Embed Size (px)
Citation preview

Electron transfer rate of redox ion controlled by electrostaticinteraction with bilayer films assembled using thiolate�/copper ion�/
carboxylate bridges
Takahiro Yamaguchi �, Rei Sakai, Kohshin Takahashi, Teruhisa Komura
Department of Chemistry and Chemical Engineering, Faculty of Engineering, Kanazawa University, 40-20 Kodatsuno 2-chome, Kanazawa 920-8667,
Japan
Received 19 August 2002; received in revised form 31 October 2002
Abstract
11-mercaptoundecanoic acid (MUA) monolayer and MUA�/copper ion�/MUA bilayer assembled using thiolate�/coppcr ion�/
carboxylate bridges on MUA monolayer electrode were prepared, and tried to control electron transfer rate of redox ions. The
soaking solution to assemble MUA on gold electrode changed from ethanolic MUA solution to 1-butanolic one, then the
differential interfacial capacitance decreased from 2.59/0.1 mF cm�2 to 1.69/0.2 mF cm�2, and electron rate constant, k0 of
[Co(phen)3]3� decreased from 20�/10�6 cm s�1 to 8.3�/10�6 cm s�1. These results show that highly ordered MUA monolayer
can be obtained only changing soaking solvent to assemble MUA, Obtained highly ordered MUA monolayer electrode was block
off completely redox anion by electrostatic repulsion and MUA film thickness. Moreover using MUA�/copper ion�/MUA bilayer
electrode, k0 of [Co(phen)3]3� decreased under 1/400 against using MUA monolayer electrode, that value become to under 0.02�/
10�6 cm s�1. This study shows that the combination of electrode surface charge and length of insulating spacers is able to control
electron transfer rate of various electroactive ions.
# 2002 Elsevier Science Ltd. All rights reserved.
Keywords: 11-Mercaptoundecanoic acid bilayer; Tris(1,10-phenanthroline)cobalt(III); Thiolate�/copper ion�/carboxylate bridges; Electron transfer
rate; Electrostatic interaction
1. Introduction
Self-assembled thiol monolayer with functionalized
terminal groups modified electrode aiming at an elec-
trochemical control of specific materials are useful as
sensor interfaces, electroanalysis, electrocatalysis, and
molecular electronic devices [1�/6]. These highly ordered
electrode surfaces may enable us to understand the basic
mechanisms of intermolecular interaction and electron
transfer at electrode j electrolyte interface. Most of the
molecular recognition systems often presented are based
on the defectsize in monolayers and on the binding
interactions (including coulombic interaction and com-
plex formation) with the terminal groups of thiol. For
example, R. M.Crooks and co-workers [7] reported that
synthesis and characterization of two-component self-
assembling monolayors that act as nanoporous mole-
cular recognition membranes. I. Willner and co-workers
[8,9] demonstrated that ionizable monolayer-electrodes
allowed the amperometric transduction of the pH
changes of a reaction medium.
On the other hand, multilayer construction by self-
assembly might be useful in creating new classes of
material possessing functional groups at controlled site
in three-dimensional arrangement. In order to attain this
kind of ordering, Sagiv and co-workers [10,11] have
shown that adsorption of a stable monolayer, followed
by alternate chemical activation and adsorption steps,
can yield organized multilayer structures without re-
course to monolayer transfer techniques. T. E. Mallouk
and co-workers [12,13] reported multilayer zirconium
phosphonate films could be prepared on silicon and gold
substrates via sequential adsorption of their zirconium
and phosphonic acid components. Organometallic mul-� Corresponding author. Fax: �/81-76-23-44800
E-mail address: [email protected] (T. Yamaguchi).
Electrochimica Acta 48 (2003) 589�/597
www.elsevier.com/locate/electacta
0013-4686/02/$ - see front matter # 2002 Elsevier Science Ltd. All rights reserved.
PII: S 0 0 1 3 - 4 6 8 6 ( 0 2 ) 0 0 7 3 2 - 6

tilayers of v -mercaptoalkanoic acids, based on the
interaction of Cu(II) ions with carboxylic acids and
thiols, were reported by A. Ulman [14]. Recently W.
Murray and co-workers [15,16] reported the controlledand reversible formation of transiently soluble alka-
nethiolate- and tiopronin-monolayer-protected cluster
aggregates was demonstrated by using Cu2��/carbox-
ylate chemistry to form cluster�/Cu2��/cluster linkages.
Thus, numerous example of multilayer construction
have been presented, but limited data on the electron
transfer kinetics at multilayer-modified electrodes have
hampered a through mechanistic understanding of thesemolecular interaction effects. To develop the promising
electrode-modification strategies for selectively detecting
a target species, we need to examine variously the effect
of the stepwisely increasing film thickness from mono-
layer to multilayer and the ionic self-assembled layer
j analyte interaction on interfacial electron transfer
kinetics.
