Embed Size (px)
Citation preview

lable at ScienceDirect
Tetrahedron 69 (2013) 8904e8913
Contents lists avai
Tetrahedron
journal homepage: www.elsevier .com/locate/ tet
Electrochemical synthesis of glycoconjugates of 3b-hydroxy-D5-steroids by using non-activated sugars and steroidal thioethers
Aneta M. Tomkiel a, Krzysztof Brzezinski a, Zenon qotowski a, Leszek Siergiejczyk a,Piotr Wa1ejko a, Stanis1aw Witkowski a, Jan Kowalski b, Jolanta P1oszy�nska b,Andrzej Sobkowiak b, Jacek W. Morzycki a,*a Institute of Chemistry, University of Białystok, Hurtowa 1, 15-399 Białystok, Polandb Faculty of Chemistry, Rzesz�ow University of Technology, P.O. Box 85, 35-959 Rzesz�ow, Poland
a r t i c l e i n f o
Article history:Received 25 May 2013Received in revised form 17 July 2013Accepted 30 July 2013Available online 15 August 2013
Keywords:GlycosylationCholesterolElectrochemical oxidationThioethersGlycoconjugates
* Corresponding author. Tel.: þ48 85 7457585; faaddress: [email protected] (J.W. Morzycki).
0040-4020/$ e see front matter � 2013 Elsevier Ltd.http://dx.doi.org/10.1016/j.tet.2013.07.106
a b s t r a c t
A new protocol for the electrochemical synthesis of glycoconjugates is presented. Thioether derivativesof cholesterol and other sterols were subjected to anodic oxidation in the presence of a sugar alcoholaffording glycoconjugates with the sugar linked to a steroid moiety by an ether bond. The isomeric 6b-3a,5a-cyclo-steroidal thioethers proved to be better sterol donors than the normal 3b-D5-steroidalthioethers.
� 2013 Elsevier Ltd. All rights reserved.
1. Introduction
In recent years our group has studied the direct electrochemicaloxidation of cholesterol under various conditions. The products ofoxidation of cholesterol, the most common steroid in the animalkingdom, are present in food due to lipid oxidation reactions in thepresence of oxygen, exposure to sunlight, food processing, etc. Suchcompounds reveal cytotoxic, apoptotic, and pro-inflammatory ac-tivity.1 Therefore, they are intensively studied by chemists andtoxicologists. From the chemical point of view cholesterol isa homoallylic alcohol with a relatively large hydrophobic moiety.The site of cholesterol electrooxidation largely depends on thereaction conditions. We have found that a reaction carried out inacetic acid as a solvent affords products of acetoxylation at the al-lylic position (C7).2 In dichloromethane, products chlorinated at thedouble bond are produced, unless an electrolyzer with separatedelectrode compartments is used.3 Then the one electron oxidationof cholesterol occurs at the oxygen atom to afford a radical cation.The subsequent cleavage of the CeO bond affords HO� and a car-bocation, which reacts with nucleophiles present in the reactionmixture, leading to various products.
x: þ48 85 7457581; e-mail
All rights reserved.
Other chemists have also investigated cholesterol electro-oxidation, but mostly using indirect methods with various media-tors. An interesting, though low-yielding, electrochemical methodof cholesterol side-chain oxidation at the tertiary position (C25)was described by the Takayama group.4 The selective reactionwas achieved with the Tl/(TFA)3-hematoporphyrin-O2-cathodicreductive system. A hydroxyl radical was suggested as an activespecies in this system. The same group has also described anodicoxidation of cholesterol in dichloromethane solution with variousadditives, leading mostly to chlorinated products.5 Takeya et al.have described6 an electrochemical system for stereoselective al-lylic hydroxylation of cholesteryl acetate with dioxygen induced byiron picolinate complexes. There is also one report on direct anodicoxidation of cholesterol at a carbon electrode in acetonitrile.7,8 Thereaction afforded cholesta-4,6-dien-3-one in a high yield. This ex-ample shows that electrochemical oxidation reactions can be usefulsince they are controllable by electrolysis conditions, including thepotential applied, and are relatively cheap due to the low cost ofelectric energy used when compared with chemical reagents.
Electrochemical reactions can also be applied to the synthesis ofglycosides.9 The first electrochemical method of synthesis of gly-cosides was developed by Noyori, who used aryl glycosides asglycosyl donors.10 The method was later improved by using arylthioglycosides instead, i.e., compounds that undergo electro-oxidation at a lower potential.11e14 Electrochemical oxidation of

A.M. Tomkiel et al. / Tetrahedron 69 (2013) 8904e8913 8905
these compounds has been shown to follow an overall EC-type, inwhich the electro-generated cation radical (electrochemical step)undergoes an irreversible carbonechalcogen bond rupture(chemical step) to produce the corresponding glycosyl cation,which reacts with alcohol. The formal oxidation potentials werefound to vary according to the identity of the chalcogenide, suchthat OPh>SPhwSTol>SePh.15 Selective electrochemical glycosyla-tion by reactivity tuning has been attempted.16 The reactions werecarried out in a suitable supporting electrolyte or in the presenceof a small amount of mediator, such as NBS, Br2,17 tris-(4-bromophenyl) ammoniumyl hexachloroantimonate18 or a support-ing electrolyte, e.g., trifluoromethanesulfonate.17
2. Results and discussion
We recently described a new electrochemical method of glyco-sylation of 3b-hydroxy-D5-steroids using non-activated sugars.19 Themethod consisted in electrooxidation of sterol (e.g., cholesterol) inthe presence of a sugar in a non-polar solvent, such as dichloro-methane. The initially formed cation radicals (steroid-OH)þ� un-dergo splitting into a steroidal carbocation and a hydroxyl radical. Inthe presence of a sugar alcohol the carbocation is trapped by a hy-droxyl group to form ethers. In such a way various glycoconjugates(including glycosides) may be prepared, though in rather low yields.There are several reasons why the reaction yields are not satisfac-tory. One is the formation of disteroidal ethers, which are formed asa result of the reaction of the starting sterol with the carbocation.Therefore, a large excess of a sugar substrate should be used to avoidthe formation of these unwanted products. Some by-products areformed due to the relatively high potential needed for the oxidationof sterols. Finally, the hydroxyl radicals or hydrogen peroxide thatare produced during sterol electrooxidation may lead to various sidereactions. The aim of this study is to improve the efficiency of theelectrochemical glycosylation method by using activated sterol de-rivatives. Since the glycosyl thioethers were successfully applied forglycosylation reactions as mentioned above, we attempted to usearyl thioether derivatives of sterols as potential donors of a steroidmoiety in the synthesis of steroid glycoconjugates.
Electrochemical oxidation of non-steroidal alkyl aryl sulfides(ArSR) has previously been studied.20,21 The products of these re-actions are remarkably dependent upon the structure of R, thepolarity of the solvent, and the supporting electrolyte. If R can yieldstabilized carbocations like benzyl or triphenylmethyl cations,extensive cleavage of the CeS bond occurs during oxidation(Scheme 1). With other R groups the corresponding sulfoxides orsulfones are obtained in good yields, especially when the
[PhCH2SPh]+ .
- H+
+ Nu-
+ Nu-
PhCHSPh
.
PhCH2SP
.
Nu
PhCH2++
PhCH2S
Scheme 1. Alternative electrooxidation p
electrolyses are performed in solvents containing water.22,23 Thealternative reaction pathways24 consist of CaeH deprotonation ofthe intermediate cation radical or the substitution of the aryl ring atthe p-position. The former reaction is strongly favored in acetic acidas a solvent and is more likely to occur in the case of the less bulkyalkyl groups, while the latter reaction can be suppressed by using p-substituted aryl thioether.25
In the case of sterol thioethers (cholesteryl aryl sulfides werechosen as model compounds), cleavage of the CeS bond is alsolikely to occur since the resulting carbocation is mesomericallystabilized due to the presence of a homoallylic double bond(Scheme 2). However, in the case of saturated steroid derivatives,the analogous cleavage is not a favored step and electrooxidationfollows a different pathway, or no reaction is observed.
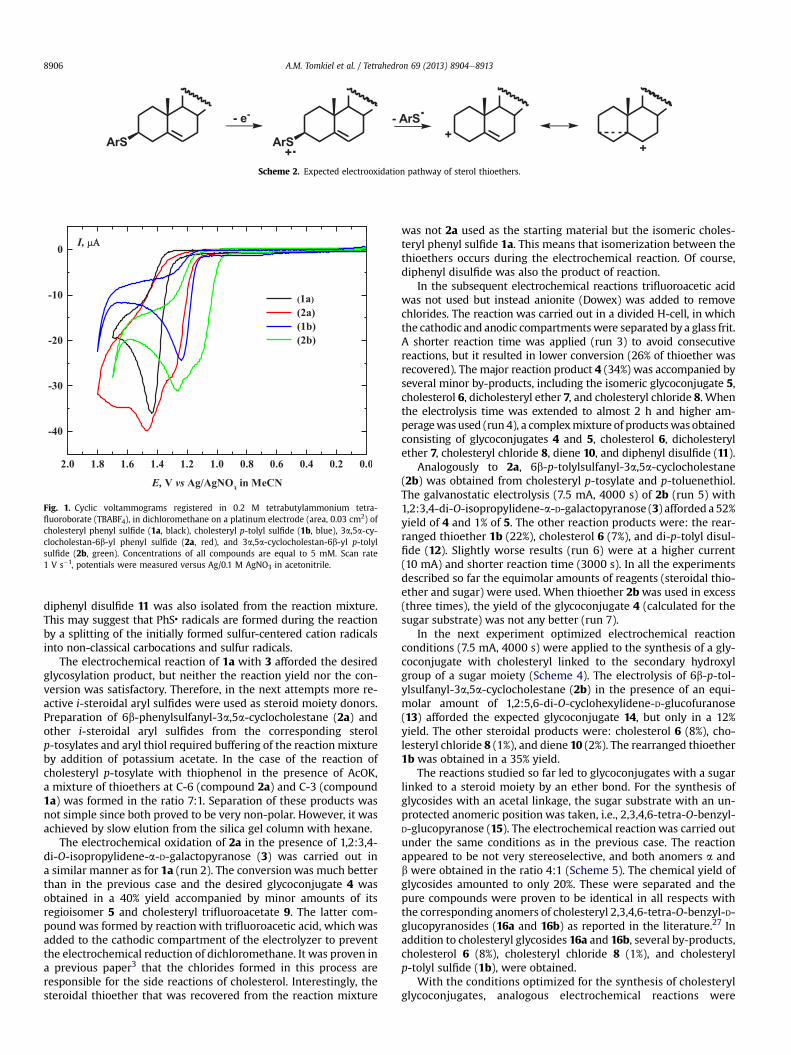
Fig. 1 indicates that all of the sterol thioesters (cholesterylphenyl sulfide 1a, cholesteryl p-tolyl sulfide 1b, 3a,5a-cyclo-cholestan-6b-yl phenyl sulfide 2a, and 3a,5a-cyclocholestan-6b-ylp-tolyl sulfide 2b), which were chosen for preparative electrolysisin this study are electroactive in the potential region where thepreparative processes occur. Cholesteryl phenyl sulfide (1a, blackcurve) and cholesteryl p-tolyl sulfide (1b, blue curve) form well-developed anodic peaks at potentials þ1.44 V and þ1.24 V (vs Ag/0.1 M AgNO3 in MeCN), respectively. 3a,5a-Cyclocholestan-6b-ylphenyl sulfide (2a, red curve) and 3a,5a-cyclocholestan-6b-yl p-tolyl sulfide (2b, green curve) give broad oxidation peaks with themain peak potentials equal toþ1.47 V andþ1.27 V for (2a) and (2b),respectively. All of the peaks are irreversible and no cathodic peaksare present in the subsequent cathodic scans. It is worth noting thatcholesteryl phenyl ether is also oxidized under the same experi-mental conditions. Its anodic peak potential is equal to 1.65 V.
