Embed Size (px)
Citation preview

Electronic structure with Turbomole
S. Boixo
Aspuru-Guzik’s group, Harvard
March 7th, 2011

1 Basis sets
2 Hückel Theory
3 Hartree Fock and beyond
4 Molecular Vibrations
5 Tests
Boixo (Harvard) Elec. Struct. Tmole App. QM 2 / 43

Introduction
During this class we are going to see how the theory that we havebeen studying in the past few lectures is applied in practice.For that, we are going to study the hydrogenation of nitrogen usingTurbomole
N2 + 3H2 → 2NH3 .
The goal is to explain the enthalpy of reaction of -18.604 kcal mol−1
(you can find this experimental value in Szabo and Ostlund).We will also see an example of an excitation energy obtained withTDDFT.As we go along, we will explain with some more detail the conceptsthat we encounter.
Boixo (Harvard) Elec. Struct. Tmole App. QM 3 / 43
1 Basis sets
2 Hückel Theory
3 Hartree Fock and beyond
4 Molecular Vibrations
5 Tests
Boixo (Harvard) Elec. Struct. Tmole App. QM 4 / 43
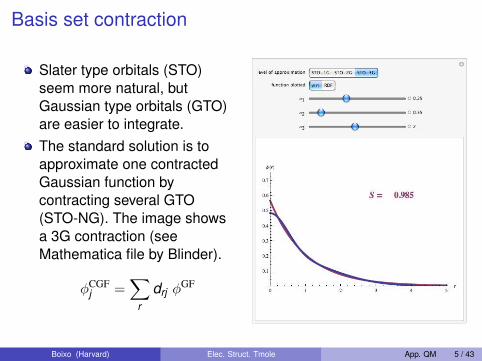
Basis set contraction
Slater type orbitals (STO)seem more natural, butGaussian type orbitals (GTO)are easier to integrate.The standard solution is toapproximate one contractedGaussian function bycontracting several GTO(STO-NG). The image showsa 3G contraction (seeMathematica file by Blinder).
φCGFj =
∑r
drj φGF
Boixo (Harvard) Elec. Struct. Tmole App. QM 5 / 43

Higher angular momentum
Also introduce primitive Gaussians of higher angular momentum,like
gpx ∝ xe−αr2
and
gdxy ∝ xye−αr2.
For convenience, when using d functions we normally add sixCartesian Gaussians (3dxx ,3dyy ,3dzz ,3dxy ,3dyz and 3dzx ) ,combinations of the usual five 3d functions(3dxy ,3dx2−y2 ,3dyz ,3dzx and 3dz2) and 3s (x2 + y2 + z2).
Boixo (Harvard) Elec. Struct. Tmole App. QM 6 / 43

Higher angular momentum
Also introduce primitive Gaussians of higher angular momentum,like
gpx ∝ xe−αr2
and
gdxy ∝ xye−αr2.
For convenience, when using d functions we normally add sixCartesian Gaussians (3dxx ,3dyy ,3dzz ,3dxy ,3dyz and 3dzx ) ,combinations of the usual five 3d functions(3dxy ,3dx2−y2 ,3dyz ,3dzx and 3dz2) and 3s (x2 + y2 + z2).
Boixo (Harvard) Elec. Struct. Tmole App. QM 6 / 43

Multiple functions for each orbital
Minimal: One basis function (STO, GTO, or CGTO) for eachatomic orbital (AO) in the atom.
Double-zeta: Two basis functions for each AO.
Triple-zeta: Three basis functions for each AO.
... and etc. for quadruple-zeta (QZ), 5Z, 6Z.
Split-valence: it uses only one basis function for each core AO,and a larger basis for the valence AO’s.
Boixo (Harvard) Elec. Struct. Tmole App. QM 7 / 43