In the first part of this work reported here, using 11-mercaptoundecanoic acid (MUA) monolayer on gold as
the ionizable terminal groups of thiol, electrochemical
characterization of MUA electrode were examined by
potential scan voltammetry and ac impedance spectro-
scopy. In addition, the effects of the coulombic interac-
tion between that monolayer and ionic redox-active
species on interfacial electron transfer rates were exam-
ined by potential scan voltammetry. In the second part,we prepared MUA�/copper ion�/MUA bilayer as-
sembled using thiolate�/copper ion�/carboxylate bridges
on gold. And we have used MUA�/copper ion�/MUA
bilayer electrode as insulating spacers, to control the
rate of interfacial electron transfer between the electrode
and tris(l,10-phenanthroline)cobalt(III).
2. Experimental
2.1. Electrode modifications
Gold thin film electrode (0.2 cm2 area, SEIKO EG &
G, QA-A9MAu) was used as working electrode. They
consisted of a 150-nm-thick Au layer (on underdepos-
ited 5 nm Ti) sputtered onto silica glass. Just prior to
surface modification, an Au electrode was electroche-mically cycled 20 times at 100 mV s�1 between 0 and 1.5
V in 0.2 M HClO4, followed by rinsing with deionized
water and then ethanol. Once cleaned, the electrode was
soaked in 1 mM 11-mercaptoundecanoic acid (MUA,
Aldrich) solution in ethanol (Kanto Chemical, for
fluorometry) or 1-butanol (Kanto Chemical, for fluor-
omerty) for 2 h (STEP 1 as shown in Scheme 1) and
rinsed thoroughly with ethanol and water to removephysically adsorbed MUA from the surfaces. Bilayer
films of MUA�/copper ion�/MUA were prepared as
follows and Scheme 1. The MUA monolayer electrode
was dipped into 1 mM ethanolic Cu(ClO4)2 solution
(STEP 2). And the resulting Cu2��/carboxylate film was
removed, rinsed with ethanol, soaked in 1 mM MUA in
ethanol (STEP 3), and then rinsed successively withethanol and deionized water. Bilayer films of MUA�/
copper ion�/mercaptopropionic acid(MPA) were also
prepared such as that of MUA�/copper ion�/MUA.
UV�/vis spectra (350�/900 nm) testing for thiolate�/
copper ion bonds were acquired using a HITACHI U-
3210 spectrophotometer using 1 cm quartz cells.
2.2. Electrochemical measurements
All of electrolyte solutions for electrochemical mea-
surements were prepared with doubly distilled water and
purged with nitrogen gas before measurements. Electro-
chemical experiments were carried out in a two com-
partment, three-electrode glass cell at room
temperature. A large Pt gauze (�/10 cm2) and an
Ag j AgCl j 3.3 M KCl electrode were used as counterand reference electrodes, respectively. All electrode
potentials are referred to Ag j AgCl electrode. Cyclic
voltammograms were obtained by HOKUTO Denko
HA-501 potentiostat coupled with HB-104 function
generator, and recorded on a YOKOGAWA 3025 X-
Y recorder. The supporting electrolyte solution con-
sisted of 0.19 M KCl and 10 mM phosphate buffer, pH
6. Main elcctroactive ion, tris(l,10-phenanthroline)co-balt(III) ([Co(phen)3]3�) was synthesized according to
published procedures [17]. Other redox ions, anthraqui-
nonce-2,6-disulfonic acid, ferric monosodium ethylene-
diammetetraacetate, hexaammineruthenium(III)
([Ru(NH3)6]3�)) and l,1?-ferrocenedimethanol, were
used without further purification.
The ac impedance of modified electrodes were mea-
sured on NF Electronic Instruments 5020 frequency-response analyzer coupled to SEIKO EG & G 263A
potentiostat. The values of impedance were determined
at five discrete frequencies per decade over the range of
104�/10�2 Hz at amplitude of 7 mV (peak to peak).
3. Results and discussion
3.1. MUA monolayer electrode
3.1.1. Electrochemical characterization of MUA
monolayer
Fig. 1 shows CVs for the reductive desorption of the
MUA in 0.19 M KCl and 10 mM borate buffer, pH 11.
Only one peak appeared at �/1.03 V in the CV for that
of the MUA. By integration of the area of the cathodic
wave corresponding to the MUA, after elimination ofbackground current, its surface coverage is calculated to
be G�/(6.99/0.6)�/10�10 mol cm�2 We considered the
effect of self-assembly on carboxylate as head group of
T. Yamaguchi et al. / Electrochimica Acta 48 (2003) 589�/597590

the MUA when soaking in e¯thanolic solution or
butanolic solution, but MUA’s surface coverage were
not changed. The MUA is strongly chemisorbed on gold
surface, yielding monomolecular film conveniently.