The study on glycosylation using aryl sulfide derivatives of ste-rols (Scheme 3) was preceded by attempts with cholesteryl phenylether. Electrooxidation of this compound with a model sugar,1,2:3,4-di-O-isopropylidene-a-D-galactopyranose (3), containingthe free OH group at the primary position, was carried out indichloromethane. However, the expected glycoconjugate 4 wasobtained only in trace amounts due to a very low conversion.
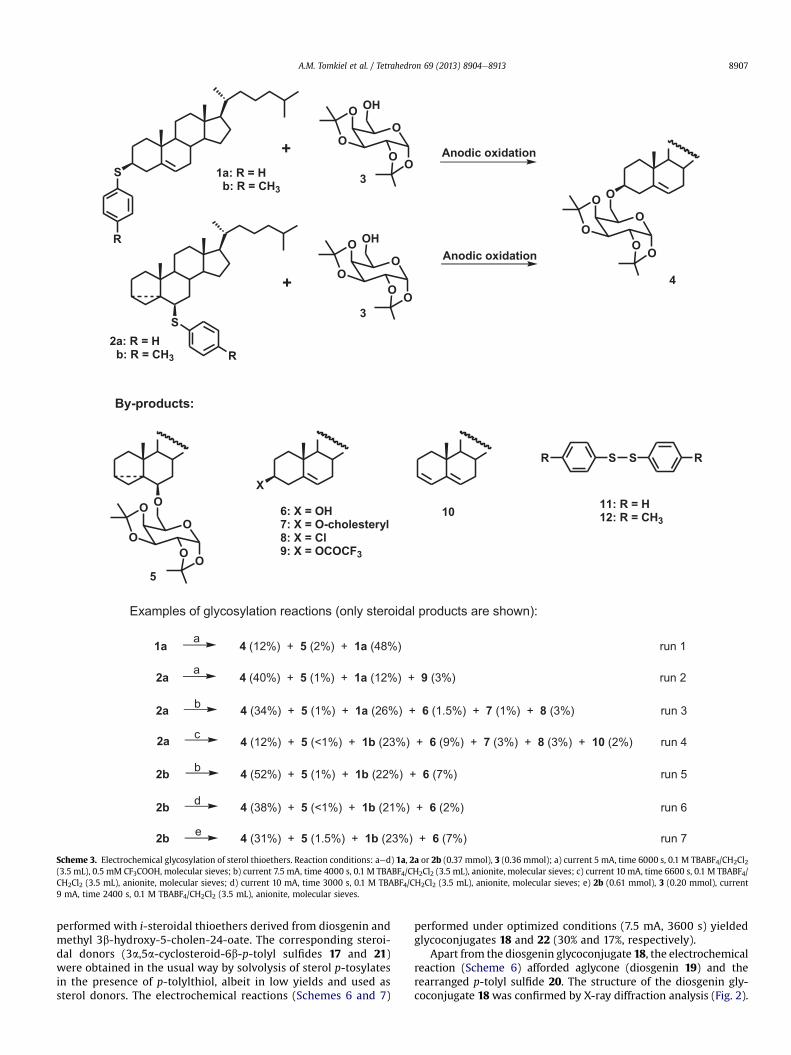
Then cholesteryl phenyl sulfide (1a) was prepared by reaction ofcholesteryl p-tosylate with ten equivalents of thiophenol in dioxaneat reflux. Electrochemical oxidation of 1a in the presence of anequimolar amount of the galactose derivative 3 (run 1) in dichloro-methane afforded product 4 (12%) along with its regioisomer 5 (2%).However, almost half of the starting cholesteryl phenyl sulfide (1a)was recovered unchanged. Isolation of the i-steroidal glycoconjugate5 is a direct proof that the non-classical carbocation is an in-termediate in the reaction. In addition to steroidal products,
- e-+ Nu
-
PhCHSPh
Nu
h
- e-
- H+
PhCHSPh
O-
+
SPh
. + Nu-
PhCH2Nu + (PhS)2
H
Nu
.- e-
- H+
PhCH2S Nu
+ H2O
- NuH
athways of benzyl phenyl sulfides.26
ArS
- ArS
.
+
+ArS
- e-
.+
Scheme 2. Expected electrooxidation pathway of sterol thioethers.
Fig. 1. Cyclic voltammograms registered in 0.2 M tetrabutylammonium tetra-fluoroborate (TBABF4), in dichloromethane on a platinum electrode (area, 0.03 cm2) ofcholesteryl phenyl sulfide (1a, black), cholesteryl p-tolyl sulfide (1b, blue), 3a,5a-cy-clocholestan-6b-yl phenyl sulfide (2a, red), and 3a,5a-cyclocholestan-6b-yl p-tolylsulfide (2b, green). Concentrations of all compounds are equal to 5 mM. Scan rate1 V s�1, potentials were measured versus Ag/0.1 M AgNO3 in acetonitrile.
A.M. Tomkiel et al. / Tetrahedron 69 (2013) 8904e89138906
diphenyl disulfide 11 was also isolated from the reaction mixture.This may suggest that PhS� radicals are formed during the reactionby a splitting of the initially formed sulfur-centered cation radicalsinto non-classical carbocations and sulfur radicals.
The electrochemical reaction of 1a with 3 afforded the desiredglycosylation product, but neither the reaction yield nor the con-version was satisfactory. Therefore, in the next attempts more re-active i-steroidal aryl sulfides were used as steroid moiety donors.Preparation of 6b-phenylsulfanyl-3a,5a-cyclocholestane (2a) andother i-steroidal aryl sulfides from the corresponding sterolp-tosylates and aryl thiol required buffering of the reaction mixtureby addition of potassium acetate. In the case of the reaction ofcholesteryl p-tosylate with thiophenol in the presence of AcOK,a mixture of thioethers at C-6 (compound 2a) and C-3 (compound1a) was formed in the ratio 7:1. Separation of these products wasnot simple since both proved to be very non-polar. However, it wasachieved by slow elution from the silica gel column with hexane.
The electrochemical oxidation of 2a in the presence of 1,2:3,4-di-O-isopropylidene-a-D-galactopyranose (3) was carried out ina similar manner as for 1a (run 2). The conversion was much betterthan in the previous case and the desired glycoconjugate 4 wasobtained in a 40% yield accompanied by minor amounts of itsregioisomer 5 and cholesteryl trifluoroacetate 9. The latter com-pound was formed by reaction with trifluoroacetic acid, which wasadded to the cathodic compartment of the electrolyzer to preventthe electrochemical reduction of dichloromethane. It was proven ina previous paper3 that the chlorides formed in this process areresponsible for the side reactions of cholesterol. Interestingly, thesteroidal thioether that was recovered from the reaction mixture
was not 2a used as the starting material but the isomeric choles-teryl phenyl sulfide 1a. This means that isomerization between thethioethers occurs during the electrochemical reaction. Of course,diphenyl disulfide was also the product of reaction.
In the subsequent electrochemical reactions trifluoroacetic acidwas not used but instead anionite (Dowex) was added to removechlorides. The reaction was carried out in a divided H-cell, in whichthe cathodic and anodic compartmentswere separated by a glass frit.A shorter reaction time was applied (run 3) to avoid consecutivereactions, but it resulted in lower conversion (26% of thioether wasrecovered). The major reaction product 4 (34%) was accompanied byseveral minor by-products, including the isomeric glycoconjugate 5,cholesterol 6, dicholesteryl ether 7, and cholesteryl chloride 8. Whenthe electrolysis time was extended to almost 2 h and higher am-peragewas used (run 4), a complexmixture of productswas obtainedconsisting of glycoconjugates 4 and 5, cholesterol 6, dicholesterylether 7, cholesteryl chloride 8, diene 10, and diphenyl disulfide (11).
Analogously to 2a, 6b-p-tolylsulfanyl-3a,5a-cyclocholestane(2b) was obtained from cholesteryl p-tosylate and p-toluenethiol.The galvanostatic electrolysis (7.5 mA, 4000 s) of 2b (run 5) with1,2:3,4-di-O-isopropylidene-a-D-galactopyranose (3) afforded a 52%yield of 4 and 1% of 5. The other reaction products were: the rear-ranged thioether 1b (22%), cholesterol 6 (7%), and di-p-tolyl disul-fide (12). Slightly worse results (run 6) were at a higher current(10 mA) and shorter reaction time (3000 s). In all the experimentsdescribed so far the equimolar amounts of reagents (steroidal thio-ether and sugar) were used. When thioether 2bwas used in excess(three times), the yield of the glycoconjugate 4 (calculated for thesugar substrate) was not any better (run 7).
