Embed Size (px)
Citation preview

Gadsby et al. – MiMM chapter – submitted May 20, 2010
1
Electrophysiological, Biochemical, and Bioinformatic Methods for Studying CFTR Channel
Gating and its Regulation
László Csanády1, Paola Vergani2, Attila Gulyás-Kovács3, & David C. Gadsby3
1Department of Medical Biochemistry, Semmelweis University, Budapest, Hungary
2Department of Neuroscience, Physiology and Pharmacology, University College London,
London, United Kingdom
3Laboratory of Cardiac/Membrane Physiology, The Rockefeller University, New York, USA
In “Cystic Fibrosis Diagnosis and Protocols, Volume I: Approaches to Study and Correct CFTRDefects.” M.D. Amaral, and K. Kunzelmann, editors. Springer, New York. 443-469.10.1007/978-1-61779-117-8_28.
Correspondence to:
David C. Gadsby
Laboratory of Cardiac/Membrane Physiology
Rockefeller University
1230 York Avenue
New York, NY 10065, USA
Tel: 212-327-8680
Fax: 212-327-7589
E-mail: [email protected]

Gadsby et al. – MiMM chapter – submitted May 20, 2010
2
Abstract
CFTR is the only member of the ABC (ATP binding cassette) protein superfamily known to
function as an ion channel. Most other ABC proteins are ATP-driven transporters, in which a
cycle of ATP binding and hydrolysis, at intracellular nucleotide binding domains (NBDs) powers
uphill substrate translocation across the membrane. In CFTR, this same ATP-driven cycle opens
and closes a transmembrane pore through which chloride ions flow rapidly down their
electrochemical gradient. Detailed analysis of the pattern of gating of CFTR channels thus offers
the opportunity to learn about mechanisms of function not only of CFTR channels, but also of
their ABC transporter ancestors. In addition, CFTR channel gating is subject to complex
regulation by kinase-mediated phosphorylation at multiple consensus sites in a cytoplasmic
regulatory domain that is unique to CFTR. Here we offer a practical guide to extracting useful
information about the mechanisms that control opening and closing of CFTR channels: on how to
plan (including information obtained from analysis of multiple sequence alignments), carry out,
and analyze electrophysiological and biochemical experiments, as well as on how to circumvent
potential pitfalls.

Gadsby et al. – MiMM chapter – submitted May 20, 2010
3
1. Introduction
1.1. Present understanding of regulation of CFTR channel gating by nucleotides
CFTR’s pair of cytoplasmic NBDs (NBD1, NBD2) are drawn together in head-to-tail
orientation (1,2), as in all other ABC proteins, by two ATP molecules bound within composite
interfacial catalytic sites, between the Walker A and B motifs of one NBD and the LSGGQ-like
ABC signature sequence of the other (3,4,5,6,7,8,9). Hydrolysis of the ATP disrupts the interface,
causing the NBDs to separate and allowing fresh ATP to bind. In CFTR, these cyclic, ATP-
driven motions of the NBDs are transmitted to the transmembrane domains (TMDs) to alternately
open and close the pore through which chloride ions flow rapidly across the membrane, down
their electrochemical gradient. Dissimilarities between NBD1 and NBD2 in CFTR, mirrored in
all members of the OAD (organic anion and drug; 10) transporter subfamily of ABC proteins to
which CFTR belongs, render only one of the composite interfacial ATP sites (the “NBD2
composite” site, incorporating the NBD2 Walker motifs) catalytically competent, while crippling
the “NBD1 composite” site by substitutions on both sides of the interface. In the absence of ATP,
phosphorylated (see 1.2., below) CFTR channels essentially remain closed (except for rare, brief,
openings; e.g., 11,12). On exposure of the cytoplasmic surface of CFTR channels to millimolar
ATP, its binding at the two interfacial sites causes NBD1-NBD2 heterodimerization and opening
of the ion pore. The average channel opening rate is a saturable function of ATP concentration
according to Michaelis Menten kinetics and is half maximal near 50 M ATP for wild-type (WT)
CFTR (13). Because this apparent affinity for activation by ATP is diminished by mutations of
either NBD that are expected to impair ATP binding, normal channel opening appears to require
ATP binding in both composite sites (12).
The pore remains open until hydrolysis of the ATP in the competent “NBD2 site” triggers
channel closure. That hydrolysis rate-limits closure is supported by prolongation of channel
openings upon interference with hydrolysis by addition of inorganic phosphate analogues such as
orthovanadate or pyrophosphate (14,15,16), or of poorly hydrolyzable ATP analogs like
AMPPNP (17,15), or by mutation of key catalytic residues (15,18,11,12,1). That the hydrolysis
occurs only in the “NBD2 site”, and not in the “NBD1 site”, is demonstrated by the findings that
only “NBD2 site” mutations prolong openings (15,18,11,12), and that photocrosslinking with
gamma-32P-8azidoATP radio-labels only the “NBD1 site”, since the 32P is released from the

Gadsby et al. – MiMM chapter – submitted May 20, 2010
4
“NBD2 site” too rapidly to be observed (19,20). The photocrosslinking experiments also
indicated that non-hydrolyzed ATP remained bound for minutes at the “NBD1 site” (19,20),
arguing that the seconds-long ATP binding and hydrolysis cycle that drives CFTR channel gating
reflects events at the “NBD2 site” (15,12,20), a conclusion further supported by the much slower
loss, after sudden ATP withdrawal, of the ability of brief exposures to pyrophosphate or
AMPPNP to elicit prolonged channel openings (21).
Because crystal structures of nucleotide-bound ABC transporters show the extracellular-
side gate in the transmembrane domains open and the cytoplasmic side closed (e.g., 7,8,9,22),
whereas both gates must be open in an ATP-bound CFTR channel to allow the observed rapid
chloride ion flow (millions of ions per sec per channel), in CFTR the cytoplasmic-side gate
would appear to have become atrophied, or uncoupled from the outer gate (23). CFTR can
therefore be considered to be a broken ABC transporter (e.g., 24,25,26,27). Detailed analysis of
the pattern of gating of CFTR channels thus offers the opportunity to learn about mechanisms of
function not only of CFTR channels, but also of their ABC transporter ancestors.
1.2. Present understanding of regulation of CFTR channel gating by phosphorylation
Before a CFTR channel can be opened by ATP, cAMP-dependent protein kinase (PKA)
must phosphorylate consensus-site serines in the ~200-residue cytoplasmic regulatory (R)
domain that links CFTR’s two homologous halves (28,29), and phosphorylation of PKC sites in
the R domain might be a prerequisite (30,31). Because cells contain millimolar levels of ATP,
enough to saturate binding at CFTR’s NBDs, phosphorylation of the R domain by PKA may be
presumed to be the principal physiological regulator of CFTR channel activity. The R domain is
unique to CFTR, and its structure and mechanism remain controversial and incompletely
understood (reviewed in 32). Evidence of structural disorder of isolated R domain (33,34), and of
graded activation of CFTR channels with increasing phosphorylation (e.g., 28,35,36), have been
interpreted as indicating redundancy among up to 9 R-domain phosphoserines, with accumulated
negative charge as the major factor. The positions of consensus serine-containing segments in
crystal structures of CFTR NBD1 extended by an R-domain fragment (37), and NMR
measurements of NBD1-R domain interactions (34,38), prompted the proposal that non-
phosphorylated R domain binds to the NBDs and interferes with ATP-mediated formation of the
NBD1-NBD2 heterodimer, and hence with CFTR channel opening; phosphorylation of the R

Gadsby et al. – MiMM chapter – submitted May 20, 2010
5
domain is proposed to release it from the NBDs, relieving the inhibition of gating. Such a simple
mechanism seems unlikely, as neither serine-containing segment is needed for channels to close
in the absence of ATP, regardless of phosphorylation status, and their deletion or mutation does
not alter the strict dependence of channel gating on phosphorylation by PKA (39). Moreover,
deletion of the entire NBD2 (40,41) or introduction of “constitutive” mutations (42), even the
CF-causing mutation G551D (43), results in channels that gate regardless of the presence of ATP
– but even this "spontaneous" activity remains strictly dependent on prior phosphorylation by
PKA. Finally, two of the phosphoserines, including the one most readily generated by PKA
(29,36,44,45), seem to have an inhibitory, rather than stimulatory, influence on channel open
probability (Po, see 3.4.2.) (46,36). A region of density interpreted as R domain that extends
between NBDs and TMDs in a cryo-EM low-resolution structure of CFTR (47) seems more
consistent with an alternative proposal, that the R domain somehow controls transmission of
information from the NBDs to the channel gates (20).

Gadsby et al. – MiMM chapter – submitted May 20, 2010
6
2. Materials
2.1. Xenopus oocyte incubation media
After injection with cRNA, oocytes are kept for 1-5 days at 18 °C in solution containing
(in mM): 83 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, 5 HEPES (pH 7.5), plus 50 g/ml Gentamycin
(48,13,12).
2.2. Solutions used for electrophysiological recordings
For recording in intact oocytes, the superfusion solution contains (in mM): 83 NaCl, 2
KCl, 1 MgCl2, 5 HEPES (pH 7.5). For excised patch recording, pipette solution contains (in mM)
138 NMDG-Cl, 2 MgCl2, 5 HEPES (pH 7.4), and bath solution contains (in mM) 138 NMDG-Cl,
2 MgCl2, 0.5 EGTA, 5 HEPES (pH 7.1); for chloride-free solutions, sulphamate or gluconate
replace chloride (48,13,12).

Gadsby et al. – MiMM chapter – submitted May 20, 2010
7
3. Methods
3.1. Heterologous expression of human CFTR in various cell types including Xenopus
oocytes
3.1.1. Expression plasmids
For expression in Xenopus oocytes, CFTR mutants are generated from pGEMHE-CFTR
(48) by mutation with Quikchange, verified by automated DNA sequencing, and in vitro
transcribed using mMessage T7 polymerase to yield cRNA. For expression in HEK293T cells,
pIRES-CFTR constructs are generated by subcloning the corresponding CFTR into the EcoRV
site of the pIRES vector (20).
3.1.2. Cell preparation
Oocytes are isolated from anaesthethized adult female Xenopus laevis, and dispersed
using collagenase, as described (13). HEK293T cells are maintained in DMEM medium
supplemented with 10% Fetal Bovine Serum, plated in 100-mm dishes and transfected with
pIRES-CFTR (20)
3.2. Biochemical analysis of CFTR regulation
3.2.1. Cysteine modification as a tool to study domain-domain interactions
To supplement the minimal structural information available for CFTR and to directly
examine changes in conformation associated with gating, sulphydryl-specific crosslinking can be
used to probe proximity of pairs of target cysteines introduced into CFTR, provided that some or
all of the 18 native Cys are first removed or shown not to interfere with the assay of target Cys
reactivity. If the C atoms of residues approach each other to within 5-8 Å, then target Cys at
those positions should form disulphide bonds upon oxidation with copper phenanthroline.
Bifunctional sulphydryl-specific reagents, e.g., based on maleimide (2) or methanethiosulphonate
(MTS; 49,50), with variable-length linkers can provide upper estimates for the minimum
separation of Cys pairs under the conditions of the assay. These reagents can be applied to
functioning CFTR channels in vivo. For example, crosslinking of several target Cys pairs across
CFTR’s head-to-tail NBD1-NBD2 heterodimer interface by BMOE (bismaleimidoethane;
reactive groups ≤8 Å apart) in intact oocytes, assessed from large gels shifts of split CFTR
channels with a target Cys in each half, has demonstrated that they must approach to within 8 Å