Since impedance spectroscopy can give much informa-
tion on the electrochemical properties of a solid j solu-
solution interface, we characterized self-assembled thiol
monolayer-modified gold electrodes using this techni-
que. Fig. 2 represents typical impedance spectra for
MUA modified Au electrode in a 0.2 M NaClO4
solution. The spectra exhibit a nearly vertical line, and
that equivalent circuit is a series circuit of ohmic
resistance R and the differential interfacial capacitance
Scheme 1. Growth of MUA multilayers on Au by adsorption of MUA and Cu2�.
Fig. 1. Cyclic voltammograms for the reductive desorption of MUA
monolayer measured at 100 mV s�1 in 0.19 M KC1 and 10 mM borate
buffer, pH 11, Solid and dotted lines represent a bare Au and a MUA
monolayer electrodes, respectively.
Fig. 2. Impedance spectra of a bare Au (o) and a MUA monolayer (+)
electrodes in 0.2 M NaClO4 solution. Electrode potential�/0.2 V.
Numerical values in the figure exhibit frequencies in Hz.
T. Yamaguchi et al. / Electrochimica Acta 48 (2003) 589�/597 591

C . Obtained impedance Z is written as
Z�Z?� jZƒ�(Rs�Rm)� j=vC (1)
where Rs is the solution resistance, Rm is the monolayerresistance, and v is the reciprocal of the angular
frequency. Thus, Rs�/Rm was determined from the
extrapolated high-frequency intercept of a complex
plane impedance plot with the real axis, and C from
the slop of the linear plot of the imaginary impedance
against 1/v at the range of 3�/0.1 kHz. Our obtained C
is a function of the film capacitance, the diffuse layer
capacitance, and the degree of protonation [18]. Table 1shows Rm and C obtained for MUA modified electrode
soaking in ethanolic solution and 1-butanolic solution in
a 0.2 M NaClO4 solution of pH 6. The resistance of
MUA monolayer calculated to be Rm�/1.5 V cm2
soaking in ethanolic solution and Rm�/1.79/0.2 V cm2
soaking in 1-butanolic solution. The Rm value soaking
in 1-butanolic solution is a little larger than that in
ethanolic solution. The differential interfacial capaci-tance at 0.2 V calculated to be C�/9.79/1.2 mF cm�2
using bare Au electrode, 2.59/0.1 mF cm�2 using MUA
electrode soaking in ethanolic solution, and 1.69/0.2 mF
cm�2 using MUA electrode soaking in 1-butanolic
solution. Both of MUA modified electrode reduced the
differential interfacial capacitance to less than that of
the base electrode. In particular, MUA modified elec-
trode soaking in 1-butanolic solution lowered thedifferential interfacial capacitance to about 1/5. We
believe that the difference of both MUA modified
electrode soaking ethanolic and butanolic solution is
considered with the protonation state of carboxyl group
when MUA is self-assembled in soaking solution.
Therefore MUA monolayer electrode prepared soaking
in 1-butanolic solution block off electrolyte ions higher
than that into ethanolic solution. Fig. 3 show thatrelationship between the differential interfacial capaci-
tance of MUA prepared soaking in ethanolic solution
and solution pH. All experimental solution is equal to
0.2 M NaClO4 solution. This result indicates that the
value of the differential interfacial capacitance of MUA
is raised at low pH values. Nevertheless, T. Kakiuchi
and co-workers [19] indicated that the transition from
fully protonated state to the fully dissociated state of the
v -carboxyl group in the self-assembled v -carboxyl
alkanethiol on Au gave rise to the increase in the
double-layer capacitance (Cdl) and Cdl in the acidic pH
range was not constant because of the possibility arisingfrom the difference in the position of the outer
Helmholtz planes between H� and Na� as counter
ions. The change in C with our experimental pH range
was barely observable, therefore, we cannot determine
clearly the pH range for deprotonated carhoxyl terminal
of MUA from C .
3.1.2. Voltammetric response of redox species at MUA
monolayer-modified electrode
Using self-assembled MUA monolayer electrode
obtained with soaking 1 mM MUA solution in 1-
butanol, we examined voltammetric behavior of various
redox-active ions in solution. All of the supportingelectrolyte solution containing redox-active ion were
adjusted to pH 6, because J. F. Smalley and co-workers
[20] reported the indirect laser-induced temperature
jump method was used to determine the surface pKaof
MUA self-assembled on Au, and that analysis gave
pKa�/5.79/0.2 at 0.1 M ionic strength. Table 2 shows
that cathodic peak potential and that current Ipc(MUA)/
Ipa(Au) of cyclic voltammograms at scan rate�/100 mVs�1 using bare Au and MUA electrode. At a bare Au
electrode, these voltammograms of anionic anthraqui-
none-2,6-disulfonic acid and ferric monosodium ethyle-
nediaminetetraacetate showed the electrochemical
quasi-reversible wave at scan rate 100 mV s�1. Never-
theless, MUA monolayer electrode interrupted inter-
facial electron transfer of these redox anion completely.