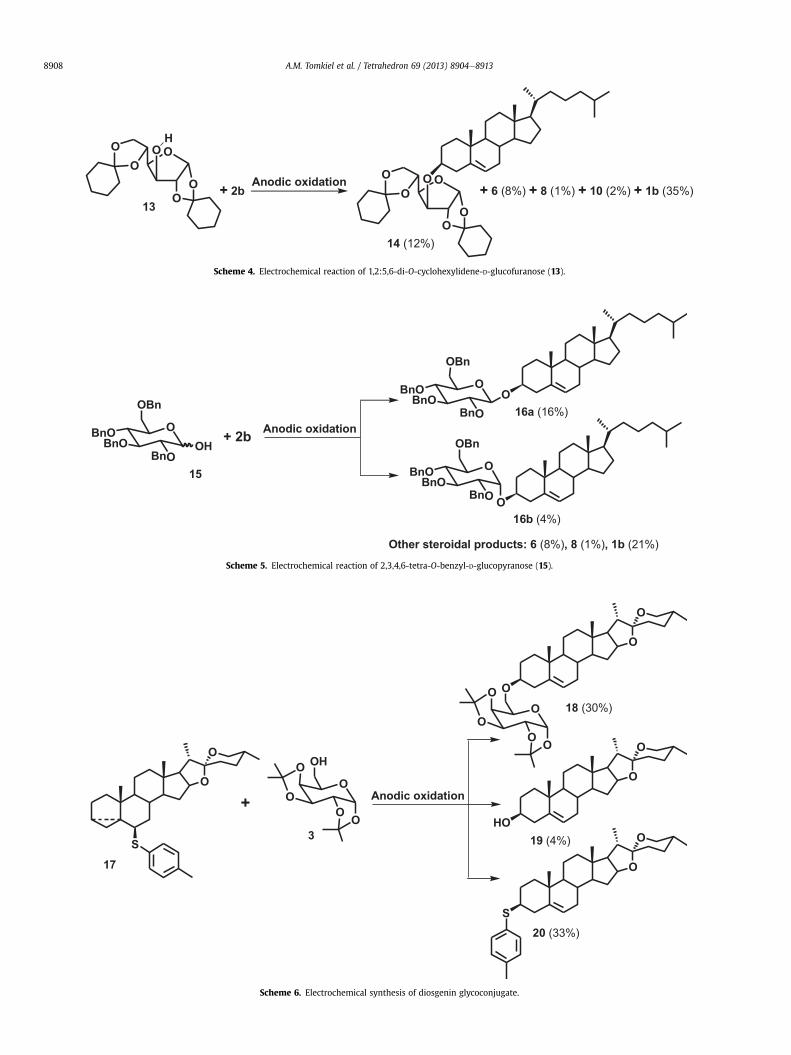
In the next experiment optimized electrochemical reactionconditions (7.5 mA, 4000 s) were applied to the synthesis of a gly-coconjugate with cholesteryl linked to the secondary hydroxylgroup of a sugar moiety (Scheme 4). The electrolysis of 6b-p-tol-ylsulfanyl-3a,5a-cyclocholestane (2b) in the presence of an equi-molar amount of 1,2:5,6-di-O-cyclohexylidene-D-glucofuranose(13) afforded the expected glycoconjugate 14, but only in a 12%yield. The other steroidal products were: cholesterol 6 (8%), cho-lesteryl chloride 8 (1%), and diene 10 (2%). The rearranged thioether1b was obtained in a 35% yield.
The reactions studied so far led to glycoconjugates with a sugarlinked to a steroid moiety by an ether bond. For the synthesis ofglycosides with an acetal linkage, the sugar substrate with an un-protected anomeric position was taken, i.e., 2,3,4,6-tetra-O-benzyl-D-glucopyranose (15). The electrochemical reaction was carried outunder the same conditions as in the previous case. The reactionappeared to be not very stereoselective, and both anomers a andb were obtained in the ratio 4:1 (Scheme 5). The chemical yield ofglycosides amounted to only 20%. These were separated and thepure compounds were proven to be identical in all respects withthe corresponding anomers of cholesteryl 2,3,4,6-tetra-O-benzyl-D-glucopyranosides (16a and 16b) as reported in the literature.27 Inaddition to cholesteryl glycosides 16a and 16b, several by-products,cholesterol 6 (8%), cholesteryl chloride 8 (1%), and cholesterylp-tolyl sulfide (1b), were obtained.
With the conditions optimized for the synthesis of cholesterylglycoconjugates, analogous electrochemical reactions were
S
O
O
O
O
O
O
OH
O
O
O
O
O
+ Anodic oxidation
O
O
O
O
O
O
OH
O
O
O
O
O
+
Anodic oxidation
S
R
3
3
4
2a: R = H
b: R = CH3
11: R = H
12: R = CH3
By-products:
5
X
6: X = OH
7: X = O-cholesteryl
8: X = Cl
9: X = OCOCF3
10
S S RR
Examples of glycosylation reactions (only steroidal products are shown):
1a 4 (12%) + 5 (2%) + 1a (48%)a
2aa
4 (40%) + 5 (1%) + 1a (12%) + 9 (3%)
2ab
4 (34%) + 5 (1%) + 1a (26%) + 6 (1.5%) + 7 (1%) + 8 (3%)
2a
b4 (52%) + 5 (1%) + 1b (22%) + 6 (7%)
R
1a: R = H
b: R = CH3
2b
c4 (12%) + 5 (<1%) + 1b (23%) + 6 (9%) + 7 (3%) + 8 (3%) + 10 (2%)
run 1
run 2
run 3
run 4
run 6
2b
d
run 5
4 (38%) + 5 (<1%) + 1b (21%) + 6 (2%)
2be
4 (31%) + 5 (1.5%) + 1b (23%) + 6 (7%) run 7
Scheme 3. Electrochemical glycosylation of sterol thioethers. Reaction conditions: aed) 1a, 2a or 2b (0.37 mmol), 3 (0.36 mmol); a) current 5 mA, time 6000 s, 0.1 M TBABF4/CH2Cl2(3.5 mL), 0.5 mM CF3COOH, molecular sieves; b) current 7.5 mA, time 4000 s, 0.1 M TBABF4/CH2Cl2 (3.5 mL), anionite, molecular sieves; c) current 10 mA, time 6600 s, 0.1 M TBABF4/CH2Cl2 (3.5 mL), anionite, molecular sieves; d) current 10 mA, time 3000 s, 0.1 M TBABF4/CH2Cl2 (3.5 mL), anionite, molecular sieves; e) 2b (0.61 mmol), 3 (0.20 mmol), current9 mA, time 2400 s, 0.1 M TBABF4/CH2Cl2 (3.5 mL), anionite, molecular sieves.
A.M. Tomkiel et al. / Tetrahedron 69 (2013) 8904e8913 8907
performed with i-steroidal thioethers derived from diosgenin andmethyl 3b-hydroxy-5-cholen-24-oate. The corresponding steroi-dal donors (3a,5a-cyclosteroid-6b-p-tolyl sulfides 17 and 21)were obtained in the usual way by solvolysis of sterol p-tosylatesin the presence of p-tolylthiol, albeit in low yields and used assterol donors. The electrochemical reactions (Schemes 6 and 7)
performed under optimized conditions (7.5 mA, 3600 s) yieldedglycoconjugates 18 and 22 (30% and 17%, respectively).
Apart from the diosgenin glycoconjugate 18, the electrochemicalreaction (Scheme 6) afforded aglycone (diosgenin 19) and therearranged p-tolyl sulfide 20. The structure of the diosgenin gly-coconjugate 18was confirmed by X-ray diffraction analysis (Fig. 2).
OAnodic oxidation O
O
O
O
O
OO
O
O
O
O
H
13
14 (12%)
+ 6 (8%) + 8 (1%) + 10 (2%) + 1b (35%)+ 2b
Scheme 4. Electrochemical reaction of 1,2:5,6-di-O-cyclohexylidene-D-glucofuranose (13).
OH
O
O
O
O
O
+Anodic oxidation
S
3
17
O
O
20 (33%)
O
O
S
19 (4%)
O
O
HO
18 (30%)
O
O
O
O
O
O
O
O
Scheme 6. Electrochemical synthesis of diosgenin glycoconjugate.
OBn
O
O
BnO
BnO
O
+ 2bAnodic oxidation
Bn
OBn
O
O
BnO
BnOO
Bn
OBn
O
O
BnO
BnO
Bn
OH
15
16b (4%)
16a (16%)
Other steroidal products: 6 (8%), 8 (1%), 1b (21%)
Scheme 5. Electrochemical reaction of 2,3,4,6-tetra-O-benzyl-D-glucopyranose (15).