Split Valence Basis Set
Apply multiple basis functions to valence AO’s and single basisfunctions to the core AO’s.
Core atomic orbitals are relatively independent of the chemicalenvironment; these states do not require to be very flexible in theirdescription. We might get the overall energy wrong, but relativeenergies should be fine.Valence atomic orbitals participate in a wide range of bondingenvironments, and require the flexibility offered by themultiple-zeta basis functions.
Boixo (Harvard) Elec. Struct. Tmole App. QM 8 / 43

Split Valence Basis Set: 6-31 G
6 indicates that each core AO is represented with 1 contractedGaussian basis function composed of 6 primitive Gaussianfunctions.31 two digits means that each valence AO is represented bytwo contracted Gaussian basis functions = double-zeta.
I 3 means that one of the valence contracted Gaussian basisfunctions contains 3 primitive Gaussian functions.
I 1 means that the other valence contracted Gaussian basisfunction contains 1 primitive Gaussian function.
Boixo (Harvard) Elec. Struct. Tmole App. QM 9 / 43

Split Valence Basis SetThe first number is the exponent, the second the linear coefficient.This is nitrogen.
n 6-31+G*6 s4173.5110000 0.0018348627.4579000 0.0139950142.9021000 0.068587040.2343300 0.232241012.8202100 0.46907004.3904370 0.36045503 s11.6263580 -0.11496102.7162800 -0.16911800.7722180 1.14585201 s0.2120313 1.0000000
Boixo (Harvard) Elec. Struct. Tmole App. QM 10 / 43
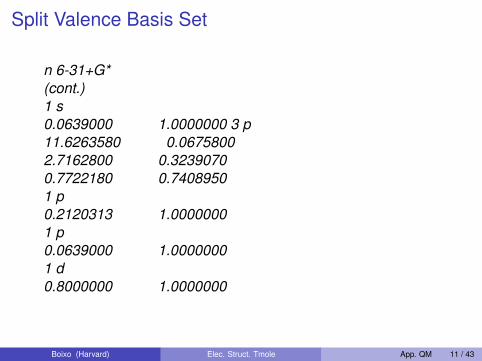
Split Valence Basis Set
n 6-31+G*(cont.)1 s0.0639000 1.0000000 3 p11.6263580 0.06758002.7162800 0.32390700.7722180 0.74089501 p0.2120313 1.00000001 p0.0639000 1.00000001 d0.8000000 1.0000000
Boixo (Harvard) Elec. Struct. Tmole App. QM 11 / 43

Polarization Functions
In molecular environments, orbitals become distorted from their atomicshapes. This is polarization. To describe the effects of polarization oneneeds to add polarization functions.
Numerical tests show that adding polarization functions is moreimportant than adding more functions to the valence electrons.Polarization functions:
usually designed with ’**’ or (d,p).
’*’ or (d) means polarization functions are added to all atoms except H and He.
’**’ or (d,p) means polarization functions are added to all atoms including H andHe.
Boixo (Harvard) Elec. Struct. Tmole App. QM 12 / 43

Polarization Functions
In molecular environments, orbitals become distorted from their atomicshapes. This is polarization. To describe the effects of polarization oneneeds to add polarization functions.
Numerical tests show that adding polarization functions is moreimportant than adding more functions to the valence electrons.Polarization functions:
usually designed with ’**’ or (d,p).
’*’ or (d) means polarization functions are added to all atoms except H and He.
’**’ or (d,p) means polarization functions are added to all atoms including H andHe.
Boixo (Harvard) Elec. Struct. Tmole App. QM 12 / 43

Diffuse Functions
Sometimes electrons are not localized close to atoms; therefore,functions that decay slowly with r are required.
Diffuse functions:
these functions are needed for situations, in which the electrons are not tightlybound to the nuclei or were long-range interactions are relevant, such as anions,metallic complexes, or excited states.
designated with ’++’ or ’aug’.
Boixo (Harvard) Elec. Struct. Tmole App. QM 13 / 43

