Embed Size (px)
Citation preview

Toxicology, 45 (1987) 213--223 Elsevier Scientific Publishers Ireland Ltd.
ENHANCEMENT BY GLUTATHIONE DEPLETION OF ETHANOL-INDUCED ACUTE HEPATOTOXICITY IN IN VIVO
VITRO AND
O. STRUBELT, M. YOUNES and R. PENTZ
lnstitut fiir Toxikologie, Medizinische Universitb't zu Liibeck, D-2400 Liibeck (F.R.G.)
(Received November 3rd, 1986) (Accepted March 13th, 1987)
SUMMARY
Ethanol at initial concentrations between 0.75 and 6 g/1 produced a dose- dependent release of the enzymes glutamic-pyruvic-transaminase and sorbitol dehydrogenase (GPT, SDH) from the isolated perfused rat liver. At the concentration of 6 g/l, it also decreased the oxygen consumption and elevated the calcium content of the isolated livers. These toxic effects of ethanol were significantly enhanced in livers, the glutathione content of which had been depleted by pretreatment with phorone. Ethanol-induced toxicity in glutathione-depleted isolated livers could be prevented both by inhibition of alcohol dehydrogenase with 4-methylpyrazole and of xanthine oxidase with allopurinol. In rats, in vivo, 1.6 g/kg ethanol injected intra- venously produced a small increase in serum GPT and SDH concentrations 4 h after its administration. This increase in enzyme activities was several- fold higher and longer lasting in rats pretreated with phorone. Glutathione depletion per se did not induce hepatotoxici ty in vitro or in vivo.
Since glutathione is involved in several lines of defense against oxidative damage, our results of an enhanced susceptibility of glutathione-depleted livers to ethanol toxicity favour the hypothesis that ethanol exerts its hepatotoxic action via an activation of molecular oxygen.
K e y words: Alcohol, Ethyl; Liver; Glutathione; Hepatotoxicity; Glutamic- pyruvic-transaminase; Sorbitol dehydrogenase
INTR ODUC TION
The mechanism of ethanol-induced liver injury is a matter of continuous debate. Recently, we have shown that ethanol at an initial concentration of
Abbreviations: GPT, glutamic-pyruvic-transaminase; GSH, reduced glutathione; GSSG, oxidized glutathione; MDA, malondialdehyde; SDH, sorbitol dehydrogcnase.
o3oo-483x/87/$03.5o © 1987 Elsevier Scientific Publishers Ireland Ltd. Printed and Published in Ireland
213

2 g/1 produces an enhanced release of enzymes from the isolated rat liver which could be abolished by an inhibition of alcohol dehydrogenase as well as of acetaldehyde dehydrogenase [1,2] . Furthermore, superoxide dismu- tase, catalase or chelation of iron ions by desferrioxamine suppressed ethanol-induced enzyme release suggesting that this toxic action of ethanol is related to an oxygen activation which presumably takes place during the metabolism of acetaldehyde [1,2] . These results are in agreement with the findings of Boveris et al. [3] on an increased hepatic superoxide production of chronically ethanol-treated rats as well with those of Sinaceur et al. [4] on an enhancement in mitochondrial superoxide production during acute ethanol intoxication of rats.
Toxic effects of oxygen radicals, however, are only expected when their rate of formation exceeds the rates of their inactivation by cellular defense mechanisms. Among those, glutathione-dependent antioxidant systems seem to play a critical role: apart from its ability to interact directly with free radicals [5] , GSH is co-substrate of both the selenium-dependent [6] and the selenium-independent glutathione peroxidases [7] . While both enzymes reduce organic hydroperoxides already formed, the selenium-dependent enzyme inactivates, in addition, hydrogen peroxide. Furthermore, gluta- thione is involved in the regeneration of the active form of a-tocopherol, which is the major lipid-soluble physiological antioxidant [8] . Indeed, a lack in the availability of glutathione renders rat livers more susceptible to oxidative damage promoted by intracellular [9] or extracellular [10] prooxidants and should also enhance ethanol-induced hepatotoxici ty if this were due to oxygen activation.
M A T E R I A L A N D M E T H O D S
Animals Male ~bistar rats (conventional animals, 320--380 g; breeder: Winkelmann,
Borchen) were used throughout. They had free access to feed (Altromin® pellets) and tap water until use. To deplete hepatic glutathione (GSH), the animals were treated with phorone (diisopropylidene acetone, 250 mg/kg in 10 ml/kg olive oil i.p.) 2 h prior to the start of the respective experiment. This dose has been shown to decrease GSH concentrations in the liver by more than 90% and in kidney and heart by 60%, while those of lung and brain remain unaffected [11] . The main advantage of phorone over other glutathione-depleting agents is the fact that xenobiotic~metabolizing enzymes are not affected upon treatment with this agent [11] .
Experiments in vitro Removal of the liver and its connection to a recirculating perfusion
system was performed as described previously [12] . The perfusion medium consisted of 250 ml Krebs-Henseleit~buffer (pH 7.4) (per 1:118 mmol NaC1, 5 mmol KC1, 1.1 mmol MgSO4, 1.2 mmol KH:PO4, 25 mmol NaHCO3). Calcium chloride (1.25 mmol/1) was added to the prewarmed perfusion
214

medium (37°C) immediately before starting the perfusion. The perfusion medium was continuously gassed with carbogen (95% 02, 5% CO2), the partial pressure of O2 amounting to 600 mmHg. Sodium taurochalate (26.7 g/l) was infused into the perfusate at a rate of 12 ml/h to stimulate bile secretion. Oxygen consumption of the isolated liver was calculated from the difference in the oxygen concentrations of the influent and the effluent perfusate using a Micro pH/Blood Gas Analyzer 413 (Instrumenta- tion Laboratory) . For further details see [ 12] .