A.M. Tomkiel et al. / Tetrahedron 69 (2013) 8904e89138908
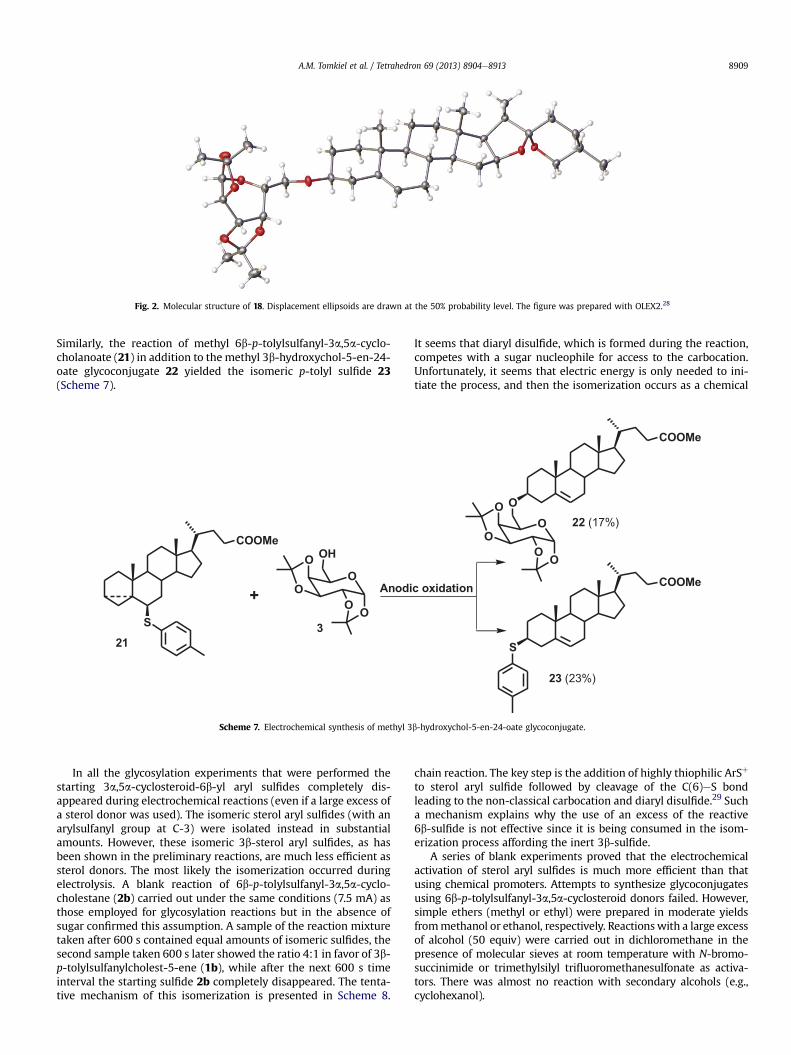
Fig. 2. Molecular structure of 18. Displacement ellipsoids are drawn at the 50% probability level. The figure was prepared with OLEX2.28
A.M. Tomkiel et al. / Tetrahedron 69 (2013) 8904e8913 8909
Similarly, the reaction of methyl 6b-p-tolylsulfanyl-3a,5a-cyclo-cholanoate (21) in addition to the methyl 3b-hydroxychol-5-en-24-oate glycoconjugate 22 yielded the isomeric p-tolyl sulfide 23(Scheme 7).
OH
O
O
O
O
O
+Anodic oxidation
S3
21
23 (23%)
S
22 (17%)O
O
O
O
O
O
COOMe
COOMe
COOMe
Scheme 7. Electrochemical synthesis of methyl 3b-hydroxychol-5-en-24-oate glycoconjugate.
In all the glycosylation experiments that were performed thestarting 3a,5a-cyclosteroid-6b-yl aryl sulfides completely dis-appeared during electrochemical reactions (even if a large excess ofa sterol donor was used). The isomeric sterol aryl sulfides (with anarylsulfanyl group at C-3) were isolated instead in substantialamounts. However, these isomeric 3b-sterol aryl sulfides, as hasbeen shown in the preliminary reactions, are much less efficient assterol donors. The most likely the isomerization occurred duringelectrolysis. A blank reaction of 6b-p-tolylsulfanyl-3a,5a-cyclo-cholestane (2b) carried out under the same conditions (7.5 mA) asthose employed for glycosylation reactions but in the absence ofsugar confirmed this assumption. A sample of the reaction mixturetaken after 600 s contained equal amounts of isomeric sulfides, thesecond sample taken 600 s later showed the ratio 4:1 in favor of 3b-p-tolylsulfanylcholest-5-ene (1b), while after the next 600 s timeinterval the starting sulfide 2b completely disappeared. The tenta-tive mechanism of this isomerization is presented in Scheme 8.
It seems that diaryl disulfide, which is formed during the reaction,competes with a sugar nucleophile for access to the carbocation.Unfortunately, it seems that electric energy is only needed to ini-tiate the process, and then the isomerization occurs as a chemical
chain reaction. The key step is the addition of highly thiophilic ArSþ
to sterol aryl sulfide followed by cleavage of the C(6)eS bondleading to the non-classical carbocation and diaryl disulfide.29 Sucha mechanism explains why the use of an excess of the reactive6b-sulfide is not effective since it is being consumed in the isom-erization process affording the inert 3b-sulfide.
A series of blank experiments proved that the electrochemicalactivation of sterol aryl sulfides is much more efficient than thatusing chemical promoters. Attempts to synthesize glycoconjugatesusing 6b-p-tolylsulfanyl-3a,5a-cyclosteroid donors failed. However,simple ethers (methyl or ethyl) were prepared in moderate yieldsfrommethanol or ethanol, respectively. Reactionswith a large excessof alcohol (50 equiv) were carried out in dichloromethane in thepresence of molecular sieves at room temperature with N-bromo-succinimide or trimethylsilyl trifluoromethanesulfonate as activa-tors. There was almost no reaction with secondary alcohols (e.g.,cyclohexanol).

C8H17
SAr
S
- e-
+. +
+
Ar
- ArS
..
2 ArS ArS-SArS
Ar
ArS-SAr
- ArS+
ArS+
C8H17
SAr
C8H17
SAr
S
+ Ar
ArS-SAr-
+
+S
Ar
ArS-SAr
- ArS+ etc.
Scheme 8. Isomerization of aryl sulfides during electrolysis.
A.M. Tomkiel et al. / Tetrahedron 69 (2013) 8904e89138910
3. Conclusions
A new electrochemical method of synthesis of sterol glyco-conjugates was developed. The method does not require activationof a sugar substrate. Sterol is activated by formation of the corre-sponding aryl sulfide. However, C-3 aryl sulfides proved to be in-sufficiently reactive in the electrochemical processes. Therefore,isomeric 3a,5a-cyclosteroid-6b-yl aryl sulfides were prepared andtested as sterol donors in the electrochemical reactions. 6b-p-Tol-ylsulfanyl-3a,5a-cyclosteroids proved superior to 6b-phenyl-sulfanyl-3a,5a-cyclosteroids in this respect. With these sulfides,moderate to good yields of glycoconjugates were achieved andlesser amounts of by-products were formed. The described elec-trochemical method is better suited for the construction of glyco-conjugates (ethers) from sugar primary alcohols than from sugarswith the hydroxyl group at the secondary or anomeric positions. Itwas proven that rapid isomerization of reactive 6b- to inert 3b-sulfides occurs during electrolysis, and for this reason conversionsachieved in many cases were unsatisfactory. Further study on theelectrochemical glycosylation of sterols is in progress.
4. Experimental section
4.1. General methods
Cyclic-voltammograms were recorded with iR compensation at25 �C using a three-electrode potentiostat (Bioanalytical SystemsModel CV-50W). The experiments were conducted in a 15-mLelectrochemical cell with an argon-purge system. The workingelectrode was a Bioanalytical Systems platinum inlay (diameter,1 mm), the auxiliary electrode a platinum mesh (contained ina glass tube with a medium porosity glass frit), and the referenceelectrode Ag/0.1 M AgNO3 in acetonitrile. The latter was containedin a Pyrex tubewith a cracked softglass tip, which was placed insidea Luggin capillary. Before each experiment the working electrodewas polished using a Buehler Micropolish Alumina Gamma 3B andBuehler Microcloth polishing cloth, rinsed with dichloromethane,and dried. The accuracy of potential measurement was equal to10 mV. All reported values of potentials and currents are means ofthree independent measurements.
The preparative electrolyses were performed with a potentio-stat/galvanostat (Princeton Applied Research Model Parstat 2273)under galvanostatic conditions using a current equal to 5e10 mAand the reaction time 2000e6000 s. The current applied was themaximum current available for the used electrolysis setup (powersupply and ohmic resistance). The reactions were monitored by
TLC and stopped when no further increase in the concentration ofglycosylation products was observed. A divided H-cell was used inwhich the cathodic and anodic compartments (3.5 mL of electro-lytes each) were separated by a glass frit. In all the measurements0.1 M solution of tetrabutylammonium tetrafluoroborate (TBABF4,from Aldrich) in dichloromethane was used as a supporting elec-trolyte. The steroid and sugar substrates were introduced into theanodic compartment together with a 0.3 g of 3A molecular sieve toeliminate traces of water, whereas anionite (1.5e2 g, Dowex 2x8,200e400 mesh, perchlorate form) was placed in the cathodiccompartment to eliminate chloride ions from forming by the re-duction of dichloromethane. The solutions in both compartmentswere stirred during electrolysis and additionally a continuousflow of argon was applied in the anodic compartment. A platinummesh was used as a cathode and a platinum plate (2�1.5 cm) wasused as an anode. All measurements were performed at roomtemperature.
The sugar substrates (1,2:5,6-di-O-cyclohexylidene-D-glucofur-anose,30 2,3,4,6-tetra-O-benzyl-D-glucopyranose,31 and 1,2:3,4-di-O-isopropylidene-a-D-galactopyranose32) were prepared accordingto known procedures.30e32
Melting points were determined on a Toledo Mettler-MP70apparatus. 1H and 13C NMR (400 and 100 MHz, respectively)spectra were recorded on a Bruker Avance II spectrometer in CDCl3solutions with TMS as the internal standard (only selected signalsin the 1H NMR spectra are reported). Infrared spectra were recor-ded on a Nicolet series II Magna-IR 550 FTIR spectrometer inchloroform solutions. Mass spectra were recorded at 70 eV witha time-of-flight (TOF) AMD-604 spectrometer with electrosprayionization (ESI) or AutoSpec Premier (Waters) (EI). Optical rotationswere measured on a PerkineElmer 141 polarimeter at the sodiumD-line (589 nm).
Merck Silica Gel 60, F 256 TLC aluminum sheets were applied forthin-layer chromatographic analysis. For a visualization of theproducts a 5% solution of phosphomolybdic acid in ethanol wasused. The reaction products were separated by column chroma-tography performed on a 70e230 mesh silica gel (J.T. Baker).
