Embed Size (px)
Citation preview

Akademisk avhandling som med tillstånd av Kungliga Tekniska Högskolan i Stockholm
framlägges till offentlig granskning för avläggande av doktorsexamen i kemi fredagen
den 2 oktober kl 13.00 i sal F3, KTH, Lindstedtsvägen 26, Stockholm. Avhandlingen
försvaras på engelska. Fakultetsopponent är Professor Frank Glorius, Westfälische
Wilhelms-Universität Münster.
Enantioenriched Cyanohydrins and
Acetoxyphosphonates – Synthesis and
Applications
Robin Hertzberg
Doctoral Thesis
Stockholm 2015

ISBN 978-91-7595-627-5
ISSN 1654-1081
TRITA-CHE-Report 2015:31
© Robin Hertzberg, 2015
Universitetsservice US AB, Stockholm

Robin Hertzberg, 2015: ”Enantioenriched Cyanohydrins and
Acetoxyphosphonates – Synthesis and Applications”, KTH Chemical Science
and Engineering, Royal Institute of Technology, SE-100 44 Stockholm,
Sweden.
Abstract
In this thesis, the synthesis of enantioenriched compounds using novel
methodologies that employ metal- and biocatalysis is described.
In the first part, the synthesis of enantioenriched cyanohydrins, which are
highly versatile synthetic intermediates, is described. A minor enantiomer
recycling methodology, which uses a catalytic system consisting of a titanium
salen dimer and a lipase, was highly successful in yielding the desired
products, often in essentially enantiopure form. Alternatively, when the minor
enantiomer recycling method gave unsatisfactory results, the same two
catalysts were used in a sequential two-step process. The minor enantiomer
recycling procedure was used to synthesize three different β-adrenergic
antagonists with very high enantiomeric excesses via the corresponding O-
acetylated cyanohydrins. With the same cyclic process, O-(α-bromoacyl)
cyanohydrins were synthesized and subsequently transformed to
aminofuranones via an intramolecular Blaise reaction. In addition, substitution
of the bromide in the O-(α-bromoacyl) cyanohydrins with different nitrogen
nucleophiles followed by reduction gave N-substituted β-amino alcohols. This
reaction sequence was applied to the synthesis of the β3-adrenergic receptor
agonist solabegron. Finally, the O-(α-bromoacyl) cyanohydrins were subjected
to a palladium catalyzed cross-coupling with a range of boronic acids. This
reaction proceeded with high yields, and was performed with enantiopure
substrates with no or only minor racemization of the resulting products.
In the second part, the first asymmetric direct addition of acylphosphonates to
aldehydes is described. This transformation is catalyzed by a tridentate Schiff
base aluminum(III) Lewis acidic complex, a Lewis base, and a Brønstedt base.
Several aromatic and aliphatic acetoxyphosphonates were isolated, in most
cases in high yields. Unfortunately, the enantioselectivity in the reaction was
only moderate. Therefore, an investigation to develop a minor enantiomer
recycling system for the synthesis of acetoxyphosphonates was initiated, but a
working cyclic process could not be found in this work.
Keywords: asymmetric synthesis, cyanohydrins, acetoxyphosphonates,
biocatalyst, titanium catalyst, minor enantiomer recycling, cross-coupling,
amino alcohols


Abbreviations
Ac Acetyl
ATHase Artificial transfer hydrogenase
aq. Aqueous
BINOL 1,1'-Bi-2-naphthol
CALA Candida antarctica lipase A
CALB Candida antarctica lipase B
CRL Candida rugosa lipase
dba Dibenzylideneacetone
ee Enantiomeric excess
GC Gas chromatography
GC-MS Gas chromatography–mass spectrometry
DABCO 1,4-Diazabicyclo[2.2.2]octane
DBU 1,8-Diazabicyclo[5.4.0]undec-7-ene
DCM Dichloromethane
DIPEA N,N-Diisopropylethylamine
DMAP 4-(Dimethylamino)pyridine
DKR Dynamic kinetic resolution
DPEPhos (Oxydi-2,1-phenylene)bis(diphenylphosphine)
DYKAT Dynamic kinetic asymmetric transformation
equiv Equivalents
GABA γ-Aminobutyric acid
h Hours
HIV Human immunodeficiency virus
HNL Hydroxynitrile Lyase
HOMO Highest occupied molecular orbital
HPLC High performance liquid chromatography
IBS Irritable bowel syndrome
KR Kinetic resolution
LUMO Lowest unoccupied molecular orbital
MAO-N Monoamine oxidase
MER Minor enantiomer recycling
MTBE tert-Butyl methyl ether
NHC N-Heterocyclic carbene
NMR Nuclear magnetic resonance
Nu Nucleophile
OAB Overactive bladder
PPL Lipase from porcine pancreas
PSL Pseudomonas cepacia lipase
r.t. Room temperature
temp Temperature
THF Tetrahydrofuran
Tf Trifluoromethanesulfonyl
TMS Trimethylsilyl
Ts p-Toluenesulfonyl
WHO World health organization

List of Publications
This thesis is based on the following papers, referred to in the text by Roman
numerals I-VI:
I. Enantioenriched ω-Bromocyanohydrin Derivatives. Improved
Selectivity by Combination of Two Chiral Catalysts Robin Hertzberg, Khalid Widyan, Berenice Heid, and Christina Moberg
Tetrahedron 2012, 68, 7680–7684
II. One-Step Preparation of O-(α-Bromoacyl) Cyanohydrins by Minor
Enantiomer Recycling: Synthesis of 4-Amino-2(5H)-furanones Robin Hertzberg, and Christina Moberg
J. Org. Chem. 2013, 78, 9174−9180
III. Minor Enantiomer Recycling: Application to Enantioselective
Syntheses of Beta Blockers Ye-Qian Wen, Robin Hertzberg, Inanllely Gonzalez, and Christina
Moberg
Chem. - Eur. J. 2014, 20, 3806–3812
IV. Enantioselective Acylphosphonylation - Dual Lewis Acid-Lewis
Base Activation of Aldehyde and Acylphosphonate Ye-Qian Wen
†, Robin Hertzberg
†, and Christina Moberg
J. Org. Chem. 2014, 79, 6172−6178 †
Authors contributed equally
V. Synthesis of the β3-Adrenergic Receptor Agonist Solabegron and
Analogous N-(2-Ethylamino)-β-amino Alcohols from O-Acylated
Cyanohydrins − Expanding the Scope of Minor Enantiomer
Recycling Robin Hertzberg, Guillermo Monreal Santiago, and Christina Moberg
J. Org. Chem. 2015, 80, 2937−2941
VI. Palladium-Catalyzed C(sp3)-C(sp
2) Cross-Couplings Between O-(α-
Bromoacyl) Cyanohydrins and Boronic Acids Robin Hertzberg and Christina Moberg
Manuscript

Paper not included in this thesis:
VII. Dual Lewis Acid-Lewis Base Catalyzed Acylcyanation of Aldehydes.
A Mechanistic Study Anna Laurell Nash, Robin Hertzberg, Ye-Qian Wen, Björn Dahlgren,
Tore Brinck, and Christina Moberg
Manuscript

Table of Contents
Abstract
Abbreviations
List of Publications
1. Introduction ..................................................................................... 1 1.1. Strategies for Obtaining Enantiopure Compounds ................................ 1
1.1.1. Kinetic Resolution ..................................................................................... 2 1.1.2. Sequential Processes ............................................................................... 4 1.1.3. Dynamic Kinetic Resolution ...................................................................... 4 1.1.4. Minor Enantiomer Recycling ..................................................................... 6
1.2. Aim of this Thesis .................................................................................. 9 2. Synthesis and Applications of O-Acylated Cyanohydrins. ............ 11
2.1. Introduction .......................................................................................... 11 2.1.1. The Importance of Cyanohydrins .............................................................11 2.1.2. Strategies for the Preparation of Enantioenriched Cyanohydrins ..............13
2.1.2.1. Biocatalytic Methods for the Preparation of Cyanohydrins ................... 13 2.1.2.2. Cooperative Catalytic Systems for the Preparation of Cyanohydrins ... 15 2.1.2.3. Minor Enantiomer Recycling for the Preparation of Cyanohydrins ....... 20
2.2. Synthesis of Enantioenriched ω-Bromocyanohydrin Derivatives ......... 22 2.2.1. Introduction ..............................................................................................22 2.2.2. Minor Enantiomer Recycling ....................................................................22 2.2.3. Sequential Process ..................................................................................26 2.2.4. Conclusions .............................................................................................27
2.3. Application of Minor Enantiomer Recycling in the Synthesis of β-
Adrenergic Antagonists ................................................................................... 27 2.3.1. Introduction ..............................................................................................27 2.3.2. Synthesis of (S)-Propranolol ....................................................................28 2.3.3. Synthesis of (R)-Dichloroisoproterenol .....................................................31 2.3.4. Synthesis of (R)-Pronethalol ....................................................................31 2.3.5. Conclusions .............................................................................................32
2.4. Synthesis and Applications of O-(α-Bromoacyl) Cyanohydrins ............ 32 2.4.1. Introduction ..............................................................................................32 2.4.2. Synthesis of O-(α-Bromoacyl) Cyanohydrins and Subsequent
Transformation to Aminofuranones ............................................................................33 2.4.2.1. Minor Enantiomer Recycling ............................................................... 35 2.4.2.2. Cyclizations to Aminofuranones .......................................................... 39
2.4.3. Functionalization of O-(α-Bromoacyl) Cyanohydrins ................................40 2.4.3.1. Synthesis of Solabegron ..................................................................... 40 2.4.3.2. C(sp
3)-C(sp
2) Cross-Coupling Between O-(α-Bromoacyl)
Cyanohydrins and Boronic Acids........................................................................... 45 2.4.4. Conclusions .............................................................................................52

3. Enantioselective Acylphosphonylation.......................................... 53 3.1. Introduction .......................................................................................... 53 3.2. Dual Activation in the Synthesis of Acetoxyphosphonates .................. 54 3.3. Minor Enantiomer Recycling ................................................................ 62 3.4. Conclusions ......................................................................................... 64
4. Concluding Remarks .................................................................... 65
Acknowledgements
Appendix
References


1
1. Introduction
Chiral molecules are essential for life on earth. Most of these molecules consist
of single enantiomers. The amino acids that build up all natural enzymes,
which catalyze chemical reactions taking place in the human body, consist
solely of L-amino acids. Organic compounds which consist of one single
stereoisomer have therefore an increasingly important role in the
pharmaceutical and agricultural industries. A large part of the pharmaceutical
drugs sold today contain one or more stereocenters, but several of these are
sold as mixtures of stereoisomers. Since the drug targets most often are
enzymes, the effect of different stereoisomers of the drug can differ to a large
extent.1 Stereoisomers of one drug might not only have different activity, but
can also have different modes of action; one stereoisomer might even be toxic
while the other gives the desired effect.2,3
The β-adrenergic antagonist propranolol (1) is an example of a chiral drug in
which the stereochemistry is vital for its biological activity (Figure 1).
Propranolol is used to treat numerous disorders and is listed in the WHO
model list of essential medicines.4 Despite the fact that the (S)-enantiomer of
this compound is over 100 times more active than the (R)-enantiomer,
propranolol is sold as a racemate, i.e. an equal mixture of both enantiomers.3,5
Another example is the pyrethroid insecticide esfenvalerate (2), which is the
(S,S)-isomer of fenvalerate and is responsible for the majority of the
insecticidal activity (Figure 1).6
Figure 1. (S)-Propranolol and esfenvalerate
1.1. Strategies for Obtaining Enantiopure Compounds
The transformation of prochiral starting material to enantioenriched product
with the aid of a chiral reagent can result in high enantiomeric excess (ee)
(Figure 2), but if the selectivity is not sufficient, separation of the unwanted
stereoisomer is necessary. The separation can be accomplished by
chromatographic techniques or by crystallization. The obvious disadvantage
with these separations is that the minor stereoisomer is taken out of the system,
thus unavoidably resulting in a reduced total yield.

2
Figure 2. Transformation of a prochiral compound (A) to enantioenriched product
(PR)
Systems that employ more than one catalyst are in many cases favorable over
single catalytic systems; fewer synthetic steps might be necessary, which
results in lower amounts of produced waste.7,8
In addition, cooperative
catalytic systems can result in higher enantiopurity of the desired products
since a reinforcing effect is obtained when two enantioselective catalysts work
in concert.9
1.1.1. Kinetic Resolution
In a kinetic resolution (KR) (Figure 3), two enantiomers of a substrate (SR and
SS) undergo a transformation with different rates under the influence of a chiral
catalyst. In the ideal case the selectivity is very high for one of the starting
enantiomers and very low for the other enantiomer (kR >> kS), such that at 50%
conversion only one of the enantiomers has been transformed to product (PR).10
Figure 3. Kinetic resolution (KR)
The stereoselectivity factor s is defined as the ratio between the rate constants
for the formation of the enantiomeric products (kR/kS). The stereoselectivity
factor can be calculated from the formulas below by using either the ee of the
substrate or the ee of the product. The stereoselectivity factor s is used for
chemocatalyzed reactions while the biochemical stereoselectivity factor, also
known as the “enantiomeric ratio”, E is often used for biocatalyzed reactions.
The equations are identical for both s and E (c is the conversion):10-13
Substrate: 𝑠=ln[(1-c)(1-ee(S))]
ln[(1-c)(1+ee(S))] Product: s=
ln[1-c(1+ee(P))]
ln[1-c(1-ee(P))]
As long as the stereoselectivity factor is >1 and the reaction is first order or
pseudo-first order in the substrate, any enantiomeric purity of the substrate can
be obtained by driving the reaction to the appropriate conversion. Thus, if the
selectivity of the KR is low, the yield of the recovered substrate will also be
low at high enantiomeric purity. The product ee is however limited to the
selectivity of the catalyst and has its maximum at the initial part of the
resolution; as the conversion increases the ee of the product decreases.9
3
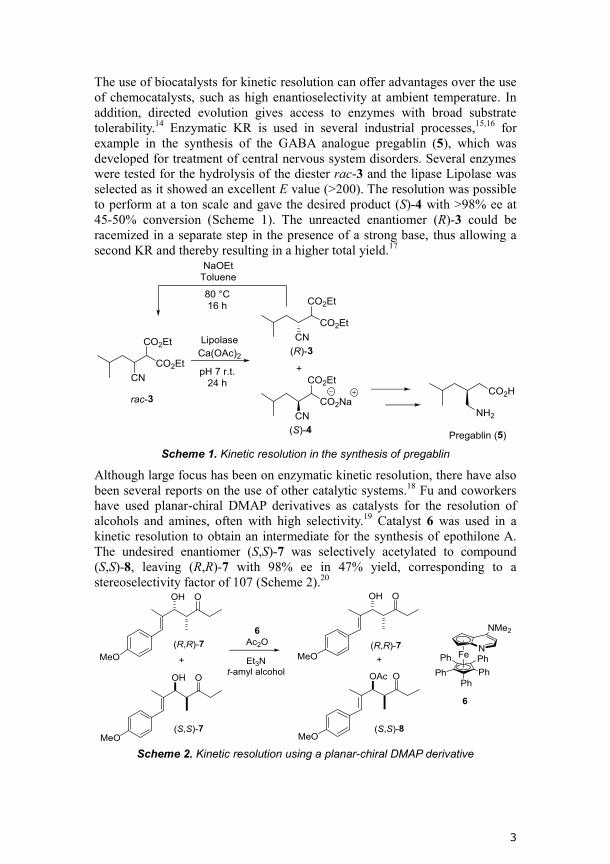
The use of biocatalysts for kinetic resolution can offer advantages over the use
of chemocatalysts, such as high enantioselectivity at ambient temperature. In
addition, directed evolution gives access to enzymes with broad substrate
tolerability.14
Enzymatic KR is used in several industrial processes,15,16
for
example in the synthesis of the GABA analogue pregablin (5), which was
developed for treatment of central nervous system disorders. Several enzymes
were tested for the hydrolysis of the diester rac-3 and the lipase Lipolase was
selected as it showed an excellent E value (>200). The resolution was possible
to perform at a ton scale and gave the desired product (S)-4 with >98% ee at
45-50% conversion (Scheme 1). The unreacted enantiomer (R)-3 could be
racemized in a separate step in the presence of a strong base, thus allowing a
second KR and thereby resulting in a higher total yield.17
Scheme 1. Kinetic resolution in the synthesis of pregablin
Although large focus has been on enzymatic kinetic resolution, there have also
been several reports on the use of other catalytic systems.18
Fu and coworkers
have used planar-chiral DMAP derivatives as catalysts for the resolution of
alcohols and amines, often with high selectivity.19
Catalyst 6 was used in a
kinetic resolution to obtain an intermediate for the synthesis of epothilone A.
The undesired enantiomer (S,S)-7 was selectively acetylated to compound
(S,S)-8, leaving (R,R)-7 with 98% ee in 47% yield, corresponding to a
stereoselectivity factor of 107 (Scheme 2).20
Scheme 2. Kinetic resolution using a planar-chiral DMAP derivative

4
1.1.2. Sequential Processes
By combining an asymmetric transformation of a prochiral substrate with a
KR, higher yield of enantiopure product compared to only a KR is possible.
When a scalemic mixture of product enantiomers is obtained after an
enantioselective reaction, the minor enantiomer is selectively transferred to a
component which can be separated from the desired product (Figure 4).21,22
The two transformations can either be performed by the same catalyst or by
two different catalysts.9
Figure 4. Sequential process
Spilling and co-workers reported the enantioselective synthesis of α-
hydroxyphosphonates using a metal-catalyzed reaction between an aldehyde
and dimethylphosphite (Scheme 3). Either the minor or the major enantiomer
of the acetylated hydroxyphosphonate was then hydrolyzed by lipases in order
to increase the ee of the desired product.23
Scheme 3. Sequential process in the synthesis of α-hydroxyphosphonates
1.1.3. Dynamic Kinetic Resolution
By combining a kinetic resolution with a simultaneous secondary process
where the substrate is continuously racemized, a theoretical yield of 100% is
possible to obtain. As the faster-reacting and slower-reacting enantiomers are
present in equal amounts, and the resolution step is practically irreversible, the
ee of the product is constant throughout the process (Figure 5).11,12
The
selectivity in a dynamic kinetic resolution (DKR) is limited to the selectivity of
the resolution catalyst. Ideally the rate of racemization should be higher than
that of the resolution (kRac > kR > kS), and the stereoselectivity factor should be
higher than 20. Alternatively, the rate of the resolution is higher than that of
the racemization (kR > kRac >> kS), but in order to have a high enantioselectivity
the resolution needs to have very high stereoselectivity (s > 200).24

5
Figure 5. Dynamic kinetic resolution (DKR)
Dynamic kinetic resolutions which employ combinations of biocatalysts and
metal catalysts have been used in the synthesis of a wide range of compounds
with high selectivity using substrates such as secondary alcohols and amines.24-
26 Bäckvall and co-workers applied DKR in the synthesis of (S)-propranolol
(1) from the corresponding β-azido alcohol 9 using Candida antarctica lipase
B (CALB or Novozyme-435) and the hydrogen transfer catalyst 11 (Scheme
4). The azido acetate 10 was obtained in 71% yield and 86% ee.
Recrystallization of the final compound resulted in almost enantiomerically
pure product.27
Scheme 4. Dynamic kinetic resolution in the synthesis of (S)-propranolol
Dynamic kinetic asymmetric transformation (DYKAT) involves a de-
racemization where the substrate equilibrates through diastereomeric
intermediates (Figure 6). Several types of DYKATs have been developed and
used in different synthetic transformations. Because the substrate isomers (SR
and SS) are converted through diastereomeric complexes (SRCat and SSCat)
they will be present in unequal amounts. If the equilibrium leading to the
major stereoisomer of the product (kREq) is faster than the equilibrium leading
to the minor stereoisomer (kSEq), the total selectivity will be higher than that of
the corresponding KR. If there is instead a mismatch case where the
equilibrium leading to the major isomer is the slowest, the selectivity will be
lower.11