Experiments in vivo We exposed a lateral taft vein in each of the rats under light ether anes-
thesia and injected 5 ml/kg 40% ethanol (i.e. 1.6 g/kg ethanol) slowly during 4--5 rain into this vein. Before this t reatment as well as 30 min and 3, 4 and 25 h after it, b lood samples were drawn by cutt ing the tip of the tail for determination of b lood ethanol and serum concentrations of GPT and SDH.
Biochemical determinations The activities of GPT and SDH were assayed using commercial kits of
Boehringer, Mannheim. Ca 2+ concentrat ions in the liver were measured colorimetrically following acid extraction also with reagent kits of Boehringer, Mannheim, whereas malondialdehyde was assayed as thio- barbituric acid-reactive material as described previously [ 13].
For glutathione determination, a piece of liver tissue was homogenized in 3% meta-phosphoric acid. After neutralization with 10 M KOH, total gluta- thione was measured according to Brehe and Butch [14] . In this specific procedure tissue extracts are added to a reagent cocktail containing GSSG- reductase, NADPH, bovine serum albumin, EDTA and Ellman's reagent at pH 7.2. The rate of conjugate formation is followed spectrophotometrical ly against a reagent blank. Oxidized glutathione (GSSG) was estimated by the same procedure after blocking reduced glutathione (GSH) with 2-vinylpyri- dine [15] . The concentrations of ethanol and acetaldehyde in the perfusate were determined by means of gas chromatography as described previously [2]. Ethanol concentrations were corrected for evaporation as determined experimentally.
Su bstances Phorone (diisopropylidene acetone) was obtained from Aldrich, Stein-
heim. Thiobarbituric acid, Ellman's reagent, 2-vinylpyridine, and taurocho- late were purchased from Sigma, Munich and glutathione reductase was a product of Boehringer, Mannheim, while all other reagents were from Merck, Darmstadt. All substances employed were of analytical grade purity.
Statistics Means + S.E.M. were calculated in the usual way. The difference between
2 means was checked with Student 's t-test or, in the case of inhomogenous variances, by the rank sum test of Wilcoxon-Mann-Whitney (U-test) taking P = 0.05 as the limit of significance.
215
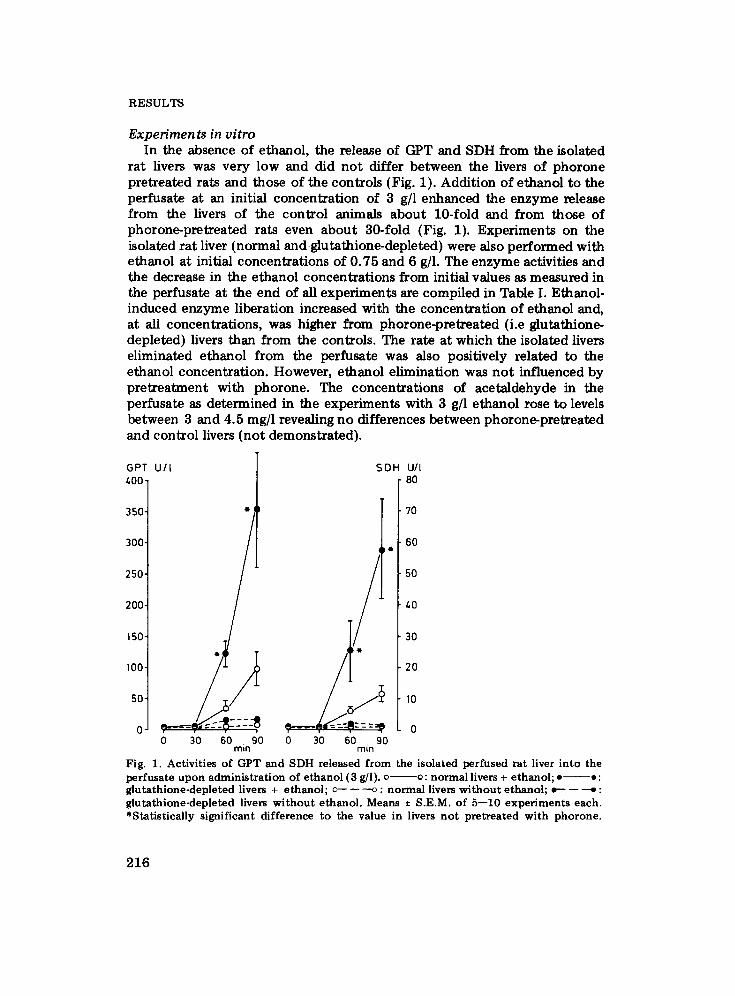
R E S U L T S
Experiments in vitro In the absence of ethanol, the release of GPT and SDH from the isolated
rat livers was very low and did not differ between the livers of phorone pretreated rats and those of the controls (Fig. 1). Addition of ethanol to the perfusate at an initial concentration of 3 g/1 enhanced the enzyme release from the livers of the control animals about 10-fold and from those of phorone-pretreated rats even abou t 30-fold (Fig. 1). Experiments on the isolated rat liver (normal and glutathione-depleted) were also performed with ethanol at initial concentrations of 0.75 and 6 g]l. The enzyme activities and the decrease in the ethanol concentrations from initial values as measured in the perfusate at the end of all experiments are compiled in Table I. Ethanol- induced enzyme liberation increased with the concentration of ethanol and, at all concentrations, was higher from phorone-pretreated (i.e glutathione- depleted) livers than from the controls. The rate at which the isolated livers eliminated ethanol from the perfusate was also positively related to the ethanol concentration. However, ethanol elimination was no t influenced by pretreatment with phorone. The concentrations of acetaldehyde in the perfusate as determined in the experiments with 3 g/1 ethanol rose to levels between 3 and 4.5 mg/1 revealing no differences between phorone-pretreated and control livers (not demonstrated).