Gadsby et al. – MiMM chapter – submitted May 20, 2010
8
of each other at some point during the gating cycle (2). In inside-out membrane patches excised
from oocytes, formation of a disulphide bond across the NBD1-NBD2 interface, between Cys at
position 1248 in the NBD2 Walker A motif and 549 in the NBD1 LSGGQ motif, prevented
closure of the crosslinked CFTR channels after withdrawal of ATP; but the channels closed when
the disulphide bonds were reduced by dithiothreitol (2), further supporting the association of
dimerized NBDs with open channels. Similarly, bifunctional MTS reagents were found, in gel-
shift assays, to crosslink NBD1 and NBD2 to cytoplasmic coupling helices that extend from the
TMDs (51,50) in a manner consistent with the “domain-swap” organization seen in the crystal
structure of Sav1866 a prokaryotic multidrug transporter orthologue (7); when tested in inside-
out patches, in contrast to the open state stabilization caused by the NBD1-NBD2 crosslink (2),
crosslinking either NBD to one of the coupling helices tended to impair channel opening (51,50).
These crosslinking experiments on CFTR have all used homology models to identify candidate
target positions for Cys substitutions.
Modification of a single introduced target Cys, while assaying function as a monitor
of reaction and its consequence, is an alternative way to probe CFTR channel structure and
mechanism. Addition of N-ethylmaleimide (NEM) to the ATP-containing solution bathing
inside-out patches irreversibly diminished the Po of CFTR channels with a single Cys introduced
into Walker A or ABC signature motif of either composite interfacial site (52). Modification of
the Walker A, but not the ABC signature, mutants became slower as ATP concentration was
raised (52) and, at each [ATP], modification was much slower in the “NBD1” than in the
“NBD2” composite site; these observations fit well with the present view of ATP-driven gating
of CFTR channels (section 1.1, above).
Further structural and mechanistic information may be obtained by determining the
gating-state dependence of modification of single Cys or of crosslinking of Cys pairs (e.g., 53).
3.2.2. Studying the kinetics of CFTR phosphorylation
Western blots of full-length (54) or split (2) CFTR, and Coomassie-stained gels or
autoradiographs of isolated R domain peptide (29,36), have all revealed mobility shifts induced
by phosphorylation with PKA. By using low [ATP] to slow phosphorylation, taking samples at
different times, and using SDS-PAGE followed by mass spectrometry of separated peptides, two-
dimensional phosphopeptide mapping, and/or point mutation to identify individual

Gadsby et al. – MiMM chapter – submitted May 20, 2010
9
phosphoserines, the kinetics of phosphorylation of individual Ser can be assessed. With this
approach, Ser768 was found to be the first one phosphorylated and phosphorylation of Ser737
was found to cause the major mobility shift (36). Kinetic analysis of several small synthetic
peptides (10-24 residues), each encompassing one (or two) of the R-domain consensus PKA
sites, showed that the peptide containing Ser768 was by far the preferred substrate for PKA (29),
consistent with the results for isolated R domain and entire CFTR (36).
A difficulty in all biochemical studies of CFTR is availability of sufficient amounts of
protein; expression and purification from Sf9 insect cells (55) and baby hamster kidney (BHK-
21) fibroblast cells (56) have proven most successful so far. The large amplification of
electrophysiological assays allows circumvention of this difficulty, as some biochemical studies
can be done in real time on CFTR channels in voltage-clamped excised membrane patches or
whole cells. For example, phosphorylation by PKA catalytic subunit or by PKC, and
dephosphorylation by specific phosphatases, of WT CFTR channels or mutants lacking one or
more consensus phosphorylation sites, can be monitored by recording current in excised patches
during direct application of exogenous kinases and/or phosphatases or inhibitors of endogenous
phosphatases (e.g., 35,57,30,58,59,31,60,36). The presence of active endogenous phosphatases in
excised patches even permits determination of PKA concentration dependence of CFTR channel
activation, assayed as Po; enhanced sensitivity to PKA in Ser768Ala mutants compared to WT
CFTR (36) confirmed the inhibitory influence of phosphoserine 768 concluded from
measurements of sensitivity to the phosphodiesterase inhibitor IBMX in intact oocytes (46). The
enhanced sensitivity to phosphorylation resulted in substantial activation of Ser768Ala CFTR
channels in resting oocytes, due to basal levels of PKA activity; phosphorylation of six Ser
(including Ser768) in the R domains of WT CFTR channels in resting oocytes was confirmed by
mass spectrometry (36).
3.3. Electrophysiological recording of CFTR currents
The possibility of following ion channel function using electrophysiological techniques
make CFTR uniquely suited for basic structure/function studies among ABC transporters.
However, some of its gating characteristics – e.g. relatively slow and non-equilibrium gating –
limit the usefulness of some techniques.
3.3.1. Recording CFTR currents in intact cells by TEVC and whole-cell patch-clamp

Gadsby et al. – MiMM chapter – submitted May 20, 2010
10
The two-microelectrode voltage-clamp (TEVC) method, and tight-seal current
recording in the whole-cell configuration, allow little or no direct modification of the intracellular
solution. However, as described above, the main factors regulating CFTR channel gating are
phosphorylation and interaction with ATP, both acting on intracellular domains. In most intact-
cell studies CFTR is activated by extracellular application of membrane penetrating forskolin,
which activates adenylate cyclase, and hence PKA, eventually resulting in an increase in whole
cell conductance. For WT CFTR expressed in oocytes, held at -50 mV, the inward current peaks
~3 minutes after forskolin addition. For CFTR mutants with low Po, conductance can increase
more slowly, e.g. reaching a 10-100 fold lower maximal conductance in 10-20 minutes. The very
indirect control of the channel gates somewhat limits the kinetic information that can be obtained
with these techniques, yet they remain useful for screening (e.g. 48,2), or studying extreme
alterations of gating kinetics (e.g. 61). In addition, whole cell current recordings can be valuable
for studying endogenous CFTR in its physiological environment, with only minimal experimental
modification (methods described in 62).
3.3.2 Recording CFTR currents in inside-out patches
The experimenter has many more possibilities if, after sealing, the patch can be
excised, so that the intracellular face of the patch is exposed. In Xenopus oocytes, this inside-out
configuration is conveniently obtained by initially “cramming” the pipette towards the centre of
the cell, before extricating it with the excised membrane patch. Because of the characteristic slow
gating of CFTR channels, relatively simple perfusion systems (e.g. available from ALA
Scientific Instruments, www.alascience.com), which allow complete exchange of the solution
bathing the intracellular (outside) face of the patch within a fraction of a second, are sufficient for
most experiments. Most of the channels present in the patch will be activated within a few
minutes after switching to a solution in which catalytic subunit of PKA (130 units/ml) and
MgATP (concentrations 2-5 mM) are present. PKA extracted from bovine heart is commercially
available (Sigma cat # P2645; note that recombinant PKA catalytic subunit, Promega cat #
V5161, was found to be much less active even using up to 300 units/ml). Millimolar [ATP] are
saturating for most mutants, the WT apparent dissociation constant being ~ 50 M (e.g. 13).
Simple solutions with high Cl-, N-methyl-D-glucamine as a counterion (to minimize cationic
conductances), lightly buffered, and containing EGTA and millimolar Mg2+ are used as bath

Gadsby et al. – MiMM chapter – submitted May 20, 2010
11
solutions, while pipettes (bathing the extracellular face) are filled with a similar solution but
lacking EGTA (e.g. 27, see 2.2, above).
In some expression systems the level of channel expression can be modulated. This
can be easily obtained when using Xenopus oocytes, by altering amounts of cRNA injected
(typically from 0.1 ng/oocyte to 10 ng/oocyte for WT CFTR), the time allowed for expression
(~12 hours to one week), the position of the patch with respect to the injection site (when
injecting within the animal pole, patches close to the injection site show higher expression). To a
lesser extent, when using mammalian cell lines, variation in expression levels - among different
cell lines and/or among clones obtained from different transfections, - can be achieved (e.g. 63).
The experimenter can thus obtain two different, complementary, sets of kinetic information:
either monitoring individual channels during steady-state gating (3.3.2.1) or recording ensemble
currents flowing through a high number of channels (3.3.2.2.).
3.3.2.1. Recording individual CFTR channel currents in inside-out patches
If the patch contains one or few (e.g. ≤8) channels the current “jumps”
corresponding to the opening and closing of individual channel gates can be detected. Since the
single-channel conductance of CFTR is small (~ 10 pS, 64), the main practical problem in
performing these experiments is keeping electrical noise as low as possible to improve the
signal/noise ratio. Practical considerations on this subject are discussed at length in excellent
texts (65,66). Most important, the electrical seal resistance between glass pipette tip and cell
membrane needs to be very large (with Xenopus oocytes, seals >100 Gcan be routinely
obtained, using freshly stripped oocytes and 0.2 m-filtered solutions) and line-frequency
interference needs to be minimized (by carefully grounding conducting objects close to the
amplifier headstage, avoiding ground loops, and shielding in a Faraday cage). In addition,
several parameters of the experimental setup can be optimized to obtain low-noise records (67).
In our labs, the patches are excised in a large fluid-filled “patch-pulling chamber”, and then
transferred to a “flow chamber” for recording (Fig. 1). Here, to minimize the capacitance of the
immersed pipette tip, the patch is immersed in a thin film of solution flowing down the walls of
the flow chamber, along the petri dish edge, and grounded (Figure 1). Luckily, CFTR’s slow
gating characteristics are here an advantage, since the recording bandwidth need not be wide and

Gadsby et al. – MiMM chapter – submitted May 20, 2010
12
filtering at 50-200 Hz does not remove events of interest for ATP- and phosphorylation-
dependent gating.
3.3.2.2 Recording macroscopic, multi-channel, CFTR currents in inside-out
patches
When studying low probability events or mutants whose gating is very slow,
accumulation of sufficient events cannot be achieved within the patch lifetime by recording from
one or a few channels. In these cases, information on channel gating regulation is most simply
obtained by recording ensemble current flow across patches containing a large number (typically
hundreds or thousands) of CFTR channels (“macropatches”). Recording from macropatches
allows relative steady-state parameters, e.g., relative Po upon sudden change in [ATP] (13), or
relative Po in ATP vs. poorly-hydrolyzable analogues (12), to be easily obtained. Alternatively,
kinetic information can be extracted from measurements of the time course of current change
following a manoeuvre that affects CFTR gating, e.g. burst duration upon washout of ATP (see
3.5, below).
3.4. Analysis of steady-state CFTR current recordings
3.4.1. Idealization of membrane currents
The first step in the process of extracting information on channel gating kinetics from
a raw current trace is to reconstruct the time series of channel opening/closing transitions. The
output of this "idealization" procedure is an ordered list of events, each event described by a pair
of numbers lk, tk; lk denotes the number of channels open during the kth event and tk its duration.
This "events list" contains in a compact form all the information the raw data carry about gating.
The simplest idealization algorithm is the half-amplitude threshold crossing technique
(68). In this procedure the baseline current and possible slow current drifts – not channel-related
– are subtracted from the raw data. Ideal conductance levels are then set to integer multiples of
the unitary current amplitude, and consecutive data points which fall within a half-amplitude
distance of a given ideal conductance level are merged into an event. For reliable application of
this simple procedure the signal-to-noise ratio must be sufficiently high to minimize half-
amplitude threshold crossings caused by noise, while individual gating transitions should remain
well resolved in time. In addition, the channels should open predominantly to a single, well