6
Figure 6. Dynamic kinetic asymmetric transformation (DYKAT) type I
1.1.4. Minor Enantiomer Recycling
In the methods described so far, the minor unwanted stereoisomer of the
product is either separated from the desired stereoisomer, which will result in a
reduced yield, or it will remain in the mixture and deplete the enantiopurity of
the product. If the minor unwanted stereoisomer (PS) instead is selectively
transformed back to the prochiral substrate (A), higher enantioselectivity and a
theoretical quantitative yield of the product are possible (Figure 7). In addition,
if enantioselective catalysts are used for both the product-forming step and the
recycling step, a reinforcing effect is obtained and the overall
enantioselectivity of the system will be higher than that of the individual steps.
Figure 7. Minor enantiomer recycling (MER). Ps is the minor enantiomer which is
selectively transformed back to prochiral substrate A. Transformation of sacrificial reagent X to low energy product Y provides the thermodynamic driving force for the
system
In this type of processes, the maximum ee is given by (E1E2-1)(E1E2+1), where
E1 and E2 are the stereoselectivity factors of the separate steps. In contrast, the
maximum ee in a DKR is dependent only on one enantioselective catalyst and
is thus given by (E-1)/(E+1).9 For a cyclic process to be feasible, the system
needs to be kept out of equilibrium and coupled to a second exergonic
reaction, which feeds the process with chemical energy and drives the system
by mass balance. This secondary process can be the transformation of a

7
sacrificial reagent to a byproduct with lower chemical potential (X → Y in
Figure 7). Without this coupled reaction, a cyclic process would not be
possible since the system would violate the principle of microscopic
reversibility, which states that the forward and reverse reaction must proceed
via the same transition state.9,28,29
Deracemizations of racemic alcohols and amines have in several cases been
accomplished by concurrent oxidation and reduction sequences. In these
systems an enzyme catalyzes the selective oxidation of one of the enantiomers
to give the prochiral ketone or imine. A second enzyme then reduces the
ketone or imine selectively to the other enantiomer (Figure 8). Whole-cells,
which sometimes contain an unknown catalytic composition, have most often
been used for the concurrent oxidation and reduction.30
Kroutil and co-workers
utilized purified enzymes in this methodology and could deracemize several
secondary alcohols to obtain enantiopure alcohols in essentially quantitative
yields. Both enantiomers of the alcohols were available by selecting the
appropriate enzyme system.31,32
A deracemization of 3H-indolines and
tetrahydroquinolines using chemocatalyzed enantioselective reduction and
oxidation was recently reported by Toste and co-workers. The
enantioselectivity of the oxidation step in this system was however very low,
but since the enantioselectivity for the reduction was high, the desired products
were obtained in high ee.33
Similarly, several procedures using an
enantioselective oxidation catalyst together with a non-selective stoichiometric
reduction reagent have been reported.34
The selectivity in these systems is,
however, dependent on only one chiral catalyst.9,11,12
Figure 8. Concurrent enantioselective oxidation and reduction
Hollmann, Turner, Ward, and co-workers recently developed a double
stereoselective cascade system employing an artificial transfer hydrogenase
(ATHase) together with a monoamine oxidase (MAO-N).35
In this system the
artificial ATHase reduces an imine (12) to an amine (13) with high
enantioselectivity to form the (R)-enantiomer. The MAO-N then selectively
oxidizes the minor (S)-enantiomer of 13 back to the starting imine (12), which
can react again. A third enzyme, a catalase, was necessary to destroy the H2O2
formed as it was thought to poison the ATHase (Scheme 5). It was observed
that the ee of the formed product (13) increased during the course of the
reaction at the same time as the conversion increased and reached a maximum,
a characteristic of a minor enantiomer recycling process. The product (R)-13

8
was obtained in enantiopure form and with >99% conversion. Identical results
were obtained when starting the reaction from the imine or the racemic amine.
Scheme 5. Synthesis of enantioenriched amine (R)-13 using an oxidation/reduction
cascade with two enantioselective catalysts
In our group a minor enantiomer recycling protocol was developed for the
synthesis of O-acylated cyanohydrins. In this system, a chiral Lewis acid
catalyzes the reaction between a prochiral aldehyde and an acyl cyanide to
form a scalemic mixture of enantiomeric O-acylated cyanohydrins. The minor
product enantiomer is then selectively hydrolyzed by a lipase to the
unprotected cyanohydrin and a carboxylic acid (Scheme 6). In the two-phase
system used, consisting of toluene and aqueous buffer, the carboxylic acid is
irreversibly deprotonated to a carboxylate ion. The transformation of acyl
cyanide to carboxylate ion is an exergonic process that provides the necessary
driving force of the system. Under the reaction conditions, the unprotected
cyanohydrin releases HCN and forms the starting aldehyde, which then reacts
again in the forward reaction.36,37
Enantiopure product can always be obtained
by allowing the lipase to hydrolyze any remaining traces of minor product
enantiomer when all of the acyl cyanide has reacted.
Scheme 6. Minor enantiomer recycling to produce O-acylated cyanohydrins
The minor enantiomer recycling methodology can result in higher selectivity
and higher yields than other methods and is therefore an attractive area of
research. Expanding the methodology used for the synthesis of O-acylated

9
cyanohydrins to other types of substrates and reactions should be possible if
appropriate conditions are found. By replacing the cyanide with other
nucleophiles, a range of different compound classes should be accessible by
this methodology. One possible type of compounds is acetoxyphosphonates
which have structures analogous to those of acylated cyanohydrins (Figure 9).
Figure 9. Targets for minor enantiomer recycling
The major challenge in achieving a successful recycling system is to find
conditions by which two enantioselective catalysts are able to function
together and reinforce each other. In addition, a sacrificial process which
provides a thermodynamic driving force of the system is required.29
1.2. Aim of this Thesis
We wanted to continue to explore the asymmetric synthesis of cyanohydrins by
using combinations of chemical catalysts and biocatalysts in either the minor
enantiomer recycling methodology or in a sequential process. We were
especially interested in synthesizing cyanohydrins which are easily
transformed to compounds with synthetic and biologic importance. In addition,
focus is on the development of new synthetic transformations of the obtained
cyanohydrins to products which otherwise can be tedious to access.
The asymmetric synthesis of acetoxyphosphonates via the direct addition of
acylphosphonates to aldehydes has previously not been described. We wanted
to explore this reaction with different enantioselective catalysts and also
investigate if the transformation could be achieved by employing the minor
enantiomer recycling methodology.

10

11
2. Synthesis and Applications of O-Acylated Cyanohydrins.
(Papers I–III and V–VI)
2.1. Introduction
2.1.1. The Importance of Cyanohydrins
Enantioenriched cyanohydrins constitute a highly important class of
compounds as they can be transformed into a wide range of valuable products.
Therefore, several different strategies for the asymmetric synthesis of
cyanohydrins have been developed.38-44
Scheme 7. Equilibrium between unprotected cyanohydrins and aldehydes
Unprotected cyanohydrins readily undergo racemization through elimination
of HCN, which results in formation of the corresponding prochiral carbonyl
compound (Scheme 7). O-Acylated cyanohydrins are not only much more
stable towards racemization and decomposition in comparison to the
unprotected cyanohydrins, they are also synthetically important.45,46
O-
Acylated cyanohydrins have found wide application in agriculture as many
pyrethroid insecticides have this type of structure (Figure 10).47,48
Many
pyrethroids are used as a mixture of stereoisomers even though it is known that
they vary in activity.6
Figure 10. Examples of O-acylated cyanohydrin insecticides
In addition, O-acylated cyanohydrins can be directly transformed to N-acylated
β-amino alcohols by reduction of the nitrile group accompanied by
intramolecular acyl transfer to the formed amino group.49
With this approach
the acyl group will be part of the final product and at the same time protect the
cyanohydrin from racemization and decomposition. Alternatively, protected or
unprotected cyanohydrins can be reduced with a hydride reagent to the free
amino alcohol, which subsequently is either alkylated or acylated (Scheme
8).43
This strategy is less attractive with regard to atom economy.

12
Scheme 8. Reduction of cyanohydrins to N-substituted β-amino alcohols
The β-amino alcohol and β-amido alcohol structural motifs are present in many
compounds with interesting biological properties (Figure 11). Examples
include the α1-adrenoreceptor agonist midodrine (16),50
the β3-adrenergic
receptor agonists solabegron (17)51,52
and mirabegron (19),53
and the β1-
adrenergic receptor agonist denopamine (18).54
Other examples are the
naturally occurring compounds (R)-tembamide (20) and (R)-aegelin (21),
which have both been used in traditional Indian medicine. In addition, the
previously mentioned β-adrenergic antagonist (S)-propranolol (1) is another
important example. Solabegron (17) has shown promising results for the
treatment of both overactive bladder (OAB) and irritable bowel syndrome
(IBS) and is currently in clinical trials.55-57
Figure 11. Biologically active compounds containing the β-amino alcohol and β-
amido alcohol structural motifs

13
2.1.2. Strategies for the Preparation of Enantioenriched Cyanohydrins
The most extensively used sources of cyanide in cyanations of carbonyl
compounds are HCN and TMSCN. HCN is extremely toxic and volatile, and
addition to aldehydes or ketones results in the unstable parent cyanohydrin. It
is therefore advantageous to instead use TMSCN as source of cyanide due to
its high reactivity and the instant protection as the silyl ether, which prevents
decomposition and racemization. However, TMSCN is also highly toxic and
volatile, and it is relatively expensive compared to that of other cyanide
sources.39,41
Cyanoformic esters have also been used to a relatively large extent
as source of cyanide while acyl cyanides have been more scarcely used. Acyl
cyanides have the advantage of producing highly stable O-acylated
cyanohydrins as products. Therefore, a method for the direct esterification of
O-TMS-cyanohydrins to give O-acylated cyanohydrins has been developed.58
2.1.2.1. Biocatalytic Methods for the Preparation of Cyanohydrins
Enzymes that catalyze the release of HCN from cyanohydrins exist naturally in
for example almonds as a protection against herbivores and microbial attack.
Hydroxynitrile Lyases (HNL’s) have been used for the highly enantioselective
synthesis of cyanohydrins by employing both aldehydes and ketones together
with an excess of HCN (Scheme 9).59-63
Scheme 9. HNL-catalyzed addition of HCN to aldehydes
Many compounds obtained by the use of these enzymes have been used in the
synthesis of pharmaceuticals, agrochemicals, and other biologically active
compounds.62
Hydroxynitrile Lyase from Hevea brasiliensis was used in the
kilogram synthesis of a β-amino alcohol (24) from furfural (22), resulting in
essentially enantiopure product (>99% ee) (Scheme 10).64
Scheme 10. Kilogram synthesis of a β-amino alcohol involving a HNL-catalyzed
HCN-addition to furfural
Candida antarctica lipase B (CALB) has been used in the kinetic resolution of
several aromatic O-acetylated cyanohydrins. In the transesterification of the
racemic compounds, the lipase is highly selectivity (E = 35–360) for the (S)-
enantiomers (Scheme 11).59,65-67
The high selectivity of CALB towards
cyanohydrins was utilized in our group for a high throughput ee analysis of O-
acetylated cyanohydrins.68,69
The enantiomeric preference of the lipase can be
predicted according to the steric properties of the substrate; these predictions
are known as Kazlauskas’ rules.70
In the active site, the (S)-enantiomer of the

14
substrate resides with the cyanide group in a pocket which has a very limited
size, and for the (R)-enantiomer to be hydrolyzed, the large aryl group needs to
fit in this pocket.71
Scheme 11. Kinetic resolution of O-acetylated cyanohydrins with CALB. The
enantiopreference of the enzyme is also shown; L represents the large substituent and M represents the medium substituent
CALB is a robust enzyme that can be used between pH 3.5 and 9.5 and the
immobilized variant can be used at temperatures up to 80 °C. CALB is also
stable in non-polar solvents as well as in more polar solvents.72
Lipases belong
to the hydrolases and follow the serine hydrolase mechanism (Scheme 12).
Scheme 12. The serine hydrolase mechanism for the hydrolysis of an ester. The
catalytic amino acids in CALB are indicated
In the active site, three amino acids are involved in the catalytic triad; serine
acts as a nucleophile and is activated by a histidine together with an aspartic or
a glutamic acid through hydrogen bonding. The reaction sequence is initiated

15
by attack of serine on the ester substrate (I) and the tetrahedral intermediate
formed is stabilized by the oxyanion hole which consists of amino acids with
hydrogen bond donor groups (II). Elimination of the alcohol product results in
an enzyme-substrate complex (III). Water subsequently attacks this complex
and again results in a tetrahedral intermediate that is stabilized by the oxyanion
hole (IV). Elimination of the acid product allows the enzyme to react again. In
the mechanism the water molecule can be replaced with another nucleophile
such as an alcohol. Depending on the reaction conditions, a lipase can work in
both directions and can thus either cleave or form an ester bond.73,74
Unprotected cyanohydrins are in equilibrium with the corresponding aldehydes
and HCN under basic conditions and thus readily racemize. By combining
enzymatic enantioselective acylation with in situ racemization, several
effective dynamic kinetic resolutions (DKR) have been developed.59
Initial
reports utilized anion-exchange resins or quinidines for the racemization and
either lipases from Pseudomonas sp. or Pseudomonas cepacia for the
transacetylation.75-78
Kanerva and co-workers expanded the scope of the
system, by employing lipase from Pseudomonas cepacia or Candida
antarctica lipase A (CALA), to more synthetically useful cyanohydrins.79-81
Hanefeld and co-workers could obtain the O-acetylated cyanohydrin (S)-28 in
97% yield and with 98% ee after four days reaction when using immobilized
CALB for the transacetylation step (Scheme 13).82,83
A disadvantage of the
method was that aliphatic substrates resulted in lower enantioselectivity than
aromatic substrates; this was believed to be due to slow racemization and/or
base-catalyzed non-selective acylation.84
Scheme 13. Dynamic kinetic resolution in the synthesis of O-acetylated
cyanohydrins using CALB
2.1.2.2. Cooperative Catalytic Systems for the Preparation of Cyanohydrins
Simultaneous activation of both the electrophile and the nucleophile occurs in
most enzymatic transformations, but can also improve the reactivity and the
enantioselectivity of chemically catalyzed organic reactions.85
Activation by a
combination of a Lewis acid and a Lewis base lowers the energy of the LUMO
of the electrophile and at the same time raises the energy of the HOMO of the
nucleophile, and thus increases the reactivity of the reagents.86
Cooperative
catalytic activation of the nucleophilic cyanide group and the electrophilic
carbonyl group has been extensively used in the asymmetric synthesis of
cyanohydrins.41,86-88
One of the earliest examples, which was reported by

16
Shibasaki and co-workers, employed the bifunctional BINOL derived catalyst
29 in the cyanosilylation of aldehydes with very good results (Scheme 14). The
aluminum center acted as a Lewis acid, activating the aldehyde, while the
phosphine oxide acted as a Lewis base and activated TMSCN.88,89
Scheme 14. Cyanosilylations catalyzed by bifunctional catalyst 29
The titanium salen dimeric complex 30 was introduced for the asymmetric
cyanosilylation of aldehydes and ketones by Belokon, North, and co-workers.
This catalyst is obtained by treating the monomeric titanium chloride-complex
with an aqueous buffer, which leads to elimination of HCl and formation of the
dimeric catalyst. High ee and 100% conversion could be obtained in the
cyanosilylation of several different aldehydes with only 0.1 mol % of catalyst
(R,R)-30 (Scheme 15).90
Reactions with ketones were slower and resulted in
products with lower enantiopurity.91
Scheme 15. Cyanosilylations catalyzed by (R,R)-30
It was also possible to use catalyst (R,R)-30 in the addition of cyanoformates to
aldehydes, which resulted in O-protected cyanohydrins. However, in this case
it was necessary to use 5 mol % of the catalyst at -40 °C in order to have
practical reaction times.92
It was later discovered that the reaction could be
accelerated, and the catalyst loading reduced, by using KCN as a co-catalyst.93
Catalyst (R,R)-30 together with KCN and different carboxylic acid anhydrides
was also used to synthesize O-acylated cyanohydrins in a one-pot protocol
(Scheme 16). Good to high enantiomeric excesses (60–93%) were observed
when employing acetic anhydride as acylating agent. The system was however
dependent on which acylating agent used; benzoic anhydride, for example,
resulted in much lower enantioselectivity.94-96
17
Scheme 16. Synthesis of O-acylated cyanohydrins using carboxylic acid anhydrides
and KCN
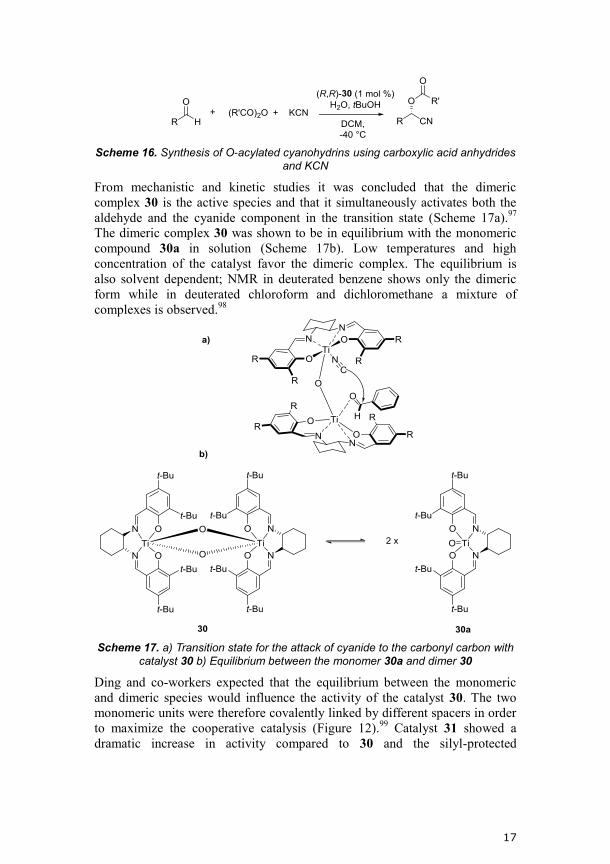
From mechanistic and kinetic studies it was concluded that the dimeric
complex 30 is the active species and that it simultaneously activates both the
aldehyde and the cyanide component in the transition state (Scheme 17a).97
The dimeric complex 30 was shown to be in equilibrium with the monomeric
compound 30a in solution (Scheme 17b). Low temperatures and high
concentration of the catalyst favor the dimeric complex. The equilibrium is
also solvent dependent; NMR in deuterated benzene shows only the dimeric
form while in deuterated chloroform and dichloromethane a mixture of
complexes is observed.98
Scheme 17. a) Transition state for the attack of cyanide to the carbonyl carbon with
catalyst 30 b) Equilibrium between the monomer 30a and dimer 30
Ding and co-workers expected that the equilibrium between the monomeric
and dimeric species would influence the activity of the catalyst 30. The two
monomeric units were therefore covalently linked by different spacers in order
to maximize the cooperative catalysis (Figure 12).99
Catalyst 31 showed a
dramatic increase in activity compared to 30 and the silyl-protected