GPT U/L Z.00'
350'
300'
250'
200'
150"
100"
50"
0
1
0 30 60 90 rain
g o
? - _ _ _ , ------ " - - - -
0 30 60 90 min
SDH U/I 80
70
6O
5O
/.0
30
20
10
0
Fig. 1. A c t i v i t i e s o f G P T a n d S D H r e l e a s e d f r o m t h e i s o l a t e d p e r f u s e d r a t l iver i n t o t h e p e r f u s a t e u p o n a d m i n i s t r a t i o n o f e t h a n o l (3 g/l) . c o: n o r m a l l ivers + e t h a n o l ; • • : g l u t a t h i o n e - d e p l e t e d l ivers + e t h a n o l ; o--- - - ---o : n o r m a l l ivers w i t h o u t e t h a n o l ; o-- - - ~ : g l u t a t h i o n e - d e p l e t e d l ivers w i t h o u t e t h a n o l . M e a n s -+ S .E .M. o f 5 - - 1 0 e x p e r i m e n t s e a c h . * S t a t i s t i c a l l y s i g n i f i c a n t d i f f e r e n c e t o t h e va lue in l ivers n o t p r e t r e a t e d w i t h p h o r o n e .
216
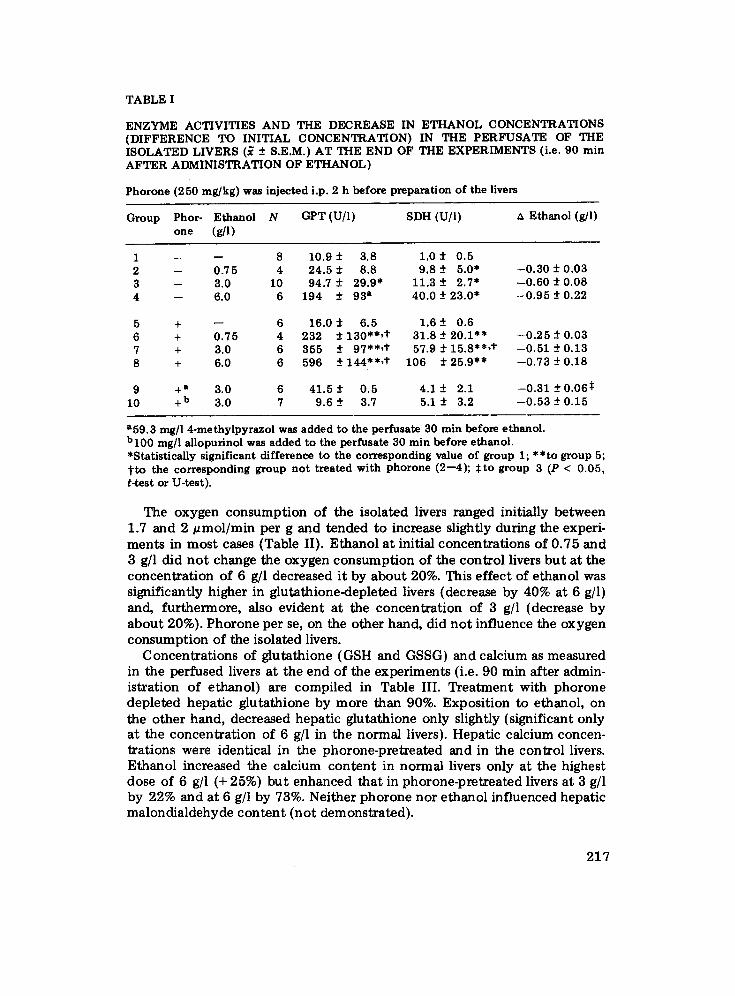
TABLE I
ENZYME ACTIVITIES AND THE DECREASE IN ETHANOL CONCENTRATIONS (DIFFERENCE TO INITIAL CONCENTRATION) IN THE PERFUSATE OF THE ISOLATED LIVERS (~ -+ S.E.M.) AT THE END OF THE EXPERIMENTS (i.e. 90 min AFTER ADMINISTRATION OF ETHANOL)
P h o r o n e ( 2 5 0 m g / k g ) was injected i .p. 2 h before preparation of the livers
Group Phor- Ethanol N GPT (U/l) SDH (U/l) ~ Ethanol (g/l) o n e (g/l)
1 -- - - 8 10.9 -+ 3.8 1.0 -+ 0.5 2 -- 0.75 4 24.5 _+ 8.8 9.8 -+ 5.0* --0.30 -+ 0.03 3 -- 3.0 10 94.7 _+ 29.9* 11.3 -+ 2.7* --0.60 -+ 0.08 4 -- 6.0 6 194 -+ 93 a 40.0 -+ 23.0* --0.95 -+ 0.22
5 + -- 6 16.0 -+ 6.5 1.6 -+ 0.6 6 + 0.75 4 232 -+ 130"*,t 31.8 -+ 20.1"* --0.25 -+ 0.03 7 + 3.0 6 355 -+ 97"*,t 57.9 -+ 15.8"*,t --0.51 -+ 0.13 8 + 6.0 6 596 _+ 144"*,t" 106 -+ 25.9** --0.73 -+ 0.18
9 +a 3.0 6 41.5 -+ 0.5 4.1 -+ 2.1 --0.31 -+ 0.065 10 +b 3.0 7 9.6 -+ 3.7 5.1 -+ 3.2 --0.53 -+ 0.15
a59.3 rag/1 4-methylpyrazol was added to the perfusate 30 min before ethanol. b l00 mg/l allopurinol was added to the perfusate 30 min before ethanol. *Statistically significant difference to the corresponding value of group 1; **to group 5; t t o the corresponding group n o t treated with phorone (2--4); $to group 3 (P < 0.05, t-test or U-test).