Gadsby et al. – MiMM chapter – submitted May 20, 2010
13
defined, conductance level. These criteria are typically met for WT CFTR under standard
conditions (e.g., (69,18,11,13)).
The half-amplitude procedure might present problems when using MOPS as an
intracellular buffer (15), for certain pore mutants (70), or for murine CFTR channels, which open
predominantly to subconductance levels (71). In such cases more elaborate algorithms are
necessary for idealization. Time-course fitting (68) and hidden Markov modeling (72,73) are
procedures that allow for successful idealization of records with frequent subconductance
openings and/or an insufficient signal-to-noise ratio; such algorithms are implemented by the free
software packages HJCFIT (http://www.ucl.ac.uk/Pharmacology/dcpr95.html) and QuB
(http://www.qub.buffalo.edu/), respectively.
3.4.2. Kinetic analysis of single-channel current traces
Channel open probability (Po), the fraction of time the pore is open, is simply obtained
as the sum of all open-time durations in the events list divided by the total recording time.
Although Po can be viewed as the physiologically relevant readout of gating in terms of chloride
transport, it provides essentially no information on mechanism. For instance, mutations might
simultaneously alter the rates of both gating transitions by up to 1000-fold with no large effect on
Po (e.g., E1371Q, (1)), emphasizing the importance of kinetic analysis for mechanistic
understanding.
WT CFTR channel gating is characterized by bursts of openings – groups of openings,
interrupted by brief closures – that are flanked by long, interburst closures. Correspondingly,
dwell-time analysis of single WT channels reveals a single open state and two distinct closed
states (69). The 3-state schemes C1-O-C2 and C1-C2-O can equally well account for such a pattern
of gating; assuming either scheme, the respective four rate constants can be estimated by
maximum likelihood fitting. Two commonly used approaches for maximum likelihood fitting of
single-channel events lists are fitting the open- and closed-time distributions (e.g., (74)), or fitting
the entire series of closed and open-time durations (75,76,77), implemented, e.g., by the MIL
subprogram of the QuB package, and by HJCFIT). In theory, the latter approach is more
powerful because it exploits the information carried by correlations between the durations of
adjacent closed and open events. However, for schemes with a single open state (or, in general,

Gadsby et al. – MiMM chapter – submitted May 20, 2010
14
when a single "gateway" state connects the sets of open and closed states), like the two schemes
above, open and closed events are uncorrelated and the two approaches therefore equivalent.
It is important to note that the schemes C1-O-C2 and C1-C2-O cannot be distinguished
in steady-state records. And, because of the short life time of state C2 (~10 ms at 25oC) relative to
C1 (~1 s at 25oC), a distinction based on the time course of current change upon sudden
addition/removal of the ligand ATP is technically not feasible. Thus, at present there is no strong
evidence to support a choice between them. Correspondingly, both schemes have been used to
interpret ATP-dependence of CFTR gating, and fitting either scheme suggests [ATP]-dependence
of a single rate: rate C1O (12) and rate C1C2, respectively (69). Because these rates are
meaningful only in the context of a particular scheme, it is customary to report only the mean
durations of open bursts and interburst closures, which are model-independent descriptive
parameters, that can be calculated from the fitted rate constants regardless of the chosen scheme.
Using this descriptive nomenclature it is generally agreed that increasing ATP concentrations
shorten the mean interburst duration in a saturable fashion, while the mean burst duration, the
mean intraburst closed time, and the mean number of intraburst closures per burst are little
influenced by ATP concentration (78,13,12,79).
An alternative approach for studying the kinetics of open bursts and interburst
closures is provided by conventional burst analysis. This method consists of ignoring brief
intraburst closures by creating a new events list of bursts and interbursts in which closures shorter
than a specified cutoff are suppressed. This cutoff is chosen based on the distribution of closed-
time durations in the original events list, and is ideally much larger than the mean duration of
intraburst closures, but much smaller than that of interburst closures. Nevertheless, because the
durations of both types of closures are exponentially distributed and therefore overlap, some
fraction of closures will be necessarily misassigned regardless of the choice of the cutoff. This
means that some fraction of intraburst closures will be erroneously kept and treated as interburst,
and some fraction of interburst closures will be erroneously misclassified as intraburst and so
eliminated. Two commonly used strategies for calculating the cutoff duration are to equalize the
probabilities of the two types of error (80) or to minimize the total probability of committing any
error (81). The merit of the first strategy is that the mean duration of reconstructed bursts
provides an undistorted estimate of the true mean burst duration, whereas the second strategy was

Gadsby et al. – MiMM chapter – submitted May 20, 2010
15
found to cause less distortion of the shape of the distribution of burst durations for a non-
equilibrium cyclic gating mechanism like that of CFTR (27).
Because ATP concentration affects bursting, but not intraburst, kinetics, restricting
kinetic analysis to a mere extraction of mean burst and interburst durations is equivalent to
modeling ATP dependent gating by a simple two-state process. Though more informative than
just extracting Po, such a simplifaction severely limits the amount of information that can be
obtained about a complex process involving binding of ATP at two distinct sites, dynamic
formation and disruption of an NBD1/NBD2 heterodimer, and hydrolysis of ATP in one of the
composite binding sites. However, by reconstructing each burst, burst analysis offers the
advantage of providing also the distributions of burst and interburst durations, not just their mean
values. These distributions carry invaluable information on the mechanism of bursts, and allow
quantitative evaluation of alternative gating schemes by fitting them to the observed distributions
using maximum likelihood. Although as a first approximation both interburst and burst durations
are reasonably fit by single-exponential distributions (18,13,12), careful fitting of large sets of
both interburst (11) and burst (27) durations have identified tell-tale deviations from single-
exponential behaviour that are indisputable signs of a highly non-equilibrium mechanism. Thus,
the clear rising phases of these distributions (see histogram of burst durations in Fig. 2B) signal
violation of microscopic reversibility (68). In addition, fitting these distributions by a cyclic
mechanism (Fig. 2A; cf., 1.1) allows estimation of the rates of conformational transitions that are
not associated with pore opening or closure (27).
3.4.3. Kinetic analysis of multi-channel current traces
Because CFTR gating is a slow process and gradual dephosphorylation by membrane-
bound phosphatases limits the length of steady-state single-channel recordings, obtaining
sufficient numbers of gating events from a patch containing only a single channel can become
challenging. In contrast, from patches with multiple (e.g., 2-8) active channels comparable
numbers of gating events are obtained in proportionately shorter duration recordings. An
ensemble of several (N) channels gating via a common mechanism but independently of each
other can be described by a single macro-system with N+1 conductance levels; transition rates
between states of the macro-scheme are functions of the single-channel transition rates.
Consequently, both maximum likelihood analysis approaches mentioned in 3.4.2. above, i.e.,

Gadsby et al. – MiMM chapter – submitted May 20, 2010
16
fitting of dwell-time distributions and fitting the entire time series, can be generalized to a multi-
channel system. However, because the macro-system has many states, the computational burden
for the latter approach becomes prohibitively large; even with improved present day (2010) PC
performances, fitting the entire time series for a record with more than 2-3 channels is still
impractical. In contrast, maximum likelihood fitting of single-channel open- and closed-time
distributions can be readily generalized to a simultaneous fitting of the ensemble of the dwell-
time distributions of all conductance levels of a multichannel patch. The computational burden of
this approach is far smaller, because once the sequence in which the events follow each other is
disregarded, the individual dwell-time distributions can be binned into histograms, resulting in a
large reduction in the size of the data set to be fitted. Moreover, there is no disadvantage to losing
correlation information between neighboring events for the simple C1-O-C2 and C1-C2-O
schemes (as noted above, 3.4.2.). Finally, a first-order correction for missed events due to limited
recording bandwidth is also readily incorporated into this approach (82). A program
implementing these procedures has been used successfully by several groups and is freely
available upon request ([email protected]).
3.5. Analysis of macroscopic current relaxations
Information on gating kinetics of single ion channels can also be obtained from the time
courses of macroscopic current relaxations in response to step changes in ligand concentration or
voltage. These time courses can be fitted with sums of exponentials, whose time constants and
fractional amplitudes are functions of the single-channel gating parameters. Non-linear least
squares fitting algorithms are included in most commercially available data acquisition software
packages (e.g., Pclamp, PulseFit).
Because, for CFTR, closing from a burst is little sensitive to ATP concentration and the
rate of opening to a burst in the absence of ATP is vanishingly small, the time constant of
macroscopic current relaxation upon sudden removal of ATP reflects the steady-state mean burst
duration (83,13,27). This technique for estimating mean burst duration has been preferentially
used for catalytic site mutants which abolish ATP hydrolysis at the composite NBD2 site, (e.g.,
K1250A) or when non-hydrolyzable ATP analogs (e.g., AMP-PNP, pyrophosphate) are applied,
because in either case burst durations are prolonged to several seconds or tens of seconds. While

Gadsby et al. – MiMM chapter – submitted May 20, 2010
17
this makes it difficult to collect sufficient numbers of steady state single-channel gating events,
macroscopic ATP-removal experiments are easy to perform, and require only moderately rapid
solution exchange (e.g., 13,12,84,85).
One limitation of this approach is that for patches excised from certain cell types removal
of PKA catalytic subunit results in a relatively abrupt shortening of mean burst durations, likely
due to rapid partial dephosphorylation of CFTR channels by membrane-bound phosphatases
(17,13). In such systems the above approach can be used to study the mean burst duration of
partially, but not of fully, phosphorylated channels. This is because sudden removal of ATP
terminates not only channel opening, but also activity of the co-applied PKA catalytic subunit,
and so full phosphorylation of the channels throughout the time course of current relaxation
cannot be guaranteed.
3.6. Thermodynamic approaches to studying the energetics of gating
3.6.1. Studying temperature dependence
Observing a chemical reaction at several different temperatures provides insight into
the energetics of the process. For an equilibrium reaction AB the standard enthalpy change
Ho between states B and A can be calculated from the van't Hoff plot, which displays the natural
logarithm of the equilibrium constant (ln Keq) as a function of the inverse of absolute temperature
(1/T). The slope of this linear plot is –Ho/(RT), where R is the gas constant (8.31 Jmol-1K-1). If
not only the equilibrium constant, but also the rates of transition between states A and B can be
measured, then temperature dependence of these rates can be used to calculate the activation
enthalpies (H‡), i.e., the standard enthalpy differences between the transition state and the stable
ground states A and B. With kAB denoting transition rate AB, plots of ln kAB (Arrhenius plot) or
ln (kAB/T) (Eyring plot) as a function of 1/T yield linear graphs with slopes –Ea/(RT) or –
H‡/(RT), respectively. Here, Ea denotes the activation energy and H‡ the enthalpy difference
between the transition state and ground state A; the two are related by the equation Ea=H‡+RT.
The corresponding parameters for the reverse step BA can be obtained analogously from
Arrhenius or Eyring plots of the reverse rate kBA (86).
Ion channels are especially well suited to such thermodynamic studies, because the
average rates of opening (ko) and closure (kc) can be readily measured in single-channel
recordings. If channel gating is an equilibrium process, then the open-closed equilibrium constant