18
cyanohydrin from benzaldehyde was obtained in almost quantitative yield and
with 96% ee after just five minutes at room temperature. Catalyst loadings
could be reduced to as low as 0.0005 mol % when longer reaction times were
used, and still with very high enantioselectivity. The catalyst could also be
used to synthesize O-acetylated cyanohydrins with high enantiopurity by
utilizing NaCN and acetic anhydride. One drawback of catalyst 31 is that the
synthesis of the required ligand is rather complicated. In contrast, the ligand
used in catalyst 30 is commercially available.100
Figure 12. Linked catalyst 31
North and co-workers coupled their previously developed synthesis of O-
acylated cyanohydrins to a lipase-catalyzed kinetic resolution (Scheme 18).101
The minor enantiomer from the product-forming reaction, which employed
(S,S)-30, KCN, and acetic anhydride, was subsequently hydrolyzed by CALB
to the unprotected cyanohydrin which could be separated from the desired
product. Initially, the DCM used in the first step was evaporated before the
enzymatic resolution, but it was later discovered that the enzyme was able to
retain its function in a mixed solvent system consisting of DCM and MTBE.
With this procedure the enantiopurity of the O-acetylated cyanohydrins
obtained in the first step was in several cases improved. However, a sequential
process such as this one involves the destruction of the minor enantiomer, and
the yield is therefore unavoidably decreased.
Scheme 18. Sequential process in the synthesis of O-acetylated cyanohydrins

19
The non-selective direct addition of acyl cyanides to carbonyl compounds was
early discovered to be catalyzed by base,102
and since then several variations of
this reaction have been developed.45,103-108
For example, N-heterocyclic
carbenes (NHCs) have been used as catalysts in the synthesis of racemic O-
acylated cyanohydrins.109
The advantage of using acyl cyanides is that both the
cyanide group and the acyl group originate from the same reagent, thereby
providing a transformation with 100% atom economy. The first asymmetric
direct addition of acyl cyanides to aldehydes was developed in our group and
employs a Lewis acid – Lewis base dual activation system (Scheme 19).110-112
The reaction is catalyzed by the titanium salen dimer 30, but in contrast to the
addition of TMSCN to aldehydes, a Lewis base is also necessary for the
reaction to proceed at low temperature. The role of the Lewis base is to
activate the acyl cyanide by adding to the carbonyl carbon thus releasing a
cyanide ion and forming a reactive acylating agent. The aldehyde is activated
by the titanium complex 30, probably in a fashion similar to that in the reaction
with TMSCN, thus allowing an enantioselective nucleophilic attack by the
cyanide ion. The resulting intermediate is then acylated by the acyl ammonium
ion. This methodology was successful for several different aldehydes and acyl
cyanides and gave the desired products in high yields and with high ee,
although some substrates gave products with lower enantioselectivity and/or
lower yield. Cyanoformates could be used in place of acyl cyanides with the
same catalytic system, also with good results.
Scheme 19. Addition of acyl cyanides to aldehydes catalyzed by 30 and Et3N
Recent mechanistic studies in our group included a Hammett correlation study
that showed that different steps are rate-determining depending on whether
electron-rich or electron-poor aldehydes are used. With the electron-rich
aldehydes the attack of cyanide on the carbonyl is the rate limiting step,
whereas with the electron-poor aldehydes the acylation step instead becomes
the slower step. It was also shown by using different combinations of
chiral/achiral base and enantiopure/racemic titanium complex 30 that acylation

20
occurs while the cyanohydrin is bound to titanium and that the unprotected
cyanohydrin most likely is not an intermediate in the reaction.113,114
Another catalytic system, consisting of a TiIV
complex of a BINOLAM-ligand
(32), has been used in the synthesis of O-aroyl cyanohydrins from aldehydes
and aroyl cyanides (Scheme 20).115,116
Several aldehydes gave the desired
products in high yields, however with moderate enantiopurity. Only aroyl
cyanides were used in the reaction as no product was formed when using
acetyl cyanide. The catalyst ligand contained a diethylaminomethyl group
which, in contrast to what is the role of the triethylamine in our dual activation
system, was proposed to act as Brønsted base which deprotonates trace
amounts of HCN present in commercial benzoyl cyanide. An aluminum
complex of ligand 32 was also used in the addition of methyl
cyanoformate117,118
and diethyl cyanophosphonate119,120
to aldehydes.
Scheme 20 Acylcyanation of aldehydes catalyzed by Ti(OiPr)4 complexed with
BINOLAM-ligand 32
2.1.2.3. Minor Enantiomer Recycling for the Preparation of Cyanohydrins
A minor enantiomer recycling procedure for the highly enantioselective
synthesis of O-acylated cyanohydrins was developed in our group (Scheme
21).37
In this system, the forward reaction between aldehyde and acyl cyanide
is catalyzed by the titanium complex (S,S)-30, while the kinetic resolution of
the resulting scalemic mixture is catalyzed by CALB. In order for the two
reactions to occur simultaneously, a mixture of a buffer phase and an organic
phase with lower density phase was used. The buffer phase is required to
ensure that the carboxylic acid formed in the hydrolysis step is deprotonated to
carboxylate ions. The buffer is also important in order to maintain the enzyme
activity which decreases at low pH.72
The acyl cyanide is added slowly into the
organic phase throughout the reaction in order to avoid hydrolysis in the
aqueous phase. Initially a tertiary amine was used as a co-catalyst, in analogy
to the dual activation procedure, but it was later discovered that the presence of
a Lewis base was not necessary in the two-phase system. Several different
aldehydes were used in the system and produced (R)-O-acylated cyanohydrins
with very high enantiomeric purity. The opposite product enantiomer was
obtained by using the enantiomeric titanium complex (R,R)-30 together with
Candida rugosa lipase (CRL).37
In addition, by using either the minor
enantiomer recycling system or a dynamic kinetic resolution, both enantiomers

21
of the O-acetylated cyanohydrin originating from (E)-2-butenal could be
obtained with the same biocatalyst.121
Scheme 21. Minor enantiomer recycling in the synthesis of O-acylated
cyanohydrins
Kinetic modelling and experimental studies of the system clearly demonstrated
that a reinforcing effect is obtained when combining two chiral catalysts.
Modelling also showed that the final ee of the product is the same regardless of
the ratio between the two catalysts, as long as the total selectivity is the same.
Interestingly, the yield is dependent on this ratio, thus showing that the yield
can be improved by varying the catalyst composition.36
Recently the reaction scope of minor enantiomer recycling was expanded to
the use of alkyl cyanocarbonates as source of cyanide.28
Titanium catalyst
(S,S)-30 was used for the product-forming reaction, and a lipase from porcine
pancreas (PPL) selectively hydrolyzed the minor product enantiomer. In this
study, the importance of having a thermodynamic driving force coupled to the
recycling was clearly demonstrated. Bovine carbonic anhydrase catalyzed the
decomposition of carbonic acid, formed in the enzymatic hydrolysis, to CO2.
In addition, a nitrogen stream was bubbled through the reaction mixture to
assure an efficient release of CO2. The required exergonic process was thus
provided by the release of CO2 from the system. When the process was
performed without either carbonic anhydrase or a nitrogen stream, the ee of the
product dramatically decreased, thus showing that when the CO2 is not
released from the system the cyclic process is halted.

22
2.2. Synthesis of Enantioenriched ω-Bromocyanohydrin Derivatives
2.2.1. Introduction
In order to demonstrate the synthetic importance of the methods developed in
our group, we wanted to synthesize the enantioenriched O-acetylated
cyanohydrins derived from 4-bromobutanal (33a) and 5-bromopentanal (33b).
The unprotected cyanohydrins have previously been synthesized by the
hydroxynitrile lyase-catalyzed HCN-addition to aldehydes 33a-b and gave the
unprotected cyanohydrins with 90–94% ee.122,123
It has been shown that the
unprotected cyanohydrins of these aldehydes can be used to synthesize 2-
cyanotetrahydrofuran (35a) and 2-cyanotetrahydropyran (35b) (Scheme 22).122
Chiral tetrahydrofurans are found in several natural products and are thus of
synthetic interest.124,125
The ω-bromocyanohydrins have also been used in the
synthesis of enantioenriched 2-substituted piperidines,126
azacycloalkan-3-
ols,127
and 2,3-disubstituted piperidines.128
Scheme 22. Retrosynthesis of 2-cyanotetrahydrofuran (35a) and 2-
cyanotetrahydropyran (35b)
2.2.2. Minor Enantiomer Recycling
We expected that the use of the minor enantiomer recycling protocol would be
beneficial for the synthesis of the O-acetylated cyanohydrins 34a and 34b from
4-bromobutanal (33a) and 5-bromopentanal (33b). We therefore initiated our
study by investigating the two individual steps of the cyclic process separately.
The forward reaction between the aldehydes 33a or 33b and acetyl cyanide,
catalyzed by titanium complex (S,S)-30, was performed in a two-phase system
consisting of toluene and pH 8 buffer (Scheme 23). To replicate the cyclic
process, three equivalents of acetyl cyanide (36) were slowly added to the
organic phase while the yield and ee were monitored by GC. As expected the
ee of the formed products remained constant throughout the reaction, although
at room temperature the reaction was only moderately selective to form the
(R)-enantiomers; 42% ee for 34a and 22% ee for 34b. During NMR analysis of
the crude reaction using 33a, we could observe the formation of the cyclized
byproduct (R)-35a. This compound was determined to have an ee of 40%,
which is almost identical to that of (R)-34a. We therefore believe that the two
products are formed either from a titanium-cyanohydrin complex or from the
free cyanohydrin.

23
Scheme 23. Forward reaction to synthesize (R)-34a and (R)-34b in the two-phase
system
We next tested some different lipases in the kinetic resolution of racemic O-
acetylated cyanohydrins 34a and 34b in the two-phase system (Scheme 24).
We followed the hydrolysis at different temperatures and pH:s by monitoring
the composition of enantiomers over time by chiral GC. CALB was highly
selective for the (S)-enantiomers of both substrates, although the hydrolysis
was slower for 34b (Figure 14) than for 34a (Figure 13). The enzyme
maintained the high selectivity when the pH was decreased to 6 at room
temperature, but when the temperature was increased to 40 °C the
enatioselectivity in the hydrolysis of 34a decreased. In contrast, hydrolysis by
Candida antarctica lipase A (CALA) was much slower and had lower
selectivity. As observed previously,37,66,129
Candida rugosa lipase (CRL)
showed a selectivity opposite of CALB for both substrates, but the rate of
hydrolysis was much slower than with CALB.
Scheme 24. Kinetic resolution of racemic O-acetylated cyanohydrins 34a and 34b

24
Figure 13. Kinetic resolution of 34a with CALB at r.t. with pH 8 buffer
Figure 14. Kinetic resolution of 34b with CALB at r.t. with pH 8 buffer
When the enzyme for the reverse step had been selected, we next continued
with the investigation of the cyclic process. Unfortunately, with aldehyde 33a
we detected large amounts of the cyclized product 35a when performing the
reaction at pH 8. This resulted in low yields of the desired product (Table 1,
entries 1 and 2), but very high ee (enantiopure product at 40 °C). Higher
temperature usually results in a faster enzymatic hydrolysis, but in the present
case also the formation of 35a was accelerated. We assumed that the
cyclization was mediated by base, and therefore the reaction was also
performed at pH 7 and 6. Indeed, lower pH gave lower amounts of byproduct;
at pH 6, 70% yield of 34a was obtained, but with only 82% ee (Table 1, entry
5). We could however obtain almost enantiopure product in 62% yield by
continued stirring of the reaction mixture after the addition of acetyl cyanide
was finished, thus hydrolyzing the remaining (S)-enantiomer (reaction profile
can be seen in Figure 15). Under these conditions cyclized product 35a was
formed in 17% yield and with 33% ee. This product had opposite absolute
configuration to that formed in the forward reaction, which indicates that the
0
10
20
30
40
50
60
0 5 10 15 20 25
%
Time (h)
(R)-38a (%)
(S)-38a (%)
0
10
20
30
40
50
60
0 5 10 15 20 25
%
Time (h)
(R)-38b (%)
(S)-38b (%)
(R)-34a (%)
(S)-34a (%)
(R)-34b (%)
(S)-34b (%)

25
cyclic product can form in the forward reaction as well as in the resolution
step.
With aldehyde 33b byproduct formation was not a problem and the desired
product was formed in high yield, although with moderate ee (Table 1, entry
7). The low selectivity can probably be explained by slow enzymatic
hydrolysis. Performing the reaction at 40 °C gave a somewhat higher ee, and
leaving the mixture to stir after complete addition of acetyl cyanide (36) gave
enantiopure (R)-34b in 67% yield (Table 1, entry 8).
Table 1. Minor enantiomer recycling with aldehydes 33a and 33b
entry aldehyde pH temp
(°C)
yielda of 34
(%)
ee of 34
(%)
1 33a 8 r.t. 46 82
2 33a 8 40 26 100
3 33a 7 r.t. 57 87b
4 33a 7 40 21 99
5 33a 6 r.t. 70 82c
6 33a 6 40 42 90d
7 33b 7 r.t. 84 61
8 33b 7 40 78 86e
aDetermined by GC. bContinued stirring for 13 h gave 50% of (R)-34a with 100% ee; cContinued stirring for 15 h gave 62% of (R)-34a with 99% ee, 17% (S)-35a with 33%
ee, and 10% 37a; dContinued stirring for 15 h gave 34% of (R)-34a with 100% ee, 28%
racemic 35a and 3% of 37a; eContinued stirring for 15 h gave 67% of (R)-34b with
>99% ee.

26
Figure 15. Reaction profile for the minor enantiomer recycling in the synthesis of
(R)-34a (Table 1, entry 5). Addition of acetyl cyanide (36) was stopped after 25 hours
2.2.3. Sequential Process
The minor enantiomer recycling process was compared to the dual activation
methodology, employing titanium catalyst (S,S)-30 and triethylamine in DCM
at -20 °C, that was developed in our group. Under these conditions the desired
products (R)-34a and (R)-34b were obtained in almost quantitative yields
(determined by GC), but with merely 81 and 80% ee, respectively. By
removing the DCM and dissolving the residue in toluene and stirring with
CALB and pH 8 buffer, essentially enantiopure products were obtained in high
yields (Scheme 25). The opposite enantiomer ((S)-34a) was obtained in
quantitative yield and 78% ee using catalyst (R,R)-30. Treatment of this
compound with CRL gave after 125 hours (S)-34a in 87% yield (determined
by GC) and 91% ee.
Scheme 25. Sequential process in the synthesis of (R)-34a and (R)-34b, and
subsequent deprotection and cyclization to give (R)-35a and (R)-35b
0
20
40
60
80
100
0 10 20 30 40
%
Time (h)
ee (%)
yield (%)

27
As the sequential procedure gave an overall better result than the minor
enantiomer recycling process it was used on a preparative scale (2.4 mmol)
and resulted in that 71% of 34a and 61% of 34b could be isolated in almost
enantiopure form. Finally (R)-34a and (R)-34b were transformed to the cyclic
compounds by initial hydrolysis to the unprotected cyanohydrins (R)-37a,b
using p-TsOH in EtOH. Cyclization using AgClO4122
gave compounds (R)-35a
and (R)-35b with no or very little racemization in moderate yields (Scheme
25).
2.2.4. Conclusions
Essentially enantiopure O-acetylated ω-bromocyanohydrins were obtained in
high yields using a sequential procedure. Minor enantiomer recycling could
also be used for the synthesis of these compounds but was less efficient due to
byproduct formation and low rate of enzymatic hydrolysis.
The highly enantioenriched cyanohydrins were transformed to (R)-2-
cyanotetrahydrofuran and (R)-2-cyanotetrahydropyran.
2.3. Application of Minor Enantiomer Recycling in the Synthesis of β-Adrenergic Antagonists
2.3.1. Introduction
We wanted to further expand the usefulness of the minor enantiomer recycling
methodology and at the same time show that the method can be used to access
compounds which are otherwise not readily available. As cyanohydrins easily
can be reduced to β-amino alcohols, we envisioned that the β-adrenergic
antagonists (S)-propranolol (1),130
(R)-dichloroisoproterenol (38),131
and (R)-
pronethalol (39)130
would all be available with a common strategy employing
enantioenriched cyanohydrins obtained via minor enantiomer recycling
(Scheme 26).
Scheme 26 Retrosynthesis to obtain enantioenriched β-adrenergic antagonists via
minor enantiomer recycling

28
Several different strategies have been used to synthesize (S)-propranolol (1),
but there are no reports which include the enantioselective addition of cyanide
to the prochiral aldehyde to obtain the corresponding cyanohydrin. (S)-
Propranolol was however synthesized by kinetic resolution of the racemic
unprotected cyanohydrin by Pseudomonas cepacia lipase. The cyanohydrin
was obtained with 96% ee at 56% conversion.132
The (R)-enantiomer of the
acylated cyanohydrin was obtained in 68% yield and merely 78% ee in a
dynamic kinetic resolution.77
The (R)-O-acetylated cyanohydrin from 3,4-dichlorobenzaldehyde, which is
the precursor to 38, has previously been synthesized in 40% yield and 93% ee
by enzymatic kinetic resolution of the racemic O-acetylated cyanohydrin.133
Addition of TMSCN to the same aldehyde gave the silyl-protected
cyanohydrin with 78% ee.134
Several successful syntheses of the cyanohydrin which is the precursor to 39
have been reported using different cyanide sources.41
2.3.2. Synthesis of (S)-Propranolol
We started by investigating the selectivity of the dual activation system in the
reaction between aldehyde 40 and acetyl cyanide (36) using catalyst (S,S)-30
and Et3N at -30 °C. The acylated cyanohydrin (S)-41 could be isolated in 69%
yield, however with only 15% ee. Using TMSCN as source of cyanide together
with catalyst (S,S)-3090
gave a slightly higher ee (Scheme 27). Preliminary
studies performed in our laboratory indicated that low selectivity is obtained in
cyanations of aldehydes containing a heteroatom in the α-position when using
catalyst 30.135
The aldehyde 40 has also been reported to produce racemic
cyanohydrin when subjected to (R)-hydroxynitrilase-catalyzed HCN-
addition.136
Scheme 27 Addition of acetyl cyanide or TMSCN to aldehyde 40 using catalyst
(S,S)-30

29
As the direct methods attempted gave low enantioselectivity, it was thought
that the minor enantiomer recycling procedure would be an advantageous
method. The racemic acetylated cyanohydrin 41 was therefore treated with
different enzymes in order to identify the optimal biocatalyst for the recycling
step. CALB was the superior enzyme and showed high selectivity for the (R)-
enantiomer (Scheme 28, Figure 16). Pseudomonas cepacia lipase (PSL) was
selective but the rate of hydrolysis was lower; both CALA and CRL were
unselective and slow.
Scheme 28. Kinetic resolution of 41 by CALB
Figure 16. Kinetic resolution of 41 with CALB at r.t. with pH 8 buffer
The forward reaction in the two-phase system, which was catalyzed by (S,S)-
30, was unselective and gave racemic product. The low selectivity was not
surprising as the dual activation methodology at low temperature with the
same catalyst also gave low ee. We still believed that it was possible to achieve
a successful recycling system as the enzymatic hydrolysis step was highly
selective.
We started by employing our standard amounts of catalysts at room
temperature, but this unfortunately gave both low yield and low ee (Table 2,
entry 1). We could improve the results somewhat by raising the temperature to
40 °C (entry 2). Addition of acetyl cyanide (36) at a lower rate allowed the
system to reach steady state and gave almost enantiopure product, although in
low yield (entries 3 and 4). Increasing the amount of titanium catalyst (S,S)-30
gave an increased yield, although with a slightly lower ee (entries 5 and 6).
0
10
20
30
40
50
60
0 5 10 15 20 25 30
%
Time (h)
Series1
Series2
(S)-41 (%)
(R)-41 (%)

