Embed Size (px)
Citation preview

Energy landscape for proteins With statistical mechanics, one can try to
understand protein folding kinetics. An extension from theories for glasses and
polymers. Energy landscape for protein folding:
Must be rough, containing many local minimum. Must exist guiding forces that stabilize the native
structure. This is the “minimum frustration” principle.
Proposed and elaborated mainly by Peter G. Wolynes.


A folding trajectory

Associative memory Hamiltonian: a knowledge-based force field
Generalizing theory for spin glasses to treat heteropolymers.
Wolynes and other people have developed a method to extract “associative memory Hamiltonian” from a database of known protein structures.
Associative memory Hamiltonian automatically have the funnel-like and rugged energy landscape.

How well does Associative memory Hamiltonian do in predicting structures?

Copyright ©2000 by the National Academy of Sciences
Hardin, Corey et al. (2000) Proc. Natl. Acad. Sci. USA 97, 14235-14240
Fig. 4. Structural alignments of Qbest structures from simulated annealing of one training set protein (434 repressor) and the three test set proteins to their x-ray structures

Copyright ©2003 by the National Academy of Sciences
Hardin, Corey et al. (2003) Proc. Natl. Acad. Sci. USA 100, 1679-1684
Fig. 1. Superpositions of Qbest structures onto the native state

II. Knowledge-based methods More and more protein structures are solved
with either X-ray or NMR. Observation: Many of them are similar either
globally or partially. (evolution?) Can we use these structure as clues?
Derive empirical force field from the database. Homology modeling; Phylogenomic inference. Teach computers to deduce structural clues.

The use of homology (R. B. Altman http://www.smi.stanford.edu/projects/helix/bmi214/5-15-01-c2.pdf )
Homology Modeling (sequences with high homology to sequences of known structure) Given a sequence with homology > 25-30% with
known structure in PDB, use known structure as starting point to create a model of the 3D structure of the sequence.
Takes advantage of knowledge of a closely related protein. Use sequence alignment techniques to establish correspondences between known “template” and unknown.
http://www.luc.edu/faculty/kolsen/lecture2/ppframe.htm

http://www.luc.edu/faculty/kolsen/lecture2/ppframe.htm

http://www.luc.edu/faculty/kolsen/lecture2/ppframe.htm

http://www.luc.edu/faculty/kolsen/lecture2/ppframe.htm

http://www.luc.edu/faculty/kolsen/lecture2/ppframe.htm

A more careful analysis can be performed with phylogenomic analysis

Phylogenomic analysis http://phylogenomic.berkeley.edu

“Train” computers to deduce rules for protein structures For secondary structures: determining α
helixes,β sheets, or loops from sequence data Neural networks: (>72% accuracy)
• B Rost: “PHD: predicting one-dimensional protein structure by profile based neural networks.” Meth. in Enzymolgy, 266, 525-539, (1996)
• Available over the web. http://cubic.bioc.columbia.edu/predictprotein
Support vector machines• Developed recently in Taiwan.• Slightly out-performed neural networks.

As an example, pattern recognition is very hard to defined with simple “rules”.. The letter “A” can also be “A” “A” “A” or .
Neural networks or SVM algorithms do not assume any rules a priori. Instead they allow the system to be “trained”, i.e. the undefined parameters are to be determined by seeing examples.
Needs a (large) training set, and a separate testing set to see how well it does.
So part of the protein structure problems can be solved by this “pattern recognition” process.
Neural networks

Secondary structure
Secondary structure prediction can be done with more sophisticated algorithms. Artificial intelligence such as neuronal
networks or support vector machines. Basically look at a local sequence and
recongnize its pattern. Usually such methods need a training set.
I.e. knowledge-based methods.


PredictProtein does more than 2º structure prediction

Figure 9-6NMR structure of protein GB1.
Pag
e 28
0
Green: residues 23-33. Cyan: residues 42-53. Chm-alpha: a new sequence
replaces green part. Chm-beta: the same new
sequence replaced cyan part. Both are structurally similar to
native GB1. The same sequence can be
either an alpha helix or a beta sheet structure, depending on their context.

Threading Instead of searching over a global range of
structure, threading uses existing protein conformations.
Still needs an energy function. Improves the speed and accuracy if:
the constrained conformation set has the target native structure (unknown), and
within this set the energy function is good enough to distinguish the native structure from others.