The o x y g e n c o n s u m p t i o n of the i so la ted livers ranged init ial ly b e t w e e n 1.7 and 2 ~ m o l / m i n pe r g and t e n d e d to increase slightly dur ing the exper i - m e n t s in m o s t cases (Table II) . E t h a n o l a t init ial c o n c e n t r a t i o n s o f 0 .75 and 3 g/1 did n o t change the o x y g e n c o n s u m p t i o n of the con t ro l livers b u t a t the c o n c e n t r a t i o n of 6 g/1 decreased i t by a b o u t 20%. This e f f ec t o f e t hano l was s ignif icant ly h igher in g lu t a th ione -dep le t ed livers (decrease by 40% a t 6 g/l) and, f u r t h e r m o r e , also ev iden t a t the c o n c e n t r a t i o n of 3 g/1 (decrease b y a b o u t 20%). P h o r o n e pe r se, on the o the r hand, did n o t in f luence the o x y g e n c o n s u m p t i o n o f the i so la ted livers.
C o n c e n t r a t i o n s of g lu t a th ione (GSH and GSSG) and ca lc ium as measu red in the pe r fused livers a t the end o f the e x p e r i m e n t s (i.e. 90 min a f te r admin- i s t ra t ion of e thano l ) are c o m p i l e d in Tab le I I I . T r e a t m e n t wi th p h o r o n e d e p l e t e d hepa t i c g lu t a th ione b y m o r e than 90%. Expos i t i on to e thanol , on the o t h e r hand , dec reased hepa t i c g lu t a th ione on ly slightly (s ignif icant on ly a t the c o n c e n t r a t i o n of 6 g/1 in the n o r m a l livers). Hepa t i c ca lc ium concen- t r a t ions were ident ica l in the p h o r o n e - p r e t r e a t e d and in the con t ro l livers. E thano l increased the ca lc ium c o n t e n t in n o r m a l livers on ly a t the highest dose of 6 g/1 (+ 25%) b u t e n h a n c e d t h a t in p h o r o n e - p r e t r e a t e d livers a t 3 g/1 by 22% and at 6 g/1 by 73%. Ne i the r p h o r o n e n o r e thano l in f luenced hepa t i c m a l o n d i a l d e h y d e c o n t e n t ( n o t d e m o n s t r a t e d ) .
217
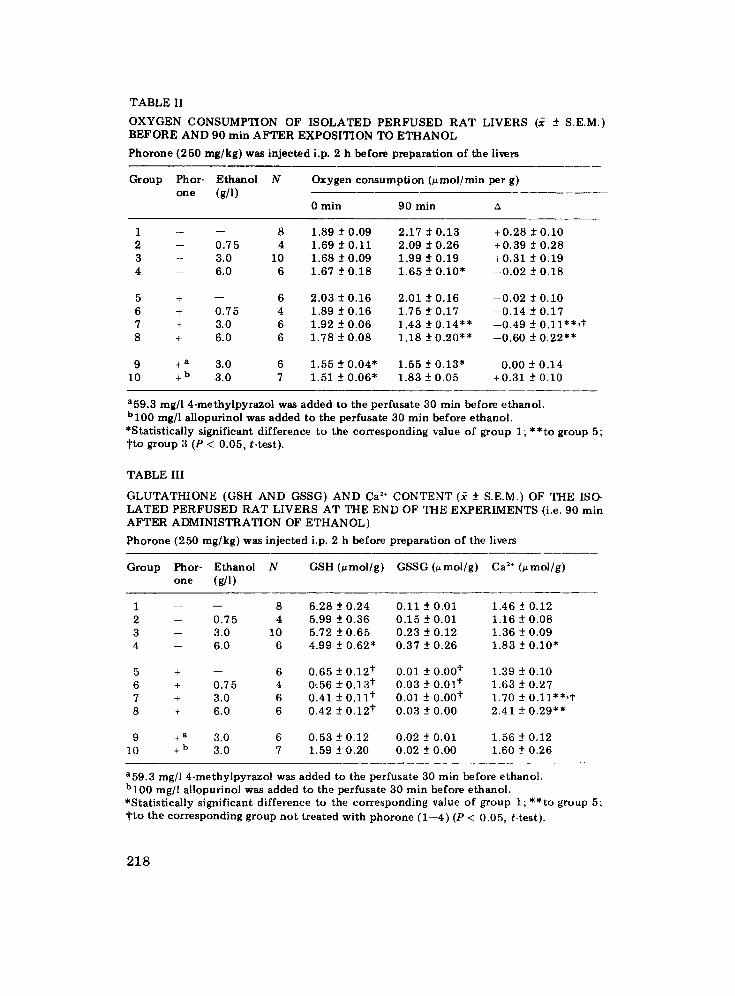
T A B L E II
O X Y G E N C O N S U M P T I O N O F I S O L A T E D P E R F U S E D R A T L I V E R S (~ ± S .E .M. ) B E F O R E A N D 90 m i n A F T E R E X P O S I T I O N T O E T H A N O L
P h o r o n e ( 2 5 0 m g / k g ) w a s i n j e c t e d i.p. 2 h b e f o r e p r e p a r a t i o n o f t h e l ivers
G r o u p P h o r - E t h a n o l N one (g/l)
Oxygen consumption (~mol/min per g)
0 m i n 9 0 m i n