Diffuse Functions
Sometimes electrons are not localized close to atoms; therefore,functions that decay slowly with r are required.
Diffuse functions:
these functions are needed for situations, in which the electrons are not tightlybound to the nuclei or were long-range interactions are relevant, such as anions,metallic complexes, or excited states.
designated with ’++’ or ’aug’.
Boixo (Harvard) Elec. Struct. Tmole App. QM 13 / 43
General Comments
Polarization functions are always required for quantitative work.
Anions must have diffusion functions.
Best resource for getting basis sets: https://bse.pnl.gov/bse/portal
ErrorsThe relative size of the errors should be:
Method > basis set > everything else.
Boixo (Harvard) Elec. Struct. Tmole App. QM 14 / 43

General Comments
Polarization functions are always required for quantitative work.
Anions must have diffusion functions.
Best resource for getting basis sets: https://bse.pnl.gov/bse/portal
ErrorsThe relative size of the errors should be:
Method > basis set > everything else.
Boixo (Harvard) Elec. Struct. Tmole App. QM 14 / 43
1 Basis sets
2 Hückel Theory
3 Hartree Fock and beyond
4 Molecular Vibrations
5 Tests
Boixo (Harvard) Elec. Struct. Tmole App. QM 15 / 43

Derivation of the Secular EquationThe Hückel Molecular Orbital (HMO) theory was developed in the1930s by Erich Hückel to explain some of the unique properties ofunsaturated aromatic hydrocarbons.Use the linear combination of atomic orbitals (LCAO)
ψi =∑µi
ciµφµ
The total energy E is given by:
E =
∫ψ∗Hψdτ∫ψ∗ψdτ
=
∫(∑
r c∗r φ∗r ) H (∑
s csφs) dτ∫(∑
r c∗r φ∗r ) (∑
s csφs) dτ
=
∑r∑
s
(c∗r cs
∫φ∗r Hφsdτ
)∑
r∑
s(c∗r cs
∫φ∗r φsdτ
)Define Srs is the overlap integral and Hrs is the resonance integral
Hrs ≡∫φ∗r Hφsdτ Srs ≡
∫φ∗r φsdτ
Boixo (Harvard) Elec. Struct. Tmole App. QM 16 / 43

Derivation of the Secular Equation
The energy E must now be minimized with respect to all of thecoefficents (variational principle). This gives
∂E∂cs
= 0 s = 1,2, . . . ,N
After some algebra, these conditions will give rise to N equations:
H11 − ES11 H12 − ES12 · · · H1N − ES1NH21 − ES21 H22 − ES22 · · · H2N − ES21
.... . .
...HN1 − ESN1 HN2 − ESN2 · · · HNN − ESNN
c1c2...
cN
=
00...0
Boixo (Harvard) Elec. Struct. Tmole App. QM 17 / 43

Derivation of the Secular Equation
From linear algebra we know that a set of N equations in N unknownshas solution only if the determinant formed from the coefficients of theunknowns is equal to zero. This is called the secular equation.∣∣∣∣∣∣∣∣∣
H11 − ES11 H12 − ES12 · · · H1N − ES1NH21 − ES21 H22 − ES22 · · · H2N − ES2N
.... . .
...HN1 − ESN1 HN2 − ESN2 · · · HNN − ESNN
∣∣∣∣∣∣∣∣∣ = 0
Boixo (Harvard) Elec. Struct. Tmole App. QM 18 / 43

HMO Assumptions
Only the π electrons are considered in the HMO theory.
Hrr is a constant (α) and is called the Coulomb integral.
Hrr =
∫φ∗r Hφr dτ = α
For bonded atoms, Hrs is also a constant (β) and is called theResonance integral.
Hrs =
∫φ∗r Hφsdτ = β for Cr and Cs bonded.
Hrs (r 6= s) is zero for Cr and Cs not bonded together.The atomic overlap is Srs = δrs.
Boixo (Harvard) Elec. Struct. Tmole App. QM 19 / 43
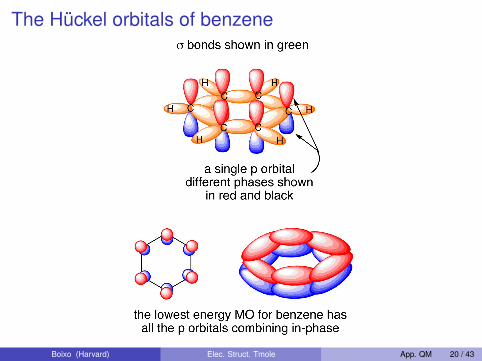
The Hückel orbitals of benzene
Boixo (Harvard) Elec. Struct. Tmole App. QM 20 / 43

