the crystal structure based on the principle that X-rays are
diffracted by crystals with different angles and intensities (Jaspert et al, 2005)
Differential scanning Calorimetry is usually more sensitive in detecting crystalline
material, but XRD is much more reliable in determining the type of polymorphs in the
dispersions because it provides structural data. (Bunjes, 2005)Assignment of polymorphic forms
in DSC curves should be supported by x-ray data.
Hot Stage Microscopy HSM can also be combined with DSC to characterize the solid
state form of the drug in the solid dispersions. (Savolaien et al, 2003). This method combines the
properties of microscopy and thermal analysis, which gives it the advantage of distinguishing
between the excipients and the drug behavior. (Jaspert et al, 2005)
Fourier Transform Infrared (FT-IR) spectroscopy can also be used to detect the solid state
of the lipid matrices or solid dosage forms, and to detect interactions between the drug and the
bulk matrix that may decrease the crystallinity of the drug. (Savolaien et al, 2002; Savolaien et
al, 2003; Jaspert et al, 2005)

15
2.2.4 Drug loading and entrapment efficiency:
The determination of drug incorporation is an important characteristic to evaluate SLMs,
high entrapment efficiency of the drug is needed to decrease the amount of SLMs to be
administered. Several factors may affect drug incorporation. The physicochemical properties of
the drug and the matrix can affect drug incorporation in the SLMs; lipid which forms highly
crystalline particles with a perfect lattice lead to drug expulsion. On the other hand lattice defects
in the lipid structure could offer space to accommodate the drug. (El-kamel et al, 2007) the
preparation method, particles size, and initial drug loading can also affect drug incorporation in
SLMs. (Jaspert et al, 2005)
The most reliable term to express drug incorporation is the entrapment efficiency (EE).
EE can be determined using ultraviolet (UV) analysis or high performance liquid
chromatography (HPLC) analysis. Where it can be calculated by the following equation:
α = (Wthe/Wact) × 100
Where Wact is the actual amount of the drug encapsulated into the SLMs and Wthe is the
theoretical amount of the drug contained in the SLM. (Jaspert et al, 2005;Sukiyaki, 2009)
The lost or unentrapped drug could also be due to the solubility of the drug in the water
phase. (El-kamel et al, 2007)

16
2.2.5 Dissolution Test:
The aim of preparing a solid dispersion is to control the release rate and the dissolution
characteristics of our drug of interest. This give dissolution tests a prime importance in assessing
the success of the approach.
Drug release is expected to be affected by several parameters, physicochemical properties
of the drug, the preparation method of SLMs, particle size, drug content and EE all can affect the
release profile of the drugs (Jaspert et al, 2005)
Drug release profile can be assessed by an in vitro dissolution test that is usually carried
out according to USP or EP guidelines using a basket or paddle stirring apparatus.(Jaspert et al,
2005) basket stirring with dialysis technique is one of the most important techniques that can be
used to assess drug release from SLMs. (D’Sousa and Deluca, 2006) In this method, the
microparticles are separated from the bulk media by a dialysis tubing device, and the release is
assessed from the outer bulk media overtime and finally assayed spectrophotometrically. (Jasper
et al, 2005; D’Sousa and Deluca, 2006)
The suspension of the microparticles is introduced in the dialysis bag, and the bag is
sealed and placed in a vessel containing the bulk media. The vessel content is agitated to increase
drug diffusion from the dialysis bag.(zahirul and khan, 1996; D’Sousa and Deluca, 2006) The
volume of the media inside the dialysis membrane should be 6-10 folds less than the bulk media,
in order to provide a driving force for the drug transport to the outside and maintain sink
condition. (D’Sousa and Deluca, 2006)

17
Dialysis method is an attractive option to study the release properties from microparticles
and other dosage form owing to its ease of sampling and media replacement without the
interference of the microparticles. (D’Sousa and Deluca, 2006)
2.2.6 Rheological behavior of the suspension
Rheological methods help in determining the settling behavior of the suspension. Brook
field viscometer with variable shear stress control can be used to evaluate viscosity of the
suspension. (El-kamel et al, 2007) data obtained from this technique can help in determining
whether changes have taken place on SLM upon storing.

18
3- Related studies:
Some studies tried to incorporate polymers in microparticles and investigated their effect
on the characteristics and the release profile of the microparticles. Albrenti et al investigated the
effect of adding muco-adhesive polymers such as chitosan, CMC and poloxamers (Lutrol F68
and F127) on the characteristics of Econazole loaded SLM with a lipid-hydrophilic matrix
(Gelucire 53/10) using spray congealing technique. The particles were characterized for their
morphology, particle size, drug loading, muco-adhesive strength and solubility. The results
showed that poloxamers improved solubility of the drug and muco-adhesive strength with a high
yield and non-aggregated microparticles. (Albrenti et al, 2008)
Savolainen et al tried to prepare controlled release tablets for felodipine, a poorly water
soluble drug, using a hydrophilic polymers and polar lipids into SLM using spray congealing
method. The microparticles were characterized using IR and Raman spectroscopy, X-ray
diffraction, hot-stage microscopy, SEM, and image analysis. Results showed that incorporation
of hydrophilic polymers widened the particles size distribution and increased the amount of
agglomerates; results also showed that addition of polymers decreased the crystallinity of
Felodipine in the SLM.( Savolainen et al, 2003)
Dharmala et al tried to increase the available concentration of phenethyl isothiocyanate
(PEITC), a tumor suppressive agent, by developing chitosan- solid lipid nanoparticles
microparticles (CMS), and incorporating efflux transporter inhibitors, such as Tamoxifen,
Verapamil and Nifedipine. In this study, SLN were prepared using Stearic acid, PEITC and
Tween 80, and then SLN were dispersed in chitosan solution containing the efflux transporter
inhibitor to form CMS. Particle size, morphology, drug loading and in vitro drug release were all

19
examined. Cytotoxicity study was also performed for the CMS with and without efflux inhibitor.
(Dharmala et al, 2008)
Dong et all tried to prepare carbamazepine- loaded enteric microparticles using
coaceravation method. Instead of SLM, the study prepared polymeric microparticles, where an
aqueous polymeric stabilizer solution were added to an organic Carbamazepine/Eudragit L 100-
55 solution, which cause phase separation and formation of coacervate droplets. Further addition
of aqueous phase will harden the droplets into microparticles. Particle size distribution,
morphology, encapsulation efficiency, yield, physical state and physical stability of the drug,
wettability, in vitro release and in vivo bioavailability were all examined. Results showed that
microparticles posses a smooth surface and dense structure with high encapsulation efficiency
and yield. The drug was in a non-crystalline state in the matrix and physically stable for 5
months at room temperature. Under sink conditions, the drug dissolution rate from the
microparticles was significantly enhanced compared to the physical mixture and to the pure
drug; the release profile of the microparticles was stable after 5 months. These results
contributed to a significantly enhanced oral bioavailability from the microparticles when
compared to the physical mixture. (Dong et al, 2006)

20
4- Objective, hypothesis and specific aims of the study:
4.1 Objective:
The objective of this study is to investigate the effect of release modulator type and
quantity on the physical properties and the release profile of carbamazepine loaded solid lipid
microparticles, prepared by melt emulsification method, using compritol as a lipid carrier, and to
prepare suspension for selected formulas and evaluate them in term of physical and drug release
properties. The release modulators that are intended to be used are pluronic-F 127, polyvinyl
alcohol, polyvinyl pyrrolidone and glyceryl monostearate.
4.2 Hypothesis:
These studies were performed based on that Polymeric release modulators improve the
solubility of carbamazepine by many mechanisms; polymorphs, particle size reduction,
formation of solid solutions and solid dispersions, and formation of eutectic mixtures. Some
work also as surfactants that increase wettability and thus increase release rate. So, it is expected
that Incorporation of polymeric release modulators is expected to increase the release rate of
carbamazepine from SLMs.
4.3 Specific aims:
1- To prepare solid lipid microparticles using melt-emulsification method.
2- To establish a correlation between the levels of the hydrophobic drug
incorporated and the physical and drug release properties of SLMs.
3- To establish a correlation between the levels of the polymer incorporated and the
physical and drug release properties of SLMs.
4- To prepare suspensions from the promising formulas and to study their physical
and drug release properties.

21
Experimental Part
1. Materials and Suppliers:
- Carbamazepine was brought from Joswe (Jordan). It is white, or almost white, crystalline
powder that is very slightly soluble in water, sparingly soluble in acetone and alcohol, melting
point 189-193°C (B.P., 2005) used as antiepileptic agent.(B.P, 2005)
- Glyceryl behenate (Compritol® 888ATO) was obtained by Gattefosse (France). Mixture of
mono-, di- and tribehenate of glycerol (18%, 52% and 28% in weight, respectively) and presents
a drop point ranging from 69 °C to 74 °C and a HLB value of 2. used for controlled-release
applications by direct compression and more recently by: hot-melt coating, melt granulation or
pelletization or the formation of solid–lipid nanoparticles (Jannin, et al, 2003)
- Poloxamer 407 (Lutrol® F127, Pluronic F127) was purchased from BASF the chemical
company (Germany). Block copolymer of ethylene oxide (EO) and propylene oxide (PO) a
surfactant used in different industrial areas such as detergents, foaming, lubrication, dispersion,
stabilization, cosmetics, inks and is extensively used in the pharmaceutical field in the form of
gel, microemulsions, nanoparticles and solid polymer blends.(Boylan et al, 1983)
- Sodium lauryl sulfate (SLS) was obtained from COGNIS KIMYA A.S. White or cream-
colored to pale yellow crystals, flakes or powder having a smooth feel a soapy, bitter taste and a
faint odor of fatty substances. SLS is used as a wetting agent, tablet lubricant anionic emulsifier,
skin cleanser and detergent in medicated shampoos.(Boylan et al, 1983)
-Carboxy methyl cellulose sodium (CMC) was supplied by Purum Fluka (Switzerland). A white
to faintly yellow, odorless, hygroscopic powder or granular material having a faint paper-like

22
taste.CMC is soluble in water in all temperatures, practically insoluble in organic solvents.
Aqueous solutions of CMC exhibit a pseudoplastic flow behavior. CMC is used as emulsifying
agent, binding agent and gel forming agent. (Boylan et al, 1983)
- D-Sorbitol was obtained by Purum Fluka (Switzerland). It is white or almost colorless,
crystalline, odorless, hygroscopic powder, with a pleasant, cooling and sweet taste (50-60 % the
sweetness of sucrose).D-sorbitol is very soluble in water, soluble in warm alcohol and glycerin,
and slightly soluble in hydrocarbons. It is used as humectants, non sugar sweetener, viscosity
agent, vehicle for oral and topical liquids, and forth prevention of cap locking in syrups and
elixirs. (Boylan et al, 1983)
-Glyceryl monostearate (GMS) was obtained from Croda, UK. it is a white to cream-colored,
wax like solid beads, flakes or powder. It is waxy to the touch and has a slight fatty odor and
taste. It is practically insoluble in water, but readily dispersible in hot water with the aid of
anionic or cationic agent, soluble in hot alcohol, ether, chloroform, benzene, hot acetone, mineral
oils and fixed oils. It is used as a non-ionic emulsifier, stabilizer, emollient, plasticizer and anti-
tack agent.(Boylan et al, 1983)
-Poly vinyl alcohol (PVA-L) was obtained from Aldrich chemistry, USA. white to cream-colored
granular powder or granules. Odorless, essentially soluble in hot and cold water, partially soluble
in some polyhydroxy compounds, certain amines and amides. Has average molecular weight of
9,000-10,000 KDalton with low viscosity. Used as suspending and/or viscosity increasing
agent.(Boylan et al, 1983)
-Poly vinyl alcohol (PVA-M) was obtained from Aldrich chemistry, USA. white to cream-
colored granular powder or granules. Odorless, essentially soluble in hot and cold water, partially

23
soluble in some polyhydroxy compounds, certain amines and amides. Has average molecular
weight of 89,000-98,000 KDalton with medium viscosity. Used as suspending and/or viscosity
increasing agent.(Boylan et al, 1983)
-poly vinyl pyrrolidone K-30 was obtained by ICN biomedicals, Aurora. It is a white to creamy
white, odorless or almost odorless, hygroscopic powder, readily soluble in water up to 60 %,
freely soluble in many organic solvents, essentially insoluble in ether, hydrocarbons, carbon
tetrachloride, ethyl acetate and mineral oils. Used as carrier for drugs, dispersing agent,
suspending or viscosity builder, tablet binder, tablet diluents and coating agent.(Boylan et al,
1983)

24
2. Equipments:
- Digital temperature controller, PolyScience, USA.
- Pharma test PTWII, Dissolution tester, Germany.
- Unicam UV2-100 UV/Vis spectrometer, Unicam, England.
- Ultra-Turrax, Homogenizer, Janke and Kunkel Ika-Turrax, Germany.
- Telestar Cryodos, Freeze drier, Spain.
- True-Sweep , Sonicator, Crest Ultrasonic , Scotch road.Trenton.
- METTLER TOLEDO DSC 823, Differential Scanning Calorimeter, Mettler, STARe software,
Switzerland.
- BINDER KBF 240, Stability chamber, BINDER, USA.
- Physica MCR 301, Rheometer, Anton Paar, Graz, Austria.
- SHIMADZU, Electronic balance, Shimadzu, Japan.
- Nylon filters 0.45μm, PETRATECH, Jordan.
-Dialysis Tubing size 9, Diameter 28.6mm, MWCO 12000-14000 Dalton, Medicall International
Ltd, London.

25
3. Methods:
3.1 Preparation of carbamazepine- loaded solid lipid microparticles:
The microparticles were prepared by melt emulsification technique. Where accurately
weighed amounts of compritol and stabilizers were melted over a water bath at 100 ºc until a
clear melt is obtained or the release modulator was well dispersed in compritol melt.
Carbamazepine is then added to the melt and the drug-containing melt is kept at the same
temperature until the drug is well dispersed. 400 ml of distilled water, which was previously
heated to boiling, is then added to the melt while it is kept in water bath at 85 ºc (10 ºc above the
melting point of the lipid), and emulsified by homogenizer at high speed with decreasing the
temperature until it reached 50°c(time needed was 34 minutes). The obtained white creamy
emulsion was then cooled to 0 ºC. Cooling was accompanied with magnetic stirring. Melting and
cooling was performed in the same water bath which is attached to a temperature controller with
a specific cooling rate of approximately 2 ºc/min.
The same procedure was followed in preparing the microparticles with different release
modulators except poly vinyl alcohol, where it was dissolved in the hot aqueous phase before
pouring it to the melt.
The obtained suspension was then frozen in a deep freezer at –70 ºC over night and then
lyophilized in a freeze dryer (-70 to – 80 C°, 0.1-0.2 mBAR) for five days to ensure dryness of
the microparticles.

26
3.2 Characterization of carbamazepine-loaded SLMs:
3.2.1 Determination of drug content:
The drug content of the microparticles (of size fraction ≤ 63μm) was determined by dissolving an
accurately weighed sample of the microparticles equivalent to 10 mg of carbamazepine in 100 ml
absolute ethanol under sonication at room temperature for one hour. Quantification by UV
spectrophotometry using Unicam UV2-100 UV/Vis spectrometer at 285 nm was performed on
the solution after filtration with 0.45μm nylon filters. The drug content was carried out in
triplicate and the results were expressed as percentages of the theoretical drug content calculated
using the following equation:
% drug content = (Real drug content/Theoretical drug content) 100 %
3.2.2 Thermal analysis:
Differential scanning calorimetry (DSC) thermograms were performed using METTLER
TOLEDO DSC 823 with an empty pan as a reference. 5 mg samples of starting materials and
prepared formulas (using size fraction ≤ 63μm) were contained in aluminum pans while scanning
was done at 10 ºC min-1 under 80 ml min-1 nitrogen flux at temperature range of 25-250 ºC.
3.2.3 In-vitro drug release (dissolution):
Dissolution of carbamazepine from the microparticles was tested using the dialysis
method with 1% SLS as dissolution medium. An accurately weighed amounts of the
microparticles (of size fraction ≤ 63μm) equivalent to 25mg carbamazepine were placed inside a
basket covered by a dialysis membrane which was previously sealed from the bottom, and 5ml of
dissolution media was also added then the membrane was wrapped around the baskets of USP I

27
dissolution apparatus (Pharma test PTWII, Dissolution tester) and attached to the apparatus from
above.
The dissolution test was performed in 900ml of 1% SLS at 37 ºC at 100 r.p.m. samples of 5ml
were withdrawn at predetermined time intervals (0.5, 1, 1.5, 2, 4, 6, 8 and 12 hour). An equal
volume of fresh media, equilibrated at the same temperature, was added after each sampling to
maintain sink conditions.
Withdrawn samples were filtered through 0.45 nylon filters and analyzed
spectrophotometrically by Unicam UV2-100 UV/Vis spectrometer at 285nm and the results were
expressed as cumulative percentages of the dissolved drug. The values expressed the mean of
three independent experiments.
3.2.4 Stability:
Samples (of size fraction ≤ 63μm) from selected formulas were stored in a closed glass
bottle in BINDER KBF 240 Stability chamber at temperature of 40º and 75% relative humidity
(RH). Dissolution testing was performed for the samples stored after 2 weeks and one month of
incubation, as described in section 3.2.3.
3.3 Preparation of suspension:
A quantity of the microparticles (of size fraction ≤ 63μm) from selected formulas was
dispersed in HCl/KCl buffer solution pH 2, containing 1.5% CMC and 30% D-sorbitol to give a
final concentration of 100mg carbamazepine /5ml of the suspension.

28
3.4 Characterization of suspension:
3.4.1 Separation fraction:
Suspensions samples were placed in 10ml graduated cylinder at room temperature and
left to settle. The separation fraction, described as the ratio of the settled height (Vu) to the
original suspension height (Vo), was measured after 2, 4, 8, 24 hours, 2, 4, days and 1 week. The
measurements were done in triplicate.
3.4.2 Rheological behavior:
The rheological properties of the prepared suspensions were measured by Physica MCR
301 rheometer using spindle no. CC27, 20 ml samples of the suspensions were equilibrated at 25
ºC prior to each measurement and the results were obtained in the form of rheograms.
3.4.3 In-vitro drug release (dissolution):
Dissolution of carbamazepine from the suspensions was performed as described in
section 3.2.3, using volumes of the suspensions containing carbamazepine loaded SLMs
equivalent to 25mg carbamazepine.
Withdrawn samples were filtered through 0.45 nylon filters and analyzed
spectrophotometrically by Unicam UV2-100 UV/Vis spectrometer at 285nm and the results were
expressed as cumulative percentages of the dissolved drug. The values expressed the mean of
three independent experiments.