In the case of neutral 1,1?ferrocenedimethanol, reversi-ble wave of the voltammogram obtained at a bare Au
electrode, however, at MUA monolayer electrode,
oxidation current decreased (Ipa(MUA)/Ipa(Au)�/0.50)
Table 1
Impedance data for various electrodes in a 0.2 M NaClO4 solution at
0.2 V
Electrode STEP 1
sol.aSTEP 3
sol.aC/mF
cm�2
Rm/Vcm2
Au �/ �/ 9.791.2 0
MUA Ethanol �/ 2.590.1 1.590.1
MUA Butanol �/ 1.690.2 1.790.2
MUA �Cu2� �MUA Butanol Ethanol 0.9590.04 1.890.2
a STEP 1 and 3 are shown in Sceme 1.
Fig. 3. Differential capacitance versus solution pH for a MUA
monolayer confined to Au surfaces in 0.2 M NaClO4 solution.
Electrode potential �/0.2 V (o) and �/0.1 V (+).
T. Yamaguchi et al. / Electrochimica Acta 48 (2003) 589�/597592
and clear anodic peak was not appeared. On the other
hand, cationic [Ru(NH3)6]3� and [Co(phen)3]3�, which
obtained the reversible wave at a bare Au electrode,
appeared reductive peak currents at more negative
potential using MUA monolayer electrode. Especially,
[Ru(NH3)6]3� showed quasi-reversible wave at scan rate
100 mV s�1 at MUA monolayer electrode. Redox ion
size may be a key factor of the redox reduction on MUA
monolayer electrode. These results indicate that electron
transfer rate of redox ion is controlled by an electro-
static interaction and the distance between negative
terminal carboxylate groups of MUA monolayer elec-
trode and redox ion. Therefore, redox cation such as
[Ru(NH3)6]3� and [Co(phen)3]3� reacted on MUA
electrode because of electrostatic attraction in spite of
arising the distance between MUA electrode and redox
cation. We selected [Co(phen)3]3� from various redox
species because of the effect of electron transfer rate
constant on MUA modified electrode using the theory
of cyclic voltammetry.
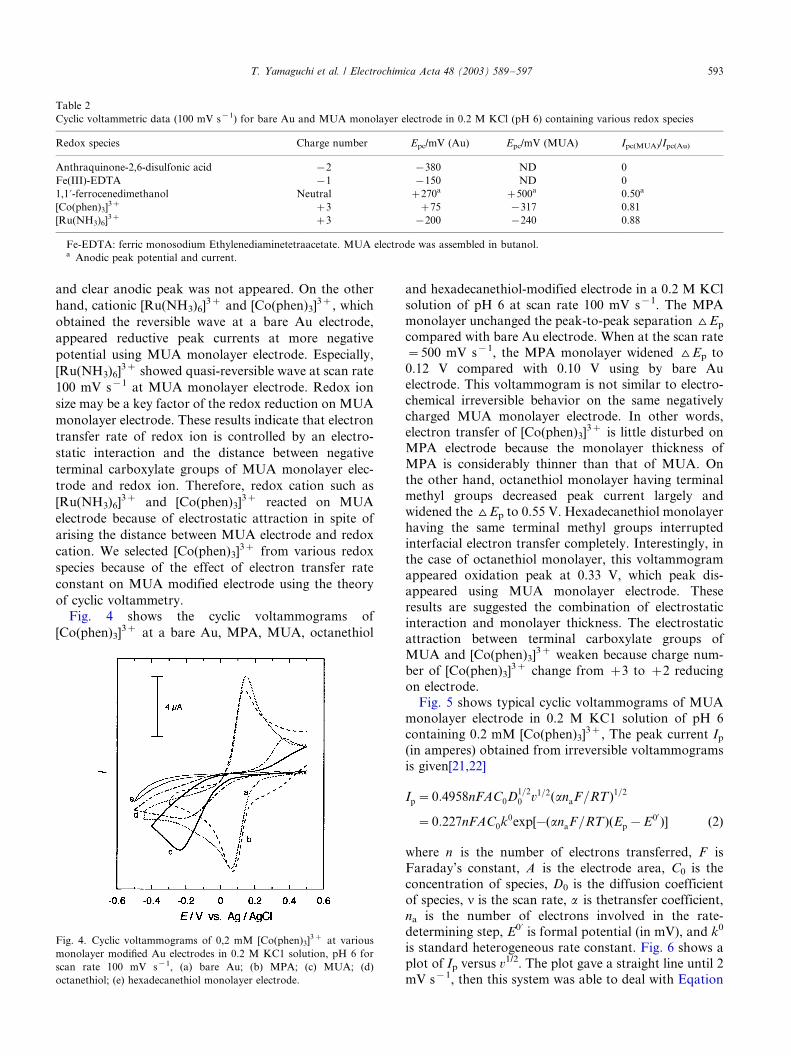
Fig. 4 shows the cyclic voltammograms of
[Co(phen)3]3� at a bare Au, MPA, MUA, octanethiol
and hexadecanethiol-modified electrode in a 0.2 M KCl
solution of pH 6 at scan rate 100 mV s�1. The MPAmonolayer unchanged the peak-to-peak separation ^Ep
compared with bare Au electrode. When at the scan rate
�/500 mV s�1, the MPA monolayer widened ^Ep to
0.12 V compared with 0.10 V using by bare Au
electrode. This voltammogram is not similar to electro-
chemical irreversible behavior on the same negatively
charged MUA monolayer electrode. In other words,
electron transfer of [Co(phen)3]3� is little disturbed onMPA electrode because the monolayer thickness of
MPA is considerably thinner than that of MUA. On
the other hand, octanethiol monolayer having terminal
methyl groups decreased peak current largely and
widened the ^Ep to 0.55 V. Hexadecanethiol monolayer
having the same terminal methyl groups interrupted