Gadsby et al. – MiMM chapter – submitted May 20, 2010
18
is given by Keq=Po/(1–Po); because Po=ko/(ko+kc), an equivalent formulation is Keq= ko/kc. Thus,
measuring Po of such a single channel at various temperatures allows construction of a van't Hoff
plot (i.e., ln [Po/(1–Po)] versus 1/T) for estimation of the standard enthalpy change between the
open and closed ground states. Similarly, measuring ko and kc at various temperatures allows
estimation of the activation enthalpies for opening and closure by construction of Arrhenius or
Eyring plots.
However, in the case of WT CFTR there is strong evidence that the gating involves
irreversible steps and therefore follows a non-equilibrium cycle (15,87,11,27), such that channels
close from a burst predominantly through a pathway different from that by which they open (see
1.2.). This circumstance has important implications for the interpretion of temperature
dependence of WT CFTR bursting kinetics. First, if the process is not at equilibrium, then the
apparent equilibrium constant Kapp= Po/(1–Po) is not a true equilibrium constant. Thus,
construction of a van't Hoff plot is meaningless, as the slope of a plot of ln Kapp versus 1/T does
not report Hoopen-closed. (Note, that if Ho for the process closedopenclosed is non-zero, then
the quantity Hoopen-closed cannot even be defined in general; statements must be limited to
comparing an open state to either the preceding or the subsequent closed state (85).) It is also
important to note, that a linear van't Hoff plot does not imply an equilibrium mechanism, as has
been argued (88). Because the apparent equilibrium constant Kapp= Po/(1–Po) is always equal to
ko/kc, the relationship ln Kapp=ln ko–ln kc also holds regardless of the mechanism. Therefore, if the
Arrhenius (or Eyring) plots for opening and closure are linear – as observed for CFTR (88,89,85)
– then the plot of ln Kapp versus 1/T will also be linear, regardless of the mechanism. Similarly,
caution must be applied to the interpretation of energetic barriers. If WT CFTR indeed exits the
open burst state through a pathway which involves ATP hydrolysis, then H‡ for opening and
closure characterize single sides of two distinct energy barriers, rather than the two sides of a
single barrier. In this case the barrier height for the reversal of the opening step must be obtained
from the temperature dependence of closing rate under conditions that prevent ATP hydrolysis
(e.g., using NBD2 composite catalytic site mutations or non-hydrolyzable ATP analogs).
Together the three H‡ values for opening, non-hydrolytic closure and normal hydrolytic closure
then allow reconstruction of the Ho profile for a partial gating cycle which involves transition
from an ATP-bound closed state through a transition state to the open state, and then from the
open state to a second, distinct, transition state for closure (85).

Gadsby et al. – MiMM chapter – submitted May 20, 2010
19
By transition state theory (e.g., 86,90) the absolute value of a transition rate k,
measured at a given temperature, provides at least an upper estimate of the activation free energy
(G‡) in the form G‡≤RTln(kBT/(kh)), where kB is Boltzmann's constant (1.38.10-23 J/oK) and h
is Planck's constant (6.63.10-34 Js). This upper estimate of G‡ (G‡
max) together with the
estimate of H‡ yields a lower estimate of the activation entropy S‡: S‡min=(H‡–G‡
max)/T
(cf., (85)) which provides additional information about the mechanisms underlying CFTR
channel gating.
3.6.2. Mutant cycle analysis
The thermodynamic mutant cycle formalism (90) can be used to detect energetic
coupling between two amino acid positions in a protein. In a generalized double mutant cycle, the
WT, two single site mutants and the double mutant form the vertices of a thermodynamic cycle.
From patch recordings of CFTR channel currents, several kinetic parameters can be measured
that can be used to characterize WT and single and double mutants in terms of the G between
two states. If the two residues do not interact, the effects of mutating one site will not depend on
whether that mutation was done in a WT or mutant (at the other site) background; i.e., mutation-
linked changes in G on parallel sides of the cycle are equal (Gint = 0). Any difference (Gint
≠ 0) signifies, and (to some extent) quantifies, energetic coupling between the two residues.
Effective dissociation constants for ATP activation of channel gating, opening rates, and
equilibrium constant between closed and open states in a non-hydrolytic background [footnote:
after reducing the cyclic gating scheme to a closed-to-open equilibrium by impairing hydrolysis,
this equilibrium constant can be obtained from Po, as Keq= Po/(1–Po)], can all be used to estimate
a mutation-induced change in free energy, in each case probing a different step of the gating
cycle. Several energetic coupling values can therefore be determined for each pair of target sites,
allowing inferences to be made about how coupling between two positions changes as the NBDs
bind ATP, and the channel opens, and transits between open and closed states, i.e., as the channel
progresses through its gating cycle (1).
Unfortunately, interpretation of data obtained using the mutant cycle formalism is
rarely straightforward. A first complicating factor relates to the large statistical variability
observed in CFTR gating measurements. Because coupling energies are obtained as sums (of
mutation-linked changes in G on parallel sides of the cycle), the errors on the individual sets of

Gadsby et al. – MiMM chapter – submitted May 20, 2010
20
measurements are summed too, resulting in coupling energies, Gint, which are not significantly
different from zero unless the mutation effects are large. A second problem arises from the
oversimplifying assumptions required to reduce kinetic data to free energy differences between
two states. Thus, it is likely that the “equilibrium constants” estimated from measurements reflect
the steady state distribution of channels among more than two underlying states; and states with
low occupancy in WT might become important in certain mutants with severe phenotypes. A
third problem can arise from the quantitative interpretation of Gint as interaction energy in the
WT when the target-site residues still form significant interactions in the single and double
mutants (90). Such interactions can usually be avoided by using alanine substitutions. However,
mutant cycles in which the substitutions form a novel interaction in the double mutant only result
in a stronger deviation from simple additivity, and this might help reveal energetic coupling when
the single mutation effects are small.
3.6.3. REFER analysis
Rate-equilibrium free energy relationship (REFER) analysis provides information on
transition state structures and has been used to study the temporal sequence in which various
regions of an ion channel protein move during a closed-open conformational transition. REFER
theory uses transition state formalism, and was developed for equilibrium reactions. The method
assumes that structural perturbations introduced into a given region of an ion channel protein
affect the free energy of the transition state for opening in proportion to their effects on the free
energy of the open ground state (both free energies measured relative to the closed ground state);
i.e., Gotransition-closed=·Go
open-closed (0≤ ≤1). Because the logarithm of the opening rate (ko)
and of the closed-open equilibrium constant (Keq) are identically sensitive to Gotransition-closed and
Goopen-closed, respectively, the slope of a plot of ln ko as a function of ln Keq for a series of
structural perturbations to a particular region of a protein (Brønsted or REFER plot) provides the
proportionality coefficient . (Because Gotransition-closed=·Go
open-closed implies Gotransition-
open=(·Goopen-closed, the slope of the Brønsted plot for channel closure is 1.) Large
values (close to 1) indicate that in the transition state the perturbed region is already near its
open-state conformation, while a value close to 0 is an indication that the region is still closed-
like in the transition state. Therefore, high values are generally interpreted to signal early, and
low values late, movement of the target region during the closed-to-open transition (92).

Gadsby et al. – MiMM chapter – submitted May 20, 2010
21
The success of REFER analysis for ligand-gated ion channels (e.g., 93,94) has
encouraged its application to CFTR gating (95,96). Unfortunately, however, interpretation of
REFER plots depends strictly on the assumption of an underlying equilibrium process. For this
reason, although the observed linearity of the REFER plots and the complementarity of the slopes
of such plots for opening and closure of CFTR channels have been argued to support an
equilibrium mechanism (96), such inferences have no theoretical foundation. Indeed, it has been
shown that both equilibrium and non-equilibrium mechanisms can result in either linear or non-
linear REFER plots, and that the complementary REFER slopes for opening and closure are a
trivial feature of all ion channels regardless of their gating mechanism; moreover, for non-
equilibrium mechanisms REFER analysis provides no information on the transition-state
structures (97). These limitations should be considered before applying REFER analysis to
CFTR.
3.7. Bioinformatic approaches to identify coevolving amino-acid positions
3.7.1. Coevolution reports on functional residue-residue interactions
Information about gating of CFTR channels can also be gleaned from the evolutionary
record in the form of correlated amino acid substitutions. The conformational changes associated
with gating are controlled by residue interactions, and interacting residues exert evolutionary
pressure on each other, such that substitutions at one position are coupled to substitutions at
another. In terms of multiple sequence alignments, this means that the variation of residues found
in one column will correlate with the variation in some other column(s). This is illustrated
schematically in Fig. 3 for evolution of positions x and y in a hypothetical protein family. In Fig.
3A, because of strong interaction between positions x and y, their substitutions are correlated,
leading to their coevolution, whereas in Fig. 3B the positions do not interact strongly, and so they
evolve independently, and hence substitutions at those positions are uncorrelated (98,99, cf.,
100).
Because CFTR belongs to the ABC protein superfamily, the large quantity of
sequence data available makes such coevolution analysis attractive. Depending on which ABC
subfamily sequences are included in the alignment, the analysis may illuminate general
mechanisms shared by the whole ABC superfamily (e.g., interactions across the NBD dimer
interface; 1) or may relate specifically to a particular subfamily. An alignment restricted to OAD

Gadsby et al. – MiMM chapter – submitted May 20, 2010
22
family sequences, for instance, may be expected to yield information about mechanism
applicable not only to CFTR but also to paralogs such as the SUR and MRP proteins.
3.7.2. Statistical approaches for coevolution prediction
In practice, given the protracted timescale of evolution, inferences about past
substitution events made from alignments of contemporary sequences are used to generate a
statistic that relates to the probability that two positions coevolved. A number of methods have
been developed for these analyses, and they use one of two basic approaches (for recent reviews,
see 98,101). In one, the coevolution statistic is derived from correlated patterns in the alignment;
methods using this approach include McBASC (102), SCA (103), ELSC (104), OMES (105),
CAPS (106), and MIp (107). In the other, an assumed evolutionary mechanism is used to
reconstruct the history of amino-acid mutations, from which correlations in the substitution
process can be extracted; this approach is used by CoMap (108,109), BMM (110), CorrMut (111)
and methods based on explicit coevolution models (112,113). This latter approach adheres
closely to the above (3.7.1.) definition of coevolution as temporally correlated substitutions, but
is subject to the uncertainty inherent in inferring past substitution and branching events from
present sequences.
3.7.3. Input data and practical aspects
A multiple sequence alignment comprises the input data for coevolution prediction.
As misalignments increase with sequence divergence, filtering out distant sequences improves
alignment quality but at the expense of discarding useful information. Calculation of BLAST E-
values (114) or HMMER log-odds likelihood scores (115), combined with estimates of local
(116) or global (117) alignment quality, can help guide this tradeoff between sequence number
and divergence. Once sequences are aligned, gaps need to be removed by deleting entire columns
and/or sequences. Ideally, the resulting multiple sequence alignment will retain hundreds of
homologs each containing hundreds of positions.
3.7.4. Validation of methods with structural contact prediction
Benchmarking, to validate the predictive performance of a method on a test set of data
under particular conditions, would ideally require a protein family for which all coevolving, and
independently evolving, position pairs are known. As no such family exists, a data set can be
simulated, which offers precise control over phylogenetic tree parameters and rates of evolution,
and allows clear distinction between coevolving and independently evolving pairs. Applicability