29
3.4.4 Stability:
The suspensions were stored in closed glass bottles in BINDER KBF 240 Stability
chamber at temperature of 40ºC and 75% RH. Dissolution testing was performed on the stored
samples of suspensions after1 month as described in section 3.2.3.
3.6 Experimental design:
3.6.2 Effect of initial drug amount on the properties of carbamazepine-loaded SLMs:
Microparticles were prepared using different contents of carbamazepine with fixed
amount of Compritol as lipid matrix and Pluronic F127 (PL-127) or poly vinyl alcohol with low
M.W (PVA-L) as release modulators. The prepared microparticles were characterized in terms
of, drug content, thermal analysis and in-vitro-drug release.
Quantity(gm)
Formula Carbamazepine Compritol (PL127) (PVA-L)
PL127-3 3 10 3 0
PL127-5 5 10 3 0
PVA -3-3 3 10 0 3
PVA -3-5 5 10 0 3
Table (1): formulas prepared to investigate the effect of initial drug amount on carbamazepine-
loaded SLMs
30
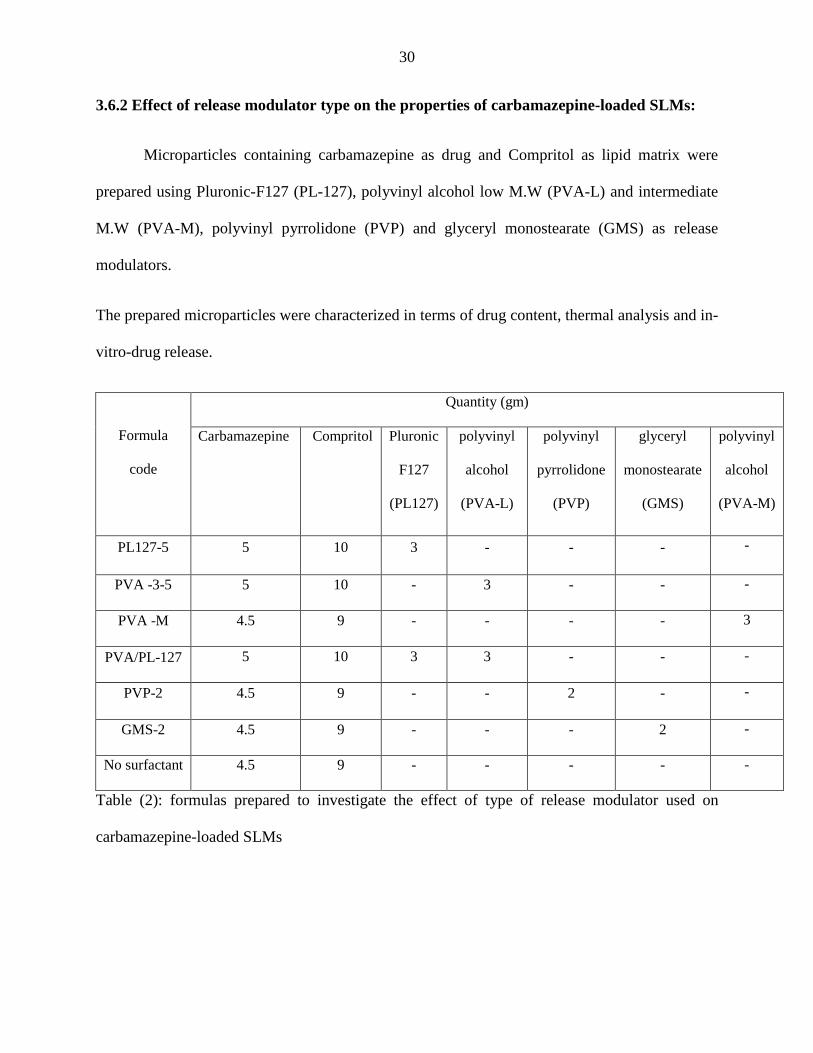
3.6.2 Effect of release modulator type on the properties of carbamazepine-loaded SLMs:
Microparticles containing carbamazepine as drug and Compritol as lipid matrix were
prepared using Pluronic-F127 (PL-127), polyvinyl alcohol low M.W (PVA-L) and intermediate
M.W (PVA-M), polyvinyl pyrrolidone (PVP) and glyceryl monostearate (GMS) as release
modulators.
The prepared microparticles were characterized in terms of drug content, thermal analysis and in-
vitro-drug release.
Formula
code
Quantity (gm)
Carbamazepine Compritol Pluronic
F127
(PL127)
polyvinyl
alcohol
(PVA-L)
polyvinyl
pyrrolidone
(PVP)
glyceryl
monostearate
(GMS)
polyvinyl
alcohol
(PVA-M)
PL127-5 5 10 3 - - - -
PVA -3-5 5 10 - 3 - - -
PVA -M 4.5 9 - - - - 3
PVA/PL-127 5 10 3 3 - - -
PVP-2 4.5 9 - - 2 - -
GMS-2 4.5 9 - - - 2 -
No surfactant 4.5 9 - - - - -
Table (2): formulas prepared to investigate the effect of type of release modulator used on
carbamazepine-loaded SLMs
31
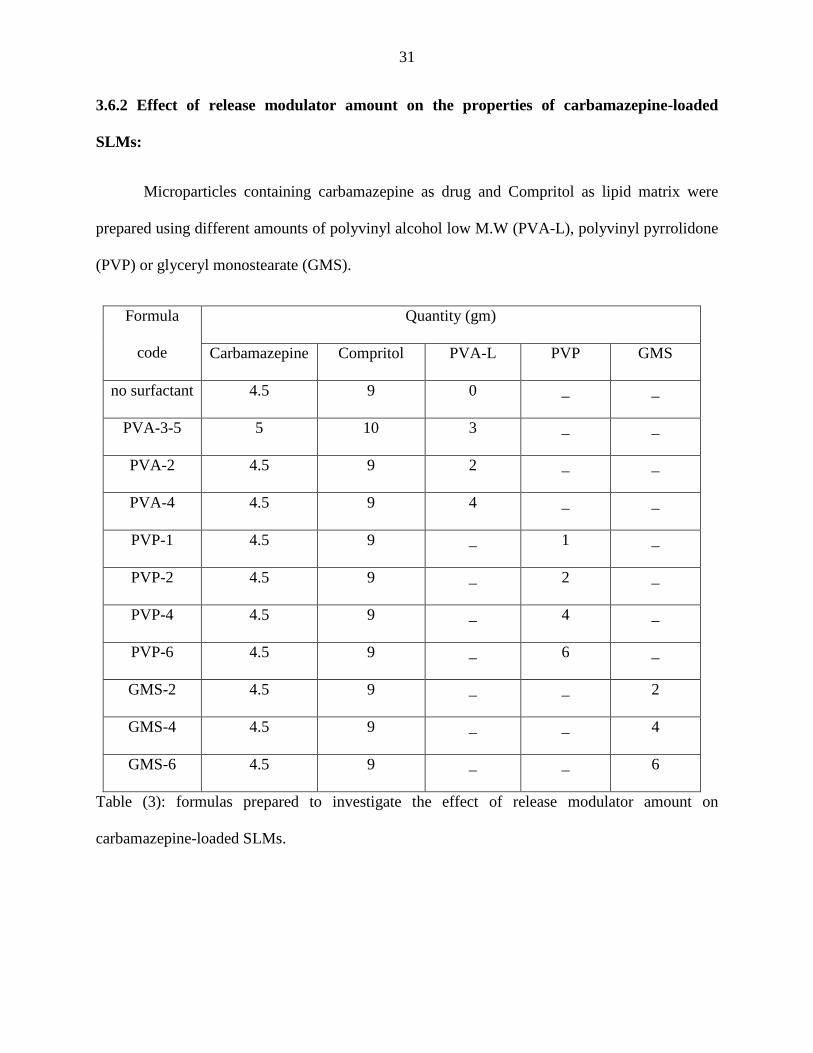
3.6.2 Effect of release modulator amount on the properties of carbamazepine-loaded
SLMs:
Microparticles containing carbamazepine as drug and Compritol as lipid matrix were
prepared using different amounts of polyvinyl alcohol low M.W (PVA-L), polyvinyl pyrrolidone
(PVP) or glyceryl monostearate (GMS).
Formula
code
Quantity (gm)
Carbamazepine Compritol PVA-L PVP GMS
no surfactant 4.5 9 0 _ _
PVA-3-5 5 10 3 _ _
PVA-2 4.5 9 2 _ _
PVA-4 4.5 9 4 _ _
PVP-1 4.5 9 _ 1 _
PVP-2 4.5 9 _ 2 _
PVP-4 4.5 9 _ 4 _
PVP-6 4.5 9 _ 6 _
GMS-2 4.5 9 _ _ 2
GMS-4 4.5 9 _ _ 4
GMS-6 4.5 9 _ _ 6
Table (3): formulas prepared to investigate the effect of release modulator amount on
carbamazepine-loaded SLMs.

32
3.6.3 Effect of method of addition of PVP as a release modulator:
Microparticles of formula code PVP-2 were prepared with the same composition and
method except that PVP was added once to the molten compritol and in the other formula PVP
was added to the aqueous phase.
The prepared microparticles were characterized in terms of drug content, thermal analysis
and in-vitro-drug release.
33
RESULTS AND DISCUSSION
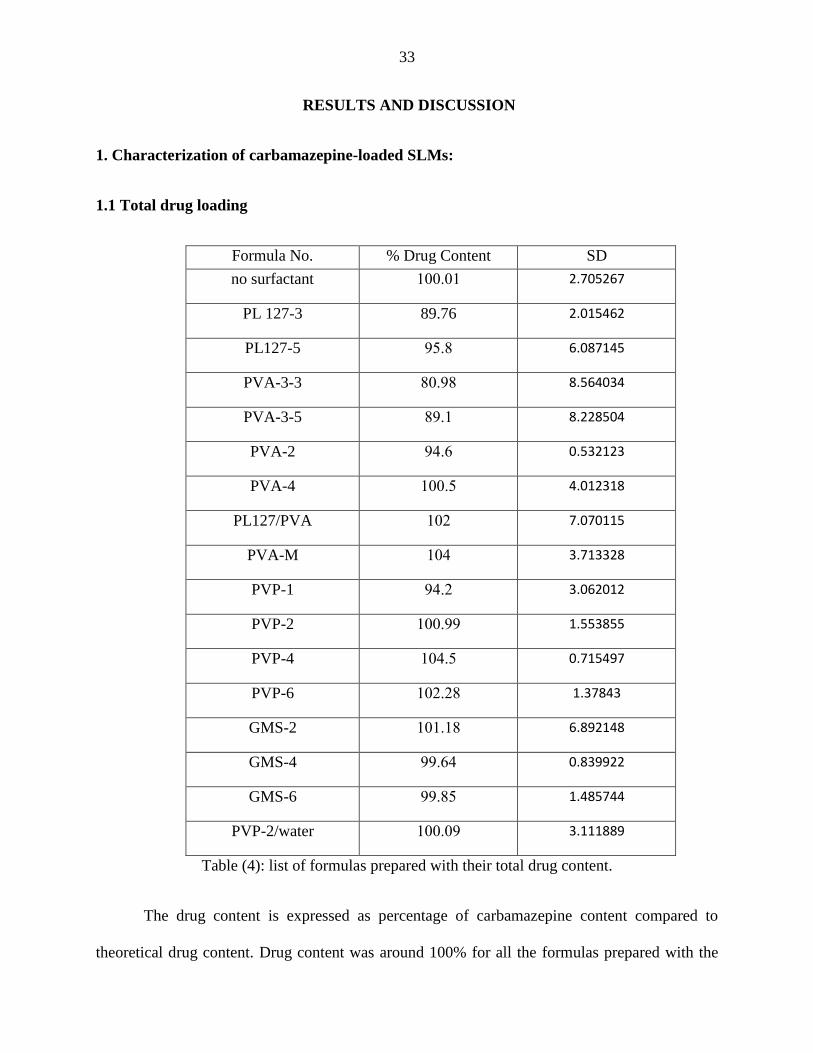
1. Characterization of carbamazepine-loaded SLMs:
1.1 Total drug loading
SD % Drug Content Formula No.
2.705267 100.01 no surfactant
2.015462 89.76 PL 127-3
6.087145 95.8 PL127-5
8.564034 80.98 PVA-3-3
8.228504 89.1 PVA-3-5
0.532123 94.6 PVA-2
4.012318 100.5 PVA-4
7.070115 102 PL127/PVA
3.713328 104 PVA-M
3.062012 94.2 PVP-1
1.553855 100.99 PVP-2
0.715497 104.5 PVP-4
1.37843 102.28 PVP-6
6.892148 101.18 GMS-2
0.839922 99.64 GMS-4
1.485744 99.85 GMS-6
3.111889 100.09 PVP-2/water
Table (4): list of formulas prepared with their total drug content.
The drug content is expressed as percentage of carbamazepine content compared to
theoretical drug content. Drug content was around 100% for all the formulas prepared with the

34
exception of PL127-5, PVA-3-3 and PVA-3-5 formulas. This may be due to loss of the material
during preparation, transfer or other procedures the emulsion was subjected to.
1.2 Thermal Analysis:
The release performance of SLMs depends highly on drug distribution in SLMs matrices. The
models of drug incorporation into SLMs are classified as homogeneous matrix, drug-enriched
shell model, and drug-enriched core models. The structure of the formed SLM depends mainly
on the chemical nature of drugs, excipients, and their interaction. The structure may be also
influenced by the production conditions. (Zhang et al, 2008)
The homogeneous matrix model refers to a drug dispersed molecularly or being presented in
amorphous clusters in the matrices of SLMs. It is considered as an ideal drug incorporation
model, and it is mainly noticed upon incorporating lipophilic drugs in SLMs with the hot
homogenization method. No phase separation between lipid and drug occurs during the
production process. (Zhang et al, 2008)
During the production of SLMs, the crystal structures are constructed to be perfect crystals or
less-ordered crystals. This depends mainly on the methods of production and the properties of
the excipients. In the perfect crystals, drug is expulsed from carrier matrices, causing drug-
entrapment efficiency to be low. Less-ordered structure is formed by the mixtures of spatially
different lipids acting as excipients, where the lipid matrix is solid but not crystalline and is kept
in an amorphous state. The DSC methods can be used to investigate the interaction between drug
and lipid matrix. (Zhang et al, 2008)
The thermal behavior of compritol, carbamazepine and the corresponding surfactants
used in preparing SLMs are illustrated in the figures below:
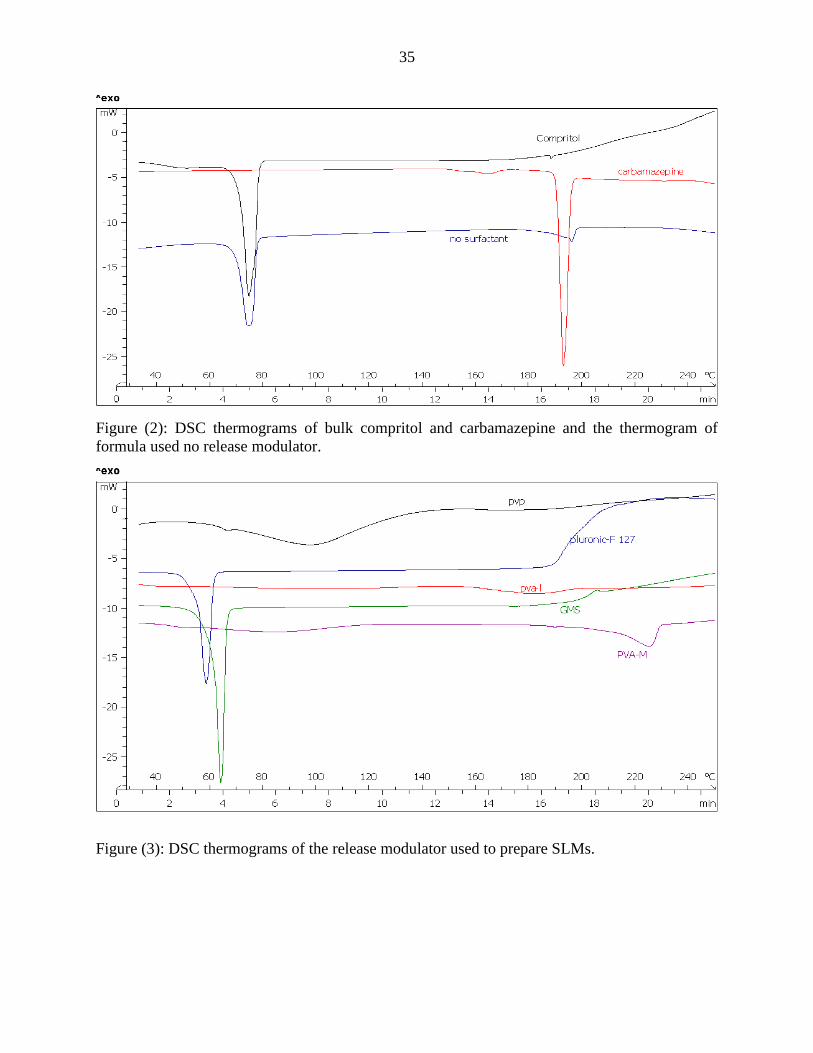
35
Figure (2): DSC thermograms of bulk compritol and carbamazepine and the thermogram of
formula used no release modulator.
Figure (3): DSC thermograms of the release modulator used to prepare SLMs.

36
Compritol showed an endothermic peak at 73.421 °C with normalized enthalpy of 115.855
mJ/mg. Carbamazepine showed a single endothermic peak at 191.692 °C with normalized
enthalpy of 103.84 mJ/mg. Lutrol-F 127 also showed an endothermic peak at 57.1653 °C with
normalized enthalpy of 107.13 mJ/mg. PVA-L showed no endothermic peaks, while PVA-M
showed an endothermic peak at 225.396 °C with a normalized enthalpy of 39.88 mJ/mg. PVP
showed also an endothermic peak at 92.476 °C with normalized enthalpy of 97.13 mJ/mg. GMS
showed an endothermic peak at 63.7152 °C with normalized enthalpy of 148.05 mJ/mg. each one
of those endothermic peaks corresponds to the melting of those materials.
DSC thermogram of no surfactant formula showed an endothermic peak at 195.147ºC
with a normalized enthalpy of 42.555 mJ/mg indicating that 40% of CBZ was found in
particulate form.
GMS and PVP as release modulators enhanced the solubility of CBZ in compritol
compared to formula prepared without any release modulator, PVA-M and PVA-L release
modulators did not enhance the solubility of CBZ in compritol as it showed similar endothermic
peaks with normalized enthalpy value of 40.73 mJ/mg, 70.912 mJ/mg, 50.17 mJ/mg and 58.33
mJ/mg in PVA-M, PVA-2, PVA-3-5 and PVA-4 respectively.
All formulas showed a peak of compritol with similar values of the normalized enthalpy in the
range of 115-136 mJ/mg and melting points between 69-74ºC.