1 -- - - 8 1 . 8 9 _ + 0 . 0 9 2 . 1 7 ± 0 . 1 3 + 0 . 2 8 _ + 0 . 1 0 2 -- 0 . 7 5 4 1 .69 ± 0 . 1 1 2 .09 _+ 0 . 2 6 + 0 . 3 9 ± 0 . 2 8 3 -- 3 .0 10 1 .68 ± 0 . 0 9 1 .99 ± 0 . 1 9 + 0 . 3 1 ± 0 . 1 9 4 - - 6 .0 6 1 .67 _+ 0 . 1 8 1 . 6 5 -+ 0 . 1 0 " - - 0 . 0 2 -+ 0 . 1 8
5 + - - 6 2 . 0 3 ± 0 . 1 6 2 .01 ± 0 . 1 6 - - 0 . 0 2 ± 0 . 1 0 6 + 0 . 7 5 4 1 .89 ± 0 . 1 6 1 .75 _+ 0 . 1 7 - - 0 . 1 4 -+ 0 . 1 7 7 + 3 .0 6 1 .92 -+ 0 . 0 6 1 .43 ± 0 . 1 4 " * - - 0 . 4 9 ± 0 . 1 1 " * , t 8 + 6 .0 6 1 . 7 8 ± 0 . 0 8 1 . 1 8 ± 0 . 2 0 * * - - 0 . 6 0 ± 0 . 2 2 * *
9 + a 3 .0 6 1 . 5 5 ± 0 . 0 4 " 1 . 5 5 _ + 0 . 1 3 " 0 . 0 0 - + 0 . 1 4 10 + b 3 .0 7 1 .51 - + 0 . 0 6 * 1 . 8 3 ± 0 . 0 5 + 0 . 3 1 -+0 .10
a59.3 mg/l 4-methylpyrazol was added to the perfusate 30 min before ethanol. bl00 mg/l allopurinol was added to the perfusate 30 rnin before ethanol. *Statistically significant difference to the corresponding value of group l;**to group 5; tto group 3 (P < 0.05, t-test).
T A B L E III
G L U T A T H I O N E ( G S H A N D G S S G ) A N D Ca 2÷ C O N T E N T (~ -+ S .E .M. ) O F T H E IS(> L A T E D P E R F U S E D R A T L I V E R S A T T H E E N D O F T H E E X P E R I M E N T S (i.e. 90 r a in A F T E R A D M I N I S T R A T I O N O F E T H A N O L )
P h o r o n e ( 2 5 0 m g / k g ) w a s i n j e c t e d i.p. 2 h b e f o r e p r e p a r a t i o n o f t he l ivers
Group Phor- Ethanol N GSH (umol/g) GSSG (umol/g) Ca 2÷ (umol/g) o n e (g/ l )
1 -- - - 8 6 . 2 8 -+ 0 . 2 4 0 . 1 1 -+ 0 .01 1 .46 ± 0 .12 2 -- 0 . 7 5 4 5 .99 -+ 0 . 3 6 0 . 1 5 -+ 0 .01 1 .16 ± 0 . 0 8 3 -- 3 .0 10 5 .72 ± 0 . 6 5 0 . 2 3 ± 0 . 1 2 1 .36 -+ 0 . 0 9 4 -- 6 .0 6 4 . 99 ± 0 . 6 2 * 0 . 3 7 ± 0 . 2 6 1 .83 -+ 0 . 1 0 "
5 + - - 6 0 . 6 5 -+ 0 . 1 2 ¢ 0 .01 ± 0 . 0 0 t 1 .39 _+ 0 . I 0 6 + 0 . 7 5 4 ff .56 -+ 0 . 1 3 ¢ 0 . 0 3 -+ 0 .01 t 1 . 6 3 _+ 0 . 2 7 7 + 3 .0 6 0 .41 ± 0 . 1 1 t 0 . 01 ± 0 . 0 0 t 1 .70 -+ O . 1 1 * * , t 8 + 6 .0 6 0 . 4 2 -+ 0 . 1 2 t 0 . 0 3 -+ 0 . 0 0 2 .41 -+ 0 . 2 9 * *
9 + a 3 .0 6 0 . 5 3 _ + 0 . 1 2 0 . 0 2 - + 0 . 0 1 1 . 5 6 ± 0 . 1 2 10 + b 3 .0 7 1 .59 -+ 0 . 2 0 0 . 0 2 -+ 0 . 0 0 1 .60 -+ 0 . 2 6
a59.3 mg/l 4-methylpyrazol was added to the perfusate 30 rain before ethanol. bl00 mg/l allopurinol was added to the perfusate 30 min before ethanol. *Statistically significant difference to the corresponding value of group 1; **to group 5; tto the corresponding group not treated with phorone (1--4) (P < 0.05, t-test).