interfacial electron transfer completely. Interestingly, in
the case of octanethiol monolayer, this voltammogramappeared oxidation peak at 0.33 V, which peak dis-
appeared using MUA monolayer electrode. These
results are suggested the combination of electrostatic
interaction and monolayer thickness. The electrostatic
attraction between terminal carboxylate groups of
MUA and [Co(phen)3]3� weaken because charge num-
ber of [Co(phen)3]3� change from �/3 to �/2 reducing
on electrode.Fig. 5 shows typical cyclic voltammograms of MUA
monolayer electrode in 0.2 M KC1 solution of pH 6
containing 0.2 mM [Co(phen)3]3�, The peak current Ip
(in amperes) obtained from irreversible voltammograms
is given[21,22]
Ip�0:4958nFAC0D1=20 v1=2(anaF=RT)1=2
�0:227nFAC0k0exp[�(anaF=RT )(Ep�E00)] (2)
where n is the number of electrons transferred, F is
Faraday’s constant, A is the electrode area, C0 is the
concentration of species, D0 is the diffusion coefficient
of species, n is the scan rate, a is thetransfer coefficient,
na is the number of electrons involved in the rate-
determining step, E0? is formal potential (in mV), and k0
is standard heterogeneous rate constant. Fig. 6 shows a
plot of Ip versus v1/2. The plot gave a straight line until 2
mV s�1, then this system was able to deal with Eqation
Table 2
Cyclic voltammetric data (100 mV s�1) for bare Au and MUA monolayer electrode in 0.2 M KCl (pH 6) containing various redox species
Redox species Charge number Epc/mV (Au) Epc/mV (MUA) Ipc(MUA)/Ipc(Au)
Anthraquinone-2,6-disulfonic acid �2 �380 ND 0
Fe(III)-EDTA �1 �150 ND 0
1,1?-ferrocenedimethanol Neutral �270a �500a 0.50a
[Co(phen)3]3� �3 �75 �317 0.81
[Ru(NH3)6]3� �3 �200 �240 0.88
Fe-EDTA: ferric monosodium Ethylenediaminetetraacetate. MUA electrode was assembled in butanol.a Anodic peak potential and current.
Fig. 4. Cyclic voltammograms of 0,2 mM [Co(phen)3]3� at various
monolayer modified Au electrodes in 0.2 M KC1 solution, pH 6 for
scan rate 100 mV s�1, (a) bare Au; (b) MPA; (c) MUA; (d)
octanethiol; (e) hexadecanethiol monolayer electrode.
T. Yamaguchi et al. / Electrochimica Acta 48 (2003) 589�/597 593

2 of irreversible reaction. To estimate k0, we plotted
ln Ipc against (Ep�/E0?) in Fig. 7. When (Ep�/E0?) is
equal to 0 in this plot, the value of ln Ipc doesn’t contain
the parameters of a and n0 and obtained k0�/8.3�/
10�6 cm s�1. In the case of bare Au electrode, we can
not determine k0 directly from the voltammogram of
[Co(phen)3]3� under 500 mV s�1 because these currents
is controlled with diffusion of redox species. Never-
theless, we estimated a rough k0 value from the
parameter L , defined as [22,23]
L�k0(RT=nFDv)1=2 (3)
for Do�/DR�/D , and a�/0.5.