37
1.3 In-vitro Drug Release:
Carbamazepine belongs to class II of the biopharmaceutical classification system.
Compounds in this category have high intestinal permeability and low water solubility.
Subsequently, the bioavailability of such compounds is limited by their solubility in water. (Nair
et al, 2002) Some of the main possibilities for improving dissolution are to increase the surface
area available for dissolution by decreasing the particle size of the solid compound and/or by
optimizing the wetting characteristics of the compound surface by using surfactants. (Leuner and
Dressman, 2000)
Many mechanisms can also be suggested to improve the solubility of carbamazepine
upon using surfactant; polymorphs, particle size reduction, formation of solid solutions and solid
dispersions, and formation of eutectic mixtures. (Leuner and Dressman, 2000) Consequently, it is
expected that addition of surfactants to CBZ-loaded SLMs will improve in vitro CBZ release.
The results of in vitro release test are shown below. The results were normalized
according to the results of drug content shown in section 1.1
38
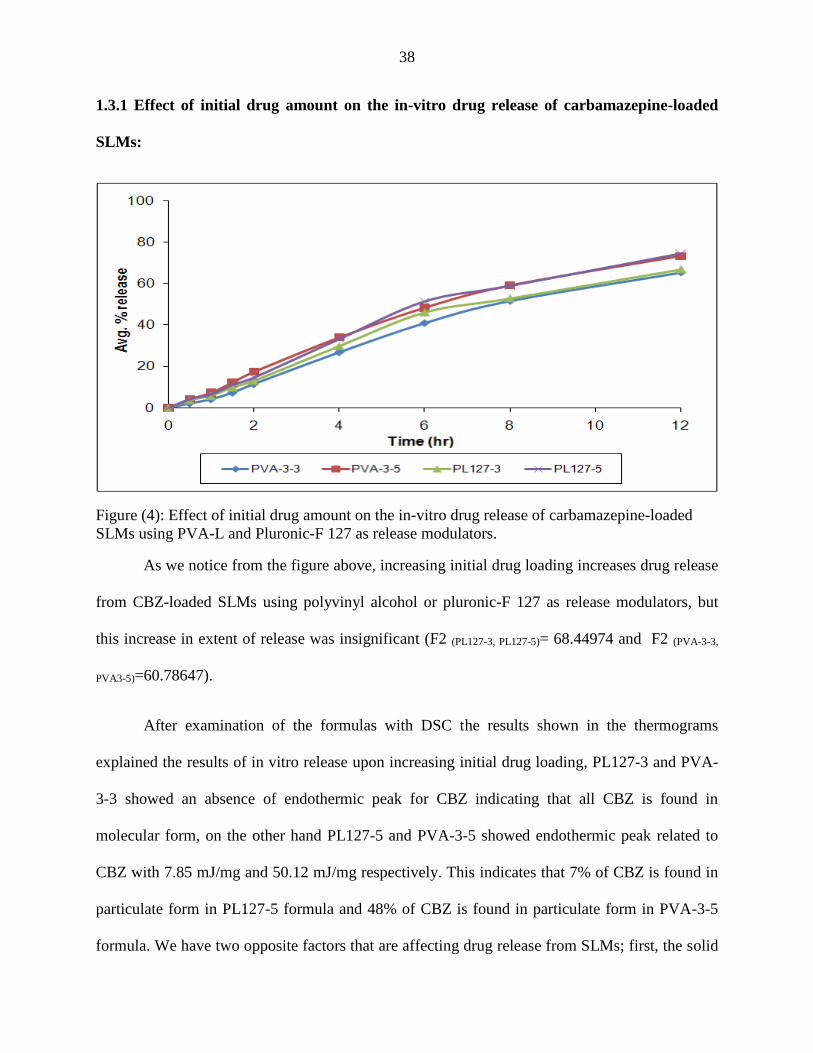
1.3.1 Effect of initial drug amount on the in-vitro drug release of carbamazepine-loaded
SLMs:
Figure (4): Effect of initial drug amount on the in-vitro drug release of carbamazepine-loaded
SLMs using PVA-L and Pluronic-F 127 as release modulators.
As we notice from the figure above, increasing initial drug loading increases drug release
from CBZ-loaded SLMs using polyvinyl alcohol or pluronic-F 127 as release modulators, but
this increase in extent of release was insignificant (F2 (PL127-3, PL127-5)= 68.44974 and F2 (PVA-3-3,
PVA3-5)=60.78647).
After examination of the formulas with DSC the results shown in the thermograms
explained the results of in vitro release upon increasing initial drug loading, PL127-3 and PVA-
3-3 showed an absence of endothermic peak for CBZ indicating that all CBZ is found in
molecular form, on the other hand PL127-5 and PVA-3-5 showed endothermic peak related to
CBZ with 7.85 mJ/mg and 50.12 mJ/mg respectively. This indicates that 7% of CBZ is found in
particulate form in PL127-5 formula and 48% of CBZ is found in particulate form in PVA-3-5
formula. We have two opposite factors that are affecting drug release from SLMs; first, the solid
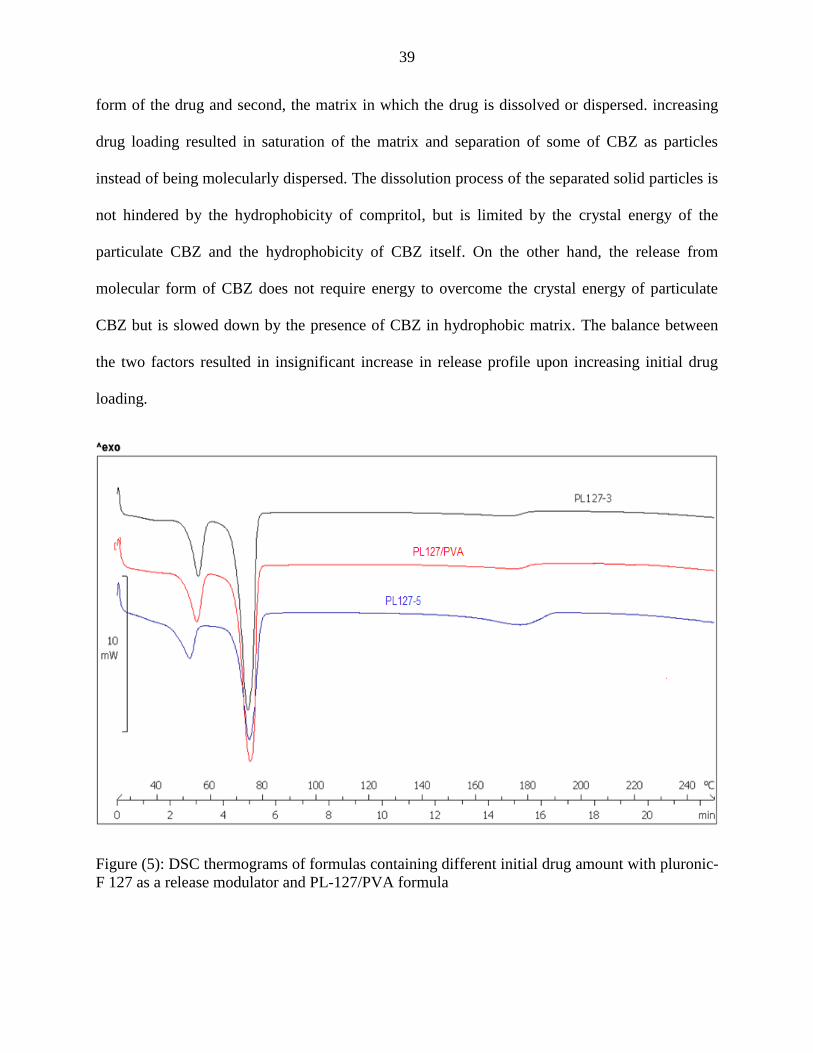
39
form of the drug and second, the matrix in which the drug is dissolved or dispersed. increasing
drug loading resulted in saturation of the matrix and separation of some of CBZ as particles
instead of being molecularly dispersed. The dissolution process of the separated solid particles is
not hindered by the hydrophobicity of compritol, but is limited by the crystal energy of the
particulate CBZ and the hydrophobicity of CBZ itself. On the other hand, the release from
molecular form of CBZ does not require energy to overcome the crystal energy of particulate
CBZ but is slowed down by the presence of CBZ in hydrophobic matrix. The balance between
the two factors resulted in insignificant increase in release profile upon increasing initial drug
loading.
Figure (5): DSC thermograms of formulas containing different initial drug amount with pluronic-
F 127 as a release modulator and PL-127/PVA formula
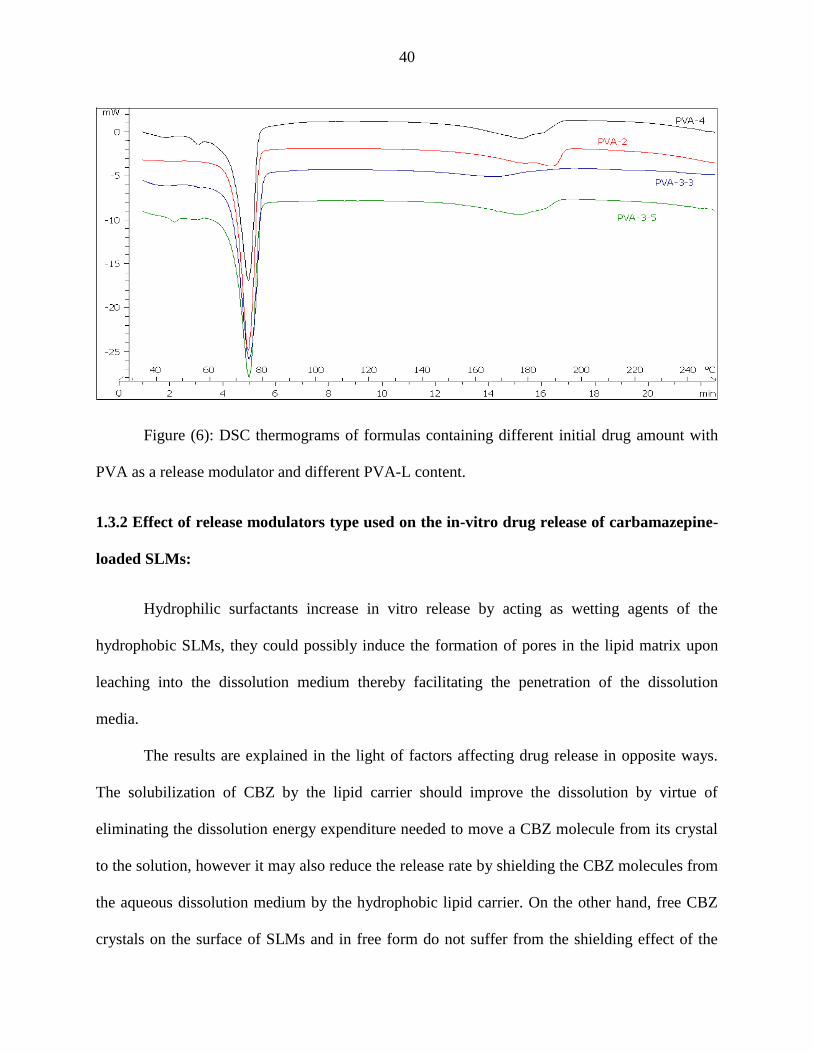
40
Figure (6): DSC thermograms of formulas containing different initial drug amount with
PVA as a release modulator and different PVA-L content.
1.3.2 Effect of release modulators type used on the in-vitro drug release of carbamazepine-
loaded SLMs:
Hydrophilic surfactants increase in vitro release by acting as wetting agents of the
hydrophobic SLMs, they could possibly induce the formation of pores in the lipid matrix upon
leaching into the dissolution medium thereby facilitating the penetration of the dissolution
media.
The results are explained in the light of factors affecting drug release in opposite ways.
The solubilization of CBZ by the lipid carrier should improve the dissolution by virtue of
eliminating the dissolution energy expenditure needed to move a CBZ molecule from its crystal
to the solution, however it may also reduce the release rate by shielding the CBZ molecules from
the aqueous dissolution medium by the hydrophobic lipid carrier. On the other hand, free CBZ
crystals on the surface of SLMs and in free form do not suffer from the shielding effect of the

41
carrier but their dissolution is not too fast because of the low intrinsic solubility of CBZ which is
attributed to both the crystal energy and hydrophobicity of CBZ.
The fastest drug release was observed with SLMs stabilized by Pluronic F127; this is an
expected result as this surfactant is characterized by a high HLP value of 21.5. In addition, the
heat of fusion of CBZ in these particles was 7.85 mJ/mg indicating that only 7% of the present
CBZ is available as particles. The high molecularity of CBZ loaded system in addition to the
high HLP value of the surfactant are probably the main reason for the fast release observed.
The second fastest release was observed in the case of SLMs prepared without a
stabilizer. This was a surprising result, however upon examining these SLMs using DSC, the
Heat of fusion of CBZ was found to be 42.55 mJ/mg meaning that around 41% of CBZ was in
particulate crystal form and was not incorporated molecularly in the SLMs. This has provided a
balance of factors that favored fast release from the system.
A similar enthalpy value near that of formula with no surfactant (40.734mJ/ mg) was
observed with the PVA-M stabilized formula. However the release from this formula was much
slower than that of the formula with no surfactants. This was not surprising considering the fact
that PVA separated from the Lipid-CBZ system as flakes upon freeze drying, these PVA
particles may have caused gelling of the system upon dissolution testing and slowed down the
release from the SLMs that are, otherwise, identical to the ones not stabilized by surfactants in
terms of the form of their CBZ content.
PVA-2 formula showed a similar peak with normalized enthalpy value of 70.91mJ/ mg
suggesting that 68% of CBZ is found as particles, and was not incorporated molecularly in the
SLMs. But this formula did not show flakes of PVA upon freeze drying. The absence of
separated PVA particles eliminates the gelling effect that may retard the release rate of CBZ
from SLMs, and this has provided a balance of factors that favored fast release from the system.

42
Another observation was that a mixture of Pluronic F127 and PVA as stabilizers
produced the slowest release although it produced a complete molecular solution of CBZ with a
heat of fusion value of zero. This is expected as a result of the imbalance of the factors described
above and the hindering of water penetration into the matrix system by the lipophilic carrier.
Another factor that will cause slow down in the release rate is possibility of gelling of the system
upon dissolution testing as a result of the flakes of PVA-L that were formed after freeze drying.
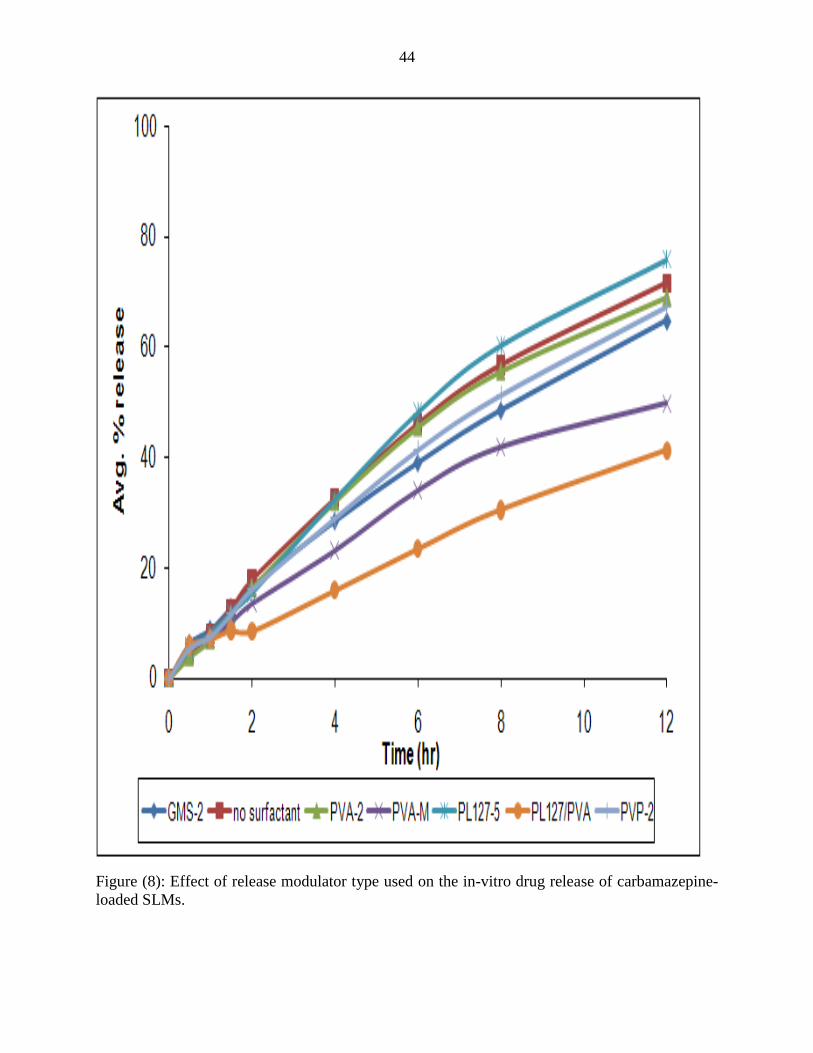
PVP-2 and GMS-2 formulas showed a linear release profile, PVP and GMS have an
enhancing effect on the release profile of CBZ from SLMs and free crystals. Result of PVP
stabilized formula comes in agreement with the results reported by Nair et al, 2002, where they
studied the effect of PVP on the dissolution of carbamazepine in the form solid dispersion and
they found that PVP dispersions can significantly improve the dissolution of carbamazepine due
to the formation of amorphous carbamazepine within the solid dispersion and the presence of
carbamazepine as very small crystallites within the dispersion.
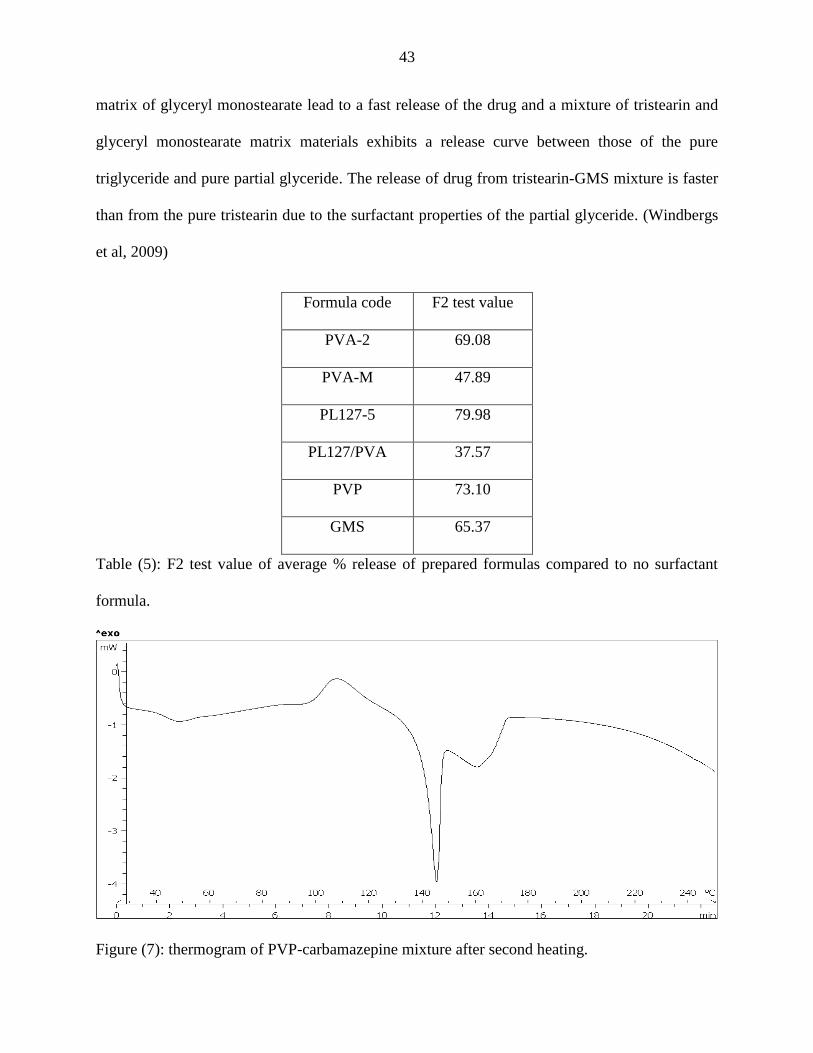
The enhancement of release from SLMs by PVP may be due to the formation of eutectic
mixture between CBZ and PVP as illustrated in figure (7). The figure showed that the mixture
undergo recrystalization from solid solution forming eutectic mixture with lower melting point
which will enhance the dissolution i.e. when a mixture consisting of a slightly soluble CBZ and
highly water soluble PVP, is dissolved in an aqueous medium, PVP will dissolve rapidly,
releasing very fine crystals of the CBZ giving a large surface area of the resulting suspension
which results in an enhanced dissolution rate.
Results of GMS-2 formula come in agreement with Windbergs et al results, where they
studied the release properties from solid lipid matrices using tristearin and glyceryl
monostearate, the results showed that the surfactant properties of the pure partial glyceride
43
matrix of glyceryl monostearate lead to a fast release of the drug and a mixture of tristearin and
glyceryl monostearate matrix materials exhibits a release curve between those of the pure
triglyceride and pure partial glyceride. The release of drug from tristearin-GMS mixture is faster
than from the pure tristearin due to the surfactant properties of the partial glyceride. (Windbergs
et al, 2009)
Formula code F2 test value
PVA-2 69.08
PVA-M 47.89
PL127-5 79.98
PL127/PVA 37.57
PVP 73.10
GMS 65.37
Table (5): F2 test value of average % release of prepared formulas compared to no surfactant
formula.
Figure (7): thermogram of PVP-carbamazepine mixture after second heating.
44
Figure (8): Effect of release modulator type used on the in-vitro drug release of carbamazepine-
loaded SLMs.