218

Inhibition of alcohol dehydrogenase by adding 4-methylpyrazole to the perfusate 30 min prior to ethanol clearly suppressed ethanol-induced enzyme release from phorone-pretreated livers (Table I). In parallel, ethanol meta- bolism was inhibited as evidenced by a 50% slower rate of ethanol elimina- tion from the perfusate (Table I). Ethanol-induced enzyme release from phorone-pretreated livers was also inhibited by administration of the xanthine oxidase inhibitor allopurinol (Table I). Allopurinol also prevented the decrease in oxygen consumption induced by ethanol in these livers (Table II) bu t did no t influence ethanol elimination (Table I).
Experiments in vivo Rats were treated with phorone (250 mg/kg i.p.) and 2 h later with
ethanol (1.6 kg/kg i.v.), controls receiving olive oil instead of phorone and saline instead of ethanol. Plasma activities of GPT and SDH as measured before and after t reatment with ethanol are compiled in Fig. 2. In saline- treated animals, the enzyme concentrations remained nearly constant during the investigation period, and no differences were evident between phorone- pretreated rats and control animals. Ethanol produced a small bu t statisti- cally significant increase in the GPT and SDH concentrations of the control animals 4 h after its administration. However, a several-fold higher increase of serum enzyme activities occurred at this time upon ethanol administration in the rats pretreated with phorone, and this increase was also evident 2 h (SDH), 3 h (both enzymes) and 25 h (GPT) after the t reatment (Fig. 2). Blood ethanol concentrations, on the other hand, did no t differ between phorone-pretreated and control rats (Fig. 2).
DISCUSSION
Hepatic enzyme release is commonly regarded as a toxic response due to an altered membrane permeability of the hepatic cell and is widely used in the detection and evaluation of liver injury in laboratory animals and in man [16] . In this respect, GPT and SDH are considered as the most sensitive and specific indicators [17--20] . An increased leakage of enzymes from the liver has also been observed upon administration of ethanol in vitro [1,2,21] and in vivo [22--25] , and was confirmed by this investigation, too. Further indices of ethanol hepatotoxic i ty became evident at the highest concentrat ion of 6 g/l, namely a decrease in the oxygen consumption and an increase in the calcium content of the isolated livers. The increase in intra- cellular calcium is reported here as a preliminary and auxiliary observation. A detailed explanation of the phenomenon was no t attempted.
The toxic effects increased markedly with livers, the glutathione content of which had been depleted by t reatment with phorone. Since glutathione depletion per se did no t at all induce hepatotoxici ty, a potent iat ion of the hepatotoxic effects of ethanol by glutathione depletion must be postulated. This potentat ion was not only evident in the isolated rat liver in vitro but also in the whole animal in vivo.
219
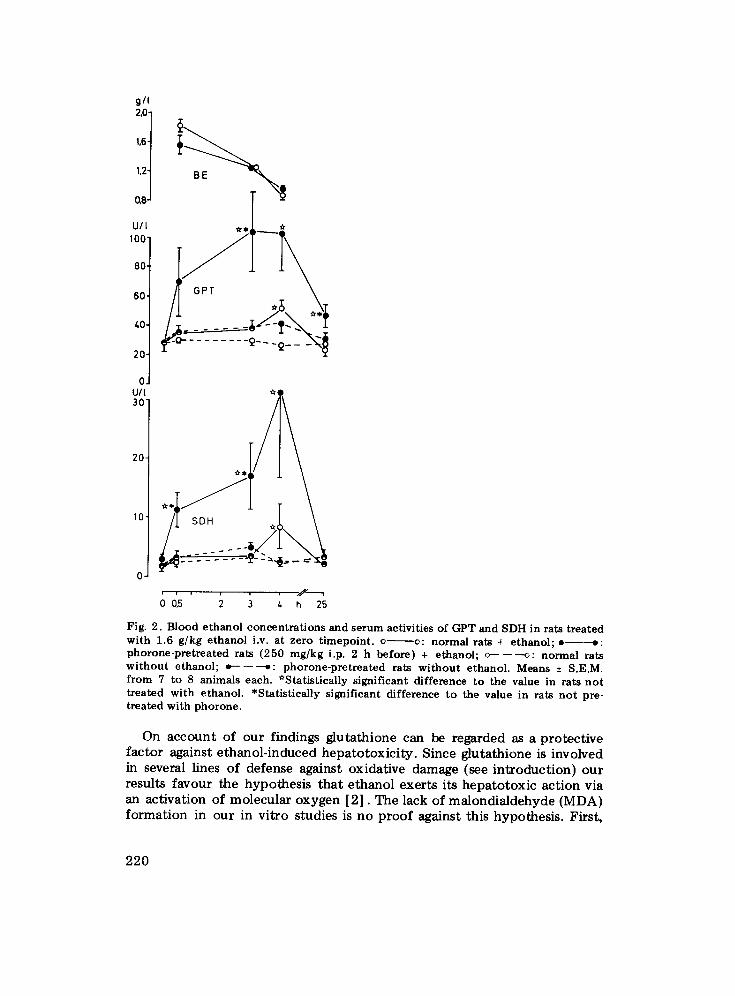
g/I 2,0j t.6
1,2
0,8
U/ 100" 80-
60-
40- 20- O. U/I 30"
2 0 -
10
.y-----.