The Hückel orbitals of benzene
Energies calculated within the Hückel approximation:∣∣∣∣∣∣∣∣∣∣∣
α − E β 0 0 0 ββ α − E β 0 0 00 β α − E β 0 00 0 β α − E β 00 0 0 β α − E ββ 0 0 0 β α − E
∣∣∣∣∣∣∣∣∣∣∣= 0
The roots of the determinant are:
E = α± 2β, α± β, α± β
Boixo (Harvard) Elec. Struct. Tmole App. QM 21 / 43
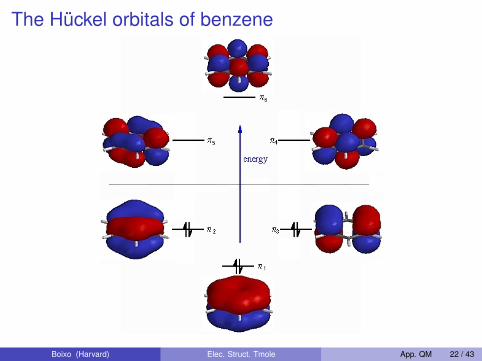
The Hückel orbitals of benzene
Boixo (Harvard) Elec. Struct. Tmole App. QM 22 / 43

Extended Hückel Theory
Ignore core electronsCompute Srs (in standard Hückel theory it was assumed to be δrs).Use empirical values for Hrr , such as ionization potentials.For the off diagonal use
Hrs =12
Crs(Hrr + Hss)Srs .
There are different choices for Crs, but typically 1.75 is taken for allpossibilities.
Boixo (Harvard) Elec. Struct. Tmole App. QM 23 / 43
1 Basis sets
2 Hückel Theory
3 Hartree Fock and beyond
4 Molecular Vibrations
5 Tests
Boixo (Harvard) Elec. Struct. Tmole App. QM 24 / 43

Scaling of Hartree-Fock
We have seen in a past lecture that in a naive implementation thescaling of the cost of Hartree-Fock is K 4, where K is the number ofGaussians used for the AOs. This comes from the electron-electroninteraction terms, which give (in chemist notation)∑
σµλν
(σµ|λν) .
Because the number of AOs scales at least linearly with the number ofatoms this gives a scaling O(N4) for N atoms.We will now see that for big linear molecules the scaling is O(N2), upto logarithmic factors [[Dyczmons 1972, paper included]].
Boixo (Harvard) Elec. Struct. Tmole App. QM 25 / 43
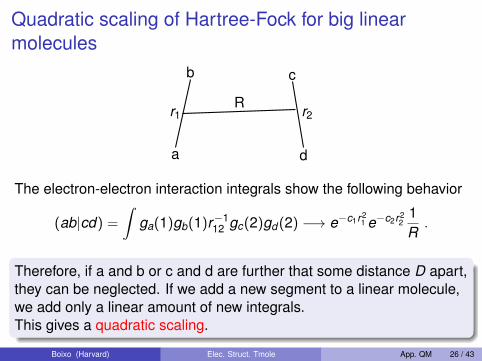
Quadratic scaling of Hartree-Fock for big linearmolecules
b
r1
a
c
d
r2R
The electron-electron interaction integrals show the following behavior
(ab|cd) =
∫ga(1)gb(1)r−1
12 gc(2)gd (2) −→ e−c1r21 e−c2r2
21R.
Therefore, if a and b or c and d are further that some distance D apart,they can be neglected. If we add a new segment to a linear molecule,we add only a linear amount of new integrals.This gives a quadratic scaling.
Boixo (Harvard) Elec. Struct. Tmole App. QM 26 / 43