45
1.3.3 Effect of release modulators amount on the in-vitro drug release of carbamazepine-
loaded SLMs:
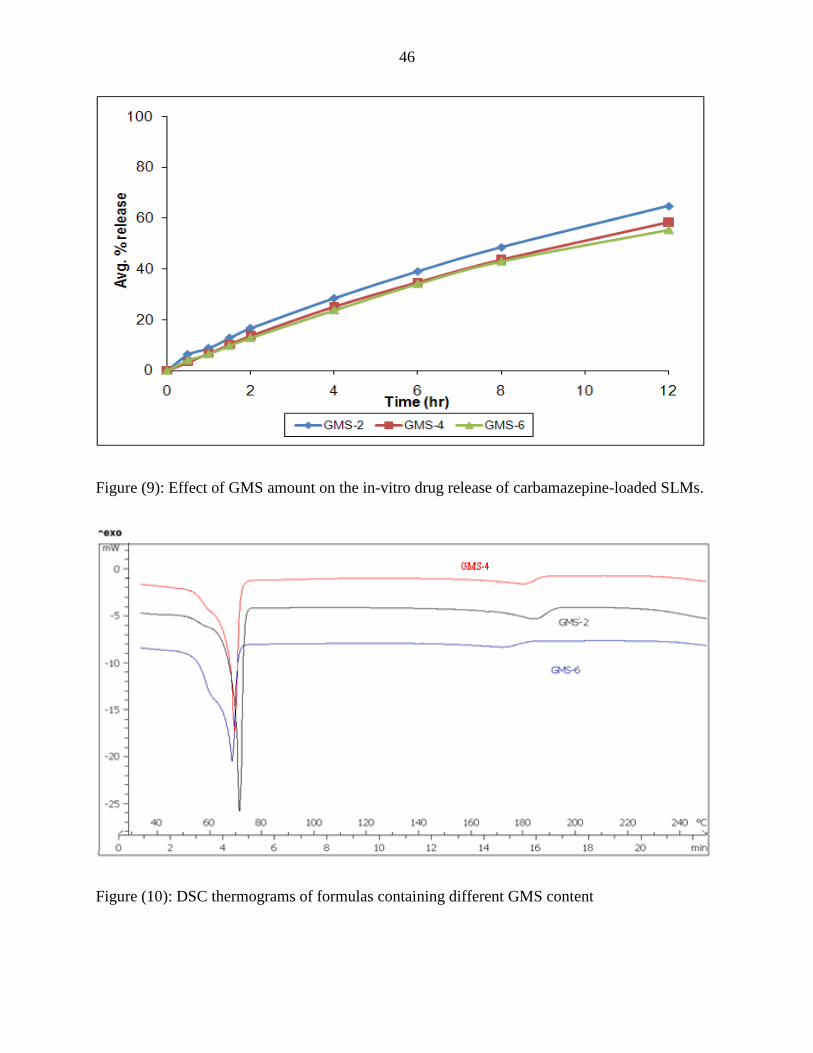
As the figure below shows, increasing GMS content lead to decrease in release rate of
CBZ from SLMs where average release after 12 hour was 64.789%, 58.445% and 55.279% for
GMS-2, GMS-4 and GMS-6 respectively.
Results of F2 test showed that this decrease was insignificant (F2 (GMS-2,GMS-
6)=64.45636), this effect could be due to the hydrophobic nature of GMS (HLB=3.5) which
makes GMS to behaves as another carrier in the SLMs with Compritol beside its function as
surfactant which enhances the solubilization of CBZ in the lipid matrix. GMS-4 and GMS-6
formulas upon examination on DSC showed all CBZ in molecular form, while GMS-2 formula
showed an endothermic peak with 19.52 mJ/mg suggesting that 19% of CBZ is found in
particulate crystalline form. The release from molecular form of CBZ is slowed down by the
presence of CBZ in hydrophobic matrix, but does not require energy to overcome the crystal
energy of particulate CBZ, while the dissolution process of the separated solid particles is not
hindered by the hydrophobicity of compritol, but is limited by the crystal energy of the
particulate CBZ and the hydrophobicity of CBZ itself. The balance between those factors
resulted in insignificant decrease in CBZ release from SLMs upon increasing the amounts of
GMS as a stabilizer.
46
Figure (9): Effect of GMS amount on the in-vitro drug release of carbamazepine-loaded SLMs.
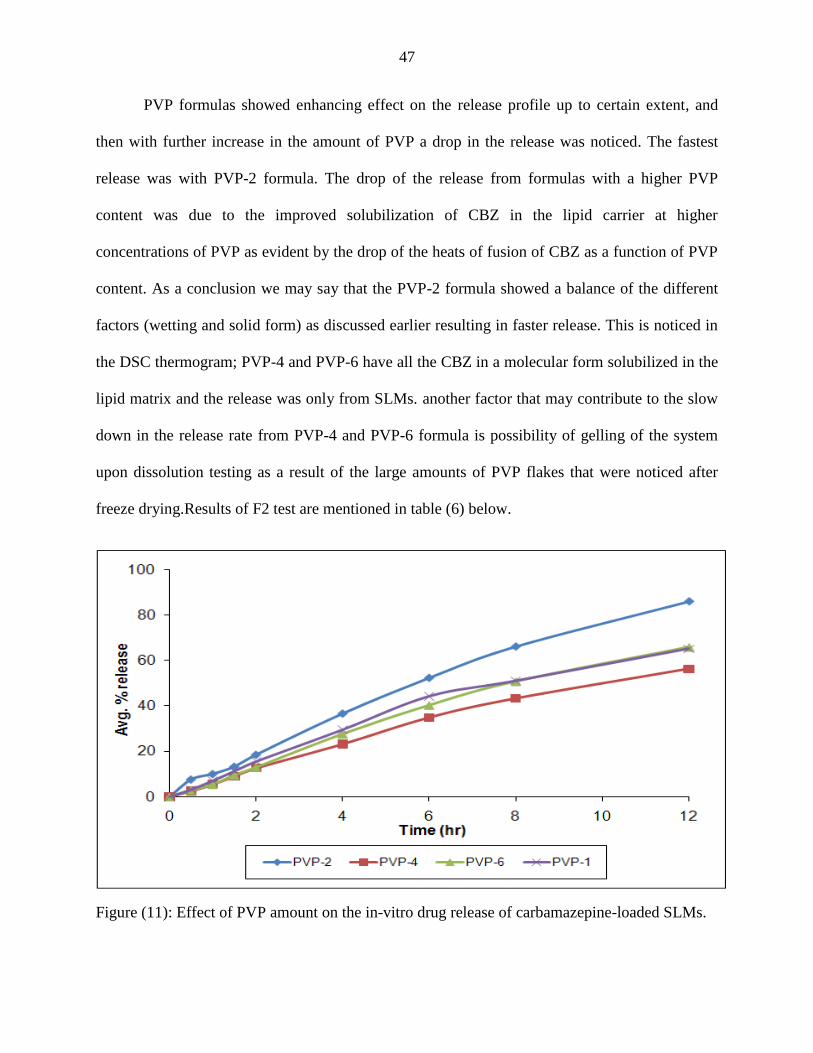
Figure (10): DSC thermograms of formulas containing different GMS content
47
PVP formulas showed enhancing effect on the release profile up to certain extent, and
then with further increase in the amount of PVP a drop in the release was noticed. The fastest
release was with PVP-2 formula. The drop of the release from formulas with a higher PVP
content was due to the improved solubilization of CBZ in the lipid carrier at higher
concentrations of PVP as evident by the drop of the heats of fusion of CBZ as a function of PVP
content. As a conclusion we may say that the PVP-2 formula showed a balance of the different
factors (wetting and solid form) as discussed earlier resulting in faster release. This is noticed in
the DSC thermogram; PVP-4 and PVP-6 have all the CBZ in a molecular form solubilized in the
lipid matrix and the release was only from SLMs. another factor that may contribute to the slow
down in the release rate from PVP-4 and PVP-6 formula is possibility of gelling of the system
upon dissolution testing as a result of the large amounts of PVP flakes that were noticed after
freeze drying.Results of F2 test are mentioned in table (6) below.
Figure (11): Effect of PVP amount on the in-vitro drug release of carbamazepine-loaded SLMs.
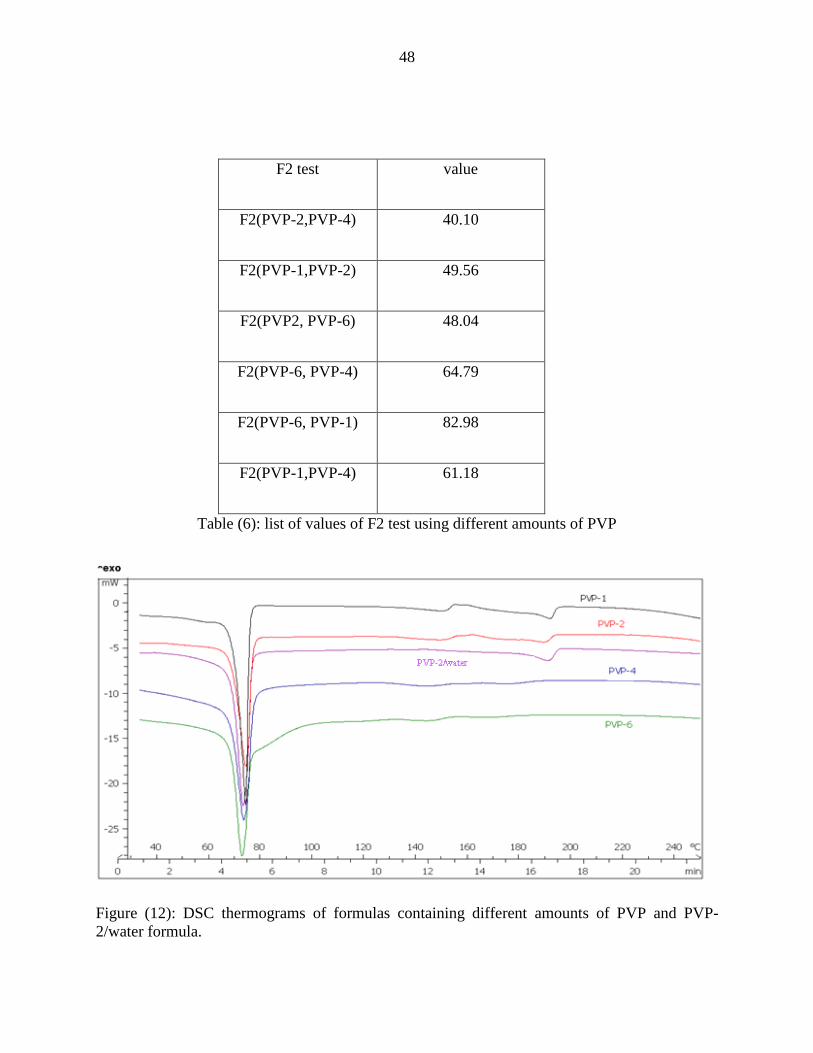
48
F2 test value
F2(PVP-2,PVP-4) 40.10
F2(PVP-1,PVP-2) 49.56
F2(PVP2, PVP-6) 48.04
F2(PVP-6, PVP-4) 64.79
F2(PVP-6, PVP-1) 82.98
F2(PVP-1,PVP-4) 61.18
Table (6): list of values of F2 test using different amounts of PVP
Figure (12): DSC thermograms of formulas containing different amounts of PVP and PVP-
2/water formula.
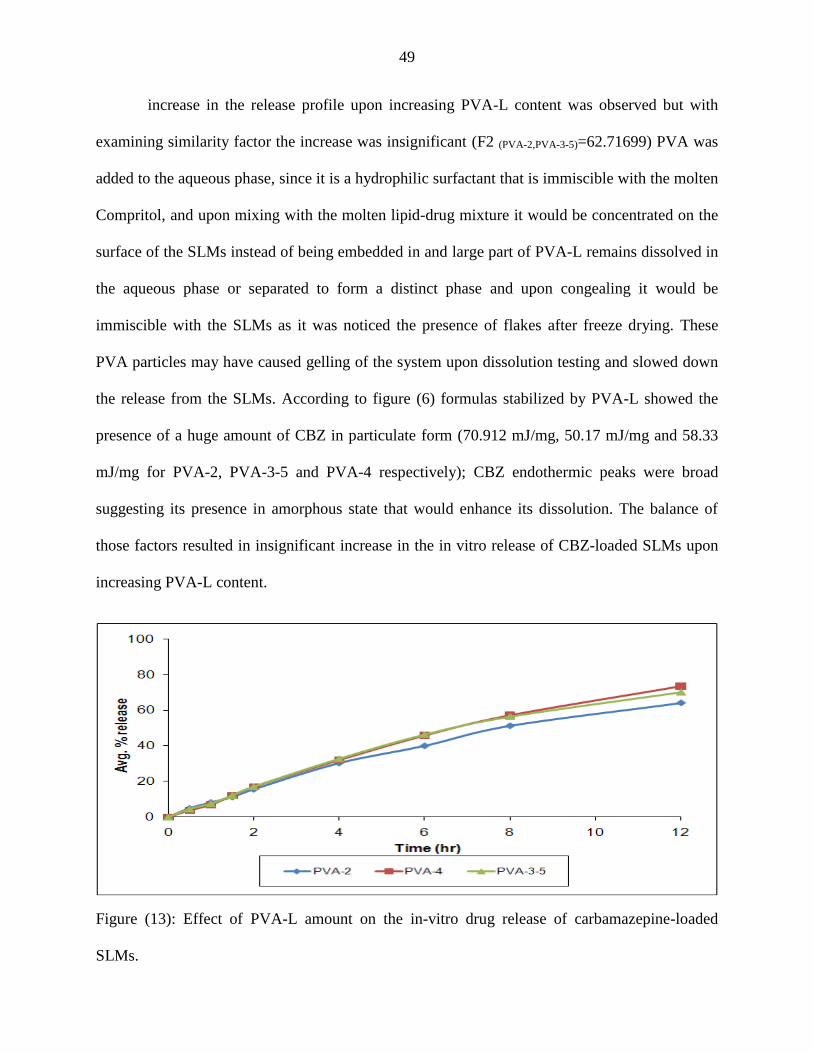
49
increase in the release profile upon increasing PVA-L content was observed but with
examining similarity factor the increase was insignificant (F2 (PVA-2,PVA-3-5)=62.71699) PVA was
added to the aqueous phase, since it is a hydrophilic surfactant that is immiscible with the molten
Compritol, and upon mixing with the molten lipid-drug mixture it would be concentrated on the
surface of the SLMs instead of being embedded in and large part of PVA-L remains dissolved in
the aqueous phase or separated to form a distinct phase and upon congealing it would be
immiscible with the SLMs as it was noticed the presence of flakes after freeze drying. These
PVA particles may have caused gelling of the system upon dissolution testing and slowed down
the release from the SLMs. According to figure (6) formulas stabilized by PVA-L showed the
presence of a huge amount of CBZ in particulate form (70.912 mJ/mg, 50.17 mJ/mg and 58.33
mJ/mg for PVA-2, PVA-3-5 and PVA-4 respectively); CBZ endothermic peaks were broad
suggesting its presence in amorphous state that would enhance its dissolution. The balance of
those factors resulted in insignificant increase in the in vitro release of CBZ-loaded SLMs upon
increasing PVA-L content.
Figure (13): Effect of PVA-L amount on the in-vitro drug release of carbamazepine-loaded
SLMs.
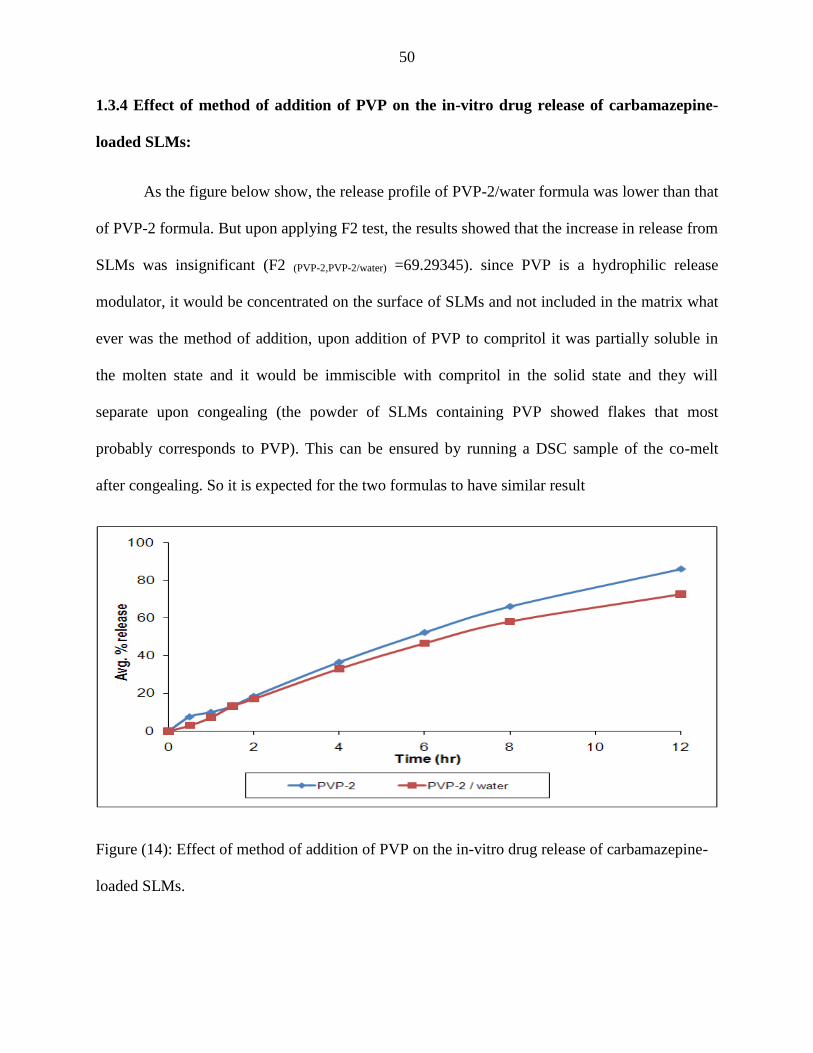
50
1.3.4 Effect of method of addition of PVP on the in-vitro drug release of carbamazepine-
loaded SLMs:
As the figure below show, the release profile of PVP-2/water formula was lower than that
of PVP-2 formula. But upon applying F2 test, the results showed that the increase in release from
SLMs was insignificant (F2 (PVP-2,PVP-2/water) =69.29345). since PVP is a hydrophilic release
modulator, it would be concentrated on the surface of SLMs and not included in the matrix what
ever was the method of addition, upon addition of PVP to compritol it was partially soluble in
the molten state and it would be immiscible with compritol in the solid state and they will
separate upon congealing (the powder of SLMs containing PVP showed flakes that most
probably corresponds to PVP). This can be ensured by running a DSC sample of the co-melt
after congealing. So it is expected for the two formulas to have similar result
Figure (14): Effect of method of addition of PVP on the in-vitro drug release of carbamazepine-
loaded SLMs.