Fig. 2. Blood e t h a n o l c o n c e n t r a t i o n s and se rum act ivi t ies of GPT and SDH in rats t r ea t ed w i t h 1.6 g /kg e t h a n o l i.v. a t ze ro t i m e p o i n t , o v: n o r m a l ra ts + e t h a n o l ; e - - - - - o : p h o r o n e - p r e t r e a t e d rats (250 m g / k g i.p. 2 h be fo re ) + e thano l ; c - - - - - - ~ : n o r m a l rats w i t h o u t e thano l ; t~-------~: p h o r o n e - p r e t r e a t e d rats w i t h o u t e thano l . Means + S.E.M. f rom 7 to 8 an imals each. *Sta t i s t ica l ly s igni f icant d i f ference to the value in ra ts n o t t r ea ted w i th e thano l . *Sta t i s t ica l ly s igni f icant d i f ference to the value in rats n o t pre- t r ea ted w i th p h o r o n e .
On account of our findings glutathione can be regarded as a protective factor against ethanol-induced hepatotoxicity. Since glutathione is involved in several lines of defense against oxidative damage (see introduction) our results favour the hypothesis that ethanol exerts its hepatotoxic action via an activation of molecular oxygen [2]. The lack of malondialdehyde (MDA) formation in our in vitro studies is no proof against this hypothesis. First,
220

MDA is a poor indicator of lipid peroxidation in intact organs, as it is rapidly metabolized [26,27] and as it readily reacts with tissue constituents [28]. Second, Miiller and Sies recently observed an increased formation of ethane, a far more sensitive indicator of lipid peroxidation, in isolated perfused rat livers upon application of ethanol [29].
On the other hand, acetaldehyde, the oxidation product of ethanol, was shown to bind covalently to tissue macromolecules, a reaction which was also suggested to mediate ethanol hepatotoxicity [30]. Glutathione inhibits this reaction [30] ; its absence, thus, would also potentiate ethanol hepato- toxicity if it were mediated by covalent binding. Ethanol has to be meta- boli~zed for both mechanisms to occur. This fact is substantiated by the prevention of ethanol toxicity to glutathione<lepleted isolated perfused livers, in our study, upon inhibition of alcohol dehydrogenase by 4-methyl- pyrazole. The inhibitory action of allopurinol, however, favors the oxygen activation hypothesis. Namely, xanthine oxidase was shown to act aerobi- cally upon acetaldehyde thereby causing the peroxidation of linolenate via formation of reactive oxygen species [31]. Consequently, the prevention of the toxic action of ethanol towards the liver by allopurinol most probably reflects a decreased oxygen activation and supports our hypothesis on the production of reactive oxygen species as a mechanism of acute ethanol hepatotoxicity [2].
ACKN OWLE DGEMENTS
The skillful technical assistance of Astrid RSbke and Cornelia Magnnssen is greatly appreciated. Thanks go also to Eva-Maria Stahl for excellent preparation of the manuscript.
REFERENCES
1 0 . Strubelt, M. Younes and R. Pentz, Ethanol-induced enzyme release from the isolated rat liver and its mechanism. Arch. Pharmacol. Suppl., 334 (1986) 151.
2 M. Younes and O. Strubelt, Alcohol-induced hepatotoxicity: a role for oxygen free Radicals. Free Rad. Res. Commun., 3 (1987) 19.
3 A. Boveris, C.G. Fraga, A.I. Varsavsky and O.R. Koch, Increased chemiluminescence and superoxide production in the liver of chronically ethanol-treated rats. Arch. Biochem. Biophys., 227 (1983) 534.
4 J. Sinaceur, C. Ribi~re, D. Sabourault and R. Nordmann, Superoxide formation in liver mitochondria during ethanol intoxication: possible role in alcohol hepatotoxic- ity, in G. Poll, K.H. Cheeseman, M.U. Dianzani and T.F. Slater (Eds.), Free Radicals in Liver Injury, IRL Press Limited, Oxford, 1985, p. 175.
5 A.W. Pryor, The role of free radical reactions in biological systems, in W.A. Pryor (Ed.), Free Radicals in Biology, Vol. 1, Academic Press, New York, 1976, p. 1.
6 L. Floh~, Glutathione peroxidase brought into focus, in W.A. Pryor (Ed.), Free Radicals in Biology, Vol. 5, Academic Press, New York, 1982, p. 223.
7 R.F. Burk, K. Noshiki, R.A. Lawrence and B. Chance, Peroxide removal by selenium- dependent and selenium-independent glutathione peroxidases in hemoglobin-free perfused rat liver. J. Biol. Chem., 253 (1978) 43.