In this case at a low scan rate until 20 mV s�1, these
voltammogram is able to consider reversible system,
then this zone boundary suggested by Matsuda andAyabe[23]is L]/15. Putting D of [Co(phen)3]3� into
3.4�/10�6 cm2 s�1, obtained rough k0 value is over
2.5�/10�2 cm s�1. The MUA monolayer electrode
lowered the k0 of [Co(phen)3]3� under 1/3000 of that at
a bare Au electrode.
3.2. Electrochemical characterization and formation of
MUA biltilayer electrode
We aimed the formation of the MUA bilayer to useMUA monolayer terminal�/COO�/ copper ion�/thiolate
bridges(Scheme 1). Firstly we tested UV�/vis spectro-
photometry of ethanolic Cu(ClO4)2 solution adding
several thiol derivatives and undecanoic acid for
thiolate�/copper ion bonds. Fig. 8 shows UV�/vis spectra
of 10 mM ethanolic Cu(ClO4)2 solution containing a
four-fold molar of MUA, MPA, octanethiol, and
undecanoic acid. Adding every thiol derivatives toethanolic Cu(ClO4)2 solution, then precipitation were
produced. On the other hand, when adding four-fold
excess of undecanoic acid not having SH terminal
group, the value of absorbance was increased a little
conversely in the case of thiol derivatives. Therefore,
these complexations are occurred between thiolate anion
and Cu2� in ethanolic solution. After filtration of these
precipitation, resulting solution were used for thisspectrophotometry. The UV�/vis spectrum of Cu(ClO4)2
in ethanolic solution was showed adsorption band at ca,
820 nm. As adding thiol derivatives, the value of that
Fig. 5. Cyclic voltammograms of 0.2 mM [Co(phen)3]3� at MUA
monolayer modified Au electrodes in 0.2 M KCl solution, pH 6. Scan
rates were 2, 5, 10, 20, 50, 100, 200, 300, and 500 mVs�1.
Fig. 6. Plot of cathodic peak currents (Ipc) versus the square root of
scan rate for 0.2 mM [Co(phen)3]3� at MUA monolayer electrode in
0.2 M KCl solution, pH 6.
Fig. 7. Logarithm of cathodic peak currents (Ipc) versus (Epc �/E0?) for
0.2 mM [Co(phen)3]3��/ at MUA monolayer electrode in 0,2 M KCl
solution, pH 6 (Fig. 5).
T. Yamaguchi et al. / Electrochimica Acta 48 (2003) 589�/597594

absorbance was decreased, and that band was disap-
peared with adding four-fold molar excess of thiol.
Cu(II) is known to oxidize thiols to disufides [24,25].
And usually, a thiolate is the preferred ligand, even
when a carboxylate group is present [26,27]. A. J. Bard
and co-workers [28] reported the self-assembly of multi-
layers on gold using v -mercaptoalkanethiols and Cu(II)
ions. X-ray photoelectron spectroscopic data confirmed
a Cu(I) oxidation state and formation of a multilayer
with intralayer disulfide bonds. We believe that these
precipitations are Cu(I) clusters with disulfide and
thiolate as ligands.
Scheme 1 shows the procedure used to prepare MUA
bilayer on electrode surfaces. The MUA monolayer,
which is the first layer of MUA bilayer, obtained
soaking in 1-butanolic solution containing 1 mM
MUA because we wanted to gain a high dense mono-
layer. We also characterized MUA multilayer electrode
using impedance spectroscopy according to Section
3.1.1. Obtained values of Rm and C for MUA bilayer
modified electrode can be compared with MUA mono-
layer electrode in Table 1. Rm for MUA bilayer was little
changed with MUA monolayer obtained soaking 1-
butanolic solution, but C for MUA bilayer decreased.
Fig. 9 shows cyclic voltammograms of bare Au, MUA
monolayer, and MUA bilayer modified electrodes in 0.2
M KC1 solution of pH 6 containing 0.2 mM
[Co(phen)3]3� In Fig. 9c, it can be seen that the presence
of MUA bilayer causes the further broadening of the
peaks, a decrease in the current density in comparison
with MUA monolayer. This voltammogram indicates
that the kinetics of electron transfer through the MUA
bilayer film has become extremely slow because of
increasing MUA film thickness. The k0 MUA bilayer
is calculated according to Equation 2 such as MUA
monolayer. A summary of the CV data and k0 for
[Co(phen)3]3� at MUA mono- and bilayer modified
electrodes obtained from soaking in ehanolic or 1-
butanolic 1 mM MUA solutions is given on Table 3.
In the case of preparing MUA bilayer electrode (STEP
3), soaking solvent used for copper ion�/MUA thiolate
linkage is ethanol better than 1-butanol against in the
case of MUA monolayer. The k0 for [Co(phen)3]3� at
MUA bilayer (STEP 1 sol.: 1-butanol, STEP 3 sol.:
ethanol) electrode is 400�/8000 times smaller than that at
MUA monolayer (STEP 1 sol.: 1-butanol) electrode.