30
Table 2. Optimization of the minor enantiomer recycling using aldehyde 40
entry Temp
(°C)
Addition
time (h)
(S,S)-30
(mol %)
CALB
(mg)
Yield
(%)a ee
(%)a
1 r.t. 25 5 20 44 41
2 40 25 5 20 50 77
3 40 50 5 20 35 98
4 40 50 5 40 11 100
5 40 50 7.5 20 34 95
6 40 50 10 20 61 96 aDetermined by GC using undecane as internal standard.
The yields in the cyclic process were low to moderate in all cases and we
therefore suspected that the starting aldehyde was not stable under the reaction
conditions. Stirring the aldehyde together with CALB and (S,S)-30 in the two-
phase system indeed substantially lowered the amount of aldehyde found in the
organic phase. The effect of varying the stirring rate of the reaction mixture
was also investigated; the yields did not appear to be affected, but higher ee:s
were observed with a faster stir rate.
The conditions in entry 6 in Table 2 were used on a preparative scale. The
desired product (S)-41 was then isolated in 59% yield and with 98% ee. This
result is an improvement in the synthesis of this cyanohydrin compared to the
kinetic resolution of the unprotected cyanohydrin, which gave 96% ee at 56%
conversion,132
and to the dynamic kinetic resolution, which gave 68% yield
and 78% ee.77
Reduction of the nitrile and ester in (S)-41 with LiAlH4 to the
amino alcohol (S)-43, followed by reductive amination with acetone and
NaBH4, afforded (S)-propranolol (1) in 56% yield over two steps with very
high ee (Scheme 29).137

31
Scheme 29. Synthesis of (S)-propranolol
2.3.3. Synthesis of (R)-Dichloroisoproterenol
When subjecting 3,4-dichlorobenzaldehyde (44) to the minor enantiomer
recycling system, we observed that the ee of the acylated cyanohydrin (R)-45
initially increased over time, as expected, but after approximately 30 hours
addition of acetyl cyanide (36) the ee started to decrease. We believe that this
decrease in ee might be explained by inhibition of CALB by acetyl cyanide.
Inhibition would result in slower hydrolysis of the minor enantiomer and a
shift in the balance between the less selective forward reaction and the reverse
resolution. By successively decreasing the rate of addition of acetyl cyanide
during the course of reaction the decrease in ee was no longer observed.
However, when performing the reaction on a preparative scale we used an
alternative approach which was less time-consuming. The acetyl cyanide was
added over eight hours which gave the product with 70% ee. Addition of an
additional batch of CALB to that already present in the reaction mixture and
hydrolysis of the remaining minor enantiomer gave (R)-45 in 54% isolated
yield and with 96% ee (Scheme 30).
It was not possible to utilize LiAlH4 for the reduction of (R)-45 as this gave the
final product 38 in merely 58% ee. To avoid racemization, the ester was
hydrolyzed to give the unprotected cyanohydrin, and subsequent reduction of
the nitrile under milder conditions with BH3·THF gave (R)-
dichloroisoproterenol (38) without any depletion of the ee.
Scheme 30. Synthesis of (R)-dichloroisoproterenol
2.3.4. Synthesis of (R)-Pronethalol
In the minor enantiomer recycling with 2-naphtylaldehyde (46) acetyl cyanide
(36) was added to the reaction mixture over 50 hours at 40 °C. After this

32
addition the ee was 91%, but by allowing the mixture to stir for an additional 5
hours and 40 minutes the ee increased to 98% and (R)-47 could be isolated in
82% yield (Scheme 31). This again showed an advantage of minor enantiomer
recycling; very high ee can be obtained by allowing the enzyme to perform a
kinetic resolution of the scalemic mixture. (R)-Pronethalol (39) was obtained
with 96% ee by using the LiAlH4 reduction followed by reductive amination.
Scheme 31 Synthesis of (R)-pronethalol
2.3.5. Conclusions
The β-adrenergic antagonists (S)-propranolol, (R)-dichloroisoproterenol, and
(R)-pronethalol were all obtained with very high enantiomeric purity using the
minor enantiomer recycling procedure. In the synthesis of the cyanohydrin
which is the precursor of (S)-propranolol, other methods showed inferior
enantioselectivity, which seems to be due to the presence of the heteroatom in
the aldehyde. When employing 3,4-dichlorobenzaldehyde in the cyclic
procedure, problems with enzyme inhibition were encountered but high ee was
obtained by allowing the enzyme to hydrolyze the remaining minor enantiomer
at the end of the reaction.
2.4. Synthesis and Applications of O-(α-Bromoacyl) Cyanohydrins
2.4.1. Introduction
Cyanohydrins functionalized with an O-(α-bromoacyl) group can undergo
several different transformations and are thus precursors to a range of
important compounds (Scheme 32). Intramolecular cyclization of O-(α-
bromoacyl) cyanohydrins results in aminofuranones and hydroxyfuranones,
which can undergo further transformations (routes a and b, Scheme 32). The
acyl chain in the O-(α-bromoacyl) cyanohydrins can be varied by either
substituting the bromo group with different nucleophiles or by performing a
cross-coupling at the α-position resulting in a new C-C bond (routes c and d,
Scheme 32). Reductions of the obtained products provide biologically active
N-substituted β-amino alcohols.
Enantioenriched O-(α-bromoacyl) cyanohydrins would be available by
employing α-bromoacyl cyanides in the minor enantiomer recycling. This

33
highly divergent approach would have a very high atom economy and would
thus allow further functionalization of the obtained products after the
enantioselective step.
Scheme 32. Possible transformations of O-(α-bromoacyl) cyanohydrins; a:
Cyclization to aminofuranones. b: Cyclization to hydroxyfuranones. c: Nucleophilic
substitution of the bromo-group. d: Cross-coupling at the α-position
2.4.2. Synthesis of O-(α-Bromoacyl) Cyanohydrins and Subsequent Transformation to Aminofuranones
The 2(5H)-furanone structural motif can be found in a range of compounds
possessing biological properties (Figure 17).138-141
Additionally, both
aminofuranones and hydroxyfuranones serve as synthetic intermediates.142,143
Examples are found in the syntheses of the antibiotic virginiamycin M2144
and
the pheromone (+)-eldanolide.145
In addition, reduction of the double bond in
the hydroxyfuranones has been used to access several interesting
compounds.146,147
Figure 17. The 2(5H)-furanone structural motif
We planned to access enantioenriched aminofuranones from O-(α-bromoacyl)
cyanohydrins obtained in the minor enantiomer recycling process. α-
Halocarbonyl compounds form zinc enolates upon treatment with Zn0, and the
addition of these zinc enolates to nitriles is known as the Blaise reaction.148
Zinc enolates of O-(α-bromoacyl) cyanohydrins react in an intramolecular
fashion and produces either aminofuranones or hydroxyfuranones, depending

34
on conditions for work-up (Scheme 32, routes a and b).149
Under strongly
acidic conditions the intermediate imine is hydrolyzed to the hydroxyfuranone,
whereas work-up with NH4Cl instead gives the aminofuranone (Scheme
33).149-151
Scheme 33. Intramolecular Blaise reaction in the synthesis of aminofuranones
Other syntheses of these types of compounds include for example the base-
mediated cyclization of O-acylated cyanohydrins.152,153
However, this is
limited to cyanohydrins obtained from ketones since deprotonation of the α-
proton would otherwise occur.
In a recent example the importance of the O-(α-bromoacyl) cyanohydrins as a
synthetic intermediate was demonstrated by Fürstner and co-workers.147
In the
synthesis of nominal gobienine A, a BINOL-derived ligand was used in the
asymmetric TMSCN addition to an aliphatic aldehyde (48) which afforded the
corresponding cyanohydrin with 89% ee. The cyanohydrin was subsequently
acylated with 2-bromopropionyl bromide in a separate step to give the O-(α-
bromoacyl) cyanohydrin 49. Intramolecular cyclization followed by acidic
work-up resulted in the desired hydroxyfuranone 50. Interestingly, the authors
could also show that by transforming the hydroxyl group to a triflate group, it
was possible to perform a palladium-catalyzed methoxycarbonylation at this
position (Scheme 34).
Scheme 34. Synthesis of nominal gobienine A which include an intramolecular
Blaise reaction

35
2.4.2.1. Minor Enantiomer Recycling
The acyl cyanides (53a,b), required for the synthesis of O-(α-bromoacyl)
cyanohydrins via the minor enantiomer recycling process, were initially
synthesized according to a literature procedure from TMSCN and the
corresponding acyl bromide.154
We showed that it was possible to replace the
expensive TMSCN with CuCN, although purification was sometimes difficult
and resulted in low yields (Scheme 35).
Scheme 35. Synthesis of acyl cyanides 53a and 53b using either TMSCN or CuCN
Initially, we attempted the synthesis of the O-(α-bromoacyl) cyanohydrins
using our dual activation catalytic system consisting of titanium catalyst (S,S)-
30 together with triethylamine. The reaction between acyl cyanide 53a and
benzaldehyde (25) gave only low conversion and low amounts of the desired
O-acylated cyanohydrin 54a (Scheme 36). This is probably due to
incompatibility between the Lewis base and the acyl cyanide.
Scheme 36. Synthesis of 54a using the dual activation system
Since the two-phase system does not require the presence of a Lewis base in
order for the reaction to proceed, it was believed to be more compatible with
the acyl cyanides compared to the dual activation system. As the acyl cyanide
53a contains a stereocenter, and is used as a racemate, four stereoisomers of
the product 54 are formed in the forward step. Remarkably, the biocatalyst
(CALB) was highly selective in hydrolyzing the two stereoisomers with (S)-
configuration at the C-O stereocenter ((S,R)-54a and (S,S)-54a), whereas no
particular selectivity was observed towards the C-Br stereocenter (Figure 18).
In the subsequent cyclization the stereocenter on the acyl group would be lost
and we were therefore satisfied with the observed selectivity.

36
Figure 18. Kinetic resolution of a scalemic mixture of 54a. CALB hydrolyzes (S,R)-
54a and (S,S)-54a selectively.
The minor enantiomer recycling procedure with acyl cyanide 53a and various
aldehydes resulted in high yields and high isomeric ratios (between
(R,R)+(R,S) and (S,R)+(S,S)) of the desired products 54a-g (Scheme 37).
Conditions were optimized for each substrate in terms of addition rate of acyl
cyanide, temperature, and catalyst amount. The amount of titanium catalyst
(S,S)-30 was increased from 5 to 10 mol % in the reaction of 4-
methoxybenzaldehyde since otherwise low yield was obtained. As was seen in
the modelling studies previously discussed, alteration of catalyst composition
can improve the yield of the reaction.36
In most cases the acyl cyanide 53a
needed to be added over 50 hours, since otherwise the system did not reach
steady state ee.
0
10
20
30
40
50
0 5 10 15 20
%
Time (h)
(S,R)-54a
(S,S)-54a
(R,R)-54a
(R,S)-54a

37
Scheme 37. Minor enantiomer recycling in the synthesis of O-(α-bromoacyl)
cyanohydrins 54a-g. Ratios between isomers (R,R)+(R,S) and (S,R)+(S,S) are shown
The product from (E)-butenal (54f) gave only moderate yield as a result of the
presence of relatively high amounts of unprotected cyanohydrin 55f, but the
desired product could still be isolated with high isomeric ratio. When
subjecting pentanal to the catalytic cycle a decrease in isomeric ratio of 54g
was observed after the usual initial increase. We previously observed this
phenomenon with another substrate and it was then attributed to enzyme
inhibition by the acyl cyanide (see section 2.3.3). We therefore performed
CALB-catalyzed kinetic resolutions of 54g with and without two equivalents
of acyl cyanide 53a present in order to investigate if any enzyme inhibition
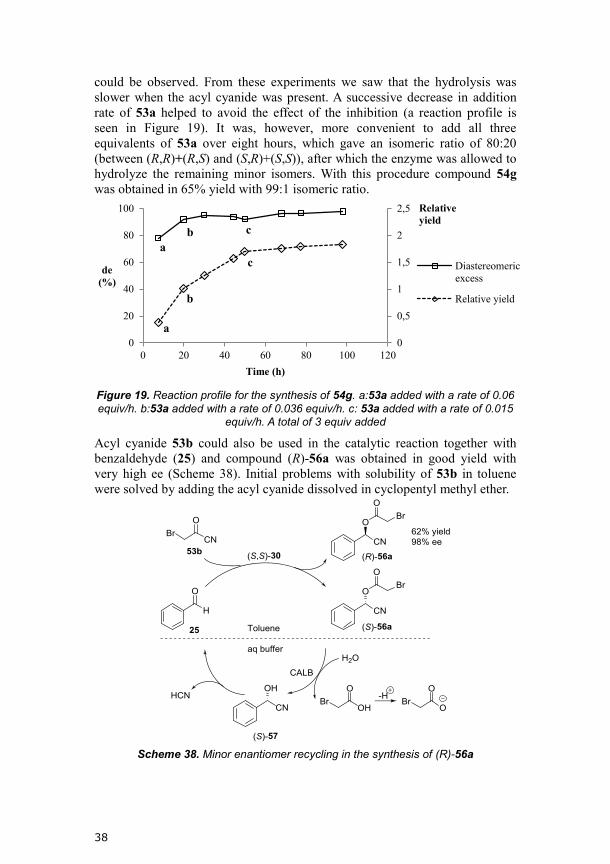
38
could be observed. From these experiments we saw that the hydrolysis was
slower when the acyl cyanide was present. A successive decrease in addition
rate of 53a helped to avoid the effect of the inhibition (a reaction profile is
seen in Figure 19). It was, however, more convenient to add all three
equivalents of 53a over eight hours, which gave an isomeric ratio of 80:20
(between (R,R)+(R,S) and (S,R)+(S,S)), after which the enzyme was allowed to
hydrolyze the remaining minor isomers. With this procedure compound 54g
was obtained in 65% yield with 99:1 isomeric ratio.
Figure 19. Reaction profile for the synthesis of 54g. a:53a added with a rate of 0.06
equiv/h. b:53a added with a rate of 0.036 equiv/h. c: 53a added with a rate of 0.015 equiv/h. A total of 3 equiv added
Acyl cyanide 53b could also be used in the catalytic reaction together with
benzaldehyde (25) and compound (R)-56a was obtained in good yield with
very high ee (Scheme 38). Initial problems with solubility of 53b in toluene
were solved by adding the acyl cyanide dissolved in cyclopentyl methyl ether.
Scheme 38. Minor enantiomer recycling in the synthesis of (R)-56a
0
0,5
1
1,5
2
2,5
0
20
40
60
80
100
0 20 40 60 80 100 120
de
(%)
Time (h)
Diastereomeric
excess
Relative yield
b
a
b
c
c
a
Relative
yield
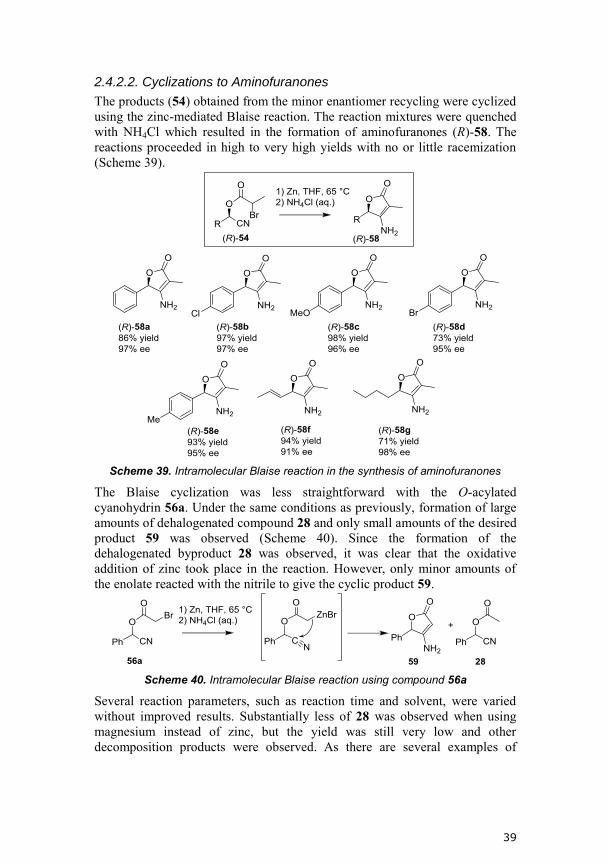
39
2.4.2.2. Cyclizations to Aminofuranones
The products (54) obtained from the minor enantiomer recycling were cyclized
using the zinc-mediated Blaise reaction. The reaction mixtures were quenched
with NH4Cl which resulted in the formation of aminofuranones (R)-58. The
reactions proceeded in high to very high yields with no or little racemization
(Scheme 39).
Scheme 39. Intramolecular Blaise reaction in the synthesis of aminofuranones
The Blaise cyclization was less straightforward with the O-acylated
cyanohydrin 56a. Under the same conditions as previously, formation of large
amounts of dehalogenated compound 28 and only small amounts of the desired
product 59 was observed (Scheme 40). Since the formation of the
dehalogenated byproduct 28 was observed, it was clear that the oxidative
addition of zinc took place in the reaction. However, only minor amounts of
the enolate reacted with the nitrile to give the cyclic product 59.
Scheme 40. Intramolecular Blaise reaction using compound 56a
Several reaction parameters, such as reaction time and solvent, were varied
without improved results. Substantially less of 28 was observed when using
magnesium instead of zinc, but the yield was still very low and other
decomposition products were observed. As there are several examples of

40
primary α-bromo esters reacting in intermolecular Blaise reactions,148
it is
possible that the cyclization with the secondary O-(α-bromoacyl) cyanohydrins
54 is accelerated by steric factors and this is why the primary compound 56a is
less reactive. In addition, the protonated aminofuranone, obtained through acid
mediated lactonization, is known to be highly unstable.150
This instability
might be one contributing factor for the low yields observed.
2.4.3. Functionalization of O-(α-Bromoacyl) Cyanohydrins
O-Acylated cyanohydrins are used as insecticides47,48
and are easily
transformed to N-substituted β-amino alcohols.49,155
Therefore, it is attractive
to have access to simple methods for the functionalization of the acyl chain in
these compounds. The disadvantage with other enantioselective syntheses of
acylated cyanohydrins is that the required acyl cyanides or acylating agents
might not be readily accessible or have unacceptable enantioselectivity.96,112
In
addition, using already functionalized acyl cyanides in the minor enantiomer
recycling could be troublesome as CALB has shown to be sensitive to
sterically demanding groups at the acyl part.69
A highly divergent strategy is
instead to use α-bromoacyl cyanides which give products that have the
possibility to react with different nucleophiles or to take part in cross-coupling
reactions after the enantioselective step (Scheme 32, routes c and d). In this
way, a variety of enantioenriched O-acetylated cyanohydrins are made
available from one common intermediate and only one single asymmetric
transformation is required.
2.4.3.1. Synthesis of Solabegron
To demonstrate the synthetic potential of O-(α-bromoacyl) cyanohydrins we
decided to synthesize the β3-adrenergic receptor agonist solabegron (17).51,52
We envisioned the synthesis to start with the preparation of a O-(α-bromoacyl)
cyanohydrin via minor enantiomer recycling, followed by substitution with an
aniline derivative and reduction of the nitrile to obtain the N-substituted β-
amino alcohol (Scheme 41). This would be the first synthesis of solabegron
that proceeded through an enantioenriched cyanohydrin. In previous syntheses
the chiral center originated either from (R)-3-chlorostyrene oxide51,156
or (R)-3-
chloromandelic acid,52,157,158
both of which are commercially available.
Scheme 41 Retrosynthetic analysis of solabegron