4.1.1. X-ray diffraction analysis of 18. Crystals suitable for X-ray dif-fractionstudywereobtainedat roomtemperaturebyslowevaporationfrom a hexane solution. The X-ray diffraction data were collected at100(2) K on a SuperNova diffractometer (Agilent) with a CCD detectorand CuKa radiation. The crystal structure was solved using directmethodswithSHELXDand refinedwithSHELXL.33All hydrogenatomswere initially located in electron-density difference maps and wereconstrained to idealized positions, with CeH¼0.95e1.00 �A and with

A.M. Tomkiel et al. / Tetrahedron 69 (2013) 8904e8913 8911
Uiso(H)¼1.5Ueq(C) for the methyl hydrogen atoms and Uiso(H)¼1.2Ueq(C) for others. The non-hydrogen atoms were refined aniso-tropically. Crystal data for (18): C39H60O8,Mr¼656.87, colorless needle,0.80�0.15�0.10 mm3, orthorhombic space group P212121,a¼10.61462(9)�A,b¼11.07419(12)�A, c¼30.5223(3)�A,V¼3587.85(6)�A3,Z¼4, rcalcd¼1.216 g cm�3, m¼0.67 mm�1, F(000)¼1432, R1¼0.0278,wR2¼0.0689, 3600 independent reflections, qmax¼68.3�, qmin¼2.9�,432 parameters, and 0 restraints. CCDC-930074 contains the supple-mentary crystallographic data for this paper. These data can be ob-tained freeof charge fromTheCambridgeCrystallographicDataCentrevia www.ccdc.cam.ac.uk/data_request/cif.
4.2. Synthesis of 3b-phenylsulfanylcholest-5-ene (1a)
Cholesteryl p-tosylate (443 mg; 1 mmol) was dissolved in di-oxane (25 mL), and thiophenol (1.1 g; 10 mmol) was added. Thereaction mixture was refluxed under argon for 4 h and, aftercooling, poured into a 1 M NaOH (200 mL) solution. The reactionproduct was exhaustively extracted with benzene (3�100 mL), thecombined extract was dried over anhydrous Na2SO4, and evapo-rated in vacuo. The crude product was purified by silica gel columnchromatography affording 283 mg (59% yield) of pure 1a (elutedwith hexane).
Compound 1a; colorless crystals; Rf¼0.39 (CH2Cl2/hexane 1:9);mp 78e80 �C (acetone); lit.34 mp 72e74 �C; [a]D20 �37.1 (c 1.0,CHCl3); lit.34 [a]D �39 (c 1.1, CHCl3); IR, nmax: 3062, 1584, 1467, 1091,1025 cm�1; 1H NMR, d (ppm): 7.42 (d, 2H, J¼8.2 Hz, HeAr), 7.28 (m,3H, HeAr), 5.34 (m, 1H, H-6), 3.05 (m, 1H, H-3a), 1.02 (s, 3H, H-19),0.95 (d, 3H, J¼6.4 Hz, H-21), 0.91 (d, 3H, J¼6.6 Hz, H-26 or H-27),0.90 (d, 3H, J¼6.7 Hz, H-26 or H-27), 0.71 (s, 3H, H-18); 13C NMR(ppm), d: 141.6 (C),134.9 (C),131.7 (CH),128.7 (CH),126.6 (CH),121.1(CH), 56.7 (CH), 56.2 (CH), 50.3 (CH), 47.3 (CH), 42.3 (C), 39.8 (CH2),39.63 (CH2), 39.62 (CH2), 39.5 (CH2), 36.8 (C), 36.2 (CH2), 35.8 (CH),31.83 (CH2), 31.80 (CH), 29.5 (CH2), 28.2 (CH2), 28.0 (CH), 24.2 (CH2),23.8 (CH2), 22.8 (CH3), 22.6 (CH3), 20.9 (CH2), 19.3 (CH3), 18.7 (CH3),11.8 (CH3); ESI MS, m/z: 501 (MNaþ, 1%); elemental analysis calcd(%) for C33H50S: C 82.78, H 10.53, S 6.70; found: C 82.49, H 10.56, S6.72.
4.2.1. Other sterol thioethers (obtained as products of electrochemicalreactions)
4.2.1.1. 3b-p-Tolylsulfanylcholest-5-ene (1b). Colorless crystals;Rf¼0.34 (CH2Cl2/hexane 1:9); mp 77e78 �C (acetone); [a]D20 �23.5(c 1.0, CHCl3); IR, nmax: 1492, 1467, 1092, 1019 cm�1; 1H NMR,d (ppm): 7.32 (d, 2H, J¼8.0 Hz, HeAr), 7.11 (d, 2H, J¼8.0 Hz, HeAr),5.31 (m,1H, H-6), 2.95 (m,1H, H-3a), 2.34 (s, 3H, H-Ar-CH3), 0.99 (s,3H, H-19), 0.93 (d, 3H, J¼6.5 Hz, H-21), 0.883 (d, 3H, J¼6.6 Hz, H-26or H-27), 0.879 (d, 3H, J¼6.6 Hz, H-26 or H-27), 0.68 (s, 3H, H-18);13C NMR (ppm), d: 141.8 (C), 136.9 (C), 132.6 (CH), 130.9 (C), 129.5(CH), 121.0 (CH), 56.8 (CH), 56.2 (CH), 50.3 (CH), 47.8 (CH), 42.3 (C),39.8 (CH2), 39.7 (CH2), 39.6 (CH2), 39.5 (CH2), 36.9 (C), 36.2 (CH2),35.8 (CH), 31.9 (CH2), 31.8 (CH), 29.6 (CH2), 28.2 (CH2), 28.0 (CH),24.3 (CH2), 23.8 (CH2), 22.8 (CH3), 22.6 (CH3), 21.1 (CH3), 20.9 (CH2),19.3 (CH3), 18.7 (CH3), 11.9 (CH3); EIMS, m/z: 492 (Mþ, 15%), 369[(M�p-CH3C6H4S)þ, 100%]; HRMS (EI): m/z calcd for C34H52S:492.3790; found 492.3773.
4.2.1.2. 3b-p-Tolylsulfanyl-25R-spirost-5-ene (20). Colorlesscrystals; Rf¼0.46 (AcOEt/hexane 5:95); mp 185e186 �C (hexane);[a]D20 �84.8 (c 0.5, CHCl3); IR, nmax: 1049, 1007, 812 cm�1; 1H NMR,d (ppm): 7.32 (d, 2H, J¼8.1 Hz, HeAr), 7.11 (d, 2H, J¼8.1 Hz, HeAr),5.30 (d, 1H, J¼4.5 Hz, H-6), 4.42 (m, 1H, H-16), 3.48 (m, 1H, H-26a),3.39 (t, 1H, J¼10.8 Hz, H-26b), 2.94 (m, 1H, H-3a), 2.34 (s, 3H,HeAreCH3), 1.01 (s, 3H, H-19), 0.98 (d, 3H, J¼7.0 Hz, H-21), 0.80 (d,3H, J¼6.8 Hz, H-27), 0.79 (s, 3H, H-18); 13C NMR (ppm), d: 141.8 (C),
136.9 (C),132.6 (CH),130.8 (C),129.6 (CH),120.8 (CH),109.3 (C), 80.8(CH), 66.8 (CH2), 62.1 (CH), 56.5 (CH), 50.2 (CH), 47.8 (CH), 41.6 (CH),40.3 (C), 39.8 (CH2), 39.63 (CH2), 39.59 (CH2), 37.0 (C), 32.0 (CH2),31.8 (CH2), 31.39 (CH2), 31.36 (CH), 30.3 (CH), 29.5 (CH2), 28.8 (CH2),21.1 (CH3), 20.7 (CH2), 19.4 (CH3), 17.1 (CH3), 16.3 (CH3), 14.5 (CH3);EIMS, m/z: 520 (Mþ, 72%), 397 [(M�p-CH3C6H4S)þ, 100%]; HRMS(EI): m/z calcd for C34H48O2S: 520.3375; found 520.3366.
4.2.1.3. Methyl 3b-p-tolylsulfanylchol-5-enoate (23). Whitesolid; Rf¼0.54 (AcOEt/hexane 1:9); [a]D20 �8.4 (c 1.0, CHCl3); IR,nmax: 1731, 1492, 1172, 1102, 1018 cm�1; 1H NMR, d (ppm): 7.32 (d,2H, J¼8.0 Hz, HeAr), 7.10 (d, 2H, J¼8.0 Hz, HeAr), 5.30 (m, 1H, H-6),3.67 (s, 3H, Hemethyl ester), 2.94 (m, 1H, H-3a), 2.34 (s, 3H,HeAreCH3), 0.99 (s, 3H, H-19), 0.94 (d, 3H, J¼6.4 Hz, H-21), 0.69 (s,3H, H-18); 13C NMR, d (ppm): 174.7 (C), 141.8 (C), 136.9 (C), 132.6(CH), 130.9 (C), 129.5 (CH), 121.0 (CH), 56.7 (CH), 55.8 (CH), 51.4(CH3), 50.3 (CH), 47.8 (CH), 42.4 (C), 39.72 (CH2), 39.64 (CH2), 39.62(CH2), 36.8 (C), 35.4 (CH), 31.80 (CH), 31.81 (CH2), 31.1 (CH2), 31.0(CH2), 29.5 (CH2), 28.1 (CH2), 24.2 (CH2), 21.1 (CH3), 20.9 (CH2), 19.3(CH3), 18.3 (CH3), 11.8 (CH3); ESI MS, m/z: 517 (MNaþ, 100%), 1011[(2MþNa)þ, 10%]; HRMS (ESI):m/z calcd for C32H46O2SNa: 517.3116;found 517.3107.
4.3. Synthesis of 6b-phenylsulfanyl-3a,5a-cyclocholestane(2a)
Cholesteryl p-tosylate (443 mg; 1 mmol) was dissolved in di-oxane (25 mL), then potassium acetate (590 mg; 6 mmol), andthiophenol (1.1 g; 10 mmol) were added. The reaction mixture wasrefluxed under argon for 6 h and, after cooling, poured into a 1 MNaOH (200 mL) solution. The reaction product was exhaustivelyextracted with benzene (3�100 mL), the combined extract wasdriedwith anhydrous Na2SO4, and evaporated in vacuo. The residuewas subjected to silica gel column chromatography. Compounds 2a(239 mg; 50%) and 1a (43 mg; 9%) were consecutively eluted withhexane.