Electron tunneling through monolayer films may dimin-
ish the electron transfer rate exponentially with the
monolayer thickness [29]. The exponentially decrease of
k0 from bare Au to MUA bilayer by way of MUA
monolayer indicate the stepwisely increase of the film
thickness. This result supports that attachment occurs
via a MUA carboxylate�/copper ion�/MUA thiolate
linkage. In spite of the growth of MUA film on Au
electrode is little effective in Rm value obtained from
impedance spectroscopy (as shown in Table 1), this
growth is effective in decreasing k0 sharply. The surface
of MUA bilayer electrode is lower density than that of
MUA monolayer, therefore, large electroactive ion such
as [Co(phen)3]3� can be blocked off, but small electro-
lyte ion cannot be done. Then Rm value of MUA bilayer
little changes against that of MUA monolayer.
In order to examine the influence of multilayer film
thickness upon electron transfer rate for [Co(phen)3]3�,
MUA�/copper ion�/MPA multilayer electrode prepared
soaking in MPA solution in place of MUA solution at
STEP 3 in Scheme 1. The chain length of MPA is much
shorter than that of MUA. Fig. 10 shows cyclic
voltammograms of bare Au, MUA monolayer, MUA-
copper ion-MPA bilayer electrode, and MUA�/Cu�/
MPA�/Cu�/MPA tri-layer electrode in 0.2 M KCl
solution of pH 6 containing 0.2 mM [Co(phen)3]3� at
Fig. 8. UV�/vis spectra for 10 mM Cu(ClO4)2 solution (a) and
containing a four-fold molar of (b) MUA, (c) undecanoic acid, (d)
MPA, and (e) octanethiol. After filtration of precipitation adding thiol
derivatives, that solution was used for this spectrophotometry.
Fig. 9. Cyclic voltammograms of modified Au electrodes in 0.2 mM
[Co(phen)3]3�, 0.2 M KCl solution (pH 6) for scan rate 10 mV s-1: (a)
bare Au; (b) Au with MUA monolayer; (c) Au with MUA�/copper
ion�/MUA bilayer.
T. Yamaguchi et al. / Electrochimica Acta 48 (2003) 589�/597 595

scan rate 2 mV s�1. Interestingly, using MUA-copper
ion-MPA bilayer electrode, that voltammetric behavior
of [Co(phen)3]3� displays quasi-reversible redox peak
pair in spite of increasing a film thickness of MUA�/
copper ion�/MPA bilayer against that of MUA mono-
layer. Similarly, we prepared MUA�/Cu�/MPA�/Cu�/
MPA tri-layer electrode to make a film thickness larger,
and measured. The voltammetric current of
[Co(phen)3]3� was increased in comparison with using
MUA�/copper ion�/MPA multilayer electrode. These
results indicate that MPA cannot self-assembled on
MUA�/Cu2� electrode surface because cohesive force
among MPA is weak. Then film thickness cannot be
increased stepwisely in spite of a repeat of STEP 2 and
STEP 3 in Scheme 1, but electrode surface charge is
growing more negative potential due to adsorption of
MPA having terminal carboxylate groups on MUA�/
Cu2� electrode surface. Therefore [Co(phen)3]3� in-
corporated in MPA multilayer film because of electro-
static binding force, and electron transfer between
adsorbed [Co(phen)3]3� and MUA�/copper ion�/MUA
bilayer electrode is easier than that between
[Co(phen)3]3� and MUA monolayer electrode through
electrostatic interaction.
4. Conclusion
This study has shown the construction of MUA
bilayer using thiolate�/copper ion�/carboxylate bridges,
electrochemical characterization of self-assembled
MUA mono- and bilayer electrode, and controlling
the electron transfer rate between redox species andmodified electrode using MUA layer as insulating and
electrostatic spacers. MUA monolayer electrode pre-
pared soaking in l-butanolic MUA solution block off
electrolyte ions higher than that in ethanolic solution
from ac impedance analysis and cyclic voltammograms
of [Co(phen)3]3�. Obtained MUA monolayer and
bilayer electrode interrupted electron transfer of redox
anion completely. On the other hand, redox cation,especially [Co(phen)3]3� is controlled by electrostatic
interaction between negative terminal groups of MUA
and redox ion, and the stepwisely increase of the length
of insulating spacers from bare Au to MUA�/copper
ion�/MUA bilayer.
References
[1] I. Willner, N. Lapidot, A. Riklin, R. Kasher, E. Zahavy, E. Katz,
J. Am. Chem. Soc. 116 (1994) 1428.
[2] D. Mandler, I. Turyan, Eleclroanalysis 8 (1996) 207.