MP2Moller Plesset perturbation theory
Moller Plesset perturbation theory is a second order perturbationtheory technique where the unperturbed Hamiltonian is defined to bethe Hartree-Fock Hamiltonian, and the perturbed Hamiltonian is theelectronic structure Hamiltonian
H = HHF + V
V =∑i<j
1rij−∑
i
vHF (i) .
We use “physics” notation for the Coulomb and exchange terms,
〈ij |kl〉 =
∫φa(1)φb(1)r−1
12 φc(2)φd (2)
〈ij ||kl〉 = 〈ij |kl〉 − 〈ij |lk〉 .
Boixo (Harvard) Elec. Struct. Tmole App. QM 27 / 43

MP2Moller Plesset perturbation theory
Moller Plesset perturbation theory is a second order perturbationtheory technique where the unperturbed Hamiltonian is defined to bethe Hartree-Fock Hamiltonian, and the perturbed Hamiltonian is theelectronic structure Hamiltonian
H = HHF + V
V =∑i<j
1rij−∑
i
vHF (i) .
We use “physics” notation for the Coulomb and exchange terms,
〈ij |kl〉 =
∫φa(1)φb(1)r−1
12 φc(2)φd (2)
〈ij ||kl〉 = 〈ij |kl〉 − 〈ij |lk〉 .
Boixo (Harvard) Elec. Struct. Tmole App. QM 27 / 43

MP2Moller Plesset perturbation theory
With this definition, the first order correction to the unperturbedHamiltonian (which is the Hartree-Fock Hamiltonian) is
E (0)0 + E (1)
0 =∑
a
εa −12
∑ab
〈ab||ab〉 ,
where εa are the orbital energies. That is, the first order perturbationgives the Hartree-Fock energy.We need to go second order, which gives the MP2 correction to theenergy.
E (2)0 =
14
∑abrs
|〈ab||rs〉|2
εa + εb − εr − εs.
The cost is O(N5).
Boixo (Harvard) Elec. Struct. Tmole App. QM 28 / 43

MP2Moller Plesset perturbation theory
With this definition, the first order correction to the unperturbedHamiltonian (which is the Hartree-Fock Hamiltonian) is
E (0)0 + E (1)
0 =∑
a
εa −12
∑ab
〈ab||ab〉 ,
where εa are the orbital energies. That is, the first order perturbationgives the Hartree-Fock energy.We need to go second order, which gives the MP2 correction to theenergy.
E (2)0 =
14
∑abrs
|〈ab||rs〉|2
εa + εb − εr − εs.
The cost is O(N5).
Boixo (Harvard) Elec. Struct. Tmole App. QM 28 / 43

Coupled ClusterThe coupled cluster method uses the ansatz for the ground state:
eT |ΦHF 〉
The cluster operator T is
T = T1 + T2 + T3 + · · ·
where the subindex indicates the number of excitations. That is
T1 =∑
i
∑a
tai ai a
†a
T2 =14
∑i,j
∑a,b
tabij ai aj a
†aa†b .
The most common method is CCSD, which includes single and doubleexcitations. CCSD(T) adds triple excitations calculated withperturbation theory.The cost of CCSD is O(N6) and CCSD(T) is O(N7).
Boixo (Harvard) Elec. Struct. Tmole App. QM 29 / 43
1 Basis sets
2 Hückel Theory
3 Hartree Fock and beyond
4 Molecular Vibrations
5 Tests
Boixo (Harvard) Elec. Struct. Tmole App. QM 30 / 43