51
1.4 Stability Study:
Instability of SLMs may arise from increase in size of the microparticles, which would
decrease the surface area exposed to dissolution media, and hence, decrease in release rate.
SLMs may also undergo rearrangement of the lipid crystal lattice to the thermodynamically
stable configurations, which may cause expulsion of the drug molecules and hence enhancement
of drug release with storage. SLMs may also undergo gelling with subsequent increase in micro-
viscosity and retard in drug release. (El-kamel et al, 2007) The causes for SLMs instability
include SLMs formulation, production conditions and storage conditions.
Stabilization of microparticles requires the presence of an energy barrier between
particles to prevent their close contact when van der Waals attraction is high. Two general
mechanisms of stabilization can be suggested. Either by electrostatic stabilization, which is based
on the charge repulsion and the formation of electrical double layers or by steric stabilization,
which is based on the steric repulsion of a thick stabilizer layer. The particles are stabilized either
by electrostatic or steric repulsion, depending on the nature of the particles and the stabilizer.
Steric forces results when nonionic surfactant or polymers with long chains are adsorbed to the
particle surfaces. A certain thickness of the adsorbed macromolecules should be found for
efficient steric stabilization. (Zhang et al, 2008)
Nonionic surfactants and polymeric surfactants provide a sufficient thickness and density
of layer on particle surface to stabilize particles. Part of their chain work as an “anchor chain”
that is adsorbed firmly on particle surfaces. Other stabilizing chains dangle in a dispersed
medium to form a stabilizer layer. When two particles approach the chains will lose
configurational entropy due to the overlap between the chains and subsequent compression of the

52
chains, hence leading to strong repulsion. The total potential of particles is sum of van der Waals
attractive potential and steric potential. (Zhang et al, 2008)
Polymeric surfactants with longer polymer chain, higher molecular weight, and better
solubility in the medium are beneficial for the particles stabilization. There should be a sufficient
amount of polymers to ensure complete coverage of the particle surface by their chains in order
to prevent any attraction between the bare areas on the particles. (Zhang et al, 2008) Polymers
also improve the physical stability of amorphous drugs in solid dispersions by increasing the Tg
of the miscible mixture, thereby reducing the molecular mobility at regular storage temperatures,
or by interacting specifically with functional groups of the drugs. (Vasconcelos et al, 2007)
Drug release from PVP-2 and GMS-2 formulas was studied after incubation under
accelerated stability conditions as mentioned in section 3.4.4. GMS-2 formula showed decrease
in release rate with increasing period of incubation in stability conditions (average % release
after 12 hour was 64.789%, 55.9% and 51.586% after preparation, after two weeks and after one
month respectively). PVP-2 formula showed decrease in release rate after two week, then the
release increased after incubation for one month (average % release after 12 hour was 67.39 %,
55.63 % and 63.17 % after preparation, after two weeks and after one month respectively).
Using F2 test, the two formulas seemed to be stable after two weeks and one month of
incubation, (F2(GMS-2, GMS-2 2weeks) =60.17, F2(PVP-2, PVP-2 2weeks)= 55.77, F2(GMS-2, GMS-2 1 month)
=65.93 and F2(PVP-2, PVP-2 1 month)= 69.84). Powder in those conditions is affected by three factors;
first, the extremely hygroscopic nature of the GMS and PVP which will increase the water
content in the powder, and thus overestimation of the weight of powder taken. It was noticed that
the powder of PVP-2 and GMS-2 formulas showed wet appearance after incubation in the
stability chamber. The second that microparticles may suffer from increase in their size, which
would decrease the surface area exposed to dissolution media, and hence, decrease in release
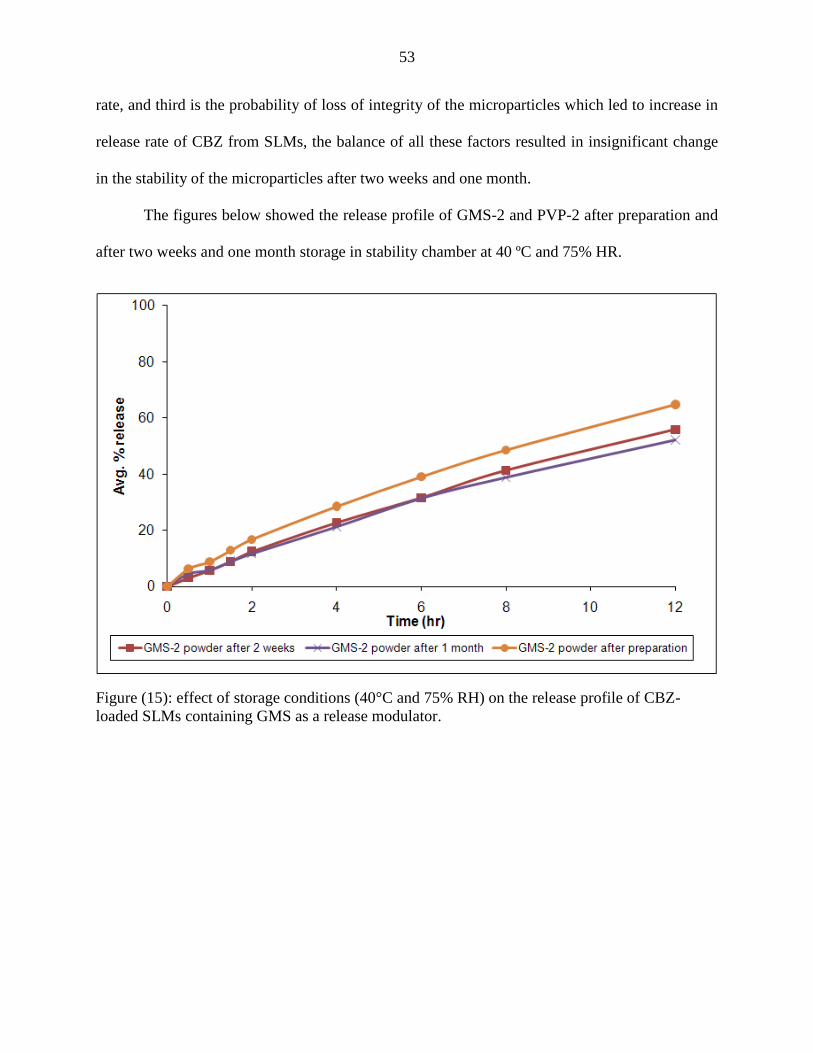
53
rate, and third is the probability of loss of integrity of the microparticles which led to increase in
release rate of CBZ from SLMs, the balance of all these factors resulted in insignificant change
in the stability of the microparticles after two weeks and one month.
The figures below showed the release profile of GMS-2 and PVP-2 after preparation and
after two weeks and one month storage in stability chamber at 40 ºC and 75% HR.
Figure (15): effect of storage conditions (40°C and 75% RH) on the release profile of CBZ-
loaded SLMs containing GMS as a release modulator.
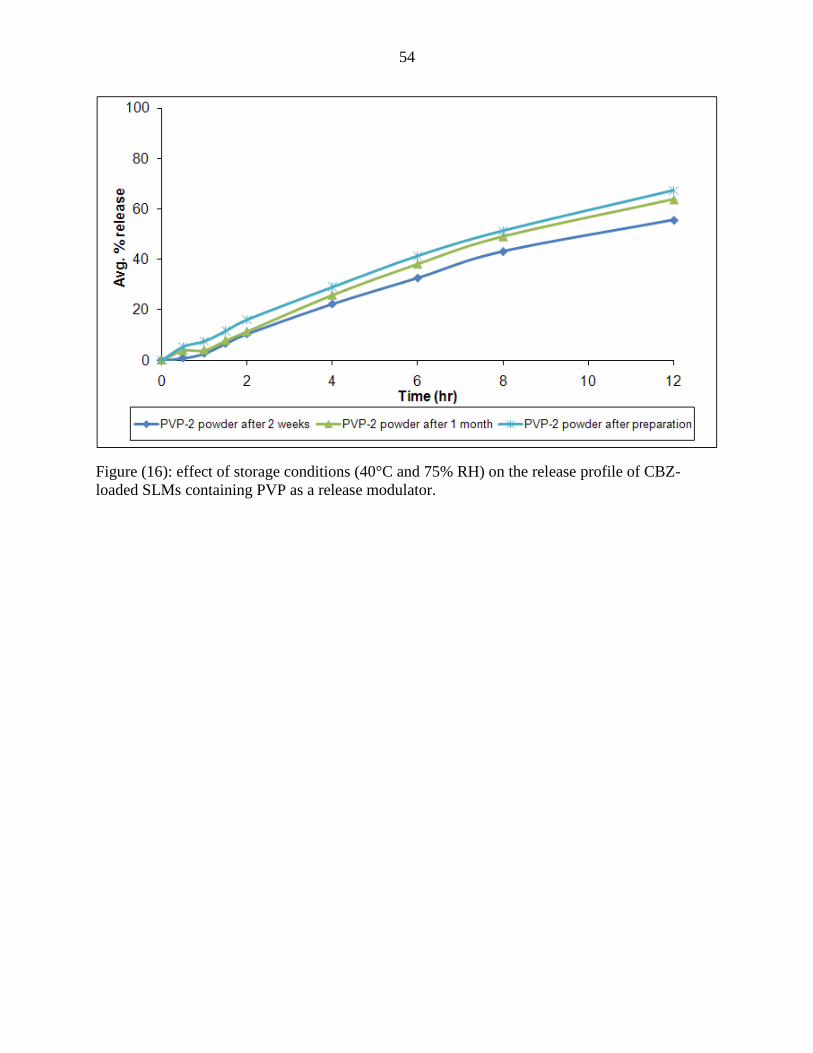
54
Figure (16): effect of storage conditions (40°C and 75% RH) on the release profile of CBZ-
loaded SLMs containing PVP as a release modulator.
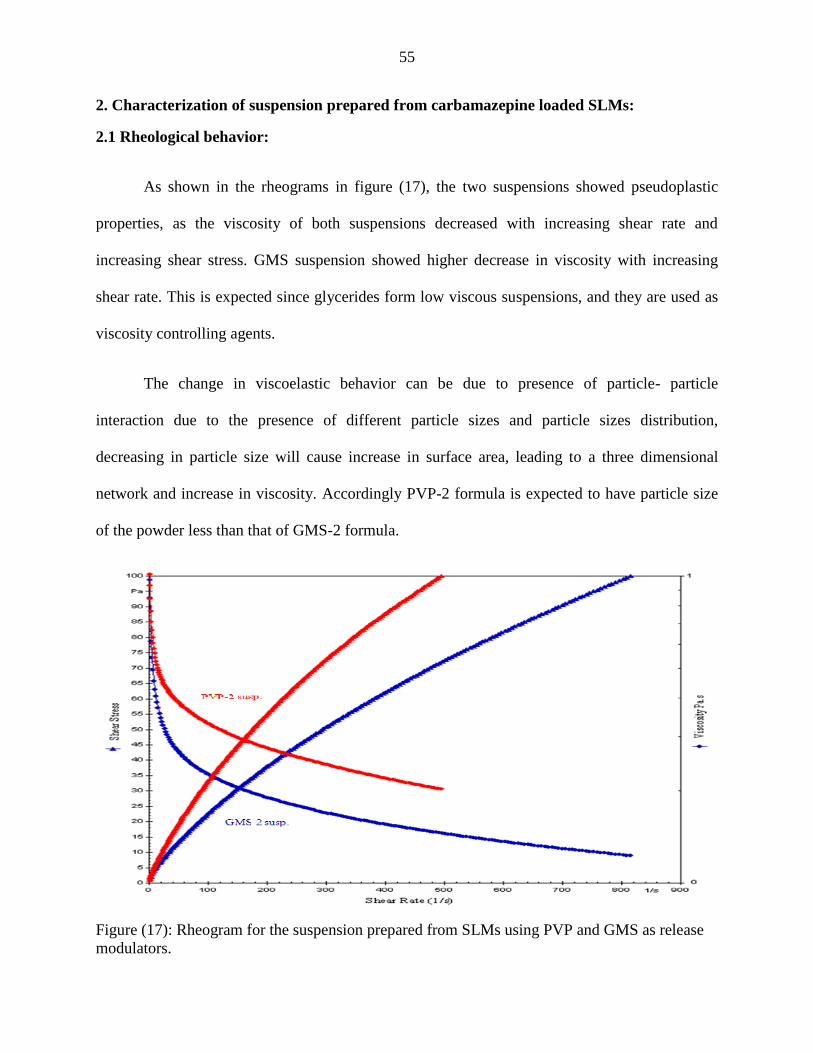
55
2. Characterization of suspension prepared from carbamazepine loaded SLMs:
2.1 Rheological behavior:
As shown in the rheograms in figure (17), the two suspensions showed pseudoplastic
properties, as the viscosity of both suspensions decreased with increasing shear rate and
increasing shear stress. GMS suspension showed higher decrease in viscosity with increasing
shear rate. This is expected since glycerides form low viscous suspensions, and they are used as
viscosity controlling agents.
The change in viscoelastic behavior can be due to presence of particle- particle
interaction due to the presence of different particle sizes and particle sizes distribution,
decreasing in particle size will cause increase in surface area, leading to a three dimensional
network and increase in viscosity. Accordingly PVP-2 formula is expected to have particle size
of the powder less than that of GMS-2 formula.
Figure (17): Rheogram for the suspension prepared from SLMs using PVP and GMS as release
modulators.