8 C.C. Reddy, R.W. Scholz, C.E. Thomas and E.J. Massaro, Vitamin E dependent
221

reduced glutathione inhibition of rat liver microsomal lipid peroxidation. Life Sci., 31 (1982) 571.
9 M. Younes and C.-P. Siegers, Mechanistic aspects of enhanced lipid peroxidation following glutathione depletion in vivo. Chem.-Biol. Interact., 34 (1981) 257.
10 C.-P. Siegers, W. Horn and M. Younes, Effect of phorone-induced glutathione deple- tion on the metabolism and hepatotoxicity of carbon tetrachloride and vinylidene chloride in rats. J. Appl. Toxicol., 5 (1985) 352.
11 M. Younes, S.C. Sharma and C.-P. Siegers, Glutathione depletion by phorone. Organ specificity and effect on hepatic microsomal mixed-function oxidase system. Drug Chem. Toxicol., 9 (1986) 67.
12 O. Strubelt, M. Younes and R. Pentz, Influence of extracellular calcium on allyl alcohol-induced hepatotoxicity. Acta Pharmacol. Toxicoi., 59 (1986) 47.
13 L.K. Dahle, E.G. Hill and R.T. Holman, The thiobarbituric acid reaction and the antoxidation of polyunsaturated fatty acid methyl esters. Arch. Biochem. Biophys., 98 (1962) 253.
14 J.E. Brehe and H.B. Burch, Enzymatic assay for glutathione. Anal. Biochem., 74 (1976) 189.
15 O.W. Griffith, Determination of glutathione and glutathione disulfide using gluta- thione reductase and 2-vinylpyridine. Anal. Biochem., 106 (1980) 207.
16 G.L. Plaa and W.R. Hewitt, Detection and evaluation of chemically induced liver injury, in A.W. Hayes (ed.), Principles and Methods of Toxicology, Raven Press, New York, 1982, p. 407.
17 O. Strubelt, H. Breining and H. Prael, Der diagnostische Aussagewert yon Serumen- zymen bei toxikologischen Untersuchungen an Ratten. Arzneimittel-Forsch., 23 (1973) 77.
18 S.J. Curtis, M. Moritz and P.J. Snodgrass, Serum enzymes derived from liver cell fractions. I. The response to carbon tetrachloride intoxication in rats. Gastroentero- logy, 62 (1972) 84.
19 G.O. Korsrud, H.C. Grice and J.M. McLaughlan, Sensitivity of several serum enzymes in detecting carbon tetrachloride-induced liver damage in rats. Toxicol. Appl. Pharma- col., 22 (1972) 474.
20 G.O. Korsrud, H.G. Grice, T.K. Goodman, J.E. Knipfel and J.M. McLaughlan, Sensi- tivity of several serum enzymes for the detection of thioacetamide-, dimethylnitro- samine- and diethanolamine-induced liver damage in rats. Toxicol. Appl. Pharmacol., 26 (1973) 299.
21 H. Bassan, J. Kendler and H.J. Zimmerman, Effect of ethionine and ethanol on the function of the perfused rat liver (35680). Proc. Soc. Exp. Biol. Med., 137 (1971) 852.
22 O. Strubelt, H. Breining and H. Wiist, Die Empfindlichkeit einiger Nachweismethoden ffir Lebersch~digungen (J131-Bengalrotprobe, Serumtransaminasen, histologischer Befund) bei experimenteller Vergiftung mit Tetrachlorkohlenstoff, Allyl- und Athyl- alkohol. Arch. Toxicol., 22 (1967) 236.
23 J. Brohult, L.A. Carlson and H. Reichard, Serum enzyme activities, cholesterin and triglycerides in serum after intake of alcohol. Scand. J. Clin. Lab. Invest. Suppl., 92 (1966).
24 E. Rubin and C.S. Lieber, Alcohol-induced hepatic injury in non alcoholic volunteers. New Engl. J. Med., 278 (1968) 869.
25 J. Takeuchi, A. Takada, R. Kanayama, N. Ohara and Y. Okumura, Effect of alcohol on the liver of rats. II. Factors contributing to elevations of plasma transaminase activities and hepatic cell necrosis following a single administration of alcohol in rats. Lab. Invest., 21 (1969) 398.
26 A.A. Horton and L. Packer, Mitochondrial metabolism of aldehydes. Biochem. J., 116 (1970) 19P.
27 J.J. Hjelle and D.R. Petersen, Metabolism of malondialdehyde by rat liver aldehyde dehydrogenase. Toxicol. Appl. Pharmacol., 70 (1983) 57.
222

28 K.S. Chio and A.L. Tappel, Inactivation of ribonuclease and other enzymes by peroxi- dizing lipids and by malondialdehyde. Biochemistry, 8 (1969) 2827.
29 A. Miiller and H. Sies, Role of alcohol dehydrogenase activity and of acetaldehyde in ethanol-induced ethane and pentane production by isolated perfused rat liver. Biochem. J., 206 (1982) 153.
30 D.J. Tuma and M.F. Sorrell, Covalent binding of acetaldehyde to hepatic proteins: role in alcoholic liver injury. Prog. Clin. Biol. Res., 183 (1984) 3.
31 E.W. Kellogg and I. Fridovich, Superoxide, hydrogen peroxide, and singiet oxygen in lipid peroxidation by a xanthine oxidase system. J. Biol. Chem., 250 (1975) 8812.
2 2 3





