41
To access the desired final compound, the bromine in the acylated cyanohydrin
needed to be substituted by an aniline derivative. When treating enantiopure
O-(α-bromoacyl) cyanohydrin (R)-56a with aniline, together with DIPEA as
base, depletion of product ee was observed. Fortunately we discovered that
aniline, which is less basic, could act as a base in the reaction. Thus, by the use
of an excess of anilines containing both electron-donating and electron-
withdrawing groups the products (R)-60a-c could be isolated in high yields
without any racemization (Scheme 42).
Scheme 42. Substitutions with aniline derivatives
Also when the secondary amines dibenzylamine and N-benzylmethylamine
were used the substituted products were obtained with good yields without any
racemization (72% yield 98% ee and 71% yield >99% ee respectively). The
use of the aliphatic dietylamine resulted in product with a decreased ee. The
primary amine propylamine did not give the desired substitution product.
Instead, the amine attacked the ester to give the unprotected cyanohydrin
which decomposed to aldehyde, which in turn reacted with a second equivalent
of amine to give imine 61. Attack on this species by HCN gave compound 62
(Scheme 43).
Scheme 43. Substitution using propylamine
The next step in the synthesis of the desired N-substituted β-amino alcohols
was the reduction of the nitrile group followed by intramolecular acyl transfer
to the formed amine. Under the previously optimized conditions for this
transformation, nickel on alumina was used as catalyst and the reaction was
performed in dioxane at 120 °C under 20 bars of hydrogen.49
Hanefeld and co-
workers applied these conditions to enantioenriched O-acetylated (S)-
mandelonitrile (28) and obtained the reduced product in 45% yield, but they

42
observed a partial racemization of the product (from 95 to 75% ee). In our
studies, we used the more readily available Raney nickel which was reported
to give similar yields as nickel on alumina. With this catalyst it was possible to
perform the reaction at a substantially lower temperature of 80 °C under 20
bars of hydrogen, and we could reduce substrates (R)-60a-b to amido alcohols
(R)-63a-b with practically no racemization, although with low yields (Scheme
44). The low yields can probably be attributed to cleavage of the benzylic C-O
bond. Reduction of 60c was also possible, but we were not able to isolate the
pure product.
Scheme 44. Catalytic hydrogenation to amido alcohols (R)-63a-b
Direct reduction of the O-(α-bromoacyl) cyanohydrins 54 or 56 would be a
more divergent strategy as only one reduction would be required. We were
unfortunately not able to obtain the desired product using the catalytic
hydrogenation. After several attempts, only reduction of the bromide was
observed (Scheme 45). Several hydride reduction agents were also attempted
for this transformation, but we could not obtain the desired products.
Scheme 45. Failed reductions of compound 54 and 56
The synthesis of solabegron was initiated with the minor enantiomer recycling
using 3-chlorobenzaldehyde (64) and acyl cyanide 53b. Unfortunately, we
observed once again a decrease in ee during addition of the acyl cyanide (see
sections 2.3.3 and 2.4.1.2). The enzymatic kinetic resolution of rac-56b
(Scheme 46) in the presence of two equivalents of acyl cyanide 53b was
extremely slow compared to the resolution without acyl cyanide added (Figure
20 and Figure 21), which indicated that CALB was inhibited by 53b.
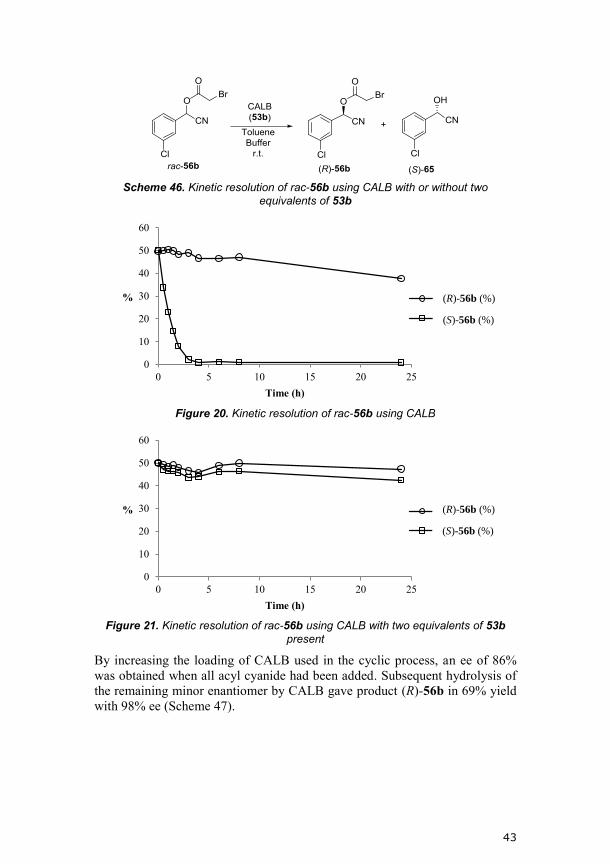
43
Scheme 46. Kinetic resolution of rac-56b using CALB with or without two
equivalents of 53b
Figure 20. Kinetic resolution of rac-56b using CALB
Figure 21. Kinetic resolution of rac-56b using CALB with two equivalents of 53b
present
By increasing the loading of CALB used in the cyclic process, an ee of 86%
was obtained when all acyl cyanide had been added. Subsequent hydrolysis of
the remaining minor enantiomer by CALB gave product (R)-56b in 69% yield
with 98% ee (Scheme 47).
0
10
20
30
40
50
60
0 5 10 15 20 25
%
Time (h)
(R)-60b (%)
(S)-60b (%)
0
10
20
30
40
50
60
0 5 10 15 20 25
%
Time (h)
(R)-60b (%)
(S)-60b (%)
(R)-56b (%)
(S)-56b (%)
(R)-56b (%)
(S)-56b (%)

44
Scheme 47. Minor enantiomer recycling in the synthesis of compound (R)-56b
Aniline derivative 69 could be synthesized in a one-pot two-step procedure in
which Pd/C catalyzed both the initial Suzuki coupling between 66 and 67 and
the subsequent reduction of the nitro group in 68 (Scheme 48).159
The
substitution employing (R)-56b, obtained in the minor enantiomer step,
proceeded in high yield to give (R)-70 with practically no racemization. Three
equivalents of the aniline derivative 69 were required in order to obtain
satisfactory yields; fortunately it was possible to recover most of the excess
amine during the purification. In the catalytic hydrogenation we could use our
conditions to obtain the reduced product (R)-71 with only a slight decrease in
enantiomeric purity, although with low yield. Next, the amide group was to be
reduced to a secondary amine without simultaneous reduction of the ester
group. This transformation was achieved without depletion of the ee by
employing two equivalents of borane dimethylsulfide complex;160
reduction of
the ester was observed if more of the reducing agent was used. Finally,
solabegron (17) was obtained by hydrolysis of the ester group in (R)-72 under
basic conditions, followed by protonation to give the carboxylic acid.52

45
Scheme 48. Synthesis of solabegron
2.4.3.2. C(sp3)-C(sp
2) Cross-Coupling Between O-(α-Bromoacyl)
Cyanohydrins and Boronic Acids
α-Halocarbonyl compounds have been extensively used in various cross-
couplings. Hartwig and co-workers generated zinc enolates from α-
bromocarbonyl compounds which successfully coupled with aryl bromides
under palladium catalysis (Scheme 49).161-163
This method successfully
employed a range of different aryl bromides in the coupling with esters and
amides to give the desired products in high isolated yields.

46
Scheme 49. Hartwig’s cross-coupling which employed zinc enolates
We expected the O-(α-bromoacyl) cyanohydrins 56 to be suitable substrates
for this transformation since they readily form the corresponding zinc enolates,
and only to a low extent react with the nitrile group. Unfortunately, the zinc
enolate derived from compound 56a did not yield the desired product in the
reaction with bromobenzene, but instead reduced compound 28 was the major
product (Scheme 50).
Scheme 50 Attempts employing compound 56a in cross-coupling via the zinc
enolate
Fu and co-workers developed an asymmetric Hiyama cross-coupling between
racemic secondary α-bromo esters and aryl silanes.164
When employing O-(α-
bromoacyl) cyanohydrin 54a in this system we could not detect the desired
product (Scheme 51).
Scheme 51. Attempts to utilize Fu’s asymmetric Hiyama cross-coupling with
compound 54a
The cross-coupling between an α-halocarbonyl compound and an organoboron
derivative was first reported by Suzuki and co-workers, and employed a
system consisting of Pd(PPh)4 together with Tl2CO3.165
Later Gooβen further
developed this transformation and showed that several boronic acids could be
coupled with simple α-bromo esters and amides in good yields.166,167
Several
modifications of reaction conditions and possible substrates that can be used
have been described.168-175
Therefore, this type of cross-coupling appeared to
be a promising alternative for the O-(α-bromoacyl) cyanohydrins.

47
The O-(α-bromoacyl) cyanohydrins contain an ester bond that is labile towards
hydrolysis and an acidic hydrogen, and therefore a Suzuki-type cross-coupling
with these substrates requires mild reaction conditions. Several natural
products were recently synthesized which included α-arylation of esters with
an adjacent stereocenter,176,177
but so far relatively simple substrates lacking
racemization-prone stereocenters have been employed in these types of cross-
couplings. This reaction is usually performed in THF or in toluene with
K2CO3, Cs2CO3, K3PO4, or KF as base and either a palladium or nickel
catalyst. We decided to use KF as a base as we assumed that metal carbonates
and phosphates would be too basic for our substrates. We started with the
cross-coupling between O-(α-bromoacyl) cyanohydrin 56a and phenylboronic
acid employing KF, Pd(OAc)2, and P(o-Tol)3 in THF.166
Unfortunately, we
detected only starting material and degradation to aldehyde and unprotected
cyanohydrin, and heating to 60 °C increased the level of degradation. In
contrast, replacing THF with toluene resulted in full conversion of 56a within
30 minutes, and only minor amounts of the protodehalogenated compound 28
could be detected (<10 %) (Scheme 52). It was essential to heat the reaction
mixture; at room temperature the reaction required approximately two days to
reach full conversion. Without phosphine ligand present, practically no
reaction occurred.
Scheme 52. Cross-coupling between 56a and phenylboronic acid
We investigated the substrate scope of the reaction using enantiopure O-(α-
bromoacyl) cyanohydrin (R)-56a in order to see if any racemization occurred
under the above conditions. Aromatic boronic acids substituted in 2-, 3-, and 4-
position with both electron-withdrawing and electron-donating groups yielded
the desired products (R)-73a-i with no or minor loss of the initial ee and with
good to high yields (Scheme 53). The O-(α-bromoacyl) cyanohydrin 56b,
obtained in the synthesis of solabegron (see section 2.4.2.1), could also be used
in the cross-coupling with phenylboronic acid with good results.
The majority of the reported cross-couplings between α-halocarbonyl
compounds and organoboron derivatives utilize several hours of reaction time.
In contrast, most of our substrates reached full conversion within 30–90
minutes. However, 3,4-dimethoxyphenylboronic acid reacted slowly and in the
initial part of the reaction the mixture turned into a thick suspension, but after
24 hours at 60 °C the desired product (R)-73g could be isolated in acceptable
yield (66%). Boronic acids containing electron-withdrawing groups reacted in
some cases sluggishly under our conditions. For example, 4-

48
ethoxycarbonylphenylboronic acid gave very low conversion to product (R)-
73h even after 20 hours at 60 °C. When we replaced Pd(OAc)2 with
Pd2(dba)3·CHCl3 we could achieve full conversion of (R)-56a after 23 hours at
60 °C and isolate (R)-73h in high yield. 4-Cyanophenylboronic acid was even
more unreactive and no product formation could be detected with Pd(OAc)2 as
a catalyst, but with Pd2(dba)3·CHCl3 we could after 24 hours at 100 °C observe
a ratio of 1:1.1 between starting material and product (according to 1H NMR).
In the cross-coupling between α-bromoesters and boronic acids, Gooβen
observed that when using Pd(OAc)2 a larger amount of biaryl product was
obtained compared to when Pd2(dba)3 was used. This was suggested to be
because the boronic acid initially reduces PdII to Pd
0;166
a reason for the slow
reaction with certain boronic acids in our system might be that the reduction of
Pd(OAc)2 is inefficient.
Scheme 53. Substrate scope for the cross-coupling reaction
Unfortunately, we were not able to obtain any product when using our
conditions with the O-(α-bromoacyl) cyanohydrin 54a which contains a
secondary bromine. Other solvents and ligands were screened but the desired
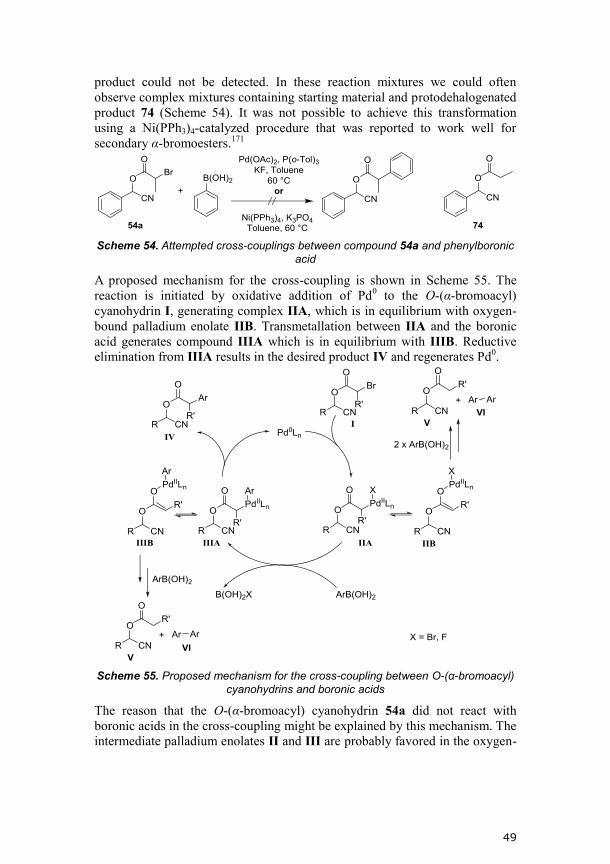
49
product could not be detected. In these reaction mixtures we could often
observe complex mixtures containing starting material and protodehalogenated
product 74 (Scheme 54). It was not possible to achieve this transformation
using a Ni(PPh3)4-catalyzed procedure that was reported to work well for
secondary α-bromoesters.171
Scheme 54. Attempted cross-couplings between compound 54a and phenylboronic
acid
A proposed mechanism for the cross-coupling is shown in Scheme 55. The
reaction is initiated by oxidative addition of Pd0 to the O-(α-bromoacyl)
cyanohydrin I, generating complex IIA, which is in equilibrium with oxygen-
bound palladium enolate IIB. Transmetallation between IIA and the boronic
acid generates compound IIIA which is in equilibrium with IIIB. Reductive
elimination from IIIA results in the desired product IV and regenerates Pd0.
Scheme 55. Proposed mechanism for the cross-coupling between O-(α-bromoacyl)
cyanohydrins and boronic acids
The reason that the O-(α-bromoacyl) cyanohydrin 54a did not react with
boronic acids in the cross-coupling might be explained by this mechanism. The
intermediate palladium enolates II and III are probably favored in the oxygen-

50
bound forms IIB and IIIB when R' ≠ H, possibly because of steric
interactions. Transmetallation followed by reductive elimination gives
protodehalogenated product V and homocoupled product VI. This hypothesis
is based on the fact that α-halocarbonyl compounds previously have been used
to promote the palladium-catalyzed homocoupling of boronic acids, and
substitution at the α-position then favored the homocoupling over the cross-
coupling.178
Since the cross-couplings between 56 and most boronic acids tested proceeded
very rapidly, we wanted to investigate if this high reactivity was general for
this type of reactions or if the structure of 56 contributed to the high rate. In
order to investigate if the presence of the nitrile group accelerated the reaction
we prepared compound 75, which contains a methyl group in place of a nitrile,
and subjected this compound to our conditions in the cross-coupling with
phenylboronic acid. In addition, since 75 is more sterically hindered then 56,
we subjected ethyl bromoacetate (77) to the same conditions (Scheme 56). All
reactions were carried out at four times lower concentration than usual (25 mM
substrate) to be able to follow the reaction over a convenient period of time.
We monitored the concentrations of substrates and products in the cross-
coupling reaction by 1H NMR using an internal standard.
Scheme 56. Variation of the structure of the α-halocarbonyl compounds in the
cross-coupling reaction
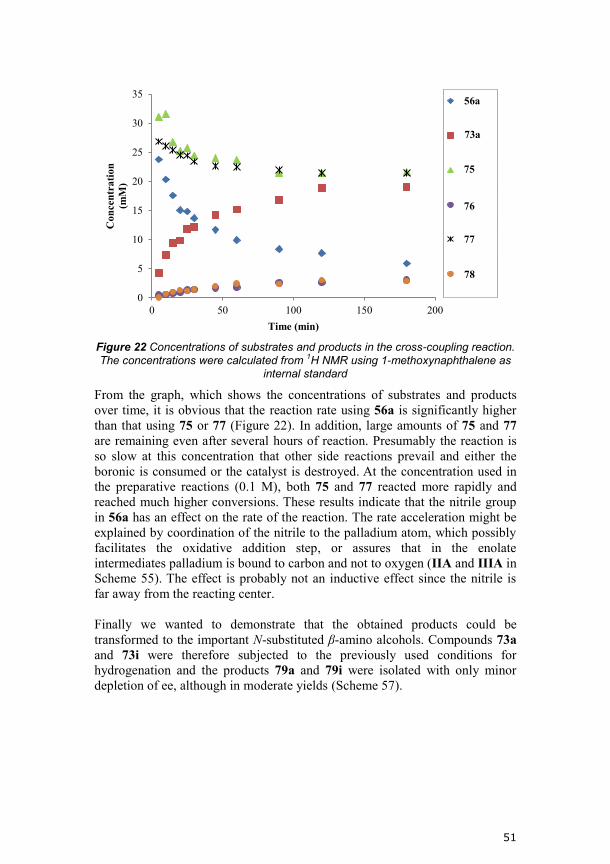
51
Figure 22 Concentrations of substrates and products in the cross-coupling reaction.
The concentrations were calculated from 1H NMR using 1-methoxynaphthalene as internal standard
From the graph, which shows the concentrations of substrates and products
over time, it is obvious that the reaction rate using 56a is significantly higher
than that using 75 or 77 (Figure 22). In addition, large amounts of 75 and 77
are remaining even after several hours of reaction. Presumably the reaction is
so slow at this concentration that other side reactions prevail and either the
boronic is consumed or the catalyst is destroyed. At the concentration used in
the preparative reactions (0.1 M), both 75 and 77 reacted more rapidly and
reached much higher conversions. These results indicate that the nitrile group
in 56a has an effect on the rate of the reaction. The rate acceleration might be
explained by coordination of the nitrile to the palladium atom, which possibly
facilitates the oxidative addition step, or assures that in the enolate
intermediates palladium is bound to carbon and not to oxygen (IIA and IIIA in
Scheme 55). The effect is probably not an inductive effect since the nitrile is
far away from the reacting center.
Finally we wanted to demonstrate that the obtained products could be
transformed to the important N-substituted β-amino alcohols. Compounds 73a
and 73i were therefore subjected to the previously used conditions for
hydrogenation and the products 79a and 79i were isolated with only minor
depletion of ee, although in moderate yields (Scheme 57).
0
5
10
15
20
25
30
35
0 50 100 150 200
Co
ncen
tra
tio
n
(mM
)
Time (min)
[S.M.]
Cyano
[Prod]
Cyano
[S.M.]10
41
[Prod]
1041
[S.M.]
1042
[Prod]
1042
56a
73a
75
76
77
78

52
Scheme 57. Catalytic hydrogenations of compounds 73a and 73i
2.4.4. Conclusions
The minor enantiomer recycling methodology was used to synthesize a variety
of O-(α-bromoacyl) cyanohydrins in high yields and with very high isomeric
ratios. When employing α-bromoacyl cyanides with a secondary bromine,
CALB was highly selective in hydrolyzing two out of the four stereoisomers.
Problems with enzyme inhibition were encountered for some substrates but the
effects could be avoided by using higher enzyme loading and allowing the
enzyme to hydrolyze the remaining minor enantiomer after the addition of acyl
cyanide was finished. The O-(α-bromoacyl) cyanohydrins were not readily
available with the dual activation system. This demonstrates that the minor
enantiomer recycling methodology can be used to access compounds which
are not otherwise easily accessible.
The O-(α-bromoacyl) cyanohydrins proved to be highly useful as synthetic
intermediates. Cyclization of these compounds, using an intramolecular Blaise
reaction, gave a range of aminofuranones in high yields and with very high ee.
The bromide in the synthesized cyanohydrins was efficiently substituted by
different amines. Subsequent reduction and intramolecular acyl transfer gave
access to N-substituted β-amino alcohols. The usefulness of this methodology
was demonstrated with the enantioselctive synthesis of the β3-adrenergic
receptor agonist solabegron.
In addition, the O-(α-bromoacyl) cyanohydrins could participate in a
palladium-catalyzed cross-coupling with a range of boronic acids to give
products in high yields, practically without any racemization. Certain boronic
acids reacted very sluggishly; replacement of the source of palladium
increased the reactivity.