Bond lengths and molecular vibrations
1 The electronic energy within the Born-Oppenheimerapproximation gives a potential energy surface.
2 The minimum of the potential energy surface gives the bondlengths.
3 Calculate the set of second derivatives of the molecular electronicenergy with respect to the 3N nuclear Cartesian coordinates
fij =1
(mimj)1/2
(∂2U∂Xi∂Xj
)4 Find the eigenvalues and eigenvectors of the matrix fij . The
eigenvalues give the frequencies and the eigenvectors the normalmodes.
Boixo (Harvard) Elec. Struct. Tmole App. QM 31 / 43
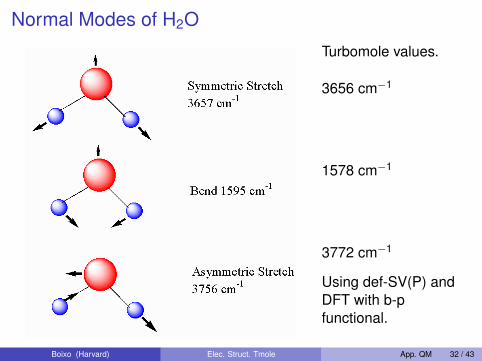
Normal Modes of H2O
Turbomole values.
3656 cm−1
1578 cm−1
3772 cm−1
Using def-SV(P) andDFT with b-pfunctional.
Boixo (Harvard) Elec. Struct. Tmole App. QM 32 / 43

1 Basis sets
2 Hückel Theory
3 Hartree Fock and beyond
4 Molecular Vibrations
5 Tests
Boixo (Harvard) Elec. Struct. Tmole App. QM 33 / 43
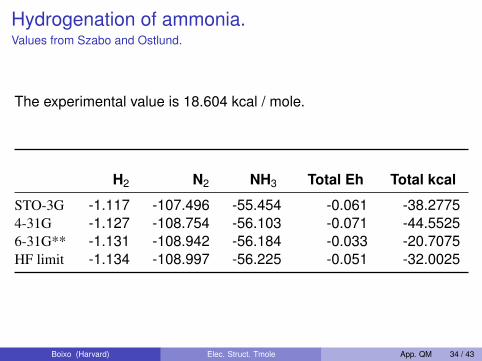
Hydrogenation of ammonia.Values from Szabo and Ostlund.
The experimental value is 18.604 kcal / mole.
H2 N2 NH3 Total Eh Total kcal
STO-3G -1.117 -107.496 -55.454 -0.061 -38.27754-31G -1.127 -108.754 -56.103 -0.071 -44.55256-31G** -1.131 -108.942 -56.184 -0.033 -20.7075HF limit -1.134 -108.997 -56.225 -0.051 -32.0025
Boixo (Harvard) Elec. Struct. Tmole App. QM 34 / 43
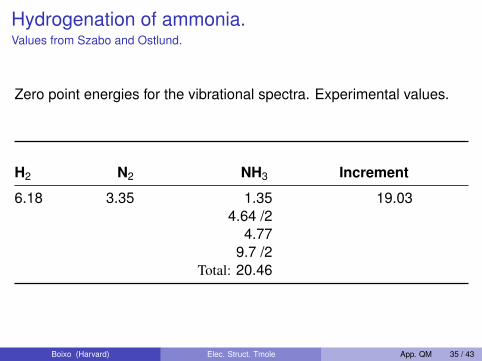
Hydrogenation of ammonia.Values from Szabo and Ostlund.
Zero point energies for the vibrational spectra. Experimental values.
H2 N2 NH3 Increment
6.18 3.35 1.35 19.034.64 /2
4.779.7 /2
Total: 20.46
Boixo (Harvard) Elec. Struct. Tmole App. QM 35 / 43
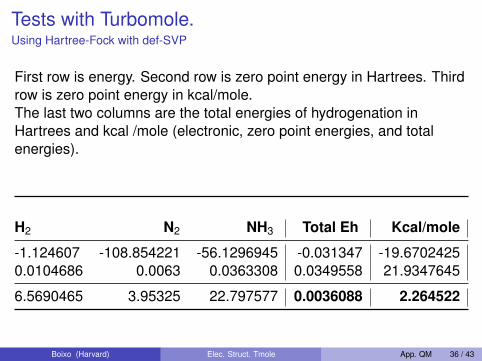
Tests with Turbomole.Using Hartree-Fock with def-SVP
First row is energy. Second row is zero point energy in Hartrees. Thirdrow is zero point energy in kcal/mole.The last two columns are the total energies of hydrogenation inHartrees and kcal /mole (electronic, zero point energies, and totalenergies).
H2 N2 NH3 Total Eh Kcal/mole
-1.124607 -108.854221 -56.1296945 -0.031347 -19.67024250.0104686 0.0063 0.0363308 0.0349558 21.9347645
6.5690465 3.95325 22.797577 0.0036088 2.264522
Boixo (Harvard) Elec. Struct. Tmole App. QM 36 / 43