Threading is often combined with other methods

Classification of protein folds Common structural motifs are often seen. They are classified
into hierarchical groups. SCOP
http://scop.mrc-lmb.cam.ac.uk/scop/ CATH
http://www.biochem.ucl.ac.uk/bsm/cath/index.html Can such knowledge helps us recognize “folds” in proteins?
(again I am quoting R. B. Altman) Fold recognition (sequences with no sequence identity (<= 30%) to
sequences of known structure. Given the sequence, and a set of folds observed in PDB, see if any of
the sequences could adopt one the known folds. Takes advantage of knowledge of existing structures, and principles by
which they are stabilized (favorable interactions).

Representatives of some popular folds(J. Mol. Graphics Modell. 19, 157, (2001))

Fold Recognition (Altman)
New sequence:MLDTNMKTQLKAYLEKLTKPVELIATLDDSAKSAEIKELL…
Library of known folds:

Fold Recognition (Altman) Library of protein structures, suitably processed
All structures Representative subset Structures with loops removed
Scoring function contact potential environmental evaluation function
Method for generating initial alignments and/or searching for better alignments.

Fold recognition server: 3D-pssmhttp://www.sbg.bio.ic.ac.uk/~3dpssm/

Additional useful links On-line course materials:
http://www.cryst.bbk.ac.uk/PPS2/course/index.html http://www.expasy.org/swissmod/course/course-index.
htm http://scpd.stanford.
edu/SOL/courses/proEd/RACMB/materials.htm PredictProtein Server:
http://www.embl-heidelberg.de/predictprotein/predictprotein.html
3D-pssm server: http://www.sbg.bio.ic.ac.uk/~3dpssm/

Folding Accessory proteins
1. Protein Disulfide Isomerases
2. Peptidyl prolyl cis-trans Isomerases
3. Chaperones


Electron micrographs - low resolution molecular structures

Diseases associated with protein structures

The story of -sheets twists
Why right-handed?

Voe
t Biochemistry
3e©
200
4 Jo
hn W
iley
& S
ons,
Inc.
Figure 8-49 Retinol binding protein.
Pag
e 25
1

Why right-handed twist?
• A question that is easier to answer for -helix. (L encounters steric hindrances).

Voe
t Biochemistry
3e©
200
4 Jo
hn W
iley
& S
ons,
Inc.
Figure 8-11 The right-handed helix.
Pag
e 22
4

References
• Chou and Scheraga PNAS (1982) 79, 7047-7051.
• Wang et. al, J. Mol. Biol. (1996) 262, 283-293.

Chou (1982)
QuickTime™ and aTIFF (LZW) decompressor
are needed to see this picture.

Voe
t Biochemistry
3e©
200
4 Jo
hn W
iley
& S
ons,
Inc.
Table 9-1 Propensities and Classifications of Amino Acid Residues for Helical and Sheet Conformations.
Pag
e 30
0

Conclusions from Chou 1982
• Intrastrand twisting (I.e. single-strand twisting) is an important factor.
• For a single peptide strand, twisted strand is more stable than a flattened strand. This is because a higher torsional energy exists
in flat structure. Also a more favorable non-bonding interaction
in the twisted structures.
• Right-handed twist is more stable than left-handed twist, due to a sum of many non-bonding interactions.

QuickTime™ and aTIFF (LZW) decompressor
are needed to see this picture.
QuickTime™ and aTIFF (LZW) decompressor
are needed to see this picture.
QuickTime™ and aTIFF (LZW) decompressor
are needed to see this picture.
QuickTime™ and aTIFF (LZW) decompressor
are needed to see this picture.
QuickTime™ and aTIFF (LZW) decompressor
are needed to see this picture.
QuickTime™ and aTIFF (LZW) decompressor
are needed to see this picture.

14 years later…
QuickTime™ and aTIFF (LZW) decompressor
are needed to see this picture.
QuickTime™ and aTIFF (LZW) decompressor
are needed to see this picture.

What was done?
• Molecular dynamics simulation, instead of previous conformational energy minimization.
• Studied poly-Ala, poly-Val and poly-Gly, each with 2 or 3 strands, and each strand has 3 or 5 residues.
• Examined effects of electrostatic vs. van der Waal interactions; effect of single vs. multiple strands; effects of solvents.