53
3. Enantioselective Acylphosphonylation.
(Paper IV)
3.1. Introduction
Derivatives of α-hydroxy phosphonic acids show biological activity for a wide
range of targets. The absolute configuration of the compounds is important for
their biological activity, and several strategies for the asymmetric synthesis of
α-hydroxy phosphonic acid derivatives have therefore been developed.179,180
The Pudovik reaction,181
which is the base-catalyzed hydrophosphonylation of
aldehydes by dialkylphosphites, has been widely used in the synthesis of both
racemic and enantioenriched hydroxyphosphonates (Scheme 58).179
Scheme 58. The Pudovik reaction
The syntheses of acetoxyphosphonates have received much less attention
despite the interesting biological properties which are shown by these types of
compounds.182,183
Racemic acetoxyphosphonates have for example been
synthesized by the in situ acetylation of hydroxyphosphonates obtained from
the MgO- or K2CO3-catalyzed addition of diethylphosphite to aldehydes.184,185
Enzymatic kinetic resolutions have been used to produce both enantioenriched
hydroxyphosphonates and acetoxyphosphonates.186
For example,
Hammerschmidt and co-workers resolved racemic acetoxyphosphonates
derived from benzaldehyde using the lipase from Rhizopus oryzae (F-AP
15).187
The enantioselectivity was claimed to be very high (E > 100), although
another group later determined the enatioselectivity factor to be merely 32 for
the same kinetic resolution.188
CALB proved to be highly enantioselective in
the acetylation of hydroxyphosphonates derived from short-chain aliphatic
aldehydes.189
Since the phosphonic group will be the larger substituent
according to Kazlauskas’ rules, the size of the group derived from the aldehyde
is highly limited (Scheme 59).186
Thus, phenyl and propyl groups were too
large to fit in the medium-size pocket.
Scheme 59. Kinetic resolution of hydroxyphosphonates by CALB

54
The high selectivity of CALB for these types of substrates was utilized in the
dynamic kinetic resolution (DKR) of racemic hydroxyphosphonates.190
Essentially enantiopure acetoxyphosphonates were obtained in high yields, but
the reaction was only investigated for substrates obtained from short aliphatic
aldehydes (Scheme 60).
Scheme 60. Dynamic kinetic resolution of hydroxyphosphonates
As previously discussed (section 1.1.2), Spilling and co-workers used a
sequential process which encompassed a lipase-catalyzed hydrolysis of the
minor enantiomer of the acetoxyphosphonates obtained from asymmetric
addition of dimethylphosphite to aldehydes.23
An alternative strategy to access acetoxyphosphonates, analogous to that used
for acylcyanations of aldehydes, would be to add acylphosphonates to
aldehydes (Scheme 61). This would be a transformation with perfect atom
economy and would avoid the use of auxiliary acylating agents.
Scheme 61. Analogy between acylcyanation and acylphosphonylation
This approach has previously been used in the samarium-catalyzed synthesis
of racemic acetoxyphosphonates, but the transformation was limited to
aromatic acylphosphonates.191
The asymmetric synthesis of
acetoxyphosphonates via addition of acylphosphonates to aldehydes had not
previously been reported.
3.2. Dual Activation in the Synthesis of Acetoxyphosphonates
In the initial attempts we used the phosphite 80192
in the reaction with
aldehydes as this type of compounds are known to form acetoxyphosphonates
even without the presence of catalyst (Scheme 62).193
However, the reaction
proved to be sluggish and was not accelerated by Lewis acid catalysis using
titanium complex 30.

55
Scheme 62. Attempts of using phosphite 80 in the synthesis of
acetoxyphosphonates
Therefore we continued the investigation with acylphosphonates in the
reaction with aldehydes catalyzed by a combination of Lewis acid and Lewis
base. When treated with nucleophiles, acylphosphonates behave as activated
carboxylic acid derivatives and eliminate a dialkylphosphoryl group.194
Primary and secondary amines are known to react with acylphosphonates to
form amides and phosphites.195
In addition, DBU has been shown to catalyze
the acylation of alcohols with acylphosphonates; aliphatic acylphosphonates
required one equivalent of DBU in order to reach acceptable yields.196
For the reaction between an aldehyde and an acylphosphonate to be initiated,
the carbon-phosphorus bond in the acylphosphonate needs to be cleaved by an
amine, resulting in a phosphite and an acylated amine. Activation of the
aldehyde by the Lewis acid would allow the phosphite to add to the carbonyl
carbon. Acylation of the resulting phosphonate would give the desired
acetoxyphosphonate and regenerate the catalyst.
Scheme 63. Acylphosphonylation of benzaldehyde
In the optimization study we employed acylphosphonate 81 together with
benzaldehyde (25) (Scheme 63). Using Et3N, DABCO, or DMAP without
Lewis acid, the formation of a tetrahedral intermediate between the base and
the acylphosphonate was observed,197
but no desired product 82a. With DBU,
which is more basic, formation of the enolate of acetylphosphonate was also
observed.198
Only trace amounts of 82a were detected when using the system
consisting of titanium catalyst 30 and a Lewis base, a catalytic system which
was highly successful in the acylcyanation of aldehydes.110,112
The N-acylated form of 1H-1,2,3-benzotriazole is known to be an efficient
acylating agent.199
The unprotected hydroxyphosphonate 83 was indeed
acylated with full conversion when treated with this acylating agent in the
presence of DBU; in the absence of base the reaction was very slow (pKa of
1H-benzotriazole is ca 8200
). Employing a catalyst system consisting of
titanium catalyst (S,S)-30, DBU, and 1H-benzotriazole (85) gave only

56
formation of byproduct 84.201
Slow addition of 81 helped to suppress the
formation of this byproduct and the desired product 82a was formed, although
with significant amounts of 84. Replacement of (S,S)-30 with the aluminum
complex 86202
as Lewis acid gave only trace amounts of product. In contrast,
the aluminum complex 87a derived from a tridentate L-valinol-based Schiff
base resulted in a highly improved result. This catalyst has previously shown
to give highly enantioenriched products in high yields in the
hydrophosphonylation of aldehydes.203
High to very high yields of
acetoxyphosphonate 82a were obtained by optimizing the amounts of 1H-
benzotriazole and DBU (Table 3). It was obvious from this optimization study
that the presence of 1H-benzotriazole was essential for the reaction as
otherwise low yields were obtained even when 50 mol % DBU was used
(entries 1–2). Higher loading of 1H-benzotriazole gave higher yield but with
slightly lower enantioselectivity (entry 5). In contrast, a larger amount of DBU
resulted in reduced yield, possibly because of enolate formation (entries 6–7).
The best enantioselectivity was observed when 20 mol % of 1H-benzotriazole
was used (entry 4). Replacing DBU with DMAP, DABCO, or DIPEA resulted
in lower yields (entries 8–10). Chiral bases had only minor effects on the
enantioselectivity; reactions in the presence of cinchonidine and cinchonine
gave ee:s of 32 and 28%, respectively.
Table 3. a Optimization of the amounts of 1H-benzotriazole (85) and DBU
entry 85
(mol %)
base
(mol %) 82a:83
(mol ratio)
yield
(%)b ee (%)c
1 0 DBU (15) 1:0.13 37 28 (R)
2 0 DBU (50) 1:0.16 38 18 (R)
3 10 DBU (15) 1:0.13 53 28 (R)
4 20 DBU (15) 1:0.05 79 38 (R)
5 50 DBU (15) 1:0 100 30 (R)
6 20 DBU (30) 1:0 36 32 (R)
7 20 DBU (50) 1:0 32 26(R)
8 20 DMAP (15) 1:0.04 63 34 (R)
9 20 DABCO (15) 1:0.09 61 38 (R)
10 20 DIPEA (15) 1:1.7 16 8 (S) aReaction conditions: benzaldehyde (0.25 mmol), 87a (10 mol %), toluene (1 mL),
room temperature; 81 (3 equiv), diluted to 200 µL with toluene, was added over 20 h
using a syringe pump. bDetermined by 1H NMR with 1-methoxynaphthalene as internal
standard. cDetermined by HPLC using chiral IA column.

57
In order to improve the enantioselectivity, we decided to vary the structure of
the Schiff base ligand (Table 4). Variation of the steric properties of the amino
alcohol part (entries 1–3 and 5) or in the 3-position of the aromatic ring
(entries 4–6) did not lead to any improved result. In addition, changing the
electronic properties of the ligand had no positive effect on the
enantioselectivity (entries 7–8). We therefore continued our investigation with
ligand 87a.
Table 4.a Catalyst optimization
entry catalyst
(10 mol %) 82a:83
(mol ratio)
yield
(%)b ee (%)c
1 87a 1:0.05 79 38 (R)
2 87b 1:0.04 48 2 (R)
3 87c 1:0.25 35 28 (R)
4 87d 1:0.31 38 12 (R)
5 87e 1:0.51 23 4 (S)
6 87f 1:0.28 13 10 (S)
7 87g 1:0 61 20 (R)
8 87h 1:0 46 32 (R) aReaction conditions: benzaldehyde (0.25 mmol), 1H-benzotriazole (20 mol %), DBU
(15 mol %) in toluene (1 mL), room temperature, 81 (3 equiv), diluted to 200 µL with
toluene, was added over 20 h using a syringe pump. bDetermined by 1H NMR with 1-
methoxynaphthalene as internal standard. cDetermined by HPLC using chiral IA
column.
Fewer equivalents of acylphosphonate 81 could be used when the addition rate
was reduced; a quantitative yield was obtained with merely 1.5 equiv, although
under these conditions the enantioselectivity was lower.

58
The enantioselectivity could be improved by performing the reaction at a lower
temperature. At 0 °C the ee of the product was 52% (Table 5), but reducing the
temperature to -10 °C gave lower yield without improvement of the
enantioselectivity. Higher loading of 1H-benzotriazole (50 mol %) was
required at 0 °C in order to maintain a high yield. In addition, catalyst 87a
proved to be insoluble in toluene below room temperature, and DCM had to be
used as solvent. We could observe that the byproduct 84 was formed in lower
amount in DCM than in toluene; instantaneous addition of 81 to the reaction
mixture at room temperature gave 35% yield after 47 hours, compared to the
reaction in toluene which gave merely 5% yield.
Table 5. aAcylphosphonylations at reduced temperature
entry temp
(°C) 85
(mol %)
DBU
(mol %) 82a:83
(mol ratio)
yield
(%)b
ee
(%)c
1 0 20 15 1:07 60 56
2 0 50 20 1:0 100 52 aReaction conditions: benzaldehyde (0.25 mmol) and 87a (10 mol %) in DCM (1 mL).
81 (3 equiv), diluted to 300 µL with DCM, was added over 45 h using a syringe pump. bDetermined by 1H NMR with 1-methoxynaphthalene as internal standard. cDetermined
by HPLC using chiral IA column.
The optimized reaction conditions were applied to aromatic aldehydes
substituted in the 2-, 3-, and 4-positions with both electron-withdrawing and
electron-donating groups, and high isolated yields of the desired products were
obtained in most cases, although with moderate enantioselectivities (Scheme
64). Propanal proved to be less reactive and the reaction had to be performed at
room temperature in order to reach acceptable yields. The
acetoxyphosphonates we obtained were shown to have R absolute
configuration, which is opposite to that obtained in the
hydroxyphosphonylation using the same ligand.203
This indicates that the
structure of the catalyst-substrate complex in the enantioselective step in our
case differs from that in the hydroxyphosphonylation.

59
Scheme 64. Substrate scope of the acylphosphonylation
We also attempted to use some other acylphosphonates in our catalytic system
(Scheme 65). Replacing the acetyl group in 81 with a benzoyl group, as in
compounds 88, gave the desired product in very low yield. Using the more
sterically demanding 89 gave the corresponding acetoxyphosphonate in good
yield, but with very low ee. Use of acylphosphonate 90204
resulted in a
product in low yield without any improvement of the enantioselectivity.
Scheme 65. Other acylphosphonates used in the acylphosphonylation of
benzaldehyde
In our proposed mechanism we believe that the reaction is initiated by the
deprotonation of 1H-benzotriazole (85) by DBU, resulting in an increase in its

60
nucleophilicity.205,206
The deprotonated benzotriazole then attacks
acylphosphonate 81, resulting in phosphite 91 and the acylated benzotriazole
(92) (Scheme 66). The formed phosphite is probably coordinated to the
aluminum center, as was proposed in the hydrophosphonylation with the same
catalyst.207
Phosphite 91 attacks the Lewis acid-activated aldehyde, resulting in
hydroxyphosphonate 93, which subsequently is acylated by 92 to obtain final
product 82. Phosphite 91 can alternatively attack an unreacted
acylphosphonate 81 to give byproduct 84. The cleavage of the
acylphosphonate by benzotriazole is supported by the presence of acetylated
benzotriazole 92 according to NMR of the crude reaction mixture.
Scheme 66. Proposed mechanism for the acylphosphonylation
In order to determine whether the acyl- and the phosphite groups in the product
are derived from the same molecule of acylphosphonate, we performed an
experiment which employed the two acylphosphonates 90 and 94.204
With one
equivalent of benzaldehyde (25) and 0.5 equivalents each of the
acylphosphonates we detected the compounds 82a and 95 by GC-MS (Scheme
67). Therefore we concluded that the acylation does not have to take place
within the same complex as the P-C bond formation.
Scheme 67. Acylphosphonylation with acylphosphonates 90 and 94
Feng and co-workers observed a strong positive nonlinear effect with catalyst
87a in the hydrophosphonylation of aldehydes, which indicated the
involvement of a dimeric species.203
Calculations showed that the reaction with

61
dimeric catalyst was thermodynamically and kinetically more favorable than
the reaction with monomeric catalyst.207
In addition, the same ligand in
complex with Et2AlBr was shown by X-ray crystallography to have a dimeric
structure.208
To gain insight into the nature of the catalyst in our system we investigated the
correlation between ligand ee (of catalyst 87a) and product (82a) ee. In
contrast to what Feng and co-workers observed,203
our reaction displayed a
weak negative nonlinear effect (Figure 23). A positive nonlinear effect might
be an indication that a heterochiral dimer is formed which is less reactive and
also more stable than the homochiral dimer. In this way, some of the racemic
catalyst will be removed from the catalytic cycle and more enantioenriched
catalyst will take part in the reaction. A negative nonlinear effect might
indicate that the heterochiral dimer is the more reactive species. However, it
cannot be definitely concluded whether our system involves a dimeric species
or not; a negative nonlinear effect might arise for other reasons such as the
competition with a non-catalyzed background reaction.209
Figure 23. Correlation between product ee and ligand ee
0
10
20
30
40
50
60
0 20 40 60 80 100
ee product
(%)
ee ligand
(%)

62
3.3. Minor Enantiomer Recycling
We anticipated that acetoxyphosphonates with higher enantiomeric purity
would be accessible by a minor enantiomer recycling procedure analogous to
that applied in the synthesis of acylated cyanohydrins (Scheme 68).
Scheme 68. Envisioned synthesis of acetoxyphosphonates via minor enantiomer
recycling
For a minor enantiomer recycling process to be successful, the forward
reaction needs to take place in the two-phase system; the aqueous phase is
required for the formation of carboxylate ions, which supply the
thermodynamic driving force for the system. Additionally, a catalyst which
hydrolyzes the minor enantiomer obtained in the forward reaction is essential.
Finally, the hydroxyphosphonate formed in the reverse step needs to
spontaneously eliminate a dialkylphosphite and give back the starting
aldehyde.
Initially an experiment was performed to investigate if the
hydroxyphosphonate formation is reversible. The hydroxyphosphonate 83 was
stirred together with one equivalent of p-tolualdehyde (96) and one equivalent
of DBU in THF; formation of benzaldehyde (25) was indeed observed, thus
demonstrating that the reaction is reversible (Scheme 69).
Scheme 69. Reversibility experiment
Unfortunately the forward reaction in the two-phase system resulted in low
yields of the desired acetoxyphosphonates. However, we discovered that
hydroxyphosphonate 83 was almost completely acetylated in the two-phase
system when in the presence of acetylated benzotriazole 92210
and DBU.
Therefore we attempted the reaction between diethyl phosphite (98) and
benzaldehyde (25) in the presence of acetylated benzotriazole 92 catalyzed by
the aluminum catalyst 87a and DBU. This resulted in low yield of the product

63
together with hydroxyphosphonate 83 (Scheme 70). Using acylphosphonate 81
in place of diethyl phosphite (98) in the two-phase system gave no improved
yield.
Scheme 70. Forward reaction in the two-phase system
For the reverse step we investigated several different enzymes for the kinetic
resolution of acetoxyphosphonates. CALB has been reported to catalyze the
acetylation of racemic hydroxyphosphonates derived from short-chain
aldehydes with high selectivity.189
In the kinetic resolution of racemic
acetoxyphosphonate 99190
using CALB or lipase from Pseudomonas cepacia,
we discovered that the substrate seemed to partly dissolve in the aqueous phase
since both enantiomers of 99 were rapidly consumed. No enantioselective
hydrolysis was observed by CALB when the organic phase was toluene, but
with MTBE as solvent we could observe some selective hydrolysis, although
still large amounts of 99 dissolved in the aqueous phase (Scheme 71). Lipase
from Pseudomonas cepacia was also slightly enantioselective in toluene.
Scheme 71. Kinetic resolution of rac-99
The kinetic resolution of aromatic acetoxyphosphonates was investigated with
several different enzymes without success. For example, the enantioselective
hydrolysis of racemic 101 or 82a with Rhizopus oryzae (F-AP 15)187
was not
successful in a two-phase system consisting of toluene and 2 M pH 7
phosphate buffer (Scheme 72).
Scheme 72. Kinetic resolution of racemic 101 and 82a

64
3.4. Conclusions
A one-step, direct addition of acylphosphonates to aldehydes in the synthesis
of enantioenriched acetoxyphosphonates was developed. The system utilized a
combination of Lewis base, Lewis acid, and Brønstedt base in a cooperative
catalytic activation. A range of aldehydes was used and gave products in most
cases in high yields. The enantioselectivity of the reaction was however only
moderate. Several derivatives of a Schiff-based ligand were synthesized and
assessed in the catalytic reaction, but without any improvement of the
enantioselectivity.
An investigation aimed at synthesizing acetoxyphosphonates via a minor
enantiomer recycling procedure was also initiated. Several difficulties were
however encountered. The forward reaction in the two-phase system was
inefficient and only low yields of the product were obtained. In addition, we
did not succeed in finding an enzyme to catalyze the reverse reaction under our
conditions.