Tests with Turbomole.
The answer is off, although looking only at the electronic energy looksgood.To get the enthalpy U + pV we need to take into accountp∆V = RT ∆n. There are two more moles of reactants than products,but the contribution is only 0.0017101 Eh. It really does not make anydifference.The main problem, looking at the data from Szabo, is the size of thebasis set. At the HF limit the result is much better.Also, N2 has a triple bond, which means a fair amount of correlation,so HF will have problems.
Boixo (Harvard) Elec. Struct. Tmole App. QM 37 / 43
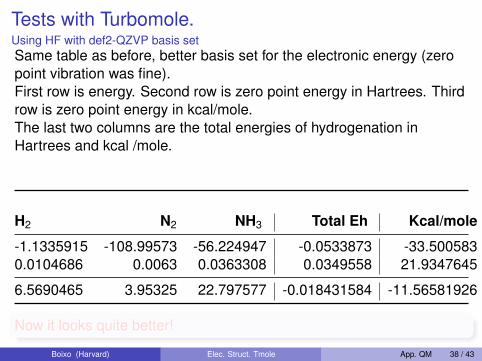
Tests with Turbomole.Using HF with def2-QZVP basis setSame table as before, better basis set for the electronic energy (zeropoint vibration was fine).First row is energy. Second row is zero point energy in Hartrees. Thirdrow is zero point energy in kcal/mole.The last two columns are the total energies of hydrogenation inHartrees and kcal /mole.
H2 N2 NH3 Total Eh Kcal/mole
-1.1335915 -108.99573 -56.224947 -0.0533873 -33.5005830.0104686 0.0063 0.0363308 0.0349558 21.9347645
6.5690465 3.95325 22.797577 -0.018431584 -11.56581926
Now it looks quite better!
Boixo (Harvard) Elec. Struct. Tmole App. QM 38 / 43
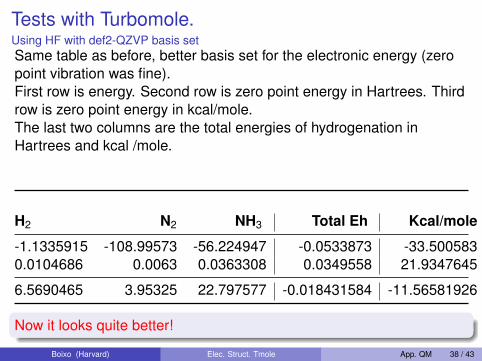
Tests with Turbomole.Using HF with def2-QZVP basis setSame table as before, better basis set for the electronic energy (zeropoint vibration was fine).First row is energy. Second row is zero point energy in Hartrees. Thirdrow is zero point energy in kcal/mole.The last two columns are the total energies of hydrogenation inHartrees and kcal /mole.
H2 N2 NH3 Total Eh Kcal/mole
-1.1335915 -108.99573 -56.224947 -0.0533873 -33.5005830.0104686 0.0063 0.0363308 0.0349558 21.9347645
6.5690465 3.95325 22.797577 -0.018431584 -11.56581926
Now it looks quite better!
Boixo (Harvard) Elec. Struct. Tmole App. QM 38 / 43