QuickTime™ and aTIFF (LZW) decompressor
are needed to see this picture.
QuickTime™ and aTIFF (LZW) decompressor
are needed to see this picture.
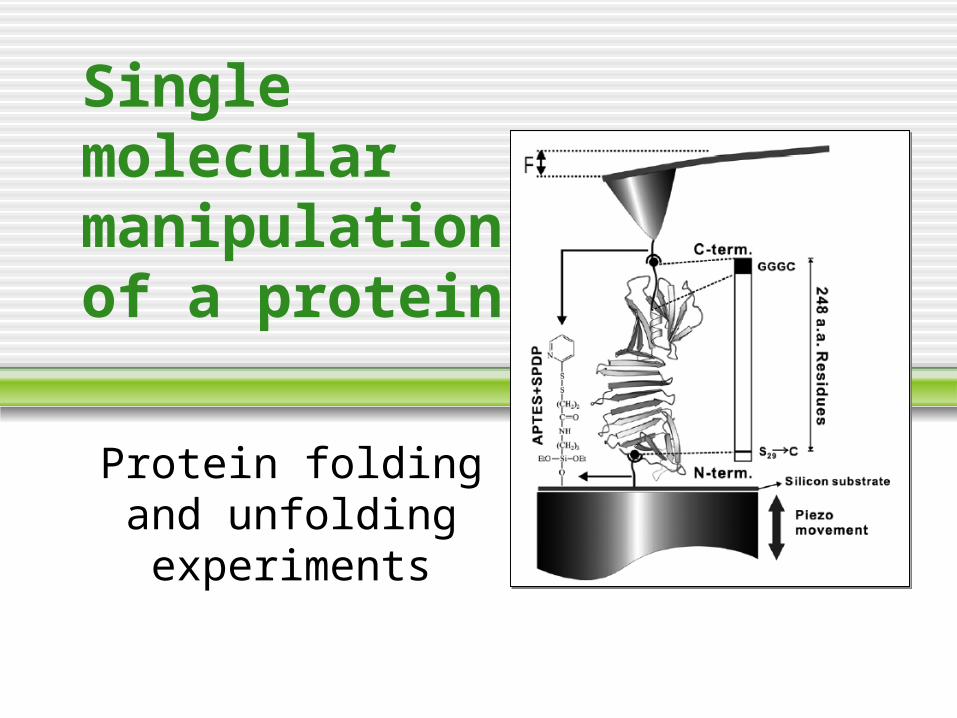
Single molecular manipulation of a protein
Protein folding and unfolding
experiments

Science 288, 143 (2000)


Bacteriorodopsin


Science 303, 1674 (2004)

Force Clamp?
• A. F. Oberhauser, P. K. Hansma, M. Carrion-Vazquez, and J. M. Fernandez, PNAS 468, 98 (2001)

Science 303, 1674 (2004)


120 pN 50 pN 120 pN
120 pN 35 pN 120 pN
120 pN 35 pN 120 pN
120 pN 23 pN 120 pN


In Conclusion
• The authors said: “Our results contradict the generally held view that folding and unfolding reactions correspond to transitions between well-defined discrete states. In contrast, we observed that ubiquitin folding occurs through a series of continuous stages that cannot be easily represented by state diagrams.”
• What do you think?


![Predicting Experimental Quantities in Protein Folding Kinetics ...ai.stanford.edu/~apaydin/recomb06.pdfplied to ligand-protein docking [17], protein folding [3,2], and RNA folding](https://img.pdfslide.net/doc/110x75/60d6bde9a1a7162f153e3cd1/predicting-experimental-quantities-in-protein-folding-kinetics-ai-apaydinrecomb06pdf.jpg)












![cnr'l - 323yfe3mr9mijtj8a3j9xqh6-wpengine.netdna-ssl.com · NE'\~ ZEP.Lp]D GEO RGE VI. ... MAGNIFYING GLASSES. D diare. Folding type in bakelite and alumi~u.:L. Amply strong enough](https://img.pdfslide.net/doc/110x75/5d1ee89088c9934c378c8400/cnrl-323yfe3mr9mijtj8a3j9xqh6-ne-zeplpd-geo-rge-vi-magnifying.jpg)