Gadsby et al. – MiMM chapter – submitted May 20, 2010
23
of the assumptions about mechanisms of (co)evolution used in the model, however, remains
uncertain. An alternative approach is to use a known physical quantity, such as spatial proximity,
to characterize position pairs. The ability of a method to predict coevolution that arose from
structural contact can then be assessed for protein conformations for which a high-resolution 3D
structure exists. In the absence of an X-ray crystal structure for CFTR, homology models (e.g.,
51,118,119) based on the structure of Sav1866 (7,8) provide a reasonable alternative (119).
Coevolution analysis has been shown to predict structural contact between amino
acids in proteins significantly (120,109,121) better than random choice. Limits to the
performance of coevolution analysis assessed by contact prediction include incomplete overlap
between functional and structural interactions, in part due to functional interactions between
spatially distant positions (connected by chains of direct, structural, interactions) that nevertheless
induce coevolution (122). In general, correction for phylogenetic relatedness of sequences (123),
instead of assuming sequence independence, improves performance (124,125,126,110,107). In
addition, coevolution analysis methods show different sensitivities to the heterogeneity of
substitution rates across positions, i.e., conserved, slowly evolving positions versus rapidly
evolving, variable positions (127,128), and the accuracy of coevolution prediction may be
improved by taking this into account.
In the case of CFTR, comparison of the results obtained from different methods of
coevolution analysis, followed by structural contact tests, can exploit the strengths and overcome
the weaknesses of individual methods, and so yield useful functional information.

Gadsby et al. – MiMM chapter – submitted May 20, 2010
24
References
1. Vergani, P., Lockless, S.W., Nairn, A.C., and Gadsby, D.C. (2005) CFTR channel opening by
ATP-driven tight dimerization of its nucleotide-binding domains. Nature 433, 876-880.
2. Mense, M., Vergani, P., White, D.M., Altberg, G., Nairn, A.C., and Gadsby, D.C. (2006) In
vivo phosphorylation of CFTR promotes formation of a nucleotide-binding domain
heterodimer. EMBO J. 25, 4728-4740.
3. Hopfner, K.P., Karcher, A., Shin, D.S., Craig, L., Arthur, L.M., Carney, J.P., and Tainer, J.A.
(2000) Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA
double-strand break repair and the ABC-ATPase superfamily. Cell 101, 789-800.
4. Locher, K.P., Lee, A.T., and Rees, D.C. (2002) The E. coli BtuCD structure: a framework for
ABC transporter architecture and mechanism. Science 296, 1038-1040.
5. Smith, P.C., Karpowich, N., Millen, L., Moody, J.E., Rosen, J., Thomas, P.J., and Hunt, J.F.
(2002) ATP binding to the motor domain from an ABC transporter drives formation of a
nucleotide sandwich dimer. Mol. Cell 10, 139-149.
6. Chen, T.Y., Chen, M.F., and Lin, C.W. (2003) Electrostatic control and chloride regulation of
the fast gating of ClC-0 chloride. J. Gen. Physiol. 122, 641-651.
7. Dawson, R.J. and Locher, K.P. (2006) Structure of a bacterial multidrug ABC transporter.
Nature 443, 180-185.
8. Dawson, R.J. and Locher, K.P. (2007) Structure of the multidrug ABC transporter Sav1866
from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 581, 935-938.
9. Oldham, M.L., Khare, D., Quiocho, F.A., Davidson, A.L., and Chen, J. (2007) Crystal
structure of a catalytic intermediate of the maltose transporter. Nature 450, 515-521.
10. Dassa, E.B.P. (2001) The ABC of ABCS: a phylogenetic and functional classification of
ABC systems in living organisms. Research in Microbiology 152, 211-229.
11. Zeltwanger, S., Wang, F., Wang, G.T., Gillis, K.D., and Hwang, T.C. (1999) Gating of cystic
fibrosis transmembrane conductance regulator chloride channels by adenosine
triphosphate hydrolysis. Quantitative analysis of a cyclic gating scheme. J. Gen. Physiol.
113, 541-554.
12. Vergani, P., Nairn, A.C., and Gadsby, D.C. (2003) On the mechanism of MgATP-dependent
gating of CFTR Cl- channels. J. Gen. Physiol. 121, 17-36.

Gadsby et al. – MiMM chapter – submitted May 20, 2010
25
13. Csanády, L., Chan, K.W., Seto-Young, D., Kopsco, D.C., Nairn, A.C., and Gadsby, D.C.
(2000) Severed channels probe regulation of gating of cystic fibrosis transmembrane
conductance regulator by its cytoplasmic domains. J. Gen. Physiol. 116, 477-500.
14. Baukrowitz, T., Hwang, T.C., Nairn, A.C., and Gadsby, D.C. (1994). Coupling of CFTR Cl-
channel gating to an ATP hydrolysis cycle. Neuron 12, 473-482.
15. Gunderson, K.L. and Kopito, R.R. (1995) Conformational states of CFTR associated with
channel gating: the role ATP binding and hydrolysis. Cell 82, 231-239.
16. Carson, M.R., Winter, M.C., Travis, S.M., and Welsh, M.J. (1995b) Pyrophosphate stimulates
wild-type and mutant cystic fibrosis transmembrane conductance regulator Cl- channels.
J. Biol. Chem. 270, 20466-20472.
17. Hwang, T.C., Nagel, G., Nairn, A.C., and Gadsby, D.C. (1994) Regulation of the gating of
cystic fibrosis transmembrane conductance regulator C1 channels by phosphorylation and
ATP hydrolysis. Proc. Natl. Acad. Sci. U S A 91, 4698-4702.
18. Carson, M.R., Travis, S.M., and Welsh, M.J. (1995a) The two nucleotide-binding domains of
cystic fibrosis transmembrane conductance regulator (CFTR) have distinct functions in
controlling channel activity. J. Biol. Chem. 270, 1711-1717.
19. Aleksandrov, L., Aleksandrov, A.A., Chang, X.B. and Riordan, J.R. (2002) The First
Nucleotide Binding Domain of Cystic Fibrosis Transmembrane Conductance Regulator Is
a Site of Stable Nucleotide Interaction, whereas the Second Is a Site of Rapid Turnover. J.
Biol. Chem. 277, 15419-15425.
20. Basso, C., Vergani, P., Nairn, A.C., and Gadsby, D.C. (2003) Prolonged nonhydrolytic
interaction of nucleotide with CFTR’s NH2-terminal nucleotide binding domain and its
role in channel gating. J. Gen. Physiol. 122, 333-348.
21. Tsai, M.F., Shimizu, H., Sohma, Y., Li, M., and Hwang, T.C. (2009) State-dependent
modulation of CFTR gating by pyrophosphate. J. Gen. Physiol. 133, 405-419.
22. Ward, A., Reyes, C.L., Yu, J., Roth, C.B., and Chang, G. (2007) Flexibility in the ABC
transporter MsbA: Alternating access with a twist. Proc. Natl. Acad. Sci. U S A 104,
19005-19010.
23. Gadsby, D.C. (2009). Ion channels versus ion pumps: the principal difference, in principle.
Nat. Rev. Mol. Cell. Biol. 10, 344-352. PMCID: PMC2742554.

Gadsby et al. – MiMM chapter – submitted May 20, 2010
26
24. Jordan, I.K., Kota, K.C., Cui, G., Thompson, C.H., and McCarty, N.A. (2008) Evolutionary
and functional divergence between the cystic fibrosis transmembrane conductance
regulator and related ATP-binding cassette transporter. Proc. Natl. Acad. Sci. U S A 105,
18865-18870.
25. Chen, T.Y. and Hwang, T.C (2008) CLC-0 and CFTR: chloride channels evolved from
transporters. Physiol. Rev. 88, 351-387.
26. Muallem, D. and Vergani, P. (2009) ATP hydrolysis-driven gating in cystic fibrosis
transmembrane conductance regulator. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 364, 247-
255.
27. Csanády, L., Vergani, P., and Gadsby, D.C. (2010) Strict coupling between CFTR's catalytic
cycle and gating of its Cl- ion pore revealed by distributions of open channel burst
durations. Proc. Natl. Acad. Sci. U S A 107, 1241-1246.
28. Cheng, S.H., Rich, D.P., Marshall, J., Gregory, R.J., Welsh, M.J., and Smith, A.E. (1991)
Phosphorylation of the R domain by camp-dependent protein kinase regulates the CFTR
chloride channel. Cell 66, 1027-1036.
29. Picciotto, M.R., Cohn, J.A., Bertuzzi, G., Greengard, P., and Nairn, A.C. (1992)
Phosphorylation of the cystic fibrosis transmembrane conductance. J. Bio. Chem. 267,
12742-12752.
30. Jia, Y., Mathews, C.J., and Hanrahan, J.W. (1997) Phosphorylation by protein kinase C is
required for acute activation of cystic fibrosis transmembrane conductance regulator by
protein kinase A. J. Biol. Chem. 272, 4978-84.
31. Chappe, V., Hinkson, D.A., Zhu, T., Chang, X.B., Riordan, J.R., and Hanrahan, J.W. (2003)
Phosphorylation of protein kinase C sites in NBD1 and the R domain control CFTR
channel activation by PKA. J. Physiol. 548, 39-52.
32. Gadsby, D.C., Vergani, P., and Csanady, L. (2006) The ABC protein turned chloride channel
whose failure causes cystic fibrosis. Nature 440, 477-483.
33. Ostedgaard, L.S., Baldursson, O., Vermeer, D.W., Welsh, M.J., and Robertson, A.D. (2000)
A functional R domain from cystic fibrosis transmembreane conductance regulator is
predominantly unstructured in solution. Proc. Natl. Acad. Sci. U S A 97, 5657-5662.