56
2.2 Separation Fraction:
Separation fraction depends on many factors; particle size, viscosity and density of the
external phase are the most important ones. By reducing particle size, increasing the viscosity,
and increasing the density of the external phase, we may decrease separation fraction.
From figure (18), we notice a significant difference between the two suspensions over the
week. GMS-2 showed a high separation fraction compared to PVP-2. It showed a remarkable
increase in separation fraction after two days with a small increase over the next five days. PVP-
2 showed an increase in separation fraction to a small extent comparing with GMS and it kept
stepping up over the next five days.
A possible explanation for these results that PVP-2 suspension is more viscous than
GMS-2 suspension as seen from the results of rheological behavior of the two suspensions, this
viscous system would form condensed network that prevent separation of particles from the
suspension. GMS-2 suspension has a high separation fraction because of the low viscosity of
such suspension that contains GMS.
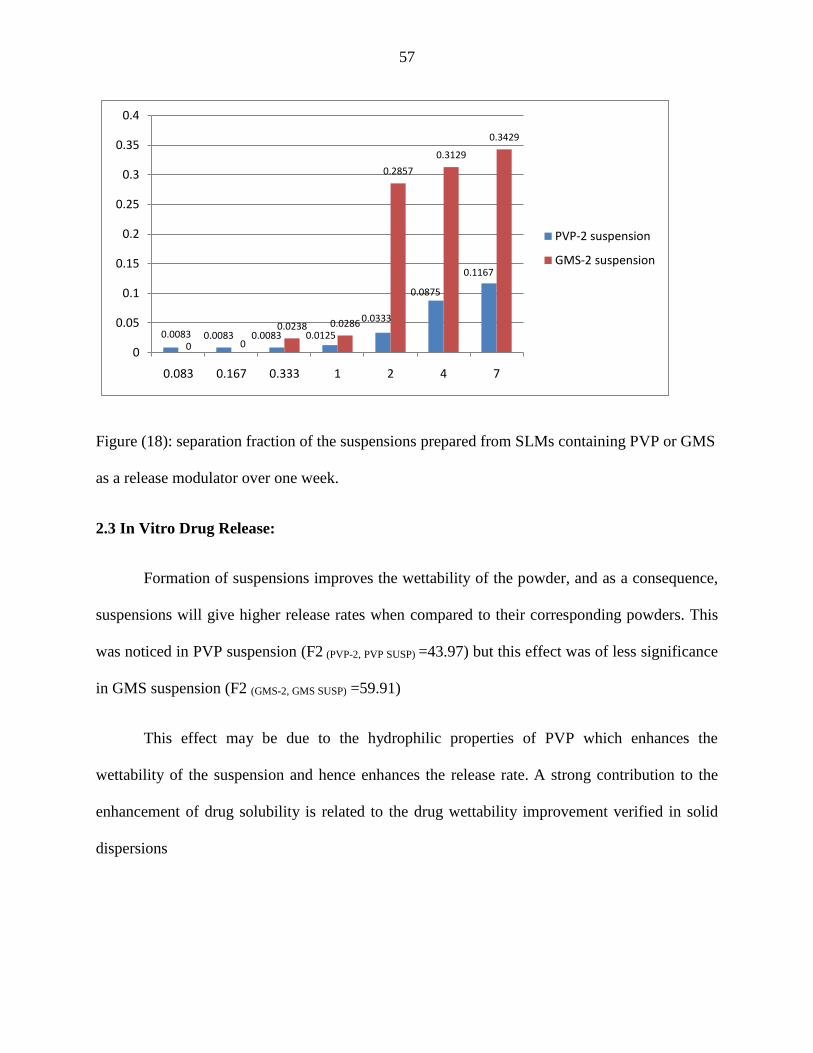
57
0.0083 0.0083 0.0083 0.0125
0.0333
0.0875
0.1167
0 0
0.0238 0.0286
0.2857
0.3129
0.3429
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
0.4
0.083 0.167 0.333 1 2 4 7
PVP-2 suspension
GMS-2 suspension
Figure (18): separation fraction of the suspensions prepared from SLMs containing PVP or GMS
as a release modulator over one week.
2.3 In Vitro Drug Release:
Formation of suspensions improves the wettability of the powder, and as a consequence,
suspensions will give higher release rates when compared to their corresponding powders. This
was noticed in PVP suspension (F2 (PVP-2, PVP SUSP) =43.97) but this effect was of less significance
in GMS suspension (F2 (GMS-2, GMS SUSP) =59.91)
This effect may be due to the hydrophilic properties of PVP which enhances the
wettability of the suspension and hence enhances the release rate. A strong contribution to the
enhancement of drug solubility is related to the drug wettability improvement verified in solid
dispersions
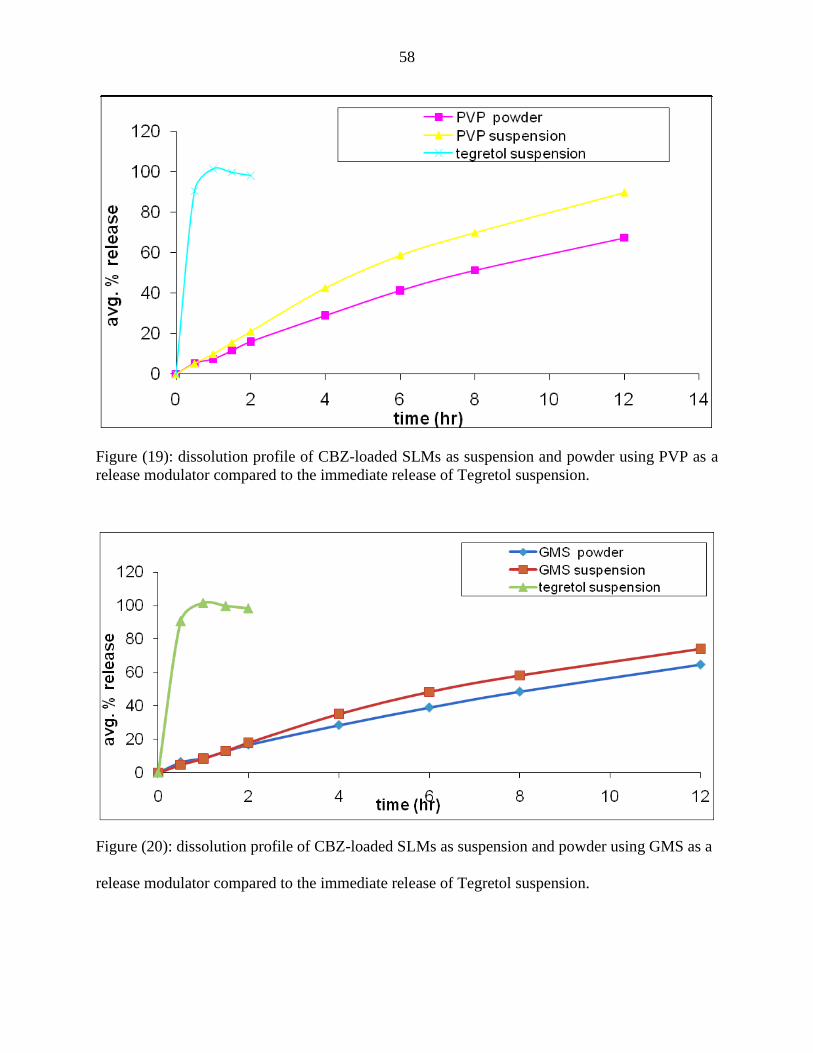
58
Figure (19): dissolution profile of CBZ-loaded SLMs as suspension and powder using PVP as a
release modulator compared to the immediate release of Tegretol suspension.
Figure (20): dissolution profile of CBZ-loaded SLMs as suspension and powder using GMS as a
release modulator compared to the immediate release of Tegretol suspension.
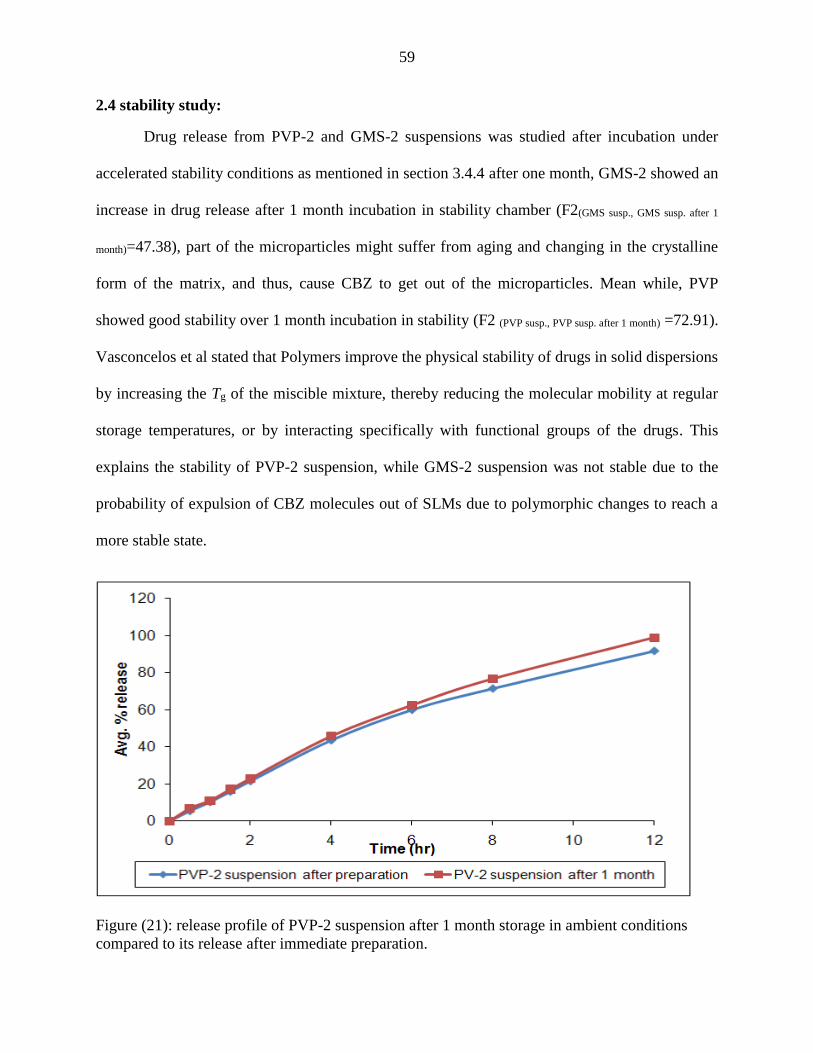
59
2.4 stability study:
Drug release from PVP-2 and GMS-2 suspensions was studied after incubation under
accelerated stability conditions as mentioned in section 3.4.4 after one month, GMS-2 showed an
increase in drug release after 1 month incubation in stability chamber (F2(GMS susp., GMS susp. after 1
month)=47.38), part of the microparticles might suffer from aging and changing in the crystalline
form of the matrix, and thus, cause CBZ to get out of the microparticles. Mean while, PVP
showed good stability over 1 month incubation in stability (F2 (PVP susp., PVP susp. after 1 month) =72.91).
Vasconcelos et al stated that Polymers improve the physical stability of drugs in solid dispersions
by increasing the Tg of the miscible mixture, thereby reducing the molecular mobility at regular
storage temperatures, or by interacting specifically with functional groups of the drugs. This
explains the stability of PVP-2 suspension, while GMS-2 suspension was not stable due to the
probability of expulsion of CBZ molecules out of SLMs due to polymorphic changes to reach a
more stable state.
Figure (21): release profile of PVP-2 suspension after 1 month storage in ambient conditions
compared to its release after immediate preparation.
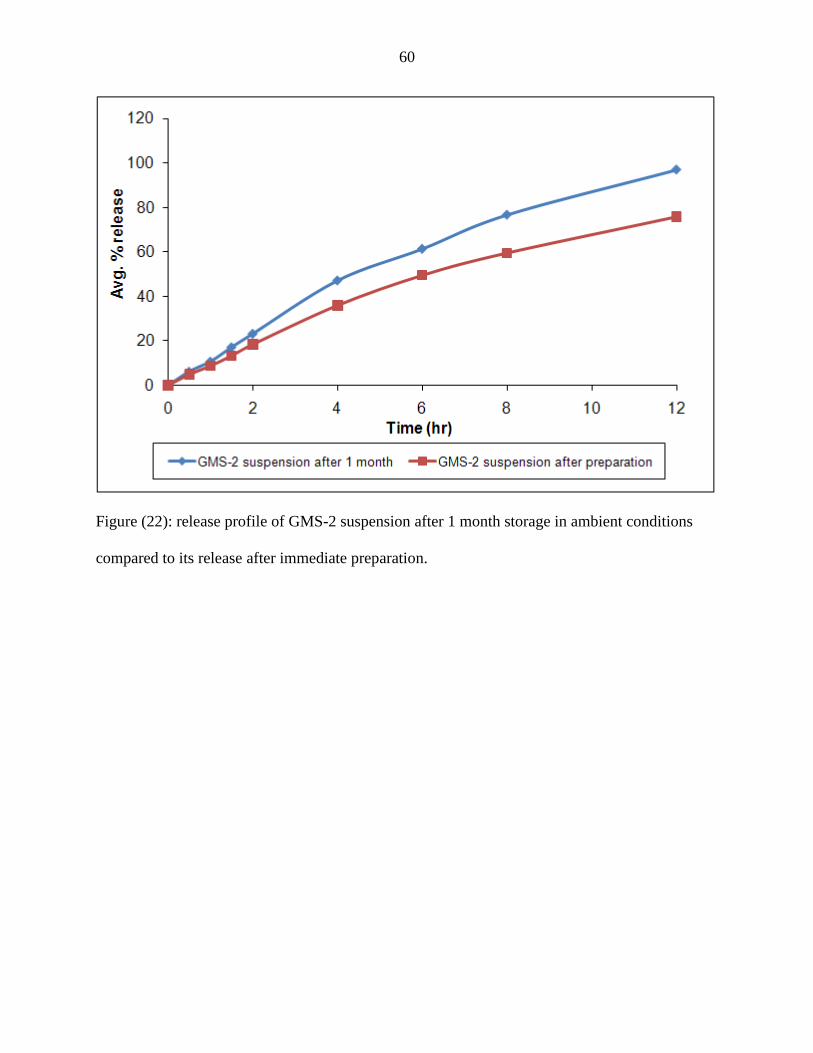
60
Figure (22): release profile of GMS-2 suspension after 1 month storage in ambient conditions
compared to its release after immediate preparation.

61
3- Mechanism of carbamazepine release:
Several mathematical models have been published, to elucidate the water and drug
transport processes and to predict the resulting drug release kinetics. The mathematical
description of the entire drug release process is rather difficult, because of the number of
physical characteristics that must be taken into consideration. drug diffusion, axial and radial
transport in a 3-dimensional system, concentration dependent diffusivities of the species, and
changing matrix dimensions, porosity and composition and many other factors that may affect
drug release kinetics. (Shoaib et al, 2006)
Each model makes certain assumptions and due to these assumptions, the applicability of
the respective models is restricted to certain drug–polymer systems. (Shoaib et al, 2006)
The zero order rates describe the systems where the drug release rate is independent of its
concentration.
C = kot
Where, K0 is zero-order rate constant expressed in units of concentration/time and t is the time.
Higuchi described the release of drugs from insoluble matrix as a square root of time
dependent process based on Fickian diffusion Equation:
Q = Kt1/ 2
Where, K is the constant reflecting the design variables of the system. Q is fraction of
drug released at time t. (Shoaib et al, 2006)
Korsmeyer derived a simple relationship which described drug release from a polymeric
system. To find out the mechanism of drug release, first 60% drug release data was fitted in
Korsmeyer–Peppas model:
Mt /M∞ = Kt n
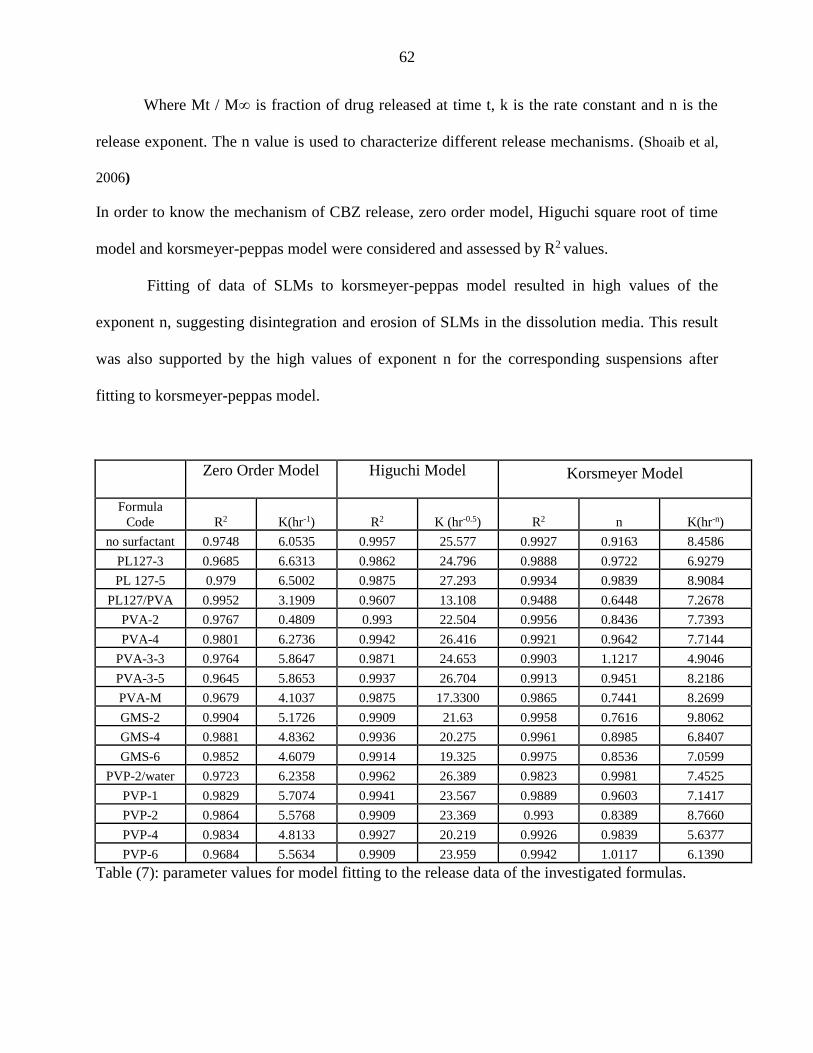
62
Where Mt / M∞ is fraction of drug released at time t, k is the rate constant and n is the
release exponent. The n value is used to characterize different release mechanisms. (Shoaib et al,
2006)
In order to know the mechanism of CBZ release, zero order model, Higuchi square root of time
model and korsmeyer-peppas model were considered and assessed by R2 values.
Fitting of data of SLMs to korsmeyer-peppas model resulted in high values of the
exponent n, suggesting disintegration and erosion of SLMs in the dissolution media. This result
was also supported by the high values of exponent n for the corresponding suspensions after
fitting to korsmeyer-peppas model.
Zero Order Model
Higuchi Model
Korsmeyer Model
Formula
Code R2 K(hr-1) R2 K (hr-0.5) R2 n K(hr-n)
no surfactant 0.9748 6.0535 0.9957 25.577 0.9927 0.9163 8.4586
PL127-3 0.9685 6.6313 0.9862 24.796 0.9888 0.9722 6.9279
PL 127-5 0.979 6.5002 0.9875 27.293 0.9934 0.9839 8.9084
PL127/PVA 0.9952 3.1909 0.9607 13.108 0.9488 0.6448 7.2678
PVA-2 0.9767 0.4809 0.993 22.504 0.9956 0.8436 7.7393
PVA-4 0.9801 6.2736 0.9942 26.416 0.9921 0.9642 7.7144
PVA-3-3 0.9764 5.8647 0.9871 24.653 0.9903 1.1217 4.9046
PVA-3-5 0.9645 5.8653 0.9937 26.704 0.9913 0.9451 8.2186
PVA-M 0.9679 4.1037 0.9875 17.3300 0.9865 0.7441 8.2699
GMS-2 0.9904 5.1726 0.9909 21.63 0.9958 0.7616 9.8062
GMS-4 0.9881 4.8362 0.9936 20.275 0.9961 0.8985 6.8407
GMS-6 0.9852 4.6079 0.9914 19.325 0.9975 0.8536 7.0599
PVP-2/water 0.9723 6.2358 0.9962 26.389 0.9823 0.9981 7.4525
PVP-1 0.9829 5.7074 0.9941 23.567 0.9889 0.9603 7.1417
PVP-2 0.9864 5.5768 0.9909 23.369 0.993 0.8389 8.7660
PVP-4 0.9834 4.8133 0.9927 20.219 0.9926 0.9839 5.6377
PVP-6 0.9684 5.5634 0.9909 23.959 0.9942 1.0117 6.1390
Table (7): parameter values for model fitting to the release data of the investigated formulas.
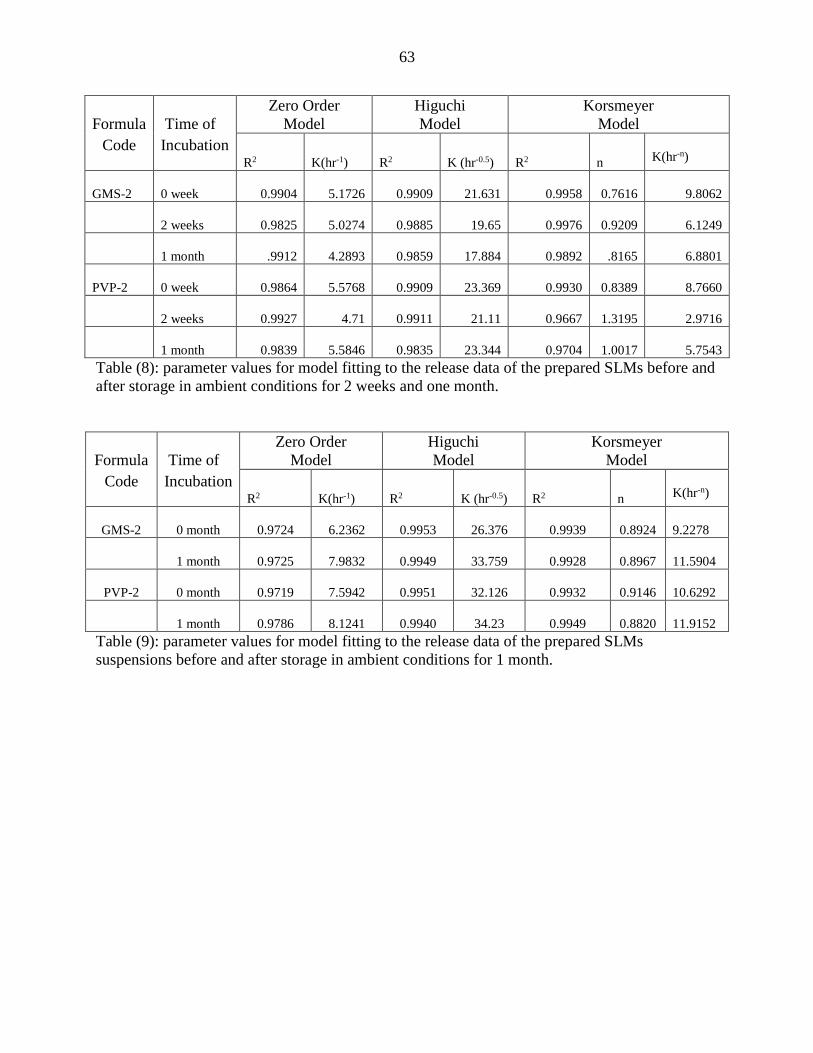
63
Formula
Code
Time of
Incubation
Zero Order
Model
Higuchi
Model
Korsmeyer
Model
R2 K(hr-1) R2 K (hr-0.5) R2 n
K(hr-n)
GMS-2 0 week 0.9904 5.1726 0.9909 21.631 0.9958 0.7616
9.8062
2 weeks 0.9825 5.0274 0.9885 19.65 0.9976 0.9209
6.1249
1 month .9912 4.2893 0.9859 17.884 0.9892 .8165
6.8801
PVP-2 0 week 0.9864 5.5768 0.9909 23.369 0.9930 0.8389
8.7660
2 weeks 0.9927 4.71 0.9911 21.11 0.9667 1.3195
2.9716
1 month 0.9839 5.5846 0.9835 23.344 0.9704 1.0017
5.7543
Table (8): parameter values for model fitting to the release data of the prepared SLMs before and
after storage in ambient conditions for 2 weeks and one month.
Formula
Code
Time of
Incubation
Zero Order
Model
Higuchi
Model
Korsmeyer
Model
R2 K(hr-1) R2 K (hr-0.5) R2 n
K(hr-n)
GMS-2 0 month 0.9724 6.2362 0.9953 26.376 0.9939 0.8924
9.2278
1 month 0.9725 7.9832 0.9949 33.759 0.9928 0.8967
11.5904
PVP-2 0 month 0.9719 7.5942 0.9951 32.126 0.9932 0.9146
10.6292
1 month 0.9786 8.1241 0.9940 34.23 0.9949 0.8820
11.9152
Table (9): parameter values for model fitting to the release data of the prepared SLMs
suspensions before and after storage in ambient conditions for 1 month.