65
4. Concluding Remarks
In this thesis, asymmetric syntheses of enantioenriched cyanohydrins and
acetoxyphosphonates have been investigated. Problems that arise when truly
enantiopure products are desired have been highlighted, and large focus has
been on the development and application of the minor enantiomer recycling
methodology. In the recycling procedure, a titanium salen dimer catalyzes the
reaction between an aldehyde and an acylcyanide to give an O-acylated
cyanohydrin, and a lipase selectively hydrolyzes the unwanted stereoisomer to
the unprotected cyanohydrin, which is in equilibrium with the aldehyde. In
several cases the enantioselectivity of the cyclic procedure was superior to that
of other available methods. The benefit of using minor enantiomer recycling
was obvious in the synthesis of the cyanohydrin that is a precursor to (S)-
propranolol. In cases where the minor enantiomer recycling process failed to
give product with sufficient enantiopurity, the remaining unwanted
stereoisomer could be subjected to a kinetic resolution with the lipase in the
same reaction mixture.
When the minor enantiomer recycling gave unsatisfactory results the same
enantioselective catalysts were used in a sequential process. The O-acylated
cyanohydrins were first obtained using a combination of the titanium salen
dimer and a Lewis base at low temperature, and subsequent enzymatic kinetic
resolution yielded practically enantiopure products.
It was demonstrated that the various cyanohydrins produced were highly
useful as synthetic intermediates. O-(α-Bromoacyl) cyanohydrins proved to be
especially versatile and could be further transformed to several biologically
active compounds. For example, a novel synthesis of the highly
enantioenriched β3-adrenergic receptor agonist solabegron was developed.
In addition, the first enantioselective acylphosphonylation of aldehydes was
developed. In analogy to the synthesis of O-acylated cyanohydrins, a
combination of a chiral Lewis acid and a Lewis base catalyzed the addition of
acylphosphonates to aldehydes. The system developed gave access to a variety
of enantioenriched acetoxyphosphonates that were obtained in high yields. The
enantioselectivity of this reaction was however only moderate. More work
concerning catalyst design and investigation of the reaction mechanism might
be necessary to improve the results. We did not succeed to find conditions
where we could use the minor enantiomer recycling in the synthesis of the
acetoxyphosphonates; the main problem was finding an efficient enzyme to
catalyze the reverse reaction. Further optimization of reaction conditions or
enzyme engineering might facilitate this step.

66
In conclusion, the work presented in this thesis provides chemists with new
tools for the asymmetric synthesis of several important types of compounds.
Since the biological activity of stereoisomers often differs, the development of
efficient syntheses of enantiopure compounds will reduce costs and
environmental impact in the manufacturing of pharmaceuticals and
agrochemicals. The continuing efforts to develop synthetic methodologies that
produce enantiopure compounds with high yields are therefore of vital interest.

67
Acknowledgements
Firstly I want to thank my supervisor, Professor Christina Moberg, for
accepting me as a Ph.D. student, always having time for discussions, and
teaching me how to conduct research. Your enthusiasm for chemistry is highly
inspiring.
I also want to thank my co-supervisor, Dr. Krister Zetterberg, for always
having useful advices during group meetings.
My appreciation also goes to Vetenskapsrådet (VR) for funding, and the Aulin
Erdtman foundation for helping me to present my work at conferences.
Thanks to Dr. Peter Dinér, Dr. Anna Laurell Nash, and Dr. Laure Theveau for
reading my thesis and giving me valuable comments.
Thanks to my co-authors: Professor Christina Moberg, Dr. Ye-Qian Wen, Dr.
Khalid Widyan, Berenice Heid, Inanllely Gonzalez, Dr. Anna Laurell Nash,
Björn Dahlgren, Professor Tore Brinck, and Guillermo Monreal Santiago.
Thanks to Professor Karl Hult and Dr. Linda Fransson for many valuable
discussions.
Many thanks to the talented students who have contributed to my research:
Berenice, Inanllely, Robin, Alexis, Tina, Lorraine, and Guillermo.
Thanks to Anna for being a great colleague and friend!
Thanks to all past and present members in the Ki-group and to everybody at
the department of organic chemistry. Special thanks to: Brinton, Juho, Anna,
Jakob, Tessie, Piret, Fredrik, Björn, Lei H, Lei W, Yan, Laure, Brian, Erik, Ye-
Qian, Hui, Youcai, Ende, Khalid, Johan, Rosalba, Antanas, and Quentin.
Thanks to Dr. Zoltán Szabó for help with NMR studies, and thanks to Dr. Ulla
Jacobsson, Henry Challis, and Lena Skowron for help with everything else!
Thanks to Professor Ingemar Kvarnström, Dr. Veronica Sandgren, and Dr.
Marcus Bäck at LiU for introducing me to research in organic chemistry.
Of course I also want to thank all of my friends for all the good times during
the years!
Till sist vill jag tacka Åsa, mina föräldrar och min bror med familj för det stöd
och den uppmuntran ni alltid gett mig.

68
Appendix
The following is a description of my contributions to Publications I to VI, as
requested by KTH:
Paper I: I contributed to the formulation of the research problem, performed
the majority of the experimental work, and wrote part of the manuscript. I
supervised the undergraduate student Berenice Heid.
Paper II: I contributed to the formulation of the research problem and shared
the writing of the manuscript. I performed the experimental work assisted by
undergraduate students Inanllely Gonzalez, Alexis Bordet, and Robin
Lafficher.
Paper III: I contributed to the formulation of the research problem, performed
some of the experimental work, and wrote part of the manuscript. I supervised
the undergraduate student Inanllely Gonzalez.
Paper IV: I contributed to the formulation of the research problem, shared the
experimental work, and wrote part of the manuscript.
Paper V: I contributed to the formulation of the research problem, shared the
experimental work, and wrote the majority of the manuscript. I supervised the
undergraduate student Guillermo Monreal Santiago.
Paper VI: I contributed to the formulation of the research problem, performed
the experimental work, and wrote the majority of the manuscript.

69
References
1 Cossy, J. R. In Comprehensive Chirality; Carreira, E. M., Yamamoto, H., Eds.; Elsevier:
Amsterdam, 2012; Vol. 1, p 1-7.
2 Islam, M.; Mahdi, J.; Bowen, I. Drug Saf. 1997, 17, 149-165.
3 Nguyen, L. A.; He, H.; Pham-Huy, C. Int. J. Biomed. Sci. 2006, 2, 85-100.
4 19th WHO Model List of Essential Medicines, April 2015.
http://www.who.int/medicines/publications/essentialmedicines/en/
5 Barrett, A. M.; Cullum, V. A. Br. J. Pharmacol. 1968, 34, 43-55.
6 Pérez-Fernández, V.; García, M. Á.; Marina, M. L. J. Chromatogr. A 2010, 1217, 968-989.
7 Denard, C. A.; Hartwig, J. F.; Zhao, H. ACS Catal. 2013, 3, 2856-2864.
8 Köhler, V.; Turner, N. J. Chem. Commun. 2015, 51, 450-464.
9 Fransson, L.; Moberg, C. ChemCatChem 2010, 2, 1523-1532.
10 Kagan, H. B.; Fiaud, J. C. In Top. Stereochem.; Eliel, E. L., Wilen, S. H., Eds.; John Wiley
& Sons, Inc.: Hoboken, NJ, USA, 1988, p 249-330.
11 Steinreiber, J.; Faber, K.; Griengl, H. Chem. – Eur. J. 2008, 14, 8060-8072.
12 Faber, K. Chem. – Eur. J. 2001, 7, 5004-5010.
13 Chen, C.-S.; Fujimoto, Y.; Girdaukas, G.; Sih, C. J. J. Am. Chem. Soc. 1982, 104, 7294-
7299.
14 Timms, N.; Daniels, A. D.; Berry, A.; Nelson, A. In Comprehensive Chirality; Carreira, E.
M., Yamamoto, H., Eds.; Elsevier: Amsterdam, 2012; Vol. 7, p 21-45.
15 Patel, R. N. In Comprehensive Chirality; Carreira, E. M., Yamamoto, H., Eds.; Elsevier:
Amsterdam, 2012; Vol. 9, p 288-317.
16 Muñoz Solano, D.; Hoyos, P.; Hernáiz, M. J.; Alcántara, A. R.; Sánchez-Montero, J. M.
Bioresour. Technol. 2012, 115, 196-207.
17 Martinez, C. A.; Hu, S.; Dumond, Y.; Tao, J.; Kelleher, P.; Tully, L. Org. Process Res. Dev.
2008, 12, 392-398.
18 Pellissier, H. Adv. Synth. Catal. 2011, 353, 1613-1666.
19 Fu, G. C. Acc. Chem. Res. 2004, 37, 542-547.
20 Bellemin-Laponnaz, S.; Tweddell, J.; Ruble, J. C.; Breitling, F. M.; Fu, G. C. Chem.
Commun. 2000, 1009-1010.
21 Wang, Y.-F.; Chen, C.-S.; Girdaukas, G.; Sih, C. J. J. Am. Chem. Soc. 1984, 106, 3695-
3696.
22 Edin, M.; Bäckvall, J.-E.; Córdova, A. Tetrahedron Lett. 2004, 45, 7697-7701.
23 Rowe, B. J.; Spilling, C. D. Tetrahedron: Asymmetry 2001, 12, 1701-1708.
24 Pàmies, O.; Bäckvall, J.-E. Chem. Rev. 2003, 103, 3247-3262.
25 Kim, C.; Park, J.; Kim, M.-J. In Comprehensive Chirality; Carreira, E. M., Yamamoto, H.,
Eds.; Elsevier: Amsterdam, 2012; Vol. 7, p 156-180.
26 Verho, O.; Bäckvall, J.-E. J. Am. Chem. Soc. 2015, 137, 3996-4009.
27 Pàmies, O.; Bäckvall, J.-E. J. Org. Chem. 2001, 66, 4022-4025.
28 Laurell Nash, A.; Widyan, K.; Moberg, C. ChemCatChem 2014, 6, 3314-3317.
29 Lente, G. Chirality 2010, 22, 907-913.

70
30 Gruber, C. C.; Lavandera, I.; Faber, K.; Kroutil, W. Adv. Synth. Catal. 2006, 348, 1789-
1805.
31 Voss, C. V.; Gruber, C. C.; Faber, K.; Knaus, T.; Macheroux, P.; Kroutil, W. J. Am. Chem.
Soc. 2008, 130, 13969-13972.
32 Voss, C. V.; Gruber, C. C.; Kroutil, W. Synlett 2010, 991-998.
33 Lackner, A. D.; Samant, A. V.; Toste, F. D. J. Am. Chem. Soc. 2013, 135, 14090-14093.
34 Turner, N. J. Chem. Rev. 2011, 111, 4073-4087.
35 Köhler, V.; Wilson, Y. M.; Dürrenberger, M.; Ghislieri, D.; Churakova, E.; Quinto, T.;
Knörr, L.; Häussinger, D.; Hollmann, F.; Turner, N. J.; Ward, T. R. Nat. Chem. 2013, 5, 93-
99.
36 Fransson, L.; Laurell, A.; Widyan, K.; Wingstrand, E.; Hult, K.; Moberg, C. ChemCatChem
2010, 2, 683-693.
37 Wingstrand, E.; Laurell, A.; Fransson, L.; Hult, K.; Moberg, C. Chem. – Eur. J. 2009, 15,
12107-12113.
38 Effenberger, F. Angew. Chem., Int. Ed. Engl. 1994, 33, 1555-1564.
39 Brunel, J.-M.; Holmes, I. P. Angew. Chem., Int. Ed. 2004, 43, 2752-2778.
40 Chen, F.-X.; Feng, X. Curr. Org. Synth. 2006, 3, 77-97.
41 North, M.; Usanov, D. L.; Young, C. Chem. Rev. 2008, 108, 5146-5226.
42 Wang, W.; Liu, X.; Lin, L.; Feng, X. Eur. J. Org. Chem. 2010, 2010, 4751-4769.
43 Gregory, R. J. H. Chem. Rev. 1999, 99, 3649-3682.
44 North, M. Tetrahedron: Asymmetry 2003, 14, 147-176.
45 Moberg, C.; Wingstrand, E. Synlett 2010, 355-367.
46 North, M.; Parkins, A. W.; Shariff, A. N. Tetrahedron Lett. 2004, 45, 7625-7627.
47 Breckenridge, C. B.; Stevens, J. T. In Principles and Methods of Toxicology; 5th ed.; Hayes,
A. W., Ed.; CRC Press: 2007, p 727-840.
48 Beckmann, M.; Haack, K.-J. Chem. Unserer Zeit 2003, 37, 88-97.
49 Veum, L.; Pereira, S. R. M.; van der Waal, J. C.; Hanefeld, U. Eur. J. Org. Chem. 2006,
1664-1671.
50 Oldenburg, O.; Kribben, A.; Baumgart, D.; Philipp, T.; Erbel, R.; Cohen, M. V. Curr. Opin.
Pharmacol. 2002, 2, 740-747.
51 Donaldson, K. H.; Shearer, B. G.; Uehling, D. E. PCT Patent WO9965877A1, 1999.
52 Uehling, D. E.; Shearer, B. G.; Donaldson, K. H.; Chao, E. Y.; Deaton, D. N.; Adkison, K.
K.; Brown, K. K.; Cariello, N. F.; Faison, W. L.; Lancaster, M. E.; Lin, J.; Hart, R.;
Milliken, T. O.; Paulik, M. A.; Sherman, B. W.; Sugg, E. E.; Cowan, C. J. Med. Chem.
2006, 49, 2758-2771.
53 Sacco, E.; Bientinesi, R.; Tienforti, D.; Racioppi, M.; Gulino, G.; D'Agostino, D.; Vittori,
M.; Bassi, P. Expert Opin. Drug Discovery 2014, 9, 433-448.
54 Ishide, T. Curr. Med. Res. Opin. 2002, 18, 407-413.
55 Ursino, M. G.; Vasina, V.; Raschi, E.; Crema, F.; De Ponti, F. Pharmacol. Res. 2009, 59,
221-234.
56 Igawa, Y.; Michel, M. C. Naunyn-Schmiedeberg's Arch. Pharmacol. 2013, 386, 177-183.
57 Ellsworth, P.; Fantasia, J. Expert Opin. Invest. Drugs 2015, 24, 413-419.
58 Norsikian, S.; Holmes, I.; Lagasse, F.; Kagan, H. B. Tetrahedron Lett. 2002, 43, 5715-5717.

71
59 Holt, J.; Hanefeld, U. Curr. Org. Synth. 2009, 6, 15-37.
60 van der Gen, A.; Brussee, J. In Stereoselective Biocatalysis; Patel, R. N., Ed.; CRC Press:
2000, p 289-320.
61 Winkler, M.; Glieder, A.; Steiner, K. In Comprehensive Chirality; Carreira, E. M.,
Yamamoto, H., Eds.; Elsevier: Amsterdam, 2012; Vol. 7, p 350-371.
62 Dadashipour, M.; Asano, Y. ACS Catal. 2011, 1, 1121-1149.
63 Effenberger, F. In Stereoselective Biocatalysis; Patel, R. N., Ed.; CRC Press: 2000, p 321-
342.
64 Purkarthofer, T.; Pabst, T.; van den Broek, C.; Griengl, H.; Maurer, O.; Skranc, W. Org.
Process Res. Dev. 2006, 10, 618-621.
65 Hanefeld, U.; Li, Y.; Sheldon, R. A.; Maschmeyer, T. Synlett 2000, 1775-1776.
66 Hanefeld, U.; Straathof, A. J. J.; Heijnen, J. J. J. Mol. Catal. B: Enzym. 2001, 11, 213-218.
67 Veum, L.; Kuster, M.; Telalovic, S.; Hanefeld, U.; Maschmeyer, T. Eur. J. Org. Chem.
2002, 1516-1522.
68 Hamberg, A.; Lundgren, S.; Penhoat, M.; Moberg, C.; Hult, K. J. Am. Chem. Soc. 2006,
128, 2234-2235.
69 Hamberg, A.; Lundgren, S.; Wingstrand, E.; Moberg, C.; Hult, K. Chem. – Eur. J. 2007, 13,
4334-4341.
70 Kazlauskas, R. J.; Weissfloch, A. N. E.; Rappaport, A. T.; Cuccia, L. A. J. Org. Chem.
1991, 56, 2656-2665.
71 Rotticci, D.; Hæffner, F.; Orrenius, C.; Norin, T.; Hult, K. J. Mol. Catal. B: Enzym. 1998, 5,
267-272.
72 Anderson, E. M.; Larsson, K. M.; Kirk, O. Biocatal. Biotransform. 1998, 16, 181-204.
73 Schmid, R. D.; Verger, R. Angew. Chem., Int. Ed. 1998, 37, 1608-1633.
74 Berglund, P.; Hult, K. In Stereoselective Biocatalysis; Patel, R. N., Ed.; CRC Press: 2000, p
633-657.
75 Inagaki, M.; Hiratake, J.; Nishioka, T.; Oda, J. J. Am. Chem. Soc. 1991, 113, 9360-9361.
76 Inagaki, M.; Hatanaka, A.; Mimura, M.; Hiratake, J.; Nishioka, T.; Oda, J. Bull. Chem. Soc.
Jpn. 1992, 65, 111-120.
77 Inagaki, M.; Hiratake, J.; Nishioka, T.; Oda, J. J. Org. Chem. 1992, 57, 5643-5649.
78 Kanerva, L. T.; Rahiala, K.; Sundholm, O. Biocatalysis 1994, 10, 169-180.
79 Paizs, C.; Toşa, M.; Majdik, C.; Tähtinen, P.; Irimie, F. D.; Kanerva, L. T. Tetrahedron:
Asymmetry 2003, 14, 619-627.
80 Paizs, C.; Tähtinen, P.; Lundell, K.; Poppe, L.; Irimie, F.-D.; Kanerva, L. T. Tetrahedron:
Asymmetry 2003, 14, 1895-1904.
81 Paizs, C.; Tähtinen, P.; Toşa, M.; Majdik, C.; Irimie, F.-D.; Kanerva, L. T. Tetrahedron
2004, 60, 10533-10540.
82 Veum, L.; Kanerva, L. T.; Halling, P. J.; Maschmeyer, T.; Hanefeld, U. Adv. Synth. Catal.
2005, 347, 1015-1021.
83 Veum, L.; Hanefeld, U. Tetrahedron: Asymmetry 2004, 15, 3707-3709.
84 Veum, L.; Hanefeld, U. Synlett 2005, 2382-2384.
85 Shibasaki, M.; Kanai, M.; Matsunaga, S.; Kumagai, N. Acc. Chem. Res. 2009, 42, 1117-
1127.