Gadsby et al. – MiMM chapter – submitted May 20, 2010
27
34. Baker, J.M., Hudson, R.P., Kanelis, V., Choy, W.H., Thibodeau, P.H., Thomas, P.J., and
Forman-Kay, J.D. (2007) CFTR regulatory region interacts with NBD1 predominantly via
multiple transient helices. Nat. Struct. Mol. Bio. 14, 738-745.
35. Chang, X.B., Tabcharani, J.A., Hou, Y.X., Jenson, T.J., Kartner, N., Alon, N., Hanrahan,
J.W. and Riordan, J.R. (1993) Protein kinase A (PKA) still activates CFTR chloride
channel after mutagenesis of all 10 PKA consensus phosphorylation sites. J. Biol. Chem.
268, 11304-11311.
36. Csanady, L., Seto-Young, D., Chan, K.W., Cenciarelli, C., Angel, B.B, Qin, J., McLachlin,
D.T., Krutchinsky, A.N., Chait, B.T., Nairn, A.C., and Gadsby, D.C. (2005b) Preferential
phosphorylation of R-domain serine 768 dampens activation of CFTR channels by PKA.
J. Gen. Physiol. 125, 171-181.
37. Lewis, H.A., Buchanan, S.G., Burley, S.K., Conners, K., Dickey, M., Dorwart, M., Fowler,
R., Gao, X., Guggino, W.B., Hendrickson, W.A., Hunt, J.F., Kearins, M.C., Lorimer, D.,
Maloney, P.C., Post, K.W., Rajashankar, K.R., Rutter, M.E., Sauder, J.M., Shriver, S.,
Thiodeau, P.H., Thomas, P.J., Zhang, M., Zhao, X., and Emtage, S. (2004) Structure of
nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator.
EMBO J. 23, 282-293.
38. Kanelis, V., Hudson, R.P, Thibodeau, P.H., Thomas, P.J., and Forman-Kay, J.D. (2010)
NMR evidence for differential phosphorylation-dependent interactions in WT an
DeltaF508 CFTR. EMBO J. 29, 263-277. PMCID: PMC2808376.
39. Csanady, L., Chan, K.W., Nairn, A.C., and Gadsby, D.C. (2005a) Functional roles of non
conserved structural segments in CFTR’s NH2-terminal nucleotide binding domain. J.
Gen. Physiol. 125, 43-55.
40. Zerhusen, B. and Ma, J. (1999) Function of the second nucleotide-binding fold in the CFTR
chloride channel. FEBS Lett. 459, 177-185.
41. Chan, K.W., Csanády, L., Nairn, A.C., and Gadsby, D.C. (1999) Deletion analysis of CFTR
channel R domain using severed molecules. Biophys. J. 76, A405.
42. Wang, W., Wu, J., Bernard, K., Li, G., Wang, G., Bevensee, M.O., et al. (2010) ATP-
independent CFTR channel gating and allosteric modulation by phosphorylation. Proc.
Natl. Acad. Sci. U S A 107, 3888-3893.

Gadsby et al. – MiMM chapter – submitted May 20, 2010
28
43. Bompadre, S.G., Sohma, Y., Li, M., and Hwang, T.C. (2007) G551D and G1349D, two CF-
associated mutations in the signature sequences of CFTR, exhibit distinct gating defects.
J. Gen. Physiol. 129, 285-98.
44. King, J.D., Jr., Fitch, A.C., Lee, J.K., McCane, J.E., Mak, D.O., Foskett, J.K., and Hallows,
K.R. (2009) AMP-activated protein kinase phosphorylation of the R domain inhibits PKA
stimulation of CFTR. Am. J. Physiol. Cell Physiol. 297, C94-C101.
45. Kongsuphol, P., Cassidy, D., Hieke, B., Treharne, K.J., Schreiber, R., Mehta, A., and
Kunzelmann, K. (2009) Mechanistic insight into control of CFTR by AMPK. J. Biol.
Chem. 284, 5645-5653.
46. Wilkinson, D.J., Strong, T.V., Mansoura, M.K., Wood, D.L., Smith, S.S., Collins, F.S., and
Dawson, D.C. (1997) CFTR activation: additive effects of stimulatory and inhibitory
phosphorylation sites in the R domain. Am. J. Physiol. 273, L127-L133.
47. Zhang, L., Aleksandrov, L.A., Zhao, Z., Birtley, J.R., Riordan, J.R., and Ford, R.C. (2009)
Architecture of the cystic fibrosis transmembrane conductance regulator protein and
structural changes associated with phosphorylation and nucleotide binding. J. Struct. Biol.
167, 242-251.
48. Chan, K.W., Csanády, L., Seto-Young, D., Nairn, A.C. and Gadsby, D.C. (2000) Severed
molecules functionally define the boundaries of the cystic fibrosis transmembrane
conductance regulator's NH2-terminal nucleotide binding domain. J. Gen. Physiol.116,
163-180.
49. Loo, T.W. and Clarke, D.M (2001) Defining the drug-binding site in the human multidrug
resistance P-glycoprotein using a methanethiosulfonate analog of verapamil, MTS-
verapamil. J. Biol. Chem. 276, 14972-14979.
50. He, L., Aleksandrov, A.A., Serohijos, A.W., Hegedus, T., Aleksandrov, L.A., Cui, L.,
Dokholyan, N.V., and Riordan, J.R. (2008) Multiple membrane-cytoplasmic domain
contacts in the cystic fibrosis transmembrane conductance regulator (CFTR) mediate
regulation of channel gating. J. Biol. Chem. 283, 26383-26390.
51. Serohijos, A.W., Hegedus, T., Aleksandrov, A.A., He, L., Cui, L., Dokholyan, N.V., and
Riordan, J.R. (2008) Phenylalanine-508 mediates a cytoplasmic-membrane domain
contact in the CFTR 3D structure crucial to assembly and channel function. Proc. Natl.
Acad. Sci. U S A 105, 3256-3261.

Gadsby et al. – MiMM chapter – submitted May 20, 2010
29
52. Cotten, J.F. and Welsh, M.J. (1998) Covalent modification of the nucleotide binding domains
of cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 273, 31873-
31879.
53. Zhang, Z.-R., Song, B. and McCarty, N.A. (2005b) State-dependent Chemical Reactivity of
an Engineered Cysteine Reveals Conformational Changes in the Outer Vestibule of the
Cystic Fibrosis Transmembrane Conductance Regulator. J. Biol. Chem. 280, 41997-
42003.
54. Seibert, F.S., Chang, X.B., Aleksandrov, A.A., Clarke, D.M., Hanrahan, J.W., and Riordan,
J.R. (1999). Influence of phosphorylation by protein kinase A on CFTR at the cell surface
and endoplasmic reticulum. Biochim. Biophys. Acta. 1461, 275-283.
55. Ramjeesingh, M., Li, C., Garami, E., Huan, L.J., Hewryk, M., Wang, Y., Galley, K., and
Bear, C.E. (1997) A novel procedure for the efficient purification of the cystic fibrosis
transmembrane conductance regulator (CFTR). Biochem. J. 327, 17-21.
56. Aleksandrov, L., Mengos, A., Chang, X., Aleksandrov, A., and Riordan, J.R. (2001)
Differential interactions of nucleotides at the nucleotide binding domains of the cystic
fibrosis transmembrane conductance regulator. J. Biol. Chem. 276, 12918-12923.
57. Berger, H.A., Travis, S.M., and Welsh, M.J. (1993) Regulation of the cystic fibrosis
transmembrane conductance regulator Cl- channel by specific protein kinases and protein
phosphatases. J. Biol. Chem. 268, 2037-2047.
58. Travis, S.M., Berger, H.A., and Welsh, M.J. (1997) Protein phosphate 2C dephosphorylates
and inactivates cystic fibrosis transmembrane conductance regulator. Proc. Natl. Acad.
Sci. U S A 94, 11055-11060.
59. Luo, J., Pato, M.D., Riordan, J.R., and Hanrahan, J.W. (1998) Differential regulation of
single CFTR channels by PP2C, PP2A, and other phosphatases. Am. J. Physiol. 274,
C1397-C1410.
60. Chappe, V., Hinkson, D.A., Howell, L.D., Evagelidis, A., Liao, J., Chang, X.B., Riordan,
J.R., and Hanrahan, J.W. (2004) Stimulatory and inhibitory protein kinase C consensus
sequences regulate the cystic fibrosis transmembrane conductance regulator. Proc. Natl.
Acad. Sci. U S A 101, 390-395.

Gadsby et al. – MiMM chapter – submitted May 20, 2010
30
61. Wilkinson, D.J., Mansoura, M.K., Watson, P.Y., Smit, L.S., Collins, F.S., and Dawson, D.C.
(1996). CFTR: the nucleotide binding folds regulate the accessibility and stability of the
activated state. J. Gen. Physiol. 107, 103-119.
62. Sheppard, D. N., Gray, M.A., Gong, X., Sohma, Y., Kogan, I., Benos, D. J., et al. (2004) The
patch-clamp and planar lipid bilayer techniques: powerful and versatile tools to
investigate the CFTR Cl- channel. J. Cyst. Fibros. 3, 101-108.
63. Thomas, P. and Smart, T.G. (2005) HEK293 cell line: A vehicle for the expression of
recombinant proteins. J. Pharmacol. Toxicol. Methods 51, 187-200.
64. Bear, C., Li, C., Kartner, N., Bridges, R., Jensen, T., Ramjeesingh, M., et al. (1992)
Purification and functional reconstitution of the cystic fibrosis transmembrane
conductance regulator (CFTR). Cell 68, 809-818.
65. Sakmann, B. and Neher, E., Eds. (1995). Single-channel recording. New York, NY, Plenum
Press, 700.
66. Ashley, R. H., Ed. (1995) Ion Channels: a practical approach. Practical Approach Series.
Oxford, Oxford University Press, 328.
67. Benndorf, K. (1995). Low-noise recording. In Single-channel recording. Sakmann, B. and
Neher, E. Eds. New York , NY, Plenum Press, 129-145.
68. Colquhoun, D. and Sigworth, F.J. (1995) Fitting and statistical analysis of single-channel
records. In Single channel recording. Sakmann, B. and Neher, E., editors. Plenum Press,
New York.
69. Winter, M.C., Sheppard, D.N., Carson, M.R., and Welsh, M.J. (1994) Effect of ATP
concentration on CFTR Cl- channels: a kinetic analysis of channel regulation. Biophys. J.
66, 1398-1403.
70. Zhang, Z.R., Cui, G., Liu, X., Song, B., Dawson, D.C., and McCarty, N.A. (2005a)
Determination of the functional unit of the cystic fibrosis transmembrane conductance
regulator chloride channel. One polypeptide forms one pore. J. Biol. Chem. 280, 458-468.
71. Lansdell, K.A., Kidd, J.F., Delaney, S.J, Wainwright, B.J., and Sheppard, D.N. (1998)
Regulation of murine cystic fibrosis transmembrane conductance regulator Cl- channels
expressed in Chinese hamster ovary cells. J. Physiol. 512 (Pt 3), 751-764.
72. Venkataramanan, L. and Sigworth, F.J. (2002) Applying hidden Markov models to the
analysis of single ion channel activity. Biophys. J. 82, 1930-1942.