64
CONCLUSION
Solid lipid microparticles loaded with CBZ were successfully prepared based on
compritol as lipid carrier using melt emulsification technique, followed by lyophilization.
Different ratios of CBZ and polymeric release modulators were used to prepare the SLMs. The
prepared SLMs were evaluated according to their physical and controlled release properties.
Suspensions from selected formulas were prepared and evaluated in terms of rheology,
separation fraction and in-vitro drug release. SLMs and their corresponding suspensions were
subjected to accelerated stability conditions and evaluated in term of in-vitro drug release.
Almost all release modulators increased the amount of CBZ incorporated in the SLMs in
molecular form compared to the SLMs containing no release modulator with the exception of
PVA-L and PVA-M. Amount of CBZ incorporated in the SLMs in molecular form was
proportional to increase in the amount of release modulator amount. A relative enhancement of
CBZ release was obtained from Pluronic F 127, PVP and GMS stabilized SLMs.
GMS showed insignificant change in the release profile of CBZ upon increasing its
amount, while PVP showed enhancing effect up to a certain limit then with further increase in
PVP amount a drop in the release rate was noticed.
Both suspensions of PVP and GMS stabilized formulas showed increase in release profile
compared to their corresponding powder. This increase was strongly noticed with formula used
PVP as a release modulator.
Stability studies showed that both formulas containing PVP or GMS as stabilizer were
stable over two weeks and one month upon storage in ambient conditions, GMS suspension
showed instability after one month storage in ambient condition, while PVP suspension was
stable under the same conditions.

65
RFERENCES:
Albertini, B., Passerini, N., Sabatino, M., Vitali, B. Brigidi, P., Rodriguez, L., 2008. Polymer–
lipid based mucoadhesive microspheres prepared by spray-congealing for the vaginal delivery of
econazole nitrate. European Journal of Pharmaceutical Sciences. 36, 591–601.
Altafullah, I., Talwara, D., Loewensona, R., Olson, K., Lockmana, L., 1988. Factors influencing
serum levels of carbamazepine and carbamazepine-lO,ll-epoxide in children. Epilepsy Res., d.
72-80
Ambrósio, A., Soares-da-Silva, P., Carvalho, C., Carvalho, A., 2001. Mechanisms of Action of
Carbamazepine and Its Derivatives, Oxcarbazepine, BIA 2-093, and BIA 2-024. Neurochemical
Research,. 27. 121–130.
Backman, E., Dahlstrom, G., Eeg-Olofsson, O., Bertler, a., 1987. The 24 hour variation of
salivary carbamazepine and carbamazepine-10,11-epoxide concentrations in children with
epilepsy. Pediatric Neurol. 3. 327-30.
British Pharmacopoeia (2005), Royal Pharmaceutical Press, London, UK.
Boylan, J., Cooper, J., Chowhan, Z., Lund, W.,ade, A., Weir, R., Yates, B., 1983, Handbook
of pharmaceutical excepients, American Pharmaceutical Association, The Pharmaceutical
Society of Great Britain.
Bunjes, H., 2005. Characterization of Solid Lipid Nano and Microparticles. Lipospheres in
Drug Targets and Delivery. 41-61.
Chambi, H., Alvim, I., Barrera-Arellano, D., Grosso, C., 2008. Solid lipid microparticles
containing water-soluble compounds of different molecular mass: Production, characterization
and release profiles. Food Research International. 41. 229–236.
Chunxi, L., Lijuan, Z., Yu, Q., 2006. Preparation and Crystal Modification of Ibuprofen-Loaded
Solid Lipid Microparticles. Chinese J. Chem. Eng., 14(4) 5. 18-52.5.
Dalpiaz, A., Mezzena, M., Scatturin, A., Scalia, S., 2007. Solid lipid microparticles for the
stability enhancement of the polar drug N6-cyclopentyladenosine. International Journal of
Pharmaceutics. 355, 81–86.
Dharmala, K., Yoo,J., Lee, C., 2008. Development of Chitosan–SLN Microparticles for
chemotherapy: In vitro approach through efflux-transporter modulation. Journal of Controlled
Release. 131, 190–197.
Dong, W., Maincent, P., Bodmeier, R., 2006. In vitro and in vivo evaluation of carbamazepine-
loaded enteric microparticles. International Journal of Pharmaceutics. 331, 84–92.

66
D’oria, C., Charcosset, C., Barresi, A., Fessi, H., 2009. Preparation of solid lipid particles by
membrane emulsification—Influence of process parameters. Colloids and Surfaces A:
Physicochem. Eng. Aspects. 338. 114–118.
El-Kamel, A., Al-Fagih, I., Alsarra, I., 2007. Testosterone solid lipid microparticles for
transdermal drug delivery. Formulation and physicochemical characterization. Journal of
Microencapsulation. 24(5), 457–475.
Erni, C., Suard, C., Freitas, S., Dreher, D., Merkle, H., Walter, E., 2002. Evaluation of cationic
solid lipid microparticles as synthetic carriers for the targeted delivery of macromolecules to
phagocytic antigen-presenting cells. Biomaterials. 23 .4667–4676.
Esposito, E., Cortesi, R., Nastruzzi, C., 2005. Production of Lipospheres for Bioactive
Compound Delivery. Lipospheres in Drug Targets and Delivery, 23-39.
Jaeghere, F., Allémann, E., Doelker, E., Gurny, R., Cerny, R., Galli, B., Steulet, A., Müller, I.,
Schütz, H., 2001. pH-Dependent Dissolving Nano- and Microparticles for Improved Peroral
Delivery of a Highly Lipophilic Compound in Dogs. AAPS Pharmsci. 3. article 8
Jannin, V., Pochard, E. and Chambin, O. (2006), Influence of poloxamers on the dissolution
performance and stability of controlled-release formulations containing Precirol® ATO 5,
International Journal of Pharmaceutics, 309(1-2), 6-15.
Jarrar, R., Buchhalter, J., 2003, Therapeutics in pediatric epilepsy, Part 1: The new antiepileptic
drugs and the ketogenic diet, Mayo Clinic, 78.3., 359-70.
Jaspart S, Piel G, Delattre L, Evrard B. 2005. Solid lipid microparticles: Formulation,
preparation, characterisation, drug release, and applications. Expert Opinion on Drug Delivery.
2, 75–87.
Karanth, H., Shenoy, V., Murthy, R., 2006. Industrially Feasible Alternative Approaches in the
Manufacture of Solid Dispersions: A Technical Report. AAPS PharmSciTech. 7 (4). Article 87.
Koennings, S., Tessmar, J., Blunk, T., Go¨ pferich, A., 2007. Confocal Microscopy for the
Elucidation of Mass Transport Mechanisms Involved in Protein Release from Lipid-based
Matrices. Pharmaceutical Research. 24. 7.1325-1335.
Kukizaki, M., 2009. Preparation of solid lipid microcapsules via solid-in-oil-in-water dispersions
by premix membrane emulsification. Chemical Engineering Journal. 151, 387–396.
Leuner, c., Dressman, j., 2000, Improving drug solubility for oral delivery using solid
dispersions, European Journal of Pharmaceutics and Biopharmaceutics. 50. 47-60.
Long, C., Zhang, L., Qian, Y., 2006. Mesoscale simulation of drug molecules distribution in the
matrix of solid lipid microparticles (SLM). Chemical Engineering Journal. 119. 99–106.

67
Motamedi, G., Meador, K., 2003. Epilepsy and cognition. Epilepsy & Behavior. 4. S25–S38.
Naima, Z., Siro, T., Juan-Manuel, G., Chantal, C., Rene, C., Jerome, D., 2000, Interactions
between carbamazepine and polyethyleneglycol (PEG)6000: characterizations of the physical,
solid dispersed and eutectic mixtures. EuropeanJournalofPharmaceuticalSciences.12.395–
404.
Nair, R., Gonen, S., Hoag, S., 2002, Influence of polyethylene glycol and povidone on the
Polymorphic transformation and solubility of carbamazepine. International Journal of
Pharmaceutics. 240.11–22.
Sanna, V., Kirschvink, N., Gustin, P., Gavini, E., Roland, I., Delattre, L., Evrard, B., 2003.
Preparation and In Vivo Toxicity Study of Solid Lipid Microparticles as Carrier for Pulmonary
Administration. AAPS PharmSciTech. 5. (2) Article 27
Savolainen, M., Khoo, C., Glad, H., Dahlqvist, C., Juppo, A., 2002. Evaluation of controlled-
release polar lipid microparticles. International Journal of Pharmaceutics. 244. 151-161.
Savolainen, M., Herder, J., Khoo, C., Lövqvist, K., Dahlqvist, C., Glad,. H., Juppo, A., 2003.
Evaluation of polar lipid–hydrophilic polymer microparticles. International Journal of
Pharmaceutics .262 . 47–62.
Saraf, S., Mishra, D., Asthana, A., Jain, R., Singh, S., Jain, N., 2005. Lipid microparticles for
mucosal immunization against hepatitis B. Vaccine. 24 . 45–56.
Saupe, A., Rades, A., 2006. Solid Lipid Nanoparticles. Nanocarrier Technologies: Frontiers
of Nanotherap., 41–50.
Shehata, G., Bateh, A., Hamed, S., Rageh, T, Elsorogy, Y., 2009. Neuropsychological effects of
antiepileptic drugs (carbamazepine versus valproate) in adult males with epilepsy.
Neuropsychiatric Disease and Treatment .5. 527–533.
Shoaib, M, Tazeen, J., Merchant, H., Yousuf, R.,2006, evaluation of drug release kinetics from
ibeuprofen matrix tablets using HPMC. Pak. J. Pharm. Sci., 19.2, 119-124.
Trotta, M., Cavalli, R., Carlotti, M., Battaglia, L., Debernardi, F., 2004. Solid lipid micro-
particles carrying insulin formed by solvent-in-water emulsion–diffusion technique.
International Journal of Pharmaceutics. 288, 281–288.
Vasconcelos, T., Sarmento, B., Costa, P., 2007, Solid dispersions as strategy to improve oral
bioavailability of poor water soluble drugs, Drug Discovery Today .12.1068-1075.
Windbergs. M., Strachan, C., Kleinebudde, P., 2009, Influence of the composition of glycerides
on thesolid-state behaviour and the dissolution profiles of solidlipid extrudates. InternationalJournalofPharmaceutics.381.184–191.
Winkler, S., Luer, M., 1998. Antiepileptic Drug Review: Part 1. Surg Neurol. 49, 449 –52.

68
Wong, H., Bendayan, R., Rauth, A., Wu, X., 2006. Simultaneous delivery of doxorubicin and
GG918 (Elacridar) by new Polymer-Lipid Hybrid Nanoparticles (PLN) for enhanced treatment
of multidrug-resistant breast cancer. Journal of Controlled Release. 116, 275–284.
Zhang, L., Liu, L., Qian, Y., Chen, Y., 2007. The effects of cryoprotectants on the freeze-drying
of ibuprofen-loaded solid lipid microparticles (SLM). European Journal of Pharmaceutics
and Biopharmaceutics. 69. 750–759.
Zhang, L., Qian, Y., Long, C., Chen, Y., 2008. Systematic Procedures for Formulation Design
of Drug-Loaded Solid Lipid Microparticles: Selection of Carrier Material and Stabilizer. Ind.
Eng. Chem. Res. 47, 6091–6100.
Zeng, K., Wang, X., Xi, Z., Yan, Y., 2009. Adverse effects of carbamazepine, phenytoin,
valproate and lamotrigine monotherapy in epileptic adult Chinese patients.clinical neurology
and neurosyrgery.
Zahirul, M., Khan, I., 1996. Dissolution testing for sustained or controlled release oral dosage
forms and correlation with in vivo data: challenges and opportunities. International Journal of
Pharmaceutics.140. 131-143.