[3] I. Rubinstein, S. Steinberg, Y. Tor, A. Shanzer, J. Sagiv, Nature
332 (1998) 426.
[4] I. Turyan, D. Mandler, Anal. Chem. 66 (1994) 58.
[5] I. Turyan, D. Mandler, Anal. Chem. 69 (1997) 894.
[6] R. Blonder, I. Willner, A.F. Buckmann, J. Am. Chem. Soc. 120
(1998) 9335.
[7] O. Chailapakul, R.M. Crooks, Langmuir 9 (1993) 884.
[8] E. Katz, M.L. Dargan, I. Willner, J. Electroanal. Chem. 426
(1996) 107.
[9] A. Doron, E. Katz, G. Tao, I. Willner, Langmuir 13 (1997) 1783.
[10] L. Sagiv, J. Netzer, J.Am. Chem. Soc. 105 (1983) 674.
[11] J. Gun, R. Iscovici, J. Sagiv, J. Colloid. Sci. 101 (1984) 201.
Table 3
Heterogeneous electron transfer rate constants as determined by cyclic voltammetry and cyclic voltammetric data of [Co(phen)3]3� for MUA
modified electrode prepared to soak in various solution
Electrode STEP 1 sol. a STEP 3 sol. a k0/10�6 cm s�1 Epc/mV Ipc/mA cm�2
Au �/ �/ �25 000 �75 23
MUA Ethanol �/ 20 �200 22
MUA �Cu2� �MUA Ethanol Ethanol 13 �303 18
MUA Butanol �/ 8.3 �317 19
MUA �Cu2� �MUA Butanol Butanol 7.0 �365 19
MUA �Cu2� �MUA Butanol Ethanol 0.02�/0.001 �530 11
a STEP 1 and 3 are shown in Scheme 1.
Fig. 10. Cyclic voltammograms of modified Au electrodes in 0.2 mM
[Co(phen)3]3�/, 0.2 M MCl solution (pH 6) for scan rate 2 mV s-1: (a)
bare Au; (b) Au with MUA�/copper ion�/MPA bilayer; (c) Au with
MUA�/monolayer; (d) Au with MUA�/Cu�/MPA�/Cu�/MPA tri-
layer.
T. Yamaguchi et al. / Electrochimica Acta 48 (2003) 589�/597596

[12] H. Lee, L.J. Kepley, H.-G. Hong, S. Akhter, T.E. Mallouk, J.
Phys. Chem. 92 (1988) 2597.
[13] H.-G. Hong, T.E. Mallouk, Langmuir 7 (1991) 2362.
[14] S.D. Evans, A. Ulman, K.E. Goppert-Berarducciand, L.J.
Gerenser, J.Am. Chem. Soc. 113 (1991) 5866.
[15] A.C. Templeton, F.P. Zamborini, W.P. Wuelfing, R.W. Murray,
Langmuir 16 (2000) 6682.
[16] F.P. Zamborini, J.F. Hicks, R.W. Murray, J. Am. Chem. Soc. 122
(2000) 4514.
[17] N. Maki, Bull. Chem. Soc. Jpn. 42 (1969) 2275.
[18] C.P. Smith, H. White, Langmuir 9 (1993) 1.
[19] T. Kakiuchi, M. Iida, S. Imabayashi, K. Niki, Langmuir 16 (2000)
5397.
[20] J.F. Smalley, K. Chalfant, S.W. Feldberg, T.M. Nahir, E.F.
Bowden, J. Phys. Chem. B 103 (1999) 1676.
[21] R.S. Nicholson, I. Shain, Anal. Chem. 36 (1964) 706.
[22] A.J. Bard, L.R. Faulkner, Electrochemical Methods Fundamen-
tals and Applications, Wiley, New York, 1980, p. 223.
[23] H. Matsuda, Y. Ayabe, Z. Elektrochem. 59 (1955) 494.
[24] L. Michaelis, M.P. Schubert, J. Am. Chem. Soc. 52 (1930) 4418.
[25] K.H. Slagle, E.E. Reid, Ind. Eng. Chem. 24 (1932) 448.
[26] H.L. Nigam, U.C. Srivastava, Inorg. Chim. Acta. 5 (1971) 338.
[27] S.T. Chow, C.A. McAuliffe, B.J. Sayle, J. Inorg. Nucl. Chem. 37
(1974) 451.
[28] M. Brust, P.M. Blass, A.J. Bard, Langmuir 13 (1997) 5602.
[29] J. Xu, H.L. Li, Y. Zhang, J. Phys. Chem. 97 (1993) 11497.
T. Yamaguchi et al. / Electrochimica Acta 48 (2003) 589�/597 597





