Excitations with TDDFT
Running Turbomole with DFT and the d-g functional on N2 (always withthe def-SV(P) basis set) you get:
1 singlet e1g excitationTotal energy: -109.0922282176429Excitation energy: 0.3523342202571314Excitation energy / eV: 9.587506051664842Excitation energy / nm: 129.3185561900520Excitation energy / cm−1: 77328.42287390638
This is the σg to πg transition, with an experimental value of 9.31 eV[Bauernschmitt et. al., 1996]. The agreement is quite good.
Boixo (Harvard) Elec. Struct. Tmole App. QM 39 / 43
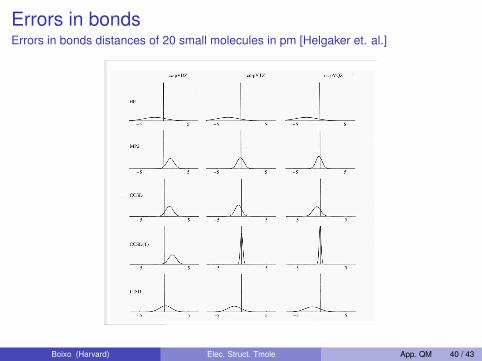
Errors in bondsErrors in bonds distances of 20 small molecules in pm [Helgaker et. al.]
Comparison of Electron Correlation Methods
Errors in bond distances of 20 small molecules in pm
Dmitrij Rappoport (Harvard U.) Electron Correlation Methods 11/12/09 Lecture 18 33 / 36
Boixo (Harvard) Elec. Struct. Tmole App. QM 40 / 43
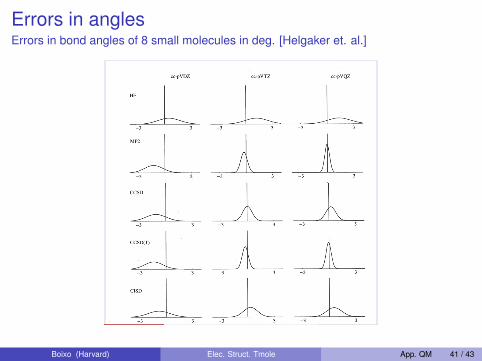
Errors in anglesErrors in bond angles of 8 small molecules in deg. [Helgaker et. al.]
Comparison of Electron Correlation Methods
Errors in bond angles of 8 small molecules in deg
Dmitrij Rappoport (Harvard U.) Electron Correlation Methods 11/12/09 Lecture 18 34 / 36
Boixo (Harvard) Elec. Struct. Tmole App. QM 41 / 43
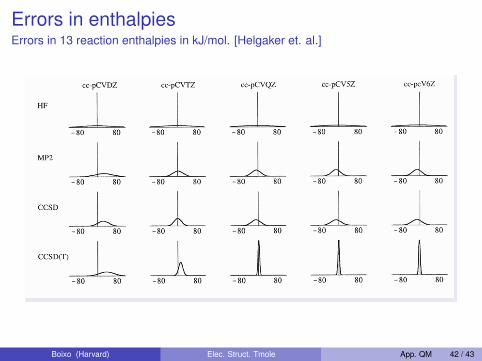
Errors in enthalpiesErrors in 13 reaction enthalpies in kJ/mol. [Helgaker et. al.]
Comparison of Electron Correlation Methods
Errors in 13 reaction enthalpies in kJ/mol
Dmitrij Rappoport (Harvard U.) Electron Correlation Methods 11/12/09 Lecture 18 35 / 36Boixo (Harvard) Elec. Struct. Tmole App. QM 42 / 43

1 Basis sets
2 Hückel Theory
3 Hartree Fock and beyond
4 Molecular Vibrations
5 Tests
Boixo (Harvard) Elec. Struct. Tmole App. QM 43 / 43





