Compound 2a; colorless oil; Rf¼0.40 (CH2Cl2/hexane 1:9); [a]D20
�3.2 (c 1.0, CHCl3); lit.34 [a]D þ34 (c 1.1, CHCl3); IR, nmax: 3062,1584,1468, 1089, 1025 cm�1; 1H NMR, d (ppm): 7.38 (d, 2H, J¼8.0 Hz,HeAr), 7.24 (m, 3H, HeAr), 2.92 (dd,1H, J1¼3.7 Hz, J2¼2.3 Hz, H-6a),1.10 (s, 3H, H-19), 0.93 (d, 3H, J¼6.5 Hz, H-21), 0.883 (d, 3H,J¼6.6 Hz, H-26 or H-27), 0.878 (d, 3H, J¼6.6 Hz, H-26 or H-27), 0.77(s, 3H, H-18), 0.68 (dd, 1H, J1¼5.2 Hz, J2¼4.2 Hz, H-4a), 0.42 (dd, 1H,J1¼8.0 Hz, J2¼5.2 Hz, H-4b); 13C NMR, d: 137.2 (C), 132.1 (CH), 128.7(CH), 126.5 (CH), 56.4 (CH), 56.0 (CH), 54.4 (CH), 47.9 (CH), 43.2(C), 42.8 (C), 40.2 (CH2), 39.5 (CH2), 36.9 (C), 36.2 (CH2), 35.8 (CH),35.5 (CH2), 33.6 (CH2), 30.6 (CH), 28.6 (CH), 28.3 (CH2), 28.0 (CH),25.3 (CH2), 24.2 (CH2), 23.9 (CH2), 22.8 (CH3), 22.7 (CH2), 22.6 (CH3),20.1 (CH3), 18.7 (CH3), 14.3 (CH2), 12.2 (CH3); EIMS, m/z: 478 (Mþ,2%), 369 [(M�CH3C6H4S)þ, 100%]; elemental analysis calcd (%) forC33H50S: C 82.78, H 10.53, S 6.70; found: C 82.51, H 10.57, S 6.71.
4.3.1. Analogous syntheses
4.3.1.1. 6b-p-Tolylsulfanyl-3a,5a-cyclocholestane (2b). Colorlessoil; Rf¼0.35 (CH2Cl2/hexane 1:9); [a]D20�10.5 (c 1.0, CHCl3); IR, nmax:3063, 1492, 1468, 1089, 1017, 812 cm�1; 1H NMR, d (ppm): 7.29 (d,2H, J¼8.0 Hz, HeAr), 7.09 (d, 2H, J¼8.0 Hz, HeAr), 2.82 (dd, 1H,J1¼3.7 Hz, J2¼2.3 Hz, H-6a), 2.33 (s, 3H, H-Ar-CH3),1.10 (s, 3H, H-19),0.93 (d, 3H, J¼6.5 Hz, H-21), 0.880 (d, 3H, J¼6.6 Hz, H-26 or H-27),0.876 (d, 3H, J¼6.6 Hz, H-26 or H-27), 0.78 (s, 3H, H-18), 0.67 (dd,1H, J1¼5.2 Hz, J2¼4.2 Hz, H-4a), 0.42 (dd, 1H, J1¼8.0 Hz, J2¼5.2 Hz,H-4b); 13C NMR, d (ppm): 136.8 (C),133.4 (C),133.0 (CH),129.5 (CH),56.3 (CH), 56.0 (CH), 55.0 (CH), 47.9 (CH), 43.2 (C), 42.8 (C), 40.2(CH2), 39.5 (CH2), 36.9 (C), 36.2 (CH2), 35.8 (CH), 35.4 (CH2), 33.6(CH2), 30.5 (CH), 28.5 (CH), 28.3 (CH2), 28.0 (CH), 25.3 (CH2), 24.2

A.M. Tomkiel et al. / Tetrahedron 69 (2013) 8904e89138912
(CH2), 23.8 (CH2), 22.8 (CH3), 22.7 (CH2), 22.6 (CH3), 21.1 (CH3), 20.1(CH3), 18.7 (CH3), 14.3 (CH2), 12.2 (CH3); EIMS, m/z: 492 (Mþ, 4%),369 [(M�p-CH3C6H4S)þ, 100%]; elemental analysis calcd (%) forC34H52S: C 82.86, H 10.63, S 6.51; found: C 82.57, H 10.66, S 6.53.
4.3.1.2. 6b-p-Tolylsulfanyl-3a ,5a-cyclo-25R-spirostane(17). Colorless crystals; Rf¼0.49 (AcOEt/hexane 5:95); mp118e121 �C (hexane); [a]D20 �86.6 (c 1.0, CHCl3); IR, nmax: 3063,1492, 1456, 1096, 1049, 898, 813 cm�1; 1H NMR, d (ppm): 7.29 (d,2H, J¼8.1 Hz, HeAr), 7.08 (d, 2H, J¼8.1 Hz, HeAr), 4.39 (m, 1H, H-16), 3.48 (m,1H, H-26a), 3.37 (t, 1H, J¼10.9 Hz, H-26b), 2.80 (dd, 1H,J1¼3.6 Hz, J2¼2.4 Hz, H-6a), 2.33 (s, 3H, H-Ar-CH3), 1.13 (s, 3H, H-19), 0.99 (d, 3H, J¼7.0 Hz, H-21), 0.90 (s, 3H, H-18), 0.80 (d, 3H,J¼6.4 Hz, H-27), 0.69 (dd, 1H, J1¼5.2 Hz, J2¼4.3 Hz, H-4a), 0.47 (dd,1H, J1¼8.0 Hz, J2¼5.2 Hz, H-4b); 13C NMR, d (ppm): 137.1 (C), 133.6(CH), 132.9 (C), 129.6 (CH), 109.3 (C), 80.8 (CH), 66.9 (CH2), 62.3(CH), 55.8 (CH), 55.1 (CH), 47.9 (CH), 43.3 (C), 41.7 (CH), 40.8 (C),40.2 (CH2), 36.7 (C), 35.2 (CH2), 33.6 (CH2), 31.8 (CH2), 31.4 (CH2),30.3 (CH), 30.0 (CH), 28.8 (CH2), 28.6 (CH), 25.3 (CH2), 22.5 (CH2),21.1 (CH3), 20.2 (CH3), 17.1 (CH3), 16.7 (CH3), 14.5 (CH3), 14.4 (CH2);EIMS, m/z: 520 (Mþ, 21%), 397 [(M�p-CH3C6H4S)þ, 100%]; HRMS(EI): m/z calcd for C34H48O2S: 520.3375; found 520.3393.
4.3.1.3. Methyl 6b-p-tolylsulfanyl-3a,5a-cyclocholanoate (21).Colorless oil; Rf¼0.55 (AcOEt/hexane 1:9); [a]D20 �35.3 (c 1.0,CHCl3); IR, nmax: 1731,1492,1438,1103, 812 cm�1; 1H NMR, d (ppm):7.28 (d, 2H, J¼7.9 Hz, HeAr), 7.08 (d, 2H, J¼7.9 Hz, HeAr), 3.67 (s,3H, Hemethyl ester), 2.82 (m, 1H, H-6), 2.32 (s, 3H, HeAreCH3),1.12 (s, 3H, H-19), 0.94 (d, 3H, J¼6.5 Hz, H-21), 0.78 (s, 3H, H-18),0.67 (dd, 1H, J1¼5.4 Hz, J2¼4.5 Hz, H-4a), 0.42 (dd, 1H, J1¼7.8 Hz,J2¼5.4 Hz, H-4b); 13C NMR, d (ppm): 174.7 (C), 136.7 (C), 133.2 (C),133.0 (CH), 129.5 (CH), 55.90 (CH), 55.89 (CH), 54.9 (CH), 51.4 (CH3),47.8 (CH), 43.1 (C), 42.7 (C), 40.1 (CH2), 36.8 (C), 35.4 (CH), 35.3(CH2), 33.6 (CH2), 31.02 (CH2), 30.96 (CH2), 30.4 (CH), 28.5 (CH), 28.1(CH2), 25.3 (CH2), 24.2 (CH2), 22.7 (CH2), 21.0 (CH3), 20.1 (CH3), 18.2(CH3), 14.2 (CH2), 12.2 (CH3); ESI MS,m/z: 1011 [(2MþNa)þ, 5%], 548[(MNaþMeOH)þ, 30%], 517 (MNaþ, 100%); HRMS (ESI):m/z calcd for(C32H46O2SNa)þ: 517.3116; found 517.3108.
4.4. Electrolysis of 6b-p-tolylsulfanyl-3a,5a-cyclocholestane(2b) in the presence of 1,2:3,4-di-O-isopropylidene-D-gal-actopyranose (3)
6b-p-Tolylsulfanyl-3a,5a-cyclocholestane (2b; 176 mg; 0.36mmol) and 1,2,3:4-di-O-isopropylidene-D-galactopyranose (3;93 mg; 0.36 mmol) were dissolved in a 0.1 M solution of tetrabu-tylammonium tetrafluoroborate in dichloromethane (3.5 mL) andintroduced into the anodic compartment together with a 0.3 g of 3Amolecular sieve to eliminate traces of water. The same supportingelectrolyte was placed in the cathodic compartment with ananionite (1 g, Dowex 2x8, 200e400 mesh, perchlorate form) addedto eliminate chloride ions from forming by the reduction ofdichloromethane. A preparative electrolysis was carried out ina divided H-cell in which the cathodic and anodic compartments(3.5 mL of electrolytes each) were separated by a glass frit undergalvanostatic conditions. A direct current 7.5 mA was run for4000 s. A platinum mesh was used as a cathode and a platinumplate (2�1.5 cm) was used as an anode. Ag/0.1 M AgNO3 in anacetonitrile electrode was used as a reference. When the electrol-ysis was completed the solvent was removed from the reactionmixture and products were separated by silica gel column chro-matography. The hexane elution afforded 3b-p-tolylsulfa-nylcholest-5-ene (1b; 38 mg; 22%). With the hexaneeethyl acetatemixture (95:5) 6b-O-(10,20:30,40-di-O-isopropylidene-a-D-gal-actopyranos-60-yl)-3a,5a-cyclocholestane (5; 2 mg; 1%) was eluted.Further elution with hexaneeethyl acetate (93:7) afforded 3b-O-
(10,20:30,40-di-O-isopropylidene-a-D-galactopyranos-60-yl)-cholest-5-ene (4; 117 mg, 52%), followed by cholesterol (6; 10 mg, 7%)eluted with hexaneeethyl acetate (9:1).