69
APPENDICES
Drug release data of different SLMs preparations.
Drug release data of SLMs after storage in accelerated stability conditions.
Drug release from suspensions before and after storage in accelerated stability conditions.
70
Data of drug release with different SLMs preparations
formula code Time Avg. % Release SD
0.5 5.297675235 0.88908752
1 8.21903391 1.071844399
1.5 11.96526707 0.290435256
PL127-5 2 15.50659718 1.602003994
4 32.45859911 3.38742345
6 48.18583962 3.575119705
8 60.30143101 3.279745073
12 75.83378078 0.811703976
0.5 4.267458408 1.169900626
1 6.617244468 2.416572682
1.5 11.16303699 4.169517959
PL127-3 2 14.64751503 6.151641391
4 33.275357 10.14354699
6 51.42075317 8.437377409
8 58.97636466 9.432042269
12 74.52496982 9.519761906
0.5 2.285000865 0.291702238
1 4.242376902 0.831717742
1.5 7.429493948 1.910833227
PVA-3-3 2 11.59579419 2.665146804
4 26.91700409 4.275631317
6 41.03611267 4.787930585
8 51.71259464 5.331507803
12 65.41665775 5.617116532
0.5 4.138792415 0.10204411
1 7.343206211 0.184513002
1.5 12.25084191 0.270894394
PVA-3-5 2 17.37417269 0.285403317
4 33.95765438 0.43440672
6 48.30710462 0.81214695
8 59.04996303 1.292051767
12 73.30776741 1.206836326
0.5 5.507371832 0.816946162
1 8.756382108 3.657715656
1.5 11.79846133 2.083061992
PVA-2 2 16.1157291 2.75231927
4 30.76120726 4.494347418
6 40.30346497 4.146101805
8 51.34302495 6.186909297
12 64.02801438 7.263844186
71
Continued
0.5 3.63986519 0.495219754
1 6.796478437 0.893537069
1.5 11.848637 1.368599646
PVA-4 2 16.71875645 1.56552102
4 31.54722547 2.500782422
6 45.00038976 3.015952165
8 55.99701951 3.573503091
12 72.06980459 4.225345845
0.5 4.127697829 0.582558542
1 8.021210168 1.277842164
1.5 12.65976321 1.780092649
no surfactant 2 17.8830504 2.61336989
4 32.59248576 4.289850169
6 46.1104888 4.54001126
8 56.71864457 4.202158943
12 71.59595868 3.87646317
0.5 3.174603175 0.090214504
1 7.072599769 0.207199578
1.5 11.26098676 0.401388602
PVP-1 2 15.50305061 0.431900973
4 29.50012515 0.785647624
6 44.17271797 1.709167652
8 50.97304148 0.987953033
12 65.07341867 0.551524333
0.5 7.711341008 0.263078843
1 10.12368558 1.058624442
1.5 13.34264874 1.136298458
PVP-2 2 18.55245968 1.053285604
4 36.66619335 0.796786017
6 52.3426678 1.468837622
8 66.08869359 1.608371353
12 85.92205978 2.121123479
0.5 2.541105542 0.511106863
1 5.428391248 0.358000447
1.5 9.024038581 0.244979087
PVP-4 2 12.57650989 0.464372238
4 23.19153279 0.817330678
6 34.9102789 2.091688555
8 43.2884041 2.626990258
12 56.33449909 3.603379301
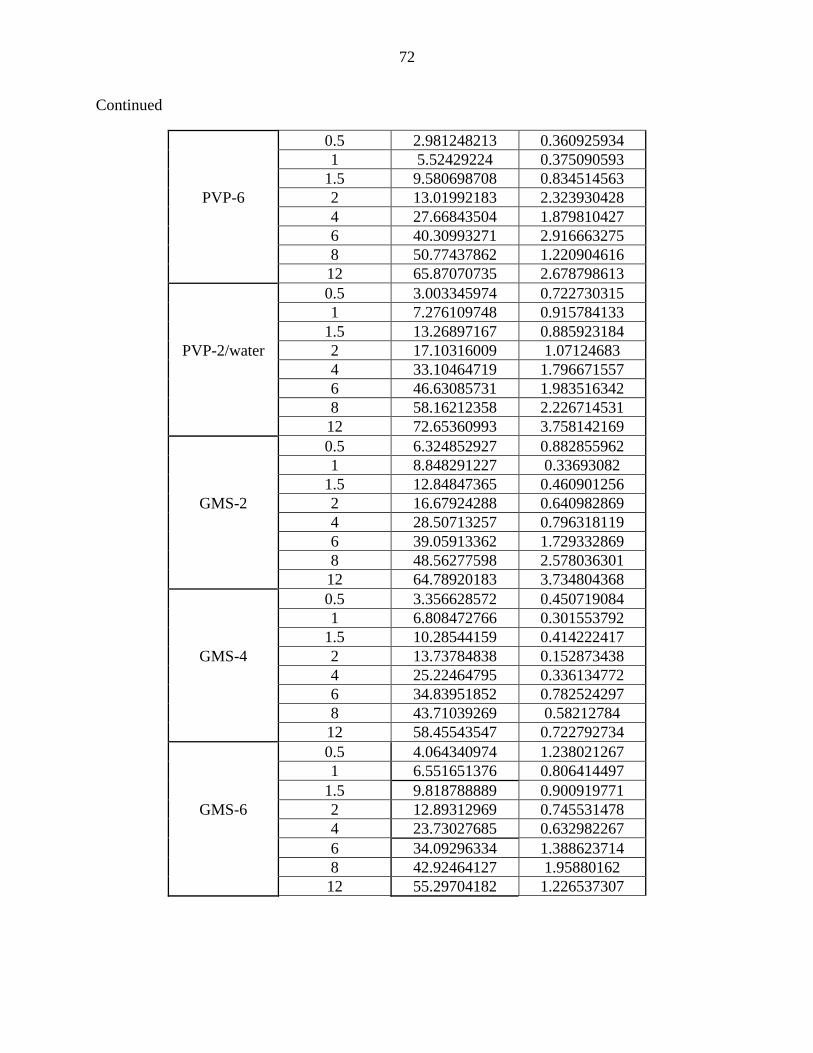
72
Continued
0.5 2.981248213 0.360925934
1 5.52429224 0.375090593
1.5 9.580698708 0.834514563
PVP-6 2 13.01992183 2.323930428
4 27.66843504 1.879810427
6 40.30993271 2.916663275
8 50.77437862 1.220904616
12 65.87070735 2.678798613
0.5 3.003345974 0.722730315
1 7.276109748 0.915784133
1.5 13.26897167 0.885923184
PVP-2/water 2 17.10316009 1.07124683
4 33.10464719 1.796671557
6 46.63085731 1.983516342
8 58.16212358 2.226714531
12 72.65360993 3.758142169
0.5 6.324852927 0.882855962
1 8.848291227 0.33693082
1.5 12.84847365 0.460901256
GMS-2 2 16.67924288 0.640982869
4 28.50713257 0.796318119
6 39.05913362 1.729332869
8 48.56277598 2.578036301
12 64.78920183 3.734804368
0.5 3.356628572 0.450719084
1 6.808472766 0.301553792
1.5 10.28544159 0.414222417
GMS-4 2 13.73784838 0.152873438
4 25.22464795 0.336134772
6 34.83951852 0.782524297
8 43.71039269 0.58212784
12 58.45543547 0.722792734
0.5 4.064340974 1.238021267
1 6.551651376 0.806414497
1.5 9.818788889 0.900919771
GMS-6 2 12.89312969 0.745531478
4 23.73027685 0.632982267
6 34.09296334 1.388623714
8 42.92464127 1.95880162
12 55.29704182 1.226537307
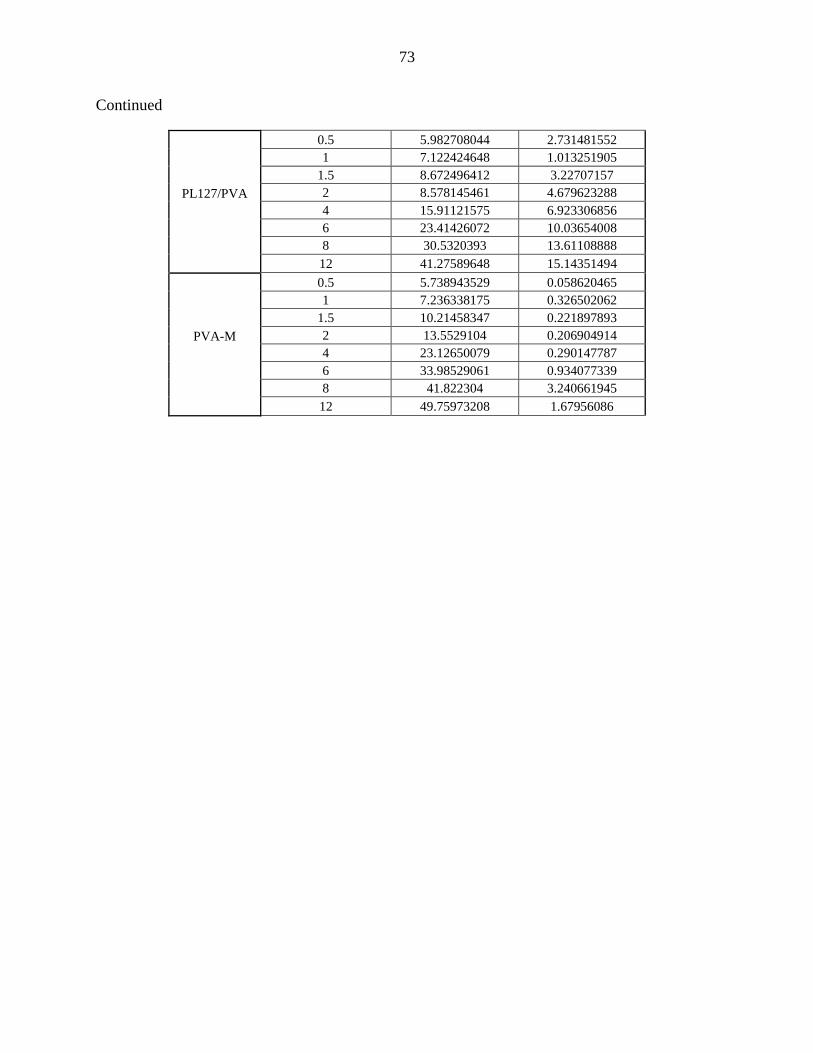
73
Continued
0.5 5.982708044 2.731481552
1 7.122424648 1.013251905
1.5 8.672496412 3.22707157
PL127/PVA 2 8.578145461 4.679623288
4 15.91121575 6.923306856
6 23.41426072 10.03654008
8 30.5320393 13.61108888
12 41.27589648 15.14351494
0.5 5.738943529 0.058620465
1 7.236338175 0.326502062
1.5 10.21458347 0.221897893
PVA-M 2 13.5529104 0.206904914
4 23.12650079 0.290147787
6 33.98529061 0.934077339
8 41.822304 3.240661945
12 49.75973208 1.67956086
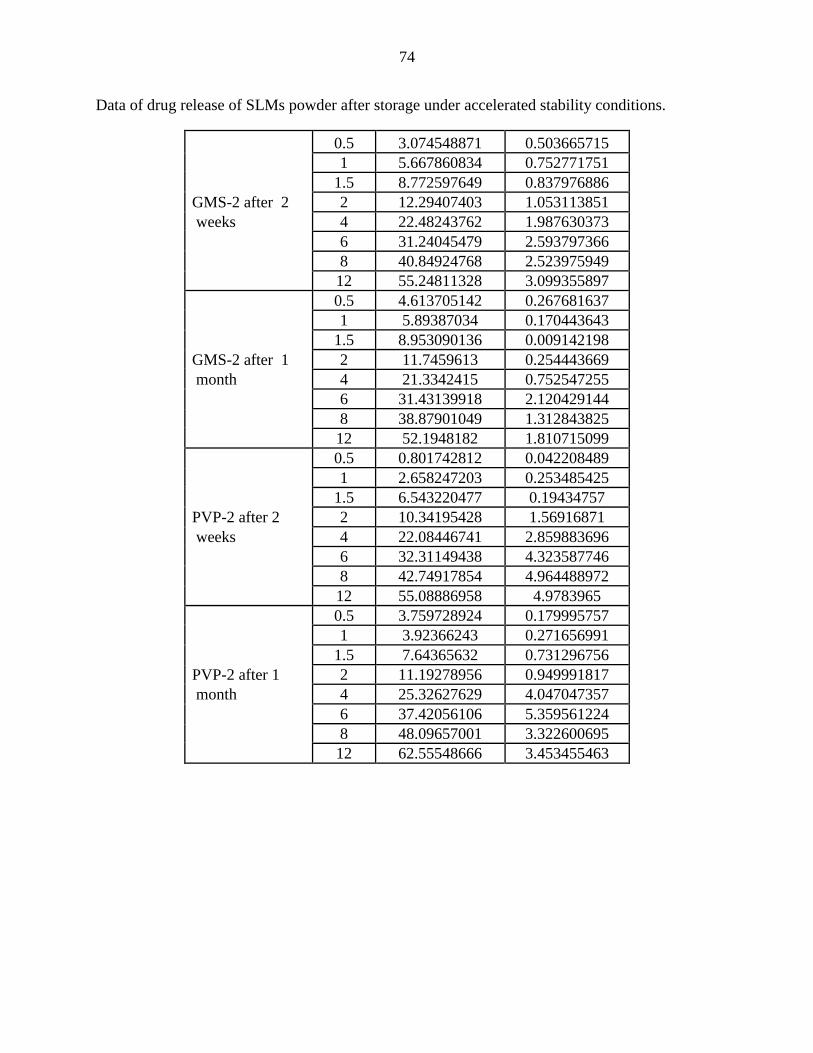
74
Data of drug release of SLMs powder after storage under accelerated stability conditions.
0.5 3.074548871 0.503665715
1 5.667860834 0.752771751
1.5 8.772597649 0.837976886
GMS-2 after 2 2 12.29407403 1.053113851
weeks 4 22.48243762 1.987630373
6 31.24045479 2.593797366
8 40.84924768 2.523975949
12 55.24811328 3.099355897
0.5 4.613705142 0.267681637
1 5.89387034 0.170443643
1.5 8.953090136 0.009142198
GMS-2 after 1 2 11.7459613 0.254443669
month 4 21.3342415 0.752547255
6 31.43139918 2.120429144
8 38.87901049 1.312843825
12 52.1948182 1.810715099
0.5 0.801742812 0.042208489
1 2.658247203 0.253485425
1.5 6.543220477 0.19434757
PVP-2 after 2 2 10.34195428 1.56916871
weeks 4 22.08446741 2.859883696
6 32.31149438 4.323587746
8 42.74917854 4.964488972
12 55.08886958 4.9783965
0.5 3.759728924 0.179995757
1 3.92366243 0.271656991
1.5 7.64365632 0.731296756
PVP-2 after 1 2 11.19278956 0.949991817
month 4 25.32627629 4.047047357
6 37.42056106 5.359561224
8 48.09657001 3.322600695
12 62.55548666 3.453455463
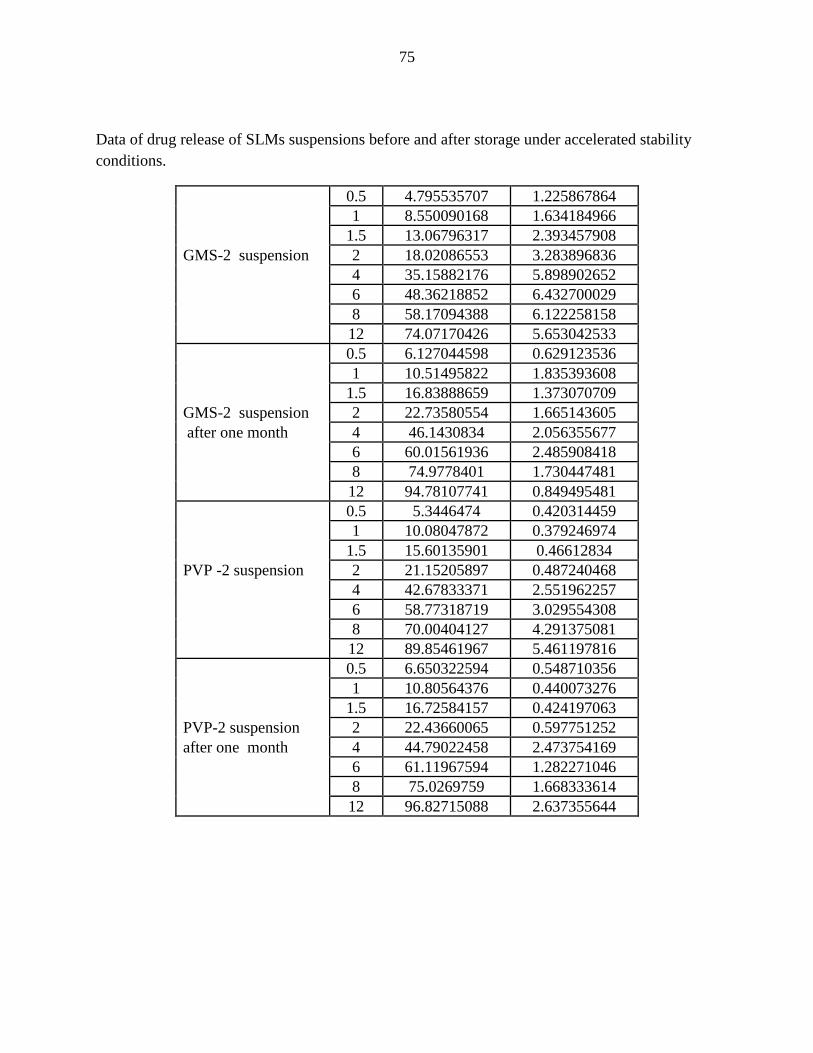
75
Data of drug release of SLMs suspensions before and after storage under accelerated stability
conditions.
0.5 4.795535707 1.225867864
1 8.550090168 1.634184966
1.5 13.06796317 2.393457908
GMS-2 suspension 2 18.02086553 3.283896836
4 35.15882176 5.898902652
6 48.36218852 6.432700029
8 58.17094388 6.122258158
12 74.07170426 5.653042533
0.5 6.127044598 0.629123536
1 10.51495822 1.835393608
1.5 16.83888659 1.373070709
GMS-2 suspension 2 22.73580554 1.665143605
after one month 4 46.1430834 2.056355677
6 60.01561936 2.485908418
8 74.9778401 1.730447481
12 94.78107741 0.849495481
0.5 5.3446474 0.420314459
1 10.08047872 0.379246974
1.5 15.60135901 0.46612834
PVP -2 suspension 2 21.15205897 0.487240468
4 42.67833371 2.551962257
6 58.77318719 3.029554308
8 70.00404127 4.291375081
12 89.85461967 5.461197816
0.5 6.650322594 0.548710356
1 10.80564376 0.440073276
1.5 16.72584157 0.424197063
PVP-2 suspension 2 22.43660065 0.597751252
after one month 4 44.79022458 2.473754169
6 61.11967594 1.282271046
8 75.0269759 1.668333614
12 96.82715088 2.637355644

76
لصلبةة الدهنية اتاثير محسنات تحرير األدوية على تحرير الدواء من الكريات الميكروي
اعداد
سماء عبد الرؤوف خالد فاخوريأ
المشرف
د. حاتم الخطيب
المشرف المشارك
د. محمد خليل
ملخص
كروية الميالكمبرتول كرياتتم استخدام طريقة االستحالب المصهور للحصول على تحضيرات مختلفة من مل ت وتشوية كمثبتاعدة محسنات تحرير لألدالصلبة المحملة بالكاربامزبين و ذلك باستخدام الدهنية
ك في بولي فينيل بيروليدون و بولي فينيل الكحول وغليسيريل مونوستيرات ، و ذلو 127بلورونك ف
اثير تقييم ت. كما تم الكرياتمحاولة لدراسة تأثيرها على الخصائص الفيزيائية و خصائص التحرر من هذه تحرر على ال نتأثير طريقة إضافة البولي فينيل بيروليدوو اربامزبين وكمية الك زيادة كمية محسن التحرر
. الكرياتمن هذه
المختارة تم تقييمها من ناحية تحرر الدواء و الجزء المنفصل من الكرياتالمعلقات المبنية على بعض المعلقات المبنية عليها لظروف فحص الكريات والمعلق و خصائص التدفق. كما تم دراسة تأثير تعرض هذه
الثباتية المتسارع.
ر خصائص التحراظهرت تحسن في وبولي فينيل بيروليدون وغليسيريل مونوستيرات 127بليورونك ف
من كريات الكمبرتول الميكروية الدهنية الصلبة
وحظ لد معين ثم الى ح خصائص التحرر طويل األمداظهر تحسن في زيادة كمية بولي فينيل بيروليدون لتحررخصائص اعدم تغير في تاظهرزيادة كمية غليسيريل مونوستيرات التحرر طويل األمد.تناقص في
.من كريات الكمبرتول الميكروية الدهنية الصلبة
ان تيرات كمن بولي فينيل بيروليدون وغليسيريل مونوس الكرياتالمعلقات المبنية على تحرر الدواء من .بولي فينيل بيروليدون نت الزيادة اكبر عند استخداماوك الكرياتمن من تحرره أسرع

77
عند استخدام بولي فينيل بيروليدون لم يتأثر الكرياتأظهر فحص الثباتية أن معدل تحرر الدواء من هذه ٍٍ من هذين فان معدل ٍٍ ٍٍ ٍٍ ٍٍ وغليسيريل مونوستيرات كمثبتات ، أما بالنسبة للمعلقات المبنية على أيٍ
من بولي فينيل بيروليدون بينما أصبح اسرع من الكرياتالمعلقات المبنية على من الدواء لم يتأثر تحررالمثبتة بواسطة غليسيريل مونوستيرات. الكرياتالمعلقات المبنية على





