Gadsby et al. – MiMM chapter – submitted May 20, 2010
31
73. Qin, F. (2004) Restoration of single-channel currents using the segmental k-means method
based on hidden Markov modeling. Biophys. J. 86, 1488-1501.
74. Sigworth, F.J. and Sine, S.M. (1987) Data transformations for improved display and fitting of
single-channel dwell time histograms. Biophys. J. 52, 1047-1054.
75. Horn, R. and Lange, K. (1983) Estimating kinetic constants from single channel data.
Biophys. J. 43, 207-223.
76. Ball, F.G. and Sansom, M.S. (1989) Ion-channel gating mechanisms: model identification
and parameter estimation from single channel recordings. Proc. R. Soc. Lond. B. Biol. Sci.
236, 385-416.
77. Qin, F., Auerbach, A., and Sachs, F. (1996) Estimating single-channel kinetic parameters
from idealized patch-clamp data containing missed events. Biophys. J. 70, 264-280.
78. Winter, M.C. and Welsh, M.J. (1997) Stimulation of CFTR activity by its phosphorylated R
domain. Nature 389, 294-296.
79. Bompadre, S.G., Ai, T., Cho, J.H., Wang, X., Sohma, Y., Li, M., and Hwang, T.C. (2005a)
CFTR gating I: Characterization of the ATP-dependent gating of a phosphorylation-
independent CFTR channel (DeltaR-CFTR). J. Gen. Physiol. 125, 361-375.
80. Magleby, K.L. and Pallotta, B.S. (1983) Burst kinetics of single calcium-activated potassium
channels in cultured rat muscle. J. Physiol. 344, 605-623.
81. Jackson, M.B., Wong, B.S., Morris, C.E., Lecar, H., and Christian, C.N. (1983) Successive
openings of the same acetylcholine receptor channel are correlated in open time. Biophys.
J. 42, 109-114.
82. Csanády, L. (2000) Rapid kinetic analysis of multichannel records by a simultaneous fit to all
dwell-time histograms. Biophys. J. 78, 785-799.
83. Weinreich, F., Riordan, J.R., and Nagel, G. (1999) Dual effects of ADP and
adenylylimidodiphosphate on CFTR channel kinetics show binding to two different
nucleotide binding sites. J. Gen. Physiol. 114, 55-70.
84. Bompadre, S.G., Cho, J.H., Wang, X., Zou, X., Sohma, Y., Li, M., and Hwang, T.C., (2005b)
CFTR gating II: Effects of nucleotide binding on the stability of open states. J. Gen.
Physiol. 125, 377-394.

Gadsby et al. – MiMM chapter – submitted May 20, 2010
32
85. Csanády, L., Nairn, A.C., and Gadsby, D.C. (2006) Thermodynamics of CFTR channel
gating: a spr-eading conformational change initiates an irreversible gating cycle. J. Gen.
Physiol. 128, 523-533.
86. Segel, I.H. (1993) Enzyme Kinetics. Behavior and Analysis of Rapid Equilibrium and
Steady-State Enzyme Systems. John Wiley & Sons Inc., New York.
87. Ishihara, H. and Welsh, M.J. (1997) Block by MOPS reveals a conformation change in the
CFTR pore produced by ATP hydrolysis. Am. J. Physiol. 273, C1278-C1289.
88. Aleksandrov, A.A. and Riordan, J.R. (1998) Regulation of CFTR ion channel gating by
MgATP. FEBS Lett. 431, 97-101.
89. Mathews, C.J., Tabcharani, J.A., and Hanrahan, J.W. (1998) The CFTR chloride channel:
nucleotide interactions and temperature-dependent gating. J. Membr. Biol. 163, 55-66.
90. Fersht, A. (2002) Structure and Mechanism in protein science. 4 ed. W.H. Freeman and
Company, New York.
91. Faiman, G. A. and Horovitz, A. (1996) On the choice of reference mutant states in the
application of the double-mutant cycle method. Protein Eng. 9, 315-316.
92. Auerbach, A. (2007) How to turn the reaction coordinate into time. J. Gen. Physiol. 130, 543-
546.
93. Chakrapani, S., Bailey, T.D., and Auerbach, A. (2004) Gating dynamics of the acetylcholine
receptor extracellular domain. J. Gen. Physiol. 123, 341-356.
94. Purohit, P., Mitra, A., and Auerbach, A. (2007) A stepwise mechanism for acetylcholine
receptor channel gating. Nature 446, 930-933.
95. Scott-Ward, T.S., Cai, Z., Dawson, E.S., Doherty, A., Da Paula, A.C., Davidson, H.,
Porteous, D.J., Wainwright, B.J., Amaral, M.D., Sheppard, D.N., and Boyd, A.C. (2007)
Chimeric constructs endow the human CFTR Cl- channel with the gating behavior of
murine CFTR. Proc. Natl. Acad. Sci. U S A 104, 16365-16370.
96. Aleksandrov, A.A., Cui, L., and Riordan, J.R. (2009) Relationship between nucleotide
binding and ion channel gating in cystic fibrosis transmembrane conductance regulator. J.
Physiol. 587, 2875-2886.
97. Csanády, L. (2009) Application of rate-equilibrium free energy relationship analysis to
nonequilibrium ion channel gating mechanisms. J. Gen. Physiol. 134, 129-136.

Gadsby et al. – MiMM chapter – submitted May 20, 2010
33
98. Galtier, N., and Dutheil, J. (2007) Coevolution within and between genes. Genome Dyn. 3, 1-
12.
99. Fitch, W.M. and Markowitz, E. (1970) An improved method for determining codon
variability in a gene and its application to the rate of fixation of mutations in evolution.
Biochem. Gene.t 4, 579-593.
100. Gutell, R.R., Larsen, N., and Woese, C.R. (1994) Lessons from an evolving rRNA: 16S
and 23S rRNA structures from a comparative perspectice. Micro. Biol. 58, 10-26.
101. Codoñer, F.M., O'Dea, S., and Fares, M.A. (2008) Reducing the false positive rate in the
non-parametric analysis of molecular coevolution. BMC Evol. Biol. 8, 106.
102. Olmea, O., and Valencia, A. (1997) Improving contact predictions by the combination of
correlated mutations and other sources of sequence information. Fold Des. 2, S25-S32.
103. Lockless, S.W., and Ranganathan, R. (1999) Evolutionarily conserved pathways of
energetic connectivity in protein families. Science 286, 295-299.
104. Dekker, J.P., Fodor, A., Aldrich, R.W., and Yellen, G. (2004) A perturbation-based
method for calculating explicit likelihood of evolutionary co-variance in multiple
sequence alignments. Bioinformatics 20, 1565-1572.
105. Kass, I., and Horovitz, A. (2002) Mapping pathways of allosteric communication in
GroEL by analysis of correlated mutations. Proteins 48, 611-617.
106. Fares, M.A., and Travers, S.A.A. (2006) A novel method for detecting intramolecular
coevolution: adding a further dimension to selective constraints analyses. Genetics 173, 9-
23.
107. Dunn, S.D., Wahl, L.M., and Gloor, G.B. (2008) Mutual information without the
influence of phylogeny or entropy dramatically improves residue contact prediction.
Bioinformatics 24, 333-340.
108. Dutheil, J., Pupko, T., Jean-Marie, A., and Galtier, N. (2005) A model-based approach for
detecting coevolving positions in a molecule. Mol. Biol. Evol. 22, 1919-1928.
109. Dutheil, J., and Galtier, N. (2007) Detecting groups of coevolving positions in a molecule:
a clustering approach. BMC Evol. Biol. 7, 242.
110. Dimmic, M.W., Hubisz, M.J., Bustamante, C.D., and Nielsen, R. (2005) Detecting
coevolving amino acid sites using Bayesian mutational mapping. Bioinformatics 21,
Suppl 1:i126-135.

Gadsby et al. – MiMM chapter – submitted May 20, 2010
34
111. Fleishman, S.J., Yifrach, O., and Ben-Tal, N.. (2004) An evolutionarily conserved
network of amino acids mediates gating in voltage-dependent potassium channels. J. Mol.
Biol. 340, 307-318.
112. Pollock, D.D., Taylor, W.R., and Goldman, N. (1999) Coevolving protein residues:
maximum likelihood identification and relationship to structure. J. Mol. Biol. 287, 187-
198.
113. Yeang, C.-H., and Haussler, D. (2007) Detecting coevolution in and among protein
domains. PLoS Comput. Biol. 3, e211.
114. Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. (1990) Basic local
alignment search tool. J. Mol. Biol. 215, 403-410.
115. Eddy, S.R. (2009) A new generation of homology search tools based on probabilistic
inference. Genome Inform. 23, 205-211.
116. Notredame, C., and Abergel, C. (2003) Using multiple alignment methods to assess the
quality of genomic data analysis. Bioinformatics and Genomes: Current Perspectives.
Horizon Scientific Press, Wymondham, UK. 30--55.
117. Lassmann, T., and Sonnhammer, E.L.L. (2005) Automatic assessment of alignment
quality. Nucleic Acids Res. 33, 7120-7128. doi:10.1093/nar/gki1020.
118. Mornon, J.P., Lehn, P., and Callebaut, I. (2009) Molecular models of the open and closed
states of the whole human CFTR protein. Cell. Mol. Life Sci. 66, 3469–3486.
119. Alexander, C., Ivetac, A., Liu, X., Norimatsu, Y., Serrano, J.R., Landstrom, A., Sansom,
M., and Dawson, D.C. (2009) Cystic fibrosis transmembrane conductance regulator:
using differential reactivity toward channel-permeant and channel-impermeant thiol-
reacgtive probes to test a molecular model for the pore. Biochemistry 48, 10078-10088.
120. Fodor, A.A., and Aldrich, R.W. (2004b) On evolutionary conservation of thermodynamic
coupling in proteins. J. Biol. Chem. 279, 19046-19050.
121. Fuchs, A., Martin-Galiano, A.J., Kalman, M., Fleishman, S., Ben-Tal, N., and Frishman,
D. (2007) Co-evolving residues in membrane proteins. Bioinformatics 23, 3312-3319.
122. Burger, L. and van Nimwegen, E. (2010) Disentangling direct from indirect co-evolution
of residues in protein alignments. PLoS Comput. Biol. 6, e1000633.
123. Felsenstein, J. (1985) Phylogenies and the comparative method. American Naturalist 125,
1.