72
86 Moberg, C. In Cooperative Catalysis; Peters, R., Ed.; Wiley-VCH Verlag GmbH & Co.
KGaA: 2015, p 35-66.
87 Ma, J.-A.; Cahard, D. Angew. Chem., Int. Ed. 2004, 43, 4566-4583.
88 Kanai, M.; Kato, N.; Ichikawa, E.; Shibasaki, M. Synlett 2005, 1491-1508.
89 Hamashima, Y.; Sawada, D.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 1999, 121, 2641-
2642.
90 Belokon, Y. N.; Caveda-Cepas, S.; Green, B.; Ikonnikov, N. S.; Khrustalev, V. N.;
Larichev, V. S.; Moscalenko, M. A.; North, M.; Orizu, C.; Tararov, V. I.; Tasinazzo, M.;
Timofeeva, G. I.; Yashkina, L. V. J. Am. Chem. Soc. 1999, 121, 3968-3973.
91 Belokon, Y. N.; Green, B.; Ikonnikov, N. S.; North, M.; Tararov, V. I. Tetrahedron Lett.
1999, 40, 8147-8150.
92 Belokon’, Y. N.; Blacker, A. J.; Clutterbuck, L. A.; North, M. Org. Lett. 2003, 5, 4505-
4507.
93 Belokon, Y. N.; Ishibashi, E.; Nomura, H.; North, M. Chem. Commun. 2006, 1775-1777.
94 Belokon, Y. N.; Gutnov, A. V.; Moskalenko, M. A.; Yashkina, L. V.; Lesovoy, D. E.;
Ikonnikov, N. S.; Larichev, V. S.; North, M. Chem. Commun. 2002, 244-245.
95 Belokon, Y. N.; Carta, P.; Gutnov, A. V.; Maleev, V.; Moskalenko, M. A.; Yashkina, L. V.;
Ikonnikov, N. S.; Voskoboev, N. V.; Khrustalev, V. N.; North, M. Helv. Chim. Acta 2002,
85, 3301-3312.
96 Belokon, Y. N.; Carta, P.; North, M. Lett. Org. Chem. 2004, 1, 81-83.
97 Belokon’, Y. N.; Green, B.; Ikonnikov, N. S.; Larichev, V. S.; Lokshin, B. V.; Moscalenko,
M. A.; North, M.; Orizu, C.; Peregudov, A. S.; Timofeeva, G. I. Eur. J. Org. Chem. 2000,
2655-2661.
98 Belokon’, Y. N.; Blacker, A. J.; Carta, P.; Clutterbuck, L. A.; North, M. Tetrahedron 2004,
60, 10433-10447.
99 Zhang, Z.; Wang, Z.; Zhang, R.; Ding, K. Angew. Chem., Int. Ed. 2010, 49, 6746-6750.
100 North, M. Angew. Chem., Int. Ed. 2010, 49, 8079-8081.
101 Belokon, Y. N.; Blacker, A. J.; Clutterbuck, L. A.; Hogg, D.; North, M.; Reeve, C. Eur. J.
Org. Chem. 2006, 4609-4617.
102 Marvel, C. S.; Brace, N. O.; Miller, F. A.; Johnson, A. R. J. Am. Chem. Soc. 1949, 71, 34-
36.
103 Hoffmann, H. M. R.; Ismail, Z. M.; Hollweg, R.; Zein, A. R. Bull. Chem. Soc. Jpn. 1990,
63, 1807-1810.
104 Scholl, M.; Lim, C.-K.; Fu, G. C. J. Org. Chem. 1995, 60, 6229-6231.
105 Okimoto, M.; Chiba, T. Synthesis 1996, 1188-1190.
106 Watahiki, T.; Ohba, S.; Oriyama, T. Org. Lett. 2003, 5, 2679-2681.
107 Baeza, A.; Nájera, C.; Retamosa, M. d. G.; Sansano, J. M. Synthesis 2005, 2787-2797.
108 Zhang, W.; Shi, M. Org. Biomol. Chem. 2006, 4, 1671-1674.
109 Zhang, J.; Du, G.; Xu, Y.; He, L.; Dai, B. Tetrahedron Lett. 2011, 52, 7153-7156.
110 Lundgren, S.; Wingstrand, E.; Penhoat, M.; Moberg, C. J. Am. Chem. Soc. 2005, 127,
11592-11593.
111 Wingstrand, E.; Lundgren, S.; Penhoat, M.; Moberg, C. Pure Appl. Chem. 2006, 78, 409-
414.

73
112 Lundgren, S.; Wingstrand, E.; Moberg, C. Adv. Synth. Catal. 2007, 349, 364-372.
113 Laurell Nash, A., Development and Studies of the Processes Involved in Minor Enantiomer
Recycling, School of Chemical Science and Engineering, KTH Royal Institute of
Technology, 2014.
114 Laurell Nash, A.; Hertzberg, R.; Wen, Y.-Q.; Dahlgren, B.; Brinck, T.; Moberg, C.
Manuscript
115 Baeza, A.; Nájera, C.; Sansano, J. M.; Saá, J. M. Tetrahedron: Asymmetry 2005, 16, 2385-
2389.
116 Baeza, A.; Nájera, C.; Sansano, J. M.; Saá, J. M. Tetrahedron: Asymmetry 2011, 22, 1282-
1291.
117 Casas, J.; Baeza, A.; Sansano, J. M.; Nájera, C.; Saá, J. M. Tetrahedron: Asymmetry 2003,
14, 197-200.
118 Baeza, A.; Casas, J.; Nájera, C.; Sansano, J. M.; Saá, J. M. Eur. J. Org. Chem. 2006, 1949-
1958.
119 Baeza, A.; Casas, J.; Nájera, C.; Sansano, J. M.; Saá, J. M. Angew. Chem., Int. Ed. 2003, 42,
3143-3146.
120 Baeza, A.; Nájera, C.; Sansano, J. M.; Saá, J. M. Chem. – Eur. J. 2005, 11, 3849-3862.
121 Laurell, A.; Moberg, C. Eur. J. Org. Chem. 2011, 3980-3984.
122 Menéndez, E.; Brieva, R.; Rebolledo, F.; Gotor, V. J. Chem. Soc., Chem. Commun. 1995,
989-990.
123 Hernández, L.; Luna, H.; Solís, A.; Vázquez, A. Tetrahedron: Asymmetry 2006, 17, 2813-
2816.
124 Kang, E. J.; Lee, E. Chem. Rev. 2005, 105, 4348-4378.
125 Wolfe, J. P.; Hay, M. B. Tetrahedron 2007, 63, 261-290.
126 Nazabadioko, S.; Pérez, R. J.; Brieva, R.; Gotor, V. Tetrahedron: Asymmetry 1998, 9, 1597-
1604.
127 Monterde, M. I.; Nazabadioko, S.; Rebolledo, F.; Brieva, R.; Gotor, V. Tetrahedron:
Asymmetry 1999, 10, 3449-3455.
128 Monterde, M. I.; Brieva, R.; Gotor, V. Tetrahedron: Asymmetry 2001, 12, 525-528.
129 Effenberger, F.; Gutterer, B.; Ziegler, T.; Eckhardt, E.; Aichholz, R. Liebigs Ann. Chem.
1991, 47-54.
130 Dukes, M.; Smith, L. H. J. Med. Chem. 1971, 14, 326-328.
131 Powell, C. E.; Slater, I. H.; LeCompte, L.; Waddell, J. E. J. Pharmacol. Exp. Ther. 1958,
122, 480-488.
132 Wang, Y.-F.; Chen, S.-T.; Liu, K. K. C.; Wong, C.-H. Tetrahedron Lett. 1989, 30, 1917-
1920.
133 van Almsick, A.; Buddrus, J.; Hönicke-Schmidt, P.; Laumen, K.; Schneider, M. P. J. Chem.
Soc., Chem. Commun. 1989, 1391-1393.
134 North, M.; Omedes-Pujol, M.; Williamson, C. Chem. – Eur. J. 2010, 16, 11367-11375.
135 Mühlhäuser, T., Enantioselective Preparation of O-Acylated Cyanohydrins from
Heteroatom-containing Aldehydes, Diploma work, KTH Royal Institute of Technology,
2013.
136 Lin, G.; Han, S.; Li, Z. Tetrahedron 1999, 55, 3531-3540.

74
137 Matsuo, N.; Ohno, N. Tetrahedron Lett. 1985, 26, 5533-5534.
138 Liu, Y.; Bae, B. H.; Alam, N.; Hong, J.; Sim, C. J.; Lee, C.-O.; Im, K. S.; Jung, J. H. J. Nat.
Prod. 2001, 64, 1301-1304.
139 Duval, R. A.; Poupon, E.; Brandt, U.; Hocquemiller, R. Biochim. Biophys. Acta, Bioenerg.
2005, 1709, 191-194.
140 Csuk, R.; Barthel, A.; Kluge, R.; Ströhl, D. Bioorg. Med. Chem. 2010, 18, 7252-7259.
141 Pérez, M.; Pérez, D. I.; Martínez, A.; Castro, A.; Gómez, G.; Fall, Y. Chem. Commun. 2009,
3252-3254.
142 Carter, N. B.; Nadany, A. E.; Sweeney, J. B. J. Chem. Soc., Perkin Trans. 1 2002, 2324-
2342.
143 Hiyama, T.; Oishi, H.; Suetsugu, Y.; Nishide, K.; Saimoto, H. Bull. Chem. Soc. Jpn. 1987,
60, 2139-2150.
144 Schlessinger, R. H.; Li, Y.-J. J. Am. Chem. Soc. 1996, 118, 3301-3302.
145 Li, Y.-J.; Ho, G.-M.; Chen, P.-Z. Tetrahedron: Asymmetry 2009, 20, 1854-1863.
146 Nishide, K.; Aramata, A.; Kamanaka, T.; Inoue, T.; Node, M. Tetrahedron 1994, 50, 8337-
8346.
147 Kondoh, A.; Arlt, A.; Gabor, B.; Fürstner, A. Chem. – Eur. J. 2013, 19, 7731-7738.
148 Prakash Rao, H. S.; Rafi, S.; Padmavathy, K. Tetrahedron 2008, 64, 8037-8043.
149 Pöchlauer, P.; Riebel, P.; Mayrhofer, H.; Wirth, I. PCT Patent US 6417377, 2002.
150 Syed, J.; Förster, S.; Effenberger, F. Tetrahedron: Asymmetry 1998, 9, 805-815.
151 Effenberger, F.; Syed, J. Tetrahedron: Asymmetry 1998, 9, 817-825.
152 Bühler, H.; Bayer, A.; Effenberger, F. Chem. – Eur. J. 2000, 6, 2564-2571.
153 Ohta, H.; Kimura, Y.; Sugano, Y. Tetrahedron Lett. 1988, 29, 6957-6960.
154 Herrmann, K.; Simchen, G. Synthesis 1979, 204-205.
155 Ikezaki, M.; Umino, N.; Gaino, M.; Aoe, K.; Iwakuma, T.; Oh-Ishi, T. Yakugaku Zasshi
1986, 106, 80-89.
156 Cooke, J. W. B.; Glover, B. N.; Lawrence, R. M.; Sharp, M. J.; Tymoschenko, M. F. PCT
Patent WO2002066418A2, 2002.
157 Zhang, C. PCT Patent WO2010118291A2, 2010.
158 Lawrence, R. M.; Millar, A. PCT Patent WO2001042195A1, 2001.
159 Dear, A. E.; Liu, H. B.; Mayes, P. A.; Perlmutter, P. Org. Biomol. Chem. 2006, 4, 3778-
3784.
160 Imanishi, M.; Itou, S.; Washizuka, K.; Hamashima, H.; Nakajima, Y.; Araki, T.;
Tomishima, Y.; Sakurai, M.; Matsui, S.; Imamura, E.; Ueshima, K.; Yamamoto, T.;
Yamamoto, N.; Ishikawa, H.; Nakano, K.; Unami, N.; Hamada, K.; Matsumura, Y.;
Takamura, F.; Hattori, K. J. Med. Chem. 2008, 51, 4002-4020.
161 Hama, T.; Liu, X.; Culkin, D. A.; Hartwig, J. F. J. Am. Chem. Soc. 2003, 125, 11176-11177.
162 Hama, T.; Culkin, D. A.; Hartwig, J. F. J. Am. Chem. Soc. 2006, 128, 4976-4985.
163 Hama, T.; Ge, S.; Hartwig, J. F. J. Org. Chem. 2013, 78, 8250-8266.
164 Dai, X.; Strotman, N. A.; Fu, G. C. J. Am. Chem. Soc. 2008, 130, 3302-3303.
165 Sato, M.; Miyaura, N.; Suzuki, A. Chem. Lett. 1989, 1405-1408.
166 Gooßen, L. J. Chem. Commun. 2001, 669-670.

75
167 Zimmermann, B.; Dzik, W. I.; Himmler, T.; Goossen, L. J. J. Org. Chem. 2011, 76, 8107-
8112.
168 Liu, X.-x.; Deng, M.-z. Chem. Commun. 2002, 622-623.
169 Lu, T.-Y.; Xue, C.; Luo, F.-T. Tetrahedron Lett. 2003, 44, 1587-1590.
170 Duan, Y.-Z.; Deng, M.-Z. Tetrahedron Lett. 2003, 44, 3423-3426.
171 Liu, C.; He, C.; Shi, W.; Chen, M.; Lei, A. Org. Lett. 2007, 9, 5601-5604.
172 Duan, Y.; Zhang, J.; Yang, J.; Deng, M. Chin. J. Chem. 2009, 27, 179-182.
173 Peng, Z.-Y.; Wang, J.-P.; Cheng, J.; Xie, X.-m.; Zhang, Z. Tetrahedron 2010, 66, 8238-
8241.
174 Lundin, P. M.; Fu, G. C. J. Am. Chem. Soc. 2010, 132, 11027-11029.
175 Molander, G. A.; Traister, K. M.; Barcellos, T. J. Org. Chem. 2013, 78, 4123-4131.
176 De Joarder, D.; Jennings, M. P. Tetrahedron Lett. 2013, 54, 3990-3992.
177 De Joarder, D.; Jennings, M. P. Eur. J. Org. Chem. 2015, 3303-3313.
178 Lei, A.; Zhang, X. Tetrahedron Lett. 2002, 43, 2525-2528.
179 Merino, P.; Marqués-López, E.; Herrera, R. P. Adv. Synth. Catal. 2008, 350, 1195-1208.
180 Kolodiazhnyi, O. I. Tetrahedron: Asymmetry 2005, 16, 3295-3340.
181 Pudovik, A. N.; Konovalova, I. V. Synthesis 1979, 81-96.
182 He, H.-W.; Yuan, J.-L.; Peng, H.; Chen, T.; Shen, P.; Wan, S.-Q.; Li, Y.; Tan, H.-L.; He,
Y.-H.; He, J.-B.; Li, Y. J. Agric. Food Chem. 2011, 59, 4801-4813.
183 Kategaonkar, A. H.; Pokalwar, R. U.; Sonar, S. S.; Gawali, V. U.; Shingate, B. B.; Shingare,
M. S. Eur. J. Med. Chem. 2010, 45, 1128-1132.
184 Kaboudin, B.; Karimi, M. Bioorg. Med. Chem. Lett. 2006, 16, 5324-5327.
185 Kaboudin, B.; Karimi, M. ARKIVOC (Gainesville, FL, U. S.) 2007, 124-132.
186 Kolodiazhnyi, O. I. Russ. Chem. Rev. 2011, 80, 883-910.
187 Li, Y.-F.; Hammerschmidt, F. Tetrahedron: Asymmetry 1993, 4, 109-120.
188 Colton, I. J.; Yin, D.; Grochulski, P.; Kazlauskas, R. J. Adv. Synth. Catal. 2011, 353, 2529-
2544.
189 Zhang, Y.; Yuan, C.; Li, Z. Tetrahedron 2002, 58, 2973-2978.
190 Pàmies, O.; Bäckvall, J.-E. J. Org. Chem. 2003, 68, 4815-4818.
191 Takaki, K.; Itono, Y.; Nagafuji, A.; Naito, Y.; Shishido, T.; Takehira, K.; Makioka, Y.;
Taniguchi, Y.; Fujiwara, Y. J. Org. Chem. 2000, 65, 475-481.
192 Müller, P.; Müller-Dolezal, H.; Stoltz, R.; Söll, H. Houben-Weyl Methods of Organic
Chemistry Vol. XII/2, 4th Edition: Phosphorus Compounds II (Derivatives of Phosphorous
and Phosphoric Acid); Thieme: Germany, 1964.
193 Okamoto, Y.; Azuhata, T. Bull. Chem. Soc. Jpn. 1984, 57, 2693-2694.
194 Breuer, E. In The Chemistry of Organophosphorus Compounds; Hartley, F. R., Ed.; John
Wiley & Sons, Ltd: 1996, p 653-729.
195 Sekine, M.; Satoh, M.; Yamagata, H.; Hata, T. J. Org. Chem. 1980, 45, 4162-4167.
196 Sekine, M.; Kume, A.; Hata, T. Tetrahedron Lett. 1981, 22, 3617-3620.
197 Katzhendler, J.; Ringel, I.; Karaman, R.; Zaher, H.; Breuer, E. J. Chem. Soc., Perkin Trans.
2 1997, 341-350.
198 Afarinkia, K.; Echenique, J.; Nyburg, S. C. Tetrahedron Lett. 1997, 38, 1663-1666.
199 Katritzky, A. R.; Suzuki, K.; Wang, Z. Synlett 2005, 1656-1665.

76
200 Katritzky, A. R.; Rachwal, S.; Hitchings, G. J. Tetrahedron 1991, 47, 2683-2732.
201 McConnell, R. L.; Coover Jr, H. W. J. Am. Chem. Soc. 1956, 78, 4450-4452.
202 Sigman, M. S.; Jacobsen, E. N. J. Am. Chem. Soc. 1998, 120, 5315-5316.
203 Zhou, X.; Liu, X.; Yang, X.; Shang, D.; Xin, J.; Feng, X. Angew. Chem., Int. Ed. 2008, 47,
392-394.
204 Maeda, H.; Takahashi, K.; Ohmori, H. Tetrahedron 1998, 54, 12233-12242.
205 Baidya, M.; Brotzel, F.; Mayr, H. Org. Biomol. Chem. 2010, 8, 1929-1935.
206 Breugst, M.; Corral Bautista, F.; Mayr, H. Chem. – Eur. J. 2012, 18, 127-137.
207 Li, W.; Qin, S.; Su, Z.; Hu, C.; Feng, X. Comput. Theor. Chem. 2012, 989, 44-50.
208 Wang, C.; Xu, C.; Tan, X.; Peng, H.; He, H. Org. Biomol. Chem. 2012, 10, 1680-1685.
209 Satyanarayana, T.; Abraham, S.; Kagan, H. B. Angew. Chem., Int. Ed. 2009, 48, 456-494.
210 Katritzky, A. R.; Huang, T.-B.; Voronkov, M. V.; Steel, P. J. J. Org. Chem. 2000, 65, 8069-
8073.






















![storage.googleapis.com€¦ · [katheryne davis] [and heirs and assigns] [john mchale] [and heirs and assigns] [ricki reese] [and heirs and assigns] [nicole phelps] [and heirs and](https://img.pdfslide.net/doc/110x75/5f06dad27e708231d41a1204/katheryne-davis-and-heirs-and-assigns-john-mchale-and-heirs-and-assigns.jpg)