4.4.1. 3b-O-(10,20:30,40-Di-O-isopropylidene-a-D-galactopyranos-60-yl)-cholest-5-ene (4).35 Colorless crystals; Rf¼0.35 (AcOEt/hexane1:9); mp 128e131 �C (hexane); [a]D20 �63.7 (c 1.0, CHCl3); IR, nmax:1070 cm�1; 1H NMR, d (ppm): 5.53 (d, 1H, J¼5.0 Hz, H-10), 5.34 (m,1H, H-6), 4.60 (dd, 1H, J1¼7.9 Hz, J2¼2.3 Hz, H-30), 4.30 (dd, 1H,J1¼5.3 Hz, J2¼2.3 Hz, H-20), 4.28 (dd, 1H, J1¼8.1 Hz, J2¼1.8 Hz, H-40),3.92 (dt, 1H, J1¼6.3 Hz, J2¼1.7 Hz, H-50), 3.68 (dd, 1H, J1¼10.1 Hz,J2¼6.3 Hz, H-60a), 3.62 (dd, 1H, J1¼10.1 Hz, J2¼6.5 Hz, H-60b), 3.21(m, 1H, H-3a), 1.54 (s, 3H, H-isopropylidene), 1.45 (s, 3H, H-iso-propylidene), 1.35 (s, 3H, Heisopropylidene), 1.33 (s, 3H,Heisopropylidene), 1.00 (s, 3H, H-19), 0.92 (d, 3H, J¼6.6 Hz, H-21),0.870 (d, 3H, J¼6.6 Hz, H-26 or H-27), 0.866 (d, 3H, J¼6.6 Hz, H-26or H-27), 0.68 (s, 3H, H-18); 13C NMR, d (ppm): 141.0 (C), 121.4 (CH),109.0 (C), 108.4 (C), 96.3 (CH), 79.7 (CH), 71.1 (CH), 70.63 (CH), 70.58(CH), 67.0 (CH), 66.7 (CH2), 56.7 (CH), 56.1 (CH), 50.1 (CH), 42.3 (C),39.7 (CH2), 39.5 (CH2), 39.0 (CH2), 37.2 (CH2), 36.8 (C), 36.1 (CH2),35.7 (CH), 31.9 (CH2), 31.8 (CH), 28.4 (CH2), 28.2 (CH2), 27.9 (CH),26.1 (CH3), 26.0 (CH3), 24.9 (CH3), 24.4 (CH3), 24.2 (CH2), 23.8 (CH2),22.8 (CH3), 22.5 (CH3), 21.0 (CH2), 19.3 (CH3), 18.7 (CH3), 11.8 (CH3);EIMS, m/z: 628 [Mþ, <1%], 368 [(M�1,2:3,4-di-O-isopropylidene-a-D-galactopyranose)þ, 100%]; elemental analysis calcd (%) forC39H64O6: C 74.48, H 10.26; found: 74.32, H 10.23.
4.4.2. 6b-O-(10,20:30,40-Di-O-isopropylidene-a-D-galactopyranos-60-yl)-3a,5a-cyclocholestane (5). White solid; Rf¼0.40 (AcOEt/hexane1:9); [a]D20�17.3 (c 0.1, CHCl3); IR, nmax: 1070; 1H NMR, d (ppm): 5.52(d, 1H, J¼5.0 Hz, H-10), 4.60 (dd, 1H, J1¼8.0 Hz, J2¼2.2 Hz, H-30), 4.35(dd, 1H, J1¼8.1 Hz, J2¼1.7 Hz, H-40), 4.30 (dd, 1H, J1¼5.0 Hz,J2¼2.3 Hz, H-20), 3.95 (m,1H, H-50), 3.62 (t,1H, J¼8.5 Hz, H-60a), 3.46(dd, 1H, J1¼8.8 Hz, J2¼5.7 Hz, H-60b), 2.91 (m, 1H, H-6), 1.56 (s, 3H,Heisopropylidene), 1.45 (s, 3H, Heisopropylidene), 1.35 (s, 6H,Heisopropylidene), 1.00 (s, 3H, H-19), 0.92 (d, 3H, J¼6.5 Hz, H-21),0.880 (d, 3H, J¼6.6 Hz, H-26 or H-27), 0.875 (d, 3H, J¼6.6 Hz, H-26or H-27), 0.72 (s, 3H, H-18), 0.58 (dd, 1H, J1¼4.9 Hz, J2¼4.2 Hz, H-4a), 0.40 (dd, 1H, J1¼7.8 Hz, J2¼4.9 Hz, H-4b); 13C NMR, d (ppm):108.8 (C), 108.4 (C), 96.3 (CH), 80.7 (CH), 71.0 (CH), 70.9 (CH), 70.5(CH), 66.8 (CH), 65.9 (CH2), 56.5 (CH), 56.4 (CH), 48.0 (CH), 43.3 (C),42.8 (C), 40.4 (CH2), 39.6 (CH2), 36.4 (C), 36.2 (CH2), 35.9 (CH), 34.4(CH2), 33.3 (CH2), 30.2 (CH), 28.3 (CH2), 28.0 (CH), 26.1 (CH3), 26.0(CH3), 25.1 (CH2), 25.0 (CH3), 24.4 (CH3), 24.2 (CH2), 23.9 (CH2),22.82 (CH3), 22.79 (CH2), 22.6 (CH3), 22.2 (CH), 19.4 (CH3), 18.7(CH3), 12.9 (CH2), 12.3 (CH3); ESI MS,m/z: 651 (MNaþ, 100%); HRMS(ESI): m/z calcd for C39H64O6Naþ: 651.4601; found 651.4614.
4.4.3. Other glycoconjugates obtained as products of the electro-chemical reactions
4.4.3.1. 3b-O-(10,20:50,60-Di-O-cyclohexylidene-a-D-glucofuranos-30-yl)-cholest-5-ene (14). Colorless crystals; Rf¼0.53 (AcOEt/hexane15:85); mp 49e52 �C (hexane); [a]D20 �17.4 (c 0.25, CHCl3); IR, nmax:1077 cm�1; 1H NMR, d (ppm): 5.89 (d, 1H, J¼3.6 Hz, H-10), 5.34 (m,1H, H-6), 4.46 (d,1H, J¼3.6 Hz, H-20), 4.30 (m,1H, H-50), 4.10 (m, 2H,H-60a, H-40), 4.04 (d, 1H, J¼3.1 Hz, H-30), 3.95 (dd, 1H, J1¼8.4 Hz,J2¼6.1 Hz, H-60b), 3.36 (m, 1H, H-3a), 1.01 (s, 3H, H-19), 0.92 (d, 3H,J¼6.5 Hz, H-21), 0.88 (d, 3H, J¼6.6 Hz, H-26 or H-27), 0.87 (d, 3H,J¼6.6 Hz, H-26 or H-27), 0.69 (s, 3H, H-18); 13C NMR, d (ppm): 140.7(C), 121.8 (CH), 112.4 (C), 109.4 (C), 105.0 (CH), 83.6 (CH), 81.4 (CH),80.1 (CH), 79.6 (CH), 72.2 (CH), 67.2 (CH2), 56.8 (CH), 56.2 (CH), 50.2(CH), 42.3 (C), 39.8 (CH2), 39.5 (CH2), 38.9 (CH2), 37.3 (CH2), 36.8 (C),36.51 (CH2), 36.48 (CH2), 36.2 (CH2), 35.78 (CH), 35.76 (CH2), 34.9(CH2), 32.0 (CH2), 31.9 (CH), 29.3 (CH2), 28.2 (CH2), 28.0 (CH), 25.2(CH2), 24.9 (CH2), 24.3 (CH2), 24.1 (CH2), 23.91 (CH2), 23.89 (CH2),

A.M. Tomkiel et al. / Tetrahedron 69 (2013) 8904e8913 8913
23.8 (CH2), 23.6 (CH2), 22.8 (CH3), 22.5 (CH3), 21.1 (CH2), 19.4 (CH3),18.7 (CH3), 11.8 (CH3); EIMS, m/z: 708 (Mþ, 1%), 368 [(M�10,20:50,60-di-O-cyclohexylidene-D-glucofuranose)þ, 100%]; elemental analysiscalcd (%) for C45H72O6: C 76.23, H 10.24; found: C 76.17, H 10.28.