Gadsby et al. – MiMM chapter – submitted May 20, 2010
35
124. Wollenberg, K.R., and Atchley, W.R. (2000) Separation of phylogenetic and functional
associations in biological sequences by using the parametric bootstrap. Proc. Nat.l Acad.
Sci. U S A 97, 3288-3291.
125. Tillier, E.R.M., and Lui, T.W.H. (2003) Using multiple interdependency to separate
functional from phylogenetic correlations in protein alignments. Bioinformatics 19, 750-
755.
126. Noivirt, O., Eisenstein, M., and Horovitz, A. (2005) Detection and reduction of
evolutionary noise in correlated mutation analysis. Protein Eng. Des. Sel. 18, 247-253.
127. Fodor, A.A., and Aldrich, R.W. (2004a) Influence of conservation on calculations of
amino acid covariance in multiple sequence alignments. Proteins 56, 211-221.
128. Martin, L.C., Gloor, G.B., Dunn, S.D., and Wahl, L.M. (2005) Using information theory
to search for co-evolving residues in proteins. Bioinformatics 21, 4116-4124.
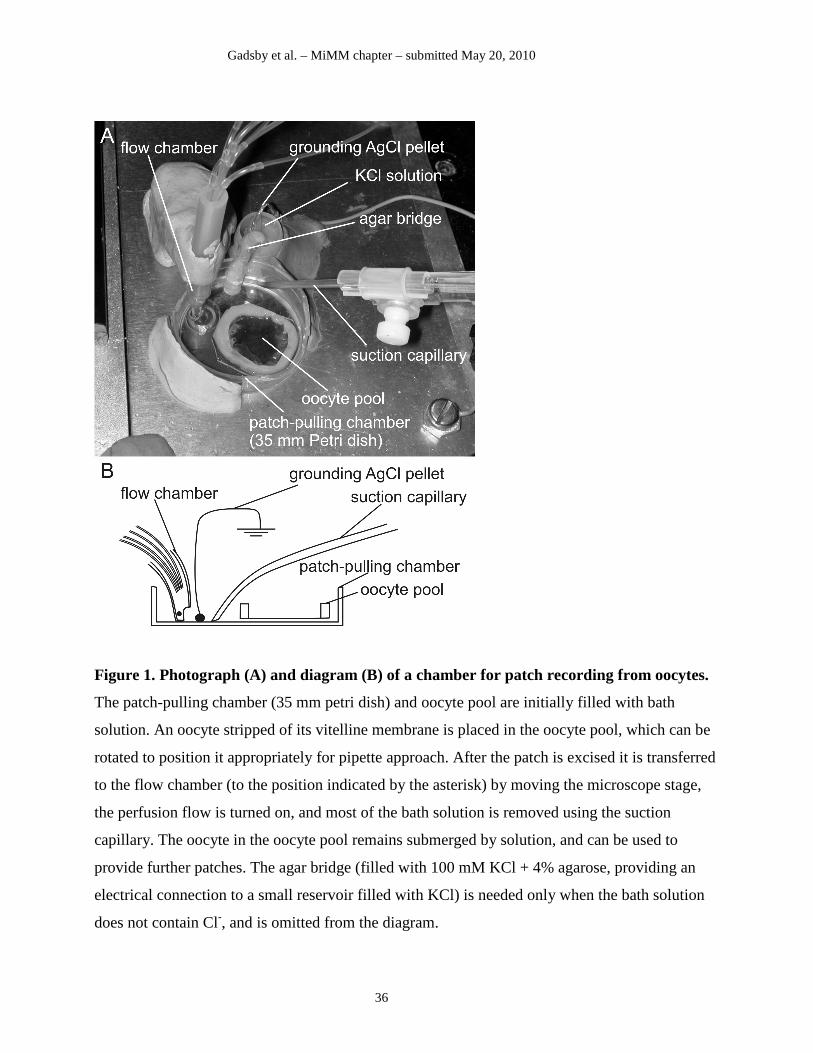
Gadsby et al. – MiMM chapter – submitted May 20, 2010
36
Figure 1. Photograph (A) and diagram (B) of a chamber for patch recording from oocytes.
The patch-pulling chamber (35 mm petri dish) and oocyte pool are initially filled with bath
solution. An oocyte stripped of its vitelline membrane is placed in the oocyte pool, which can be
rotated to position it appropriately for pipette approach. After the patch is excised it is transferred
to the flow chamber (to the position indicated by the asterisk) by moving the microscope stage,
the perfusion flow is turned on, and most of the bath solution is removed using the suction
capillary. The oocyte in the oocyte pool remains submerged by solution, and can be used to
provide further patches. The agar bridge (filled with 100 mM KCl + 4% agarose, providing an
electrical connection to a small reservoir filled with KCl) is needed only when the bath solution
does not contain Cl-, and is omitted from the diagram.
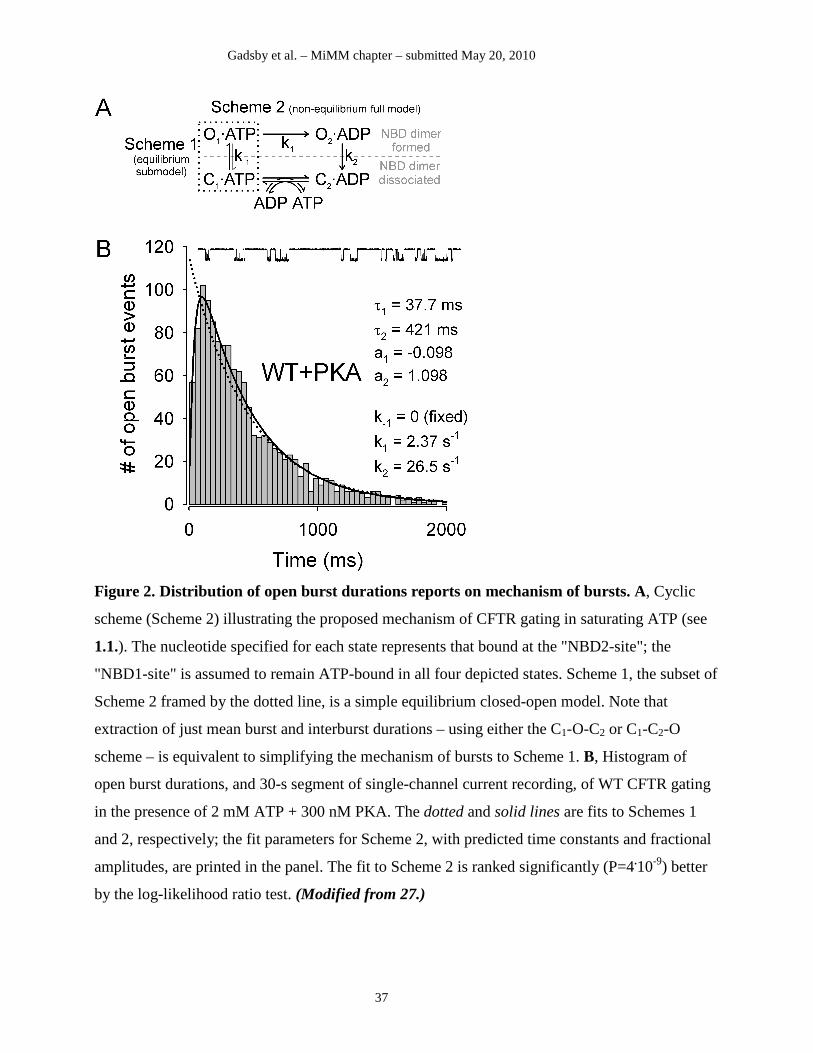
Gadsby et al. – MiMM chapter – submitted May 20, 2010
37
Figure 2. Distribution of open burst durations reports on mechanism of bursts. A, Cyclic
scheme (Scheme 2) illustrating the proposed mechanism of CFTR gating in saturating ATP (see
1.1.). The nucleotide specified for each state represents that bound at the "NBD2-site"; the
"NBD1-site" is assumed to remain ATP-bound in all four depicted states. Scheme 1, the subset of
Scheme 2 framed by the dotted line, is a simple equilibrium closed-open model. Note that
extraction of just mean burst and interburst durations – using either the C1-O-C2 or C1-C2-O
scheme – is equivalent to simplifying the mechanism of bursts to Scheme 1. B, Histogram of
open burst durations, and 30-s segment of single-channel current recording, of WT CFTR gating
in the presence of 2 mM ATP + 300 nM PKA. The dotted and solid lines are fits to Schemes 1
and 2, respectively; the fit parameters for Scheme 2, with predicted time constants and fractional
amplitudes, are printed in the panel. The fit to Scheme 2 is ranked significantly (P=4.10-9) better
by the log-likelihood ratio test. (Modified from 27.)
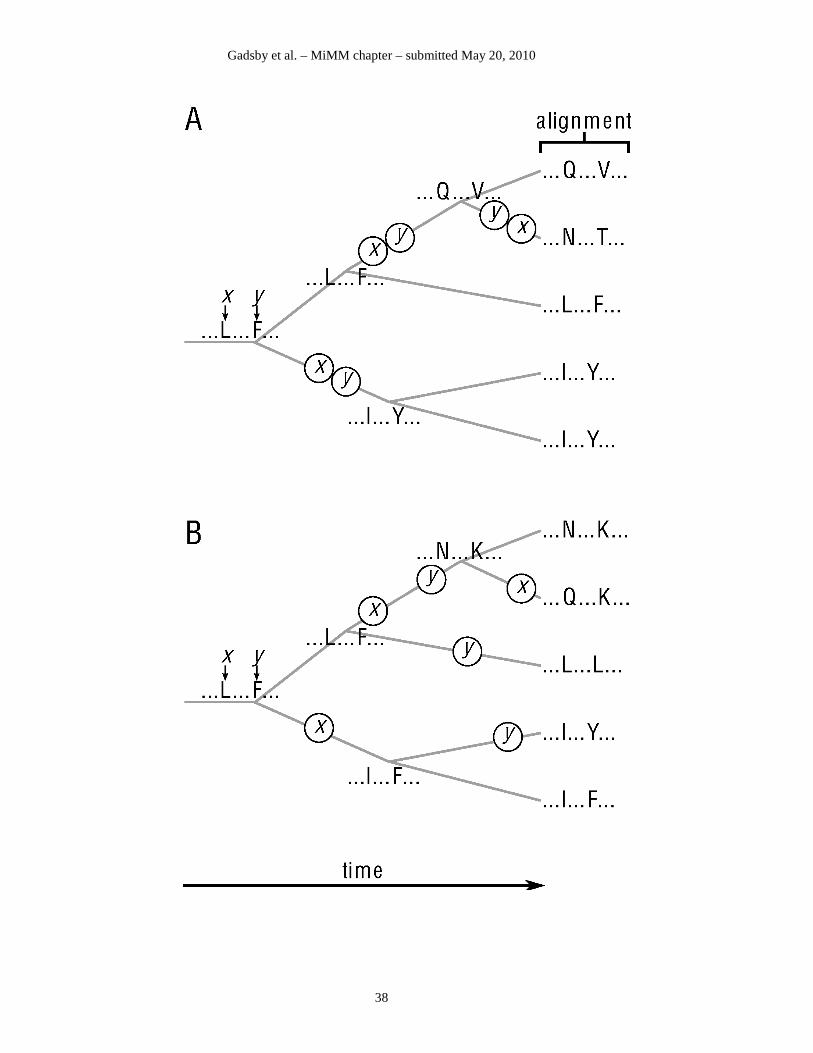
Gadsby et al. – MiMM chapter – submitted May 20, 2010
38

Gadsby et al. – MiMM chapter – submitted May 20, 2010
39
Figure 3. Signals of coevolution report on functional residue-residue interactions. A, Strong
interaction between positions x and y induces coevolution. B, Negligibly weak interaction lets
positions x and y evolve independently. In both cases an ancestral sequence (left) evolves along
the same phylogenetic tree, whose bifurcations represent gene duplication or speciation events.
Only two sequence positions, x and y, are considered, the rest are indicated by dots. Encircled x
and y characters mark substitution events at that position. As a result of substitutions, a sequence
at the right (more recent) node of a branch may differ from the left (earlier) node at one or both
positions. The process results in the set of contemporary sequences on the right, organized into a
multiple alignment. Though substitutions occur stochastically in both cases, only in case A do
they correlate temporally between the two positions. Since the protracted timescale of evolution
precludes direct observation of substitution events, the challenge is to distinguish between the
two cases A and B indirectly, using the alignment as the only available input data.





