4.4.3.2. 3b-O-(10,20:30,40-Di-O-isopropylidene-a-D-galactopyr-anos-60-yl)-(25R)-spirost-5-ene (18). Colorless crystals; Rf¼0.27(AcOEt/hexane 1:9); mp 179e180 �C (hexane), [a]D20 �92.1 (c 1.0,CHCl3); IR, nmax: 1456, 1069 cm�1; 1H NMR, d (ppm): 5.54 (d, 1H,J¼5.0 Hz, H-10), 5.34 (m,1H, H-6), 4.60 (dd, 1H, J1¼7.9 Hz, J2¼2.3 Hz,H-30), 4.40 (m, 1H, H-16), 4.30 (dd, 1H, J1¼5.0 Hz, J2¼2.4 Hz, H-20),4.28 (dd, 1H, J1¼8.1 Hz, J2¼2.1 Hz, H-40), 3.93 (dt, 1H, J1¼6.3 Hz,J2¼1.6 Hz, H-50), 3.68 (dd,1H, J1¼10.1 Hz, J2¼6.2 Hz, H-60a), 3.63 (dd,1H, J1¼10.1 Hz, J2¼6.5 Hz, H-60b), 3.48 (m, 1H, H-26a), 3.38 (t, 1H,J¼10.8 Hz, H-26b), 3.22 (m, 1H, H-3a), 1.55 (s, 3H,Heisopropylidene), 1.45 (s, 3H, Heisopropylidene), 1.35 (s, 3H,Heisopropylidene), 1.34 (s, 3H, Heisopropylidene), 1.02 (s, 3H, H-19), 0.98 (d, 3H, J¼6.9 Hz, H-21), 0.80 (d, 3H, J¼6.0 Hz, H-27), 0.79 (s,3H, H-18); 13C NMR, d (ppm): 141.1 (C), 121.1 (CH), 109.3 (C), 109.1(C), 108.5 (C), 96.4 (CH), 80.8 (CH), 79.7 (CH), 71.1 (CH), 70.7 (CH),70.6 (CH), 67.1 (CH), 66.82 (CH2), 66.80 (CH2), 62.1 (CH), 56.5 (CH),50.1 (CH), 41.6 (CH), 40.3 (C), 39.8 (CH2), 39.0 (CH2), 37.2 (CH2), 37.0(C), 32.1 (CH2), 31.8 (CH2), 31.44 (CH), 31.39 (CH2), 30.3 (CH), 28.8(CH2), 28.4 (CH2), 26.1 (CH3), 26.0 (CH3), 24.9 (CH3), 24.4 (CH3), 20.8(CH2), 19.4 (CH3), 17.1 (CH3), 16.3 (CH3), 14.5 (CH3); ESI MS,m/z: 679(MNaþ, 100%); HRMS (ESI): m/z calcd for C39H60O8Na: 679.4186;found 679.4198.
4.4.3.3. Methyl 3b-O-(10,20:30,40-di-O-isopropylidene-a-D-gal-actopyranos-60-yl)-chol-5-enoate (22). White solid; Rf¼0.38(AcOEt/hexane 2:8); [a]D20 �60.3 (c 1.0, CHCl3); IR, nmax: 1731,1070 cm�1; 1H NMR, d (ppm): 5.53 (d, 1H, J¼5.0 Hz, H-10), 5.35 (m,1H, H-6), 4.60 (dd, 1H, J1¼8.0 Hz, J2¼2.4 Hz, H-30), 4.30 (dd, 1H,J1¼5.1 Hz, J2¼2.4 Hz, H-20), 4.28 (dd, 1H, J1¼8.2 Hz, J2¼1.8 Hz, H-40),3.94 (dt, 1H, J1¼6.3 Hz, J2¼1.7 Hz, H-50), 3.69 (dd, 1H, J1¼10.1 Hz,J2¼6.4 Hz, H-60a), 3.67 (s, 3H, Hemethyl ester), 3.63 (dd, 1H,J1¼10.1 Hz, J2¼6.4 Hz, H-60b), 3.22 (m, 1H, H-3a), 1.55 (s, 3H,Heisopropylidene), 1.46 (s, 3H, Heisopropylidene), 1.36 (s, 3H,Heisopropylidene), 1.34 (s, 3H, Heisopropylidene), 1.01 (s, 3H, H-19), 0.94 (d, 3H, J¼6.5 Hz, H-21), 0.70 (s, 3H, H-18); 13C NMR,d (ppm): 174.7 (C), 141.2 (C), 121.4 (CH), 109.2 (C), 108.5 (C), 96.5(CH), 79.8 (CH), 71.3 (CH), 70.9 (CH), 70.8 (CH), 67.2 (CH), 66.9(CH2), 56.8 (CH), 55.9 (CH), 51.4 (CH3), 50.3 (CH), 42.5 (C), 39.9(CH2), 39.2 (CH2), 37.3 (CH2), 36.9 (C), 35.4 (CH), 32.00 (CH), 31.98(CH2), 31.14 (CH2), 31.11 (CH2), 28.5 (CH2), 28.1 (CH2), 26.2 (CH3),26.1 (CH3), 25.0 (CH3), 24.5 (CH3), 24.3 (CH2), 21.1 (CH2), 19.4 (CH3),18.4 (CH3), 11.9 (CH3); ESI MS, m/z: 653 (MNaþ, 100%); HRMS (ESI):m/z calcd for (C37H58O8Na)þ: 653.4029; found 653.4041.
Acknowledgements
Financial support from the Polish National Science Center(UMO-2011/01/B/ST5/06046) is gratefully acknowledged. The pur-chase of an X-ray diffractometer was sponsored by the EuropeanFund for Regional Development as part of the Operational Program
Development of Eastern Poland 2007e2013 (project No.POPW.01.03.00-20-034/09-00).
Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.tet.2013.07.106.
References and notes
1. Otaegui-Arrazola, A.; Men�endez-Carre~no, M.; Ansorena, D.; Astiasar�an, I. FoodChem. Toxicol. 2010, 48, 3289e3303.
2. Kowalski, J.; P1oszy�nska, J.; Sobkowiak, A.; Morzycki, J. W.; Wilczewska, A. Z. J.Electroanal. Chem. 2005, 585, 275e280.
3. Kowalski, J.; qotowski, Z.; Morzycki, J. W.; P1oszy�nska, J.; Sobkowiak, A.;Wilczewska, A. Z. Steroids 2008, 73, 543e548.
4. Maki, S.; Konno, K.; Takayama, H. Tetrahedron Lett. 1997, 38, 7067e7070.5. Maki, S.; Konno, K.; Ohba, S.; Takayama, H. Tetrahedron Lett. 1998, 39,
3541e3542.6. Okamoto, I.; Funaki, W.; Nakaya, K.; Kotani, E.; Takeya, T. Chem. Pharm. Bull.
2004, 52, 756e759.7. Hosokawa, Y. Y.; Hakamata, H.; Murakami, T.; Aoyagi, S.; Kuroda, M.; Mimaki, Y.;
Itoh, A.; Morosawa, S.; Kusu, F. Electrochim. Acta 2009, 54, 6412e6416.8. Hosokawa, Y. Y.; Hakamata, H.; Murakami, T.; Kusu, F. Tetrahedron Lett. 2010, 51,
129e132.9. Nokami, T.; Saito, K.; Yoshida, J. Carbohydr. Res. 2012, 363, 1e6.
10. Noyori, R.; Kurimoto, I. J. Org. Chem. 1986, 51, 4320e4322.11. Mallet, J.-M.; Meyer, G.; Yvelin, F.; Jutand, A.; Amatore, C.; Sinay, P. Carbohydr.
Res. 1993, 244, 237e246.12. Amatore, C.; Jutand, A.; Mallet, J.-M.; Meyer, G.; Sinay, P. J. Chem. Soc., Chem.
Commun. 1990, 718e719.13. Balavoine, G.; Gref, A.; Fischer, J.-C.; Lubineau, A. Tetrahedron Lett. 1990, 31,
718e719.14. Suzuki, S.; Matsumoto, K.; Kawamura, K.; Suga, S.; Yoshida, J. Org. Lett. 2004, 6,
3755e3758.15. France, R. R.; Rees, N. V.; Wadhawan, J. D.; Fairbanks, A. J.; Compton, R. G. Org.
Biomol. Chem. 2004, 2, 2188e2194.16. France, R. R.; Compton, R. G.; Davis, B. G.; Fairbanks, A. J.; Rees, N. V.; Wad-
hawan, J. D. Org. Biomol. Chem. 2004, 2, 2195e2202.17. Tanaka, N.; Ohnishi, F.; Uchihata, D.; Torii, S.; Nokami, J. Tetrahedron Lett. 2007,
48, 7383e7387.18. Drouin, L.; Compton, R. G.; Fairbanks, A. J. J. Phys. Org. Chem. 2008, 21, 516e522.19. Morzycki, J. W.; qotowski, Z.; Siergiejczyk, L.; Wa1ejko, P.; Witkowski, S.; Ko-
walski, J.; P1oszy�nska, J.; Sobkowiak, A. Carbohydr. Res. 2010, 345, 1051e1055.20. Uneyama, K.; Torii, S. Tetrahedron Lett. 1971, 12, 329e332.21. Torii, S.; Uneyama, K.; Lida, K.; Sasaki, K. Tetrahedron Lett. 1972, 13, 4513e4516.22. Uneyama, K.; Torii, S. J. Org. Chem. 1972, 37, 367e369.23. Humffray, A. A.; Houghton, D. S. Electrochim. Acta 1972, 17, 1435e1445.24. Baciocchi, E.; Rol, C.; Scamosci, E.; Sebastiani, G. V. J. Org. Chem. 1991, 56,
5498e5502.25. Wendt, H. Angew. Chem., Int. Ed. Engl. 1982, 21, 256e270.26. Viertler, H.; Gruber, J.; Pardini, V. L. In Organic Electrochemistry, 4th ed.; Lund, H.,
Hammerich,O., Eds.;MarcelDekker:NewYork,NY, 2001;Chapter17, pp621e668.27. Vankayalapati, H.; Singh, G.; Tranoy, I. Tetrahedron: Asymmetry 2001, 12,
1373e1381.28. Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J.
Appl. Crystallogr. 2009, 42, 339e341.29. Suga, S.; Matsumoto, K.; Ueoka, K.; Yoshida, J. J. Am. Chem. Soc. 2006, 128,
7710e7711.30. Helferich, B.; Portz, W. Chem. Ber. 1953, 86, 604e612.31. Koto, S.; Morishima, N.; Miyata, Y.; Zen, S. Bull. Chem. Soc. Jpn. 1976, 49,
2639e2640.32. Preparative Carbohydrate Chemistry; Hanessian, S., Ed.; Marcel Dekker: New
York, NY, 1997; pp 21e22.33. Sheldrick, G. M. Acta Crystallogr., Sect. A 2008, 64, 112e122.34. Shoppee, C. W.; Richards, H. C.; Summers, H. R. J. Chem. Soc. 1956, 4817e4821.35. This compound was described in our previous paper,19 but the physical and
spectroscopic data were erroneously given therein for the 30 ,40-mono-O-iso-propylidene derivative.





























